|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea000013119b*\id |
|
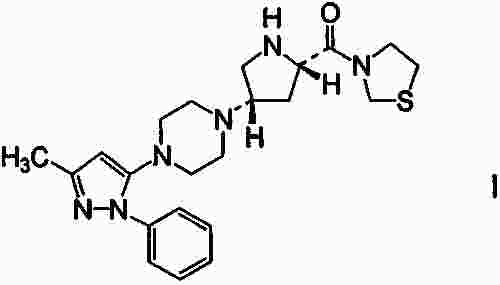
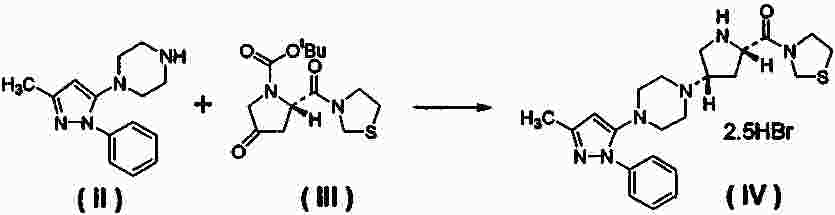
Термины запроса в документе Реферат Настоящее изобретение относится к 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидину (соединение I), который можно использовать как ингибитор дипептидилпептидазы-IV, имеет превосходные свойства стабильности и гигроскопичности, воспроизводимую структуру кристалла, и к способу его получения. Формула [0001] Соль 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина с 2,0 экв. бромисто-водородной кислоты, 2,5 экв. бромисто-водородной кислоты, 2 экв. малеиновой кислоты, 2 экв. толуолсульфоновой кислоты, 2 экв. безиловой кислоты, 2 экв. соляной кислоты, 2,5 экв. соляной кислоты, 2 экв. нафталин-1-сульфоновой кислоты, 2 экв. нафталин-2-сульфоновой кислоты, 2 экв. камфорсульфоновой кислоты, с фумаровой кислотой, серной кислотой, янтарной кислотой, L-винной кислотой или лимонной кислотой или ее гидрат. [0002] Соль 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина с 2,0 экв. бромисто-водородной кислоты, 2,5 экв. бромисто-водородной кислоты, 2 экв. малеиновой кислоты, 2 экв. толуолсульфоновой кислоты, 2,5 экв. соляной кислоты, 2 экв. нафталин-1-сульфоновой кислоты, 2 экв. метилсульфоновой кислоты, 3 экв. метилсульфоновой кислоты или 2 экв. нафталин-2-сульфоновой кислоты или ее гидрат. [0003] 2,5 экв. Гидробромид 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина или его гидрат. [0004] Соединение по п.3, рентгенодифракционный спектр которого характеризуется пиками, соответствующими углам 2 θ:5,4, 13,4 и 14,4 ° (каждый ±0,2 °), или его гидрат. [0005] Гидрат по п.4, который представляет собой 1,0-2,0 экв. гидрат. [0006] Соединение по п.3, рентгенодифракционный спектр которого имеет пики, соответствующие углам 2 θ:5,4, 13,4, 14,4, 22,6 и 26,5 ° (каждый ±0,2 °), или его гидрат. [0007] Гидрат по п.6, который представляет собой 1,0-2,0 экв. гидрат. [0008] Соединение по п.3, которое имеет растворимость в воде от 7 мг/мл до 2 г/мл при температуре внешней среды, или его гидрат. [0009] Соединение по п.8, которое имеет растворимость в воде не менее чем 20 мг/мл при 37 °С, или его гидрат. [0010] Соединение по п.8, которое имеет растворимость в воде 7 мг/мл при рН 9-12, или его гидрат. [0011] Соединение по п.3, которое имеет гигроскопичность не более чем 6% при измерении при 25 °С, или его гидрат. [0012] Соединение по п.11, которое имеет гигроскопичность 5% при измерении при относительной влажности в пределах диапазона 0-50% при 25 °С, или его гидрат. [0013] Соединение по п.11, которое имеет гигроскопичность 2% при измерении при относительной влажности в пределах диапазона 5-90% при 25 °С, или его гидрат. [0014] Способ получения 2,5 экв. гидробромида 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина или его гидрата, включающий удаление 1,1-диметилэтилоксикарбонила из 3-{(2S,4S)-1-(1,1-диметилэтилоксикарбонил)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина действием бромисто-водородной кислоты с одновременным образованием соли. [0015] Способ получения 2,5 экв. гидробромида 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина или его гидрата, включающий кристаллизацию 2,5 экв. гидробромида 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина из воды и/или растворителя, выбранного из этанола, 1-пропанола, 2-пропанола, этилацетата и ацетона. [0016] 1,0-2,0 экв. Гидрат 2,5 экв. гидробромида 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина. Полный текст патента Область техники, к которой относится изобретение Настоящее изобретение относится к новой соли 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина, которая может быть применена как ингибитор дипептидилпептидазы-IV (в дальнейшем обозначаемой как DPP-IV), и к ее сольвату. DPP-IV ингибиторы ингибируют инактивацию глюкагоноподобного пептида-1 (в дальнейшем обозначаемого как GLP-1) в плазме, и потенцируют его инкретиновое действие. Следовательно, они могут быть применены в качестве терапевтических препаратов при диабете и подобном и исследуются и разрабатываются как лекарственные средства, потенциально эффективные для лечения диабета, особенно диабета типа 2 (см. ссылки на патенты 1-6 и непатентную ссылку 1). Ряд соединений был описан в качестве производных тиазолидина, которые могут быть применены (см. ссылку на патент 7). Заслуживает внимания как пример соединений, описанных в упомянутой работе, 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидин (в дальнейшем обозначаемый как соединение I). В то время как соединение I описано в форме соли с 3 экв. гидрохлорида, эта соль имеет фармацевтически нежелательные свойства в отношении стабильности и гигроскопичности, и, как было найдено, воспроизводимое получение в той же форме является трудным. В частности, для удовлетворения нормативных требований к разработке фармацевтических продуктов необходимы воспроизводимые методы получения соединения. Следовательно, эти свойства, наблюдаемые для соединения I в форме соли с 3 экв. гидрохлорида, считаются неблагоприятными для разработки фармацевтических продуктов. Кроме того, в то время как упомянутая работа (ссылка на патент 7) впервые описывает отдельные соли соединения I и других производных тиазолидина в качестве соединений по примерам, не было найдено обсуждения полиморфного кристалла любого из соединений по примерам. Способность вещества кристаллизоваться в двух или более видах кристаллических структур известно как полиморфизм, и индивидуальные кристаллические формы называются полиморфным кристаллом. Различные полиморфные кристаллы отдельного соединения иногда обнаруживают полностью различные свойства в отношении сохранения стабильности, растворимости и подобные. Такое различие в свойствах может приводить к различию в эффективности действия. С точки зрения таких различий, исследование индивидуальных полиморфных кристаллов и смесей полиморфных кристаллов является особенно полезным для разработки фармацевтических продуктов. Существуют многочисленные обозначения полиморфных кристаллов в зависимости от номенклатуры, такие как форма А, форма В, форма I, форма II, форма α, форма β и подобные. В этих обозначениях, термин "тип" (тип А и т.