|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea000014898b*\id |
|
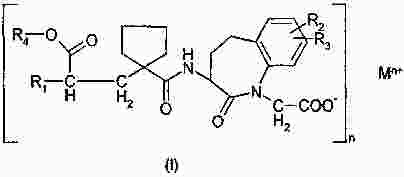
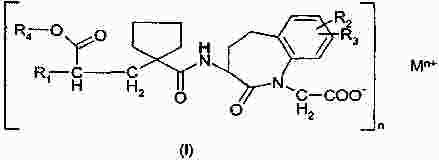
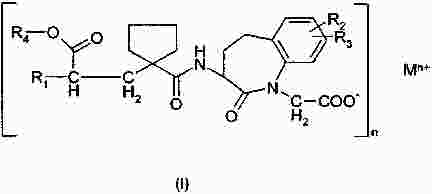
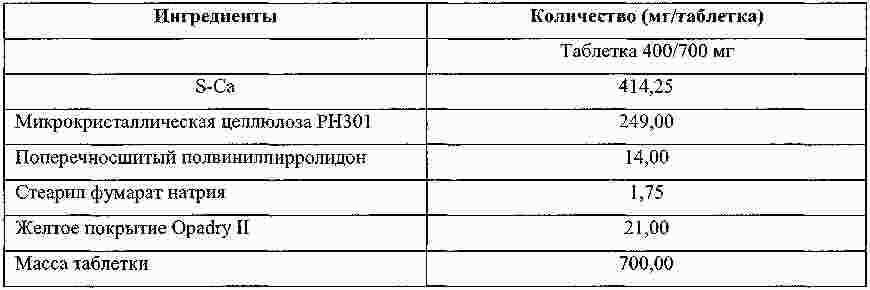
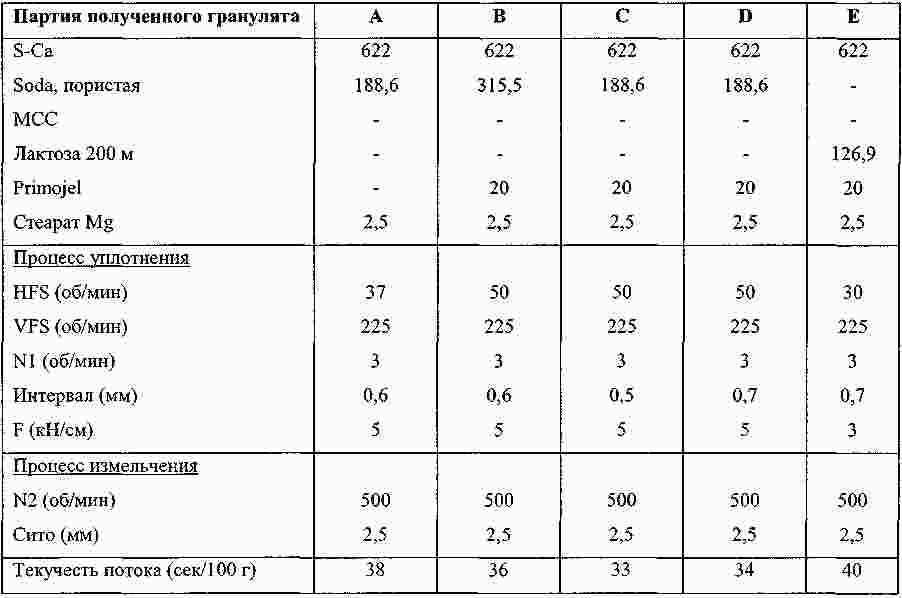
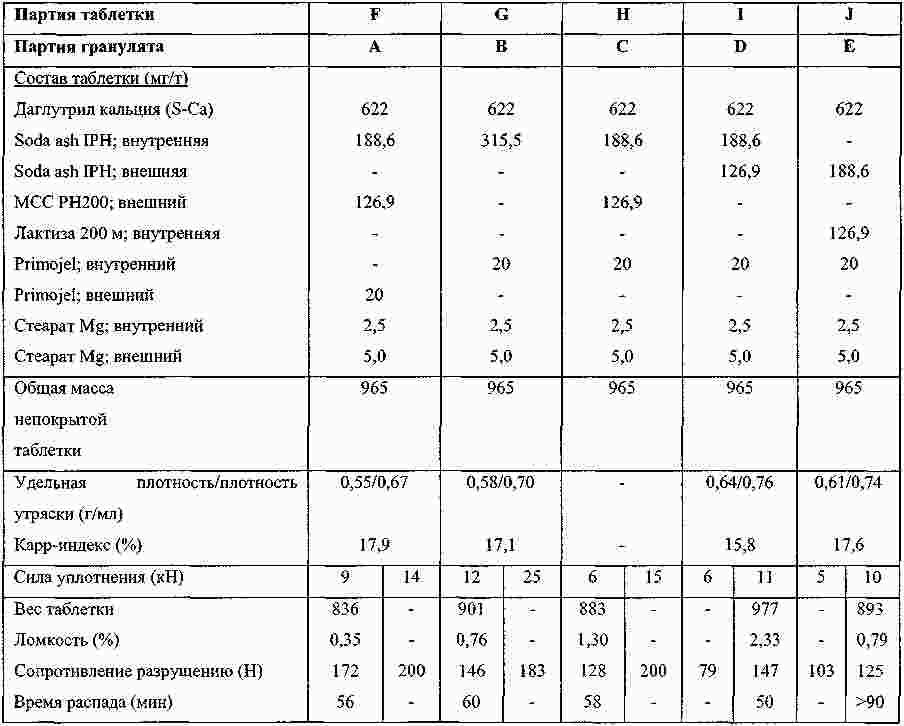
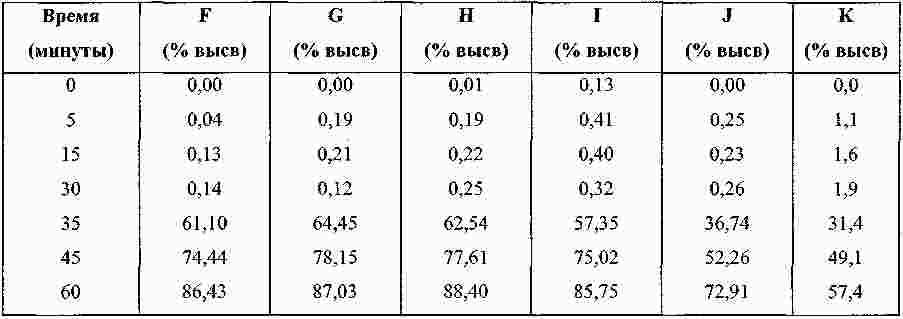
Термины запроса в документе Реферат Изобретение относится к пероральной композиции производных бензазепин-1-ацетиловой кислоты, включающей: а) указанное активное соединение в количестве от 10 до 80% от общего веса лекарственной формы; б) по меньшей мере 10 вес.% щелочного соединения или смеси щелочных соединений; в) необязательно включает вспомогательные материалы в количестве от 1 до 45% от общего веса лекарственной формы. Кроме того, изобретение имеет отношение к определенной выше пероральной композиции, включающей карбонат натрия со специфическим размером частиц и/или площадью поверхности в качестве щелочного соединения. Формула [0001] Пероральная фармацевтическая лекарственная форма из активного соединения общей формулы [0002] Лекарственная форма по п.1, в которой щелочное соединение выбрано из группы, состоящей из неорганических и органических щелочных соединений, таких как бикарбонат натрия, бикарбонат калия, карбонат натрия, карбонат калия, цитрат натрия, трис-буфер, триэтаноламин, щелочные гидроксиды, такие как гидроксид натрия, гидроксид калия или гидроксид магния, щелочные фосфаты, такие как гидрофосфат калия и меглюмин или смеси этих щелочных соединений. [0003] Пероральная лекарственная форма из активного вещества общей формулы [0004] Лекарственная форма по п.3, где лекарственная форма не содержит поверхностно-активное вещество. [0005] Лекарственная форма по п.3 или 4, в которой карбонат натрия имеет гранулометрический состав, при котором более чем 98% частиц имеют размер меньше чем 500 мкм, более чем 60% частиц меньше чем 160 мкм и более чем 30% частиц меньше чем 63 мкм. [0006] Лекарственная форма по п.5, в которой карбонат натрия имеет гранулометрический состав, при котором около 99,8% частиц имеют размер меньше чем 500 мкм, около 80% частиц меньше чем 160 мкм и около 40% частиц меньше чем 63 мкм. [0007] Лекарственная форма по любому из пп.3-6, в которой карбонат натрия имеет специфическую площадь поверхности больше чем 1,0 м2/г. [0008] Лекарственная форма по п.7, в которой карбонат натрия имеет специфическую площадь поверхности больше чем 1,5 м2/г. [0009] Лекарственная форма по п.8, в которой карбонат натрия имеет специфическую площадь поверхности около 2,0 м2/г. [0010] Лекарственная форма по любому из пп.3-9, в которой карбонат натрия присутствует в количестве по меньшей мере 20 вес.% лекарственной формы. [0011] Лекарственная форма по любому из пп.1-10, в которой М представляет собой кальций в его 2+ форме. [0012] Лекарственная форма по любому из пп.1-11, характеризующаяся тем, что количество щелочного соединения составляет больше чем 55 вес.%, предпочтительно больше чем 60 вес.%. [0013] Лекарственная форма по любому из пп.1-12, характеризующаяся тем, что активным веществом является кальциевая соль 3-[[[1-[2-(этоксикарбонил)-4-фенилбутил]циклопентил]карбонил]амино]-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-ацетиловой кислоты в ее 3S,2'R форме. [0014] Лекарственная форма по любому из пп.1-13 в форме гранул, уплотненных таблеток или капсул. [0015] Способ получения лекарственной формы по любому из пп.1-14, включающий следующие стадии: [0016] Способ получения лекарственной формы по любому из пп.1-14, включающий следующие стадии: [0017] Фармацевтическая лекарственная форма по любому из пп.1-14, где лекарственная форма имеет растворимость по меньшей мере 50% за 5 мин, измеренную с использованием конфигурации прибора 2 USP при скорости вращения барабана 50 об./мин при 37 °С и при рН 6,8. [0018] Фармацевтическая лекарственная форма по п.17, где лекарственная форма имеет растворимость по меньшей мере 65% за 15 мин, измеренную с использованием конфигурации прибора 2 USP при скорости вращения барабана 50 об./мин при 37 °С и при рН 6,8. [0019] Фармацевтическая лекарственная форма по п.17, где лекарственная форма имеет растворимость по меньшей мере 75% за 30 мин, как измерено с использованием конфигурации прибора 2 USP при скорости вращения барабана 50 об./мин при 37 °С