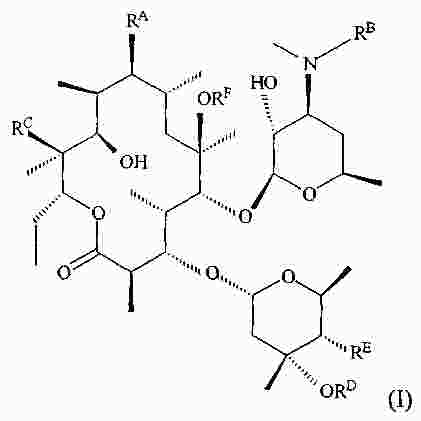
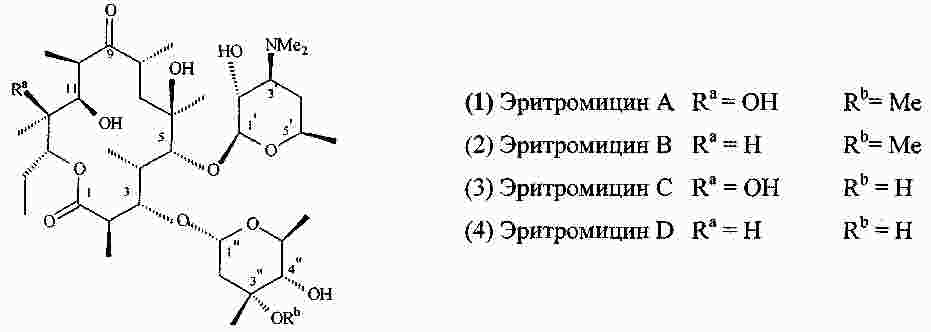
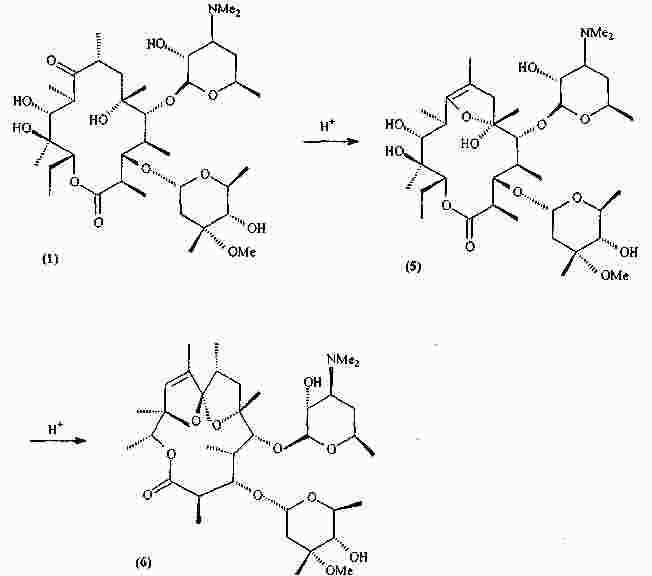
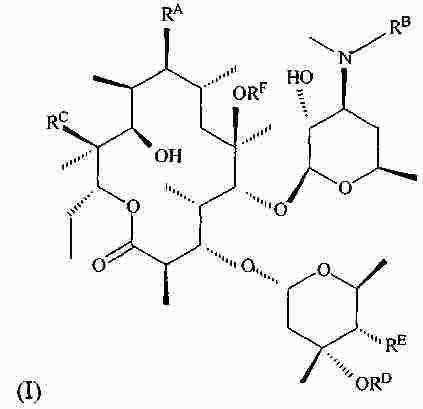
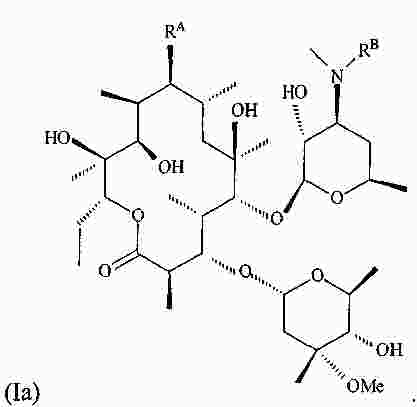
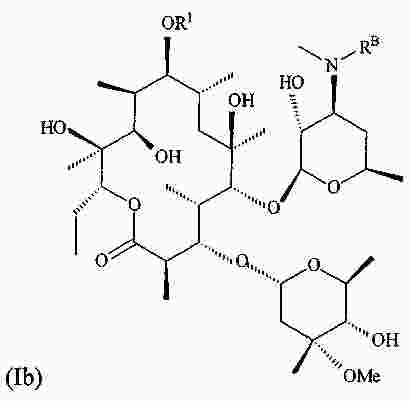
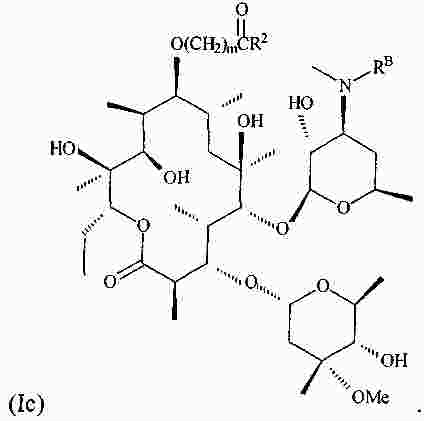
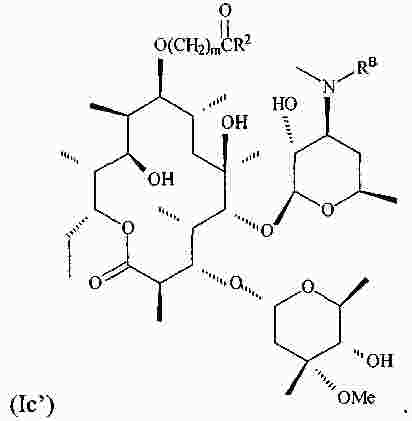
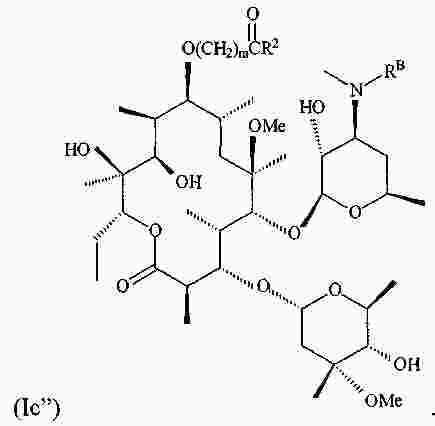
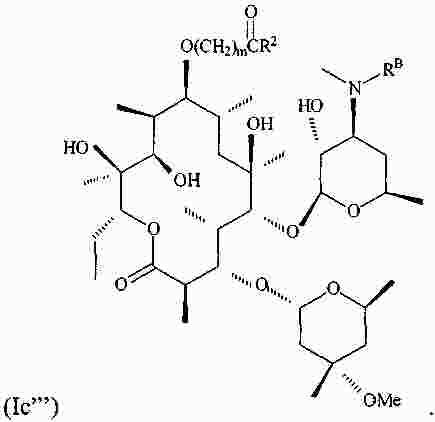
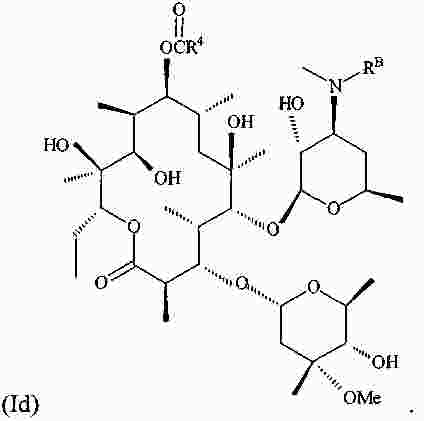
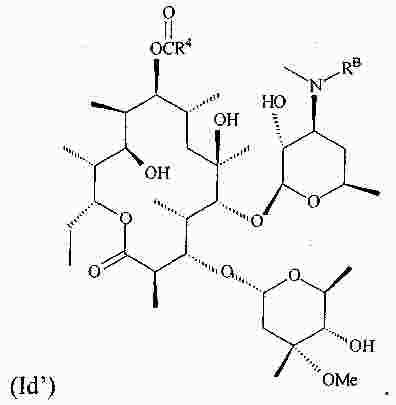
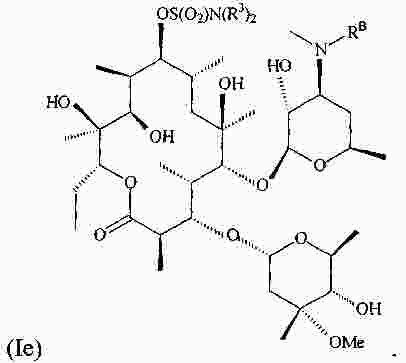
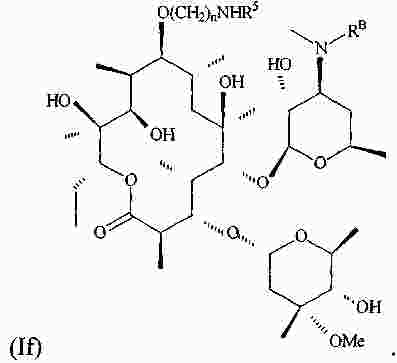
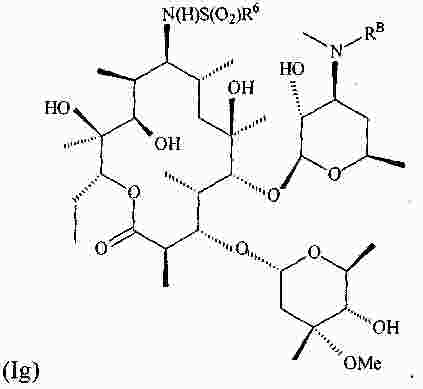
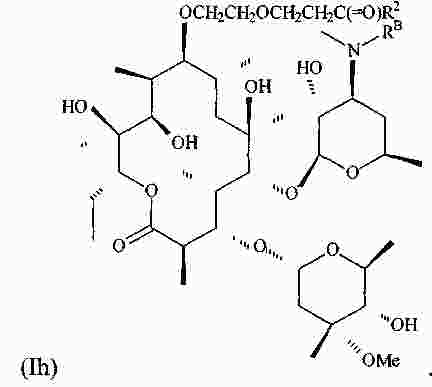
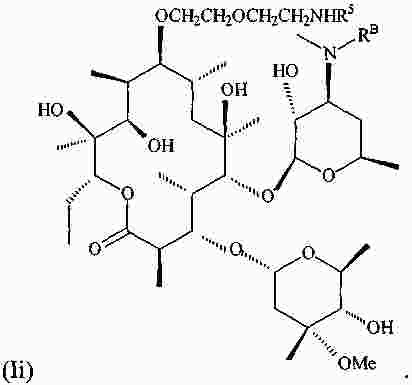
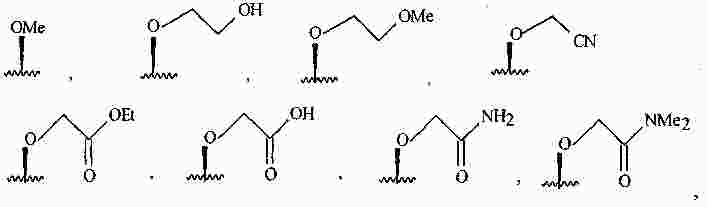
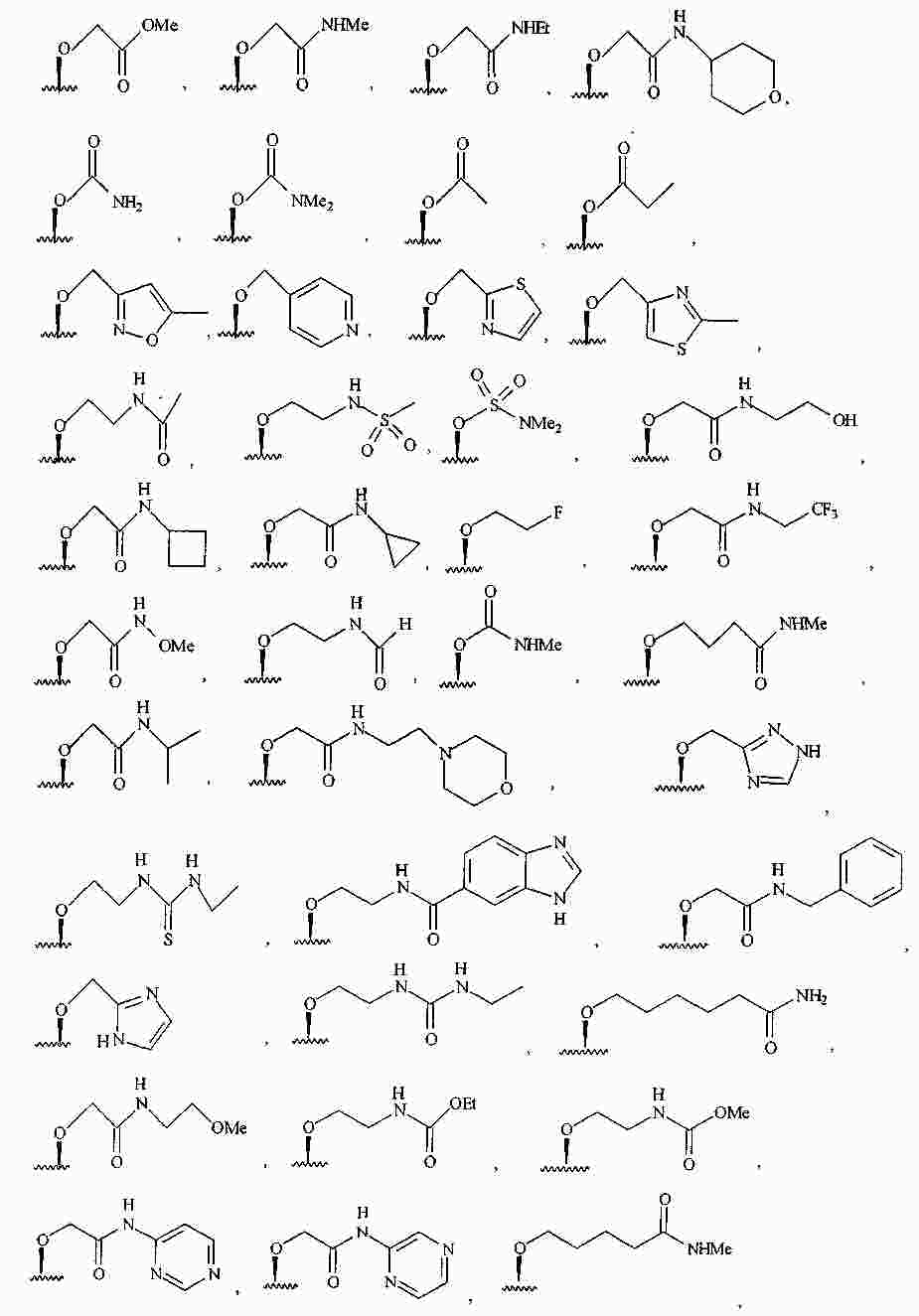
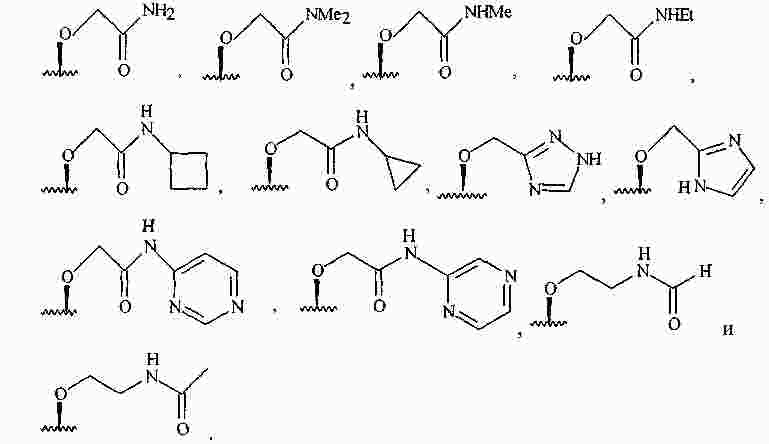
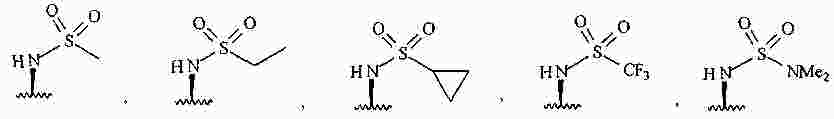
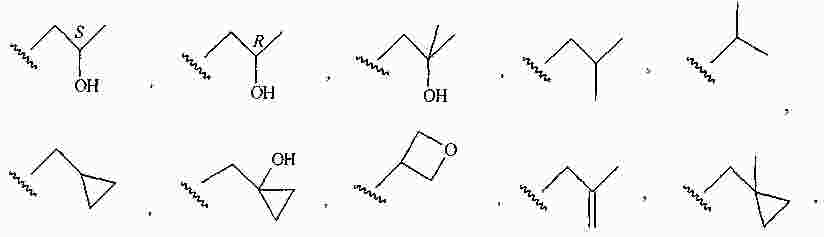
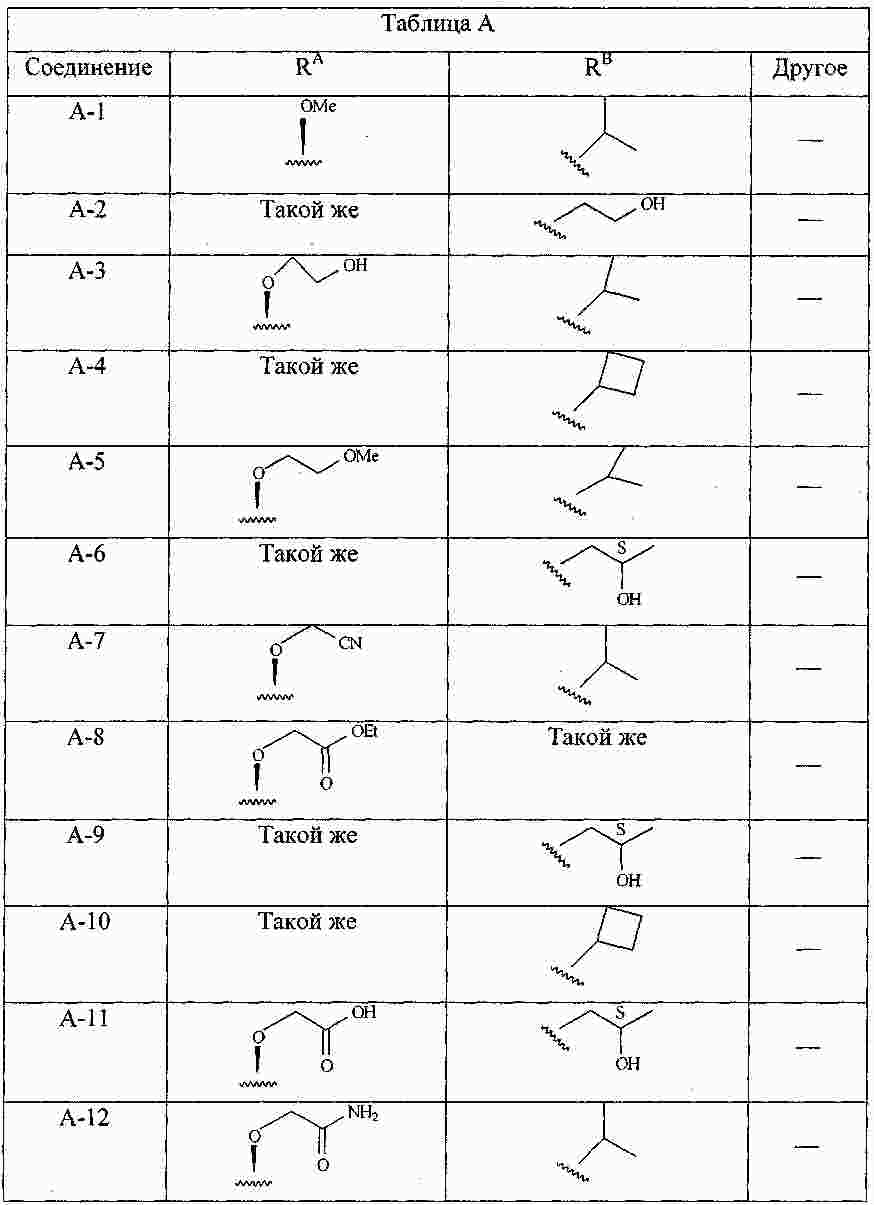
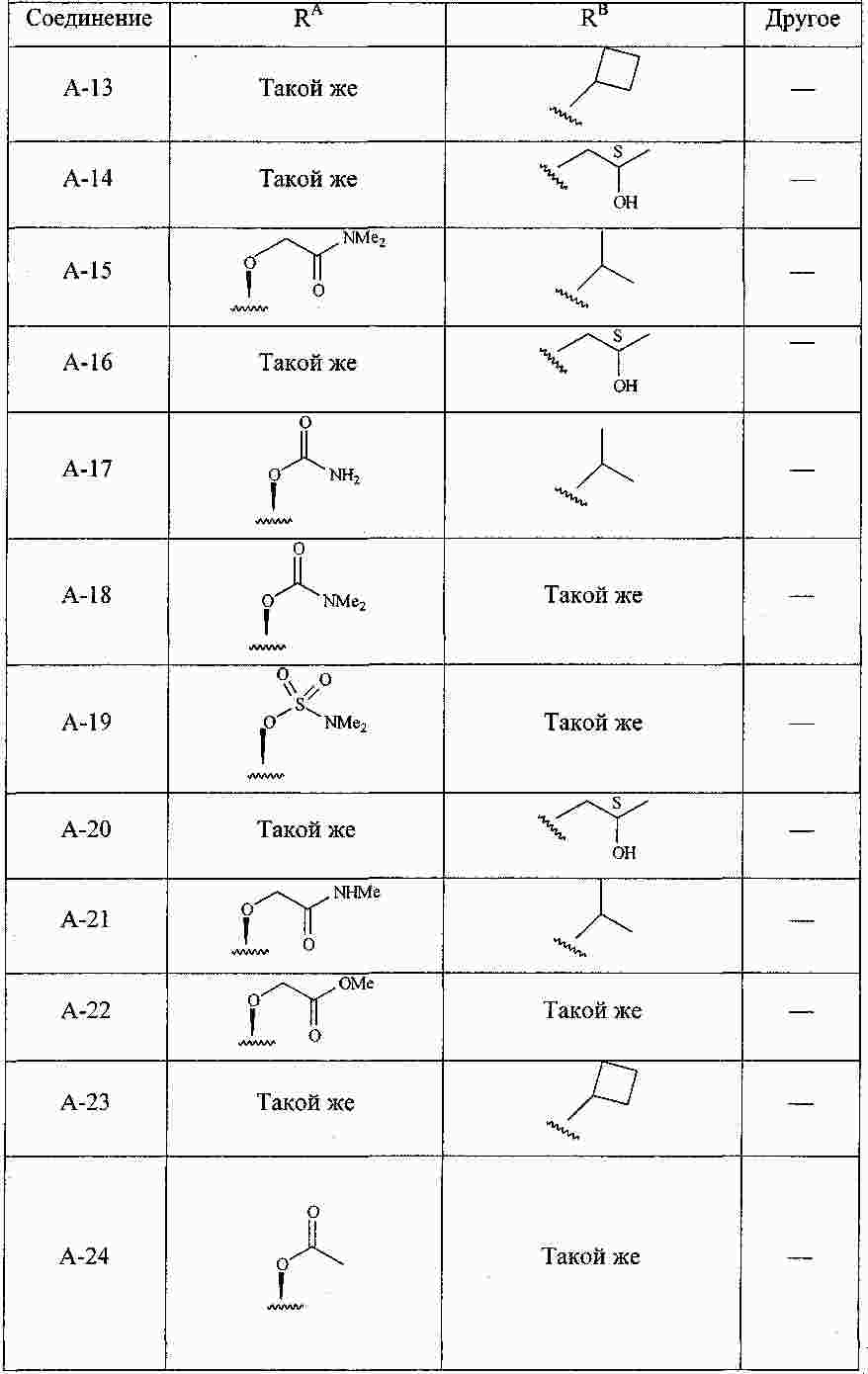
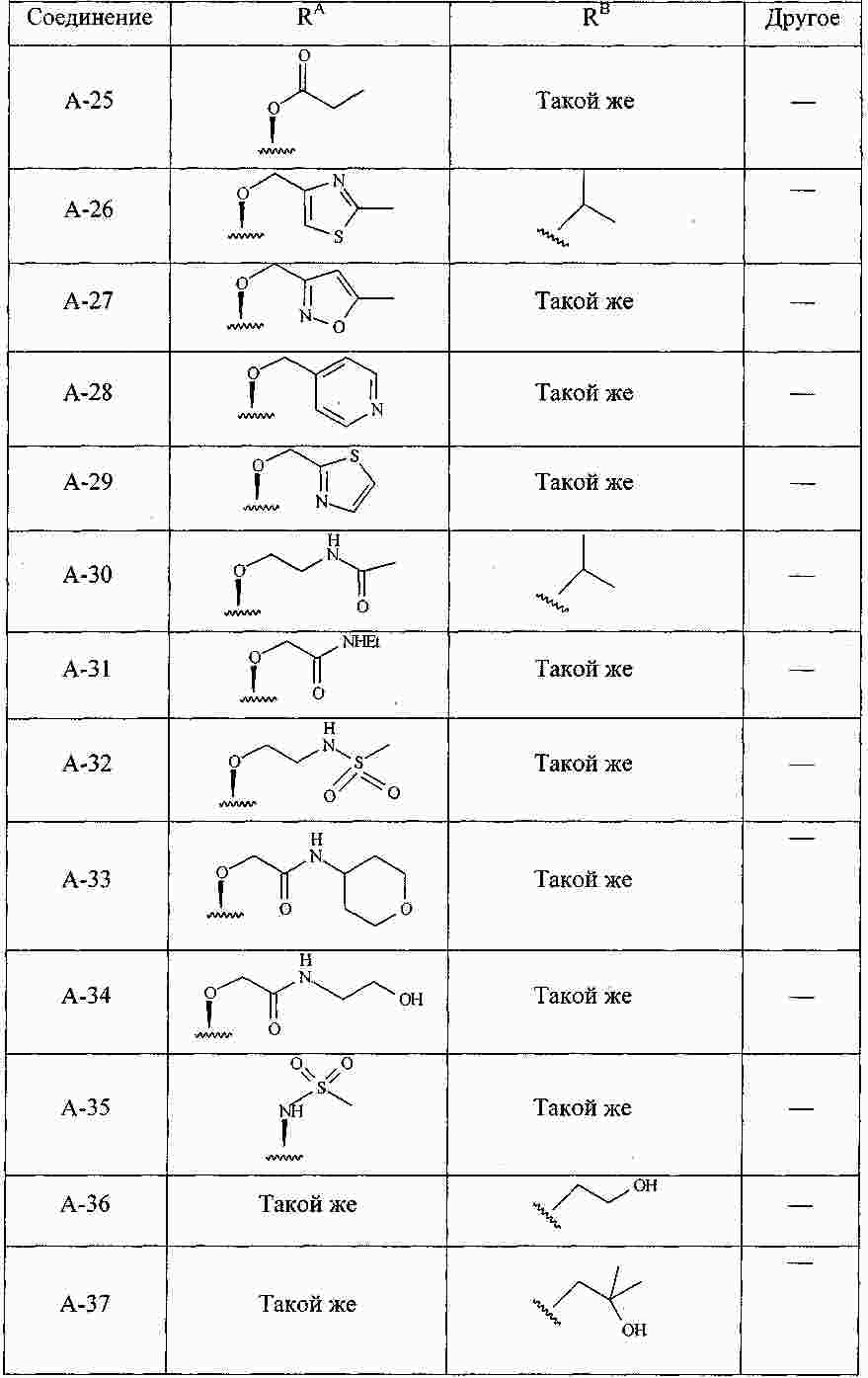
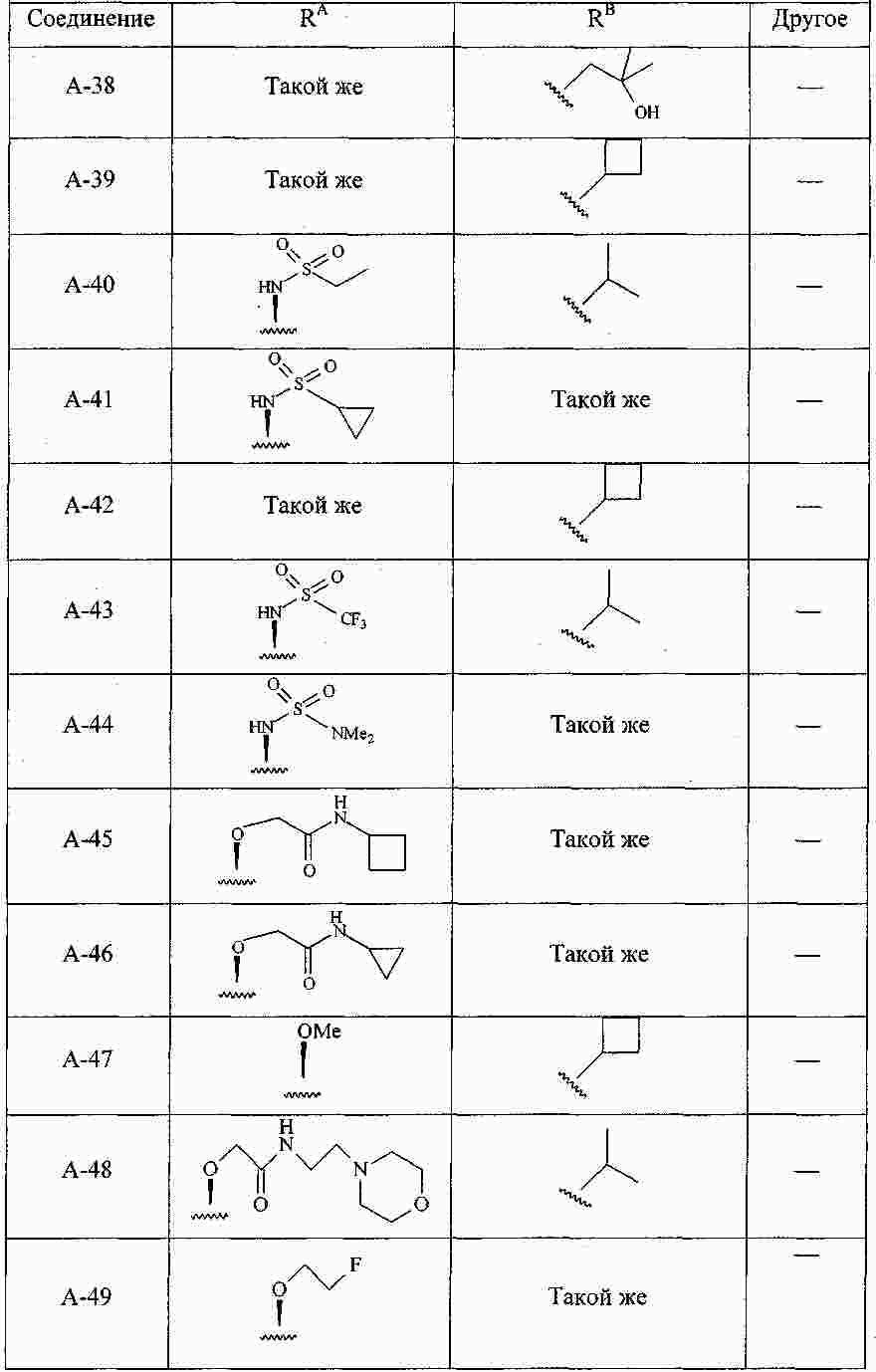
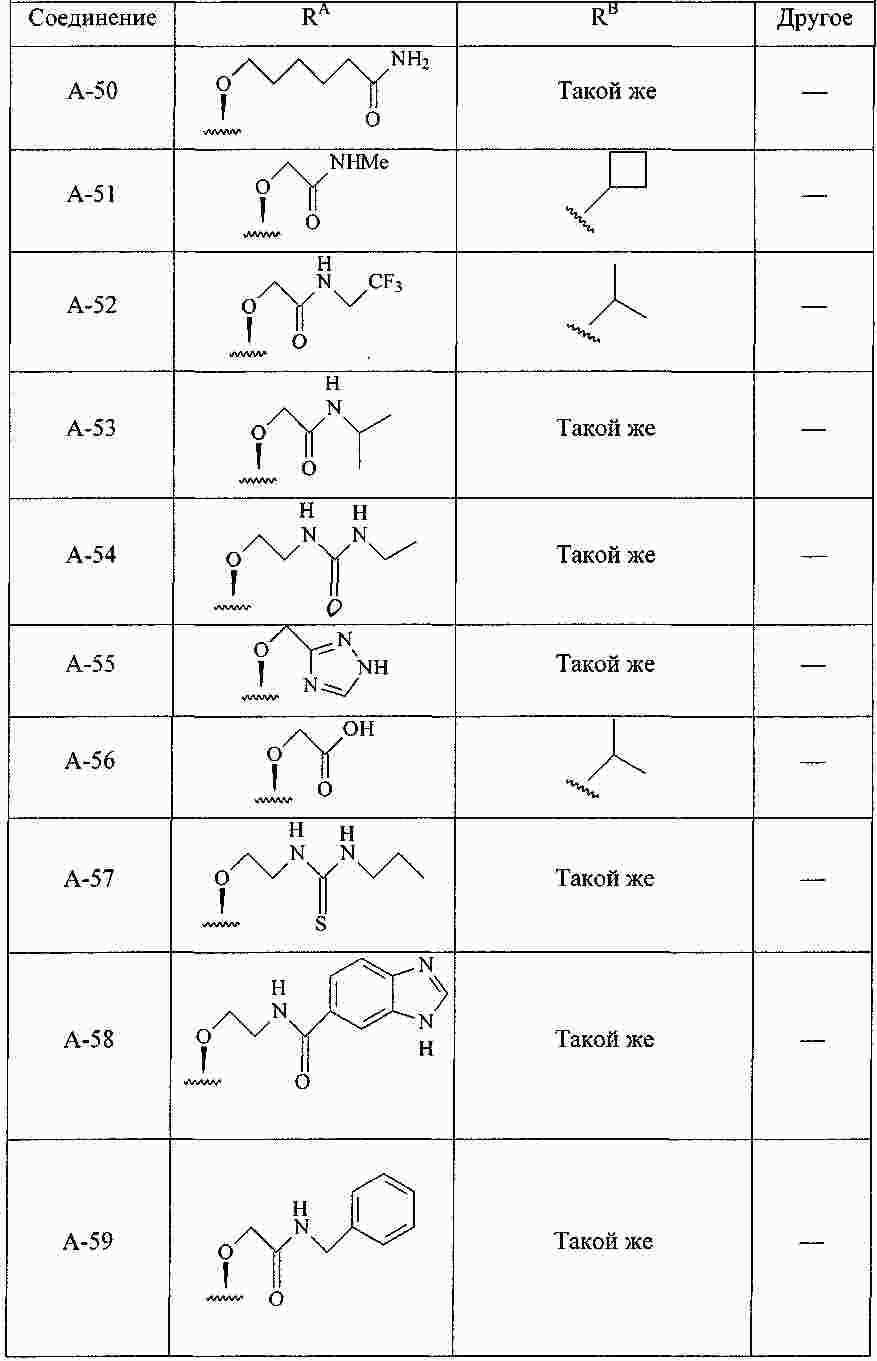
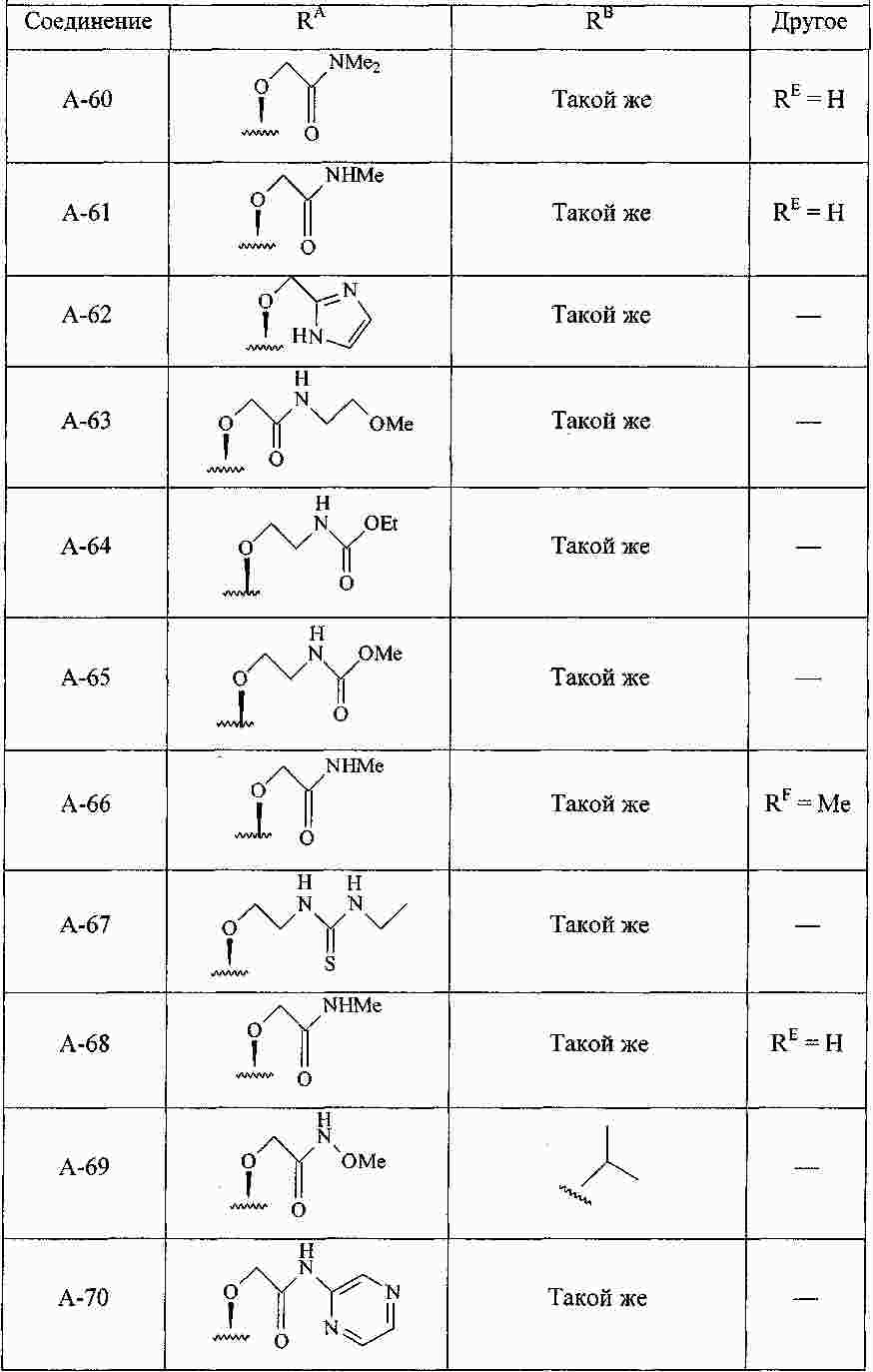
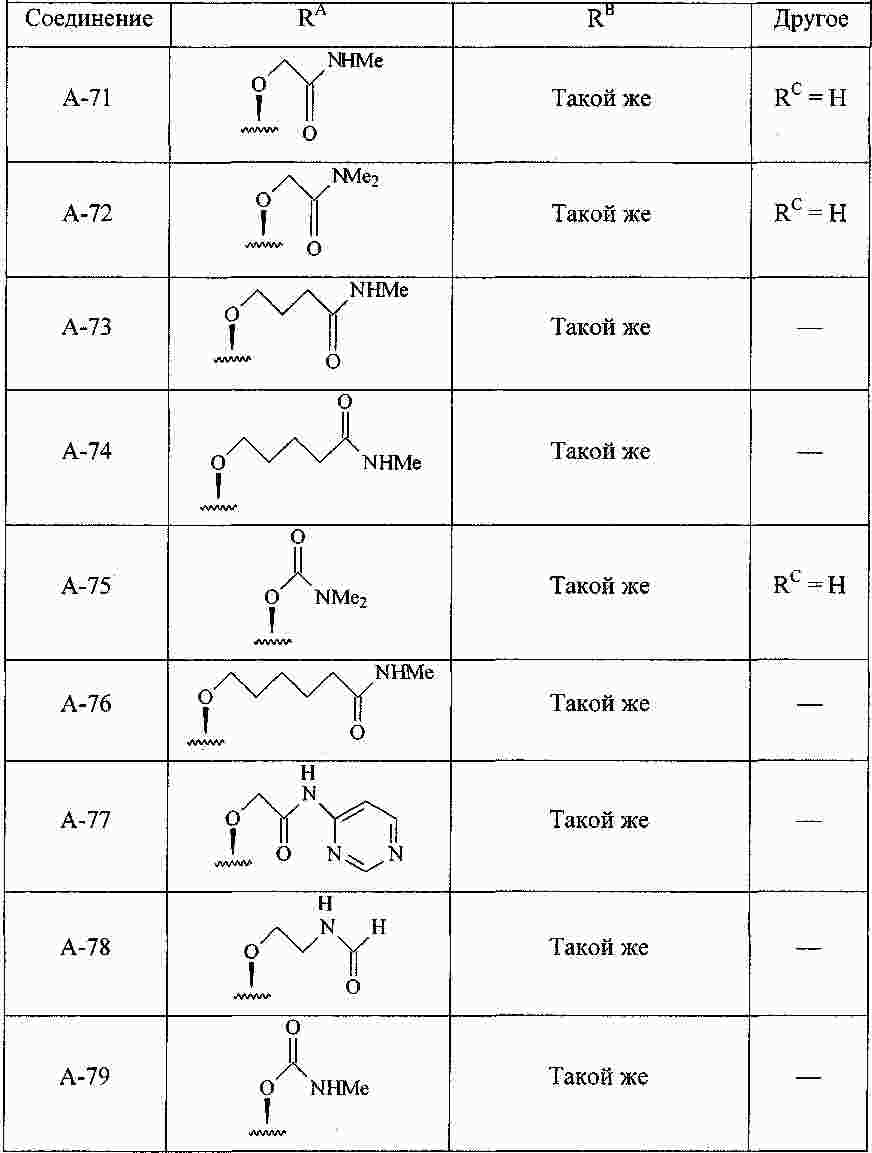
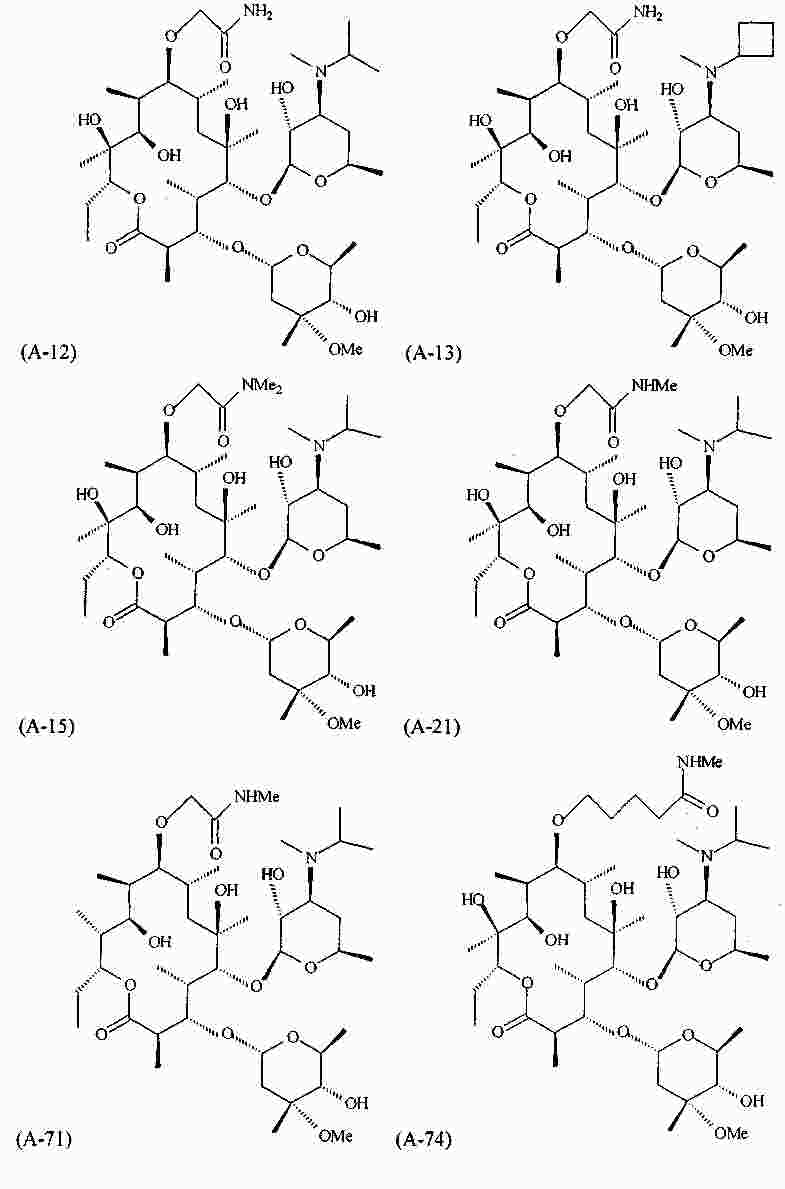
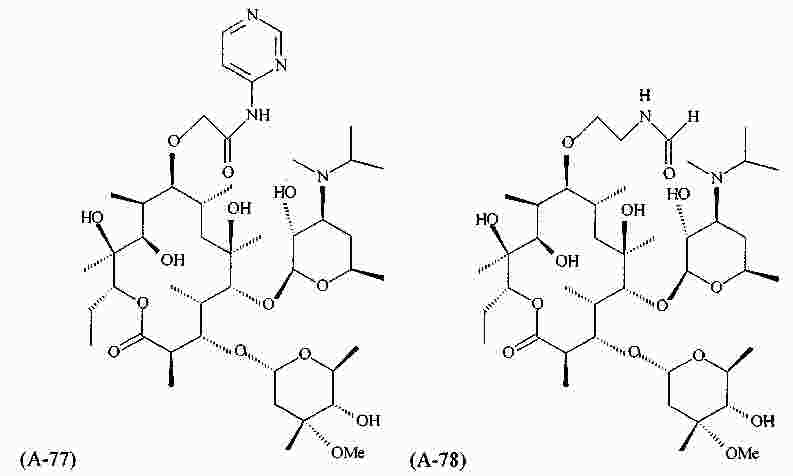
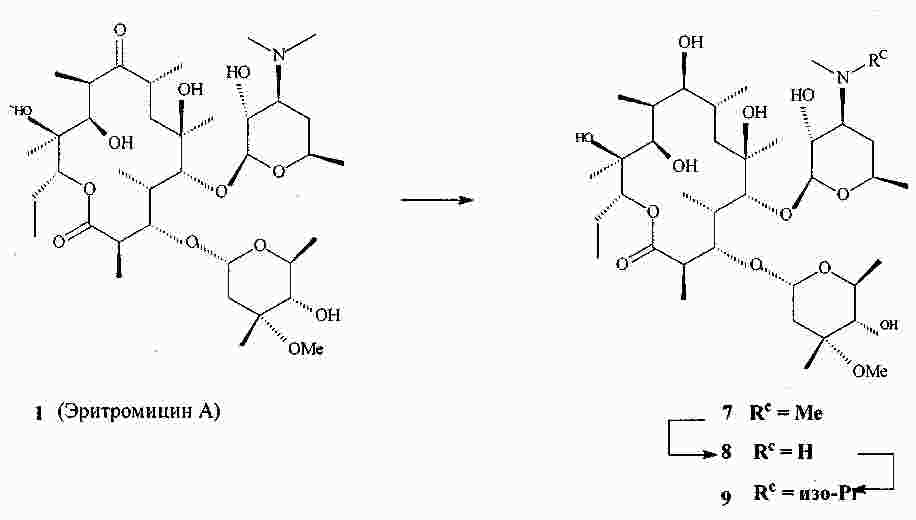
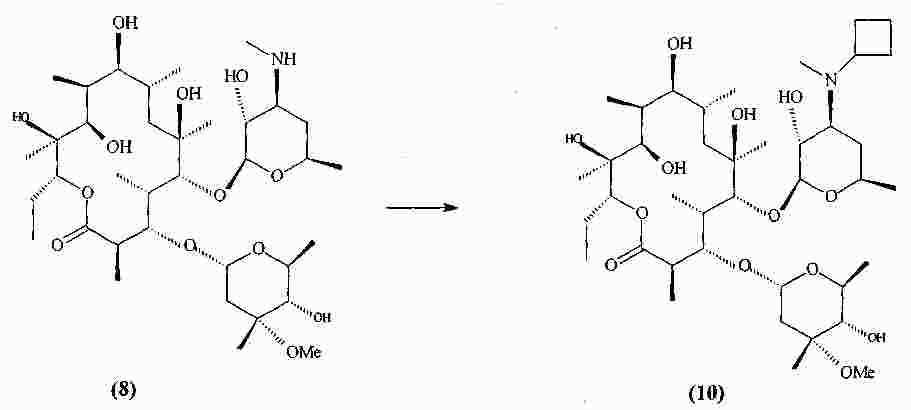
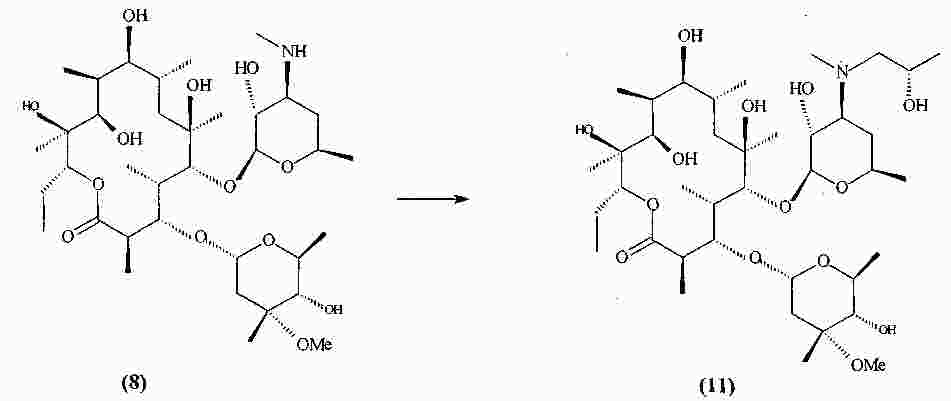
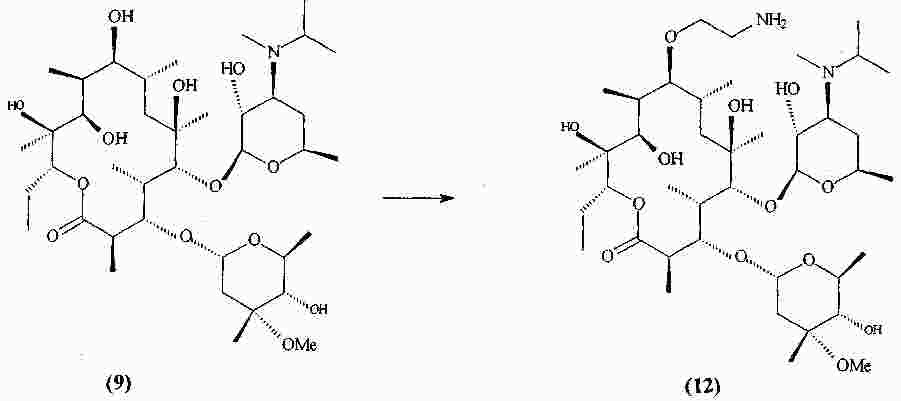
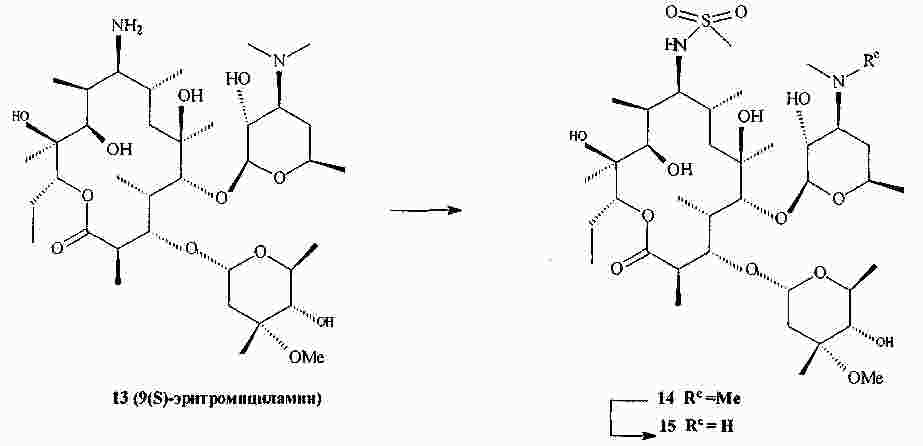
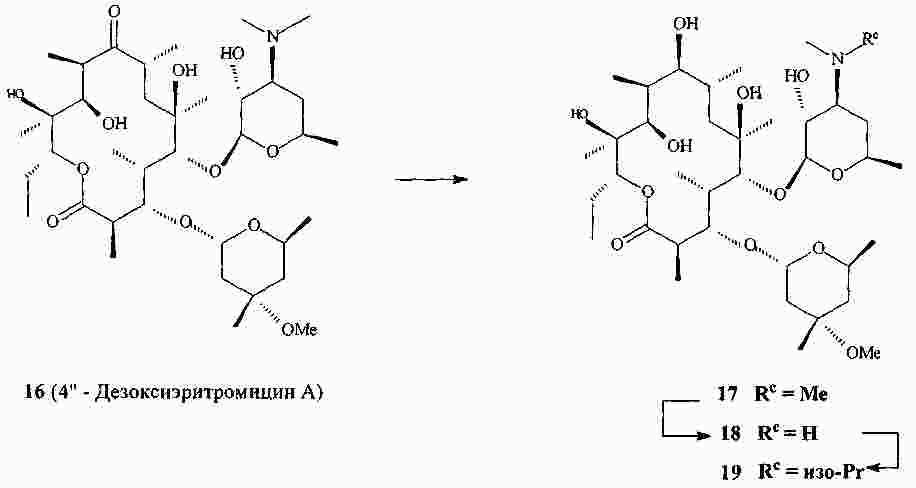
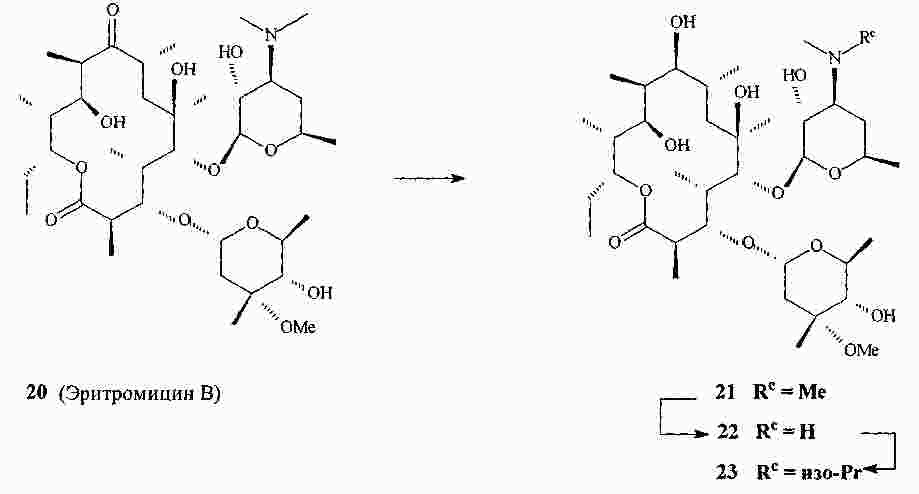
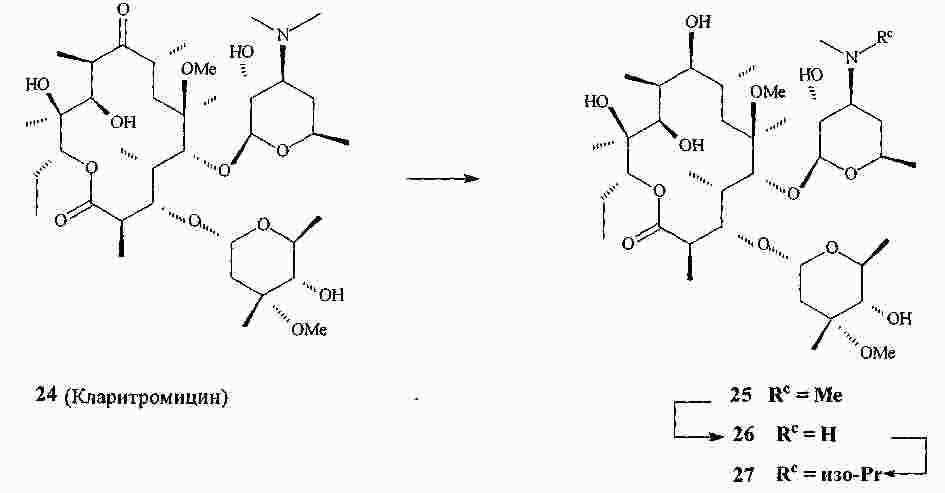
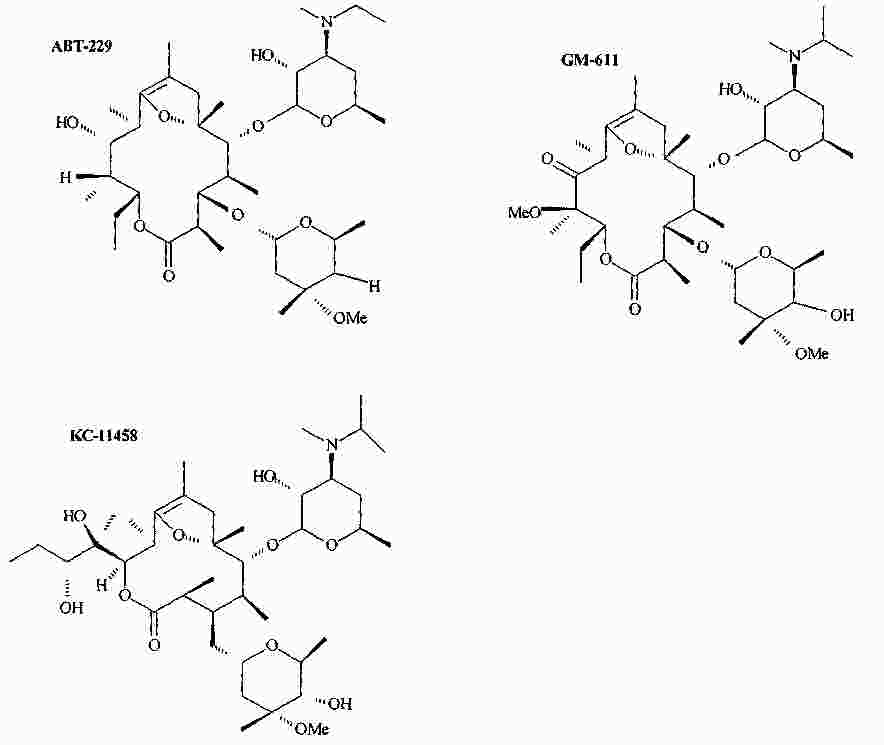
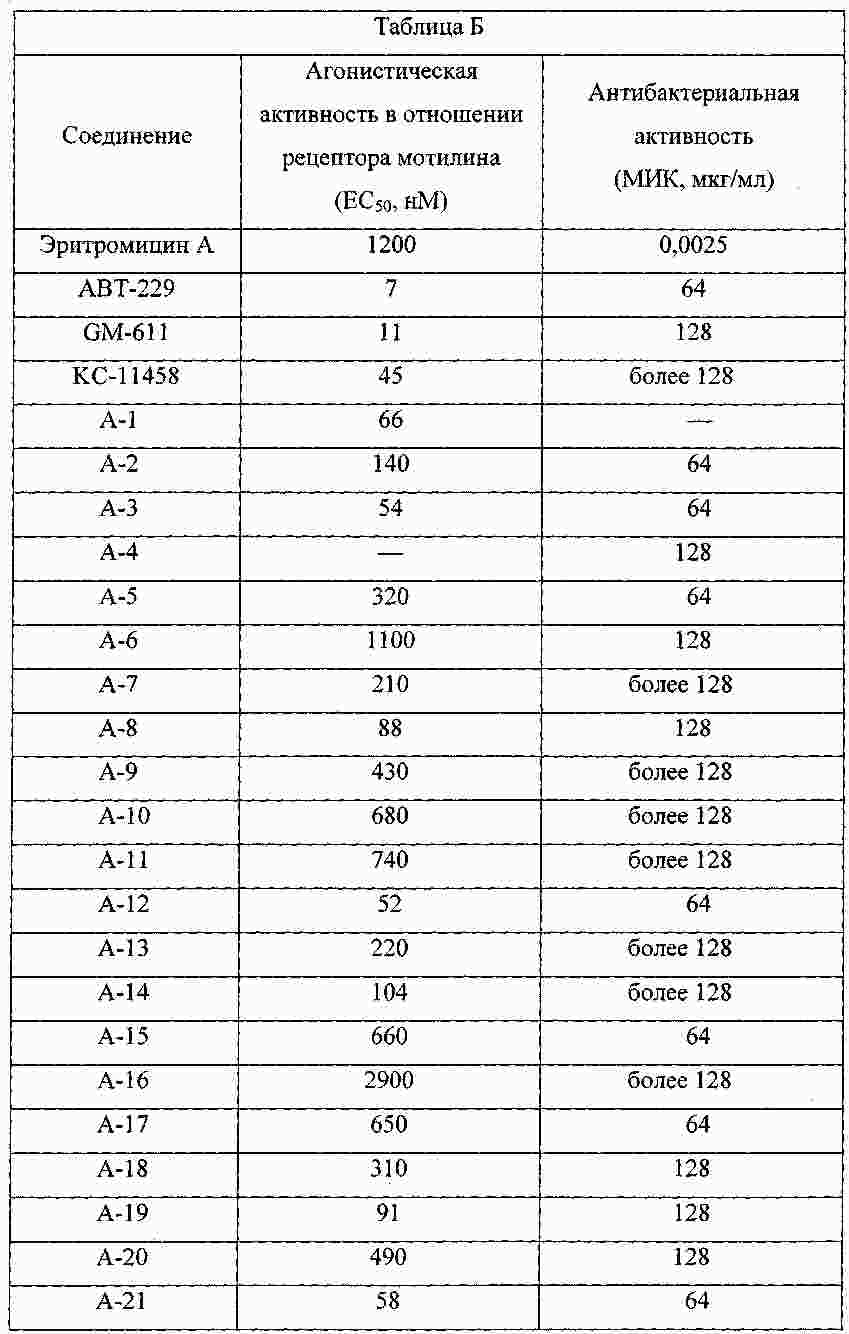
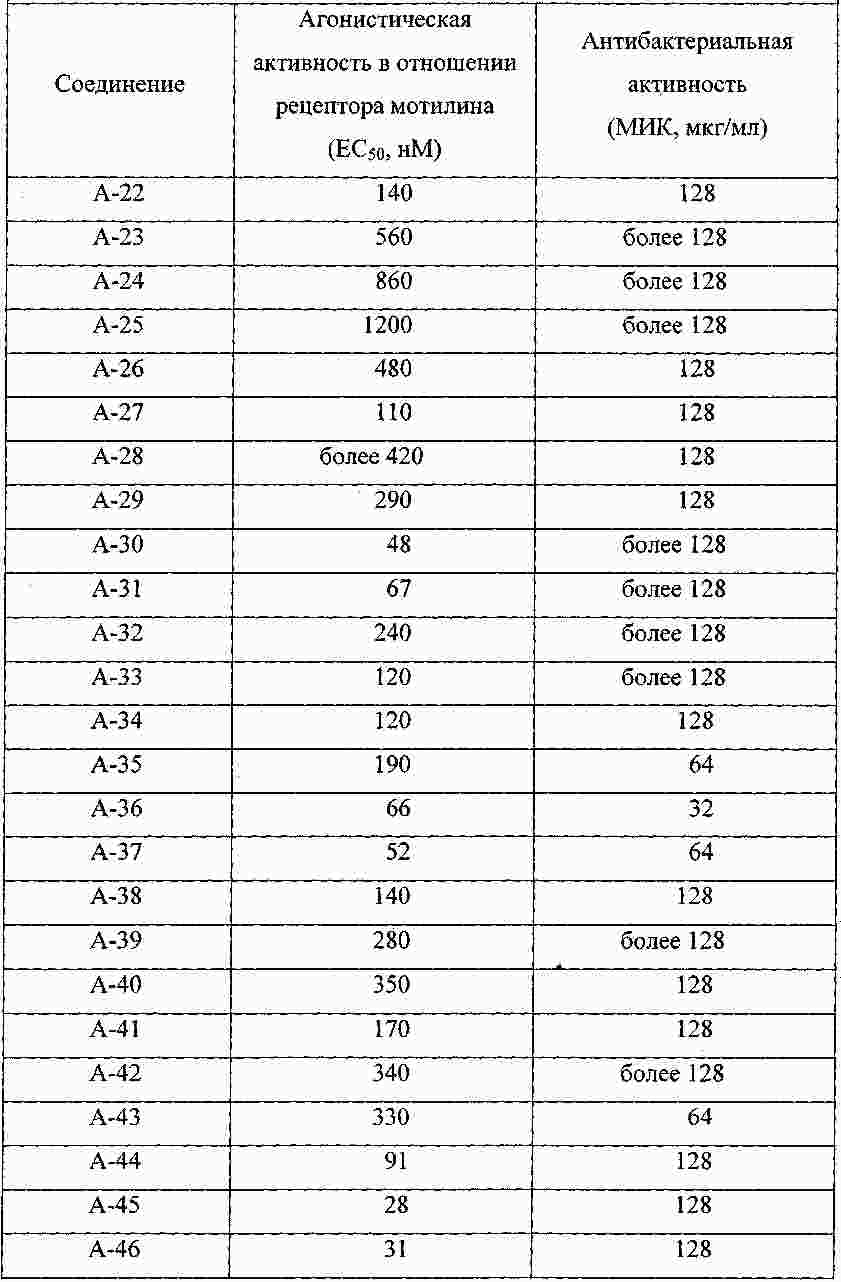
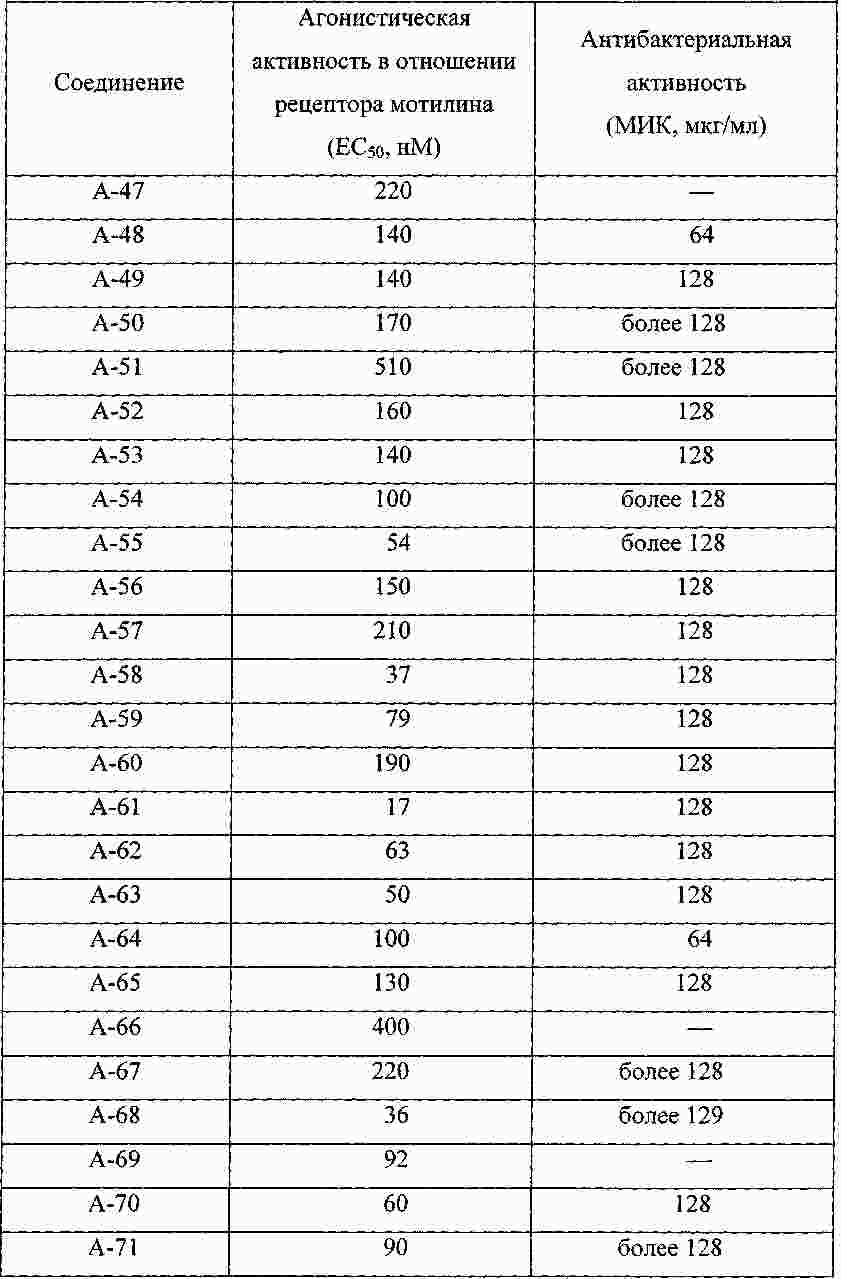
|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea000014711b*\id |
|
Термины запроса в документе Реферат Предложены соединения, имеющие структуру согласно формуле (I), где R A , R B , R C , R D , R E и R F являются такими, как определено в описании, полезные в качестве прокинетических агентов; содержащая их фармацевтическая композиция, их применение, способ лечения заболевания со сниженной моторикой желудочно-кишечного тракта и способ индукции сокращения ткани. Формула [0001] Соединение, имеющее структуру, представленную формулой (I) [0002] Соединение по п.1, имеющее структуру, представленную формулой (Ia) [0003] Соединение по п.1, где RA выбран из группы, состоящей из [0004] Соединение по п.1, где RA выбран из группы, состоящей из [0005] Соединение по п.1, где RA выбран из группы, состоящей из [0006] Соединение по п.5, где RB представляет собой [0007] Соединение по п.1, имеющее структуру, представленную формулой Ib, Ic, Ic', Ic", Ic'", Id, Id', Ie, If, Ig, Ih или Ii [0008] Соединение по п.1, где RB выбран из группы, состоящей из этила, н-пропила, н-бутила, 2-бутила, [0009] Соединение по п.1, где RB выбран из группы, состоящей из [0010] Соединение по п.1, имеющее структуру, представленную формулой А-12, А-13, А-15, А-21, А-71, А-74, А-77 или А-78 [0011] Способ лечения заболевания со сниженной моторикой желудочно-кишечного тракта, включающий введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по п.1. [0012] Способ по п.11, где заболевание выбрано из группы, состоящей из гастропареза, гастроэзофагеальной рефлюксной болезни, анорексии, стаза желчного пузыря, послеоперационной паралитической непроходимости кишечника, склеродермы, псевдонепроходимости кишечника, гастрита, рвоты и хронической констипации (инертная толстая кишка). [0013] Фармацевтическая композиция, содержащая соединение по п.1 и эксципиент. [0014] Способ индукции сокращения ткани, способной к сократительному ответу на мотилин, включающий приведение этой ткани в контакт с соединением по п.1 в количестве, эффективном для индукции такого сокращения. [0015] Способ по п.14, где ткань представляет собой человеческую ткань. [0016] Применение соединения по п.1 для изготовления лекарственного средства для лечения заболевания со сниженной моторикой желудочно-кишечного тракта. Полный текст патента Область изобретения Данное изобретение относится к агентам для лечения нарушений моторики желудочно-кишечного тракта и к способам их получения и применению. Моторика желудочно-кишечного тракта (ЖКТ) регулирует упорядоченное движение перевариваемого материала через кишечник для обеспечения адекватной абсорбции питательных веществ, электролитов и жидкостей. Надлежащий переход содержимого ЖКТ через пищевод, желудок, тонкую кишку и толстую кишку зависит от регионального контроля внутрипросветного давления и ряда сфинктеров, которые регулируют его поступательное движение и предотвращают обратное забрасывание. Нормальная моторика ЖКТ может быть нарушена под влиянием ряда обстоятельств, включая заболевание или хирургическое вмешательство. Нарушения моторики желудочно-кишечного тракта включают гастропарез и гастроэзофагеальную рефлюксную болезнь (ГЭРБ). Гастропарез, симптомы которого включают желудочное расстройство, изжогу, тошноту и рвоту, представляет собой отсроченное опорожнение желудка. ГЭРБ относится к различным клиническим проявлениям рефлюкса содержимого желудка и двенадцатиперстной кишки в пищевод. Наиболее распространенными симптомами являются изжога и дисфазия с известными также случаями кровопотери из-за эрозии пищевода. Другие примеры расстройств ЖКТ, в которые вовлечена нарушенная моторика ЖКТ, включают анорексию, стаз желчного пузыря, послеоперационную паралитическую непроходимость кишечника, склеродерму, псевдонепроходимость кишечника, синдром раздраженного кишечника, гастрит, рвоту и хроническую констипацию (инертная толстая кишка). Мотилин представляет собой пептидный гормон из 22 аминокислот, секретируемый эндокринными клетками в слизистой оболочке желудочно-кишечного тракта. Его связывание с рецептором мотилина в ЖК-тракте стимулирует моторику ЖКТ. Для лечения расстройств ЖКТ было предложено введение терапевтических агентов, которые действуют в качестве агонистов мотилина (прокинетические агенты). Эритромицины представляют собой семейство макролидных антибиотиков, получаемых в результате ферментации актиномицетов Saccharopolyspora erythraea. Эритромицин А, широко используемый антибитик, является наиболее распространенным и важным членом этого семейства (в эритромицине 16-членное лактонное кольцо именуется макролактонной частью молекулы или агликоном, а гликозидные остатки, присоединенные к атомам углерода в положениях 3 и 5, именуются остатками кладинозы и дезозаминозы, соответственно). Побочные эффекты эритромицина А включают тошноту, рвоту и дискомфорт в брюшной полости. Эти эффекты были приписаны активности агониста мотилина эритромицина А (1) и, кроме того, продукта его первичной катализируемой кислотой деградации (5) (продукт вторичной деградации, спирокеталь (6), является неактивным). Стимулируемые обнаружением активности агониста мотилина у эритромицина А и продукта его деградации 5 исследователи приложили усилия для открытия новых мотилидов, как были названы макролиды с прокинетической активностью. Многие исследователи сконцентрировались на создании новых аналогов эритромицина либо посредством постферментационной химической трансформации продуцируемого естественным путем эритромицина, либо посредством модификации (включая генную инженерию) ферментационного процесса. Иллюстративные описания, относящиеся к мотилидам, включают: Omura et al., US 5008249 (1991) и US 5175150 (1992); Harada et al., US 5470961 (1995); Freiberg et al., US 5523401 (1996); US 5523418 (1996); US 5538961 (1996) и US 5554605 (1996); Lartey et al., US 5578579 (1996); US 5654411 (1997); US 5712253 (1998) и US 5834438 (1998); Koga et al., US 5658888 (1997); Miura et al., US 5959088 (1998); Premchandran et al., US 5922849 (1999); Keyes et al., US 6084079 (2000); Ashley et al., US 2002/0025936 A1 (2002); Ashley et al., US 2002/0094962 A1 (2002); Carreras et al., US 2002/0192709 A1 (2002); Ito et al., JP 60-218321 (1985) (соответствующий номер реферата в Chemical Abstracts - 104:82047); Santi et al., US 2004/138150 A1 (2004); Carreras et al., US 2005/0113319 A1 (2005); Carreras et al., US 2005/0119195 A1 (2005); Liu et al., US 2005/0256064 A1 (2005); Omura et al., J. Antibiotics 1985, 38, 1631-2; Faghih et al., Biorg. & Med. Chem. Lett., 1998, 8, 805-810; Faghih et al., J. Med. Chem., 1998, 41, 3402-3408; Faghih et al., Synlett., Jul. 1998, 751 и Lartey et al., J. Med. Chem., 1995,38, 1793-1798. Также потенциально имеющими отношение к настоящему изобретению являются соединения со скелетом эритромицина, имеющие дериватизированный эфирный кислород или азот в положении 9, даже если такие соединения не являются агонистами мотилина, иллюстративными описаниями являются: Krowicki et al., US 3855200 (1974); Radobolja et al., US 3939144 (1976); Kobrehel et al., US 3983103 (1976); Toscano, US 4588712 (1986); Agouridas et al., US 5444051 (1995); Agouridas et al., US 5561118 (1996); Agouridas et al., US 5770579 (1998); Asaka et al., US 6169168 Bl (2001); Kobrehel et al., DE 2402200 (1974); Pliva Pharmaceuticals, GB 1416281 (1975); Pliva Pharmaceuticals, GB 1461032 (1977); Asaga et al., JP 2002/241391 (2002); Ryden et al., J. Med. Chemistry, 1973, 16 (9), 1059-1060; Naperty et al., Roczniki Chemii, 1977, 51 (6), 1207-10; Kobrehel et al., Eur. J. Med. Chemistry, 1978, 13 (1), 83-7; Egan et al., J. Antibiotics, 1978, 31 (1), 55-62; Matijasevic et al., Croatica Chemica Acta, 1980, 53 (3), 519-24; Radobolja et al., Croatica Chemica Acta, 1985, 58 (2), 219-25; Hunt et al., J. Antibiotics, 1989, 42 (2), 293-298; Myles et al., J. Org. Chem., 1990, 55, 1636-1648. Описания всех вышеуказанных документов включены в данное описание посредством ссылок. В первом аспекте изобретения предложено соединение, полезное в качестве прокинетического агента, имеющее структуру, представленную формулой (I) и его фармацевтически приемлемые соли, сольваты и гидраты, где (A) R A представляет собой: (1) OR 1 ; (2) O(CH 2 ) m C(=O)R 2 ; (3) OC(=O)R 4 ; (4) OS(O 2 )N(R 3 R 3A ); (5) O(CH 2 ) n NHR 5 ; (6) N(H)S(O 2 )R 6 ; (7) OCH 2 CH 2 OCH 2 CH 2 C(=O)R 2 или (8) OCH 2 CH 2 OCH 2 CH 2 NHR 5 ; (Б) R B выбран из группы, состоящей из С 2 -С 4 алкила, С 3 -С 4 алкенила или С 3 -С 4 алкинила, 3- или 4-членной циклоалифатической группировки и 3- или 4-членной гетероциклоалифатической группировки, причем каждый член этой группы возможно замещен одним или более заместителями, выбранными из группы, состоящей из ОН, CN и галогена; (B) R C представляет собой Н или ОН; (Г) R D представляет собой Н или Me; (Д) R E представляет собой Н или ОН и (Е) R F представляет собой Н или Me; где R 1 представляет собой С 1 -С 4 алкил, возможно замещенный ОН, CN, группой О(С 1 -С 3 алкил), галогеном, арилом, циклоалифатической группировкой, гетероарилом или гетероциклоалифатической группировкой, причем указанные арил, циклоалифатическая группировка, гетероарил и гетероциклоалифатическая группировка возможно замещены С 1 -С 4 алкилом; R 2 представляет собой OR 3 , N(R 3 R 3A ), С 1 -С 4 алкил, (СН 2 ) n OH или С 2 -С 4 галогеноалкил; R 3 представляет собой Н, С 1 -С 4 алкил или (CH 2 ) n OH; R 3A представляет собой Н, С 1 -С 4 алкил, (CH 2 ) n OH, (CH 2 ) n O(С 1 -С 2 алкил), С 2 -С 4 галогеноалкил, С 1 -С 4 алкил(арил), С 1 -С 4 алкил(гетероарил), О(С 1 -С 4 алкил), гетероарил или где X представляет собой N или СН; Y представляет собой О, S, NH, N(С 1 -С 3 алкил), СН 2 или связь; каждый р представляет собой (1) независимо 1 или 2, когда X представляет собой СН 2 ; (2) 2, когда X представляет собой N и Y не является СН 2 или связью; и (3) независимо 1 или 2, когда X представляет собой N