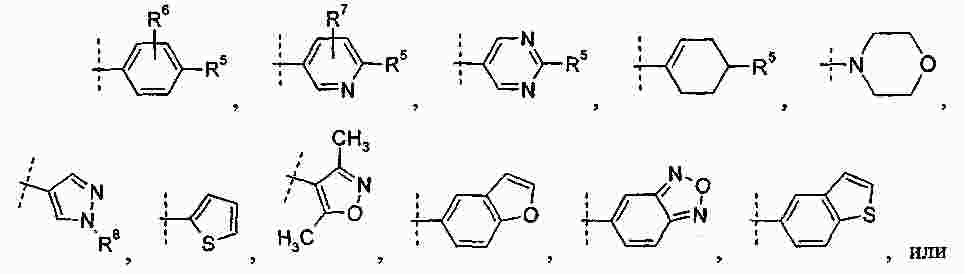
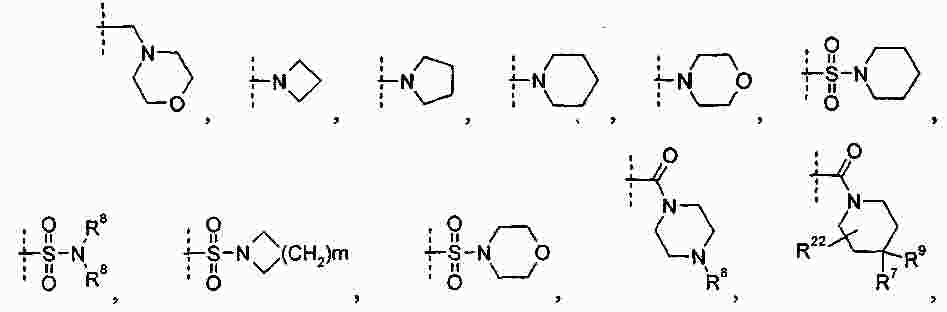
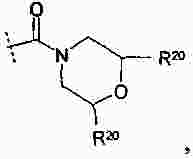
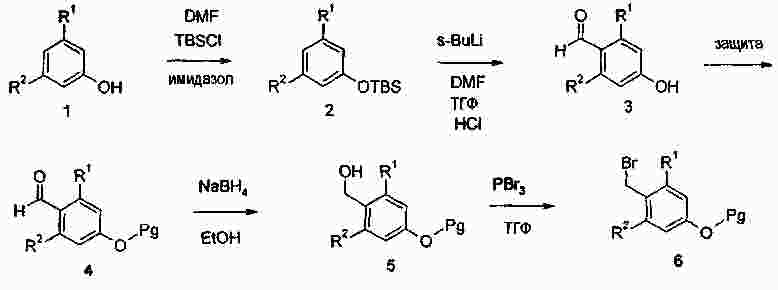
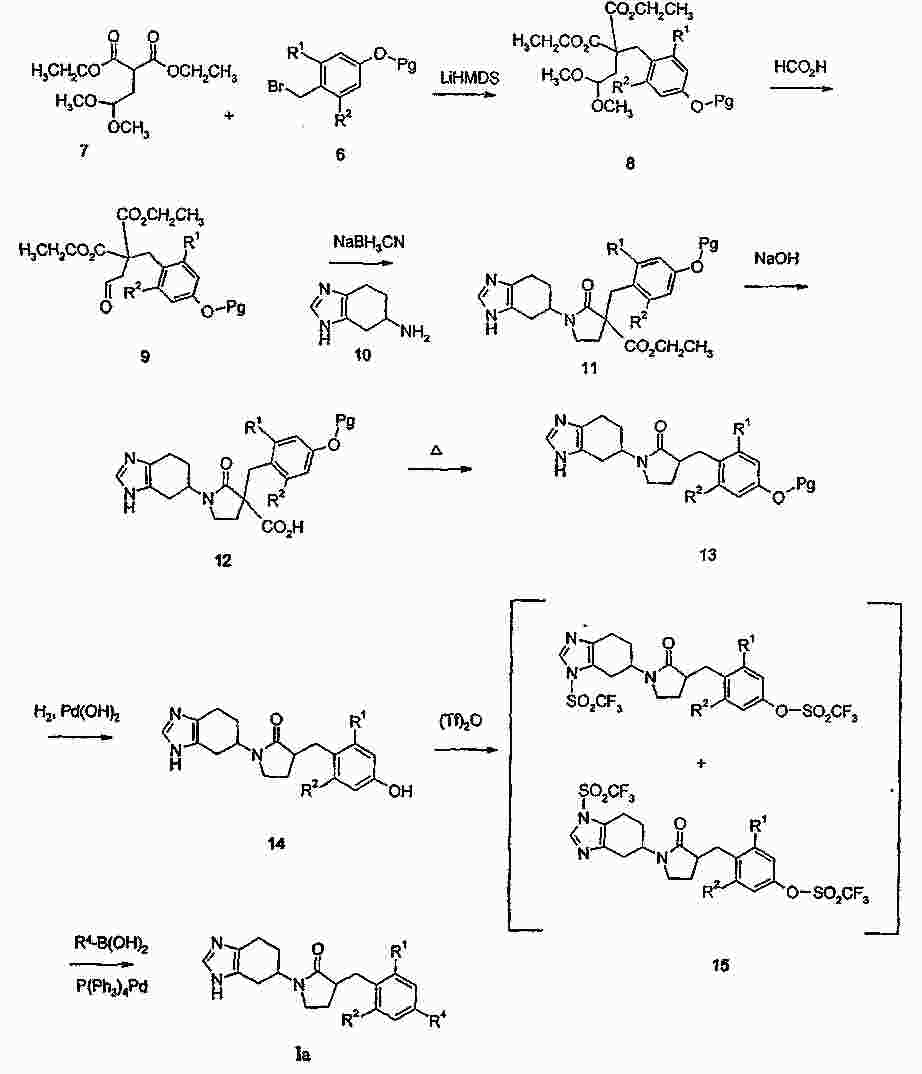
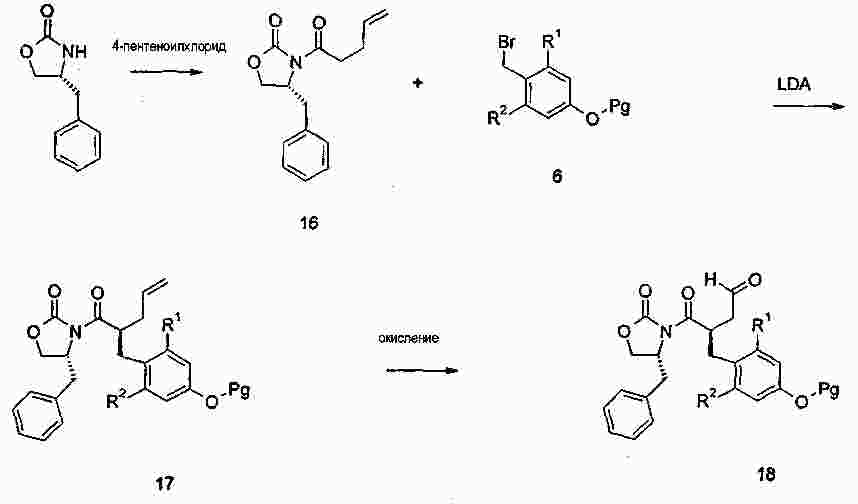
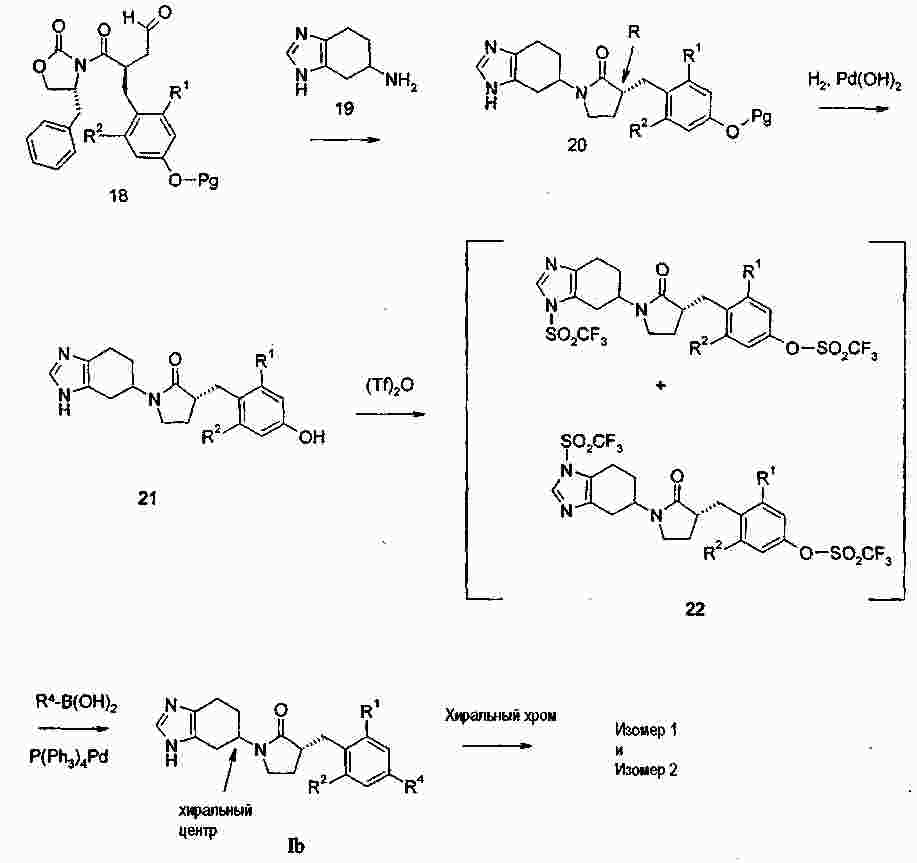
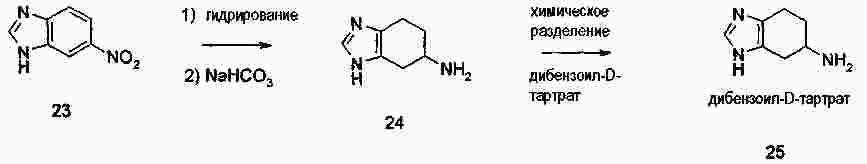
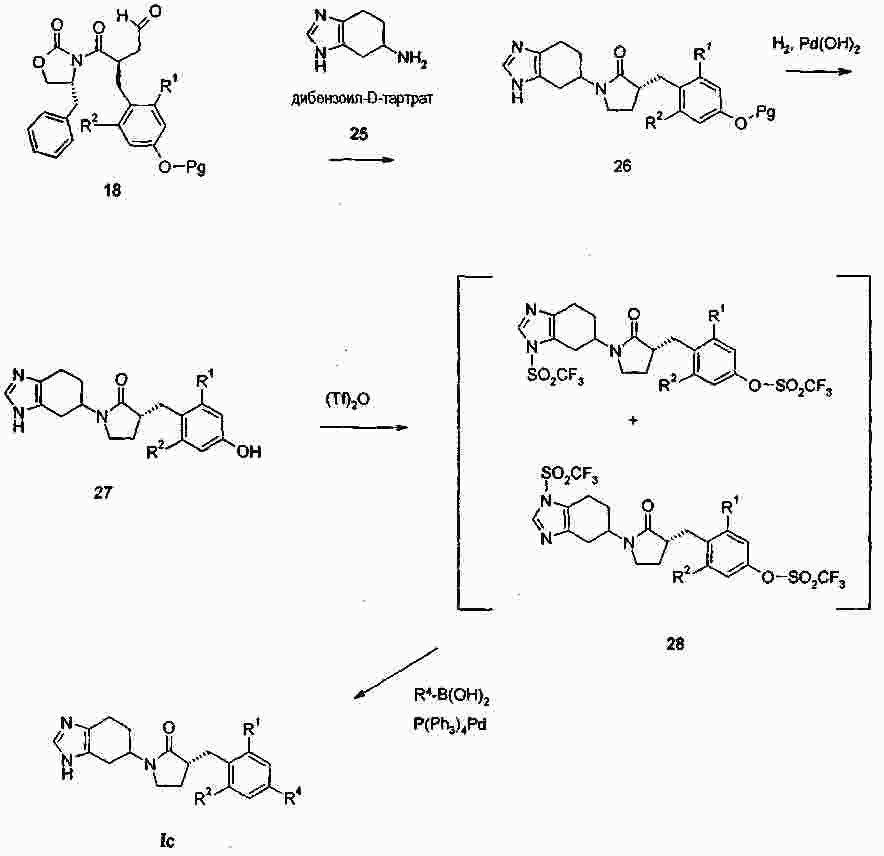
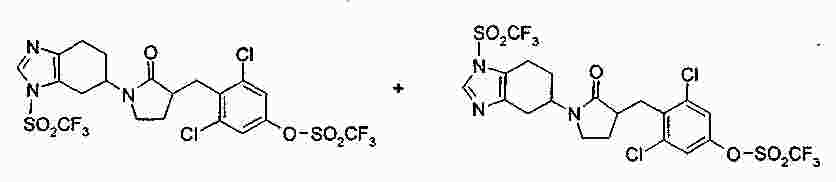
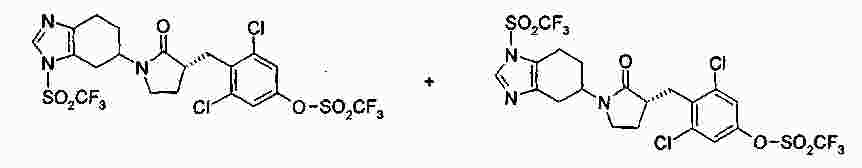
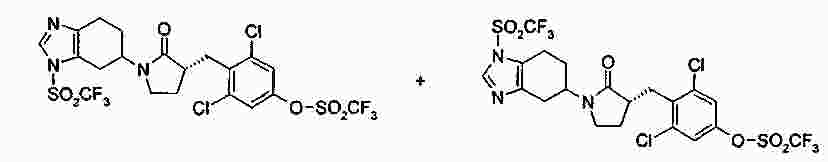
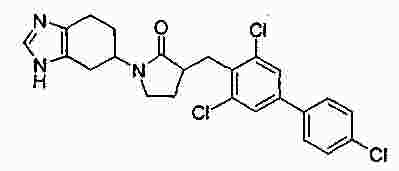
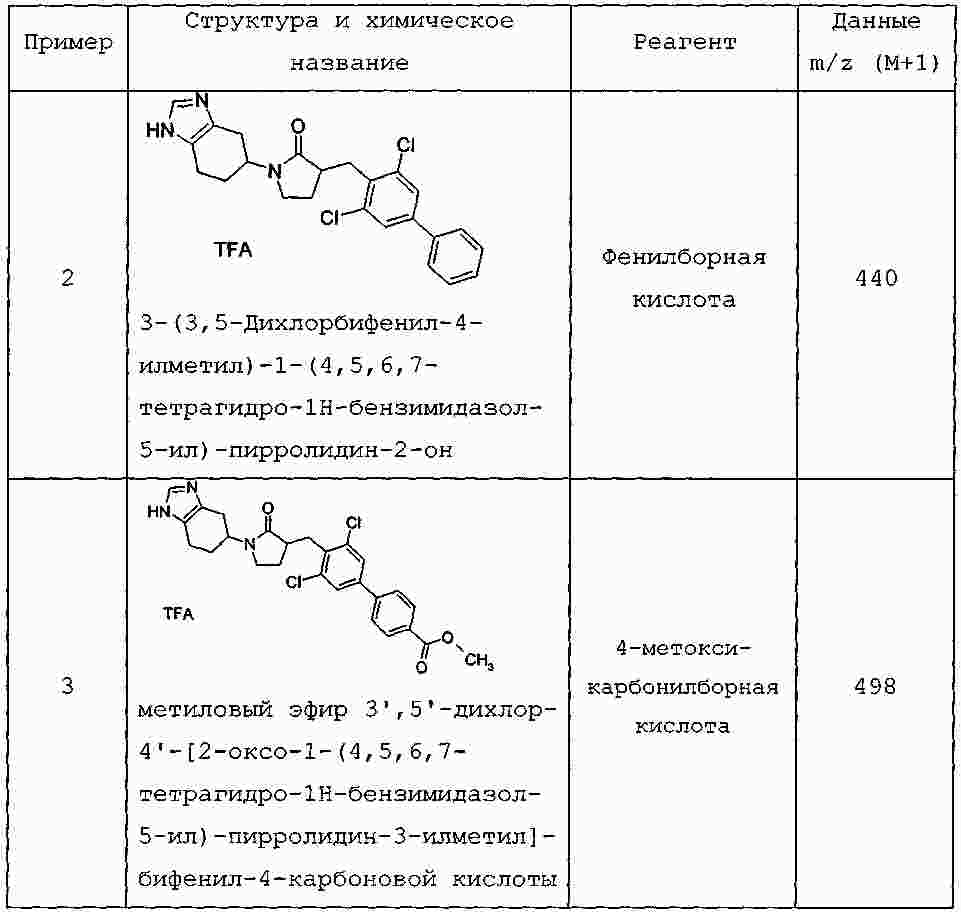
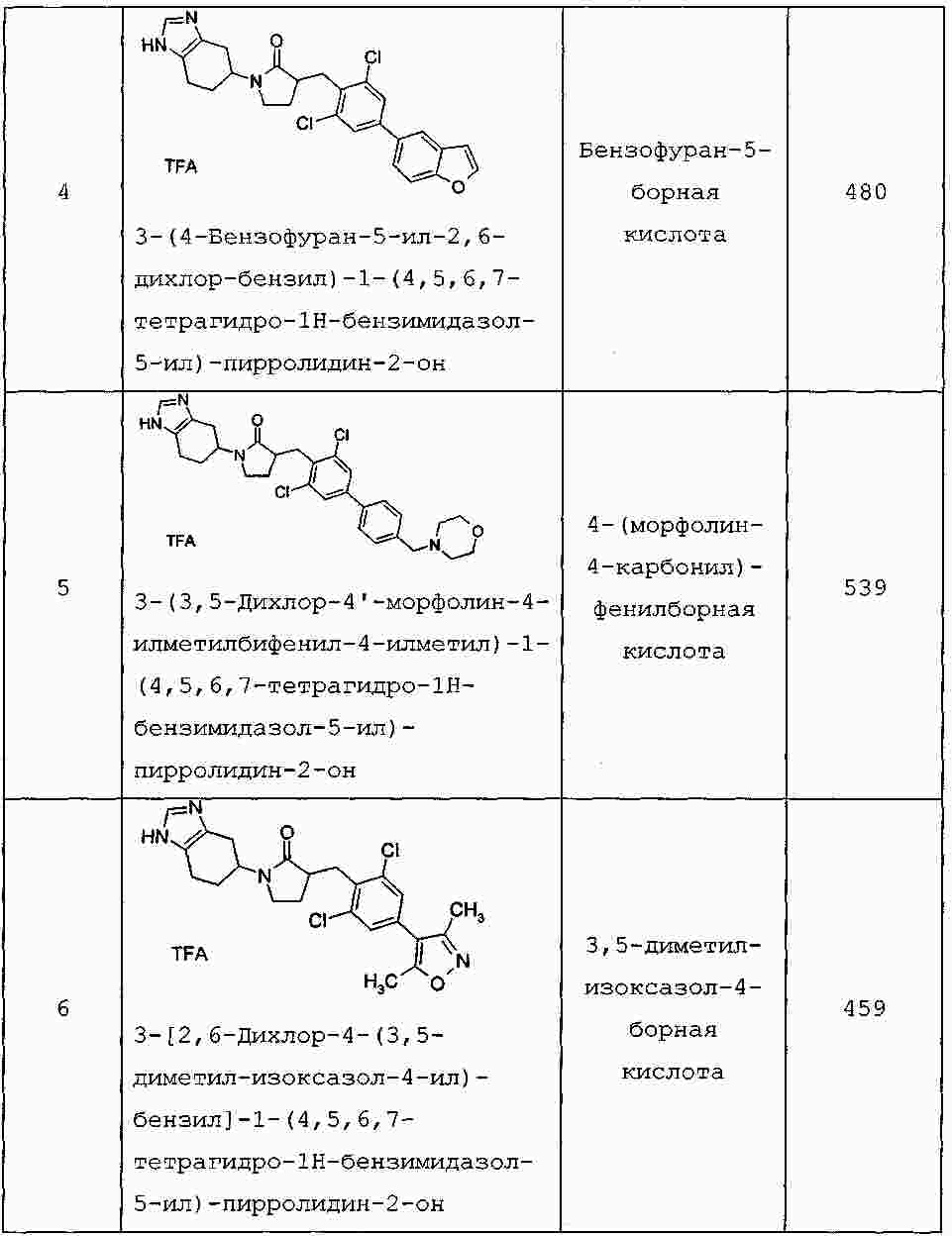
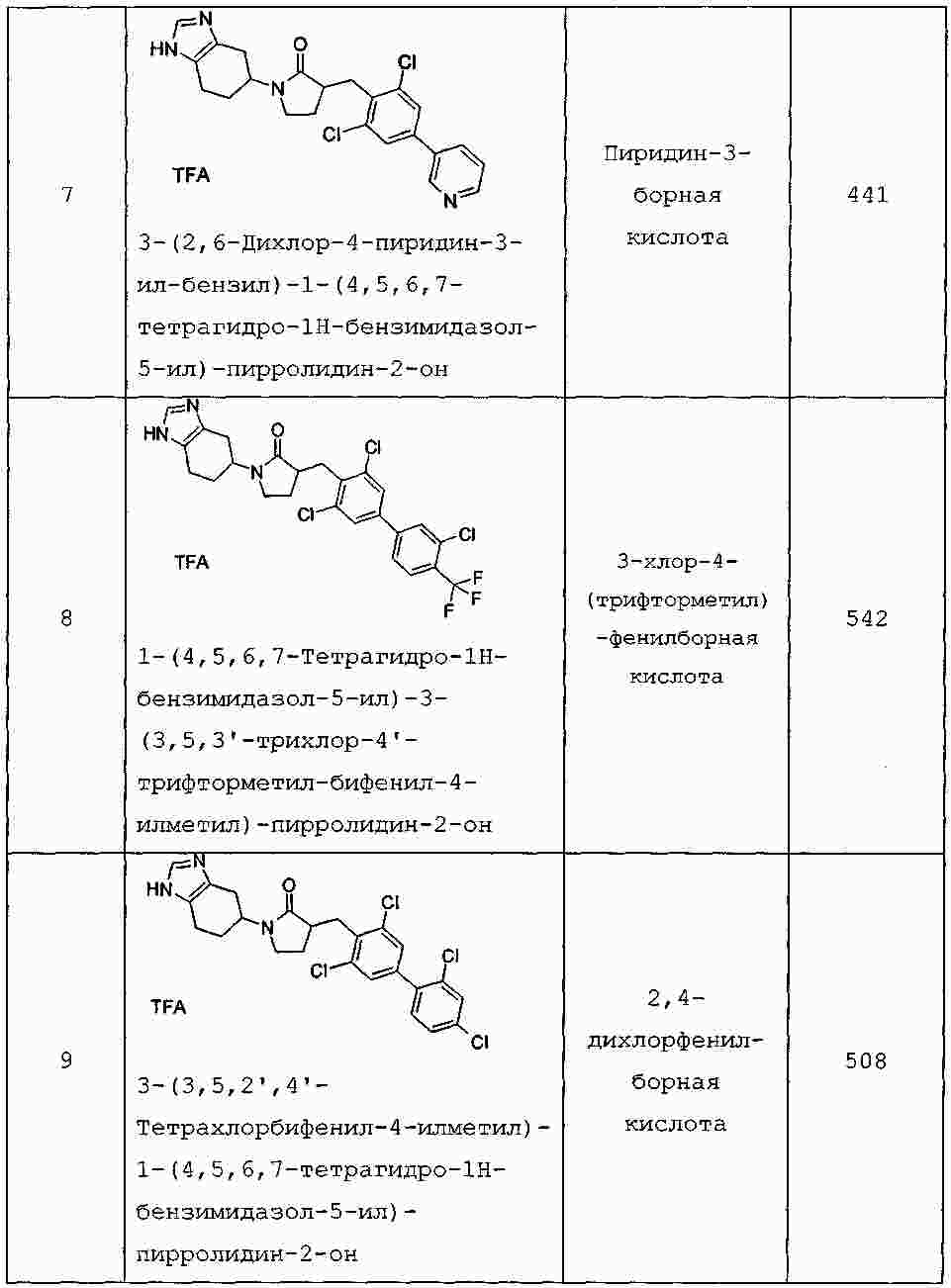
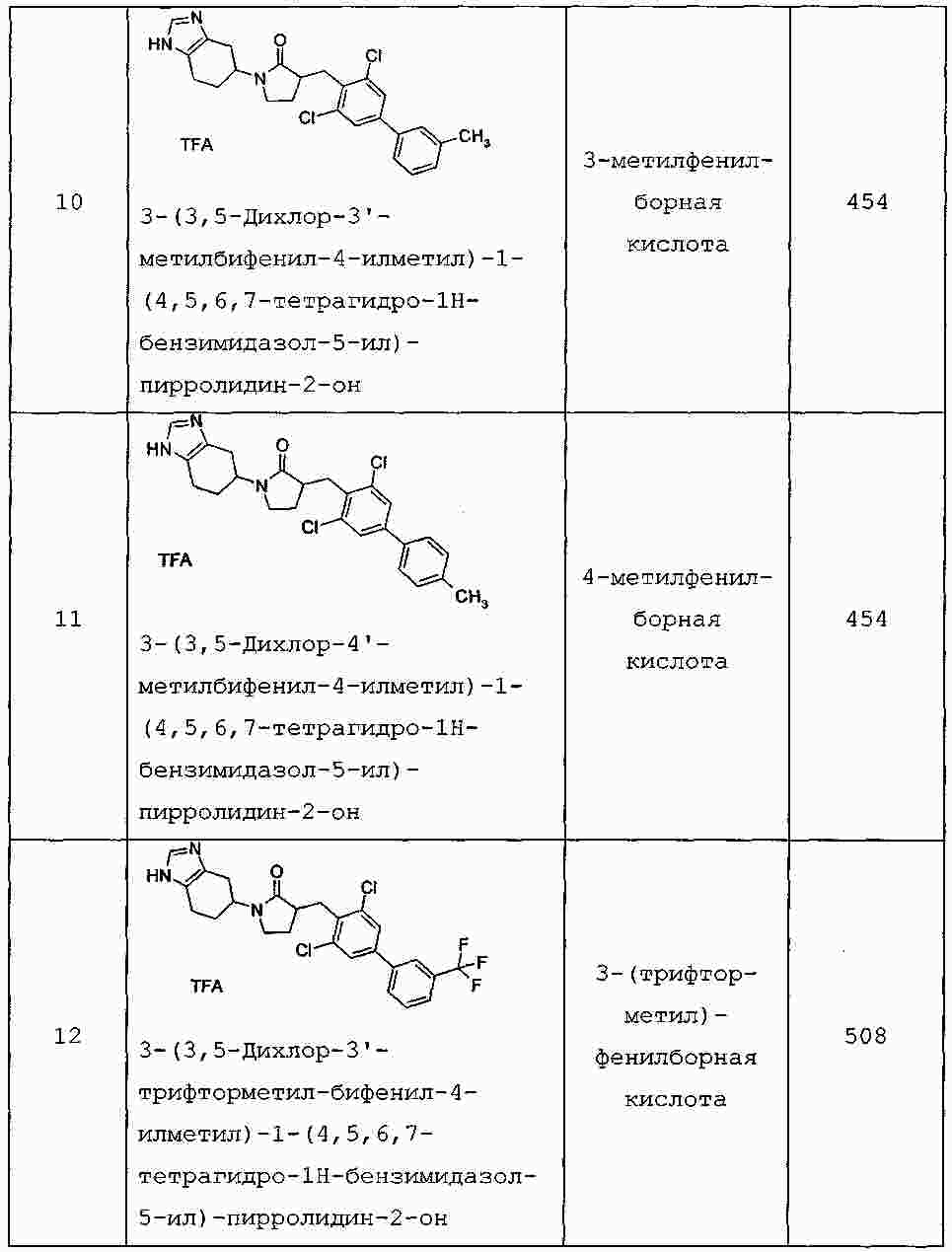
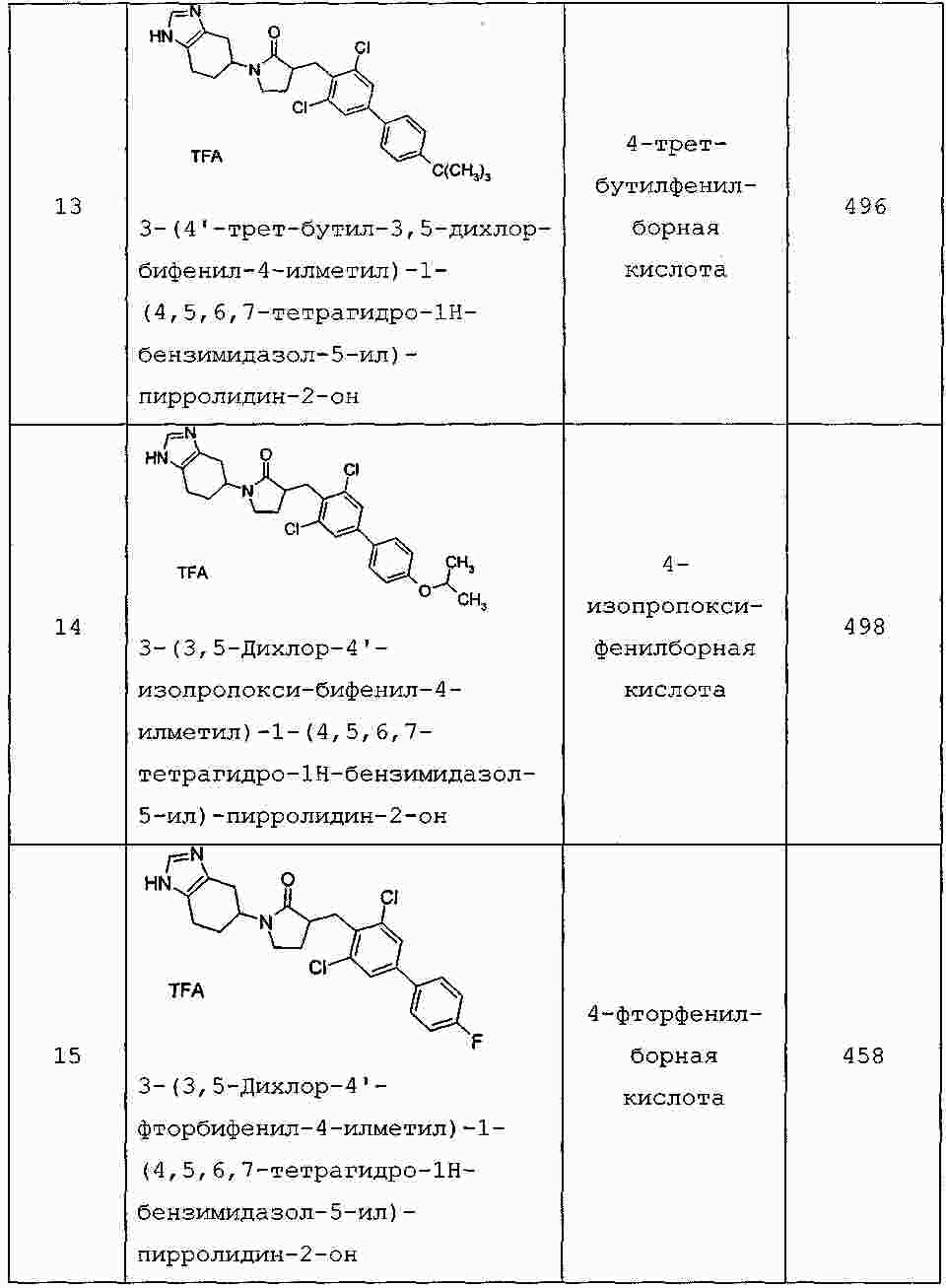
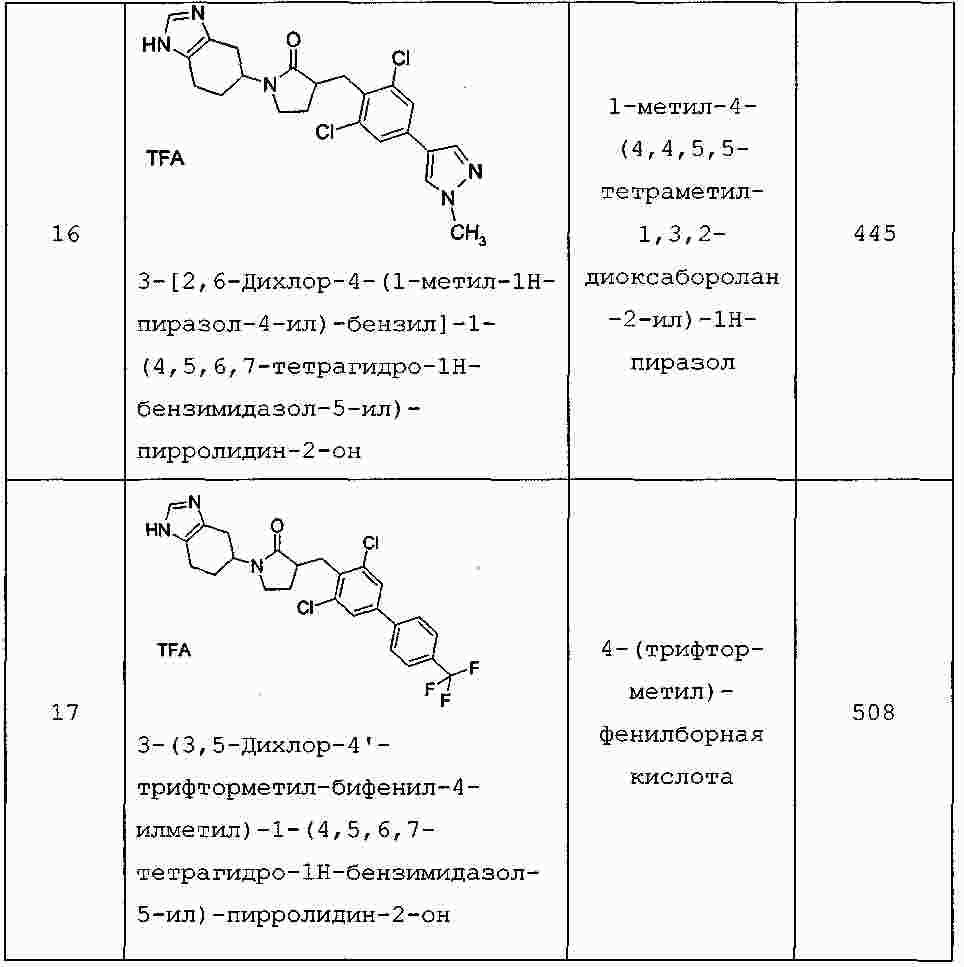
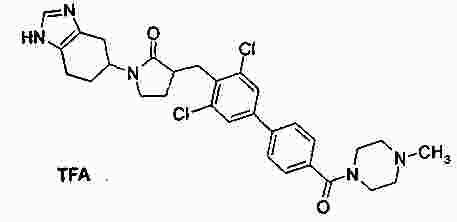
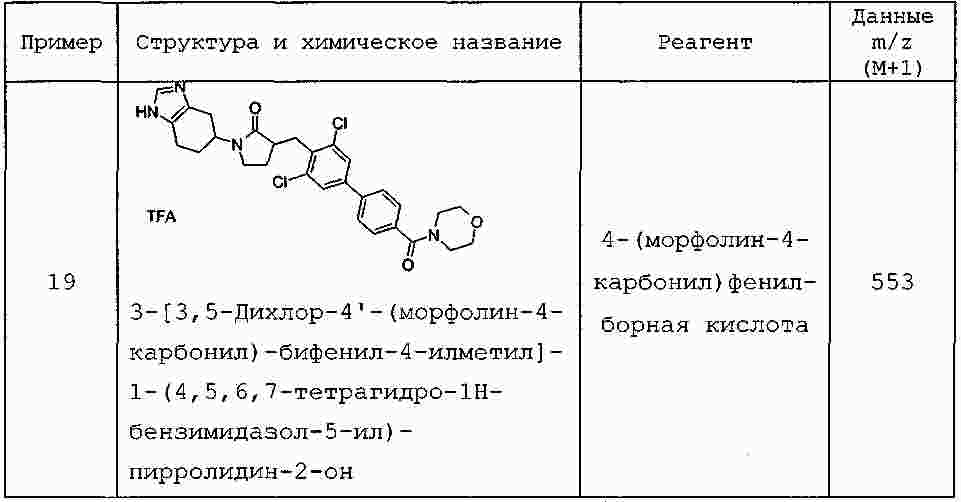
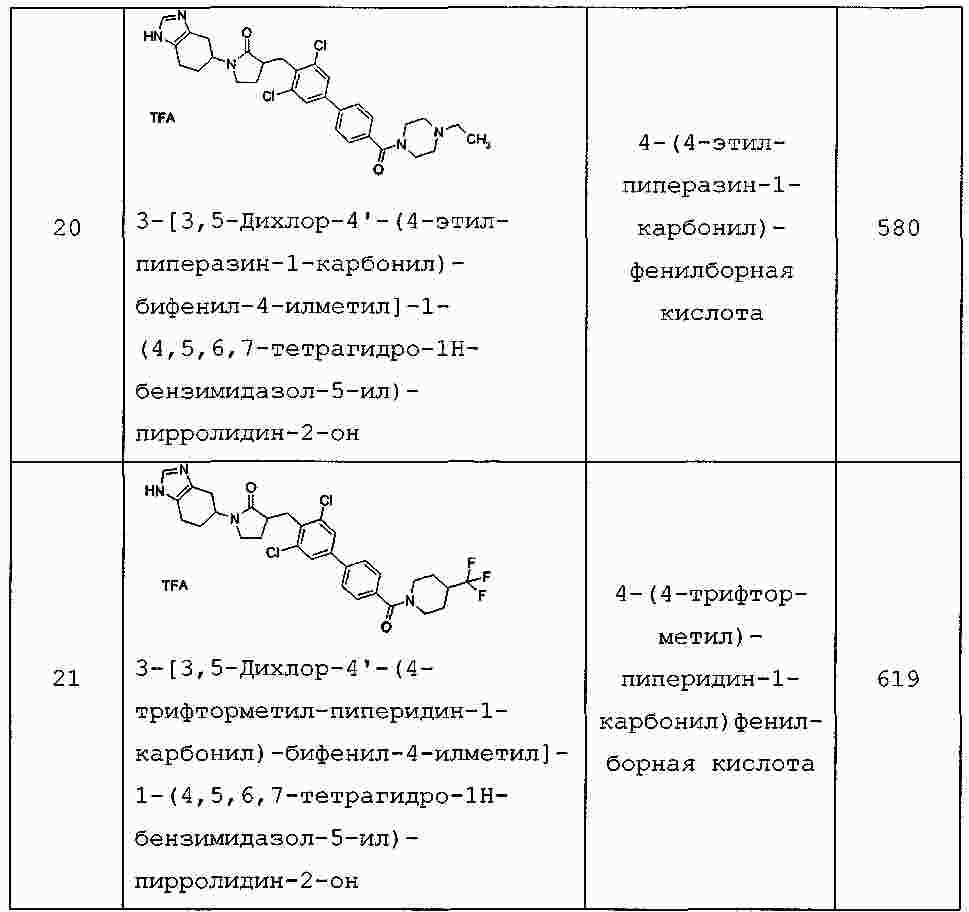
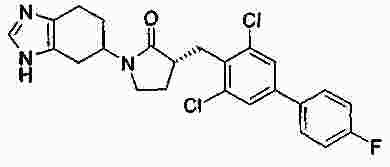
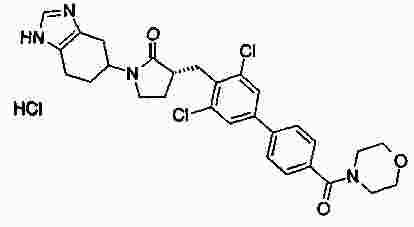
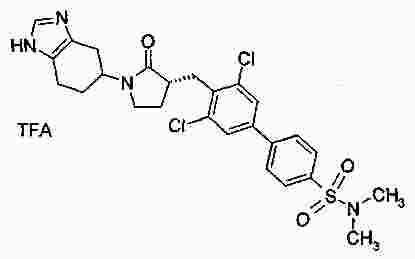
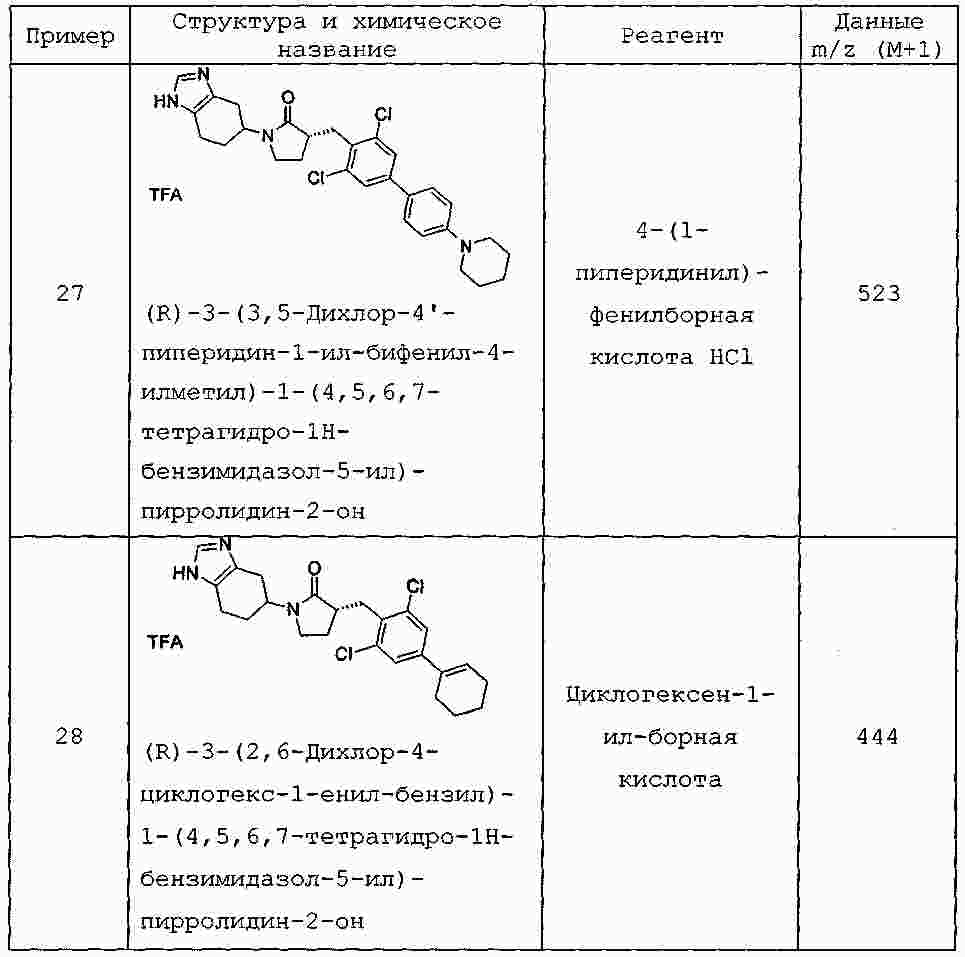
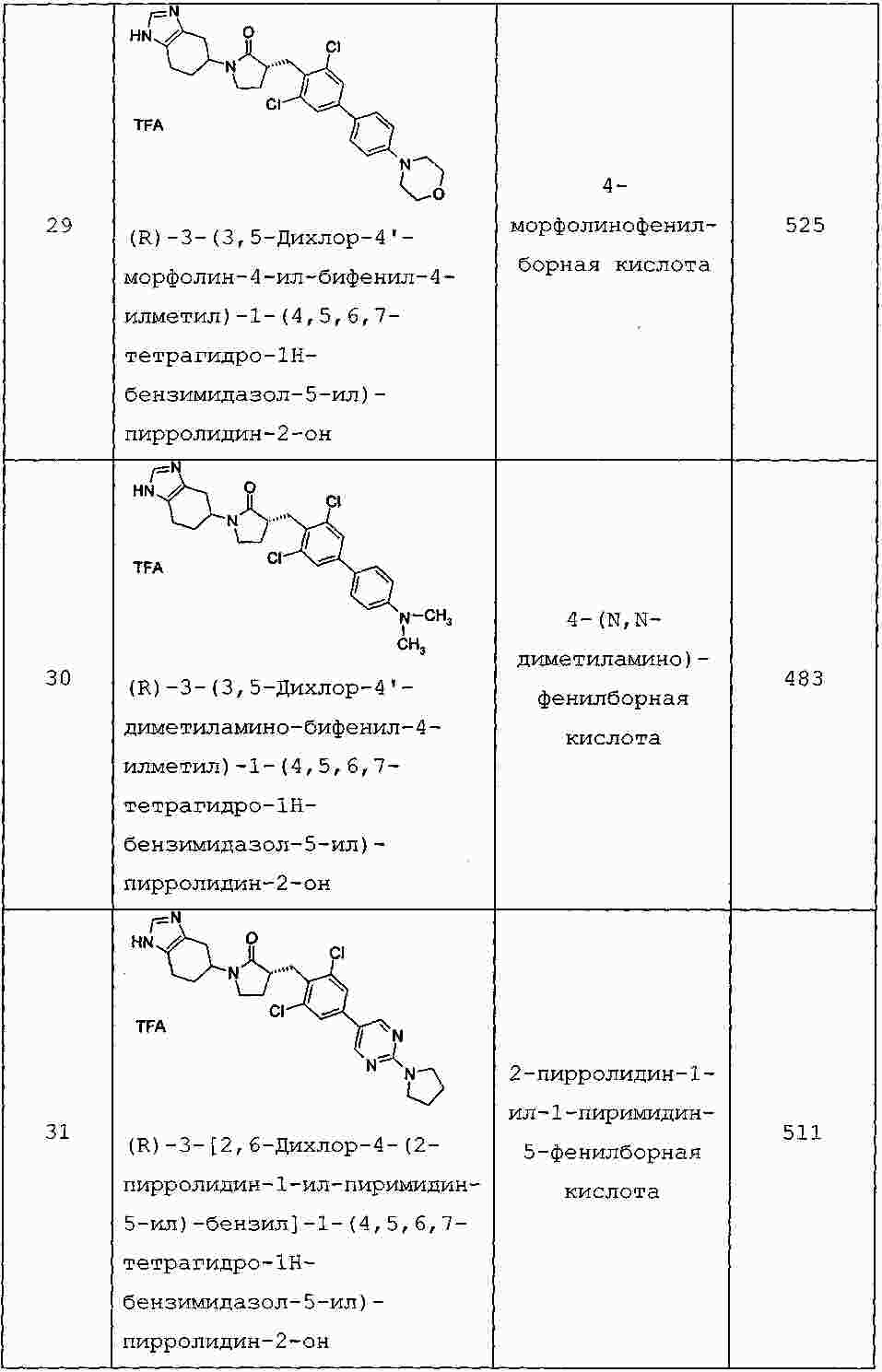
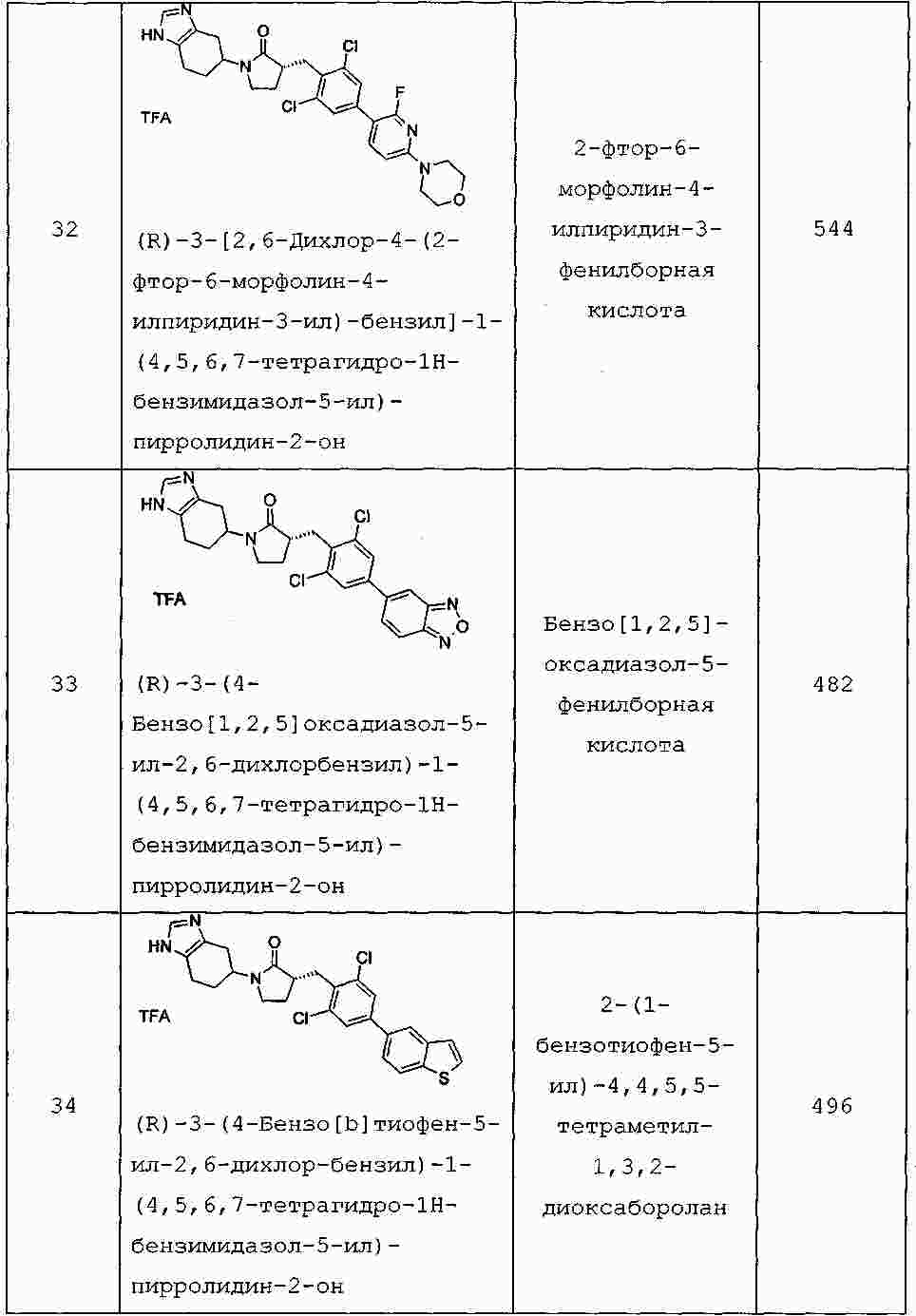
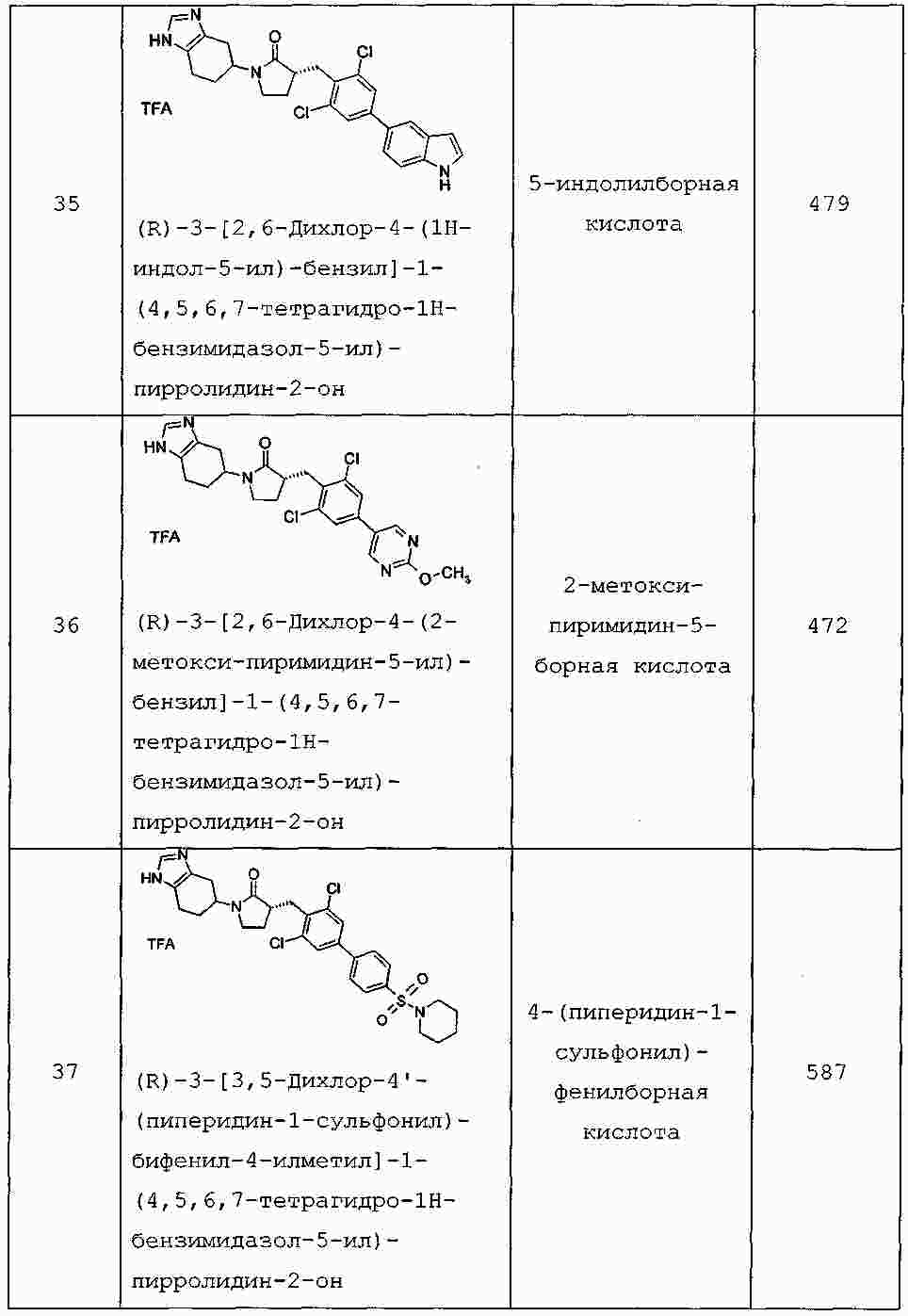
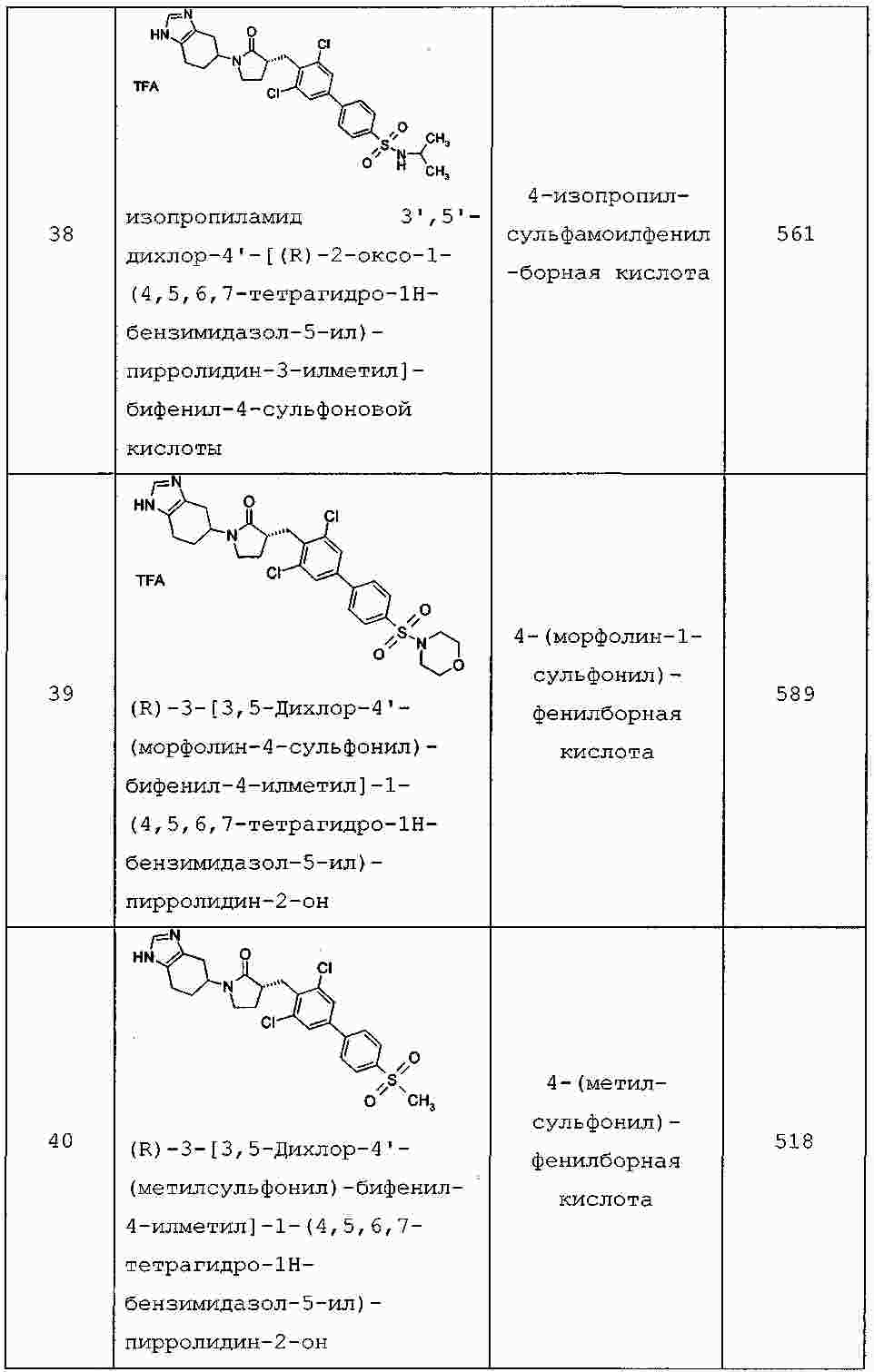
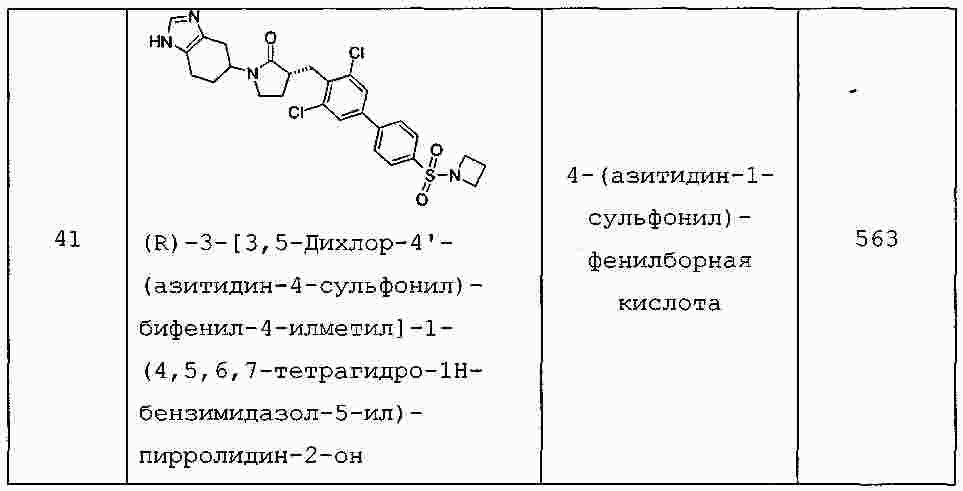
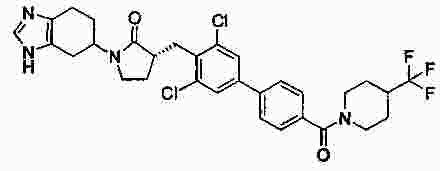
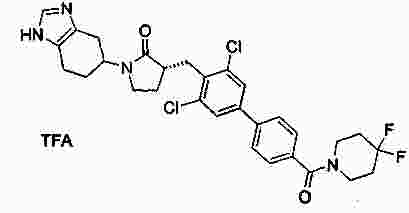
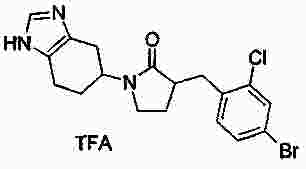
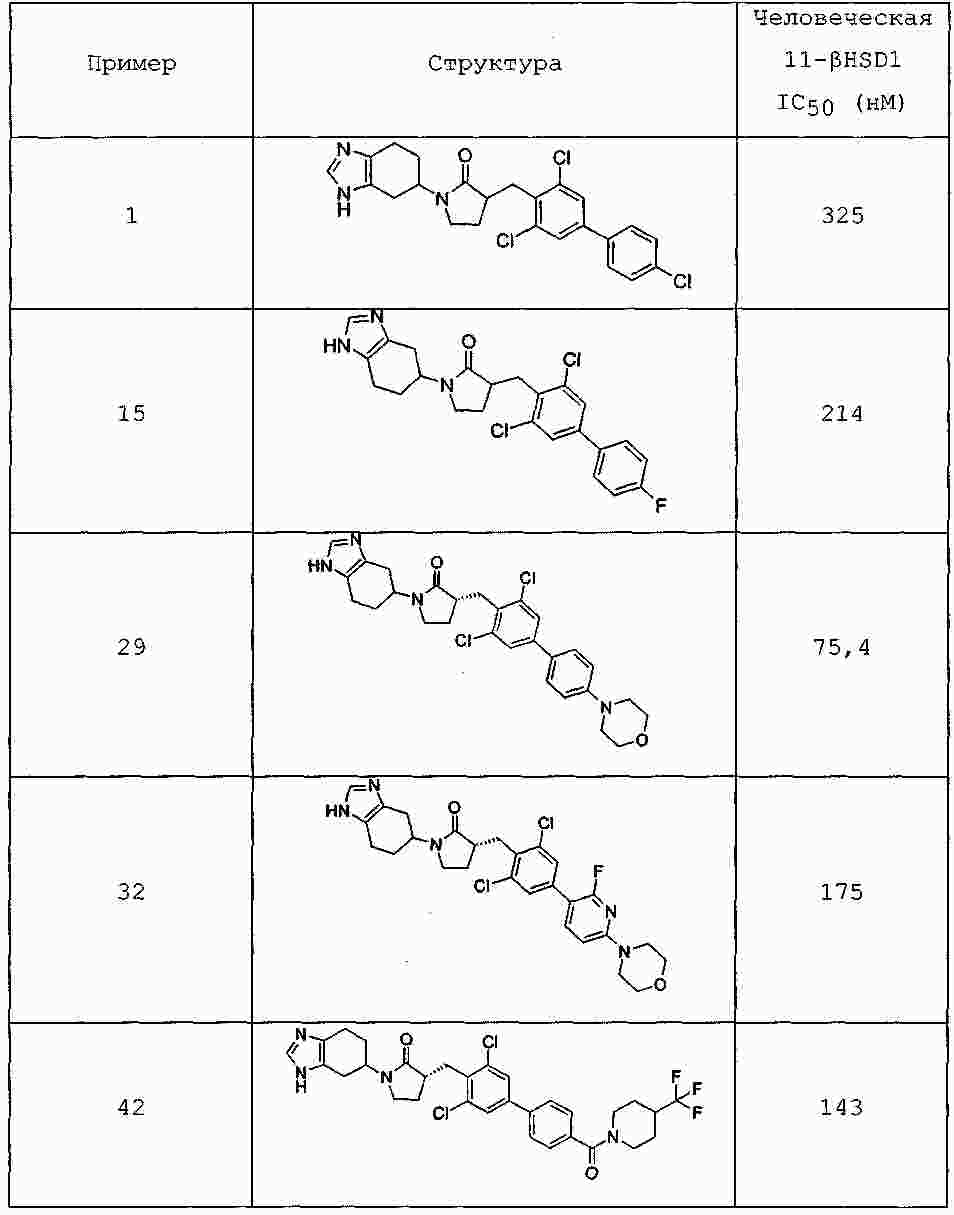
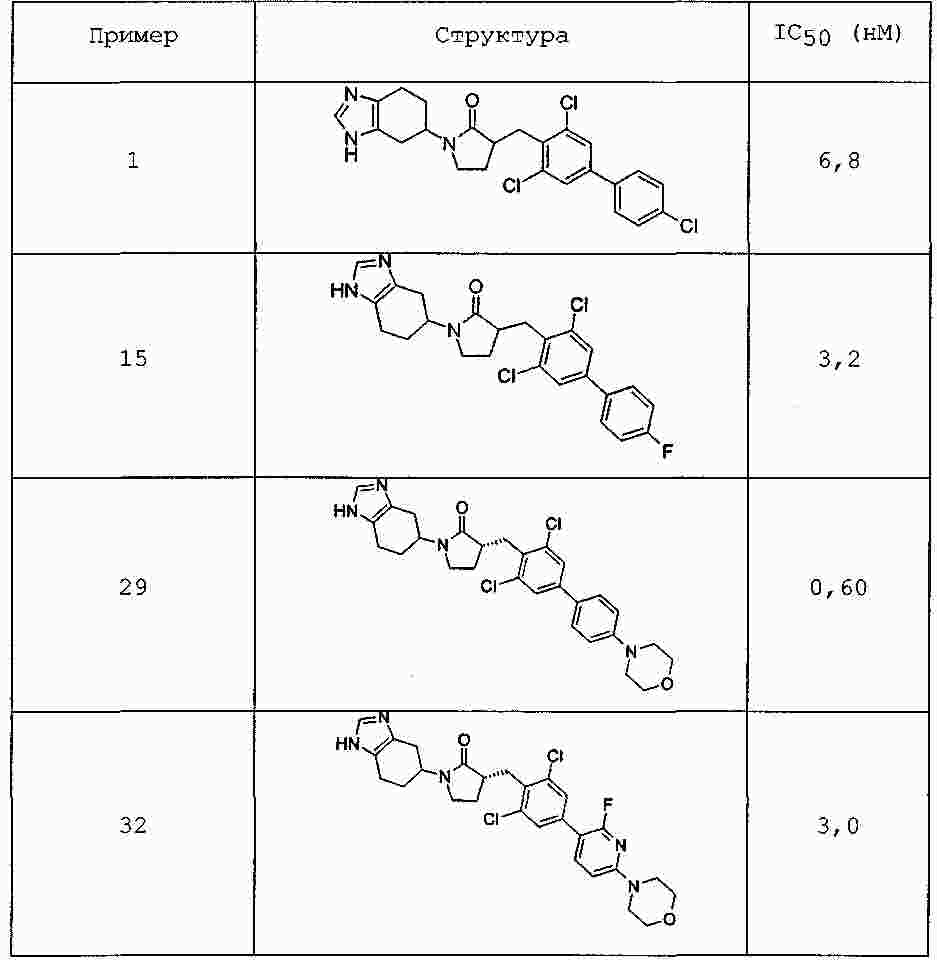
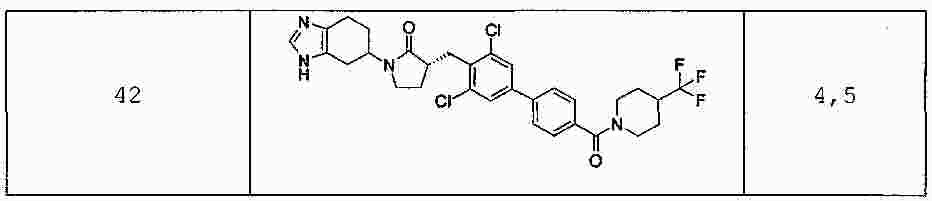
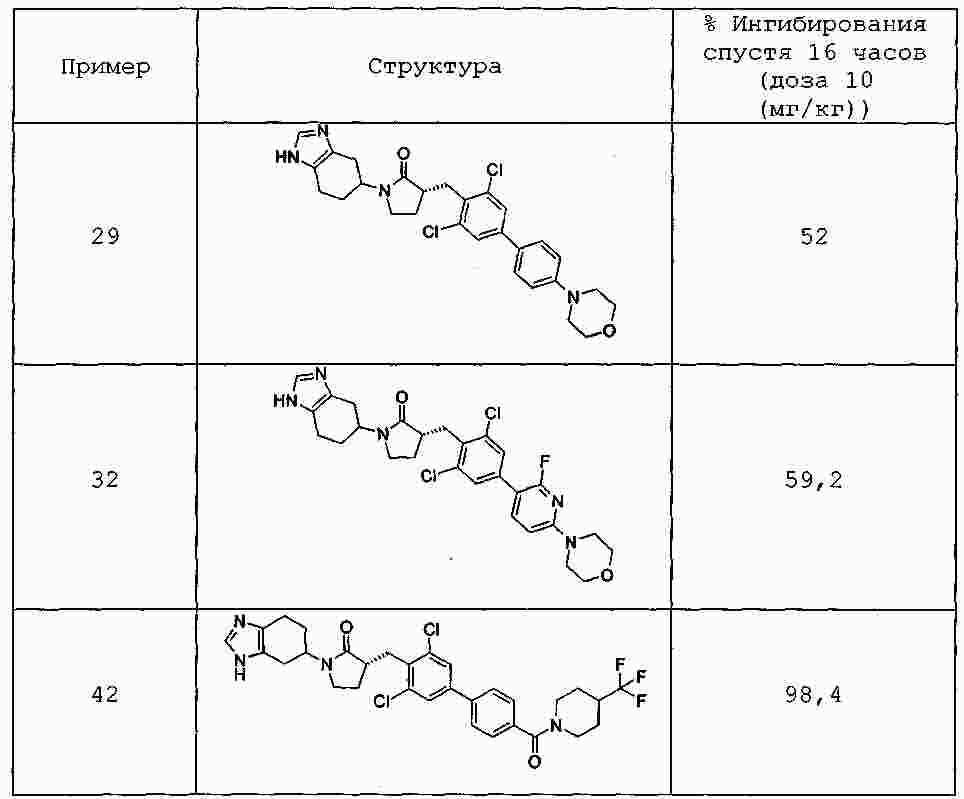
|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea000014717b*\id |
|
Термины запроса в документе Реферат Настоящее изобретение относится к новым соединениям, отвечающим формуле (I) обладающим антагонистической активностью по отношению к 11- β-HSD первого типа, а также к способу приготовления таких соединений. В другом примере реализации настоящее изобретение относится к фармацевтическим композициям, включающим соединения, отвечающие формуле (I), а также к способам применения соединений и композиций для лечения диабета, гипергликемии, ожирения, гипертонии, гиперлипидемии, синдрома нарушения обмена веществ и других состояний, связанных с активностью 11- β-HSD первого типа. Формула [0001] Соединение, структура которого представлена формулой [0002] Соединение по п.1, структура которого представлена формулой [0003] Соединение по п.1 или 2, в котором R1 и R2 представляют собой хлор, или фармацевтически приемлемая соль указанного соединения. [0004] Соединение по любому из пп.1-3, в котором R3 представляет собой водород, или фармацевтически приемлемая соль указанного соединения. [0005] Соединение по любому из пп.2-4, в котором R0 представляет собой или фармацевтически приемлемая соль указанного соединения. [0006] Соединение по любому из пп.2-4, в котором R0 представляет собой или фармацевтически приемлемая соль указанного соединения. [0007] Соединение по любому из пп.1-6, в котором R4 представляет собой и R7 представляет собой водород, или фармацевтически приемлемая соль указанного соединения. [0008] Соединение по любому из пп.1-6, в котором R4 представляет собой и R6 представляет собой водород, или фармацевтически приемлемая соль указанного соединения. [0009] Соединение по любому из пп.1-8, в котором R5 представляет собой и R8 представляет собой -(C1-С3)алкил (необязательно, имеющий в качестве заместителей от 1 до 3 атомов галогена), или фармацевтически приемлемая соль указанного соединения. [0010] Соединение по любому из пп.1-8, в котором R5 представляет собой хлор или фтор, или фармацевтически приемлемая соль указанного соединения. [0011] Соединение по любому из пп.1-8, в котором R5 представляет собой фтор, или фармацевтически приемлемая соль указанного соединения. [0012] Соединение по п.1, которое представляет собой (R)-3-(3,5-дихлор-4'-морфолин-4-илбифенил-4-илметил)-1-(4,5,6,7-тетрагидро-1Н-бензоимидазол-5-ил)пирролидин-2-он, или фармацевтически приемлемая соль указанного соединения. [0013] Соединение по п.1, которое представляет собой 3-(3,5-дихлор-4'-фторбифенил-4-илметил)-1-(4,5,6,7-тетрагидро-1Н-бензоимидазол-5-ил)пирролидин-2-он, или фармацевтически приемлемая соль указанного соединения. [0014] Соединение по п.1, которое представляет собой 3-[R]-[3,5-дихлор-4'-(4-трифторметилпиперидин-1-карбонил)бифенил-4-илметил]-1-(4,5,6,7-тетрагидро-3Н-бензоимидазол-5-ил)пирролидин-2-он, или фармацевтически приемлемая соль указанного соединения. [0015] Соединение по п.1, выбранное из группы, включающей [0016] Фармацевтическая композиция, включающая соединение по любому из пп.1-15 или фармацевтически приемлемую соль указанного соединения и фармацевтически приемлемый носитель. [0017] Применение соединения по любому из пп.1-15 или фармацевтически приемлемой соли указанного соединения для приготовления лекарственного средства. [0018] Промежуточное соединение для приготовления соединения по любому из пп.12, 13 или 14, причем указанное промежуточное соединение представляет собой Полный текст патента По заявке на данный патент испрашивается приоритет согласно предварительной заявке на патент США № 60/745321 от 21 апреля 2006 г. Настоящее изобретение относится к соединениям, представляющим собой ингибиторы 11- β-гидроксистероид дегидрогеназы 1 типа (11- β-HSD1), к фармацевтическим композициям, содержащим такие соединения, и применению таких соединений и композиций для лечения организма человека или животного, а также к новым промежуточным соединениям, которые можно применять для получения ингибиторов. Настоящие соединения демонстрируют эффективное и селективное ингибирование 11- β-HSD1, и в этом качестве их можно применять при лечении чувствительных к модуляции 11- β-HSD1 нарушений, например диабета, метаболического синдрома, расстройства познавательной способности и т.п. Глюкокортикоиды, функционирующие в печени, жировой ткани и в мышцах, являются важными регуляторами метаболизма глюкозы, липидов и белков. Хронический избыток глюкокортикоидов связан с резистентностью к инсулину, висцеральным ожирением, гипертонией и дислипидемией, которые также представляют собой классические признаки метаболического синдрома. Соединение 11- β-HSD1 катализирует преобразование неактивного кортизона в активный кортизол и, как полагают, вовлечено в развитие метаболического синдрома. Данные, полученные для грызунов и человека, свидетельствуют о связи 11- β-HSD1 с метаболическим синдромом. Данные позволяют предположить, что лекарственный препарат, специфично ингибирующий 11- β-HSD1 у пациентов с диабетом 2 типа, снижает уровень глюкозы в крови, замедляя глюконеогенез в печени, снижает центральное ожирение, улучшает атерогенный липопротеиновый фенотип, уменьшает кровяное давление и снижает резистентность к инсулину. Действие инсулина в мышцах должно усиливаться, и секреция инсулина бета-клетками островков также может возрастать. Данные, полученные при исследовании животных и человека, также свидетельствуют о том, что избыток глюкокортикоидов нарушает познавательную функцию. Недавние результаты указывают на то, что инактивация 11- β-HSD1 усиливает функционирование памяти как у человека, так и у мышей. Было показано, что ингибитор 11- β-HSD карбеноксолон улучшает когнитивную функцию у здоровых пожилых людей и у людей, страдающих диабетом 2 типа, а инактивация гена 11- β-HSD1 предотвращает обусловленные старением повреждения у мышей. Недавно было показано, что селективное ингибирование 11- β-HSD1 фармацевтическим агентом улучшает сохранение информации в памяти у мышей. В последние годы появился ряд публикаций, сообщающих об агентах, ингибирующих 11- β-HSD1. См. международную публикацию заявки WO 2004/056744, описывающую адамантилацетамиды в качестве ингибиторов 11- β-HSD, международную публикацию WO 2005/108360, в которой в качестве ингибиторов 11- β-HSD описаны производные пирролидин-2-она и пиперидин-2-она, и международную публикацию WO 2005/108361, в которой в качестве ингибиторов 11- β-HSD описаны производные адамантилпирролидин-2-она. Несмотря на существование ряда способов лечения заболеваний, в которые вовлечена 11- β-HSD1, современные способы лечения имеют ряд недостатков, в том числе низкую или недостаточную эффективность, неприемлемые побочные эффекты и противопоказания для некоторых групп пациентов. Таким образом, сохраняется потребность в улучшении способов лечения с применением альтернативных или усовершенствованных фармацевтических агентов, которые ингибируют 11- β-HSD1 и, таким образом, могут оказаться полезными при лечении заболеваний, при которых ингибирование 11- β-HSD1 может оказывать благоприятный эффект. Настоящее изобретение обеспечивает такой вклад в данную область техники благодаря тому, что был обнаружен новый класс соединений, оказывающих сильное и селективное ингибирующее действие на 11- β-HSD1. Отличительной особенностью настоящего изобретения являются представленные конкретные структуры и их активность. Сохраняется потребность в новых способах лечения диабета, метаболического синдрома и расстройств познавательной способности, и задачей настоящего изобретения является удовлетворение данной и других потребностей. Настоящее изобретение относится к соединению, структура которого представлена формулой (I) или к фармацевтически приемлемой соли указанного соединения, где R 1 представляет собой -Н, -галоген, -О-СН 3 (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена) или -СН 3 (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 2 представляет собой -Н, -галоген, -О-СН 3 (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена) или -СН 3 (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 3 представляет собой -Н или -галоген; R 4 представляет собой -ОН, галоген, цианогруппу, -(С 1 -С 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -(C 1 -С 6 )алкокси (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -SCF 3 , -С(О)О(С 1 -С 4 )алкил, -(С 3 -C 8 )циклоалкил, -О-фенил-С(O)O-(C 1 -C 4 )алкил, -СН 2 -фенил, -NHSO 2 -(C 1 -C 4 )алкил, -NHSO 2 -фенил(R 21 )(R 21 ), -(C 1 -C 4 )алкил-С(О)N(R 10 )(R 11 ), в которых пунктирной линией обозначено положение присоединения R 4 в формуле (I); R 5 представляет собой -Н, -галоген, -(С 1 -С 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -С(О)ОН, С(O)O-(C 1 -C 4 )алкил, -С(О)-(C 1 -C 4 )алкил, -О-(C 1 -C 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -SO 2 -(C 1 -C 4 )алкил, -N(R 8 )(R 8 ), в которых пунктирной линией обозначено положение присоединения R 5 ; где m имеет значения 1, 2 или 3; n имеет значения 0, 1 или 2 и где если n равен 0, то "(СН 2 )n" представляет собой связь; R 6 представляет собой -Н, -галоген, -CN или -(С 1 -С 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 7 представляет собой -Н, -галоген или -(С 1 -С 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 8 в каждом случае независимо представляет собой -H, -(C 1 -С 6 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -С(О)-(C 1 -С 6 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -С(О)-(С 3 -C 8 )циклоалкил, -S(О 2 )-(С 3 -C 8 )циклоалкил или -S(О 2 )-(C 1 -С 3 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 9 представляет собой -Н или -галоген; каждый из R 10 и R 11 независимо представляет собой -Н или -(C 1 -C 4 )алкил, или R 10 и R 11 вместе с атомом азота, к которому они присоединены, образуют пиперидинил, пиперазинил или пирролидинил; R 20 в каждом случае независимо представляет собой -Н или -(C 1 -С 3 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 21 в каждом случае независимо представляет собой -Н, -галоген или -(C 1 -С 3 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 22 в каждом случае независимо представляет собой -Н или -(С 1 -С 6 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 23 в каждом случае независимо представляет собой -Н, -(C 1 -С 4 )алкил или -С(О)О-(С 1 -С 4 )алкил. Настоящее изобретение относится к соединениям, отвечающим формуле (I), которые могут быть использованы в качестве мощных и селективных ингибиторов 11- β-HSD1. Кроме того, настоящее изобретение относится к фармацевтической композиции, которая включает соединение, отвечающее формуле (I), или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или наполнитель. Кроме того, настоящее изобретение относится к способу лечения синдрома нарушения обмена веществ и связанных с ним нарушений, который включает введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения, отвечающего формуле (I), или фармацевтически приемлемой соли указанного соединения. Согласно одному из примеров реализации настоящее изобретение относится к соединениям, отвечающим формуле (I), или фармацевтически приемлемым солям указанных соединений, как подробно описано выше. Несмотря на то что для лечения пригодны все соединения, предлагаемые согласно настоящему изобретению, некоторые из этих соединений оказались особенно интересными и поэтому являются предпочтительными. Ниже перечислены некоторые группы предпочтительных соединений. Другой пример реализации изобретения относится к соединению, структура которого представлена формулой (Ia) или фармацевтически приемлемой соли указанного соединения, где R 0 представляет собой R 1 представляет собой -галоген; R 2 представляет собой -галоген; R 3 представляет собой -Н или -галоген; R 4 представляет собой в которых пунктирной линией обозначено положение присоединения R 4 в формуле (Ia); R 5 представляет собой -Н, -галоген, -(С 1 -С 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -С(О)ОН, -С(О)О-(C 1 -C 4 )алкил, -С(О)-(C 1 -C 4 )алкил, -О-(С 1 -С 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -SO 2 -(C 1 -C 4 )алкил, -N(R 8 )(R 8 ), в которых пунктирной линией обозначено положение присоединения R 5 ; где m имеет значения 1, 2 или 3; n имеет значения 0, 1 или 2 и где если n имеет значение 0, то "(СН 2 )n" представляет собой связь; R 6 представляет собой -Н, -галоген, -CN или -(С 1 -С 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 7 представляет собой -Н, -галоген или -(C 1 -C 