и при рН 6,8. [0020] Фармацевтическая лекарственная форма по п.17, где лекарственная форма имеет растворимость по меньшей мере 50% за 5 мин, 65% за 15 мин и 75% за 30 мин, измеренную с использованием конфигурации прибора 2 USP при скорости вращения барабана 50 об./мин при 37 °С и при рН 6,8. [0021] Фармацевтическая лекарственная форма по пп.17-20, где лекарственная форма имеет растворимость менее чем 5% за 30 мин, измеренную с использованием конфигурации прибора 2 USP при скорости вращения барабана 50 об./мин при 37 °С и при рН 2,0. Полный текст патента Настоящее изобретение относится усовершенствованной пероральной лекарственной форме из активного соединения с общей формулой где R 1 выбирают из: (1) (C 1 -C 6 )алкокси(C 1 -C 6 )алкил, который необязательно замещен (C 1 -C 6 )алкокси; (2) фенил-(C 1 -C 6 )алкил и фенилокси-(C 1 -C 6 )алкил, где фенильная группа необязательно замещается (C 1 -C 6 )алкил, (C 1 -C 6 )алкокси или галогеном; и (3) нафтил-(C 1 -C 6 )алкил; R 2 и R 3 оба независимо представляют собой водород или галоген, R 4 представляет собой биолабильную эфиробразующую группу, М представляет собой водород или ион металла, предпочтительно двухвалентный ион металла, а n 1, 2 или 3. Эти соединения и их соли, и биолабильные эфиры охватываются объемом охраны настоящего изобретения и являются мощными ингибиторами ECE/NEP и описаны у Waldeck и др., US 5677297 и ЕР 0733642. Соединения бензазепин-N-уксусной кислоты, применяемые в настоящем изобретении, известны из ЕР 0733642, 0830863, WO 00/48601 и 01/03699 и могут быть получены при помощи методов, описанных в упомянутых US 5677297 и ЕР 0733642. Эти патенты посвящены указанным соединениям и собственно их физиологически приемлемым солям, и применению соединений при сердечной недостаточности. WO 03/059939 описывает специфические соли этих соединений, особенно соли кальция. ЕР 0830863, WO 00/48601 и 01/03699 описывают применение вышеупомянутых соединений в области гастроинтестинального кровообращения для лечения гипертензии и лечения и профилактики кардиальных повреждений, вызванных адриамицином и сходными противоопухолевыми препаратами соответственно. Многие активные соединения, включая вышеупомянутые соединения с формулой (I), обладают очень плохой водорастворимостью. При введении этих активных соединений в тело они часто обладают плохой биодоступностью из-за плохой растворимости в пищеварительном соке. Чтобы решить эту проблему, было разработано несколько методов, таких как микронизация, включение в циклодекстрины, использование водорастворимых носителей, использование твердотельного измельчения (WO 00/00179) или использование твердых растворов или нанокристаллических или аморфных форм активного соединения. WO 03/068266 описывает композицию пероральной лекарственной формы твердого раствора соединений с формулой (I), имеющую увеличенную биодоступность по сравнению с упомянутыми активными соединениями в традиционно составленной форме. Кроме того, эта лекарственная форма имеет хорошие характеристики биодоступности, ее недостаток состоит в том, что она формируется путем сплавления смеси, приводящего к некоторым ограничениям: она должна быть сформирована либо в капсулу, либо в таблетку методом экструзии сплава. Более того, размер лекарственной формы будет слишком большим для больших дозировок. WO 2006/067150 (без предварительной публикации) описывает пероральную лекарственную форму с быстрым высвобождением соединения формулы (I), включающую активное соединение в количестве, достигающем 60% от общего веса лекарственной формы, по меньшей мере 10 вес.% щелочного соединения или смеси щелочных соединений, от 0,1 до 10 вес.% одного или более поверхностно-активных веществ и необязательно вспомогательные материалы в количестве от 1 до 45% от общего веса лекарственной формы. В частности, хорошая биодоступность активного соединения достигается, когда докузат натрия используется в качестве поверхностно-активного вещества. Целью настоящего изобретения является обеспечить альтернативную пероральную лекарственную форму для соединения с формулой (I) с существенным увеличением биодоступности по сравнению с упомянутыми активными соединениями в традиционно составленной форме, как определено выше, которая является достаточно стабильной для коммерческого использования и которая также может быть использована для