и Y представляет собой СН 2 или связь; и q представляет собой: (1) 0, 1, 2 или 3, когда X представляет собой СН, и (2) 2 или 3, когда X представляет собой N; R 4 представляет собой N(R 3 R 3A ) или С 1 -С 4 алкил; R 5 представляет собой S(О 2 )(С 1 -С 4 алкил), С(=О)(С 1 -С 4 алкил), С(=О)арил, С(=О)(гетероарил), С(=О)Н или С(=W)NH(С 1 -С 4 алкил), где W представляет собой О или S; R 6 представляет собой С 1 -С 4 алкил, циклобутил, циклопропил, CF 3 или N(R 3 R 3A ); m представляет собой 1, 2, 3, 4, 5 или 6 и n представляет собой независимо в каждом случае 2, 3 или 4. В другом аспекте данного изобретения предложен способ лечения заболевания со сниженной моторикой желудочно-кишечного тракта, включающий введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению. В другом аспекте изобретения предложена фармацевтическая композиция содержащая соединение по данному изобретению и эксципиент. В еще одном аспекте изобретения предложен способ индуцирования сокращения ткани, способной к сократительному ответу на мотилин, включающий приведение такой ткани в контакт с соединением по данному изобретению в количестве, эффективном для индукции таких сокращений. В еще одном аспекте изобретения предложено применение соединения по данному изобретению для изготовления лекарственного средства для лечения заболевания со сниженной моторикой желудочно-кишечного тракта. Определения "Алифатическая" означает углеводородную неароматическую группировку с прямой или разветвленной цепью, которая является насыщенной или ненасыщенной, имеющую определенное количество атомов углерода (например С 3 алифатическая, С 1 -С 5 алифатическая или от C 1 до С 5 алифатическая, причем последние два выражения являются синонимами для обозначения алифатической группировки, имеющей от 1 до 5 атомов углерода), или, в случае когда количество атомов углерода не указано, от 1 до 4 атомов углерода (от 2 до 4 атомов углерода в случае ненасыщенной алифатической группировки). "Алкил" означает насыщенную алифатическую группировку с тем же самым принципом обозначения количества атомов углерода. Так, в качестве иллюстрации, C 1 -С 4 алкильные группировки включают метил, этил, пропил, изопропил, изобутил, трет-бутил, 1-бутил, 2-бутил и тому подобное, но ими не ограничиваются. "Алкенил" означает алифатическую группировку, имеющую по меньшей мере одну двойную связь углерод-углерод, с тем же самым принципом обозначения количества атомов углерода. Так, в качестве иллюстрации С 2 -С 4 алкенильные группировки включают этенил (винил), 2-пропенил (аллил или проп-2-енил), цис-1-пропенил, транс-1-пропенил, Е- (или Z-)-2-бутенил, 3-бутенил, 1,3-бутадиенил (бут-1,3-диенил) и тому подобное, но ими не ограничиваются. "Алкинил" означает алифатическую группировку, имеющую по меньшей мере одну тройную связь углерод-углерод, с тем же самым принципом обозначения количества атомов углерода. Так, в качестве иллюстрации С 2 -С 4 алкинильные группы включают этинил (ацетиленил), пропаргил (проп-2-инил), 1-пропинил, бут-2-инил и тому подобное, но ими не ограничиваются. "Циклоалифатическая группировка" означает насыщенную или ненасыщенную неароматическую углеводородную группировку, имеющую от 1 до 3 колец, каждое из которых содержит от 3 до 8 (предпочтительно от 3 до 6) атомов углерода. "Циклоалкил" означает циклоалифатическую группировку, в которой каждое кольцо является насыщенным. "Циклоалкенил" означает циклоалифатическую группировку, в которой по меньшей мере одно кольцо содержит по меньшей мере одну двойную связь углерод-углерод. "Циклоалкинил" означает циклоалифатическую группировку, в которой по меньшей мере одно кольцо содержит по меньшей мере одну тройную связь углерод-углерод. Так, в качестве иллюстрации, циклоалифатические группировки включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогептил, циклооктил и адамантил, но не ограничиваются ими. Предпочтительными циклоалифатическими группировками являются циклоалкильные группировки, особенно циклопропил, циклобутил, циклопентил и циклогексил. "Гетероциклоалифатическая группировка" означает циклоалифатическую группировку, по меньшей мере в одном кольце которой вплоть до трех (предпочтительно 1 или 2) атомов углерода заменены гетероатомом, независимо выбранным из N, О или S, где N или S возможно могут быть окислены и N возможно может быть кватернизирован. Аналогично "гетероциклоалкил", "гетероциклоалкенил" и "гетероциклоалкинил" означают циклоалкильную, циклоалкенильную или циклоалкинильную группировки соответственно, в которых по меньшей мере одно из их колец модифицировано указанным образом. Примеры гетероциклоалифатической группировки включают азиридинил, азетидинил, 1,3-диоксанил, оксетанил, тетрагидрофурил, пирролидинил, пиперидинил, пиперазинил, тетрагидропиранил, тетрагидротиопиранил, тетрагидротиопиранилсульфон, морфолинил, тиоморфолинил, тиоморфолинилсульфоксид, тиоморфолинилсульфон, 1,3-диоксоланил, тетрагидро-1,1-диоксотиенил, 1,4-диоксанил, тиетанил и тому подобное. "Алкокси", "арилокси", "алкилтио" и "арилтио" означают -О(алкил), -О(арил), -S(алкил) и -S(арил), соответственно. Примерами являются метокси, фенокси, метилтио и фенилтио, соответственно. "Галоген" или "галогено" означает фтор, хлор, бром или иод. "Арил" означает углеводородную группировку, содержащую моно-, би- или трициклическую кольцевую систему, где каждое кольцо имеет от 3 до 7 атомов углерода и по меньшей мере одно кольцо является ароматическим. Кольца в такой кольцевой системе могут быть конденсированы друг с другом (как в нафтиле) или могут быть связаны друг с другом (как в бифениле) и могут быть конденсированы или связаны с неароматическими кольцами (как в инданиле или циклогексилфениле). В качестве дополнительной иллюстрации арильные группировки включают, без ограничения ими, фенил, нафтил, тетрагидронафтил, инданил, бифенил, фенантрил, антраценил и аценафтил. "Гетероарил" означает группировку, содержащую моно-, би- или трициклическую кольцевую систему, где каждое кольцо имеет от 3 до 7 атомов углерода и по меньшей мере одно кольцо является ароматическим кольцом, содержащим от 1 до 4 гетероатомов, независимо выбранных из N, О или S, где N и S возможно могут быть окислены и N возможно может быть кватернизирован. Такое содержащее по меньшей мере один гетероатом ароматическое кольцо может быть конденсировано с другими типами колец (как в бензофураниле или тетрагидроизохинолиле) или непосредственно соединено с другими типами колец (как в фенилпиридиле или 2-циклопентилпиридиле). В качестве допонительной иллюстрации гетероарильные группировки включают пирролил, фуранил, тиофенил (тиенил), имидазолил, пиразолил, оксазолил, изоксазолил, тиазолил, изотиазолил, триазолил, тетразолил, пиридил, N-оксопиридил, пиридазинил, пиримидинил, пиразинил, хинолинил, изохинолинил, хиназолинил, циннолинил, хинозалинил, нафтиридинил, бензофуранил, индолил, бензотиофенил, оксадиазолил, тиадиазолил, фенотиазолил, бензимидазолил, бензотриазолил, дибензофуранил, карбазолил, дибензотиофенил, акридинил и тому подобное. Если указано, что группировка может быть замещена, например, посредством использования терминов "замещенный или незамещенный" либо "возможно замещенный" в выражениях типа "замещенный или незамещенный С 1 -С 5 алкил" или "возможно замещенный гетероарил", то это означает, что такая группировка может иметь один или более независимо выбранных заместителей, предпочтительно в количестве от одного до пяти, более предпочтительно в количестве один или два. Для получения соединений, которые являются химически стабильными и которые могут быть синтезированы способами, известными в данной области, такими как способы, раскрытые в данном описании, специалистом могут быть выбраны заместители и варианты замещения, подходящие для группировки, к которой этот заместитель присоединен. "Арилалкил", "(гетероциклоалифатическая группировка)алкил", "арилалкенил", "арилалкинил", "биарилалкил" и тому подобные термины означают соответственно алкильные, алкенильные или алкинильные группировки, замещенные арильной, гетероциклоалифатической, биарильной и т.д. группировками, соответственно с открытой (несвязанной) валентностью на алкильной, алкенильной или алкинильной группировке, например, как в бензиле, фенетиле, N-имидазоилэтиле, N-морфолиноэтиле и тому подобном. И наоборот, "алкиларил", "алкенилциклоалкил" и тому подобное означает арильную, циклоалкильную и т.д. группировку, соответственно замещенную алкильной, алкенильной и т.д. группировкой, соответственно, например, как в метилфениле (толиле) или аллилциклогексиле. "Гидроксиалкил", "галогеноалкил", "алкиларил", "цианоарил" и тому подобное означает алкильную, арильную и т.д. группировку, соответственно замещенную одним или более из числа указанных заместителей (гидроксил, галогено и т.д., соответственно). В качестве иллюстрации, допустимые заместители включают, без ограничения ими, алкил (особенно метил или этил), алкенил (особенно аллил), алкинил, арил, гетероарил, циклоалифатическую группировку, гетероциклоалифатическую группировку, галогено (особенно фтор), галогеноалкил (особенно трифторметил), гидроксил, гидроксиалкил (особенно гидроксиэтил), циано, нитро, алкокси, -О(гидроксиалкил), -О(галогеноалкил) (особенно -OCF 3 ), -О(циклоалкил), -О(гетероциклоалкил), -О(арил), алкилтио, арилтио, =O, =NH, =N(алкил), =NOH, =NO(алкил), -С(=О)(алкил), -С(=О)Н, -СО 2 Н, -C(=O)NHOH, -С(=О)О(алкил), -С(=О)О(гидроксиалкил), -C(=O)NH 2 , -С(=O)NH(алкил), -С(=О)N(алкил) 2 , -ОС(=О)(алкил), -ОС(=О)(гидроксиалкил), -ОС(=О)О(алкил), -ОС(=О)О(гидроксиалкил), -OC(=O)NH 2 , -ОС(=O)NH(алкил), -ОС(=О)N(алкил) 2 , азидо, -NH 2 , -NH(алкил), -N(алкил) 2 , -NH(арил), -NH(гидроксиалкил), -NHC(=О)(алкил), -NHC(=O)H, -NHC(=O)NH 2 , -NHC(=O)NH(алкил), -NHC(=O)N(алкил) 2 , -NHC(=NH)NH 2 , -OSO 2 (алкил), -SH, -S(алкил), -S(арил), -S(циклоалкил), -S(=O)алкил, -SO 2 (алкил), -SO 2 NH 2 , -SO 2 NH(алкил), -SO 2 N(алкил) 2 и тому подобное. Если замещенная группировка представляет собой алифатическую группировку, то предпочтительными заместителями являются арил, гетероарил, циклоалифатическая группировка, гетероциклоалифатическая группировка, галогено, гидроксил, циано, нитро, алкокси, -О(гидроксиалкил), -О(галогеноалкил), -О(циклоалкил), -О(гетероциклоалкил), -О(арил), алкилтио, арилтио, =O, =NH, =N(алкил), =NOH, =NO(алкил), -СО 2 Н, -C(=O)NHOH, -С(=О)О(алкил), -С(=О)О(гидроксиалкил), -C(=O)NH 2 , -С(=О)NH(алкил), -С(=О)N(алкил) 2 , -ОС(=О)(алкил), -ОС(=О)(гидроксиалкил), -ОС(=О)О(алкил), -ОС(=О)О(гидроксиалкил),-OC(=O)NH 2 , -ОС(=О)NH(алкил), -ОС(=О)N(алкил) 2 , азидо, -NH 2 , -NH(алкил), -N(алкил) 2 , -NH(арил), -NH(гидроксиалкил), -NHC(=O)(алкил), -NHC(=O)H, -NHC(=O)NH 2 , -NHC(=О)NH(алкил), -NHC(=О)N(алкил) 2 , -NHC(=NH)NH 2 , -OSO 2 (алкил), -SH, -S(алкил), -S(арил), -S(циклоалкил), -S(=О)алкил, -SO 2 (алкил), -SO 2 NH 2 , -SO 2 NH(алкил) и -SO 2 N(алкил) 2 . Более предпочтительными заместителями являются галогено, гидроксил, циано, нитро, алкокси, -О(арил), =O, =NOH, =NO(алкил), -ОС(=О)(алкил), -ОС(=О)О(алкил), -OC(=O)NH 2 , -ОС(=О)NH(алкил), -ОС(=О)N(алкил) 2 , азидо, -NH 2 , -NH(алкил), -N(алкил) 2 , -NH(арил), -NHC(=О)(алкил), -NHC(=O)H, -NHC(=O)NH 2 , -NHC(=О)NH(алкил), -NHC(=О)N(алкил) 2 и -NHC(=NH)NH 2 . Если замещенная группировка представляет собой циклоалифатическую группировку, гетероциклоалифатическую группировку, арильную или гетероарильную группировку, то предпочтительными заместителями являются алкил, алкенил, алкинил, галогено, галогеноалкил, гидроксил, гидроксиалкил, циано, нитро, алкокси, -О(гидроксиалкил), -О(галогеноалкил), -О(циклоалкил), -О(гетероциклоалкил), -О(арил), алкилтио, арилтио, -С(=О)(алкил), -С(=О)Н, -СО 2 