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 8 в каждом случае независимо представляет собой -Н, -(C 1 -С 6 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -C(О)-(C 1 -С 6 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -С(О)-(C 3 -C 8 )циклоалкил, -S(O 2 )-(С 3 -С 8 )циклоалкил или -S(О 2 )-(C 1 -С 3 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 9 представляет собой -Н или -галоген; R 20 в каждом случае независимо представляет собой -H или -(C 1 -С 3 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 21 в каждом случае независимо представляет собой -Н, -галоген или -(C 1 -С 3 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 22 в каждом случае независимо представляет собой -Н или -(C 1 -С 6 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 23 в каждом случае независимо представляет собой -Н, -(C 1 -С 4 )алкил или -С(О)О-(С 1 -С 4 )алкил. Другой пример реализации изобретения относится к соединению, структура которого представлена формулой (Ia) или фармацевтически приемлемой соли указанного соединения, где R 0 представляет собой R 1 представляет собой -хлор, -фтор или -бром; R 2 представляет собой -хлор, -фтор или -бром; R 3 представляет собой -Н или -галоген; R 4 представляет собой в которых пунктирной линией обозначено положение присоединения R 4 в формуле (Ia); R 5 представляет собой -Н, -галоген, -(C 1 -C 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -С(О)ОН, -С(О)О-(C 1 -C 4 )алкил, -С(О)-(C 1 -C 4 )алкил, -О-(C 1 -С 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -SO 2 -(C 1 -C 4 )алкил, -N(R 8 )(R 8 ), в которых пунктирной линией обозначено положение присоединения R 5 ; где m имеет значения 1, 2 или 3; R 6 представляет собой -H, -галоген, -CN или -(C 1 -C 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 7 представляет собой -Н, -галоген или -(C 1 -C 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 8 в каждом случае независимо представляет собой -Н, -(C 1 -С 6 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -C(О)-(C 1 -C 6 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -С(О)-(С 3 -C 8 )циклоалкил, -S(О 2 )-(С 3 -C 8 )циклоалкил или -S(О 2 )-(C 1 -С 3 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 9 представляет собой -Н или -галоген; R 20 в каждом случае независимо представляет собой -Н или -(C 1 -С 3 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 21 в каждом случае независимо представляет собой -Н, -галоген или -(C 1 -С 3 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 22 в каждом случае независимо представляет собой -Н или -(C 1 -С 6 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 23 в каждом случае независимо представляет собой -Н, -(C 1 -С 4 )алкил или -С(О)О-(С 1 -С 4 )алкил. Другой пример реализации изобретения относится к соединению, структура которого представлена формулой (Ia) или фармацевтически приемлемой соли указанного соединения, где R 0 представляет собой R 1 представляет собой -хлор, -фтор или -бром; R 2 представляет собой -хлор, -фтор или -бром; R 3 представляет собой -Н или -галоген; R 4 представляет собой R 5 представляет собой -Н, -галоген, -(С 1 -С 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -С(О)ОН, -С(O)O-(С 1 -С 4 )алкил, -С(О)-(C 1 -C 4 )алкил, -О-(С 1 -С 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -SO 2 -(C 1 -C 4 )алкил, -N(R 8 )(R 8 ), в которых пунктирной линией обозначено положение присоединения R 5 ; где m имеет значения 1, 2 или 3; R 6 представляет собой -Н, -галоген, -CN или -(С 1 -С 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 7 представляет собой -Н, -галоген или -(С 1 -С 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 8 в каждом случае независимо представляет собой -Н, -(C 1 -С 6 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -С(О)-(C 1 -С 6 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -С(О)-(С 3 -С 8 )циклоалкил, -S(O 2 )-(С 3 -С 8 )циклоалкил или -S(O 2 )-(C 1 -С 3 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 9 представляет собой -Н или -галоген; R 20 в каждом случае независимо представляет собой -H или -(C 1 -С 3 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 21 в каждом случае независимо представляет собой -Н, -галоген или -(C 1 -С 3 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 22 в каждом случае независимо представляет собой -Н или -(C 1 -С 6 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 23 в каждом случае независимо представляет собой -Н, -(C 1 -С 4 )алкил или -С(О)О-(C 1 -C 4 )алкил. Другой пример реализации изобретения относится к соединению, структура которого представлена формулой (Ia) или фармацевтически приемлемой соли указанного соединения, где R 0 представляет собой R 1 представляет собой -хлор, -фтор или -бром; R 2 представляет собой -хлор, -фтор или -бром; R 3 представляет собой -Н или -галоген; R 4 представляет собой R 5 представляет собой в которых пунктирной линией обозначено положение присоединения R 5 ; R 6 представляет собой -Н, -галоген, -CN или -(С 1 -С 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 7 представляет собой -Н, -галоген или -(C 1 -C 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 8 в каждом случае независимо представляет собой -Н, -(С 1 -С 6 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -С(О)-(C 1 -С 6 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -С(О)-(С 3 -С 8 )циклоалкил, -S(O 2 )-(С 3 -С 8 )циклоалкил или -S(О 2 )-(C 1 -С 3 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 9 представляет собой -Н или -галоген; R 20 в каждом случае независимо представляет собой -Н или -(C 1 -С 3 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 21 в каждом случае независимо представляет собой -Н, -галоген или -(C 1 -С 3 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 22 в каждом случае независимо представляет собой -Н или -(C 1 -С 6 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 23 в каждом случае независимо представляет собой -Н, -(C 1 -С 4 )алкил или -С(О)О-(C 1 -C 4 )алкил. Другой пример реализации изобретения относится к соединению, структура которого представлена формулой (Ia) или фармацевтически приемлемой соли указанного соединения, где R 0 представляет собой R 1 представляет собой -хлор, -фтор или -бром; R 2 представляет собой -хлор, -фтор, или -бром; R 3 представляет собой -Н или -галоген; R 4 представляет собой R 5 представляет собой где m имеет значения 1, 2 или 3; R 6 представляет собой -Н, -галоген, -CN или -(С 1 -С 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 7 представляет собой -H, -галоген или -(C 1 -C 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 8 в каждом случае независимо представляет собой -Н, -(C 1 -С 6 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -C(О)-(C 1 -С 6 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -С(О)-(С 3 -С 6 )циклоалкил, -S(О 2 )-(С 3 -C 8 )циклоалкил или -S(О 2 )-(C 1 -С 3 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена). Другой пример реализации изобретения относится к соединению, структура которого представлена формулой (Ia) или фармацевтически приемлемой соли указанного соединения, где R 0 представляет собой R 1 представляет собой -хлор, -фтор или -бром; R 2 представляет собой -хлор, -фтор или -бром; R 3 представляет собой -Н или -галоген; R 4 представляет собой R 5 представляет собой R 6 представляет собой -Н, -галоген, -CN, или -(C 1 -C 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 7 представляет собой -Н, -галоген, или -(С 1 -С 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена); R 8 в каждом случае независимо представляет собой -Н, -(C 1 -С 6 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -C(О)-(C 1 -С 6 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена), -С(О)-(C 3 -C 8 )циклоалкил, -S(O 2 )-(С 3 -С 8 )циклоалкил или -S(О 2 )-(C 1 -С 3 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена). Существуют и другие примеры реализации настоящего изобретения, представляющие собой частные случаи описанных выше примеров реализации, ограниченные перечисленными ниже предпочтительными аспектами. В частности, каждый из предпочтительных аспектов, описанных ниже, может быть использован в сочетании с каждым из вышеперечисленных примеров реализации, и полученная конкретная комбинация будет представлять собой другой пример реализации изобретения, в котором предпочтительный изменяемый параметр имеет более узкое значение по сравнению с исходным предпочтительным параметром. Предпочтительно примеры реализации настоящего изобретения могут быть представлены структурой формулы в которой R 0 представляет собой Предпочтительно R 0 представляет собой Предпочтительно R 0 представляет собой Предпочтительно R 1 представляет собой -галоген. Предпочтительно R 1 представляет собой -СН 3 . Предпочтительно R 1 представляет собой -хлор, -фтор или -бром. Предпочтительно R 1 представляет собой -хлор. Предпочтительно R 1 представляет собой -фтор. Предпочтительно R 1 представляет собой -бром. Предпочтительно R 2 представляет собой -галоген. Предпочтительно R 2 представляет собой -СН 3 . Предпочтительно R 2 представляет собой -хлор, -фтор или -бром. Предпочтительно R 2 представляет собой -хлор. Предпочтительно R 2 представляет собой -фтор. Предпочтительно R 2 представляет собой -бром. Предпочтительно R 1 представляет собой -хлор и R 2 представляет собой -хлор. Предпочтительно R 3 представляет собой -Н. Предпочтительно R 3 представляет собой -галоген. Предпочтительно R 4 представляет собой Предпочтительно R 5 представляет собой -N(R 8 )(R 8 ), Предпочтительно R 6 представляет собой -Н. Предпочтительно R 6 представляет собой -галоген. Предпочтительно R 6 представляет собой -(С 1 -С 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена). Предпочтительно R 7 представляет собой -Н. Предпочтительно R 7 представляет собой -галоген или -(С 1 -С 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена). Предпочтительно R 7 представляет собой -галоген. Предпочтительно R 7 представляет собой -(С 1 -С 4 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена). Предпочтительно R 8 в каждом случае независимо представляет собой -H. Предпочтительно R 8 в каждом случае независимо представляет собой -(C 1 -С 3 )алкил. Предпочтительно R 8 в каждом случае независимо представляет собой -СН 3 . Предпочтительно R 9 представляет собой -Н. Предпочтительно R 9 представляет собой -галоген. Предпочтительно R 7 представляет собой -фтор и R 9 представляет собой -фтор. Другой пример реализации изобретения относится к соединению, структура которого представлена формулой (Ia), или к фармацевтически приемлемой соли указанного соединения, где R 0 представляет собой R 1 представляет собой -хлор; R 2 представляет собой -хлор; R 3 представляет собой -Н; R 4 представляет собой в которых пунктирной линией обозначено положение присоединения R 4 в формуле (Ia); R 5 представляет собой -Н, -хлор, -фтор, -СН 3 , -CF 3 , -С(СН 3 ) 3 , -СН(СН 3 ) 2 , -O-С(СН 3 ) 2 , -С(О)О-СН 3 , -N(-СН 3 )(-СН 3 ), в которых пунктирной линией обозначено положение присоединения R 5 ; где m равен 1, 2 или 3; R 6 представляет собой -H, -хлор, -фтор, -бром, -CH 3 , CF 3 ; R 7 представляет собой -Н, -хлор, -фтор, -бром; R 8 в каждом случае независимо представляет собой -Н или -СН 3 , -СН 2 -СН 3 , -С(СН 3 ) 3 , -СН(СН 3 ) 2 ; R 9 представляет собой -Н или -хлор, -фтор, -бром; R 20 в каждом случае независимо представляет собой -Н, -СН 3 ; R 22 в каждом случае независимо представляет собой -H. Предпочтительные примеры реализации настоящего изобретения представляют собой соединения, отвечающие следующим формулам: (R)-3-(3,5-дихлор-4'-морфолин-4-илбифенил-4-илметил)-1-(4,5,6,7-тетрагидро-1Н-бензимидазол-5-ил)пирролидин-2-он, (R)-3-[2,6-дихлор-4-(2-фтор-6-морфолин-4-илпиридин-3-ил)бензил]-1-(4,5,6,7-тетрагидро-1Н-бензимидазол-5-ил)пирролидин-2-он и 3-(R)-[3,5-дихлор-4'-(4-трифторметилпиперидин-1-карбонил)бифенил-4-илметил]-1-(4,5,6,7-тетрагидро-3Н-бензимидазол-5-ил)пирролидин-2-он или 3-(3,5-дихлор-4'-фторбифенил-4-илметил)-1-(4,5,6,7-тетрагидро-1H-бензимидазол-5-ил)пирролидин-2-он; или стереоизомеры указанных соединений или фармацевтически приемлемые соли указанных соединений. Еще один пример реализации настоящего изобретения относится к описанному в настоящем патенте способу приготовления новых промежуточных соединений, которые могут быть полезны для приготовления ингибиторов 11- β-HSD1, представленных формулой (I), и примеров реализации, рассмотренных в настоящем описании. У пациентов, страдающих диабетом 2 типа, часто развивается "инсулинорезистентность", которая приводит к развитию аномального гомеостаза глюкозы и гипергликемии, которые, в свою очередь, приводят к повышенной заболеваемости и ранней смертности. Аномальный гомеостаз глюкозы связан с ожирением, повышенным кровяным давлением и изменениями в липидном, липопротеиновом и аполипопротеиновом метаболизме. У пациентов, страдающих диабетом 2 типа, весьма высока вероятность развития нарушений сердечно-сосудистой системы, например атеросклероза, коронарной болезни сердца, инсульта, заболеваний периферических сосудов, гипертонии, нефропатии, ретинопатии и патологий нервной системы. Таким образом, терапевтический контроль гомеостаза глюкозы, липидного метаболизма, ожирения и гипертонии важен при регулировании и лечении сахарного диабета. У многих пациентов с инсулинорезистентностью, но у которых тем не менее не развивается диабет 2 типа, также высока вероятность развития "синдрома X" или "метаболического синдрома". Синдром нарушения обмена веществ характеризуется инсулинорезистентностью, сопровождаемой внутрибрюшным ожирением, гиперинсулинемией, высоким кровяным давлением, низкими уровнями HDL, высокими уровнями VLDL, гипертонией, атеросклерозом, коронарной болезнью сердца и хронической почечной недостаточностью. У этих пациентов высок риск развития перечисленных выше сердечно-сосудистых нарушений либо у них может развиться сахарный клинический диабет. Поскольку предлагаемые соединения способны ингибировать 11- β-HSD1, их можно применять для лечения