д.) может быть применен взамен термина "форма". В любом варианте обе системы обозначения применяются для обозначения того же. Однако не всегда легко найти различные полиморфные кристаллы определенного соединения. Если присутствие отдельного полиморфного кристалла признано и его характеристики считаются предпочтительными, возникает необходимость в разработке способа обеспечения полиморфного кристалла в большом количестве в виде отдельных кристаллов. Найти способ получения отдельного кристалла или, по существу, отдельного кристалла определенного полиморфного кристалла не легко, и требуются интенсивные исследования. Патент 1: WO 97/040832. Патент 2: WO 98/019998. Патент 3: патент США № 5939560. Патент 4: WO 01/055105. Патент 5: WO 02/002560. Патент 6: WO 02/062764. Патент 7: WO 02/014271. Непатентная ссылка 1: J. Med. Chem., 47(17), 4135-4141 (2004). Проблема настоящего изобретения состояла в нахождении, в отношении соединения I, соединения, имеющего превосходные свойства в плане стабильности, растворимости, гигроскопичности, биодоступности и подобного, пригодных для получения фармацевтических продуктов и воспроизводимой структуры кристалла, а также в разработке способа его получения. Авторы настоящего изобретения получили соли соединения I с моно-, ди- и трехосновными кислотами, и охарактеризовали кристаллы индивидуальных солей и их гидратов, и нашли новые соли соединения I, имеющие предпочтительные свойства в плане стабильности и гигроскопичности. Далее авторы провели интенсивные исследования и нашли стабильный промышленный способ получения новой соли по настоящему изобретению, что привело к завершению настоящего изобретения. Соответственно объектом настоящего изобретения являются соли 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина с 2,0 экв. бромисто-водородной кислоты, 2,5 экв. бромисто-водородной кислоты, 2 экв. малеиновой кислоты, 2 экв. толуолсульфоновой кислоты, 2 экв. безиловой кислоты, 2 экв. соляной кислоты, 2,5 экв. соляной кислоты, 2 экв. нафталин-1-сульфоновой кислоты, 2 экв. нафталин-2-сульфоновой кислоты, 2 экв. камфорсульфоновой кислоты, с фумаровой кислотой, серной кислотой, янтарной кислотой, L-винной кислотой или лимонной кислотой или их гидраты. Ещё одним объектом настоящего изобретения являются соли 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина с 2,0 экв. бромисто-водородной кислоты, 2,5 экв. бромисто-водородной кислоты, 2 экв. малеиновой кислоты, 2 экв. толуолсульфоновой кислоты, 2,5 экв. соляной кислоты, 2 экв. нафталин-1-сульфоновой кислоты, 2 экв. метилсульфоновой кислоты, 3 экв. метилсульфоновой кислоты или 2 экв. нафталин-2-сульфоновой кислоты или их гидраты. Другим объектом настоящего изобретения является 2,5 экв. гидробромид 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина или его гидрат, предпочтительно 1,0-2,0 экв. гидрат. Указанное соединение или его гидрат имеет рентгенодифракционный спектр, который характеризуется пиками, соответствующими углам 2 θ: 5,4 °, 13,4 ° и 14,4 ° (каждый ±0,2 °). В предпочтительном варианте указанное соединение или его гидрат характеризуется рентгенодифракционным спектром, который имеет пики, соответствующие углам 2 θ: 5,4 °, 13,4 °, 14,4 °, 22,6 ° и 26,5 ° (каждый ±0,2 °). В предпочтительном варианте 2,5 экв. гидробромид 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина имеет растворимость в воде от 7 мг/мл до 2 г/мл при температуре внешней среды. Предпочтительно растворимость указанного соединения в воде составляет не менее чем 20 мг/мл при 37 °С. Предпочтительно растворимость указанного соединения в воде составляет 7 мг/мл при рН 9-12. Предпочтительно 2,5 экв. гидробромид 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина имеет гигроскопичность не более чем 6% при измерении при 25 °С. Еще более предпочтительно, если указанное соединение имеет гигроскопичность 5% при измерении при относительной влажности в пределах диапазона 0-50% при 25 °С. И наиболее предпочтительно, если указанное соединение имеет гигроскопичность 2% при измерении при относительной влажности в пределах диапазона 5-90% при 25 °С. Ещё одним объектом изобретения является способ получения 2,5 экв. гидробромида 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина, или его гидрата, включающий удаление 1,1-диметилэтилоксикарбонила из 3-{(2S,4S)-1-(1,1-диметилэтилоксикарбонил)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина действием бромисто-водородной кислоты, с одновременным образованием соли. Другим объектом изобретения является способ получения 2,5 экв. гидробромида 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина или его гидрата, включающий кристаллизацию 2,5 экв. гидробромида 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина из воды и/или растворителя, выбранного из этанола, 1-пропанола, 2-пропанола, этилацетата и ацетона. Объектом настоящего изобретения также является 1,0-2,0 экв. гидрат 2,5 экв. гидробромида 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина. Соли соединения I, их гидраты и их новые полиморфные кристаллы имеют одно или несколько свойств, выбранных из улучшенной стабильности, улучшенной гигроскопичности (расплывание в адсорбированной