Н, -C(=O)NHOH, -С(=О)О(алкил), -С(=О)О(гидроксиалкил), -C(=O)NH 2 , -С(=О)]NH(алкил), -C(=O)N(алкил) 2 , -ОС(=О)(алкил), -ОС(=О)(гидроксиалкил), -ОС(=О)О(алкил), -ОС(=О)О(гидроксиалкил), -OC(=O)NH 2 , -OC(=O)NH(алкил), -OC(=O)N(алкил) 2 , азидо, -NH 2 , -NH(алкил), -N(алкил) 2 , -NH(арил), -NH(гидроксиалкил), -NHC(=О)(алкил), -NHC(=O)H, -NHC(=O)NH 2 , -NHC(=O)NH(алкил), -NHC(=О)N(алкил) 2 , -NHC(=NH)NH 2 , -OSO 2 (алкил), -SH, -S(алкил), -S(арил), -S(циклоалкил), -S(=O)алкил, -SO 2 (алкил), -SO 2 NH 2 , -SO 2 NH(алкил) и -SO 2 N(алкил) 2 . Более предпочтительными заместителями являются алкил, алкенил, галогено, галогеноалкил, гидроксил, гидроксиалкил, циано, нитро, алкокси, -О(гидроксиалкил), -С(=О)(алкил), -С(=О)Н, -СО 2 Н, -C(=O)NHOH, -С(=О)О(алкил), -С(=О)О(гидроксиалкил), -C(=O)NH 2 , -С(=О)NH(алкил), -С(=О)N(алкил) 2 , -ОС(=О)(алкил), -ОС(=О)(гидроксиалкил), -ОС(=О)О(алкил), -ОС(=О)О(гидроксиалкил), -OC(=O)NH 2 , -ОС(=О)NH(алкил), -ОС(=О)N(алкил) 2 , -NH 2 , -NH(алкил), -N(алкил) 2 , -NH(арил), -NHC(=О)(алкил), -NHC(=O)H, -NHC(=O)NH 2 , -NHC(=О)NH(алкил), -NHC(=O)N(алкил) 2 и -NHC(=NH)NH 2 . Когда указан диапазон, как в выражениях "(от C 1 до С 5 )алкил" или "от 5 до 10%", то такой диапазон включает конечные точки или границы диапазона. За исключением случаев, когда конкретно указаны отдельные стереоизомеры (например, посредством жирной или пунктирной связи по соответствующему стереоцентру в структурной формуле, посредством описания двойной связи как имеющей Е или Z конфигурацию в структурной формуле или путем использования обозначающей стереохимию номенклатуры), все стереоизомеры включены в объем изобретения, как в виде чистых соединений, так и в виде их смесей. Если не указано иного, все индивидуальные энантиомеры, диастереомеры, геометрические изомеры и их комбинации охватываются настоящим изобретением. "Фармацевтически приемлемая соль" означает соль соединения, пригодную для фармацевтической композиции. Если соединение имеет одну или более основных функциональных групп, такая соль может представлять собой соль присоединения кислоты, такую как сульфат, гидробромид, тартрат, мезилат, малеат, цитрат, фосфат, ацетат, памоат (эмбонат), гидроиодит, нитрат, гидрохлорид, лактат, метилсульфат, фумарат, бензоат, сукцинат, мезилат, лактобионат, суберат, тозилат и тому подобное. Если соединение имеет одну или более кислотных группировок, то указанная соль может представлять собой такую соль, как соль кальция, соль калия, соль магния, соль меглумина, соль аммония, соль цинка, соль пиперазина, соль трометамина, соль лития, соль холина, соль диэтиламина, соль 4-фенилциклогексиламина, соль бензатина, соль натрия, соль тетраметиламмония и тому подобное. Полиморфные кристаллические формы и сольваты также входят в объем данного изобретения. В предпочтительном воплощении изобретения R C представляет собой ОН, R D представляет собой Me, R E представляет собой ОН и R F представляет собой Н, что соответствует соединению, имеющему структуру, представленную формулой Ia. Такое соединение может быть получено из эритромицина А, легко доступного вещества, как описано здесь далее. В предпочтительном воплощении соединения формулы Ia имеют структуру, представленную формулой Ib В другом предпочтительном воплощении соединения формулы Ia имеют структуру, представленную формулой Ic В другом предпочтительном воплощении соединения формулы Ia имеют структуру формулы Ic' В другом предпочтительном воплощении соединения формулы Ia имеют структуру формулы Ic" В другом предпочтительном воплощении соединения формулы Ia имеют структуру формулы Ic'" В другом предпочтительном воплощении соединения формулы Ia имеют структуру, представленную формулой Id В другом предпочтительном воплощении соединения формулы Ia имеют структуру, представленную формулой Id' В другом предпочтительном воплощении соединения формулы Ia имеют структуру, представленную формулой Ie В другом предпочтительном воплощении соединения формулы Ia имеют структуру, представленную формулой If В другом предпочтительном воплощении соединения формулы Ia имеют структуру, представленную формулой Ig В другом предпочтительном воплощении соединения формулы Ia имеют структуру, представленную формулой Ih В другом предпочтительном воплощении соединения формулы Ia имеют структуру, представленную формулой Ii В вышеприведенных формулах с Ia по Ii разные группы R A , R B , R 1 , R 2 и т.д., где они присутствуют, имеют такие значения, как они определены в отношении формулы I в разделе «Краткое изложение сущности изобретения », за исключением случаев, когда указано иное. Группы R A , содержащие эфирный кислород в положении 9, могут быть выбраны из группы, состоящий из Предпочтительно такие группы R A выбраны из группы, состоящей из Особенно предпочтительно, чтобы такие предпочтительные группы R A были скомбинированы с R B , представляющим собой R C , представляющим собой Н или ОН, R D , представляющим собой Me, R E , представляющим собой Н или ОН, и R F , представляющим собой Н или Me. Предпочтительные группы R A , содержащие азот в положении 9, выбраны из группы, состоящей из Предпочтительные группы R B выбраны из группы, состоящей из этила, н-пропила, н-бутила, 2-бутила, Более предпочтительно группы R B выбраны из группы, состоящей из В предпочтительном воплощении R 3 представляет собой Н или Me в OR 3 и R 3 представляет собой Н в R 3 R 3A . Методики модификации дезозаминнной диметиламиногруппы в эритромициновых соединениях для замены одной из присутствующих в природном соединении метальных групп другой группой R B представлены, например, в Ashley et al., US 6750205 B2 (2004); Ashley et al., US 2002/0094962 A1 (2002); Santi et al., US 2004/0138150 A1 (2004); Carreras et al., US 2005/0113319 A1 (2005); Carreras et al., US 2005/0119195 A1 (2005) и Liu et al., US 2005/0256064 A1 (2005), описания которых включены посредством ссылки. Если алкильная группа является замещенной, она предпочтительно замещена по β-, γ- или δ-углероду, в противоположность α-углероду. Конкретные примеры соединений по данному изобретению согласно формуле I представлены в табл. А (если не указано иного в колонке "Другое", R C представляет собой ОН, R D представляет собой Me, R E представляет собой ОН и R F представляет собой Н). Предпочтительно соединения по данному изобретению формул I, Ia, Ib, Ic, Ic', Ic", Ic'", Id, Id', Ie, If, Ig, Ih и Ii по стереохимическим центрам в положениях 2, 3, 4, 5, 6, 8, 10, 11, 12 и 13 в макролидном кольце, по стереохимическим центрам в положениях 1', 2', 3' и 5' в дезозаминном остатке и по стереохимическим центрам в положениях 1", 3", 4" и 5" в кладинозном остатке имеют стереохимию эритромицина А. Особенно предпочтительными соединениями по данному изобретению являются соединения А-12, А-13, А-15, А-21, А-71, А-74, А-77 и А-78, чьи полные структурные изображения являются следующими: Специалисту понятно, что релевантым к разработке мотилидов является целый ряд параметров. Во-первых, эволюция продукции эритромицина в естественным образом продуцирующих его организмах определялась его антибактериальной эффективностью, а не его прокинетической эффективностью. Соответственно, остается обширное поле для оптимизации взаимосвязи структура-активность в отношении активности агониста мотилина. Во-вторых, на практике наличие у мотилида антибактериальной активности является нежелательным. ЖК тракт является местом обитания для большой популяции бактерий, воздействие на которых мотилида, обладающего антибактериальной активностью, может индуцировать развитие у них устойчивости к эритромициновым антибиотикам. Либо обладающий антибактериальной активностью мотилид может привести к гибели полезных кишечных бактерий. Таким образом, мотилид желательно должен быть модифицирован таким образом, чтобы он обладал повышенной прокинетической активностью, и чтобы у него отсутствовала антибактериальная активность. В-третьих, у созданных к настоящему времени мотилидов имеется общий недостаток, заключающийся в их предрасположенности к десенсибилизации рецептора мотилида, что означает, что после начальной дозы последующие дозы мотилида вызывают более слабый ответ или вообще его не вызывают (тахифилаксия). В-четвертых, проблемы стабильности и биодоступности, о чем свидетельствует быстрая деградация эритромицина А в желудке и отсутствие активности у его вторичного продукта деградации. В-пятых, имеются сообщения о наличии у некоторых соединений эритромицинового ряда нежелательных проаритмических эффектов, включая пролонгацию QT интервала и индукцию желудочковой аритмии. Желательно ограничение этих эффектов до приемлемого уровня. Таким образом, существует настоятельная необходимость в разработке новых мотилидов, у которых будут сбалансированы требующиеся характеристики. В дополнение к вышеперечисленным факторам, фактором, который также должен быть принят во внимание, является биодоступность. Желательно, чтобы прокинетический агент обладал быстрой биодоступностью, давая возможность пациенту принимать его незадолго перед приемом пищи, а не за несколько часов, что должно быть удобным для пациента. Кроме того, прокинетический агент не должен задерживаться в организме, а напротив, он должен быстро выводиться из системы после того, как осуществит свое действие, т.е. он должен иметь короткий период полувыведения. В другом аспекте настоящего изобретение предложены способы использования соединений по данному изобретению в лечении сниженной моторики желудочно-кишечного тракта. В целом способы использования соединений по настоящему изобретению включают введение нуждающемуся в этом субъекту терапевтически эффективного количества соединения по настоящему изобретению. Иллюстративные примеры расстройств, которые можно лечить соединениями по изобретению, включают, без ограничения ими, гастропарез, гастроэзофагеальную рефлюксную болезнь, анорексию, стаз желчного пузыря, послеоперационную паралитическую непроходимость кишечника, склеродерму, псевдонепроходимость кишечника, гастрит, рвоту и хроническую констипацию (инертная толстая кишка), в особенности гастропарез и гастроэзофагеальную рефлюксную болезнь. Субъектом может являться человек или другое млекопитающее. Терапевтически эффективное количество может быть выражено как общая суточная доза соединения или соединений по данному изобртению, и оно может быть введено субъекту в разовой или в разделенных дозах. Общая суточная доза может представлять собой количество, например, от примерно 0,01 до примерно 10 мг/кг массы тела, или более традиционно, от примерно 0,1 до примерно 2 мг/кг массы тела. Композиции разовой дозы могут содержать такие количества или их доли, чтобы обеспечить суточную дозу. В общем схемы введения согласно настоящему изобретению включают введение нуждающемуся в таком лечении субъекту от примерно 10 до примерно 1000 мг соединения(ий) по настоящему изобретению в сутки в разовой или разделенных дозах. Как правило, соединение по изобретению составляет часть фармацевтической композиции или препарата, которые могут находиться в любой приемлемой форме, такой как твердая, полутвердая или жидкая форма. Как правило, фармацевтический препарат содержит одно или более соединений по изобретению в качестве активного ингредиента и фармацевтически приемлемый носитель или эксципиент. Как правило, активный ингредиент находится в смеси с органическим или неорганическим носителем или эксципиентом, подходящим для наружного, энтерального или парентерального применения. Активный ингредиент может быть скомбинирован, например, с обычными нетоксичными фармацевтически приемлемыми носителями для таблеток, пилюль, капсул, суппозиториев, пессариев, растворов, эмульсий, суспензий и любой другой