разнообразных состояний и нарушений, состояние при которых может быть улучшено за счет ингибирования 11- β-HSD1. В настоящем описании такие нарушения и состояния называются "диабетическими нарушениями" и "нарушениями, вызванными метаболическим синдромом". Специалист в данной области техники способен идентифицировать "диабетические нарушения" и "нарушения, вызванные метаболическим синдромом" в соответствии с активностью 11- β-HSD1, проявляемой либо в патофизиологии нарушения, либо в гомеостатической реакции на нарушение. Таким образом, предлагаемые соединения могут быть использованы, например, для предотвращения, лечения или облегчения заболеваний или состояний либо симптомов или последствий, связанных с "диабетическими нарушениями" и "нарушениями, вызванными метаболическим синдромом". Неограничивающие примеры "диабетических нарушений" и "нарушений, вызванных метаболическим синдромом" включают диабет, диабет 1 типа, диабет 2 типа, гипергликемию, гиперинсулинемию, прекращение деятельности бета-клеток, усиленную функцию бета-клеток при восстановлении ответной реакции первой фазы, гипергликемию после приема пищи, предотвращение апоптоза, нарушенную гликемию натощак (IFG), синдром нарушения обмена веществ, гипогликемию, гипер/гипокалиемию, нормализацию уровней глюкагона, улучшение отношения LDL/HDL (липопротеинов низкой/высокой плотности), уменьшение потребления пищи между регулярными приемами пищи, нарушение потребления пищи, снижение массы тела, синдром поликистозных яичников (PCOS), ожирение как следствие заболевания диабетом, латентный аутоиммунный диабет взрослых (LADA), инсулит, пересадку островковой ткани, педиатрический диабет, диабет, обусловленный беременностью, отсроченные осложнения диабета, микро/макроальбуминурию, нефропатию, ретинопатию, нейропатию, изъязвление ступней при диабете, сниженную перистальтику кишечника из-за введения глюкагона, синдром укороченной тонкой кишки, антидиарейные эффекты, повышение желудочной секреции, снижение кровотока, эректильную дисфункцию, глаукому, послеоперационный стресс, уменьшение повреждения ткани органов, вызванного реперфузией тока крови вследствие ишемии, ишемическое повреждение сердца, сердечную недостаточность, застойную сердечную недостаточность, инсульт, инфаркт миокарда, аритмию, преждевременную смерть, антиапоптоз, заживление ран, нарушение толерантности к глюкозе (IGT), синдромы инсулинорезистентности, синдром нарушения обмена веществ, синдром X, гиперлипидемию, дислипидемию, гипертриглицеридимию, гиперлипопротеинемию, гиперхолестеринемию, артериосклероз, включая атеросклероз, глюкагоному, острый панкреатит, сердечно-сосудистые заболевания, гипертонию, гипертрофию сердца, желудочно-кишечные нарушения, ожирение, диабет как следствие ожирения, диабетическую дислипидемию и т.д. Таким образом, настоящее изобретение также относится к способу лечения "диабетических нарушений" и "нарушений, вызванных метаболическим синдромом", который также включает снижение или устранение одного или нескольких нежелательных побочных эффектов, имеющихся у существующих способов лечения. Кроме того, настоящее изобретение относится к соединению, отвечающему формуле (I), или к фармацевтически приемлемой соли указанного соединения, или фармацевтическому составу, который включает соединение, отвечающее формуле (I), или фармацевтически приемлемую соль указанного соединения и фармацевтически приемлемый носитель, разбавитель или наполнитель, применяемым для ингибирования активности 11- β-HSD1; применяемым для ингибирования клеточной ответной реакции, медиатором которой является активность 11- β-HSD1, у млекопитающего; применяемым для снижения гликемического уровня у млекопитающего; применяемым для лечения заболевания, вызываемого избыточной активностью 11- β-HSD1; применяемым для лечения диабетических и других нарушений, вызванных метаболическим синдромом у млекопитающего; и применяемым для лечения диабета, синдрома нарушения обмена веществ, ожирения, гипергликемии, атеросклероза, ишемической болезни сердца, инсульта, нейропатии и для заживления ран. Таким образом, способы, предлагаемые согласно настоящему изобретению, включают профилактическое и терапевтическое введение соединения, представленного формулой (I). Кроме того, настоящее изобретение относится к применению соединения, отвечающего формуле (I), или фармацевтически приемлемой соли указанного соединения для изготовления лекарственного средства, пригодного для ингибирования активности 11- β-HSD1; для изготовления лекарственного средства, пригодного для ингибирования клеточной ответной реакции, медиатором которой является активность 11- β-HSD1, у млекопитающего; для изготовления лекарственного средства, пригодного для снижения гликемического уровня у млекопитающего; для изготовления лекарственного средства, пригодного для лечения заболевания, вызываемого избыточной активностью 11- β-HSD1; для изготовления лекарственного средства, пригодного для лечения диабетических и других нарушений обмена веществ у млекопитающего; и для изготовления лекарственного средства, пригодного для предотвращения или лечения диабета, синдрома нарушения обмена веществ, ожирения, гипергликемии, атеросклероза, ишемической болезни сердца, инсульта, нейропатии и плохого заживления ран. Настоящее изобретение также относится к способу лечения состояний, вызываемых избыточной активностью 11- β-HSD1 у млекопитающего; к способу ингибирования активности 11- β-HSD1 у млекопитающего; к способу ингибирования клеточной ответной реакции, медиатором которой является активность 11- β-HSD1, у млекопитающего; к способу снижения гликемического уровня у млекопитающего; к способу лечения диабетических и других нарушений обмена веществ у млекопитающего; к способу предотвращения или лечения диабета, синдрома нарушения обмена веществ, ожирения, гипергликемии, атеросклероза, ишемической болезни сердца, инсульта, нейропатии и плохого заживления ран; причем указанные способы включают введение млекопитающему, нуждающемуся в таком лечении, такого количества соединения, отвечающего формуле (I), или его фармацевтически приемлемой соли, или фармацевтического состава, который включает соединение, отвечающее формуле (I), или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или наполнитель, которое пригодно для ингибирования активности 11- β-HSD1. Кроме того, настоящее изобретение относится к фармацевтическому составу, который содержит соединение, отвечающее формуле (I), или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или наполнитель, пригодному к применению, направленному на ингибирование активности 11- β-HSD1; к применению, направленному на ингибирование клеточных ответных реакций, медиатором которых является активность 11- β-HSD1; к применению, направленному на снижение гликемического уровня у млекопитающего; к применению, направленному на лечение диабетических и других нарушений, вызванных метаболическим синдромом у млекопитающего; и к применению, направленному на предотвращение или лечение диабета, синдрома нарушения обмена веществ, ожирения, гипергликемии, атеросклероза, ишемической болезни сердца, инсульта, нейропатии и плохого заживления ран. В соответствии с другим аспектом настоящего изобретения осуществляют введение предлагаемых соединений в сочетании с одним или несколькими другими активными веществами в любых подходящих соотношениях. Указанные другие активные вещества могут быть выбраны, например, из противодиабетических средств, средств, предназначенных для лечения ожирения, гипотензивных средств, средств, предназначенных для лечения осложнений, вызываемых или связанных с диабетом, и средств, предназначенных для лечения осложнений и нарушений, вызываемых или связанных с ожирением. Ниже представлен список нескольких групп таких сочетаний. Следует понимать, что каждое из указанных средств может быть использовано в сочетании с другими указанными средствами, что позволяет получать дополнительные сочетания. Так, в других примерах реализации настоящего изобретения предлагаемые соединения можно вводить в сочетании с одним или несколькими противодиабетическими средствами. Подходящие противодиабетические средства включают инсулин, аналоги и производные инсулина, например, описанные в патентной заявке ЕР 792290 (Novo Nordisk A/S), например N ε B29 -тетрадеканоил-дез-(В30)-человеческий инсулин, в патентных заявках ЕР 214826 и ЕР 705275 (Novo Nordisk A/S), например Asp B28 -человеческий инсулин, в патенте США 5504188 (Eli Lilly), например Lys B28 Pro B29 -человеческий инсулин, в патентной заявке ЕР 368187 (Aventis), например производные Lantus ®, GLP-1 и GLP-1, подобные описанным в международной патентной заявке WO 98/08871 (Novo Nordisk A/S), а также гипогликемические средства, активные при пероральном приеме. Гипогликемические средства, активные при пероральном введении, предпочтительно включают имидазолины, сульфонилмочевины, бигуанидины, меглитиниды, оксадиазолидиндионы, тиазолидиндионы, активаторы инсулина, средства, усиливающие секрецию инсулина, например глимепирид, ингибиторы α-глюкозидазы, средства, воздействующие на АТФ-зависимые калиевые каналы β-клеток, например вещества, открывающие калиевые каналы, например, описанные в заявках WO 97/26265, WO 99/03861 и WO 00/37474 (Novo Nordisk A/S), или митиглинид, или блокиратор калиевых каналов, например, BTS-67582, натеглинид, антагонисты глюкагона, например, описанные в заявках WO 99/01423 и WO 00/39088 (Novo Nordisk A/S и Agouron Pharmaceuticals, Inc.), антагонисты GLP-1, ингибиторы DPP-IV (дипептидил-пептидазы-IV), ингибиторы РТРазы (протеин-тирозин-фосфатазы), ингибиторы ферментов печени, участвующие в стимуляции глюконеогенезиса и/или глюкогенолиза, модуляторы абсобции глюкозы, активаторы глюкокиназы (GK), например, описанные в заявках WO 00/58293, WO 01/44216, WO 01/83465, WO 01/83478, WO 01/85706, WO 01/85707 и WO 02/08209 (Hoffman-La Roche) или описанные в заявках WO 03/00262, WO 03/00267 и WO 03/15774 (AstraZeneca), ингибиторы GSK-3 (гликоген-синтазы киназы-3), соединения, изменяющие липидный обмен, например противолипидемические средства, например, ингибиторы HMG СоА (статины), соединения, снижающие потребление пищи, лиганды PPAR (рецептор активатора пролиферации пероксисом), включающие подтипы PPAR-альфа, PPAR-гамма и PPAR-дельта, и агонисты RXR (ретиноидных X рецепторов), например, ALRT-268, LG-1268 или LG-1069. В другом примере реализации предлагаемые соединения вводят в сочетании с инсулином или аналогом или производным инсулина, например N ε B29 -тетрадеканоил-дез-(В30)-человеческим инсулином, Asp B28 -человеческим инсулином, Lys B28 Pro B29 -человеческим инсулином, Lantus ® или смешанным препаратом, включающим одно или несколько указанных соединений. В еще одном примере реализации настоящего изобретения предлагаемые соединения вводят в сочетании с сульфонилмочевиной, например глибенкламидом, глипизидом, толбутамидом, хлоропамидемом, толазамидом, глимепридом, гликазидом и глибуридом. В другом примере реализации настоящего изобретения предлагаемые соединения вводят в сочетании с бигуанидом, например метформином. И в еще одном примере реализации настоящего изобретения предлагаемые соединения вводят в сочетании с меглитинидом, например репаглинидом или натеглинидом. В еще одном примере реализации настоящего изобретения предлагаемые соединения вводят в сочетании с активатором инсулина на основе тиазолидиндиона, например с троглитазоном, циглитазоном, пиоглитазоном, розиглитазоном, изаглитазоном, дарглитазоном, энглитазоном, CS-011/CI-1037 или Т 174 или соединениями, описанными в заявках WO 97/41097, WO 97/41119, WO 97/41120, WO 00/41121 и WO 98/45292 (Dr. Reddy's Research Foundation). В еще одном примере реализации настоящего изобретения предлагаемые соединения могут быть введены в сочетании с активатором инсулина, таким как, например, GI 262570, YM-440, МСС-555, JTT-501, AR-H039242, KRP-297, GW-409544, CRE-16336, AR-H049020, LY510929, МВХ-102, CLX-0940, GW-501516, или соединениями, описанными в заявках WO 99/19313, WO 00/50414, WO 00/63191, WO 00/63192, WO 00/63193, например рагаглитазаром (NN 622 или (-)DRF 2725) (Dr. Reddy's Research Foundation) и WO 00/23425, WO 00/23415, WO 00/23451, WO 00/23445, WO 00/23417, WO 00/23416, WO 00/63153, WO 63196, WO 00/63209, WO 00/63190 и WO 00/63189 (Novo Nordisk A/S). В другом примере реализации настоящего изобретения предлагаемые соединения вводят в сочетании с ингибитором α-глюкозидазы, например воглибозом, эмиглитатом, миглитолом или акарбозом. В другом примере реализации настоящего изобретения предлагаемые соединения вводят в сочетании со средством, воздействующим на АТФ-зависимые калиевые канальцы β-клеток, например толбутамидом, глибенкламидом, глипизидом, гликазидом, BTS-67582 или репаглинидом. В еще одном примере реализации настоящего изобретения предлагаемые соединения могут быть введены в сочетании с натеглинидом. В еще одном примере реализации настоящего изобретения предлагаемые соединения вводят в сочетании с антилипидемическим средством или антигиперлипидемическим средством, например холестирамином, колестиполом, клофибратом, гемфиброзилом, ловастатином, правастатином, симвастатином, питавастатином, розувастатином, пробуколом, декстротироксином, фенофибратом или аторвастином. В еще одном примере реализации настоящего изобретения предлагаемые соединения вводят в сочетании с соединениями, понижающими потребление пищи. В другом примере реализации настоящего изобретения предлагаемые соединения вводят в сочетании с несколькими (более одного) из вышеуказанных соединений, например, в сочетании с метформином и сульфонилмочевиной, например глибуридом; сульфонилмочевиной и акарбозом; натеглинидом и метформином; репаглинидом и метформином; акарбозом и метформином; сульфонилмочевиной метформином и троглитазоном; инсулином и сульфонилмочевиной; инсулином и метформином; инсулином, метформином и сульфонилмочевиной; инсулином и троглитазоном; инсулином и ловастатином. Общие термины, используемые для описания рассматриваемых соединений, имеют