получения лекарственных форм с высоким содержанием активного вещества с разумным размером. Другой целью настоящего изобретения является обеспечить лекарственную форму, которая может быть получена с использованием обычных процедур и оборудования для получения, чтобы не требовалось больших инвестиций. Эта цель может быть достигнута посредством настоящего изобретения при помощи усовершенствованной пероральной лекарственной формы из активного соединения с общей формулой где R 1 выбирают из группы, состоящей из (C 1 -C 6 )алкокси(С 1 -C 6 )алкила, который может быть замещен (C 1 -C 6 )алкокси, фенил-(C 1 -C 6 )алкила и фенилокси-(C 1 -C 6 )алкила, где фенильная группа может быть замещена (C 1 -C 6 )алкил, (C 1 -C 6 )алкокси или галогеном, и нафтил-(C 1 -C 6 )алкила, R 2 и R 3 оба независимо представляют собой водород или галоген, R 4 представляет собой биолабильную эфиробразующую группу, М представляет собой водород или ион металла, предпочтительно двухвалентный ион металла. n - 1, 2 или 3; включая: а) указанное активное соединение в количестве от 10 до 80% от общего веса лекарственной формы; б) по меньшей мере 10% по весу щелочного соединения или смеси щелочных соединений; в) необязательно включает вспомогательные материалы в количестве от 1 до 45% от общего веса лекарственной формы, при условии, что лекарственная форма не содержит поверхностно-активное вещество. М предпочтительно выбирают из группы, состоящей из Li+, Са 2+ , Mg 2+ и Zn 2+ , а наиболее предпочтительно Са 2+ . (C 1 -C 6 )алкил определяется, как прямая или разветвленная алкильная группа, состоящая из от 1 до 6 атомов углерода. (C 1 -C 6 )алкокси определяется как прямая или разветвленная алкильная группа, состоящая из от 1 до 6 атомов углерода. R1 предпочтительно представляет собой фенилэтил, R 2 и R 3 предпочтительно представляют собой водород, a R 4 предпочтительно представляет собой этил. В рамках настоящего изобретения подходящие R 4 группы, которые могут образовывать биолабильные эфиры, включают низшие алкильные группы, фенильные или группы фенил-низший алкил, которые необязательно замещены по фенильному кольцу низшим алкилом или низшей алкиленовой цепочкой, связанной с двумя соседними атомами углерода, диоксоланилметильные группы, которые необязательно замещены по диоксолановому кольцу низшим алкилом, или С 2 -С 6 алканоилоксиметильные группы, которые необязательно замещены по оксиметильной группе низшим алкилом. Там, где группа R 4 , формирующая биолабильный эфир, представляет собой низший алкил, она предпочтительно может являться неразветвленной алкильной группой с от 1 до 4, предпочтительно 2, атомами углерода. Там, где группа, формирующая биолабильный эфир, представляет собой необязательно замещенную группу фенил-низший алкил, ее алкиленовая цепочка может содержать от 1 до 3, предпочтительно 1, атом углерода. Там, где фенильное кольцо замещено низшей алкиленовой цепочкой, она может содержать от 3 до 4, в частности 3, атома углерода. Особенно подходящими фенилсодержащими заместителями R 4 являются фенил, бензил или инданил. Там, где R 4 является необязательно замещенной алканоилоксиметильной группой, ее алканоилгруппа может содержать от 2 до 6, предпочтительно от 3 до 5 атомов углерода и предпочтительно является разветвленной, и может быть, например, пивалоилоксиметил радикалом (трет-бутилкарбонилоксиметил радикал). Предпочтительным соединением является кальциевая соль 3[[[1-[2-(этоксикарбонил)-4-фенилбутил]циклопентил]карбонил]амино]-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-ацетиловой кислоты. Наиболее предпочтительным соединением является указанное соединение в его 3S,2'R форме, известной также, как даглутрил кальция или кальций SLV 306. Это соединение носит название Соединение S-Ca, соответствующая кислота 3[[[1-[2-(этоксикарбонил)-4-фенилбутил]циклопентил]карбонил]амино]-2,3,4,5-тетрагидро-2-оксо-1Н-1-бензазепин-1-ацетиловая кислота, также известная как даглутриловая, или SLV306 носит название Соединение S-H. Активное соединение с формулой (I) обычно используется в количестве от около 10 до 80 вес.%, более предпочтительно в количестве от 15 до 75 вес.%, даже более предпочтительно в количестве от 20 до 65 вес.% и наиболее предпочтительно в количестве от около 45 до 65 вес.