влаге), быстрое выделение из растворителя и упрощенный способ получения, которые способствуют использованию соединения I в качестве фармацевтического продукта. Фиг. 1 показывает результаты измерения порошковой рентгенограммы соединения, обозначенного в заголовке примера 4, где ось Y показывает интенсивность дифракции и ось абсцисс показывает угол дифракции (2 θ). Фиг. 2 показывает результаты измерений порошковой рентгенограммы соединения примера 5(1), где ось Y показывает интенсивность дифракции и ось абсцисс показывает угол дифракции (2 θ). Фиг. 3 показывает результаты измерений порошковой рентгенограммы соединения примера 5(2), где ось Y показывает интенсивность дифракции и ось абсцисс показывает угол дифракции (2 θ). Фиг. 4 показывает результаты измерений порошковой рентгенограммы соединения примера 5(3), где ось Y показывает интенсивность дифракции и ось абсцисс показывает угол дифракции (2 θ). Фиг. 5 показывает результаты измерений гигроскопичности соединения, обозначенного в заголовке примера 3, где -о- отображает адсорбцию воды в соединении при влажности по оси абсцисс и - •- отображает десорбцию воды из соединения при влажности по оси абсцисс. 3-{(2S,4S)-4-[4-(3-Метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил} тиазолидин (соединение I) показан ниже Соединение I в форме соли с 3 экв. гидрохлорида может быть получено согласно способу синтеза, описанному как пример 222 в WO 02/14271. Это соединение может быть конвертировано в свободное основание с использованием приемлемого основания. В качестве применяемого основания могут быть упомянуты карбонаты щелочного металла или щелочно-земельного металла (бикарбонат натрия, карбонат натрия, карбонат калия и т.д.), гидроксиды щелочного металла или щелочно-земельного металла (гидроксид натрия, гидроксид калия и т.д.) и подобные. Соединение I может быть получено, например, путем добавления соединения из примера 222 к водному раствору любого основания, упомянутого выше, и экстрагирования смеси углеводородным растворителем (бензол, толуол и т.д.), галогенированным углеводородным растворителем (дихлорметан, дихлорэтан, хлорформ, тетрахлорид углерода и т.д.), этилацетатом и подобными. Кроме того, соединение I в форме соли с 2,5 экв. гидробромида также может быть получено согласно следующей схеме: Соединение I (IV) может быть получено в форме соли с 2,5 экв. гидробромида введением 3-[(2S)-1-(1,1-диметилэтилоксикарбонил)-4-оксопирролидин-2-илкарбонил]тиазолидина (III) в восстановительное аминирование с 1-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазином (II) или его солью и затем удаления 1,1-диметилэтилоксикарбонила из полученного соединения действием бромисто-водородной кислоты. Восстановительное аминирование проводится с применением от около 0,5 до 10 моль, предпочтительно от около 1 до 2 моль соединения, представленного формулой (III) и от около 0,5 до 10 моль, предпочтительно от около 1 до 2 моль металлгидридного комплексного соединения (композитного водородного соединения, такого как боргидрид натрия, цианоборгидрид натрия, триацетоксиборгидрид натрия и подобные; диборан и т.д.), обоих в расчете на 1 моль соединения, представленного формулой (II) или его солью, в инертном растворителе и, где необходимо, в присутствии кислотного катализатора (уксусная кислота, паратолуолсульфокислота, комплекс борон трифторид эфират-диэтиловый эфир и т.д.). В качестве инертного растворителя могут быть упомянуты спирты (метанол, этанол, 1-пропанол, 2-пропанол (в дальнейшем обозначаемый как ИПС), бутанол и т.д.), нитрилы (ацетонитрил, пропионитрил и т.д.), амиды (формамид, N,N-диметилформамид, N,N-диметилацетамид, 1,3-диметил-2-имидазолидинон и т.д.), простые эфиры (диэтиловый эфир, диизопропиловый простой эфир, трет-бутилметиловый эфир, 1,4-диоксан, тетрагидрофуран (в дальнейшем обозначаемый как THF) и т.д.), галогенированные углеводороды (дихлорметан, хлорформ, 1,2-дихлорэтан и т.д.), углеводороды (н-гексан, циклогексан, бензол, толуол и т.д.), смешанный растворитель из любого из них и подобные. Температура реакции составляет от -20 до 200 °С, предпочтительно от 0 до 80 °С, и время реакции составляет от около 0,5 до 96 ч, предпочтительно от 0,5 до 24 ч. Путем введения в реакцию соединения I в форме соли с 2,5 экв. гидробромида может быть получен, без выделения и очистки, конечный продукт этой реакции с от 1 до 20 моль, предпочтительно от около 2,5 до 5 моль бромисто-водородной кислоты на 1 моль соединения, представленного формулой (II), или его соли, в воде, спирте (метанол, этанол, ИПС и т.д.), простом эфире (THF, диоксан и т.д.), галогенированном углеводороде (дихлорметан, дихлорэтан, хлорформ и т.д.), этилацетате, ацетонитриле и подобных, или смешанном растворителе из любого из них. Температура реакции составляет от -20 до 200 °С, предпочтительно от 0 до 100 °С, и время реакции составляет от около 0,5 до 48 ч, предпочтительно от 0,5 до 24 ч. После реакции осадок собирали фильтрованием, что дает соль, представленную формулой (IV). В настоящем изобретении гидрохлорид, гидробромид, нитрат, мезилат, малеат, тозилат, безилат, нафталин-1-сульфонат, нафталин-2-сульфонат, галлат, (+)-камфорсульфонат, (-)-камфорсульфонат, фумарат, сульфат, сукцинат, L-тартрат, этандисульфонат, цитрат или фосфат соединения I (в дальнейшем также обозначаемый как "соль по настоящему изобретению") являются оптически чистыми и, например, оптическая чистота (2S,4S)-энантиомера не менее чем 90% энантиомерного избытка (в дальнейшем обозначается как е.е.), предпочтительно не менее чем 95% е.е, более предпочтительно не менее чем 99% е.