обычно приписываемое им значение. В соответствии с настоящим описанием термины ''(С 1 -С 3 )алкил", "(С 1 -С 4 )алкил" или "(С 1 -С 6 )алкил" относятся к неразветвленным или разветвленным насыщенным алифатическим группам, содержащим указанное количество атомов углерода, например к метилу, этилу, н-пропилу, изопропилу, н-бутилу, изобутилу, втор-бутилу, трет-бутилу и подобным им радикалам. Термин "(С 1 -С 6 )алкокси" представляет собой С 1 -С 6 -алкильную группу, присоединенную через атом кислорода и включает такие фрагменты, как, например, метоксигруппа, этоксигруппа, н-пропоксигруппа, изопропоксигруппа и подобные им группы. Термин "галоген" означает фтор, хлор, бром и йод. Термин "(С 3 -С 8 )циклоалкил" означает насыщенное или частично насыщенное карбоциклическое кольцо, включающее от 3 до 8 атомов углерода, обычно от 3 до 7 атомов углерода. Неограничивающие примеры (С 3 -С 8 )циклоалкильных радикалов включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и подобные им радикалы. В настоящем описании термин "необязательно имеющий заместители" или "необязательные заместители" означает, что рассматриваемые группы либо не имеют замещений, либо имеют замещения одним или несколькими указанными заместителями. Если рассматриваемые группы имеют замещения несколькими (более одного) заместителями, то заместители могут быть как одинаковыми, так и различными. Кроме того, использование терминов "независимо", "независимо являются" и "независимо выбраны из" означает, что рассматриваемые группы могут быть как одинаковыми, так и различными. Некоторые из определенных выше терминов могут появляться в структурных формулах более одного раза, и при таком появлении каждый термин должен быть определен независимо от других. Следует понимать, что примеры пациентов, попадающих под обозначение "пациент", включают морских свинок, собак, кошек, крыс, мышей, хомяков и приматов, включая человека. Предпочтительно пациенты включают людей. Термин "пациент" включает сельскохозяйственных животных. Сельскохозяйственные животные представляют собой животных, выращиваемых для получения пищевых продуктов. Примерами сельскохозяйственных животных являются жвачные или "жвачные животные", например коровы, быки, телки, бычки, овцы, буйволы, бизоны, козы и антилопы. Другие примеры сельскохозяйственных животных включают свиней и птицу (домашнюю птицу), например кур, уток, индюшек и гусей. Предпочтительно пациент, подвергаемый лечению, представляет собой млекопитающее, в частности человека. В настоящем описании термины "лечение", "терапия" и "лечить" включают их общепринятое значение, т.е. действия, направленные на предотвращение, снижение риска возникновения или развития данного состояния или заболевания, предотвращение, ограничение, облегчение течения, улучшение состояния, замедление развития, остановку развития, отсрочку начала возникновения или обращение вспять прогрессирования или снижение тяжести заболевания или состояния у пациента, а также удержание на существующем уровне и/или лечение существующих характеристик заболевания, нарушения или патологического состояния, рассмотренного в настоящем описании, включая смягчение или облегчение симптомов или осложнений, или вылечивание или устранение заболевания, нарушения или состояния. В зависимости от необходимости, предлагаемые способы включают медикаментозное лечение и/или профилактическую терапию. В соответствии с настоящим описанием термин "терапевтически эффективное количество" означает количество соединения, предлагаемого согласно настоящему изобретению, способное облегчить симптомы различных рассмотренных в настоящем описании патологических состояний. Разумеется, конкретная доза соединения, вводимая в соответствии с настоящим изобретением, должна быть определена в зависимости от конкретных обстоятельств, влияющих на пациента, включающих, например, тип вводимого соединения, способ введения, состояние здоровья пациента и тип патологического состояния, подвергаемого лечению. Термин "композиция" означает фармацевтический состав и предназначен для определения фармацевтического продукта, включающего активный ингредиент (ингредиенты), включающий соединение (соединения), отвечающее формуле (I), и инертный ингредиент (ингредиенты), составляющие носитель. Соответственно фармацевтические составы, предлагаемые согласно настоящему изобретению, включают любые составы, приготовленные смешиванием соединения, предлагаемого согласно настоящему изобретению, и фармацевтически приемлемого носителя. Термин "по существу чистый" относится к чистой кристаллической форме соединения, включающей более приблизительно 90% нужной кристаллической формы, и предпочтительно более приблизительно 95% нужной кристаллической формы. Термин "подходящий растворитель" относится к любому растворителю или смеси растворителей, инертному по отношению к происходящей реакции, который в достаточной степени растворяет реагенты, создавая среду, в которой протекает нужная реакция. Термин "стандартная лекарственная форма" означает физически дискретные единицы, пригодные в качестве единичных дозировок, вводимых людям и другим животным, отличным от человека, и при этом каждая единица содержит заранее определенное количество активного материала, которое по расчетам должно вызвать желаемый терапевтический эффект, соединенное с подходящим фармацевтически приемлемым носителем. Соединения, предлагаемые согласно настоящему изобретению, могут иметь один или несколько хиральных центров и могут существовать в виде разнообразных стереоизомерных конфигураций. Вследствие существования указанных хиральных центров соединения, предлагаемые согласно настоящему изобретению, могут существовать в виде рацематов, в виде индивидуальных энантиомеров или смесей энантиомеров, а также в виде диастереомеров и смесей диастереомеров. Все указанные рацематы, энантиомеры, диастереомеры и их смеси как разделенные, так и частично разделенные или неразделенные смеси включены в область, защищаемую настоящим изобретением. Для примеров, представленных в настоящем описании, если имеется молекула, которая содержит хиральный центр или центры известной конфигурации, то ее стереохимию указывают в названии и в структурном представлении молекулы. Если стереохимия молекулы неизвестна или не определена, то ее стереохимию не указывают в названии и в структурном представлении молекулы. Примеры реализации настоящего изобретения включают рассмотренные ниже примеры, и, несмотря на то, что рассматриваемый пример может отвечать хиральной или конформационной форме или представлять собой соль этой формы, другие примеры реализации настоящего изобретения включают все другие стереоизомерные или конформационные формы описываемых примеров, а также из фармацевтически приемлемые соли. Указанные примеры реализации включают любые выделенные энантиомеры, диастереомеры и/или конформеры рассматриваемых структур, а также любые смеси, содержащие более одной формы. Кроме того, диастереомеры могут образовываться, если в молекуле присутствует двойная связь или полностью или частично насыщенная циклическая система, или более одного центра асимметрии, или связь с ограниченным вращением вокруг нее. Следует понимать, что любые диастереомеры, в виде разделенных, чистых или частично разделенных диастереомеров или их смесей, также включены в область, защищаемую настоящим изобретением. Кроме того, некоторые из соединений, предлагаемых согласно настоящему изобретению, могут существовать в виде различных таутомерных форм, и в этом случае следует понимать, что любые таутомерные формы, которые могут образовывать указанные соединения, также включены в область, защищаемую настоящим изобретением. В настоящем описании термин "обогащение энантиомером" означает повышение доли одного энантиомера по сравнению с другим. Имеющийся удобный способ выражения достигаемого обогащения энантиомером представляет собой концепцию энантиомерного избытка или "ее", в которой, как было показано, используют следующее уравнение: где Е 1 представляет собой количество первого энантиомера, а Е 2 представляет собой количество второго энантиомера. Таким образом, если исходное отношение двух энантиомеров равно 50:50, т.е. равно отношению энантиомеров в рацемической смеси, и при этом может быть достигнуто энантиомерное обогащение, достаточное для получения конечного отношения, равного 70:30, то значение ее по отношению к первому энантиомеру будет составлять 40%. В то же время если конечное отношение будет равно 90:10, то значение ее по отношению к первому энантиомеру будет составлять 80%. Предпочтительными являются значения ее, превышающие 90%, еще более предпочтительными являются значения ее, превышающие 95%, и наиболее предпочтительными являются значения ее, превышающие 99%. Специалист в данной области техники может легко определить значение энантиомерного обогащения при помощи стандартных методик и процедур, например, посредством газовой хроматографии или жидкостной хроматографии высокого разрешения с использованием хиральной колонки. Принцип выбора соответствующей хиральной колонки, элюента и условий, необходимых для разделения энантиомерной пары, хорошо известен специалистам в данной области техники. Кроме того, конкретные стереоизомеры и энантиомеры соединений, отвечающих формуле (I), могут быть приготовлены специалистом в данной области техники при помощи хорошо известных методик и способов, например, подобных описанным в публикации J. Jacques et al., "Enantiomers, Racemates, and Resolutions", John Wiley and Sons. Inc., 1981, и E.L. Eliel, S.H. Wilen," Stereochemistry of Organic Compounds'', (Wiley-Interscience 1994), и в Европейской патентной заявке № EP-A-838448 от 29 апреля 1998 г. Примеры способов разделения включают способы перекристаллизации или хиральной хроматографии. Соединения, отвечающие формуле (I), могут быть приготовлены специалистом в данной области техники при помощи различных методик, некоторые из которых указаны в методиках и схемах, рассмотренных ниже. Точный порядок операций, которые требуются для приготовления соединений, отвечающих формуле (I), зависит от конкретного синтезируемого соединения, исходного соединения и относительной подвижности замещающих фрагментов. Необходимые реактивы или исходные материалы обычно доступны специалистам в данной области техники, а материалы, не имеющие широкого распространения на рынке, могут быть легко синтезированы специалистами в данной области техники по стандартным методикам, обычно применяемым в данной области техники, или по методикам и процедурам, описанным ниже. Нижеследующие схемы, синтезы, примеры и процедуры приведены для лучшего понимания настоящего изобретения и никоим образом не ограничивают область его применения. Специалисты в данной области техники должны понимать, что в предложенных методиках могут быть сделаны различные изменения, которые не изменяют объема и сущности настоящего изобретения. Все публикации, упомянутые в настоящем описании, доступны по уровню специалистам в той области техники, к которой принадлежит настоящее изобретение. Оптимальное время проведения реакций, указанных в схемах, синтезах, примерах и процедурах, может быть определено при помощи традиционных хроматографических способов отслеживания хода реакции. Кроме того, реакции, применяемые согласно настоящему изобретению, предпочтительно проводить в инертной атмосфере, например под аргоном, азотом. Выбор растворителя обычно некритичен, если применяемый растворитель инертен по отношению к происходящей реакции и растворяет реагенты в достаточной степени для осуществления реакции. Предпочтительно, соединения выделяют и очищают перед их использованием в последующих реакциях. Некоторые соединения могут кристаллизоваться из реакционных растворов в процессе образования, и затем их отделяют фильтрованием, или реакционный растворитель может быть удален экстракцией, испарением или декантацией. При необходимости, промежуточные соединения и готовые продукты, отвечающие формуле (I), могут быть дополнительно очищены при помощи традиционных методик, например, при помощи перекристаллизации или хроматографии на твердых носителях, например силикагеле или оксиде алюминия. Опытные специалисты должны понимать, что не все заместители совместимы со всеми реакционными условиями. В такие соединения на определенном этапе синтеза при помощи способов, известных в данной области техники, могут быть введены защитные группы. Если не указано особо, термины и сокращения, используемые в предлагаемых схемах, синтезах, примерах и процедурах, имеют свои обычные значения. Например, в настоящем описании следующие термины имеют указанное значение: "psi" означает фунты на квадратный дюйм; "ТСХ (TLC)" означает тонкослойную хроматографию; "ЖХВР (HPLC)" означает жидкостную хроматографию высокого разрешения; "R f " означает коэффициент удержания; "R t " означает время удержания; " δ" означает части на миллион в сторону уменьшения напряженности магнитного поля относительно сигнала тетраметилсилана; ''МС (MS)" означает масс-спектрометрию, если не указано особо, термин "наблюдаемая масса (Observed Mass)" означает [М+Н]; "MS(APCi) означает хемоионизационную масс-спектрометрию при атмосферном давлении; "УФ (UV)" означает ультрафиолетовую спектрометрию" '' 1 H ЯМР (NMR)" означает спектрометрию протонного ядерного магнитного резонанса; "LCMS" означает жидкостную хроматографию-масс-спектрометрию; "GC/MS" означает газовую хроматографию/масс-спектрометрию. "IR" означает инфракрасную спектрометрию; при этом в ИК-спектрах указаны только максимумы поглощения интересующих фрагментов, а не все максимумы поглощения. "RT" означает комнатную температуру. "ТГФ (THF)" означает тетрагидрофуран; "LAH" означает литийалюминийгидрид; "LDA" означает литийдиизопропиламид; "ДМСО (DMSO)" означает диметилсульфоксид; "ДМФА (DMF)" означает диметилформамид; "EtOAc" означает этилацетат; "Pd-C'' означает палладий на угле; "ДХМ (DCM)" означает дихлорметан; "DMAP" означает диметиламинопиридин; "LiHMDS" означает литийгексаметилдисилисан; "ТФУК (TFA)" означает