%. Активное соединение используется или может необязательно использоваться в микронизированной форме. Следующие определения приведены, чтобы способствовать пониманию конкретных терминов, используемых в рамках настоящей заявки. ″Относительно стабильный для коммерческого использования ″ обозначает приемлемую химическую и физическую стабильность в период хранения в течение по меньшей мере одного года в условиях окружающей среды, предпочтительно по меньшей мере 2 лет, даже более предпочтительно по меньшей мере 3 лет и наиболее предпочтительно по меньшей мере 5 лет. ″Приемлемая химическая стабильность ″ обозначает не более 5% разрушения активного вещества в течение периода хранения, предпочтительно не более 3% и наиболее предпочтительно не более 1%. ″Приемлемая физическая стабильность ″ обозначает отсутствие существенного изменения внешнего вида, отсутствие разломов таблетки во время извлечения из упаковки по окончании периода хранения и отсутствие более чем 20% изменения распадаемости. Термин ″микронизированный ″ относится к размеру частиц, при котором касательно объема более чем 95% частиц меньше чем 75 мкм. Поверхностно-активные вещества определяют как молекулы с хорошо выраженными полярными и неполярными участками, которые позволяют им агрегировать в растворах, чтобы сформировать мицеллы. В зависимости от природы полярного участка поверхностно-активные вещества могут быть неионными, анионными. Катионными и цвиттерионными. Примерами неионных гидрофильных поверхностно-активных веществ являются полиоксиэтиленовые эфиры сорбитана, кремофоры и полоксамеры. Примерам анионных поверхностно-активных веществ являются лаурил саркозинат, докузат и фармацевтически приемлемые соли докузата, такие как докузат кальция, докузат натрия и докузат калия. Щелочное соединение выбирают из группы, состоящей из неорганических и органических щелочных соединений, таких как бикарбонат натрия, бикарбонат калия, карбонат натрия, карбонат калия, цитрат натрия, трис-буфер, триэтаноламин, щелочные гидроксиды, такие как гидроксид натрия, гидроксид калия или гидроксид магния, щелочные фосфаты, такие как гидрофосфат калия и меглюмин. Также могут быть использованы смеси этих щелочных соединений. Предпочтительными щелочными соединениями являются бикарбонат натрия, бикарбонат калия, карбонат натрия, карбонат калия и карбонат кальция. Даже более предпочтительным щелочным соединением является карбонат натрия. Предпочтительно щелочная система присутствует в количестве более чем 10 вес.% состава без покрытия, более предпочтительно более чем 20 вес.% или присутствует в количестве более чем 25, 30, 40 45, 55 или 60 вес.% состава без покрытия. Термин ″состав без покрытия ″ обозначает лекарственную форму до нанесения дополнительного покровного(ых) вещества(в). В случае использования карбоната он предпочтительно используется в количестве 25% от общего веса лекарственной формы без покрытия или присутствует в количестве по меньшей мере 45 вес.% или присутствует в количестве более чем 50, 55 или 60 вес.% лекарственной формы без покрытия. В следующем предпочтительном варианте изобретение относится к улучшенной пероральной фармацевтической композиции активного вещества с общей формулой где R 1 выбирают из группы, состоящей из (C 1 -C 6 )алкокси(C 1 -С 6 )алкила, который может быть замещен (C 1 -C 6 )алкокси, фенил-(C 1 -C 6 )алкила и фенилокси-(C 1 -C 6 )алкила, где фенильная группа может быть замещена (C 1 -C 6 )алкил, (C 1 -С 6 )алкокси или галогеном, и нафтил-(C 1 -C 6 )алкила, R 2 и R 3 оба независимо представляют собой водород или галоген, R 4 представляет собой биолабильную эфиробразующую группу, М представляет собой водород или ион металла, предпочтительно двухвалентный ион металла. n - 1, 2 или 3; включая: а) указанное активное соединение в количестве от 10 до 80% от общего веса лекарственной формы; б) по меньшей мере 10 вес.