е. Форма соли по настоящему изобретению не является специально ограниченной, и соль может представлять собой масло, аморфную форму или кристалл. Предпочтительной формой соли является кристалл. Как соль в форме кристалла могут быть упомянуты 2,0 экв. гидрохлорид, 2,5 экв. гидрохлорид, 2 экв. гидробромид, 2,5 экв. гидробромид, 2 экв. мезилат, 3 экв. мезилат, 2 экв. тозилат, 2 экв. безилат, 2 экв. нафталин-1-сульфонат, 2 экв. нафталин-2-сульфонат, 2 экв. (+)-камфорсульфонат, 2 экв. малеат, 2 экв. фумарат, 2 экв. L-тартрат и подобные. Эти соли также могут быть охарактеризованы пиками на рентгенодифракционном спектре. В настоящем изобретении полиморфный кристалл 2,0 экв. гидробромида обозначали как форму А, форму В или форму С. Кроме того, полиморфный кристалл 2 экв. тозилата обозначали как форму А, форму В или форму С, полиморфный кристалл 2 экв. безилата обозначали как форму А или форму В, полиморфный кристалл 2 экв. малеата обозначали как форму А или форму В, и полиморфный кристалл 2 экв. фумарата обозначали как форму А или форму В. Сольват соли по настоящему изобретению может присутствовать в виде полу-, моно-, ди-, три-, тетра-, пента-, гексасольватов и подобных. Растворитель, примененный для кристаллизации, такой как спирт (метанол, этанол, ИПС и т.д.), альдегид, кетон (ацетон и т.д.) или сложный эфир (этилацетат и т.д.) и т.д, и вода, содержащиеся в этих растворителях, могут быть включены в кристаллическую решетку. В общем, невозможно предсказать, включается ли растворитель в сольват или нет в течение кристаллизации и после стадии получения. Это зависит от комбинации соединения, условий получения и различных взаимодействий с выбранным растворителем, особенно водой. Кроме того, стабильность кристаллической и аморфной формы соли определенного соединения или его сольвата могут быть подтверждены только реальным измерением величин. Соль по настоящему изобретению может представлять собой сольват с растворителем (вода, органический растворитель и т.д.) или несольватированную форму. Другими словами, соль по настоящему изобретению может представлять собой гидрат или негидратированную форму. Когда она представляет собой гидрат, количество гидратирующей воды может варьироваться в зависимости от различных условий. Она предпочтительно является не более чем 2,0 экв. гидратом, более предпочтительно 1,0-2,0 экв. гидратом. Соль по настоящему изобретению может содержать растворитель, безопасный для млекопитающих (фармацевтически, фармакологически или физиологически приемлемая соль и т.д.), или сольват с растворителем. "Растворитель" является выбранным из тех, что попадают под допустимую дневную экспозицию ("PDE") выше 10 мг/день в "ICH-рекомендации по остаточным растворителям Q3C" и/или тех, что попадают под класс 3 в "ICH-рекомендации по остаточным растворителям Q3C". В качестве специфических могут быть упомянуты этанол, 1-пропанол, ИПС, 1-бутанол, 2-бутанол, 1-пентанол, уксусная кислота, метилацетат, этилацетат, пропилацетат, изопропилацетат, н-бутилацетат, изобутилацетат, муравьиная кислота, этилформиат, ацетон, метилэтилкетон, метилизобутилкетон, гептан, пентан, диэтиловый эфир, трет-бутилметиловый эфир, THF, анизол, кумен, диметилсульфоксид и подобные. Из этих растворителей этанол является предпочтительным. Содержание "растворителя" составляет не более чем 50000 м.д., предпочтительно не более чем 5000 м.д. Соль по настоящему изобретению может быть получена согласно способу, известному per se. Например, соль по настоящему изобретению может быть получена путем реагирования соединения I с органической кислотой или неорганической кислотой, выбранной из соляной кислоты, бромисто-водородной кислоты, азотной кислоты, метилсульфоновой кислоты, малеиновой кислоты, толуолсульфоновой кислоты, безиловой кислоты, нафталин-1-сульфоновой кислоты, нафталин-2-сульфоновой кислоты, галловой кислоты, (+)-камфорсульфоновой кислоты, (-)-камфорсульфоновой кислоты, фумаровой кислоты, серной кислоты, янтарной кислоты, L-винной кислоты, этандисульфоновой кислоты, лимонной кислоты и фосфорной кислоты. Эта реакция обычно проводится в инертном растворителе или без растворителя. В качестве "инертного растворителя" могут быть упомянуты вода, спирты (метанол, этанол, 1-пропанол, ИПС, бутанол и т.д.), кетоны (ацетон, метилэтилкетон и т.д.), нитрилы (ацетонитрил, пропионитрил и т.д.), амиды (формамид, N,N-диметилформамид, N,N-диметилацетамид, 1,3-диметил-2-имидазолидинон и т.д.), простые эфиры (диэтиловый эфир, диизопропиловый эфир, трет-бутилметиловый эфир, 1,4-диоксан, THF и т.д.), сложные эфиры (этилформиат, этилацетат, пропилацетат и т.д.), галогенированные углеводороды (дихлорметан, хлорформ, 1,2-дихлорэтан и т.д.), углеводороды (н-гексан, циклогексан, бензол, толуол и т.д.), сульфоксиды (диметилсульфоксид и т.д.), полярные растворители (сульфолан, гексаметилфосфотриамид и т.д.), растворитель, смешанный из любых из них, и подобные. Из этих растворителей вода, смешанные растворители воды и спирта (смешанный растворитель воды и метанола, смешанный растворитель воды и этанола, смешанный растворитель воды и 1-пропанола, смешанный растворитель воды и ИПС и т.д.) являются предпочтительными. "Инертный растворитель" обычно применяется в количестве от 1 до 100% мас./об., предпочтительно от 2 до 50% мас./об. относительно соединения I. Температура реакции обычно составляет от -20 °С до температуры кипения растворителя, предпочтительно от 0 °С до температуры кипения растворителя. Время реакции обычно составляет от около 1 мин до 24 ч, предпочтительно от около 10 мин до 6 ч, более предпочтительно от около 30 мин до 3 ч. Полученная таким образом соль может быть выделена и очищена из реакционной смеси согласно способам разделения, известным per se (концентрирование, концентрирование под пониженным давлением, экстракция растворителем, кристаллизация, перекристаллизация, фазовый переход, хроматография и т.