трифторуксусную кислоту; "EDAC" означает гидрохлорид N-этил-N'-(3-диметиламинопропил)карбодиимида; "НОВТ" означает 1-гидроксибензотриазол; "Bn-9-BBN" означает бензил-9-борабицикло[3.3.1]нонан; "Pd(dppf)Cl 2 " означает [1,1'-бис-(дифенилфосфино)ферроцен)дихлорпалладий(II); "EDCI" означает гидрохлорид N-этил-N'-(3-диметиламинопропил)карбодиимида; "DBU" означает 1,8-диазабицикло[5.4.0]ундецен-7, ''TBSCl'' означает трет-бутилдиметилсиланилоксиметилхлорид, ''NBS" означает N-бромсукцинимид; "TsOH" означает п-толуолсульфокислоту; "ДХЭ (DCE)" означает дихлорэтан; "DAST" означает трифторид (диэтиламино)серы; "ЕА/H" означает смесь этилацетата/гексанов; "Pd 2 (dba) 3 " означает бис-(дибензилиденацетон)палладий; "BINAP" означает 2,2'-бис-(дифенилфосфино-1,1'-бинафталин); "NMP" означает N-метилпирролидин; "TMSCN" означает триметилсилилцианид; "TBAF" означает тетрабутиламмонийфторид; "Tf 2 O" означает трифторметансульфоновый ангидрид; "TBSO" означает трет-бутилдиметилсиланилокси; "OTf" означает трифторметансульфонат, MeTi(Oi-Pr) 3 означает триизопропоксид метилтитана; "BBr 3 " означает трибромид бора; "PBr 3 " означает трибромид трехвалентного фосфора; "Pd(PPh 3 ) 4 " означает тетракис(трифенилфосфин)палладий(0); "ОАс" означает ацетат; "DME" означает диметилэтан; "Et 2 O" означает диэтиловый эфир; "(Ph 3 P) 4 Pd" означает тетракис(трифенилфосфин)палладий(0); "DMFDMA" означает диметилацеталь N,N-диметилформамида; "Et 3 N" означает триэтиламин; "tBu" означает трет-бутил; "DIPEA" означает диизопропилэтиламин; "EDC" означает гидрохлорид(3-диметиламинопропил)-3-этилкарбодиимида; "НОАс" означает уксусную кислоту; "boc" означает трет-бутоксикарбонил. В структуре "Ph" означает фенил; "Me" означает метил; "Et" означает этил; "Bn" означает бензил; "МеОН" означает метанол; "OTf" означает трифторметансульфонат, "TIPSO" означает триизопропилсиланилокси, "TBSO" означает трет-бутилдиметилсиланилокси. Рассмотренные в описании примеры приведены для иллюстрации настоящего изобретения и никоим образом не ограничивают область его применения. Названия соединений, указанных в примерах и синтезах, образованы программами AutoNom 2.2 в ChemDraw Ultra, или AutoNom 2000 в MDL ISIS/Draw, версия 2.5 SP1, предоставляемой MDL Information Systems, Inc., или предоставлены Chemical Abstracts Services. Спектры 1 H ЯМР получали в указанных растворителях на спектрометре Varian INOVA 400 МГц. Для проведения жидкостной хроматографии-масс-спектрометрии (LCMS) применяли прибор Agilent HP1100, оборудованный масс-спектрометром (Agilent MSD SL). В качестве неподвижной фазы использовали Waters Xterra C18 (2,1 ×50 мм, 3,5 мкм); стандартный способ включал использование градиента 5-100% ацетонитрила/метанола (50:50) с добавлением 0,2% формиата аммония в течение 3,5 мин, затем выдерживали при 100% В в течение 0,5 мин при температуре колонки, равной 50 °С, и расходе, равном 1,0 мл/мин. Другой стандартный способ включал использование градиента 5-100% ацетонитрила/метанола (50:50) с добавлением 0,2% формиата аммония в течение 7,0 мин, затем выдерживали при 100% В в течение 1,0 мин при температуре колонки, равной 50 °С, и расходе, равном 1,0 мл/мин. Дополнительный масс-спектрометрический анализ, проводимый на Agilent MSD (циклический прибор), представлял собой стандартный проточно-инъекционный анализ (FIA) в отсутствии колонки при скорости потока 80% МеОН с добавлением 6,5 мМ ацетата аммония, равной 0,5 мл/мин, выдерживаемой в течение 30 с. В соответствии со схемой А в необязательно замещенный фенол (1) вводят защитную группу (например, TBSCl) с получением соединения 2 и затем соединение 2 превращают в альдегид (3). Осуществляют реакцию соединения 3 с соединением, содержащим защитную группу (Pg) и уходящую группу (Lg) с образованием простого эфира 4. Pg может представлять собой -СН 3 или -CH 2 -фенил, a Lg может представлять собой метилсульфонатную или галогеногруппу. Предпочтительно соединение Lg-Pg представляет собой I-СН 3 или Br-СН 2 -фенил. Полученный альдегид восстанавливают с образованием спирта (5) и затем превращают в соединение 6. Предпочтительно соединение 5 галогенируют при помощи PBr 3 с образованием 2-бромметильного производного. Способы введения и снятия защитных групп в соединениях, которые образуют соединения, отвечающие формуле (I), и в других соединениях хорошо известны специалистам в данной области техники и описаны в литературе (см., например: Greene and Wuts, Protective Groups in Organic Synthesis, Third Edition, John Wiley and Sons Inc., 1999). В соответствии со схемой В соединение, отвечающее формуле (Ia), получают, сначала осуществляя реакцию соединения 7 с соединением 6 (схема А) с образованием соединения 8. Затем диметоксипроизводное 8 превращают в альдегид (9). Соединение 9 вводят в реакцию с тетрагидробензимидазол-5-иламином (10) с образованием рацемата лактама (11), который затем превращают в соединение 13. С соединения 13 снимают защиту, получая фенол (14). Соединение 14 превращают в пару изомеров (15), путем осуществления реакции с трифторметансульфоновым ангидридом. Затем проводят реакцию сочетания смеси 15 с производным борной кислоты (R 4 -B(OH) 2 ) в присутствии катализатора, например тетракис(трифенилфосфен)палладия(0). На схеме С показан стереоселективный синтез, позволяющий получить промежуточное соединение 18. Соединение 16 получают ацилированием коммерчески доступного (R)-4-бензилоксазолидин-2-она 4-пентеноилхлоридом. Затем его алкилируют необязательно замещенным соединением 6 (см. схему А) с образованием соединения 17. Соединение 17 окисляют с образованием промежуточного альдегида 18 под действием озона и трифенилфосфина или тетраоксида осмия и окислителя, например, метаперйодата натрия. В соответствии со схемой D промежуточное соединение (18) превращают в лактам 20, находящийся в "R" конфигурации. Затем производят снятие защиты с образованием соединения 21, которое затем подвергают триметансульфонированию (трифторметансульфоновым ангидридом) с образованием пары изомеров (22). Затем смесь 22 вводят в реакцию сочетания с производным борной кислоты (R 4 -B(OH) 2 ) в присутствии катализатора, например тетракис(трифенилфосфен)палладия(0). В соответствии со схемой Е нитробензимидазол (23) гидрируют с образованием рацемической смеси 4,5,6,7-тетрагидро-3Н-бензимидазол-5-иламинов (24). Затем, смесь 25 разделяют фракционной кристаллизацией с использованием дибензоил-D-тартрата (Siegfried Schwarz, Walter Schunack; Arch. Pharm. Vol. 312, с. 933-939, 1979 г.). В соответствии со схемой F промежуточное соединение (18) вводят в реакцию с разделенным соединением 25 (схема Е), получая лактам 26, который находится в "R" конфигурации. Затем производят снятие защиты с образованием соединения 27, которое затем подвергают триметансульфонированию (трифторметансульфоновым ангидридом) с образованием пары изомеров (28). Затем смесь 28 вводят в реакцию сочетания с производным борной кислоты (R 4 -B(OH) 2 ) в присутствии катализатора, например тетракис(трифенилфосфен)палладия(0). Синтез 1. 2,6-Дихлор-4-гидрокси-бензальдегид. 3,5-Дихлорфенол (1 кг, 6,13 моль) растворяют в 3 л диметилформамида (ДМФА) и охлаждают до 0 °С. Добавляют имидазол (918,74 г, 6,75 моль), а затем добавляют трет-бутилдиметилсилилхлорид (1017,13 г, 6,75 моль). Смесь нагревают до комнатной температуры и перемешивают в течение 15 мин. Выливают в воду (6 л) и экстрагируют эфиром (4 л). Органический слой 2 раза промывают водой, 10%-ным водным раствором хлорида лития, затем солевым раствором и сушат над сульфатом натрия. Фильтруют и концентрируют в вакууме, получая трет-бутил-(3,5-дихлорфенокси)диметилсилан (1700 г) в виде масла. трет-Бутил-(3,5-дихлорфенокси)диметилсилан (425 г, 1,5 моль) растворяют в 4 л сухого тетрагидрофурана и охлаждают до -68 °С. Медленно добавляют 1,1 эквивалента втор-бутиллития (103,1 г, 1,61 моль) при -68 °С (~1,75 ч). По окончании добавления реакционную смесь перемешивают при -70 °С в течение 30 мин. Добавляют диметилформамид (168,5 г, 2,3 моль) и реакционную смесь перемешивают при -70 °С в течение 1 ч. Добавляют 1 М соляную кислоту в воду (3,5 л) и реакционную смесь оставляют нагреваться до комнатной температуры. Реакционную смесь выливают в эфир (5 л), промывают водой, затем солевым раствором. Сушат над сульфатом натрия и концентрируют в вакууме до получения оранжевого твердого вещества. Размешивают с холодным дихлорметаном и фильтруют, выделяя 250 г (80 %) бледно-желтого твердого вещества. Синтез 2. 2,6-Дихлор-4-метокси-бензальдегид. 2,6-Дихлор-4-гидрокси-бензальдегид (120 г, 628,24 ммоль) и карбонат калия (173,65 г, 1256,5 ммоль) помещают в 900 мл диметилформамида и обрабатывают йодметаном (107 г, 753,9 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 3 ч. Отфильтровывают твердые вещества и выливают в 6 л воды. Отфильтровывают твердые вещества, несколько раз промывают водой, сушат на воздухе и растворяют в этилацетате. Промывают водой, а затем добавляют солевой раствор и затем сушат над сульфатом натрия. Фильтруют и концентрируют в вакууме приблизительно до объема 100 мл; при этом из раствора начинают выпадать твердые вещества. Фильтруют и затем фильтрат концентрируют с образованием второй порции твердых веществ. Промывают гексаном, объединяют все твердые вещества и сушат в вакууме, получая 112,3 г беловатого твердого вещества. 1 Н ЯМР (400 МГц, CDCl 3 ) δ 10,41 (с, 1Н), 6,90 (с, 2Н), 3,87 (с, 3Н). Синтез 3. 2,6-Дихлор-4-бензилокси-бензальдегид. Смесь 2,6-дихлор-4-гидроксибензальдегида (250 г, 1,3 моль) и карбоната калия (361,8 г, 2,62 моль) в 2 л диметилформамида обрабатывают бензилбромидом (268,64 г, 1,57 моль). Реакционную смесь перемешивают при комнатной температуре в течение 1 ч. Отфильтровывают твердые вещества и выливают в 12 л воды. Отфильтровывают твердое вещество, несколько раз промывают водой, сушат на воздухе и растворяют в этилацетате. Сушат над сульфатом магния, фильтруют и концентрируют в вакууме до ~1,5 л. Оставляют на ночь, после чего фильтруют. Твердое вещество промывают минимальным количеством гексана и сушат в вакууме. Фильтрат концентрируют в вакууме и размешивают с гексаном, получая вторую порцию продукта, которую объединяют с первой порцией и получают 245 г белых кристаллов. Повторяя, получают третью порцию, равную 80 г, в виде слегка коричневатого порошка (общий выход: 88%). 1 H ЯМР (400 МГц, ДМСО-d 6 ) δ 10,26 (с, 1Н), 7,43 (м, 5Н), 7,28 (с, 2Н), 5,25 (с, 2Н). Синтез 4. (2,6-Дихлор-4-метоксифенил)метанол. 2,6-Дихлор-4-метоксибензальдегид (112 г, 546 ммоль) суспензируют в 1500 мл этанола и охлаждают на ледяной бане до 7 °С. Порциями добавляют боргидрид натрия (20,67, 546 ммоль), получая раствор. Ледяную баню убирают и перемешивают в течение 2 ч. Реакционную смесь осторожно добавляют в насыщенный раствор хлорида аммония (~4 л) и перемешивают до полного тушения. Экстрагируют дихлорметаном (3 ×1 л) и объединенные органические экстракты сушат над сульфатом натрия. Фильтруют и концентрируют в вакууме с получением 113 г слегка коричневатого твердого вещества. 1 H ЯМР (400 МГц, CDCl 3 ) δ 6,86 (с, 2Н), 4,86 (с, 2Н), 3,78 (с, 3Н), 2,07 (с, 1H). Синтез 5. (2,6-Дихлор-4-бензилоксифенил)метанол. Указанное в заголовке соединение готовили практически в соответствии с синтезом 4. ЯМР (ДМСО-d 6 ) δ 7,38 (м, 4Н), 7,33 (м, 1Н), 7,12 (с, 2H), 5,14 (с, 2Н), 5,05 (т, 1Н), 4,59 (д, 2Н). Синтез 6. 2-Бромметил-1,3-дихлор-5-метоксибензол. (2,6-Дихлор-4-метоксифенил)метанол (113 г, 545,76 ммоль) растворяют в 1200 мл сухого ТГФ и охлаждают до 0 °С в атмосфере азота. В атмосфере азота добавляют PBr 3 (59,1 г, 218,3 ммоль) и перемешивают при 0 °С в течение 30 мин. Выливают в насыщенный водный NaHCO 3 и экстрагируют EtOAc. Сушат и концентрируют в вакууме, получая 129,4 г продукта в виде беловатого твердого вещества. ЯМР (CDCl 3 ) δ 6,88 (с, 2Н), 4,73 (с, 2Н), 3,79 (с, 3Н). Синтез 7. 2-Бромметил-1,3-дихлор-5-бензилоксибензол. Указанное в заголовке соединение готовили практически в соответствии с синтезом 6 с выходом 89%. ES MS (m/z): 347 (М+1). Синтез 8. 4,5,6,7-Тетрагидро-3Н-бензимидазол-5-иламина дигидрохлорид. В стеклянную колбу Парра емкостью 500 мл добавляют 5% Rh/C (5,215 г). Колбу продувают азотом и 5% Rh/C смачивают 3н. HCl (50 мл). Затем в продутую колбу добавляют 6-нитро-1Н-бензимидазол (10,354 г, 0,0635 моль) и 3н. HCl (100 мл). Колбу Парра герметизируют и реакционный сосуд продувают азотом (3Х) и водородом (3Х). Затем водородом нагнетают давление в реакционной смеси (60 фунтов/кв.дюйм, что составляет примерно 0,41 МПа), герметизируют сосуд, но по мере необходимости добавляют водород для поддержания давления, равного 60 фунтов/кв.дюйм (0,41 МПа); реакционную смесь перемешивают и нагревают до 80 °С. Реакцию продолжают в течение 24 ч, затем нагревание снимают и реакционную смесь оставляют охлаждаться до температуры окружающей среды. Избыток водорода выпускают из сосуда, сосуд продувают азотом (3Х) и фильтруют реакционную смесь, удаляя катализатор - 5%Rh/C. Фильтрат концентрируют досуха на роторном испарителе, а затем в высоком вакууме. Твердый остаток растворяют в 20 мл метанола, нагревают приблизительно до 60 °С и добавляют дополнительное количество метанола (приблизительно до 10 мл или по необходимости) до полного растворения остатка. Раствор охлаждают до комнатной температуры и добавляют эфир (40 мл). Выделяется белое твердое вещество, которое фильтруют и сушат в высоком вакууме с образованием продукта (8 г). 