% карбоната натрия, имеющего гранулометрический состав, при котором более чем 97% частиц меньше чем 500 мкм, более чем 40% частиц меньше чем 160 мкм и более чем 10% частиц меньше чем 63 мкм; в) необязательно включает вспомогательные материалы в количестве от 1 до 45% от общего веса лекарственной формы. В этом предпочтительном варианте щелочная система включает карбонат натрия, имеющий меньший размер частиц, чем обычный карбонат натрия, который имеет гранулометрический состав (определенный при помощи гранулометрического анализа и основанный на весе), при котором по большей мере 25% частиц меньше чем 160 мкм. Этот карбонат кальция также имеет большую специфичную площадь поверхности, нежели чем обычный карбонат натрия, который имеет специфичную площадь поверхности (определено при помощи стандартного метода измерения площади БЭТ) порядка 0,2 м 2 /г. Как отмечено выше, карбонат натрия, используемый в предпочтительном варианте, имеет гранулометрический состав (определенный при помощи гранулометрического анализа и основанный на весе), при котором более чем 97% частиц меньше чем 500 мкм, более чем 40% частиц меньше чем 160 мкм и более чем 10% частиц меньше чем 63 мкм. В даже более предпочтительной модификации более чем 98% частиц меньше чем 500 мкм, более чем 60% частиц меньше чем 160 мкм и более чем 30% частиц меньше чем 63 мкм. Специфическая площадь поверхности предпочтительно больше чем 1 м 2 /г и более предпочтительно больше чем 1,5 м 2 /г. Наиболее предпочтительным карбонатом натрия является специальный карбонат натрия, продаваемый Solva SA как Soda Ash IPH. У этого типа карбоната натрия обычно 99,8% частиц меньше чем 500 мкм, 80% частиц меньше чем 160 мкм и 40% частиц меньше чем 63 мкм и этот тип карбоната натрия имеет специфическую площадь поверхности 2 м 2 /г. Неожиданно авторы настоящего изобретения обнаружили, что использование в составе щелочного соединения в лекарственной форме, одного или в смеси, даже без какого-либо поверхностно-активного вещества предотвращает образование труднорастворимого геля в кислом желудочном соке, увеличивая, таким образом, растворимость SLV-306 практически, как показано во время in vitro исследований растворения на двухфазной модели растворения (см. Пример 1а), которая показывает как улучшение растворимости in vivo, так и улучшение биодоступности. Особенно при использовании карбоната натрия со средним размером частиц 100 мкм и специфической площадью поверхности 2 м 2 /г (такого как Soda Ash IPH), получается лекарственная форма с хорошей растворимостью in vivo и хорошей биодоступностью. Кроме того, ожидается, что составы обладают хорошей стабильностью при хранении. В случае если используется вышеупомянутый карбонат со специфическим средним размером частиц и поверхностной площадью, то он предпочтительно используется в количестве по меньшей мере 15% от общего веса лекарственной формы без покрытия, более предпочтительно в количестве по меньшей мере 18%, даже более предпочтительно в количестве по меньшей мере 20%, или он присутствует в количестве более чем 25, 30, 40, 50 или 60 вес.% лекарственной формы без покрытия. Специфические твердые щелочные соединения, такие как бикарбонаты и карбонаты, как описано выше, часто используются в сочетании с твердыми кислотными соединениями (например, лимонная кислота, винная кислота, адипиновая кислота, фумаровая кислота, янтарная кислота, аскорбиновая кислота, никотиновая кислота, сахарин, аспирин, яблочная кислота, дигидрофосфат натрия, дигидропирофосфат натрия, дигидроцитрат натрия и гидроцитрат натрия) в шипучих составах. В настоящем изобретении состав предпочтительно не содержит кислого компонента. Лекарственная форма необязательно включает вспомогательные вещества в количестве до 45% от общего веса лекарственной формы и предпочтительно от 1 до 45% от общего веса лекарственной формы. Примеры этих вспомогательных веществ включают, но не ограничиваются: а) связующие вещества, включающие, но не ограничивающиеся, акациевую камедь, альгиновую кислоту и их соли, производные целлюлозы, метилцеллюлозу, гидроксиэтил целлюлозу, гидроксипропил целлюлозу, полиэтиленгликоль, камеди, полисахаридные кислоты, гидроксипропил метилцеллюлозу, желатин, поливинилпирролидон, сополимер поливинилпирролидон/винил ацетат, полиметакрилаты, гидроксипропилметилцеллюлозу, крахмал, прежелатинизированный крахмал, этилцеллюлозу, трагантовую камедь, декстрин, микрокристаллическую целлюлозу, сахарозу или глюкозу и тому подобное; б) разрыхлители, включающие, но не ограничивающиеся, крахмалы, прежелатинизированный кукурузный крахмал, прежелатинизированный крахмал, целлюлозы, поперечно-сшитую карбоксиметилцеллюлозу, кросповидон, поперечно-сшитый поливинилпирролидон, альгинатный комплекс кальция или натрия, глины, альгинаты, камеди или натриевый гликолят крахмала и любые разрыхлители, используемые в приготовлении таблеток; в) наполнители, включающие, но