д.). Соль соединения I может быть получена в форме кристалла кристаллизацией полученной таким образом соли. В качестве способа кристаллизации могут быть упомянуты способы, известные per se, и могут быть упомянуты кристаллизация из раствора, кристаллизация из пара, кристаллизация из расплавленной формы (см. A.S. Myerson Ed., Handbook of Industrial Crystallization Second Edition, Butterworth-Heinemann 2002). Как способ "кристаллизации из раствора" могут быть упомянуты способ концентрирования, способ отжигания, реакционные способы (диффузионный способ или электролитический способ), способ гидротермального роста, флюсовый способ и подобные. В качестве применяемого растворителя, может быть упомянут растворитель, подобный вышеуказанному "инертному растворителю". В качестве способа "кристаллизация из пара" могут быть упомянуты газификационные способы (способ запаянной трубки или способ газового потока), способ газофазной реакции, способ химической транспортировки и подобные. В качестве способа для "кристаллизации из расплавленной формы" могут быть упомянуты способы нормального замораживания (способ выращивания кристаллов методом вытягивания, способ температурного градиента или способ Бриджмена (Bridgman)), способы зонной плавки (способ зонного выравнивания или способ плавающей зоны), специальные способы роста (VLS-способ или способ эпитаксиального выращивания из жидкой фазы) и подобные. Для кристаллизации соли соединения I применялись выпадение кристалла в осадок путем охлаждения раствора, содержащего соль соединения I, растворенную в нем нагреванием до обычно от 40 °С до температуры кипения применяемого растворителя, или выпадение кристалла в осадок путем добавления плохого растворителя к раствору, содержащему соль соединения I, растворенную в нем (особенно концентрированный раствор), и подобные. Как способ анализа полученного кристалла обычно применяется способ рентгеноструктурного анализа. Измеренные результаты рентгеноструктурного анализа представлены на оси Y, показывающей интенсивность дифракции, и оси абсцисс, показывающей углы дифракции (2 θ), где величины 2 θ имеют разброс в пределах определенного диапазона, даже когда измеряется та же кристаллическая форма. Являясь специфическим, разброс в ±0,2 ° представляет собой обычный диапазон. Несколько большая ошибка может быть получена в зависимости от условий измерения, и подобного. Для сравнения кристаллических форм, основанных на величинах 2 θ, специалисты в данной области сравнивают кристаллические формы с учетом разброса. Кроме того, соль и ее сольват по настоящему изобретению могут обнаруживать некоторый разброс в угле дифракции в зависимости от содержания воды, что также включается в объем настоящего изобретения. Соль и ее сольват по настоящему изобретению (в дальнейшем для простоты обозначаемые как соль по настоящему изобретению) имеют превосходную стабильность и, следовательно, допускают долгосрочную консервацию при комнатной температуре. Кроме того, так как они не требуют усложненного оперирования на стадии получения и консервации, и проведение получения является легким, они могут быть применены как большие объемы фармацевтических продуктов. С точки зрения высокой растворимости соли по настоящему изобретению в воде, форма дозировки, имеющая более высокую степень свободы, может разрабатываться как рецептура для инъекции. Когда соль по настоящему изобретению применяется как фармацевтический агент, соль по настоящему изобретению смешивают с фармацевтически приемлемым носителем (наполнитель, связущее, дезинтегрирующий агент, модификатор, отдушка, эмульгатор, разжижитель, ускорители растворения и т.д.), что дает фармацевтическую композицию или рецептуру (таблетка, драже, капсула, гранула, порошок, сироп, эмульсия, эликсир, суспензия, раствор, инъекция, капельная инфузия, суппозиторий и т.д.), которые могут быть назначены перорально или парентерально. Фармацевтическая композиция может быть переработана в рецептуру по традиционному способу. В настоящем описании под парентеральным подразумеваются подкожная инъекция, внутривенная инъекция, внутримышечная инъекция, интраперитонеальная инъекция, инфузия и подобные. Рецептура для инъекции может быть получена способом, известным в технике. Суппозитории для ректального назначения могут быть приготовлены смешиванием лекарственного средства с приемлемым наполнителем и подобным. В качестве формы дозировки твердого вещества для перорального введения могут быть упомянуты те, что упомянуты выше, такие как порошок, гранула, таблетка, драже, капсула и подобные. В качестве жидкости для перорального введения могут быть упомянуты фармацевтически приемлемая эмульсия, сироп, эликсир, суспензия, раствор и подобные. Дозу соли по настоящему изобретению определяли с учетом возраста, массы тела, общего состояния здоровья, пола, диеты, времени введения, способа введения, величины клиренса, комбинации лекарственных средств и тяжести состояния, из-за которого пациенты получают лечение, а затем и других факторов. Соль по настоящему изобретению обнаруживает меньшую токсичность и может быть применена безопасно. В то время как дневная доза варьирует в зависимости от состояния и массы тела пациентов, вида соли, пути назначения и подобного, она составляет, например, от 0,01 до 100 мг/кг массы тела/день, предпочтительно от 0,05 до 50 мг/кг массы тела/день, для парентерального введения подкожным, внутривенным, внутримышечным или ректальным путем, и от 0,01 до 100 мг/кг массы тела/день, предпочтительно от 0,05 до 50 мг/кг массы тела/день для перорального введения, которое предпочтительно проводить один раз или за несколько порций в день. Настоящее изобретение объясняется в деталях ниже путем отсылки к ссылочному примеру и примерам, которые не должны рассматриваться как ограничивающие настоящее изобретение. Пока не указано иное, безводный сульфат натрия или безводный сульфат магния применяли для высушивания органического раствора для экстракции. Колоночную хроматографию проводили с применением силикагеля, производимого FUJI SILYSIA CHEMICAL LTD. Для термического анализа (DSC) указаны температура (начальная величина) в точке пересечения расширения линейной части до плавления с росширением линейной части в процессе плавления на термической кривой и температура (величина, соответствующая верху пика) при точке перегиба вблизи точки плавления на термической кривой. Порошковая рентгенограмма дифракционных полос (XRD) показывает характеристические пики при углах 2 θ ( ±0,2 °). 