1 H ЯМР (D 2 O) δ 1,80-2,11 (1Н), 2,09-2,22 (1Н), 2,60-2,79 (4Н), 3,01-3,15 (1Н), 3,61-3,75 (1Н), 8,41 (1Н). Синтез 9. Диэтиловый эфир 2-(4-бензилокси-2,6-дихлорбензил)-2-(2,2-диметоксиэтил)малоновой кислоты. Диэтиловый эфир 2-(2,2-диметоксиэтил)малоновой кислоты (4,5 г, 16,3 ммоль) в 50 мл ТГФ охлаждают до -78 °С и к полученной смеси по каплям добавляют гексаметилсиланамид лития (1М, 17 мл). Спустя 15 мин добавляют 5-бензилокси-2-бромметил-1,3-дихлорбензол (17,9 ммол, 6,2 г) в 10 мл ТГФ. Полученную смесь нагревают до температуры окружающей среды, подкисляют 1н. HCl и экстрагируют этилацетатом. Органические слои концентрируют и очищают при помощи колоночной хроматографии с нормальной фазой, в результате чего получают указанное в заголовке соединение в виде белого твердого вещества (7,0 г, 80%). Синтез 10. Диэтиловый эфир 2-(4-бензилокси-2,6-дихлорбензил)-2-(2-оксоэтил)малоновой кислоты. К раствору диэтилового эфира 2-(4-бензилокси-2,6-дихлорбензил)-2-(2,2-диметоксиэтил)малоновой кислоты (10,0 г, 18,5 ммоль) (синтез 9) в 95 мл ацетона добавляют 10 мл муравьиной кислоты. Спустя 24 ч концентрируют реакционную смесь, получая диэтиловый эфир 2-(4-бензилокси-2,6-дихлорбензил)-2-(2-оксоэтил)малоновой кислоты в виде желтого маслянистого продукта (6,7 г, 80%). Синтез 11. Этиловый эфир 3-(4-бензилокси-2,6-дихлорбензил)-2-оксо-1-(4,5,6,7-тетрагидро-3Н-бензимидазол-5-ил)пирролидин-3-карбоновой кислоты. Цианоборгидрид натрия (3,6 г, 57,4 ммоль) добавляют порциями к раствору диэтилового эфира 2-(4-бензилокси-2,6-дихлорбензил)-2-(2-оксоэтил)малоновой кислоты (6,7 г, 14,4 ммоль) (синтез 10) и дигидрохлориду 4,5,6,7-тетрагидро-3Н-бензимидазол-5-иламина (6,0 г, 28,7 ммоль) (синтез 1) в 70 мл метанола. Спустя 3 ч добавляют 4 мл уксусной кислоты и нагревают до 60 °С в течение 16 ч. Большую часть метанола удаляют на роторном испарителе. Полученную смесь гасят насыщенным раствором бикарбоната и экстрагируют этилацетатом. Экстракты сушат над сульфатом натрия, концентрируют и очищают при помощи колоночной хроматографии с нормальной фазой (0-10% метанола в этилацетате), получая этиловый эфир 3-(4-бензилокси-2,6-дихлорбензил)-2-оксо-1-(4,5,6,7-тетрагидро-3Н-бензимидазол-5-ил)пирролидин-3-карбоновой кислоты в виде белого твердого вещества (3 г). Синтез 12. 3-(4-Бензилокси-2,6-дихлорбензил)-1-(4,5,6,7-тетрагидро-3Н-бензимидазол-5-ил)пирролидин-2-он. Этиловый эфир 3-(4-бензилокси-2,6-дихлорбензил)-2-оксо-1-(4,5,6,7-тетрагидро-3Н-бензимидазол-5-ил)пирролидин-3-карбоновой кислоты (3 г, 5,5 ммоль) (синтез 11) растворяют в 10 мл метанола и затем добавляют 10 мл 2н. NaOH. Полученную смесь перемешивают в течение 16 ч, концентрируют и затем добавляют диоксан и уксусную кислоту, получая 3-(4-бензилокси-2,6-дихлорбензил)-2-оксо-1-(4,5,6,7-тетрагидро-3Н-бензимидазол-5-ил)пирролидин-3-карбоновую кислоту, которую не выделяют. Смесь кипятят с обратным холодильником в течение ночи, гасят водой и экстрагируют этилацетатом. Экстракты промывают насыщенным раствором бикарбоната и концентрируют с получением продукта, 3-(4-бензилокси-2,6-дихлорбензил)-1-(4,5,6,7-тетрагидро-3Н-бензимидазол-5-ил)пирролидин-2-она (2,6 г). MS: m/z (M+1)=470. Синтез 13. 3-(2,6-Дихлор-4-гидроксибензил)-1-(4,5,6,7-тетрагидро-1Н-бензимидазол-5-ил)пирролидин-2-он. К продутому азотом раствору 3-(4-бензилокси-2,6-дихлорбензил)-1-(4,5,6,7-тетрагидро-1Н-бензимидазол-5-ил)пирролидин-2-она (4,3 г, 9,2 ммоль) (синтез 12) в 50 мл этилацетата и 15 мл метанола добавляют гидроксид палладия. Затем нагнетают сжатый азот приблизительно до достижения давления, равного 22 фунта/кв.дюйм (что составляет примерно 0,15 МПа), и перемешивают в течение 16 ч. Полученную смесь фильтруют и концентрируют, получая 3-(2,6-дихлор-4-гидроксибензил)-1-(4,5,6,7-тетрагидро-1Н-бензимидазол-5-ил)пирролидин-2-он в виде твердого продукта (2 г). MS: m/z (M+1)=380. Синтез 14. 3,5-Дихлор-4-[2-оксо-1-(3-трифторметансульфонил-4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)пирролидин-3-илметил]фениловый эфир трифторметансульфоновой кислоты и 3,5-дихлор-4-[2-оксо-1-(1-трифторметансульфонил-4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)пирролидин-3-илметил]фениловый эфир трифторметансульфоновой кислоты 3-(2,6-Дихлор-4-гидроксибензил)-1-(4,5,6,7-тетрагидро-3Н-бензимидазол-5-ил)пирролидин-2-он (0,5 г) (синтез 13) растворяют в ДХМ (2 мл) и пиридине (2 мл). Раствор охлаждают до 0 °С на ледяной бане и по каплям при перемешивании добавляют трифторметансульфоновый ангидрид (0,5 мл). Затем реакционную смесь оставляют при -30 °С в течение ночи в морозильнике. Реакционную смесь разбавляют этилацетатом (50 мл). Промывают 1М водной HCl (2 ×50 мл), солевым раствором (50 мл). Слой этилацетата сушат над сульфатом натрия и концентрируют на роторном испарителе. Сушка остатка в высоком вакууме приводит к получению смеси 3,5-дихлор-4-[2-оксо-1-(3-трифторметансульфонил-4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)пирролидин-3-илметил]фенилового эфира трифторметансульфоновой кислоты и 3,5-дихлор-4-[2-оксо-1-(1-трифторметансульфонил-4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)пирролидин-3-илметил]фенилового эфира трифторметансульфоновой кислоты в виде желтого пенистого твердого вещества (650 мг). Вводят в следующую реакцию без очистки. MS: m/z (M+1)=643. Синтез 15. (R)-4-Бензил-3-пент-4-еноилоксазолидин-2-он. Трехгорлую круглодонную колбу емкостью 12 л, снабженную механической мешалкой, внутренним детектором температуры/вводом N 2 и 1-литровой капельной воронкой, продувают азотом в течение 20 мин, и затем в нее помещают (R)-4-бензил-2-оксазолидинон (250 г, 1,41 моль). Разбавляют тетрагидрофураном (ТГФ) (1,8 л) и охлаждают на бане из сухого льда/ацетона до достижения внутренней температуры, равной -74 °С. В капельную воронку через полую трубку вводят 1,6М раствор н-бутиллития в гексане (970 мл, 1,552 моль) и добавляют к раствору оксазолидинона с такой скоростью, чтобы внутренняя температура не превышала -65 °С. По окончании прибавления реакционную смесь оставляют перемешиваться в охлаждающей бане в течение 30 мин. В капельную воронку вносят 4-пентеноилхлорид (175 мл, 1,585 моль) и добавляют по каплям к раствору аниона в течение 25 мин. Реакционную смесь перемешивают в течение 45 мин в охлаждающей бане. Охлаждающую баню удаляют и реакционную смесь перемешивают 18 ч по мере того, как она медленно нагревается до комнатной температуры. Смесь разбавляют 1N водной соляной кислотой (1,5 л) и диэтиловым эфиром (1 л). Слои разделяют и органическую фазу промывают водой (2 ×1 л), затем солевым раствором (1 л). Объединенные промывные воды экстрагируют эфиром (1 л). Объединенные органические фазы сушат над безводным сульфатом магния, фильтруют и концентрируют, получая 390 г светло-коричневого масла. Полученный материал очищают хроматографией на силикагеле, используя смесь гексанов:этилацетата, и получают 345 г (94,5%) прозрачного желтого масла. Синтез 16. (R)-4-Бензил-3-[(S)-2-(4-бензилокси-2,6-дихлорбензил)пент-4-еноил]оксазолидин-2-он. Смесь (R)-4-бензил-3-пент-4-еноилоксазолидин-2-она (345 г, 1,33 моль) и ТГФ (1,8 л) перемешивают в трехгорлой круглодонной кобле емкостью 12 л, снабженной внутренним детектором температуры/вводом азота и капельной воронкой, в атмосфере азота и охлаждают до -75 °С. В капельную воронку вводят 1М LiHMDS (1,6 л) и добавляют с такой скоростью, чтобы внутренняя температура не превышала -60 °С. По окончании прибавления реакционную смесь оставляют перемешиваться при -25 °С в течение 30 мин, а затем охлаждают приблизительно до -60 °С. В этот момент порциями, в течение 5 мин добавляют твердый 2-бромметил-1,3-дихлор-5-бензилоксибензол. По окончании прибавления реакционный сосуд переносят в ацетоновую баню, находящуюся при -10 °С, и внутреннюю температуру реакционной смеси поддерживают ниже 10 °С в течение 1 ч. Смесь охлаждают до 0 °С, затем гасят 2 л 1н. водной соляной кислоты. Смесь переносят в 22-литровую делительную воронку и разбавляют 2,5 л воды и 2 л эфира. Слои разделяют и водный слой экстрагируют эфиром. Объединенную органическую фазу сушат над безводным сульфатом магния, фильтруют и концентрируют, получая 800 г плотного масла. Очищают хроматографией на силикагеле, используя смесь гексанов:этилацетата; получают 597 г, (86%) бесцветного масла. Синтез 17. (R)-4-((R)-4-Бензил-2-оксо-оксазолидин-3-ил)-3-(4-бензилокси-2,6-дихлорбензил)-4-оксобутиральдегид. Смесь (R)-4-бензил-3-[(S)-2-(4-бензилокси-2,6-дихлорбензил)пент-4-еноил]оксазолидин-2-она (100 г, 190,68 ммоль) и дихлорметана (800 мл) охлаждают до -74 °С. Озон, получаемый на генераторе озона А-113 со скоростью 75%, барботируют через реакционную смесь в токе воздуха со скоростью 5 кубических футов в минуту (примерно 0,14 куб.м/мин) до окрашивания раствора в синий цвет (приблизительно 3 ч). Добавляют трифенилфосфин (60 г, 228,8 ммоль) в виде раствора в 200 мл дихлорметана и реакционную смесь оставляют перемешиваться при протекании реакции при комнатной температуре в течение ночи. Раствор концентрируют в вакууме и очищают хроматографией на силикагеле, используя градиент 20-50% этилацетата в гексане, получая 82,1 г (82%) продукта в виде белой пены. MS (m/z): 526 (М+). Альтернативная процедура для приготовления (R)-4-((R)-4-бензил-2-оксо-оксазолидин-3-ил)-3-(4-бензилокси-2,6-дихлорбензил)-4-оксо-бутиральдегида. Смесь (R)-4-бензил-3-[(S)-2-(4-бензилокси-2,6-дихлорбензил)пент-4-еноил]оксазолидин-2-она (0,96 г, 1,8 ммоль), ТГФ (21 мл) и воды (7 мл) обрабатывают 2,5% тетраоксидом осмия в трет-бутаноле (46 мг, 0,18 ммоль). Добавляют перйодат натрия (1,17 г, 5,5 ммоль), и реакционную смесь перемешивают 4 ч при комнатной температуре. Реакционную смесь гасят водой и экстрагируют этилацетатом. Органическую фазу промывают 1N водным тиосульфатом натрия, затем солевым раствором. Органический слой сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Сырой материал очищают хроматографией на силикагеле, используя смесь гексанов:этилацетата для элюирования чистого продукта. Фракции, содержащие продукт, концентрируют в вакууме, получая 0,46 г (48%) желаемого продукт. MS (m/z): 52 6 (М+). Синтез 18. 3-[R]-(4-Бензилокси-2,6-дихлорбензил)-1-(4,5,6,7-тетрагидро-3H-бензимидазол-3-ил)пирролидин-2-он. Соединение, полученное в синтезе 10 (5,25 г), добавляют к суспензии гидрохлорида имидазолоциклогексил-3-амина (2 г) (синтез 8) в метаноле (30 мл). Смесь перемешивают при комнатной температуре в течение 30 мин. Добавляют цианоборгидрид натрия (1 г), и смесь перемешивают в течение 2 ч. Реакционную смесь нагревают при 50 °С в течение 2 ч. Реакционную смесь разбавляют этилацетатом (50 мл) и полученную смесь промывают водой (50 мл). Слой этилацетата сушат над сульфатом натрия и концентрируют на роторном испарителе, получая сырое вещество. Сырое вещество очищают испарительной хроматографией на силикагеле, элюируя градиентом 0-5% метанола в ДХМ с образованием продукта в виде смеси диастереомеров 3-(4-бензилокси-2,6-дихлорбензил)-1-(4,5,6,7-тетрагидро-3H-бензимидазол-3-ил)пирролидин-2-она (4,5 г). MS: m/z=470 (M+1). Синтез 19. 3-[R]-(2,6-Дихлор-4-гидроксибензил)-1-(4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)пирролидин-2-он. В толстостенном сосуде растворяют 3-(4-бензилокси-2,6-дихлорбензил)-1-(4,5,6,7-тетрагидро-3H-бензимидазол-3-ил)пирролидин-2-он (2,5 г) (синтез 18) в метаноле (50 мл) и этилацетате (30 мл). Добавляют катализатор - гидроксид палладия (500 мг) и реакционную смесь перемешивают в атмосфере водорода (30 фунтов/кв.дюйм (примерно 0,20 МПа)) при 40 °С в течение ночи. Реакционную смесь фильтруют через целит. Фильтрат концентрируют, получая продукт, 3-[R]-(2,6-дихлор-4-гидроксибензил)-1-[R/S]-(4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)пирролидин-2-он в виде смеси белых твердых веществ (1,6 г). MS: m/z=379 (M+1). Синтез 20. 3,5-Дихлор-4-[2-оксо-1-(3-трифторметансульфонил-4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)пирролидин-3-[R]илметил]фениловый эфир трифторметансульфоновой кислоты и 3,5-дихлор-4-[2-оксо-1-(1-трифторметансульфонил-4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)пирролидин-3-[R]илметил]фениловый эфир трифторметансульфоновой кислоты (13) 3-[R]-(2,6-Дихлор-4-гидроксибензил)-1-(4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)пирролидин-2-он (1,6 г) (синтез 19) растворяют в ДХМ (20 мл) и пиридине (5 мл). Раствор охлаждают до -78 °С и по каплям при перемешивании добавляют трифторсульфоновый ангидрид (3 мл). Затем реакционную смесь оставляют на 48 ч в морозильнике. Реакционную смесь разбавляют этилацетатом (150 мл). Промывают 1М водной HCl (2 ×50 мл), солевым раствором (50 мл). Слой этилацетата сушат над сульфатом натрия и концентрируют на роторном испарителе. Остаток сушат в высоком вакууме, получая смесь 3,5-дихлор-4-[2-оксо-1-(3-трифторметансульфонил-4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)пирролидин-3-илметил]фенилового эфира трифторметансульфоновой кислоты и 3,5-дихлор-4-[2-оксо-1-(1-трифторметансульфонил-4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)пирролидин-3-илметил]фенилового эфира трифторметансульфоновой кислоты в виде твердого вещества (2,01 г). Смесь используют в следующей операции без очистки. MS: m/z (M+1)=643. Синтезы 21 и 22. Хиральная очистка 3-[R]-(4-бензилокси-2,6-дихлорбензил)-1-(4,5,6,7-тетрагидро-3H-бензимидазол-3-ил)пирролидин-2-она. Диастереоизомерную смесь 3-(4-бензилокси-2,6-дихлорбензил)-1-(4,5,6,7-тетрагидро-3H-бензимидазол-3-ил)пирролидин-2-она (2 г) (синтез 18) очищают на колонке Chiralpak AD-H (0,46 ×15 см), элюируя смесью 60:40:0,2 3А этанола/гептана/ДМЭА (скорость течения=0,6 мл/мин) с образованием: изомера 1 (синтез 21), время удержания 5,4 мин, 922 мг и изомера 2 (синтез 22), время удержания 7,1 мин, 866 мг. Изомер 2 используют в синтезе 23. Синтез 23. 4,5,6,7-Тетрагидро-3Н-бензимидазол-5-иламин-дибензоил-D-винная кислота. (Siegfried Schwarz, Walter Schunack; Arch. Pharm. Vol. 312, с. 933-939, 1979 г.) Дигидрохлорид 4,5,6,7-тетрагидро-1Н-бензимидазол-5-иламина (42 г, 200 ммоль) (синтез 8) растворяют в 350 мл Н 2 О. К полученному раствору порциями добавляют бикарбонат натрия (50 г, 600 ммоль) до прекращения вспенивания. Для полного поглощения соли может потребоваться нагревание до 60 °С. Конечный рН равен 10. Полученную водную смесь концентрируют и сушат на вакуумном насосе в течение ночи. Добавляют теплый этанол (400 мл), перемешивают до разрушения твердых кусков соли и фильтруют. Повторяют экстракцию еще раз. Для достаточного разрушения твердых веществ может понадобиться механическое перемешивание или перемешивание в течение ночи. Этанольные экстракты концентрируют до 800 мл и добавляют 400 мл H 2 O и дибензил-D-винную кислоту (75 г, 210 ммоль). Твердый осадок образуется в течение 10-15 мин. Полученную смесь оставляют перемешиваться в течение ночи. Твердые вещества отфильтровывают, получая 4,5,6,7-тетрагидро-3Н-бензимидазол-5-иламин-дибензоил-D-тартрат, который перекристаллизовывают растворением материала в метаноле (350 мл) и нагревании при 50-60 °С в конической колбе при постоянном перемешивании. Добавляют дополнительное количество метанола, необходимое для растворения всего материала. Добавляют воду (20 мл), пока смесь горячая. Смесь охлаждают при перемешивании до комнатной температуры, в результате чего образуется суспензия белого твердого вещества. Твердое вещество отфильтровывают и сушат, получая обогащенную соль аминоциклогексилимидазол-дибензил-D-винной кислоты. Перекристаллизацию повторяют еще три раза, получая обогащенный материал. Выход=25,4 г, ее ~92%. Синтез 24. 