формы, пригодной для использования. Эксципиенты, которые могут быть использованы, включают носители, поверхностно-активные агенты, загустители или эмульгаторы, твердые связующие вещества, вспомогательные средства для образования дисперсий или суспензий, солюбилизаторы, красители, корригенты, покрытия, разрыхлители, смазывающие вещества, подсластители, консерванты, изотонические агенты и их комбинации. Выбор и использование приемлемых эксципиентов раскрыты в Gennaro, ed., Remington: The Science and Practice of Pharmacy, 20th Ed. (Lippincott Williams & Wilkins 2003), описание которого включено посредством ссылки. Практическая реализация данного изобретения может быть дополнительно раскрыта с помощью следующих примеров, которые представлены только в качестве иллюстрации, а не для ограничения. Пример 1. Синтез промежуточного соединения 9. Промежуточное соединение 9 (N-дезметил-N-изопропил-(9S)-дигидроэритромицин А), используемое в синтезе ряда соединений по данному изобретению, было синтезировано следующим образом. Промежуточное соединение 9 также было описано в Santi et al., US 2004/0138150 A1 (2004), описание которого включено посредством ссылки. (9S)-Дигидроэритромицин А (7). Эритромицин А (1) (20,0 г, 27,3 ммоль) растворяли в смеси 2-пропанол - диэтиловый эфир (1:1 об./об., 400 мл) и охлаждали до 0 °С, двумя аликвотами добавляли боргидрид натрия (2,1 г, 54,5 ммоль). Смесь затем нагревали до комнатной температуры (КТ) и перемешивали при КТ в течение 3 ч. Избыток боргидрида устраняли добавлением фосфатного буфера с рН 6,0, затем добавляли триэтаноламин (80 мл). После перемешивания в течение 2 ч смесь экстрагировали EtOAc (300 мл × 4), сушили над MgSO 4 . Сырое вещество очищали посредством хроматографии на силикагеле, используя смесь 2:1 гексан-ацетон с 1% триэтиламина, и получали чистый продукт 7 (17,2 г, выход 86%). Альтернативно может быть использована следующая процедура. В 10-литровую трехгорлую круглодонную колбу, снабженную механической мешалкой и внутренней термопарой, загружали метил-трет-бутиловый эфир (2400 мл) и эритромицин А (400 г, 545 ммоль, 1,0 экв.). К этой суспензии добавляли МеОН (800 мл). Этот раствор перемешивали до тех пор, пока он не становился прозрачным (прибл. 5-15 мин). Раствор охлаждали с помощью ледяной бани до достижения внутренней температуры 2 °С. Затем одной порцией добавляли твердый NaBH 4 (30,9 г, 816 ммоль, 1,5 экв.). Полученную суспензию перемешивали при 0 °С в течение 1 ч, причем все это время раствор оставался прозрачным. Через 1 ч при 0 °С ледяную баню удаляли. Смесь оставляли нагреваться до 22 °С и перемешивали еще в течение 3 ч. Смесь постепенно становилась непрозрачной. Согласно результатам TLC (тонкослойная хроматография, 10% МеОН в CH 2 Cl 2 , пластины силикагеля 60F, предварительно обработанные аммиаком для нейтрализации любой кислотности в силикагеле) реакция была завершена. Избыток NaBH 4 устраняли путем осторожного добавления ацетона (120 мл; экзотермическая реакция: ацетон добавляли с такой скоростью, чтобы внутренняя температура поддерживалась ниже 30 °С) и фосфатного буфера (5%, рН 6,0, 120 мл). Реакционная среда приобретала вид прозрачного раствора с некоторым количеством белого осадка. Для облегчения распада комплекса эритромицин-бор добавляли триэтаноламин (400 мл) и раствор перемешивали в течение 1 ч. После добавления насыщенного раствора NaHCO 3 (3200 мл) смесь экстрагировали EtOAc (3 ×2000 мл). Объединенные экстракты промывали один раз водой и один раз рассолом (каждый раз по 2000 мл), сушили над твердым Na 2 SO 4 . После удаления растворителя сырой продукт сушили в вакуумной печи (16 ч, 50 °С). Получали белое твердое вещество (416 г, т.пл. (точка плавления) 182-185 °С), которое было пригодно для использования на следующей стадии без дополнительной очистки. N-Дезметил-(9S)-дигидроэритромицин А (8). Смесь (9S)-дигидроэритромицина А 7 (17,2 г, 23,4 ммоль) и ацетата натрия (9,75 г, 119 ммоль) в смеси метанол-вода (8:2 об./об., 400 мл) перемешивали при 50 °С. Затем двумя аликвотами с 30-минутным интервалом добавляли иод (7,25 г, 28,6 ммоль). В ходе реакции небольшими порциями добавляли 3 н. NaOH (7,9 мл). Завершение реакции определяли посредством анализов с помощью тонкослойной хроматографии. После удаления большей части растворителя смесь трижды экстрагировали EtOAc и сушили над Na 2 SO 4 . Получали сырой продукт 8 (15,6 г) в виде желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. Может быть использована следующая альтернативная процедура. В шестилитровую трехгорлую круглодонную колбу, снабженную механической мешалкой и внутренней термопарой, загружали МеОН (2000 мл), соединение 7 из предыдущего примера (150 г, теоретически 197 ммоль, 1,0 экв.) и трис(гидроксиметил)аминометан (119 г, 5 экв.). Эту смесь нагревали до достижения внутренней температуры 55 °С, в ходе чего все вещества растворялись. Осторожно добавляли иод (75 г, 1,5 экв.) с такой скоростью, чтобы не допустить повышения внутренней температуры выше 60 °С за счет слабой экзотермичности реакции. Эту смесь перемешивали при 55 °С в течение 5 ч. TLC (15% МеОН в CH 2 Cl 2 , пластины силикагеля, как описано выше) показала завершение реакции. Реакционную смесь охлаждали до комнатной температуры. Для удаления избытка иода использовали насыщенный тиосульфат натрия до тех пор, пока не исчезала характерная окраска иода. Смесь концентрировали путем удаления примерно половины МеОН, следя за тем, чтобы не удалить его слишком много - это вызывает осаждение продукта при последующем добавлении водного раствора, а осадок трудно поддается растворению при последующих экстракциях. Концентрат разбавляли водным NaHCO 3 (1500 мл) и экстрагировали CH 2 Cl 2 (3 ×1000 мл). Объединенные органические слои промывали один раз водой (1500 мл), затем сушили над Na 2 SO 4 . Сырой продукт 8 (113 г, т.пл. 118-123 °С) получали после удаления растворителя и сушки в вакуумной печи (16 ч, 50 °С). Это вещество было пригодно для использования в последующих процедурах синтеза без дополнительной очистки. Промежуточное соединение 9. Смесь вышеуказанного сырого продукта 8 (2,50 г, 3,41 ммоль), диизопропилэтиламина (6,1 мл, 10 эквивалентов), 2-иодпропана (10,2 мл, 30 эквивалентов) в CH 3 CN (50 мл) нагревали на бане 70 °С в течение 24 ч. Добавляли Н 2 О и насыщенный NaHCO 3 , раствор экстрагировали три раза EtOAc, сушили над MgSO 4 . Сырой продукт очищали на колонке SiO 2 (смесь 3:1 гексан-ацетон, 1% TEA (тетраэтиламин)) с получением сырого продукта 9 (1,80 г, выход 75% за 2 стадии), m/z: 765,0 ([М + Н] + ). Для получения промежуточного соединения 9 может быть использована следующая альтернативная процедура. В литровой трехгорлой круглодонной колбе с помощью магнитной мешалки перемешивали раствор продукта 8 (30 г, 41,5 ммоль, 1,0 экв.) в МеОН (150 мл) и ацетоне (30 мл). Добавляли уксусную кислоту (3,5 мл, 62,2 ммоль, 1,5 экв.), затем добавляли NACNBH 3 (5,25 г, 83,3 ммоль, 2 экв.). Этот раствор нагревали на масляной бане и перемешивали при температуре бани 50 °С в течение 4 ч. О завершении реакции судили по результатам TLC (смесь 1:1 гексан-ацетон). После того как смесь охлаждали до комнатной температуры, осторожно добавляли фосфатный буфер (5%, рН 6,0, 60 мл) (быстрое выделение Н 2 ) для того, чтобы погасить избыток боргидрида. Затем добавляли триэтаноламин (100 мл). Смесь перемешивали при КТ в течение 1 ч. Раствор выливали в насыщенный раствор NaHCO 3 (500 мл) и полученную смесь экстрагировали EtOAc (2 ×800 мл). Объединенные экстракты промывали один раз рассолом (600 мл), сушили над Na 2 SO 4 , фильтровали и концентрировали. Сырой продукт (31,8 г) получали в виде белого твердого вещества после сушки в высоком вакууме в течение 16 ч. В зависимости от чистоты продукта-предшественника 8 имелась или отсутствовала необходимость в очистке перед последующим использованием. Если очистка была необходима, сырое промежуточное соединение 9 растворяли в ацетонитриле (100 мл) при нагревании, затем по каплям при продолжающемся нагревании добавляли воду (100 мл) до помутнения. Мутную смесь оставляли охлаждаться до КТ, фильтровали и подвергали вакуумной сушке при 50 °С в течение 16 ч, что обеспечивало получение чистого промежуточного соединения 9 (19 г, 24,9 ммоль, выход 47% из эритромицина А, т.пл. 127-130 °С) в виде белого твердого вещества. Пример 2. Синтез соединений из промежуточного соединения 9. Соединение А-1. Гидрид натрия (60%-ная дисперсия в минеральном масле, 12,5 мг) помещели в сухую колбу, промывали один раз пентаном (5 мл) и суспендировали в диметоксиэтане (2 мл). К этой суспензии добавляли раствор промежуточного соединения 9 (200 мг, 0,262 ммоль) в диметоксиэтане (2 мл). После перемешивания при КТ в течение 10 мин добавляли метилиодид (2M b трет-бутил-метиловом эфире, 0,16 мл) в диметоксиэтане (1 мл). Смесь перемешивали при КТ в течение ночи. Реакцию гасили добавление насыщенного водного раствора NaHCO 3 , экстрагировали три раза CH 2 Cl 2 и сушили над MgSO 4 . Сырой продукт очищали на колонке с силикагелем (смесь 4:1 гексаны-ацетон, 1% триэтиламина) с получением соединения А-1 (130 мг) в виде белого твердого вещества, m/z: 779,0 ([М + Н] + ); ESI TOF MS (времяпролетная масс-спектрометрия с электрораспылительной ионизацией) m/z 778,5311, рассч. для C 40 H 76 NO 13 ([M + Н] + ) 778,5340. Соединение А-3. Смесь промежуточного соединения 9 (80 мг, 0,105 ммоль) и KOtBu (17,6 мг, 1,5 экв.) в тетрагидрофуране (THF, 4 мл) перемешивали при КТ в течение 30 мин). Добавляли насыщенный раствор этиленоксида в THF (1 мл) и реакционную смесь перемешивали в течение 2 ч. LC-MS (сочетание жидкостной хроматографии и масс-спектрометрии) показало наличие смеси исходного вещества и продукта. Чистое соединение А-3 (17,5 мг) получали после аналогичной обработки и очистки, как описано выше, m/z: 808,6 ([М + Н] + ). Соединение А-5. Соединение А-5 получали способом, аналогичным способу получения соединения А-3, но с 2-бромэтил-метиловым эфиром в качестве алкилирующего агента, m/z: 823,0 ([М + Н] + ); ESI TOF MS m/z 822,5533, рассч. для C 42 H 80 NO 14 ([M + Н] + ) 822,5573. Соединение А-7. Использовали способ, аналогичный способу, использованному для получения соединения А-3, но с 2-хлорацетонитрилом в качестве алкилирующего агента. m/z: 804,0 ([М + Н] + ); ESI TOF MS m/z 803,5278, рассч. для C 41 H 75 N 2 O 13 ([M + H] + ) 803,5264. Соединение А-8. Использовали способ, аналогичный способу, использованному для получения соединения А-3, но с этилбромацетатом в качестве алкилирующего агента, m/z: 851,0 ([М + Н] + ); ESI TOF MS m/z 850,5499, рассч. для C 43 H 80 NO 15 ([M + Н] + ) 850,5523. Соединение А-12. К раствору промежуточного соединения 9 (276 мг, 0,362 ммоль) и бромацетамида (60 мг, 0,435 ммоль, 1,2 экв.) в 1,2-диметоксиэтане (4 мл) добавляли KOtBu (1,0 М в THF, 0,54 мл, 1,5 экв.). Полученную мутную смесь перемешивали при КТ в течение 3 ч, затем разбавляли EtOAc (50 мл) и раствором NaHCO 3 (10 мл). Органическую фазу промывали рассолом (10 мл), а водную фазу экстрагировали EtOAc (2 ×10 мл). Объединенные органические слои сушили над Na 2 SO 4 , фильтровали и концентрировали. Остаток очищали хроматографией на колонке с силикагелем (от 10 до 95% ацетона в гексанах, с 1% триэтиламина) с получением соединения А-12 (220 мг, 73%) в виде белого твердого вещества, m/z: 822,0 ([М + Н] + ); ESI TOF MS m/z 821,5385, рассч. для C 41 H 77 N 2 O 14 ([M + Н] + ) 821,5369. Соединение А-15. Использовали способ, аналогичный способу, использованному для получения соединения А-12, за исключением того, что бромацетамид заменяли 2-хлор-N,N-диметилацетамидом. m/z: 850,0 ([М + Н] + ); ESI TOF MS m/z 849,5673, рассч. для C 43 H 81 N 2 O 14 ([M + Н] + ) 849,5682. Соединение А-17. Использовали модифицированную версию способа получения соединения А-12. К раствору промежуточного соединения 9 (256 мг, 0,335 ммоль) в 1,2-диметоксиэтане (2 мл) добавляли KOtBu (1,0 М в THF, 1,06 мл, 3,0 экв.). Полученную смесь перемешивали при комнатной температуре в течение 10 мин, а затем ее охлаждали до -78 °С. Добавляли трихлорацетилизоцианат (0,096 мл, 2,4 экв.). Реакционную смесь медленно нагревали до КТ в течение 3 ч. Трихлорацетильную группу подвергали гидролизу в ходе такой же водной обработки, которая описана для соединения А-12, с получением соединения А-17. m/z: 808,0 ([М + Н] + ); ESI TOF MS m/z 807,5212, рассч. для C 40 H 75 N 2 O 14 ([M + Н] + ) 807,5213. Соединение А-18. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с диметилкарбамоилхоридом вместо бромацетамида. m/z: 836,0 ([М + Н] + ); ESI TOF MS m/z 835,5533, рассч. для C 42 H 79 N 2 O 14 ([М + Н] + ) 835,5526. Соединение А-19. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с диметилсульфамоилхлоридом вместо бромацетамида. m/z: 872,0 ([М + Н] + ); ESI TOF MS m/z 871,5218, рассч. для C 41 H 78 N 2 O 15 S ([M + Н] + ) 871,5196. Соединение А-21. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с 2-бром-N-метилацетамидом вместо бромацетамида. m/z: 836 ([М + Н] + ), 678; ESI TOF MS m/z 835,5498, рассч. для C 42 H 78 N 2 O 14 ([М + Н] + ) 835,5526. 13 С ЯМР (CDCl 3 ) δ 177.3, 170.3, 101.8, 94.4, 94.2, 83.7, 77.6, 77.4, 77.1, 75.5, 74.2, 72.7, 72.6, 69.9, 69.7, 69.3, 65.6, 61.8, 52.5, 49.2, 44.0, 43.6, 38.1, 34.3, 32.7, 32.6, 32.2, 30.9, 25.5, 22.1, 22.0, 21.5, 21.1, 21.0, 20.2, 19.1, 17.4, 16.6, 14.3, 12.9, 11.2, 9.0 м.д. Может быть использована следующая альтернативная процедура. В пятилитровой трехгорлой круглодонной колбе, снабженной механической мешалкой и внутренней термопарой, на ледяной бане охлаждали раствор промежуточного соединения 9 (156,7 г, 205 ммоль), N-метил-бромацетамида (37,4 г, 246 ммоль, 1,2 экв.) в сухом THF (1800 мл). При перемешивании при внутренней температуре 0 °С под азотом одной партией добавляли твердый трет-бутоксид калия (25,3 г, 226 ммоль, 1,1 экв.). Смесь перемешивали при 0 °С в течение 1 ч. Завершение реакции определяли посредством TLC (смесь 1:2 гексан-ацетон, силикагель 60 F, предварительная обработка аммиаком). Реакционную смесь гасили путем добавления насыщенного раствора NaHCO 3 (300 мл). Реакционную смесь распределяли между разбавленным раствором NaHCO 3 (2500 мл) и EtOAc (1500 мл). Водный слой экстрагировали этилацетатом (2 × 1500 мл). Объединенные органические слои сушили над Na 2 SO 4 . Сырой продукт (178,1 г) получали в виде светло-желтого твердого вещества, которое затем очищали на колонке с силикагелем (2800 г силикагеля, 60 F, 20-40% ацетона в гексане, 1% триэтиламина) с получением соединения А-21 (135 г, выход 79%). Для удаления следовых количеств растворителей и триэтиламина продукт повторно растворяли в дихлорметане и сушили в роторном испарителе (4 цикла), затем сушили в вакуумной печи (16 ч, 50 °С) с получением конечного продукта (т.пл. 106-108 °С). Возможно получение известного реагента N-метилбромацетамида следующим образом. В 10-литровую трехгорлую круглодонную колбу, снабженную механической мешалкой и внутренней термопарой, загружали THF (3200 мл), метиламин (2 М раствор в THF, 692 мл, 1,38 моль, 1,5 экв.), NaHCO 3 (155 г, 1,845 моль, 2 экв.) и триэтиламин (128,2 мл, 922 ммоль, 1,0 экв. ). Суспензию охлаждали с помощью бани сухой лед-ацетон до внутренней температуры -70 °С. По каплям при перемешивании добавляли 2-бромацетилбромид (79,8 мл, 922 ммоль, 1,0 экв.). После завершения добавления баню с сухим льдом удаляли. Смесь нагревали до комнатной температуры. Полученную желтую суспензию гасили насыщенным NaHCO 3 (3200 мл) и экстрагировали этилацетатом (2 ×3200 мл). Объединенные органические фазы промывали насыщенным хлоридом аммония (2000 мл) и рассолом (2000 мл), сушили над Na 2 SO 4 . После концентрирования в вакууме окрашенный в красный цвет сырой продукт (82 г) растворяли в CH 2 Cl 2 (100 мл) и пропускали через набивку силикагеля (1600 г), элюируя смесью 50% этилацетат/гексан. Фракции, содержащие продукт (TLC со смесью 30% этилацетат/гексан, визуализация иодом), объединяли и концентрировали в вакууме (примечание 1) с получением чистого продукта в виде низкоплавкого твердого вещества (77,5 г, выход 55%). Соединение А-22. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с метилбромацетатом в качестве алкилирующего агента, m/z: 836,5 ([М + Н] + ); ESI TOF MS m/z 836,5343, рассч. для C 33 H 50 NO 8 ([М + Н] + ) 836,5366. Соединение А-26. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с 4-(иодметил)-2-метилтиазолом в качестве алкилирующего агента, m/z: 876,0 ([М + Н] + ), ESI TOF MS m/z 875,5310, рассч. для C 44 H 79 N 2 O 13 S ([M + Н] + ) 875,5297. Соединение А-27. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с 3-(бромметил)-5-метилизоксазолом в качестве алкилирующего агента, m/z: 860,0 ([М + Н] + ), ESI TOF MS m/z 859,5494, рассч. для C 44 H 79 N 2 O 14 ([M + Н] + ) 859,5526. Соединение А-28. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с 4-(бромметил)пиридином в качестве алкилирующего агента, m/z: 856,0 ([М + Н] + ), ESI TOF MS m/z 855,5613, рассч. для C 45 H 79 N 2 O 13 ([M + Н] + ) 855,5577. Соединение А-29. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с 2-(иодметил)тиазолом в качестве алкилирующего агента, m/z: 862,0 ([М + Н] + ), ESI TOF MS m/z 861,5181, рассч. для C 43 H 77 N 2 O 13 S ([M + Н] + ) 861,5141. Соединение A-31. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с 2-бром-N-этилацетамидом в качестве алкилирующего агента, m/z: 850 ([М + Н] + ). Соединение А-33. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с 2-бром-N-(4-тетрагидропиранил)ацетамидом в качестве алкилирующего агента, m/z: 906 ([М + Н] + ); ESI TOF MS m/z 905,5957, рассч. для C 46 H 84 N 2 O 15 ([M + Н] + ) 905,5946. Соединение А-34. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с 2-бром-N-[2-(трет-бутиддиметилсилилокси)этил]ацетамидом в качестве алкилирующего агента. Этот 9-алкилированный продукт (0,101 г, 0,104 ммоль) растворяли в THF (1,0 мл) и охлаждали до 0 °С. Добавляли фторид тетрабутиламмония (0,020 г, 0,114 ммоль, 1,1 экв.) и раствор перемешивали при 0 °С в течение 2,5 ч, после чего добавляли NaHCO 3 (15 мл). Органическую фазу экстрагировали EtOAc (3 ×15 мл), объединяли, промывали рассолом (25 мл), сушили (Na 2 SO 4 ) и концентрировали при пониженном давлении. В результате колоночной хроматографии (диоксид кремния, 55% ацетон-гексан, 1% триэтиламина) получали соединение А-34 (0,063 г) в виде белого твердого вещества; m/z: 866 ([М + Н] + ); ESI TOF MS m/z 865,5655, рассч. для C 43 H 80 N 2 O 15 ([M + H] + ) 865,5632. Соединение A-45. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с 2-бром-N-циклобутилацетамидом в качестве алкилирующего агента, m/z: 876 ([М + Н] + ), 718; ESI TOF MS m/z 874,5833, рассч. для C 45 H 83 N 2 O 14 ([M + Н] + ) 874,5839. Соединение А-46. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с 2-бром-N-циклопропилацетамидом в качестве алкилирующего агента, m/z: 862 ([М + Н] + ), 703; ESI TOF MS m/z 861,5695, рассч. для C 44 H 81 N 2 O 14 ([M + Н] + ) 861,5682. Соединение А-48. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с 2-бром-N-(2-морфолино)этилацетамидом в качестве алкилирующего агента, m/z: 934,6 ([М + Н] + ). Соединение А-49. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с 1-иод-2-фторэтаном в качестве алкилирующего агента, m/z: 811,0 ([М + Н] + ); ESI TOF MS m/z 810,5374, рассч. для C 39 H 74 NO 14 ([М + Н] + ) 810,5385. Соединение А-50. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с 6-бромгексанамидом в качестве алкилирующего агента, m/z: 877,6 ([М + Н] + ); ESI TOF MS m/z 877,5995, рассч. для C 44 H 80 NO 15 ([M + Н] + ) 877,5999. Соединение А-52. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с 2-бром-N-(трифторэтил)ацетамидом в качестве алкилирующего агента, m/z: 904 ([М + Н] + ), ESI TOF MS m/z 903,5385, рассч. для C 43 H 77 N 2 O 14 F 3 ([M + Н] + ) 903,5400. Соединение А-53. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с 2-бром-N-изопропилацетамидом в качестве алкилирующего агента, m/z: 864 ([М + Н] + ), ESI TOF MS m/z 863,5818, рассч. для C 44 H 82 N 2 O 14 ([M + Н] + ) 863,5839. Соединение А-55. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с 3-хлорметил-2-тритил-1,2,4-триазолом в качестве алкилирующего агента. Метанольный раствор (6 мл), содержащий исходный алкилированный продукт (170 мг), пиридин гидрохлорид (7 мг) и паратолуолсульфонат пиридиния (10 мг), выдерживали при 50 °С в течение ночи с перемешиванием. Реакцию гасили насыщенным водным раствором NaHCO 3 (20 мл) и экстрагировали смесью хлороформ/метанол (5/1) (20 мл, 3 ×). Объединенные органические экстракты сушили над сульфатом натрия. В результате флэш-хроматографии на силикагеле (смесь 100:10:0,5 CH 2 CCl 2 :MeOH:NH 4 OH) получали соединение А-55 в виде белого твердого вещества (35 мг), m/z: 846,0 ([М + Н] + ). Соединение А-59. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с N-бензилбромацетамидом в качестве алкилирующего агента вместо бромацетамида. m/z: 912 ([М + Н] + ), 754; ESI TOF MS m/z 911,5813, рассч. для C 48 H 82 N 2 O 14 ([M + Н] + ) 911,5839. Соединение А-62. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с 2-хлорметилимидазолом гидрохлоридом в качестве алкилирующего агента вместо бромацетамида. m/z: 845,0 ([М + Н] + ). Соединение А-63. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с N-(2-метокси)этилбромацетамидом в качестве алкилирующего агента вместо бромацетамида. m/z: 879,6 ([М + Н] + ). Соединение А-69. Использовали способ, аналогичный способу, использованному для получения соединения А-12, взаимодействие в масштабе 0,085 ммоль с 2-(триметилсилил)этиловым эфиром бромуксусной кислоты давало 9-O-уксусной кислоты 2-(триметилсилил)этиловый эфир (0,045 г, 57%), который растворяли в N,N-диметилформамиде (DMF, 1,0 мл) и охлаждали до 0 °С, после чего добавляли фторид тетрабутиламмония (0,015 г, 0,059 ммоль, 1,2 экв.). Раствор перемешивали при 0 °С в течение 5 ч, а затем добавляли этил-(3-диметиламино)пропилкарбодиимид (0,014 г, 0,074 ммоль, 1,5 экв.), гидроксибензотриазол (0,013 г, 0,098 ммоль, 2,0 экв.) и гидрохлорид метоксиламина (0,008 г, 0,098 ммоль, 2,0 экв.). Раствор перемешивали при комнатной температуре в течение 18 ч, а затем разбавляли EtOAc (15 мл) и промывали NaHCO 3 (15 мл) и рассолом (15 мл). Органические фазы сушили (Na 2 SO 4 ) и концентрировали при пониженном давлении. В результате колоночной хроматографии (диоксид кремния, 30 →50% ацетон-гексан, 1% триэтиламина) получали соединение А-69 (0,009 г, 22%) в виде белого твердого вещества, m/z: 852 ([М + Н] + ), 754; ESI TOF MS m/z 851,5490, рассч. для C 42 H 78 N 2 O 15 ([М + Н] + ) 851,5475. Соединение А-70. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с N-пиразилбромацетамидом в качестве алкилирующего агента вместо бромацетамида. m/z: 900 ([М + Н] + ), 742; ESI TOF MS m/z 899,5563, рассч. для C 45 H 78 N 4 O 14 ([М + Н] + ) 899,5587. Соединение А-73. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с N-метил-3-бромпропионамидом в качестве алкилирующего агента вместо бромацетамида. m/z: 864 ([М + Н] + ), 706; ESI TOF MS m/z 863,5814, рассч. для C 44 H 82 N 2 O 14 ([M + Н] + ) 863,5839. Соединение А-74. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с N-метил-5-бромвалериламидом в качестве алкилирующего агента вместо бромацетамида. m/z: 878 ([М + Н] + ), 720; ESI TOF MS m/z 877,5978, рассч. для C 45 H 84 N 2 OH ([M + Н] + ) 877,5995. Соединение А-76. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с N-метил 6-бромгексаноиламидом в качестве алкилирующего агента вместо бромацетамида. m/z: 892 ([M + Н] + ); 734; ESI TOF MS m/z 891,6127, рассч. для C 46 H 86 N 2 O 14 ([M + Н] + ) 891,6152. Соединение А-77. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с N-пирамидинилбромацетамидом в качестве алкилирующего агента вместо бромацетамида. m/z: 922 ([М + Na] + ), 900 ([М + Н] + ), 742; ESI TOF MS m/z 899,5552, рассч. для C 45 H 78 N 4 O 14 ([M + Н] + ) 899,5587. Соединение А-79. Трет-бутоксид калия (0,17 мл 1 М раствора в THF, 0,167 ммоль, 1,5 экв.) добавляли к раствору промежуточного соединения 9 (0,085 г, 0,111 ммоль, 1,0 экв.) в диметоксиэтане (1,0 мл). Этот раствор перемешивали при КТ в течение 10 мин, затем добавляли карбонилдиимидазол (0,022 г, 0,134 ммоль, 1,2 экв.). Этот раствор перемешивали при комнатной температуре в течение 1 ч, после чего добавляли метиламин (0,024 мл 33%-ного раствора в EtOH, 0,134 ммоль, 1,2 экв.). Полученный раствор перемешивали при КТ в течение 1,5 ч, затем выливали в NaHCO 3 (25 мл) и экстрагировали EtOAc (4 ×20 мл). Объединенные органические фазы сушили (Na 2 SO 4 ) и концентрировали при пониженном давлении. В результате колоночной хроматографии (диоксид кремния, 30% ацетон-гексан, 0,5% Et 3 N) получали соединение А-79 (0,010 г, 11%) в виде белого твердого вещества, m/z: 822 ([М + Н] + ), 664; ESI TOF MS m/z 821,5339, рассч. для C 41 H 76 N 2 O 14 ([M + Н] + ) 821,5369. Пример 3. Соединение А-2. Соединение А-2. 98-Дигидроэритромицин А7 метилировали, как описано выше в отношении соединения А-1, используя 2-иодэтанол. Дезозаминную группировку полученного 9-метоксипродукта подвергали дезметилированию и алкилировали с получением соединения А-2. m/z: 780,5 ([М + Н] + ); ESI TOF MS m/z 780,5104, рассч. для C 39 H 74 NO 14 ([M + Н] + ) 780,5113. Пример 4. Промежуточное соединение 10. Промежуточное соединение 10 (N-дезметил-N-циклобутил-(9S)-дигидроэритромицин А) использовали в синтезе соединений по данному изобретению. Смесь N-дезметил-(9S)-дигидроэритромицина А-8 (4,96 г, 6,87 ммоль), циклобутанона (1,03 мл, 2 экв.), цианоборгидрида натрия (863 мг, 2 экв.) и НОАс (1,57 мл, 4 экв.) в метаноле (40 мл) перемешивали при 50 °С в течение 4 ч. Добавляли воду, а затем триэтаноламин (20 мл). После перемешивания в течение 2 ч смесь экстрагировали три раза EtOAc, сушили над MgSO 4 . Сырой продукт очищали с использованием колонки с SiO 2 (смесь от 3:1 до 2:1 гексан-ацетон, 1% TEA) с получением чистого промежуточного соединения 10 (3,70 г), m/z: 777,0 ([М + Н] + ). Пример 5. Синтез соединений из промежуточного соединения 10. Соединение А-4. Использовали способ, аналогичный способу, использованному для получения соединения А-3, но с промежуточным соединением 10 в качестве исходного вещества, m/z: 820,6 ([М + Н] + ). Соединение А-10. Использовали способ, аналогичный способу, использованному для получения соединения А-3, но с промежуточным соединением 10 в качестве исходного вещества и этилбромацетатом в качестве алкилирующего агента, m/z: 863,0 ([М + Н] + ); ESI TOF MS m/z 862,5523, рассч. для C 44 H 80 NO 15 ([M + Н] + ) 862,5515. Соединение A-13. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с промежуточным соединением 10 в качестве исходного вещества, m/z: 834,0 ([М + Н] + ); ESI TOF MS m/z 833,5348, рассч. для C 42 H 77 N 2 O 14 ([M + Н] + ) 833,5369. Соединение А-23. Использовали способ, аналогичный способу, использованному для получения соединения А-22, но с промежуточным соединением 10 в качестве исходного вещества, m/z: 849,0 ([М + Н] + ); ESI TOF MS m/z 848,5366, рассч. для C 43 H 78 NO 15 ([M + Н] + ) 848,5367. Соединение A-24. К раствору промежуточного соединения 10 (100 мг, 0,127 ммоль) в этилацетате (10 мл) добавляли уксусный ангидрид (61 мкл, 0,65 ммоль, 5 экв.) и K 2 CO 3 . Эту смесь перемешивали при КТ в течение ночи. Реакционную смесь разбавляли EtOAc (100 мл), затем промывали насыщенным водн. NaHCO 3 (3 ×50 мл), сушили над Na 2 SO 4 , фильтровали и выпаривали досуха. Продукт (95 мг) получали после колоночной хроматографии на силикагеле (от 5 до 35% ацетона в гексанах, 1% триэтиламина). Этот продукт затем растворяли в метаноле (3 мл) и нагревали при 50 °С в течение ночи. Растворитель удаляли и после колоночной хроматографии на силикагеле (от 5 до 35% ацетона в гексанах, 1% триэтиламина) получали соединение А-24 (80 мг). m/z: 819,0 ([М + Н] + ). Соединение А-25. Использовали тот же протокол, что и для соединения А-24, за исключением того, что уксусный ангидрид заменяли пропионовым ангидридом, m/z: 833,0 ([М + Н] + ). Соединение A-47. Использовали способ, аналогичный способу, использованному для получения соединения А-1, но с промежуточным соединением 10 вместо промежуточного соединения 9. m/z: 791,0 ([М + Н] + ); ESI TOF MS m/z 790,5311, рассч. для C 41 H 76 NO 13 ([M + Н] + ) 790,5301. Соединение А-51. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с промежуточным соединением 10 в качестве исходного вещества и 2-бром-N-метилацетамидом в качестве алкилирующего агента, m/z: 848,0 ([М + Н] + ); ESI TOF MS m/z 847,5529, рассч. для C 43 H 79 N 2 O 14 ([М + Н] + ) 847,5526. Пример 6. Промежуточное соединение 11. Промежуточное соединение 11 (N-дезметил-N-(2-гидроксипропил)-(9S)-дигидроэритромицин А) использовали в синтезе соединений по данному изобретению. Раствор N-дезметил-(9S)-дигидроэритромицина А 8 (см. пример 1, 357 мг, 0,494 ммоль) и (S)-пропиленоксида (0,35 мл, 10 экв.) в метаноле (10 мл) перемешивали при КТ в течение 24 ч. Завершение реакции определяли посредством TLC. После выпаривания растворителя сырой продукт очищали на колонке с силикагелем (от 5 до 45% ацетона в гексане, 1% триэтиламина) с получением чистого промежуточного соединения 11 (271 мг, 70%). m/z: 781,0 ([М + Н] + ); ESI TOF MS m/z 780,5099, рассч. для C 39 H 74 NO 14 ([M + Н] + ) 780,5104. Пример 7. Синтез соединений из промежуточного соединения 11. Соединение А-6. Использовали способ, аналогичный способу, использованному для получения соединения А-3, но с промежуточным соединением 11 в качестве исходного материала и 2-бромэтилметиловым эфиром в качестве алкилирующего агента, m/z: 839,0 ([М + Н] + ); ESI TOF MS m/z 838,5489, рассч. для C 42 H 80 NO 15 ([M + Н] + ) 838,5522. Соединение А-9. Использовали способ, аналогичный способу, использованному для получения соединения А-3, но с промежуточным соединением 9 в качестве исходного материала и этилбромацетатом в качестве алкилирующего агента, m/z: 867,0 ([М + Н] + ); ESI TOF MS m/z 866,5433, рассч. для C 43 H 80 NO 16 ([M + Н] + ) 866,5472. Соединение А-14. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с промежуточным соединением 11 в качестве исходного материала, m/z: 838,0 ([М + Н] + ); ESI TOF MS m/z 875,4834, рассч. для C 41 H 76 N 2 O 15 K ([M + K] + ) 875,4877. Соединение А-16. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с промежуточным соединением 11 в качестве исходного материала и 2-хлор-N,N-диметилацетамидом в качестве алкилирующего агента, m/z: 866,0 ([М + Н] + ); ESI TOF MS m/z 865,5630, рассч. для C 43 H 81 N 2 O 15 ([M + H] + ) 865,5632. Соединение А-20. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с промежуточным соединением 11 в качестве исходного материала и диметилсульфамоилхлоридом вместо бромацетамида. m/z: 888,0 ([М + Н] + ); ESI TOF MS m/z 887,5151, рассч. для C 41 H 79 N 2 O 16 S ([M + Н] + ) 887,5145. Пример 8. Соединение А-11. Соединение А-11. К раствору соединения А-9 (80 мг, 0,0923 ммоль) в МеОН (3,0 мл) добавляли NaOH (1,0 М в Н 2 О, 0,1 мл). Реакционную смесь перемешивали при КТ в течение ночи, а затем при 50 °С в течение 4 ч. LC/MS показало, что все исходное вещество было израсходовано и целевой продукт являлся единственным обнаруживаемым продуктом. Растворитель удаляли при пониженном давлении и полученное твердое вещество лиофилизировали с получением соединениия А-11 (79 мг, 0,092 ммоль, 99%) в виде его натриевой соли, m/z: 839,0 ([М + Н] + ); ESI TOF MS m/z 838,5176, рассч. для C 41 H 76 NO 16 ([M + Н] + ) 838,5159. Пример 9. Промежуточное соединение 12. Промежуточное соединение 12 (9-дигидро-9-О-(2-аминоэтил)-N-дезметил-N-изопропилэритромицин А) использовали в синтезе ряда соединений по данному изобретению. К раствору промежуточного соединения 9 (55 мг, 0,072 ммоль) в THF (2,4 мл), добавляли гидробромид бромэтиламина (43 мг, 0,209 ммоль, 2,9 экв.), а затем гидроксид калия (38 мг, 0,684 ммоль, 9,5 экв.). Этот раствор перемешивали при комнатной температуре в течение 20 ч, после чего разбавляли EtOAc (15 мл) и промывали NaHCO 3 (15 мл). Водную фазу экстрагировали EtOAc (3 × 15 мл) и объединенные органические фазы сушили (MgSO 4 ), а затем концентрировали при пониженном давлении. В результате колоночной хроматографии (диоксид кремния, смесь 35% ацетон-гексан, 1% триэтиламина) получали промежуточное соединение 12 (23 мг, 40%) в виде белого твердого вещества; m/z: 808 ([М + Н] + ), 649. Пример 10. Синтез соединений из промежуточного соединения 12. Соединение А-30. К раствору промежуточного соединения 12 (50 мг, 0,062 ммоль) в CH 2 Cl 2 (1,0 мл) при КТ добавляли пиридин (0,010 мл, 0,124 ммоль, 2,0 экв.), а затем уксусный ангидрид (0,007 мл, 0,074 ммоль, 1,2 экв.). Этот раствор перемешивали при КТ в течение 2,5 ч, затем добавляли водн. NaHCO 3 (15 мл). После экстракции CH 2 Cl 2 (3 ×15 мл) органические фазы объединяли, промывали рассолом (30 мл), сушили (Na 2 SO 4 ) и концентрировали при пониженном давлении. В результате колоночной хроматографии (диоксид кремния, 50% ацетон-гексан, 1% триэтиламина) получали смесь целевых N-ацетильных и 2',N-диацетильных соединений, которые растворяли в метаноле (2 мл) и перемешивали при 50 °С в течение 3 ч. После охлаждения растворитель концентрировали с получением соединения А-30 (0,030 г, 57%) в виде белого твердого вещества; m/z: 850 ([М + Н] + ), ESI TOF MS m/z 849,5682, рассч. для C 43 H 81 N 2 O 14 ([M + Н] + ) 849,5682. Соединение А-32. К раствору промежуточного соединения 12 (75 мг, 0,093 ммоль) в CH 2 Cl 2 (1,0 мл) при КТ добавляли пиридин (0,015 мл, 0,186 ммоль, 2,0 экв.), затем метансульфонилхлорид (0,009 мл, 0,112 ммоль, 1,2 экв.). Этот раствор перемешивали при КТ в течение 2 ч, а затем добавляли водн. NaHCO 3 (20 мл). После экстракции CH 2 Cl 2 (3 ×20 мл) органические фазы объединяли, сушили (MgSO 4 ) и концентрировали при пониженном давлении. В результате колоночной хроматографии (диоксид кремния, 30% ацетон-гексан, 1% триэтиламина) получали соединение А-32 (0,045 мг, 55%) в виде белого твердого вещества; m/z: 886 ([М + Н] + ), 728; ESI TOF MS m/z 885,5321, рассч. для C 42 H 81 N 2 O 15 S ([M + Н] + ) 885,5352. Соединение А-54. К раствору промежуточного соединения 12 (0,080 г, 0,099 ммоль, 1,0 экв.) в CH 2 Cl 2 (1,0 мл) при КТ добавляли этилизоцианат (0,014 г, 0,016 мл, 0,198 ммоль, 2,0 экв.). Этот раствор перемешивали при комнатной температуре в течение 16 ч, затем добавляли дополнительное количество этилизоцианата (0,022 г, 0,025 мл, 0,316 ммоль, 3,2 экв.) и перемешивали при КТ в течение 4 ч. Этот раствор выливали в водн. NaHCO 3 (15 мл). После экстракции CH 2 Cl 2 (3 ×15 мл) объединенные органические фазы сушили (MgSO 4 ) и концентрировали при пониженном давлении. В результате колоночной хроматографии (диоксид кремния, от 35 до 50% ацетон-гексан, 1% триэтиламина) получали соединение А-54 (0,019 г) в виде белого твердого вещества; m/z: 879 ([М + Н] + ); ESI TOF MS m/z 878,5954, рассч. для C 44 H 83 N 3 O 14 ([M + Н] + ) 878,5948. Соединение А-57. К раствору промежуточного соединения 12 (0,075 г, 0,094 ммоль, 1,0 экв.) в CH 2 Cl 2 (1,0 мл) добавляли пропилизотиоцианат (0,014 г, 0,015 мл, 