1 Н-ЯМР измеряли на ядерно-магниторезонансном спектрометре на 300 МГц. Химические сдвиги 1 Н-ЯМР выражены как относительные δ величины в частях на миллион (м.д.) с использованием тетраметилсилана (TMS) в качестве внутреннего стандарта. Спин-спиновые константы показывают наблюдаемую мультиплетность в герцах (Гц), используя обозначения s (синглет), d (дублет), t (триплет), m (мультиплет) и подобные. Интенсивность оптической плотности в инфракрасной (IR) спектроскопии выражены с применением обозначений с. (сильная), ср. (средняя) и сл. (слабая). В то время как обозначенные в заголовке примера соединения в нижеследующих ссылочном примере и примерах представлены в виде несольватированных форм, они могут принимать форму сольватов (особенно, гидратов) в зависимости от условий во время получения и подобного. Ссылочный пример 1. 3-{(2S,4S)-4-[4-(3-Метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидин. 3-{(2S,4S)-1-(1,1-Диметилэтилоксикарбонил)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидин (25,45 г, синтезирован согласно соединению, описанному в примере 222 из WO 02/14271) растворяли в дихлорметане (200 мл). При комнатной температуре добавляли трифторуксусную кислоту (50 мл) и смесь перемешивали в течение 19 ч. Реакционную смесь концентрировали под пониженным давлением и к остатку добавляли насыщенный водный раствор бикарбоната натрия. Смесь экстрагировали хлороформом. Экстракт промывали насыщенным солевым раствором и высушивали. Растворитель упаривали под пониженным давлением. Остаток очищали методом колоночной хроматографии на силикагеле с получением указанного в заголовке соединения в виде твердого вещества (19,28 г, выход 93%). Пример 1. 2,5 экв. Гидрохлорид 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина. (1) Соединение (2,50 г), полученное в ссылочном примере 1, растворяли в THF (100 мл). При комнатной температуре добавляли этилацетатный раствор (3,0 мл, 4 моль/л) соляной кислоты и смесь перемешивали в течение 1 ч. Осадок собирали фильтрованием и высушивали под пониженным давлением при 50 °С с получением твердого вещества (2,69 г). (2) Продукт (300 мг), упомянутый выше, растворяли в смешанном растворителе воды (150 мкл) и этанола (1,0 мл) нагреванием и раствор перемешивали в течение 1 ч с охлаждением льдом. Осадок собирали фильтрованием и высушивали под пониженным давлением при 50 °С с получением указанного в заголовке соединения в виде кристаллов (144 мг, выход 48%). XRD: 5,2 °, 14,3 °, 16,2 °, 21,8 °, 25,2 °. Аналитически вычислено для C 22 H 30 N 6 OS 2,3HCl ∙2H 2 O: С, 48,35; Н, 6,69; N, 15,38; найдено: С, 48,02; Н, 6,60; N, 15,20. Пример 2. 2,0 экв. Гидрохлорид 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина. Соединение, обозначенное в заголовке примера (60 мг), полученное в примере 1, суспендировали в этилацетате (3,0 мл), суспензию нагревали с обратным холодильником в течение 13 ч и позволяли охладиться до комнатной температуры. Осадок собирали фильтрованием и высушивали в теплом воздухе при 40 °С с получением указанного в заголовке соединения в виде кристаллов (50 мг, выход 85%). XRD: 5,0 °, 14,8 °, 21,0 °, 21,5 °, 25,2 °. Аналитически вычислено для C 22 H 30 N 6 OS ∙2,0HCl ∙Н 2 О: С, 51,05; Н, 6,62; N, 16,24; найдено: С, 50,89; Н, 6,58; N, 16,12. Пример 3. 2,5 экв. Гидробромид 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина. Соединение (5,09 г), полученное в ссылочном примере 1, растворяли в этаноле (50,9 мл). При температуре кипения с обратным холодильником добавляли 48% бромисто-водородную кислоту (5,03 г) и смесь охлаждали до комнатной температуры в течение около 1 ч при перемешивании, далее перемешивали при комнатной температуре в течение 1 ч. Осадок собирали фильтрованием, промывали этанолом (5 мл) и высушивали в теплом воздухе при 45 °С с получением указанного в заголовке соединения в виде кристаллов (6,76 г). Точка плавления: 202,0 °С (разложение). ИК-спектр (KBr): 3600-3300 (с.), 3116-2850 (с.), 2800-2400 (с.), 1647 (с.), 1592 (ср.), 1572 (ср.), 1496 (ср.), 1450 (ср.), 1385 (ср.), 1361 (сл.), 768 (ср.), 692 (сл.). Пример 4. 