3-[R]-(4-Бензилокси-2,6-дихлорбензил)-1-(4,5,6,7-тетрагидро-3H-бензимидазол-3-ил)пирролидин-2-он. Вещество, полученное в синтезе 17 (4,87 г), добавляют к суспензии 4,5,6,7-тетрагидро-3H-бензимидазол-5-иламин-дибензоил-D-винной кислоты (синтез 23) в метаноле (20 мл). Смесь перемешивают при комнатной температуре в течение 30 мин. Добавляют цианоборгидрид натрия (0,75 г) и смесь перемешивают в течение 2 ч. Реакционную смесь нагревают при 50 °С в течение 2 ч. Реакционную смесь разбавляют ДХМ (200 мл) и смесь промывают насыщенным раствором бикарбоната натрия (2 ×100 мл) и затем водой (100 мл). Слой ДХМ сушат над сульфатом натрия и концентрируют на роторном испарителе, получая сырое вещество. Сырое вещество очищают испарительной хроматографией на силикагеле, элюируя градиентом 0-5% метанола в ДХМ, получая указанное в заголовке соединение (3,35 г). MS: m/z (M+1)=470. Синтез 25. 3-[R]-(2,6-Дихлор-4-гидроксибензил)-1-(4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)пирролидин-2-он. В толстостенном сосуде для проведения реакций под давлением растворяют 3-[R]-(4-Бензилокси-2,6-дихлорбензил)-1-(4,5,6,7-тетрагидро-3H-бензимидазол-3-ил)пирролидин-2-он (3,35 г) (синтез 24) в метаноле (30 мл) и этилацетате (20 мл). Добавляют катализатор - гидроксид палладия (500 мг) и реакционную смесь перемешивают в атмосфере водорода (30 фунтов/кв.дюйм, примерно 0,20 МПа) при 40 °С в течение 4 ч и затем при комнатной температуре в течение ночи. Реакционную смесь фильтруют через целит. Фильтрат концентрируют, получая указанное в заголовке соединение в виде белого твердого вещества (2,69 г). MS: m/z=379 (M+1). Синтез 26. 3,5-Дихлор-4-[2-оксо-1-(3-трифторметансульфонил-4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)пирролидин-3-[R]илметил]фениловый эфир трифторметансульфоновой кислоты и 3,5-дихлор-4-[2-оксо-1-(1-трифторметансульфонил-4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)пирролидин-3-[R]илметил]фениловый эфир трифторметансульфоновой кислоты 3-[R]-(2,6-дихлор-4-гидроксибензил)-1-(4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)пирролидин-2-он (синтез 25) (2,5 г) растворяют в ДХМ (10 мл) и пиридине (10 мл). Раствор охлаждают до -78 °С и по каплям при перемешивании добавляют трифторсульфоновый ангидрид (4 мл). Реакционную смесь оставляют постоять в течение ночи. Реакционную смесь разбавляют этилацетатом (150 мл). Промывают 1М водной HCl (2 ×100 мл), солевым раствором (100 мл). Слой этилацетата сушат над сульфатом натрия и концентрируют на роторном испарителе. Остаток сушат в высоком вакууме, получая смесь указанных в заголовке соединений в виде желтого пенистого твердого вещества (3,4 г). Синтез 27. Этил-2-(4-бром-2-хлорбензил)-4-оксобутаноат. К раствору этил-2-(4-бром-2-хлорбензил)пент-4-еноата (21 г, 63 ммоль), 2,5 мас.% OsO 4 (64 г, 6,3 ммоль) в ТГФ (400 мл) и воде (160 мл) добавляют перйодат натрия (41 г, 190 ммоль) и перемешивают в течение 2 ч. Реакционную смесь экстрагируют этилацетатом, органический слой промывают раствором тиосульфата натрия и солевым раствором. Сушат над сульфатом натрия, фильтруют и концентрируют. Остаток очищают на колонке силикагелем, получая указанное в заголовке соединение (15,9 г, 75%) в виде бесцветного масла. Синтез 28. (4R,5S)-(цис)-3-Пент-4-еноил-4,5-дифенилоксазолидин-2-он. (4R,5S)-(+)-цис-4,5-дифенил-2-оксазолидинон (2,06 г, 8,62 ммоль) растворяют в ТГФ (100 мл) и охлаждают до -78 °С. Добавляют n-BuLi (5,66 мл, 9,05 ммол, 1,6М раствор в гексане) и перемешивают в течение 30 мин. Затем добавляют пент-4-еноилхлорид (1,53 г, 12,93 ммоль) и продолжают перемешивание раствора в течение одного часа. Добавляют воду (100 мл) и водный слой экстрагируют этилацетатом (3 ×200 мл). Органические слои объединяют и сушат Na 2 SO 4 , фильтруют, концентрируют и очищают испарительной колоночной хроматографией (на силикагеле, 20-40% EtOAc-гексан), получая 1,42 г (51%) указанного в заголовке соединения в виде белого твердого вещества. Синтез 29. 3-Бензил-1-циклогексилпирролидин-2-он. 1-Циклогексилпирролидин-2-он (400 мг, 2,4 ммоль) помещают в ТГФ (30 мл) и охлаждают до -78 °С. Медленно добавляют LDA (2,0 М, 2,4 мл, 4,8 ммоль) и перемешивают в течение 15 мин. Добавляют бензилбромид (1,23 г, 7,2 ммоль) и перемешивают в течение 3 ч. Гасят хлоридом аммония и экстрагируют дихлорметаном. Сушат над сульфатом натрия, фильтруют и концентрируют. Очищают на силикагеле (20-50% этилацетат в гексанах), получая 432 мг (70%) указанного в заголовке соединения. Масс-спектр (apci) m/z=258,2 (M+H). Синтез 30. (4R,5S)-(цис)-3-[2-(S)-(2-Хлор-6-фторбензил)пент-4-еноил]-4,5-дифенилоксазолидин-2-он. Используя процедуру синтеза 29, (4R,5S)-(цис)-3-пент-4-еноил-4,5-дифенилоксазолидин-2-он (1,48 г, 6,61 ммоль) алкилируют 2-бромметил-1-хлор-3-фтор-бензолом, получая 1,28 г (62%) указанного в заголовке соединения в виде белого твердого вещества. Синтез 31. (4R,5S)-(цис)-3-(2-(R)-Хлор-6-фторбензил)-4-оксо-4-(2-оксо-4,5-дифенилоксазолидин-3-ил)бутиральдегид. (4R,5S)-(цис)-3-[2-(S)-(2-хлор-6-фторбензил)пент-4-еноил]-4,5-дифенилоксазолидин-2-он (1,28 г, 2,75 ммоль) растворяют в дихлорметане (100 мл) и охлаждают до 0 °С. В раствор при перемешивании барботируют озон до появления синего окрашивания. Раствор продолжают перемешивать в течение одного часа, затем через смесь барботируют азотом до исчезновения синего окрашивания. Добавляют Me 2 S (0,85 г, 13,75 ммоль) и раствор перемешивают в течение 6 ч. Растворитель отгоняют под уменьшенным давлением и остаток очищают колоночной хроматографией (на силикагеле, 20-40% EtOAc-гексан), получая 0,45 г (35%) указанного в заголовке соединения в виде бесцветного масла. Синтез 32. 3-(R)-(2-Хлор-6-фторбензил)-1-циклогексилпирролидин-2-он. (4R,5S)-(цис)-3-(2-(R)-хлор-6-фторбензил)-4-оксо-4-(2-оксо-4,5-дифенилоксазолидин-3-ил)бутиральдегид (0,45 г, 0,97 ммоль) растворяют в ТГФ (50 мл). Добавляют циклогексиламин (0,19 г, 1,94 ммоль) и уксусную кислоту (0,12 г, 1,94 ммоль) при комнатной температуре в атмосфере азота. Раствор перемешивают в течение одного часа и затем добавляют триацетоксиборгидрид натрия (0,82 г, 3,88 ммоль). Реакционную смесь перемешивают в течение ночи и затем добавляют воду (50 мл). Водный слой экстрагируют дихлорметаном (3 ×100 мл). Органические слои объединяют и сушат Na 2 SO 4 , фильтруют, концентрируют и очищают испарительной колоночной хроматографией (на силикагеле, 20-50% EtOAc-гексан), получая 0,26 г (88%) указанного в заголовке соединения в виде бесцветного масла. Масс-спектр (ионный впрыск): m/z=310,1, 312,2 (М+1). Пример 1. 1-(4,5,6,7-Тетрагидро-3H-бензимидазол-5-ил)-3-(3,5,4'-трихлорбифенил-4-илметил)пирролидин-2-он 3,5-Дихлор-4-[2-оксо-1-(3-трифторметансульфонил-4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)пирролидин-3-илметил]фениловый эфир трифторметансульфоновой кислоты и 3,5-дихлор-4-[2-оксо-1-(1-трифторметансульфонил-4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)пирролидин-3-илметил]фениловый эфир трифторметансульфоновой кислоты (синтез 14, 0,15 г, 0,23 ммоль) растворяют в 2 мл диоксана и к полученному раствору добавляют 4-хлорфенилборную кислоту (45 мг, 0,28 ммоль) и 0,5 мл 2М карбоната натрия. Смесь продувают азотом в течение 1 мин и добавляют тетракис(трифенилфосфен)палладий(0) (27 мг, 0,02 ммоль). Реакционную смесь закупоривают и нагревают, используя микроволновый нагреватель, в течение 45 мин при 90 °С. Реакционную смесь наносят на колонку SCX и промывают метанолом (два объема колонки). Затем, промывают 2н. NH 3 в метаноле, используя два объема колонки, получая получистый продукт. Далее его очищают, используя ЖХВР или хроматографию с нормальной фазой, получая 1-(4,5,6,7-тетрагидро-3Н-бензимидазол-5-ил)-3-(3,5,4'-трихлорбифенил-4-илметил)пирролидин-2-он в виде белого твердого вещества (60,0 мг, 44%). MS: m/z (M+1)=476. Примеры, указанные в табл. 1, готовят практически, как описано в примере 1, с тем лишь исключением, что 4-хлорфенилборную кислоту заменяют реагентом, указанным в колонке 3. Очистка соединений с использованием жидкостной хроматографии высокого разрешения с обращенной фазой приводит к получению солей указанных соединений и трифторуксусной кислоты. Пример 18. 3-[3,5-Дихлор-4'-(4-метилпиперазин-1-карбонил)бифенил-4-илметил]-1-(4,5,6,7-тетрагидро-1Н-бензимидазол-5-ил)пирролидин-2-он 3,5-Дихлор-4-[2-оксо-1-(3-трифторметансульфонил-4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)пирролидин-3-илметил]фениловый и 3,5-дихлор-4-[2-оксо-1-(1-трифторметансульфонил-4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)пирролидин-3-илметил]фениловый эфиры трифторметансульфоновой кислоты (синтез 14), (0,15 г, 0,23 ммоль) растворяют в 2 мл диоксана (0,15 г, 0,23 ммоль). К полученному раствору добавляют 4-(4-метилпиперазин-1-ил)фенилметанонборную кислоту (69 мг, 0,28 ммоль) и 0,5 мл 2М карбоната натрия. Смесь продувают азотом в течение 1 мин и добавляют тетракис(трифенилфосфин)палладий(0) (27 мг, 0,02 ммоль). Реакционную смесь укупоривают и нагревают, используя микроволновый нагреватель, в течение 45 мин при 90 °С. Реакционную смесь наносят на колонку SCX и промывают метанолом (два объема колонки). Затем, промывают 2н. NH 3 в метаноле, используя два объема колонки, получая получистый продукт. Далее его очищают, используя ЖХВР или хроматографию с нормальной фазой, получая 3-[3,5-дихлор-4'-(4-метилпиперазин-1-карбонил)бифенил-4-илметил]-1-(4,5,6,7-тетрагидро-1Н-бензимидазол-5-ил)пирролидин-2-он (110 мг). MS: m/z (M+1)=566. Примеры, указанные в табл. 2, готовят практически, как описано в примере 18, с тем лишь исключением, что 4-(4-метилпиперазин-1-ил)фенилметанонборную кислоту заменяют реагентом, указанным в колонке 3. Очистка соединений с использованием жидкостной хроматографии высокого разрешения с обращенной фазой приводит к получению солей указанных соединений и трифторуксусной кислоты. Примеры 22 и 23. 3-[R]-[4-(4-Фторфенил-2,6-дихлорбензил)-1-(4,5,6,7-тетрагидро-3H-бензимидазол-3-ил)пирролидин-2-он Во флаконе нагревают смесь 3,5-дихлор-4-[2-оксо-1-(3-трифторметансульфонил-4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)пирролидин-3-илметил]фенилового и 3,5-дихлор-4-[2-оксо-1-(1-трифторметансульфонил-4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)пирролидин-3-илметил]фенилового эфиров трифторметансульфоновой кислоты (850 мг) (синтез 20), 4-фторфенилборной кислоты (221 мг), тетракистрифенилфосфинпалладия (16 мг) и насыщенного раствора бикарбоната натрия (1 мл) в 10 мл диметилэтана при 90 °С. Реакционную смесь фильтруют через колонку SCX (10 г, Varion) с этилацетатом (10 мл). Продукт элюируют 1М аммиаком в метаноле и концентрируют досуха на роторном испарителе, получая продукт в виде сырого вещества (4 00 мг). Полученный материал очищают при помощи хиральной очистки на колонке Chiralpak AD-H (4,6 ×150 мм), элюируя смесью 60:40:0,2 3А этанола/гептана/ДМЭА (скорость течения =0,6 мл/мин), получая: пример 22 (изомер 1, MS: m/z (M+1)=458, Rt. 5,8 мин, ее > 99%, 187 мг), пример 23 (изомер 2, MS: m/z (M+1)=458, Rt. 9,7 мин, ее > 99%, 164 мг). Примеры 24 и 25. (R)-3-[3,5-Дихлор-4'-(морфолин-4-карбонил)бифенил-4-илметил]-1-(4,5,6,7-тетрагидро-1Н-бензимидазол-5-ил)пирролидин-2-он гидрохлорид Примеры 24 и 25 могут быть приготовлены практически, как описано в примерах 22 и 23, с тем лишь исключением, что 4-фторфенилборную кислоту заменяют 4-(морфолин-4-карбонил)фенилборной кислотой. Хиральная очистка приводит к получению: примера 24 (изомер 1, MS: m/z (M+1)=553, Rt. 7,3 мин, примера 25 (изомер 2, MS: m/z (M+1)=553, Rt. 16,6 мин. Пример 26. 1-(4,5,6,7-Тетрагидро-3H-бензимидазол-5-ил)-3-[R]-2,6-дихлор-4-(4-N,N-диметилсульфонилфенил)фенилпирролидин-2-он Вещество, полученное в синтезе 26 (0,15 г, 0,23 ммоль), растворяют в 2 мл диоксана. К полученному раствору добавляют 4-N,N-диметилсульфонилфенилборную кислоту (45 мг, 0,28 ммоль) и 0,5 мл 2М карбоната натрия. Смесь продувают азотом в течение 1 мин и добавляют тетракис(трифенилфосфин)палладий(0) (27 мг, 0,02 ммоль). Реакционную смесь укупоривают и нагревают, используя микроволновый нагреватель, в течение 45 мин при 90 °С. Реакционную смесь наносят на колонку SCX и промывают метанолом (два объема колонки). Затем, промывают 2н. NH 3 в метаноле, используя два объема колонки, получая получистый продукт. Далее его очищают, используя ЖХВР или хроматографию с нормальной фазой, получая 1-(4,5,6,7-тетрагидро-3H-бензимидазол-5-ил)-3-[R]-2,6-дихлор-4-(4-N,N-диметилсульфонилфенил)фенилпирролидин-2-он в виде белого твердого вещества (мг, 44%). MS: m/z=547 (M+1). Примеры, указанные в табл. 3, готовят практически, как описано в примере 26, с тем лишь исключением, что 4-N,N-диметилсульфонилфенилборную кислоту заменяют реагентом, указанным в колонке 3. Пример 42. 