не ограничивающиеся, лактозу, карбонат кальция, фосфат кальция, двухосновный фосфат кальция, сульфат кальция, микрокристаллическую целлюлозу, целлюлозный порошок, декстрозу, декстраты, декстран, крахмалы, прежелатинизированный крахмал, сахарозу, ксилитол, лактитол, маннитол, сорбитол и тому подобное; г) смазывающие вещества, включающие, но не ограничивающиеся, стеарат магния, гидроксид кальция, тальк, коллоидный диоксид кремния, стеарил фумарат натрия, гидрогенезированное растительное масло, стеариновую кислоту, глицерил бегенат, стеараты магния, кальция и натрия, воски, Stearowet, борную кислоту, бензоат натрия, ацетат натрия, DL-лейцин, полиэтиленгликоли, олеат натрия или лаурил сульфат натрия и тому подобное; д) стабилизаторы, включающие, но не ограничивающие, антиоксидантами, буферными агентами, кислотами и тому подобное; е) увлажнители, включающие, но не ограничивающиеся, олеиновую кислоту, глицерил моностеарат, сорбитан моноолеат, сорбитан монолаурат, триэтаноламин олеат, полиоксиэтилен сорбитан моноолеат, полиоксиэтилен сорбитан монолаурат, олеат натрия или лаурил сульфат натрия и тому подобное; ж) растворители, такие как лактоза, крахмал, маннитол, сорбитол, декстроза, микрозаттелитная целлюлоза, двухосновный фосфат кальция, наполнители, основанные на сахарозе, сахарная пудра, моногидрат одноосновного сульфата кальция, дигидрат сульфата кальция, тригидрат лактата кальция, декстраты, инозитол, гидролизованные частицы зерновых, амилоза, порошкообразная целлюлоза, карбонат кальция, глицин или бентонит и тому подобное; з) вещества, препятствующие прикреплению или глиданты, включающие, но не ограничивающиеся, коллоидный оксид кремния, тальк, кукурузный крахмал, DL-лейцин, лаурил сульфат натрия и стеараты магния, кальция или натрия и тому подобное; и) фармацевтически совместимые носители, включающие, но не ограничивающиеся, акациевую камедь, желатин, коллоидный диоксид кремния, глицерофосфат кальция, лактат кальция, мальтодекстрин, глицерин, силикат магния, казеинат натрия, соевый лецитин, хлорид натрия, фосфат кальция, гидрофосфат калия, стеароил лактилат натрия, каррагенан, моноглицерид, диглицерид или прежелатинизированный крахмал и тому подобное. В предпочтительном варианте лекарственная форма содержит по меньшей мере 10 вес.% карбоната натрия, имеющего специфический гранулометрический состав и/или площадь поверхности, как описано выше, лекарственная форма может также содержать поверхностно-активное вещество в качестве вспомогательного материала. Окончательная лекарственная форма представлена предпочтительно в форме гранул, прессованных таблеток или капсул. Лекарственная форма, описанная выше, может быть получена с использованием традиционных процедур и оборудования для приготовления. Поэтому другим аспектом настоящего изобретения является обеспечить способ получения, такой как описано выше, лекарственной формы, включающий следующие стадии: а) смешивание активного соединения с формулой I с щелочным соединением или смесью щелочных соединений и необязательно с одним или более вспомогательными материалами; б) уплотнение смеси; в) размалывание и просеивание гранул, полученных при указанном уплотнении и необязательное смешивание указанных просеянных гранул с одним или более вспомогательными материалами; и г) необязательное уплотнение смеси в таблетки с последующим необязательным покрытием и/или необязательным заполнением капсул смесью. В другом варианте изобретения лекарственную форму готовят при помощи способа органической грануляции, включающего следующие стадии: а) смешивание активного соединения с одним или более вспомогательными материалами; б) грануляция указанной смеси при помощи органического растворителя; в) удаление органического растворителя, чтобы получить гранулы; г) размалывание и просеивание гранул и смешивание указанных просеянных гранул с оставшейся порцией вспомогательных материалов; д) необязательное уплотнение смеси в таблетки с последующим необязательным покрытием и/или необязательным заполнением капсул смесью. В способе органической грануляции может быть использовано несколько органических растворителей. Примерами являются метил-т-бутиловый эфир (МТБЭ), дихлорметан и этилацетат. Когда лекарственные формы настоящего изобретения представлены в форме таблеток, эти таблетки имеют времена распадаемости от 5 до 90 