2,5 экв. Гидробромид 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина (альтернативный способ синтеза соединения, обозначенного в заголовке примера 3). (1) К суспензии триацетоксиборгидрида натрия (13,68 кг) в толуоле (300 л) добавляли ацетат 1-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазина (15,00 кг) и 3-[(2S)-1-(1,1-диметилэтилоксикарбонил)-4-оксопирролидин-2-илкарбонил]тиазолидин (14,90 кг) и смесь перемешивали при комнатной температуре в течение 2,5 ч. К реакционной смеси добавляли по каплям воду (90 л) и смесь перемешивали в течение 0,5 ч. Толуольный слой отделяли, промывали последовательно 5% водным раствором бикарбоната натрия (90 л) и водой (90 л) и концентрировали досуха под пониженным давлением. К остатку добавляли ИПС (224 л) и добавляли по каплям 48% бромисто-водородную кислоту (25,08 кг) при около 80 °С, смесь кипятили с обратным холодильником в течение 2,5 ч. Реакционной смеси позволили охладиться и перемешивали при около 60 °С в течение 1,5 ч, при около 40 °С в течение 2 ч и затем при комнатной температуре в течение 2 ч. Осадок собирали фильтрованием, промывали ИПС (30 л) и высушивали в теплом воздухе с получением указанного в заголовке соединения в виде твердого вещества (29,76 кг, выход 91%). (2) К твердому веществу (28,00 кг), полученному в (1), добавляли этанол (168 л), твердое вещество растворяли нагреванием. Раствор фильтровали горячим. Реакционную емкость промывали этанолом (28 л), фильтрат и смывы объединяли. При 67 °С добавляли воду (3 л), смеси позволили охладиться и перемешивали при 49 °С в течение 1 ч и затем при 20-15 °С в течение 1 ч. Осадок собирали фильтрованием, промывали этанолом (28 л) и высушивали в теплом воздухе с получением указанного в заголовке соединения в виде кристаллов (25,84 кг, выход 92%). XRD: 5,4 °, 13,4 °, 14,4 °, 22,6 °, 26,5 °. Пример 5. 2,0 экв. Гидробромид 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина. (1) Соединение, обозначенное в заголовке примера (130 г), полученное в примере 4, добавляли к воде (260 мл) при комнатной температуре и соединение растворяли перемешиванием. Осадок собирали фильтрованием и высушивали с получением 3,5 экв. гидрата кристаллической формы А (53,58 г) соединения, обозначенного в заголовке примера. XRD: 5,7 °, 7,7 °, 11,3 °, 16,2 °, 17,0 °. (2) Гидрат кристаллической формы А (8,5 г), полученный в (1), добавляли к этанолу (100 мл, содержащему 2% воды) при 28-30 °С и гидрат растворяли перемешиванием. Осадок собирали фильтрованием и высушивали с получением гидрата кристаллической формы В (4,56 г) соединения, указанного в заголовке примера. XRD: 5,2 °, 10,4 °, 19,1 °, 19,8 °, 20,7 °. (3) Гидрат кристаллической формы А (8,5 г), полученный в (1), добавляли к этанолу (100 мл, содержащему 2% воды) при 15-18 °С и гидрат растворяли перемешиванием. Осадок собирали фильтрованием и высушивали с получением гидрата кристаллической формы С (6,21 г) указанного в заголовке соединения. XRD: 5,5 °, 13,4 °, 14,3 °, 21,4 °, 26,7 °. Пример 7. 3 экв. Мезилат 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина. (1) Соединение (2,64 г), полученное в ссылочном примере 1, растворяли в THF (25 мл). При комнатной температуре добавляли метансульфоновую кислоту (1,32 мл) и смесь перемешивали в течение 1,5 ч. Осадок собирали фильтрованием и высушивали под пониженным давлением с получением кристаллов (3,52 г, выход 80%). (2) Вышеуказанные кристаллы (1,76 г) растворяли в этаноле (10 мл) нагреванием и раствор перемешивали при комнатной температуре в течение 17 ч. Осадок собирали фильтрованием и высушивали под пониженным давлением с получением указанного в заголовке соединения в виде кристаллов (1,26 г, выход 72%). DSC: 193-197 °С. Аналитически вычислено для C 22 H 30 N 6 OS ∙3CH 4 O 3 S ∙Н 2 О: С, 40,97%; Н, 6,05%; N, 11,47%; найдено: С, 41,05%; Н, 5,72%; N, 11,48%. Пример 8. 2 экв. Мезилат 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирро-лидин-2-илкарбонил}тиазолидина. (1) Соединение (7,96 г), полученное в ссылочном примере 1, растворяли в ИПС (60 мл). При комнатной температуре добавляли раствор метансульфоновой кислоты (3,59 г) в ИПС (20 мл) и смесь перемешивали в течение 2 ч. Осадок собирали фильтрованием и высушивали под пониженным давлением с получением твердого вещества (9,03 г, выход 78%). (2) Вышеуказанное твердое вещество (1000 мг) суспендировали в ацетонитриле (20 мл) и суспензию нагревали с обратным холодильником в течение 30 мин, затем позволили охладиться до комнатной температуры. Осадок собирали фильтрованием с получением твердого вещества (847 мг). Твердое вещество (813 мг) суспендировали в ацетонитриле (16 мл), суспензию нагревали с обратным холодильником в течение 1 ч и позволили охладиться до комнатной температуры. Осадок собирали фильтрованием и высушивали под пониженным давлением с получением указанного в заголовке соединения в виде кристаллов (690 мг, выход 72%). DSC: 213-216 °С. Аналитически вычислено для C 22 H 30 N 6 OS ∙2CH 4 O 3 S ∙0,5Н 2 О: С, 45,92; Н, 6,26; N, 13,39; найдено: С, 45,96; Н, 6,17; N, 13,37. Пример 9. 2 экв. Тозилат 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидин. (1) Соединение (5,28 г), полученное в ссылочном примере 1, растворяли в ИПС (30 мл). При комнатной температуре добавляли моногидрат толуолсульфоновой кислоты (4,94 г) и смесь перемешивали в течение 1,5 ч. Осадок собирали фильтрованием и высушивали под пониженным давлением с получением кристаллической формы А указанного в заголовке соединения (7,84 г, выход 82%). XRD: 5,3 °, 6,0 °, 14,8 °, 16,4 °, 20,8 °. Аналитически вычислено для C 22 H 30 N 6 OS ∙2C 7 H 8 O 3 S ∙0,25H 2 O: С, 55,76%; Н, 6,04%; N, 10,84%; найдено: С, 55,71%; Н, 6,06%; N, 10,80%. (2) Упомянутый выше продукт (1,5 г) растворяли в воде (20 мл) нагреванием и раствор перемешивали при комнатной температуре в течение 1 ч. Осадок собирали фильтрованием и высушивали под пониженным давлением с получением кристаллической формы В указанного в заголовке соединения (1,2 г, выход 80%). XRD: 5,7 °, 11,4 °, 14,0 °, 18,2 °, 19,7 °. Аналитически вычислено для C 22 H 30 N 6 OS ∙2C 7 H 8 O 3 S ∙0,5Н 2 О: С, 55,43%; Н, 6,07%; N, 10,77%; найдено: С, 55,14%; Н, 6,09%; N, 10,73%. (3) Конечный продукт (1,4 г), полученный в (1), суспендировали в ИПС (100 мл), суспензию нагревали с обратным холодильником в течение 1 ч и позволили охладиться до комнатной температуры. Осадок собирали фильтрованием и высушивали под пониженным давлением с получением кристаллической формы С указанного в заголовке соединения (1,1 г, выход 81%). DSC: 227-230 °С. XRD: 4,7 °, 5,7 °, 11,3 °, 19,8 °, 21,4 °. Аналитически вычислено для C 22 H 30 N 6 OS ∙2C 7 H 8 O 3 S: С, 56,08%; Н, 6,01%; N, 10,90%; найдено: С, 55,83%; Н, 6,11%; N, 10,87%. Пример 10. 