3-[R]-[3,5-Дихлор-4'-(4-трифторметилпиперидин-1-карбонил)бифенил-4-илметил]-1-(4,5,6,7-тетрагидро-3Н-бензимидазол-5-ил)пирролидин-2-он Вещество, полученное в синтезе 26 (0,10 г, 0,16 ммоль), растворяют в 2 мл диоксана. К полученному раствору добавляют (4-хлоркарбонилфенил)борный ангидрид (52 мг, 0,31 ммоль), гидрохлорид 4-трифторметилпиперидина (76 мг, 0,40 ммоль) и 0,5 мл 2М карбоната натрия. Смесь продувают азотом в течение 1 мин и добавляют тетракис(трифенилфосфин)палладий(0) (23 мг, 0,02 ммоль). Реакционную смесь укупоривают и нагревают, используя микроволновый нагреватель, в течение 30 мин при 110 °С. Реакционную смесь наносят на колонку SCX и промывают метанолом (два объема колонки). Затем, промывают 2н. NH 3 в метаноле, используя два объема колонки, получая получистый продукт. Далее его очищают, используя ЖХВР или хроматографию с нормальной фазой, получая 3-(3,5-дихлор-4'-морфолин-4-илбифенил-4-илметил)-1-(4,5,6,7-тетрагидро-1Н-бензимидазол-5-ил)пирролидин-2-он (43 мг). MS: m/z=625 (M+1). Пример 43. (R)-3-[3,5-Дихлор-4'-(4,4-дифторпиперидин-1-карбонил)бифенил-4-илметил]-1-(4,5,6,7-тетрагидро-1Н-бензимидазол-5-ил)пирролидин-2-он Соединение примера 43 может быть приготовлено практически, как описано в примере 42, с тем лишь исключением, что гидрохлорид 4-трифторметилпиперидина заменяют 4,4-дифторпиперидином. MS: m/z (M+1)=587. Пример 44. 3-(4-Бром-2-хлорбензил)-1-(4,5,6,7-тетрагидро-3Н-бензо[d]имидазол-5-ил)пирролидин-2-он, ТФУК соль Указанное в заголовке соединение получают в виде белого твердого вещества с выходом 0,52 г (45%), используя процедуру синтеза 32, и используя в качестве реагентов этил-2-(4-бром-2-хлорбензил)-4-оксобутаноат (0,95 г, 2,85 ммоль) и 4,5,6,7-тетрагидро-3Н-бензо[d]имидазол-5-амин (0,49 г, 2,85 ммоль). MS (режим APCI-pos) m/z (отн. интенсивность) 408,1 (80), 410,1 (100). В следующем разделе описан ферментативный и функциональный анализ, применяемый для оценки действия соединений, предлагаемых согласно настоящему изобретению. Ферментативный анализ 11 β-HSD типа 1. Активность человеческой 11 β-HSD типа 1 измеряли при помощи флуоресцентного анализа, оценивая выработку НАДФН (восстановленный никотинамидадениндинуклеотидфосфат). Твердые соединения растворяют в ДМСО до концентрации 10 мМ. Двадцать микролитров каждого раствора затем переносили в колонку 96-луночного полипропиленового планшета марки Nunc, где производят дальнейшее разбавление в 50 раз с последующим двукратным титрованием, десять раз в поперечном направлении планшета, с добавлением дополнительного количества ДМСО и использованием автоматизированной системы Tecan Genesis 200. Затем планшеты переносили в систему Tecan Freedom 200, снабженную 96-луночной головкой Tecan Temo и планшетным анализатором Ultra 384. Реагенты помещают в 96-луночные полипропиленовые планшеты Nunc и индивидуально распределяют по черным 96-луночным планшетам High Efficiency от Molecular Devices (емкость 40 микролитров на лунку (мкл/лунку)): 9 мкл/лунку субстрата (2,22 мМ НАДФ, 55,5 мкМ Кортизола, 10 мМ Трис, 0,25% Prionex, 0,1% Тритона Х100), 3 мкл/лунку воды в лунки, содержащие соединение, или 3 мкл в контрольные и стандартные лунки, 6 мкл/лунку рекомбинантного человеческого фермента 11 β-HSD первого типа, 2 мкл/лунку раствора соединения. Для окончательного определения процентного ингибирования добавляли ряд лунок, которые представляли собой минимум и максимум определения: один набор содержал субстрат с 667 мкМ карбеноксолона (фон), а другой набор содержал субстрат и фермент, но не содержал тестируемого соединения (максимальный сигнал). Конечные концентрации в ДМСО составляли 0,5% для всех соединений, контрольных и стандартных. Затем планшеты на 15 с помещали в шейкер (встряхивающее устройство) механической рукой Tecan, а затем закрывали и помещали в накопитель для инкубации в течение 3 ч при комнатной температуре. По завершении инкубации механическая рука извлекала каждый планшет по отдельности из накопителя и помещала их в положение, в котором производили добавление 5 мкл/лунку раствора карбеноксолона с концентрацией 250 мкМ для прекращения ферментной реакции. Затем планшеты встряхивали еще в течение 15 с, а затем помещали в анализатор планшетов Ultra 384 (355ЕХ/460ЕМ), при помощи которого определяли флуоресценцию НАДФН. Данные, полученные для соединений, исследованных в испытании 11- βHSD1, указаны ниже. Соединения, предлагаемые согласно настоящему изобретению, можно исследовать на селективность по отношению к 11- βHSD2 при помощи теста, аналогичного описанному для 11- βHSD1, но в котором используют фермент 11- βHSD2. Испытание с использованием фермента 11- βHSD2 может быть проведено при помощи способов, рассмотренных в настоящем описании, или способов, известных в данной области техники. Исследование клеток гладкой мускулатуры аорты человека. Первичные клетки гладкой мускулатуры аорты человека (AoSMC) культивировали в питательной среде, содержащей 5% FBS при количестве пассажей, равном 6, а затем гранулировали путем центрифугирования и суспендировали при плотности 9 ×10 4 клеток/мл в среде для количественного определения, содержащей 0,5% FBS и 12 нг/мл hTNF α для стимулирования экспрессии 11- β-HSD1. Клетки высевали на 96-луночные аналитические планшеты для выращивания тканевой культуры с концентрацией 100 мкл/лунку (9 ×10 3 клеток/лунку) и инкубировали в течение 48 ч при 37 °С, 5% CO 2 . После индуцирования клетки инкубировали в течение 4 ч при 31 °С, 5% СО 2 в среде для анализа, содержащей тестируемые соединения, а добавляли 10 мкМ кортизона, растворенного в среде для анализа, при концентрации 10 мкл/лунку, и инкубировали в течение 16 ч при 37 °C, 5% СО 2 . Среду из каждой лунки переносят на планшет для последующего анализа кортизола методом конкурентного флуоресцентного иммуноанализа с временным резонансным разрешением. В растворе конъюгат аллофикоцианина и (АРС)-кортизол и свободный кортизол конкурируют за связывание комплекса мышиное антитело к кортизону/европий (Eu)-анти мышиный IgG. Более высокие уровни свободного кортизола приводят к снижению переноса энергии от европия-IgG к комплексу АРС-кортизол, что снижает флуоресценцию АРС. Интенсивности флуоресценции европия и АРС измеряют, используя LJL Analyst AD. Возбуждение европия и АРС измеряют, используя возбуждение при 360 нм и эмиссионные фильтры на 615 нм и 650 нм, соответственно. Параметры временного разрешения для европия составляли: время интегрирования - 1000 мкс, задержка - 200 мкс. Параметры АРС составляли: время интегрирования - 150 мкс, задержка - 50 мкс. Интенсивности флуоресценции, измеренные для АРС, модифицировали делением на значение флуоресценции Eu (АРС/Eu). Это отношение затем использовали для определения неизвестной концентрации кортизола, которое осуществляли путем интерполяции с использованием стандартной кривой для кортизола, приведенной в соответствии с 4-параметрическим логистическим уравнением. Указанные концентрации затем использовали для определения активности соединений путем построения зависимости концентрации от % ингибирования, приведенной в соответствии с 4-параметрической кривой и показывающей IC 50 . Все рассмотренные в настоящем описании примеры проявляли активность в анализе клеток гладкой мускулатуры человеческой аорты, причем во всех случаях IC 50 составляла менее 300 нМ. Данные для некоторых соединений, полученные при анализе гладкомышечных клеток человеческой аорты, показаны ниже. Экспресс-анализ конверсии кортизона in vivo. В общем случае, соединения вводили мышам перорально, и спустя определенное время после введения соединения, мышам делали подкожную инъекцию кортизона, и спустя еще некоторое время у животных брали кровь на анализ. Отделяли сыворотку крови и анализировали методом жидкостной хроматографии-масс-спектрометрии/масс-спектрометрии, устанавливая уровень кортизона и кортизола, а затем устанавливали среднее содержание кортизола и процентное замедление в каждой группе испытуемых мышей. В частности, Harlan Sprague Dawley были поставлены самцы мышей C57BL/6, средняя масса тела которых составляла 25 г. Точные массы определяли сразу после получения мышей, и затем мышей делили на группы на основании близких масс. Различные дозы соединений готовили в смесях 1 мас.%, гидроксиэтилцеллюлозы (НЕС), 0,25 мас.%, полисорбата 80, 0,05 мас.%, противовспенивателя Dow Corning #1510-US на основании средней массы, которую принимали равной 25 г. Соединения вводили перорально, по 200 мкл каждому животному, а затем одному животному вводили подкожно 200 мкл кортизона в отношении 30 мг/кг спустя 1-24 ч после введения соединения. Спустя 10 мин после стимулирования кортизоном, каждое животное умерщвляли путем помещения на 1 мин в камеру, заполненную СО 2 , затем отбирали кровь путем пункции сердца в пробирки, для отделения сыворотки. После полного сворачивания крови в пробирках, их центрифугировали при 2500 ×g, 4 °С в течение 15 мин, сыворотку переносили в лунки 96-луночных планшетов (блок пробирок Corning Inc, Costar #4410, 1,2 мл, полипропилен) и замораживали планшеты при -20 °С до начала анализа методом жидкостной хроматографии-масс-спектрометрии/масс-спектрометрии. Для анализа образцы сыворотки размораживали и осаждали белок добавлением ацетонитрила, содержащего внутренний стандарт d4-кортизола. Образцы перемешивали вихревым образом (на вортексе) и центрифугировали. Жидкость над осадком удаляли и сушили в токе теплого азота. Экстракты вновь растворяли в смеси метанол/вода (1:1) и вводили в систему для жидкостной хроматографии-масс-спектрометрии/масс-спектрометрии. Уровни кортизона и кортизола оценивали в режиме селективного мониторинга реакции с последующей ACPI ионизацией на тройном квадрупольном масс-спектрофотометре. Данные экспресс-анализа конверсии кортизона in vivo, полученные для некоторых соединений, приведены ниже Фармацевтически приемлемые соли и общие способы их приготовления известны в данной области техники. См., например, P. Stahl, et al., Handbook of pharmaceutical salts: properties, selection and use, (VCHA/Wiley-VCH, 2002); S.M. Berge, et al., "Pharmaceutical Salts", Journal of Pharmaceutical Sciences, Vol. 66, № 1, January 1977. Соединения, предлагаемые согласно настоящему изобретению, предпочтительно изготавливают в виде фармацевтических составов, вводимых различными способами. Наиболее предпочтительными являются составы для перорального введения. Такие фармацевтические составы и способы их приготовления хорошо известны в данной области техники. См., например, Remington: The science and practice of pharmacy (A. Gennaro, et al., eds., 19 th ed., Mack Publishing Co., 1995). Конкретная доза соединения, отвечающего формуле (I), или его фармацевтически приемлемой соли, необходимая для получения эффективного количества, определяемого согласно настоящему изобретению, зависит от конкретных обстоятельств и состояний, подвергаемых лечению. Выбор дозы, способа введения и частоты введения лучше всего поручить лечащему врачу. В общем случае, приемлемые и эффективные дозировки для перорального или парентерального введения составляют приблизительно от 0,1 мг/кг/сутки приблизительно до 10 мг/кг/сутки, что означает приблизительно от 6 мг до 600 мг, и чаще от 30 мг до 200 мг для человека. Такие дозировки вводят пациенту, нуждающемуся в лечении, от одного до трех раз в сутки или с такой частотой, которая требуется для лечения заболевания, выбираемого из заболеваний, рассмотренных в настоящем описании. Специалист в области приготовления рецептур может легко выбрать нужную форму и способ введения в зависимости от конкретных характеристик выбранного соединения, нарушения или состояния, подвергаемого лечению, стадии развития нарушения или состояния или других важных факторов. (Remington's Pharmaceutical Sciences, 18th Edition, Mack Publishing Co. (1990)). Заявляемые соединения могут быть введены при помощи ряда способов. При проведении лечения пациента, страдающего рассмотренным в настоящем описании заболеванием или склонного к развитию рассмотренного в настоящем описании заболевания, соединение, отвечающее формуле (I), или его фармацевтически приемлемая соль могут быть введены в любой форме или любым способом, который позволяет сделать эффективное количество указанного соединения или соли биодоступным, включая пероральное и парентеральное введение. Например, активные соединения могут быть введены ректально, перорально, ингаляцией или подкожным, внутримышечным, внутривенным, трансдермальным, интраназальным, ректальным, офтальмологическим, топическим, сублингвальным, буккальным либо другим способом. Для лечения рассмотренных в настоящем описании нарушений предпочтительным является пероральный способ введения. В тех случаях, когда пероральный способ введения невозможен или не является предпочтительным, состав может быть приготовлен в форме, пригодной для парентерального введения, например внутривенного, внутрибрюшинного или внутримышечного введения. |
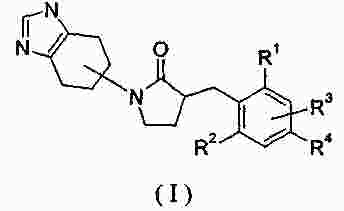
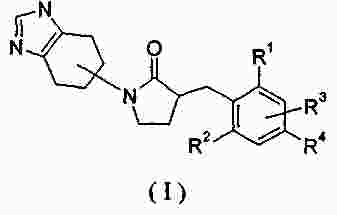
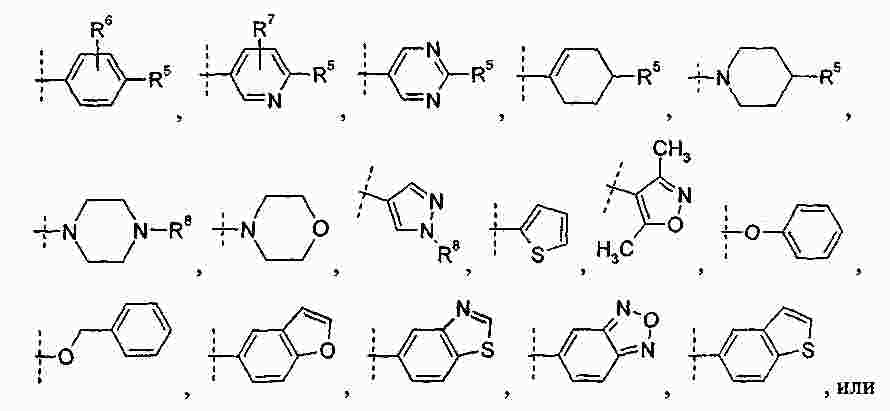
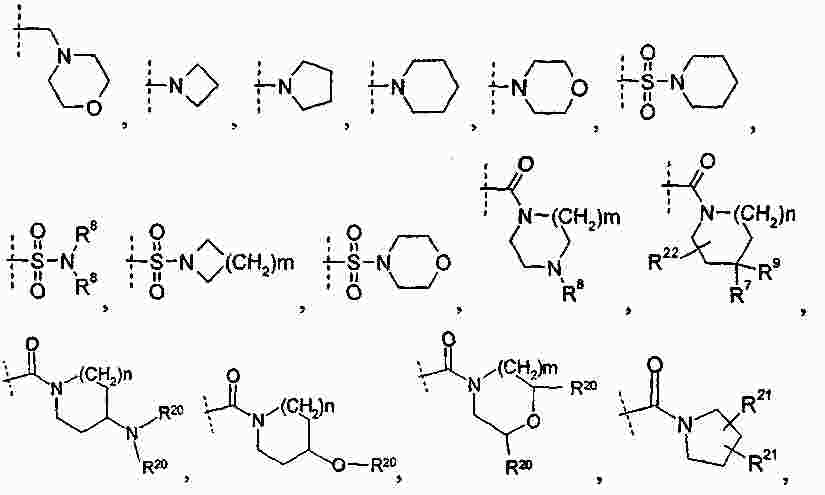
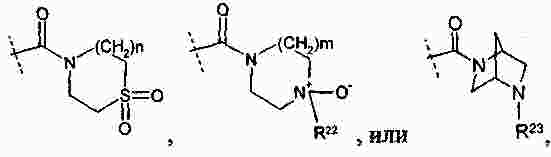
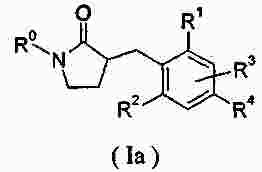
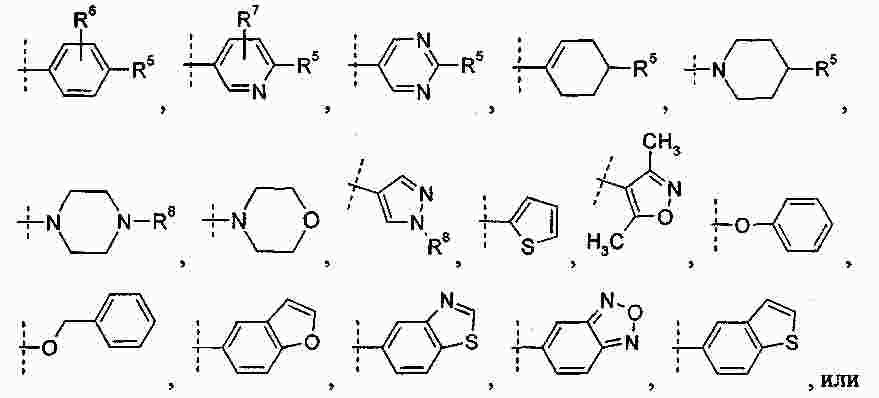
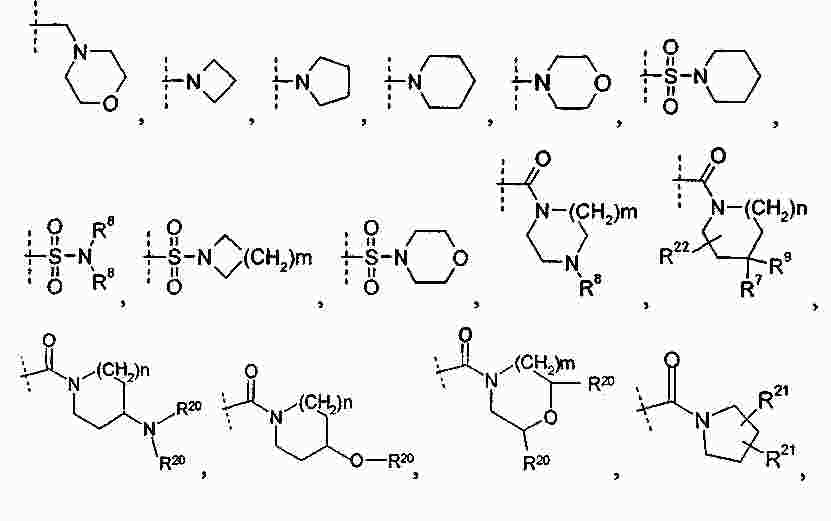
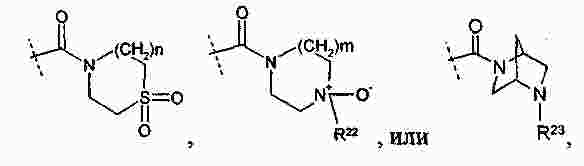
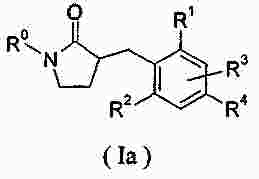
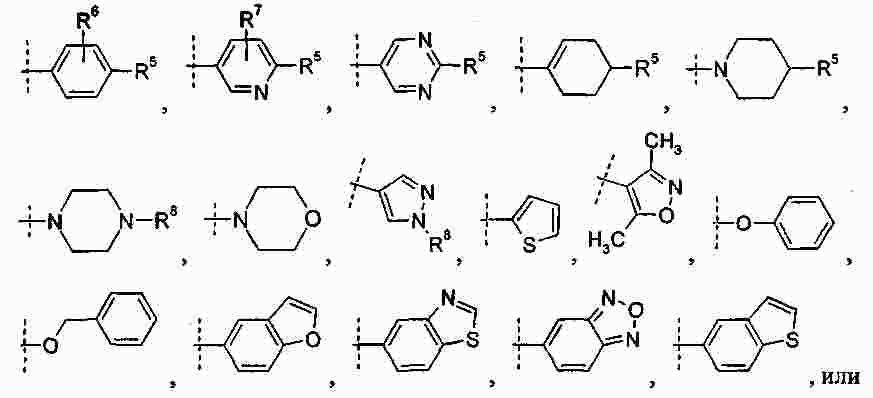
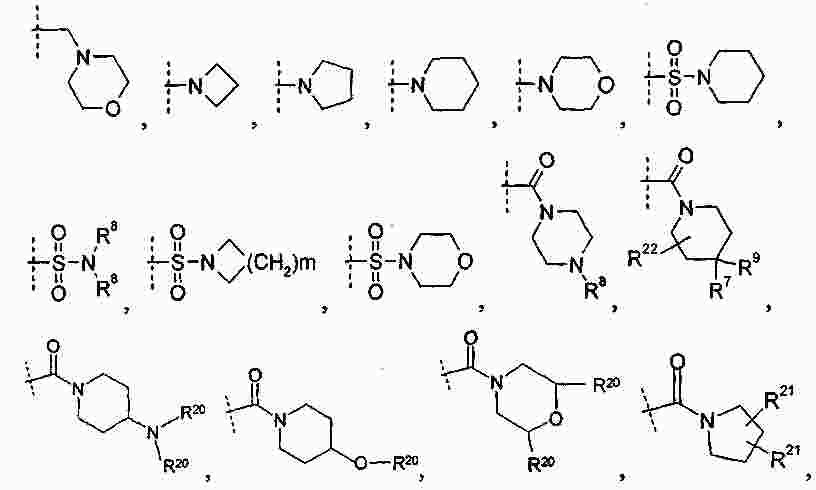
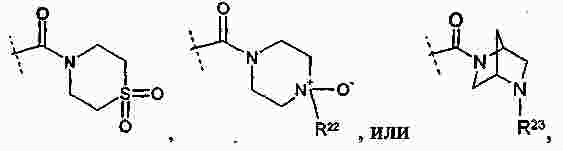
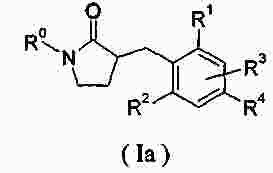
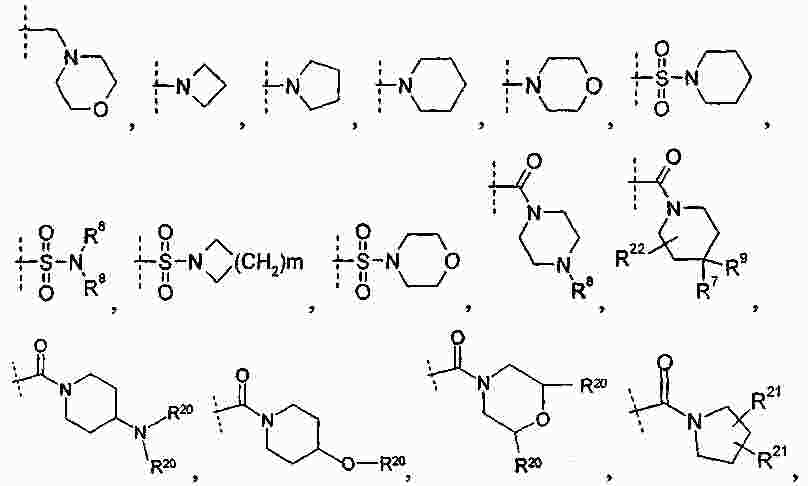
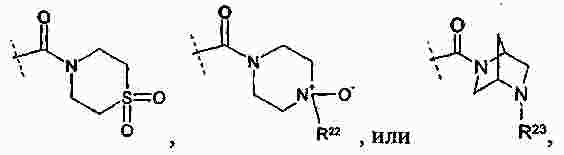
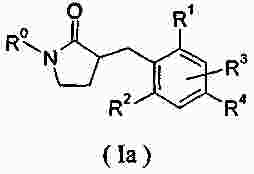
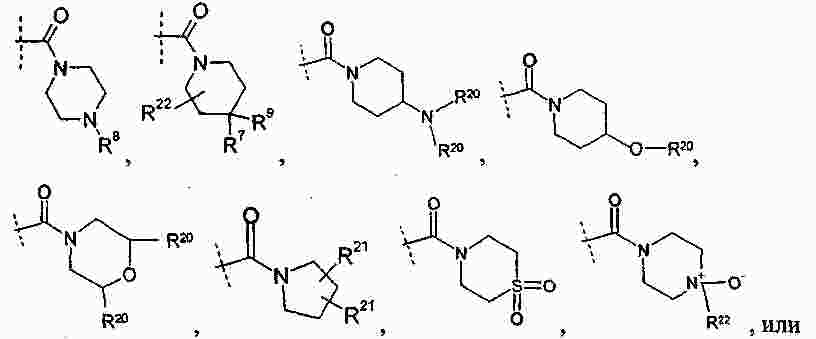
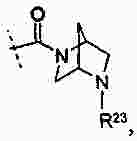
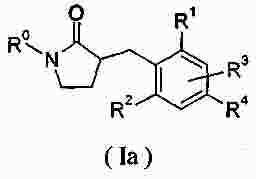
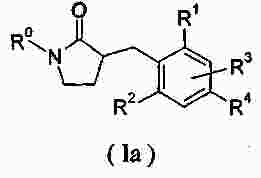
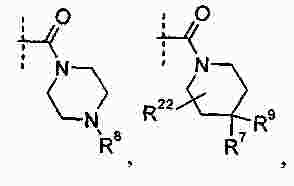
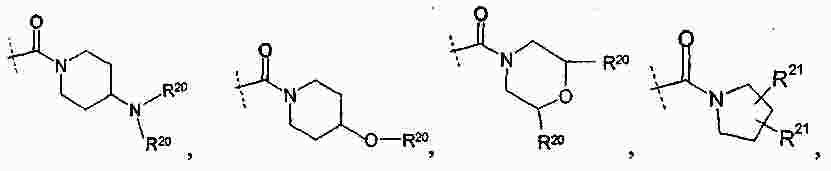
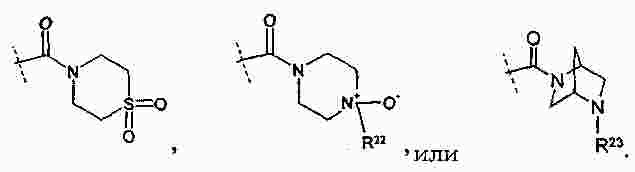 Предпочтительно R 5 представляет собой
Предпочтительно R 5 представляет собой 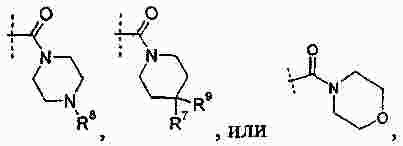 и R 8 представляет собой -(C 1 -С 3 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена). Предпочтительно R 5 представляет собой галоген. Предпочтительно R 5 представляет собой хлор. Предпочтительно R 5 представляет собой фтор.
и R 8 представляет собой -(C 1 -С 3 )алкил (необязательно имеющий в качестве заместителей от 1 до 3 атомов галогена). Предпочтительно R 5 представляет собой галоген. Предпочтительно R 5 представляет собой хлор. Предпочтительно R 5 представляет собой фтор.