мин. Предпочтительно времена распадаемости составляют меньше 60 мин, а наиболее предпочтительно они составляют меньше 45 мин. Лекарственные формы с короткими распадаемостями могут быть приготовлены, используя пористый карбонат натрия, такой как доступный в Soda Ash IPH). Различные дополнительные стадии также могут быть частью процесса, такие как сушка, измельчение, просеивание, смешивание и упаковка, но эти стадии не являются существенными характеристиками для получения лекарственной формы по настоящему изобретению. С другой стороны, изобретение обеспечивает состав, имеющий благоприятный профиль высвобождения. Поэтому настоящее изобретение, как описано выше, также относится к пероральным фармацевтическим композициям, где композиция имеет растворимость по меньшей мере 50% за 5 мин, как измерено с использованием 2 конфигурации прибора USP при скорости вращения барабана 50 об./мин при 37 °С и при рН 6,8. Растворимость через 5 мин предпочтительно составляет по меньшей мере 55% и даже более предпочтительно 60%. Через 15 мин растворимость составляет по меньшей мере 65%, предпочтительно по меньшей мере 70% и более предпочтительно 75%. Через 30 мин растворимость составляет по меньшей мере 75%, предпочтительно по меньшей мере 80% и более предпочтительно по меньшей мере 85%. При рН 2,0 фармацевтическая композиция по настоящему изобретению не высвобождает активное соединение существенно. Меньше чем 5% активного соединения высвобождается за 30 мин, как измерено с использованием 2 конфигурации прибора USP при скорости вращения барабана 50 об./мин при 37 °С и при рН 2,0. Предпочтительно высвобождается меньше чем 2% и наиболее предпочтительно меньше чем 1%. Следующие примеры предназначены, чтобы дополнительно проиллюстрировать изобретение более подробно, поэтому эти примеры не подразумевают ограничения изобретения каким бы то ни было способом. Пример 1. Материалы, оборудование и методы. Материалы. S-Ca готовили по рецепту, приведенному в примерах 2 и 3 WO 03/059939, начиная с кислоты, приготовленной по примеру 2 из ЕР 0733642. Soda Ash IPH (также обозначенная как Soda, пористая или пористая сода) были получены от Solvay SA Брюссель, Бельгия. Все вспомогательные вещества полностью имеются в продаже. Для роллерного уплотнения использовали роллер для уплотнения Fitzpatrick типа IR, оборудованный Fitzmill L1A. Настройки роллерного уплотнения были следующими: Скорость вращения горизонтального ходового винта (HFS в об./мин) Скорость вращения вертикального ходового винта (VFS в об./мин) Скорость вращения роллеров (N1 в об./мин) Интервал (d в мм) Сила уплотнения (F в кН/см) Настройки Fitzmill были следующими: Скорость вращения ударных ножей (N2 в об./мин) Размер ячеек (в мм) а) Описание метода двухфазного растворения in vitro. Двухфазное растворение проводилось при помощи 2 конфигураций USP-прибора. Скорость вращения барабана составляла 50 об./мин, а температура ячеек (а также среды растворения) поддерживалась на уровне 37 °С при помощи оборудования Vankel VK7010. Растворение лекарственной формы начиналось в 500 мл 0,1 М соляной кислоты (4,2 мл концентрированной соляной кислоты (HCl) в 500 мл воды) (фаза 1). Образцы отбирались через 0, 5, 15 и 30 мин. Через 30 мин к фазе 1 добавляли 500 мл 1 М фосфатного буфера (32,4 г дигидрофосфата натрия (NaH 2 PO 4 ) и 124,8 г гидрофосфата натрия (Na 2 HPO 4 ) в 1000 мл воды). Добавление фосфатного буфера изменяло рН среды растворения с рН 1 в фазе 1 до рН 6,8 в фазе 2. Во время теста растворения рН обеих фаз оставался неизменным. Образцы отбирались через 35, 45 и 60 мин. Все образцы фильтровали через Pall Zymark Acrodisc PSF, GxF/GHP, 0,45 мкм или Millipore Miller-FH (гидрофобный PTFE 0,45) фильтр. Количество растворенного даглутрила в отфильтрованных образцах было проанализировано при помощи УФ-измерений при 240 нм, используя внешнюю стандартизацию. В более раннем сравнительном исследовании с солью кальция соединения SLV306 (S-Ca) было показано, что этот метод двухфазного растворения in vitro хорошо соответствовал результатам in vivo. б) описание методов для характеристики пористого карбоната натрия. Гранулометрический состав пористого карбоната натрия был измерен посредством механической системы. Образец около 70 г продукта взвешивали и помещали в верхнее сито просеивающей машины (автоматическое устройство, которое может переносить