0,141 ммоль, 1,5 экв.) и этот раствор перемешивали при КТ в течение 18 ч. Раствор выливали в NaHCO 3 (15 мл) и органические фазы экстрагировали CH 2 Cl 2 (3 ×15 мл). Объединенные органические фазы сушили (MgSO 4 ) и концентрировали при пониженном давлении. В результате колоночной хроматографии (диоксид кремния, 50% ацетон-гексан, 0 →1% триэтиламин) получали соединение А-57 (0,032 г, 38%) в виде белого твердого вещества, m/z: 909 ([М + Н] + ), 751; ESI TOF MS m/z 908,5905, рассч. для C 45 H 85 N 3 O 13 S ([M + Н] + ) 908,5889. Соединение А-58. К раствору этил-(3-диметил)пропилкарбодиимида (0,023 г, 0,121 ммоль, 1,3 экв.) и гидроксибензотриазола (0,025 г, 0,186 ммоль, 2,0 экв.) в THF (1,0 экв.) при 0 °С добавляли 5-бензимидазолкарбоновую кислоту (0,018 г, 0,112 ммоль, 1,2 экв.). Этот раствор перемешивали при 0 °С в течение 15 мин, а затем добавляли промежуточное соединение 12 (0,075 г, 0,093 ммоль, 1,0 экв.). Через 1 ч при 0 °С раствор нагревали до КТ и перемешивали в течение 1 ч. Добавляли DMF (диметилформамид, 0,5 мл) и полученную смесь перемешивали при КТ в течение 3 ч. После разбавления EtOAc (40 мл) раствор промывали NaHCO 3 (2 ×30 мл) и рассолом (30 мл), после чего сушили (Na 2 SO 4 ) и концентрировали при пониженном давлении. В результате колоночной хроматографии (диоксид кремния, 70 →90% ацетон-гексан, 1% триэтиламина) получали соединение А-58 (0,042 г, 48%) в виде белого твердого вещества, m/z: 952 ([М + Н] + ), 794; ESI TOF MS m/z 951,5898, рассч. для C 49 H 82 N 4 O 14 ([M + Н] + ) 951,5900. Соединение А-64. К раствору промежуточного соединения 12 (0,080 г, 0,099 ммоль, 1,0 экв.) в CH 2 Cl 2 (1,0 мл) добавляли пиридин (0,016 г, 0,016 мл, 0,198 ммоль, 2,0 экв.), а затем этилхлорформиат (0,013 г, 0,011 мл, 0,119 ммоль, 1,2 экв.). Этот раствор перемешивали при КТ в течение 3 ч, после чего добавляли дополнительное количество этилхлорформиата (0,013 г, 0,011 мл, 0,119 ммоль, 1,2 экв.) и перемешивали в течение 1 ч. Раствор выливали в NaHCO 3 (15 мл) и органические фазы экстрагировали CH 2 Cl 2 (3 ×15 мл). Объединенные органические фазы сушили (Na 2 SO 4 ) и концентрировали при пониженном давлении. В результате колоночной хроматографии (диоксид кремния, 50% ацетон-гексан, 1% Et 3 N) получали соединение А-64 (0,030 г, 34%) в виде белого твердого вещества; m/z: 880 ([М + Н] + ); ESI TOF MS m/z 879,5796, рассч. для C 44 H 8 2N 2 O 15 ([M + Н] + ) 879,5788. Соединение А-65. Использовали способ, аналогичный способу, использованному для получения соединения А-64, но с метилхлорформиатом вместо этилхлорформиата. m/z: 866 ([М + H] + ); ESI TOF MS m/z 865,5630, рассч. для C 43 H 80 N 2 O 15 ([М + Н] + ) 856,5632. Соединение А-67. Использовали способ, аналогичный способу, использованному для получения соединения А-57, но с этилизотиоцианатом вместо пропилизотиоцианата. m/z: 895 ([М + Н] + ); ESI TOF MS m/z 894,5724, рассч. для C 44 H 83 N 3 O 13 S ([М + Н] + ) 894,5719. Соединение А-78. К раствору промежуточного соединения 12 (0,150 г, 0,186 ммоль, 1,0 экв.) в DMF (2,0 мл) при 0 °С добавляли диметиламино-пропилэтилкарбодиимид (0,079 г, 0,409 ммоль, 2,2 экв.) и гидроксибензотриазол (0,050 г, 0,372 ммоль, 2,0 экв.), а затем муравьиную кислоту (0,017 г, 0,014 мл, 0,372 ммоль, 2,0 экв.). Этот раствор перемешивали при 0 °С в течение 30 мин и при комнатной температуре в течение 3 ч, после чего распределяли между EtOAc (25 мл) и NaHCO 3 (25 мл). Водную фазу экстрагировали EtOAc (25 мл) и объединенные органические фазы промывали водой (35 мл), NaHCO 3 (35 мл) и рассолом (40 мл), после чего сушили (Na 2 SO 4 ) и концентрировали при пониженном давлении. В результате колоночной хроматографии (диоксид кремния, 40% ацетон-гексан, 1% триэтиламина) получали соединение А-78 (0,072 г, 46%) в виде белого твердого вещества, m/z: 836 ([М + Н] + ), 678; ESI TOF MS m/z 835,5501, рассч. для C 42 H 78 N 2 O 14 ([М + Н] + ) 835,5526. Пример 11. Промежуточное соединение 15. Промежуточное соединение 15 использовали в синтезе ряда соединений по данному изобретению. К раствору 9(S)-эритромициламина 13 (15,8 г, 21,5 ммоль; см., например, Massey et al., J. Med. Chem., 1974, 17 (1), 105-107) в CH 2 Cl 2 (60 мл) добавляли диизопропилэтиламин (14,8 мл, 85,0 ммоль), затем метансульфоновый ангидирид (6,45 г, 37,0 ммоль) в CH 2 Cl 2 (35 мл) при -10 °С в течение 1 ч и перемешивание продолжали в течение еще 1,5 ч при этой температуре. Реакционную смесь гасили путем добавления насыщенного NaHCO 3 (100 мл) и Na 2 CO 3 (10% в Н 2 О, 20 мл). Полученную смесь перемешивали в течение 10 мин при КТ. Органический слой отделяли и водный слой экстрагировали EtOAc (2 ×20 мл). Объединенные органические слои сушили над MgSO 4 /K 2 CO 3 , фильтровали через тонкую набивку K 2 CO 3 и концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии (от 5 до 70% ацетона в гексанах, 1% триэтиламина) с получением 9,9 г (12,2 ммоль, 56%) чистого соединения 14 в виде белого твердого вещества. ESI TOF MS m/z 813,4770, рассч. для C 38 H 73 N 2 O 14 S ([M + Н] + ) 813,4740. К перемешиваемому раствору соединения 14 (86,8 мг, 0,107 ммоль) и ацетата натрия (43,9 мг, 0,535 ммоль, 5,0 экв.) в смеси МеОН/Н 2 О (4:1, 2 мл) добавляли иод (29,8 мг, 0,117 ммоль, 1,1 экв.) при 50 °С. Затем по каплям в течение 1 ч добавляли 0,1 н. раствор NaOH (1,17 мл, 0,117 ммоль, 1,1 экв.). Перемешивание продолжали в течение 2 ч при той же температуре. Добавляли NaOH (0,1 мл, 0,1 н.) и I 2 (3 мг) и реакционную смесь перемешивали в течение 1 ч. Реакционную смесь концентрировали до приблизительно 200 мкл и разбавляли CH 2 Cl 2 (10 мл) и насыщенным NaHCO 3 (5 мл). Водный слой экстрагировали CH 2 Cl 2 (3 ×5 мл). Объединенные органические фазы промывали разбавленным Na 2 S 2 O 3 (5 мл), Н 2 О (5 мл) и сушили над MgSO 4 . Раствор фильтровали и растворитель удаляли при пониженном давлении. В результате очистки колоночной хроматографией (от 0 до 5% МеОН в CH 2 Cl 2 , 2% триэтиламина) получали промежуточное соединение 15 в виде белого твердого вещества (70 мг, 84%). Пример 12. Синтез соединений из промежуточного соединения 15. Соединение А-35. К раствору промежуточного соединения 15 (35 мг, 0,044 ммоль) в CH 3 CN (400 мкл) добавляли диизопропилэтиламин (76,3 мкл, 0,44 ммоль, 10,0 экв.) и 2-иодпропан (65,7 мкл, 0,66 ммоль, 15,0 экв.). Растворитель удаляли при пониженном давлении и остаток очищали посредством колоночной хроматографии (от 5 до 70% ацетона в гексанах, 1% триэтиламина) с получением соединения А-35 (24 мг, 65%). m/z: 842,0 ([М + Н] + ); ESI TOF MS m/z 841,5093, рассч. для C 40 H 77 N 2 O 14 S ([M + Н] + ) 841,5090. Соединение А-36. Использовали способ, аналогичный способу, использованному для получения соединения А-35, но с 2-иодэтанолом в качестве алкилирующего агента. m/z: 844,0 ([М + Н] + ); ESI TOF MS m/z 843,4894, рассч. для C 40 H 75 N 2 O 15 S ([М + Н] + ) 843,3883. Соединение А-37. К раствору промежуточного соединения 15 (120 мг, 0,15 ммоль) в СН 3 ОН (1,2 мл) добавляли 2,2-диметилоксиран (133 мкл, 1,5 ммоль, 10 экв.). Реакционную смесь перемешивали при 50 °С в течение ночи, а затем концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии (от 5 до 50% ацетона в гексанах, 1% триэтиламина) с получением соединения А-37 (73 мг, 54%) в виде белого твердого вещества, m/z: 872,0 ([М + Н] + ); ESI TOF MS m/z 871,5171, рассч. для C 41 H 79 N 2 O 15 S ([M + Н] + ) 871,5196. Соединение А-38. Использовали способ, аналогичный способу, использованному для получения соединения А-35, но с 1-иод-2-метилпропаном в качестве алкилирующего агента, m/z: 856,0 ([М + Н] + ); ESI TOF MS m/z 855,5186, рассч. для C 41 H 79 N 2 O 14 S ([M + Н] + ) 855,5247. Соединение А-39. К раствору промежуточного соединения 13 (240 мг, 0,30 ммоль.), NaCNBH 3 (43,4 мг, 0,69 ммоль, 2,3 экв.) и уксусной кислоты (69 мкл, 1,2 ммоль, 4,0 экв.) в МеОН (2,0 мл) добавляли циклобутанон (45 мкл, 0,6 ммоль, 2,0 экв.). Реакционную смесь перемешивали при КТ в течение ночи и разбавляли EtOAc (30 мл), Na 2 CO 3 (10%, 5 мл) и насыщенным NaHCO 3 (10 мл), рассолом (10 мл). Водный слой экстрагировали ЕЮ Ас (2х10 мл). Объединенные органические фазы сушили над Na 2 SO 4 , фильтровали и концентрировали. Остаток очищали посредством колоночной хроматографии (от 5 до 50% ацетона в гексанах, 1% триэтиламина) с получением соединения А-39 в виде белого твердого вещества (106 мг, 42%). m/z: 854,0 ([М + Н] + ); ESI TOF MS m/z 853,5090, рассч. для C 41 H 77 N 2 O 14 S ([M + Н] + ) 853,5090. Пример 13. Синтез промежуточного соединения 19. Промежуточное соединение 19, 4"-дезоксианалог промежуточного соединения 9, было синтезировано из 4"-дезоксиэритромицина А (16), используя процедуру, аналогичную использованной для получения промежуточного соединения 9: m/z: 779 ([М + Н] + ), 621; ESI TOF MS m/z 778,5345, рассч. для C 40 H 76 NO 13 ([M + Н] + ) 778,5311. Пример 14. Синтез соединений из промежуточного соединения 19. Соединение А-60. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с промежуточным соединением 19 в качестве исходного вещества и с N,N-диметилбромацетамидом в качестве алкилирующего агента вместо бромацетамида. m/z: 833,6 ([М + Н] + ). Соединение А-61. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с промежуточным соединением 19 в качестве исходного вещества и с N-метилбромацетамидом в качестве алкилирующего агента вместо бромацетамида. m/z: 819,6 ([М + Н] + ). Соединение А-68. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с промежуточным соединением 19 в качестве исходного вещества, m/z: 806,0 ([М + Н] + ), ESI TOF MS m/z 805,5410, рассч. для C 41 H 77 N 2 O 13 ([M + Н] + ) 805,5420. Пример 15. Синтез промежуточного соединения 23. Промежуточное соединение 23, эритромицин В-аналог промежуточного соединения 9, было синтезировано из эритромицина В (20) с использованием процедуры, аналогичной использованной для получения промежуточного соединения 9: m/z: 748,5 ([М + Н] + ). ESI TOF MS m/z 748,5225, рассч. для C 39 H 74 NO 12 ([M + Н] + ) 748,5206. Пример 16. Синтез соединений из промежуточного соединения 23. Соединение А-71. Раствор трет-бутоксида калия (1M в THF, 0,98 мл, 0,98 ммоль) добавляли к раствору промежуточного соединения 23 (490 мг, 0,66 ммоль) в безводном диметоксиэтане (6 мл) в инертной атмосфере и перемешивали при комнатной температуре в течение 10 мин. Добавляли N-метилбромацетамид (120 мг, 0,79 ммоль) и реакционную смесь перемешивали в течение 30 мин. TLC анализ показал завершение потребления исходного вещества, избыток реагентов гасили путем добавления насыщенного раствора NaHCO 3 и смесь экстрагировали EtOAc. Объединенные органические слои сушили с помощью Mg 2 SO 4 и концентрировали при пониженном давлении. В результате флэш-хроматографии с использованием гексана и ацетона с 2% Et 3 N получали целевой продукт. ESI TOF MS m/z 819,5572, рассч. для C 42 H 79 N 2 O 13 ([М + Н] + ) 819,5577. 13 С ЯМР (CDCl 3 ). 177.6, 170.7, 102.2, 94.8, 93.4, 84.8, 77.7, 77.4, 75.7, 74.6, 72.8(2), 70.7, 70.0, 69.4, 65.6, 62.2, 52.6, 49.3, 43.7, 43.1, 38.8, 34.7, 32.8(2), 31.0, 25.5, 24.4, 21.5, 21.2, 21.1, 20.4, 19.9, 17.6, 12.7, 11.7, 9.8, 9.7, 9.2. Соединение А-72. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с промежуточным соединением 23 в качестве исходного вещества и с N,N-диметилбромацетамидом в качестве алкилирующего агента вместо бромацетамида. ESI TOF MS m/z 833,5699, рассч. для C 43 H 81 N 2 O 13 ([M + Н] + ) 833,5733. Соединение А-75. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с промежуточным соединением 23 в качестве исходного вещества и с N,N-диэилкарбамоилхлоридом в качестве алкилирующего агента вместо бромацетамида. ESI TOF MS m/z 819,5548, рассч. для C 42 H 79 N 2 O 13 ([M + Н] + ) 819,5577. Пример 17. Соединения с R F , представляющим собой метил. Промежуточное соединение 27, 6-O-метильный аналог промежуточного соединения 9, получали из соединения 24 (6-O-метил-эритромицин А, также известный как кларитромицин), используя процедуры, аналогичные использованным для получения промежуточного соединения 9: m/z: 779 ([М + Н] + ), 621; ESI TOF MS m/z 778,5345, рассч. для C 40 H 76 NO 13 ([M + Н] + ) 778,5311. Соединение А-66. Использовали способ, аналогичный способу, использованному для получения соединения А-12, но с промежуточным соединением 27 в качестве исходного вещества и с N-метил-бромацетамидом в качестве алкилирующего агента вместо бромацетамида. m/z: 850,0 ([М + Н] + ); ESI TOF MS m/z 849,5710, рассч. для C 43 H 81 N 2 O 14 ([M + Н] + ) 849,5682. Пример 18. Синтез других соединений. Соединение А-40. Способ, аналогичный использованному для получения промежуточного соединения 15, использовали для получения этансульфонамида, который затем деметилировали и реалкилировали с помощью изопропилиодида, как описано выше в отношении соединения А-38, с получением соединения А-40. m/z: 856,0 ([М + Н] + ). Соединение А-41. Способ, аналогичный использованному для промежуточного соединения 15, использовали для получения циклопропансульфонамида, который затем деметилировали и реалкилировали с помощью изопропилиодида, как описано выше, с получением соединения А-41. m/z: 868,0 ([М + Н] + ). Соединение А-42. Использовали способ, аналогичный использованному для соединения А-41, но дезметильное промежуточное соединение подвергали взаимодействию с циклобутаноном в условиях восстановительного аминирования, как описано выше, с получением соединения А-42. m/z: 880,0 ([M + Н] + ). Соединение А-43. Способ, аналогичный использованному для получения соединения А-39, использовали для получения трифторметансульфонамида, который затем деметилировали и реалкилировали с помощью изопропилиодида, как описано выше, с получением соединения А-43. m/z: 896,0 ([М + Н] + ). Соединение А-44. Использовали способ, аналогичный использованному для соединения А-40, но с диметилсульфамоилхлоридом. m/z: 872,0 ([М + Н] + ); ESI TOF MS m/z 871,5218, рассч. для C 41 H 79 N 2 O 15 S ([M + Н] + ) 871,5196. Соединение А-56. К раствору соединения А-22 (62 мг, 0,074 ммоль, 1,0 экв.) в СН 3 ОН (1 мл) добавляли NaOH (1,0 н., 0,078 мл, 1,05 экв.). Эту реакционную смесь перемешивали при КТ в течение 2 дней, затем концентрировали и остаток лиофилизировали со смесью трет-BuOH/H 2 O (93:7) с получением соединения А-56 (60 мг, 0,071 ммоль, 96%) в форме натриевой соли, m/z: 823,0 ([М + Н] + ); ESI TOF MS m/z 822,5214, рассч. для C 41 H 75 NO 15 ([M + Н] + ) 822,5223. Специалисту в данной области техники понятно, что вышеприведенные методики синтеза могут быть применены при внесении необходимых изменений для получения других соединений по данному изобретению, включая те, в которых R C , R D , R E и R F не являются ОН, Me, ОН и Н, соответственно, используя альтернативные известные и/или имеющиеся в продаже вещества-предшественники. Соединения, в которых R F представляет собой Me, может быть получено из кларитромицина (6-О-метилэритромицин A, Biaxin ™; Watanabe et al., US 4331803 (1982)). Соединения, где R C и R D не являются ОН и Me, соответственно, могут быть получены с использованием предшественников эритромицинов В, С или D. Соединения, в которых R E представляет собой Н, могут быть получены путем удаления группы 4"-ОН из эритромицина, например, как показано в Lartey et al., US 5578579 (1996). Пример 19. Анализ на основе ткани на активность агониста мотилина. Активность агониста мотилина соединений по данному изобретению оценивали с помощью анализа на основе ткани, используя анализ сократительной способности на основе ткани двенадцатиперстной кишки кролика, в общем следуя процедуре согласно Depoortere et al., J. Gastrointestinal Motility, 1, 150-159 (1989), описание которого включено посредством ссылки. Вкратце, в этом методе измеряют способность соединения индуцировать сокращения в ткани двенадцатиперстной кишки кролика, которая несет рецепторы мотилина и способна к сократительному ответу на мотилин. Полоски двенадцатиперстной кишки кролика тестировали и оценивали для использования в данном анализе следующим образом. Сегменты двенадцатиперстной кишки кролика, 20-30 см дистально по отношению к привратнику желудка, вырезали в продольном направлении. Слизистую оболочку удаляли и из сегментов вырезали полоски продольной гладкой мышцы размером 2 ×2 ×15 мм. Эти полоски погружали в оксигенированный раствор Кребса при 37 °С с напряжением 1,5 г и ауксотонически измеряли сокращения. Полоски, демонстрирующие сильную регулярную фазовую активность (амплитуда 0,3 г, FFT пик при 0,3-0,4 Гц, более чем в три раза более сильный по сравнению с другими пиками) и быстрые воспроизводимые ответы на 1 мкМ карбахола (ССН) (пиковое сокращение менее чем 30 с, более чем 3 × фазовая амплитуда) оценивали для использования в этом анализе; полоски, не удовлетворявшие вышеуказанным критериям, отбрасывали. Затем карбахол отмывали путем двукратной смены буфера в ванне для органов. Полоски снова промывали в пределах 20 ±5 мин после сокращения в ответ на карбахол. После этой последней промывки в пределах 10 ±5 мин начинали исследование доза-ответ. Каждое тестируемое соединение растворяли в диметилсульфоксиде (DMSO) до конечной концентрации 10 мМ. Готовили серию из семи 10Х серийных разведений в воде так, чтобы концентрация седьмого серийного разведения составляла 1,0 ×10 -6 мМ. Использовали серийные разведения соединений с первого по пятое, начиная с 200 мкл наиболее разбавленного раствора. После каждого использования делали перерыв на 2 ±0,5 мин до тех пор, пока ответ не становился стабильным, а затем использовали следующую дозу (следующее серийное разведение с более высокой концентрацией). Дозу увеличивали с 10-кратными возрастаниями до тех пор, пока не наблюдали небольшой ответ. Следующие дозы увеличивали с возрастаниями от 2 до 5 раз, до тех пор, пока не получали максимальный ответ. Через 2 ±0,5 мин после последнего добавления лекрственного средства на полоски воздействовали 1 мкМ карбахола. ЕС 50 (концентрация, дающая половину максимального эффекта) рассчитывали следующим образом. Для каждого данного базовое напряжение вычитали из индуцированного соединением напряжения. Точки данных нормализовали против ответа, полученного для 1 мкМ карбахола в конце эксперимента. Концентрацию соединения наносили на графике против ответа и обрабатывали посредством следующего уравнения: где R представляет собой сократительный ответ, R max представляет собой максимальный сократительный ответ и С представляет собой концентрацию соединения. R и R max выражают как долю сокращения под действием 1 мкМ карбахола и диапазона от 0 до 1. Результаты представлены в табл. Б ниже. Также следующим образом может быть установлена и подтверждена ЕС 90 (концентрация, дающая 90% максимального эффекта): ЕС 90 сначала аппроксимируют как десятикратную EC 50 . Точность этой аппроксимации затем проверяют с помощью кривой доза - ответ. Отобранные полоски двенадцатиперстной кишки обрабатывают дозой, равной 0,25 ЕС 90 . После получения максимального ответа (2 ±0,5 мин) дозу увеличивают в четыре раза. Через 2 ±0,5 мин полоски обрабатывают карбахолом в дозе 1 мкМ. Разница между этими двумя дозами должна находиться в диапазоне 10-20%. Вторую группу отобранных полосок двенадцатиперстной кишки обрабатывали дозой, равной ЕС 90 . После получения максимального ответа (2 ±0,5 мин) дозу увеличивали в два раза. Через 2 ±0,5 мин эти полоски обрабатывали карбахолом в дозе 1 мкМ. Разница между этими двумя дозами должна составлять менее 10%. Таким образом, соединения по данному изобретению могут быть использованы для индукции сокращения ткани, несущей рецепторы мотилина, которая способна к сократительному ответу на мотилин. Индукция таких сокращений может иметь благоприятные эффекты в плане стимуляции моторики ЖКТ. Эта ткань может представлять собой ткань млекопитающего, такую как ткань кролика или человека, особенно ткань ЖКТ. Пример 20. Оценка антибактериальной активности. Антибактериальную активность соединений по данному изобретению оценивали путем измерения их минимальных ингибирующих концентраций (МИК) против Streptococcus pneumoniae АТСС 6301 (штамм, чувствительный к эритромицину А), используя серийные разведения в 96-луночных титрационных микропланшетах. Желательно, чтобы соединения обладали низкой антибактериальной активностью. Результаты представлены в табл. Б ниже. В табл. Б суммированы данные для соединений по данному изобретению. Также представлены сравнительные данные для эритромицина А, АВТ-229, GM-611 и KC-11458. Последние три соединения представляют собой опытные мотилиды от Abbott Laboratories, Chugai и Solvay, соответственно. Пример 21. Модель хронического дозирования для оценки тахифилаксии. В этом примере описывается, как можно оценить тахифилаксию (снижение ответа после первоначального введения; эффект десенситизации в ответ на агонистическое действие соединения) соединений по дпнному изобретению. Полоски двенадцатиперстной кишки кролика готовили, как описано выше, и обрабатывали тестируемым соединением в дозе, составляющей его ЕС 90 концентрацию. Регистрируют сокращение. Когда достигается пиковая сила сокращения, добавляют карбахол (1 мкМ) и регистрируют любое дополнительное сокращение. Результирующее сокращение выражают как долю сокращения, вызываемого 1 мкМ карбахола. Тестируемое соединение и карбахол отмывали путем двукратной смены омывающего раствора. Процедуру повторяют в моменты времени 30, 60 и 90 мин после первоначального дозирования. Тахифилаксию рассчитывали как процент первоначального сокращения, сохранившйся после четвертой дозы тестируемого соединения. Соединение, демонстрирующее низкую тахифилаксию, будет иметь высокую ценность. Тахифилаксия = 100% × (сокращение после 4-й дозы)/(сокращение после первоначальной дозы). Пример 22. Ингибирование hERG канала. Проаритмические эффекты эритромицина и родственных соединений характеризовали по их ингибированию калиевого канала, кодируемого геном hERG (человеческий ether-a-go-go related gene). Ингибирующее действие в отношении hERG канала соединений по данному изобретению можно оценить, используя методику, о которой сообщается в Stanat et al., Mol. Cellular Biochem., 2003, 254, 1-7, "Characterization of the Inhibitory Effects of Erythromycin and Clarithromycin on the HERG Potassium Channel". Ингибирование может быть выражено как % ингибирования при концентрации тестируемого соединения, равной 30 мкМ. Желательно, чтобы соединения имели низкий % ингибирования. Предшествующее подробное описание изобретения включает фрагменты, которые касаются главным образом или исключительно отдельных частей или аспектов изобретения. Следует понимать, что это предназначено для ясности и удобства, что конкретный признак может быть релевантным не только для того фрагмента, в котором он раскрыт, и что данное описание включает все соответствующие комбинации информации, присутствующей в разных фрагментах. Аналогично, хотя к конкретным воплощениям изобретения относятся разные описания, следует понимать, что там, где конкретный признак описан в контексте частного воплощения, такой признак также может быть использован в соответствующей степени в контексте другого воплощения, в комбинации с другим признаком или в изобретении в целом. Далее, хотя настоящее изобретение было подробно раскрыто в контексте определенных предпочтительных воплощений, данное изобретение не ограничено такими предпочтительными воплощениями. Объем изобретения определяется прилагаемой формулой изобретения. |