сочетание горизонтальных движений и резких вертикальных движений на набор сит, например, просеивающая машина ROTAP или AS 200 RETSCH), включающей сита с ячейками в 0,5, 0,25, 0,16, 0,125, 0,1, 0,063 и 0,045 мм. Процедура просеивания проводилась в течение около 15 мин. Содержимое каждого сита взвешивали и подсчитывали массу частиц, имеющих размер частиц меньше чем 500 мкм, массу частиц, имеющих размер частиц меньше чем 160 мкм, и массу частиц, имеющих размер частиц меньше чем 63 мкм. Специфическая площадь поверхности пористого карбоната натрия была измерена по стандартному методу измерения площади БЭТ. в) Описание других физических методов. Текучесть потока была измерена в латунной трубе с ∅ выходного отверстия 8 мм. Текучесть потока выражали в с/100 г. Гранулометрический состав вещества, уплотненного роллером, получали в результате ручного просеивания с использованием 0,25, 0,50, 0,71, 0,85, 1,0 и 2,0 мм сит. Было подсчитано количество (%) <0,25 мм, количество (%)> 1,0 мм и d (50%). Удельная плотность и плотность утряски смешанного гранулята определяли следующим образом: гранулят в количестве от 100 до 150 г засыпали в градуированный цилиндр; определяли заполненный объем. После 1200 встряхиваний вновь определяли заполненный объем. Карр-индекс подсчитывали из удельного объема и объема утряски. Сопротивление таблеток разрушению определяли при помощи разрушения пяти таблеток в измерителе твердости Шленигера. Был описан средний уровень. Ломкость была определена на 20 таблетках с использованием измерителя ломкости Эрвека. Условиями испытания было 10 мин при 40 об./мин. Ломкость выразили в % уменьшения веса таблеток. Распадаемость была протестирована на одной таблетке с использованием воды и среды растворения. Пример 2. Приготовление традиционно составленной покрытой таблетки S-Ca. Процедура. 1) S-Ca уплотнили и пропустили через 1,0 мм сито. 2) Вещество со стадии (1) смешали с микрокристаллической целлюлозой РН301, поперечно-сшитым полвинилпирролидоном и стеарил фумаратом натрия до получения однородной смеси. 3) Вещество со стадии (2) уплотнили, используя уплотнительную машину для таблеток. 4) Таблетки со стадии (3) покрыли на подходящем оборудовании для нанесения покрытий. Пример 3. Приготовление непокрытых таблеток S-Ca, содержащих Soda Ash IPH Процедура. 1) S-Ca, Soda Ash IPH, стеарат магния и натриевый гликолят крахмала (primojel) просеивали через #40 сито. 2) S-Ca, Soda Ash IPH (обозначенные как внутренние) и часть (обознаенную как внутренняя) стеарата магния, и необязательно натриевый гликолят крахмала (Primojel ®) (обозначенный как внутренний) и/или ранее просеянная лактоза смешивались для получения однородной смеси. 3) Вещество со стадии (2) смешивали при помощи роллерного уплотнителя при указанных настройках. 4) Перемешанный материал измельчали и просеивали через сито размером 2,5 мм. 5) Были измерены текучесть потока и гранулярный состав. 2. Формирование таблетки Процедура. 6) Материал со стадии (4) смешивали с оставшимся количеством стеарата магния (внешнего), натриевого гликолята крахмала (Primojel ®) (внешнего), Soda Ash IPH (внешней) или микрокристаллической целлюлозы (внешней). 7) Измеряли удельную плотность и плотность утряски и считали Карр-индекс. 8) Материал со стадии (6) уплотняли с использованием машины для уплотнения при указанной силе уплотнения. 9) Были измерены ломкость, сопротивление разрушению и время распада. Пример 4. Сравнительное исследование растворения для лекарственной формы SLV306 с Soda Ash IPH и традиционно сформированной таблетки Сравнительное исследование растворения по методу, описанному в примере 1, (метод а) проводилось на одной партии традиционно сформированных таблеток (таблетка К, приготовленная, как описано в примере 2) и пяти 600/965 партиях соли кальция из SLV306 (S-Ca), приготовленного по примеру 3 (см. указатели партии в таблице) (таблетка F, уплотненная при 9 кН, таблетка G, уплотненная при 12 кН, таблетка Н, уплотненная при 6 кН, таблетка I, уплотненная при 11 кН, и таблетка J, уплотненная при 10 кН). Профиль высвобождения этих лекарственных форм приведен в форме таблицы ниже и изображен на фигуре ( Заключением этого исследования является то, что была приготовлена лекарственная форма S-Ca с большой лекарственной нагрузкой, имеющая целесообразный размер и подходящий профиль высвобождения. |