2 экв. Безилат 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина. (1) Соединение (4,36 г), полученное в ссылочном примере 1, растворяли в ИПС (70 мл). При комнатной температуре добавляли безиловую кислоту (3,78 г) и смесь перемешивали в течение 1 ч. Осадок собирали фильтрованием и высушивали под пониженным давлением с получением кристаллической формы А указанного в заголовке соединения (6,05 г, выход 80%). XRD: 5,7 °, 8,9 °, 19,4 °, 20,2 °, 21,6 °. 1 Н-ЯМР (ДМСО-d 6 ): δ 1,82-2,10 (1Н, м), 2,17 (3Н, с), 2,60-4,20 (16Н, м), 4,11-4,72 (3Н, м), 5,91 (1Н, с), 7,31-7,35 (7Н, м), 7,45-7,50 (2Н, м), 7,59-7,62 (4Н, м), 7,75 (2Н, д, J=7,8 Гц). (2) Упомянутый выше продукт (1,81 г) растворяли в этаноле (25 мл) нагреванием и раствор перемешивали при комнатной температуре в течение 1 ч. Осадок собирали фильтрованием и высушивали под пониженным давлением с получением кристаллической формы В указанного в заголовке соединения (1,25 г, выход 69%). XRD: 5,6 °, 6,7 °, 19,3 °, 22,9 °, 23,2 °. Аналитически вычислено для C 22 H 30 N 6 OS ∙2C 6 H 6 O 3 S: С, 54,97%; Н, 5,70%; N, 11,31%; найдено: С, 54,67%; Н, 5,61%; N, 11,25%. Пример 11. 2 экв. Нафталин-1-сульфонат 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина. (1) Соединение (4,01 г), полученное в ссылочном примере 1, растворяли в THF (80 мл). Раствор нафталин-1-сульфоновой кислоты (4,11 г) в THF (40 мл) добавляли при комнатной температуре и смесь перемешивали в течение 3 ч. Осадок собирали фильтрованием и высушивали под пониженным давлением с получением твердого вещества (5,96 г, выход 75%). (2) Вышеуказанное твердое вещество (500 мг) растворяли в этаноле (25 мл) нагреванием и раствор кипятили с обратным холодильником в течение 30 мин и позволили охладиться до комнатной температуры. Осадок собирали фильтрованием и высушивали под пониженным давлением с получением указанного в заголовке соединения в виде кристаллов (445 мг, выход 89%). DSC: 184-189 °С. Аналитически вычислено для C 22 H 30 N 6 OS ∙2C 10 H 8 O 3 S ∙0,25Н 2 О: С, 59,52; Н, 5,52; N, 9,92; найдено: С, 59,32; Н, 5,46; N, 9,88. Пример 12. 2 экв. Нафталин-2-сульфонат 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина. (1) Соединение (2,57 г), полученное в ссылочном примере 1, растворяли в этилацетате (50 мл). Раствор моногидрата нафталин-2-сульфоновой кислоты (2,86 г) в этилацетате (25 мл) добавляли при комнатной температуре и смесь перемешивали в течение 12 ч. Осадок собирали фильтрованием и высушивали под пониженным давлением с получением твердого вещества (4,60 г, выход 91%). (2) Вышеуказанное твердое вещество (500 мг) растворяли в этаноле (25 мл) нагреванием и раствор перемешивали при комнатной температуре. Осадок собирали фильтрованием и высушивали под пониженным давлением с получением указанного в заголовке соединения в виде кристаллов (372 мг, выход 74%). DSC: 205-211 °С. Аналитически вычислено для C 22 H 30 N 6 OS ∙2C 10 H 8 O 3 S ∙0,75Н 2 О: С, 58,89; Н, 5,59; N, 9,81; найдено: С, 58,96; Н, 5,49; N, 9,76. Пример 14. 2 экв. (+)-Камфорсульфонат 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина. (1) Соединение (3,00 г), полученное в ссылочном примере 1, растворяли в смешанном растворителе из THF (72,5 мл) и трет-бутилметилового эфира (52,5 мл). При комнатной температуре добавляли (+)-камфорсульфоновую кислоту (3,25 г) и смесь перемешивали в течение 5 ч. Осадок собирали фильтрованием и высушивали под пониженным давлением с получением твердого вещества (5,65 г, выход 90%). (2) Вышеуказанное твердое вещество (650 мг) растворяли в смешанном растворителе из этанола (7,0 мл) и диэтилового эфира (15,0 мл) нагреванием и раствор перемешивали в течение ночи при комнатной температуре. Осадок собирали фильтрованием и высушивали в теплом воздухе с получением указанного в заголовке этанолсодержащего соединения в виде кристаллов (380 мг, выход 58%). TG/DTA: 142-156 °C, 200-205 °C. Аналитически вычислено для C 22 H 30 N 6 OS ∙2C 10 H 16 O 4 S ∙0,22С 2 Н 6 О ∙2,5H 2 O: С, 53,66; Н, 7,29; N, 8,86; найдено: С, 53,82; Н, 7,27; N, 8,88. Пример 15. 2 экв. (-)-Камфорсульфонат 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина. Соединение (3,00 г), полученное в ссылочном примере 1, растворяли в смешанном растворителе (70 мл, THF/трет-бутилметиловый эфир = 1:3). При комнатной температуре добавляли раствор (-)-камфорсульфоновой кислоты (3,25 г) в смешанном растворителе (THF/трет-бутилметиловый эфир = 1:3) и смесь перемешивали в течение 1 ч. Осадок собирали фильтрованием и высушивали под пониженным давлением с получением указанного в заголовке соединения в виде твердого вещества (5,66 г, выход 91%). Пример 16. 2 экв. Малеат 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина. (1) Соединение (1,70 г), полученное в ссылочном примере 1, растворяли в этаноле (50 мл). При комнатной температуре добавляли малеиновую кислоту (0,98 г) и смесь перемешивали в течение 1 ч. Осадок собирали фильтрованием и высушивали под пониженным давлением с получением кристаллической формы А указанного в заголовке соединения (1,87 г, выход 71%). Аналитически вычислено для C 22 H 30 N 6 OS ∙2С 4 Н 4 О 4 : С, 54,70%; Н, 5,81%; N, 12,76%; найдено: С, 54,42%; Н, 5,76%; N, 12,57%. XRD: 8,6 °, 15,8 °, 17,8 °, 18,6 °, 23,4 °. (2) Вышеуказанный кристалл (3,0 г) растворяли в воде (15 мл) нагреванием и раствор перемешивали при комнатной температуре в течение 1 ч. Осадок собирали фильтрованием и высушивали под пониженным давлением с получением кристаллической формы В указанного в заголовке соединения (1,83 г, выход 61%). XRD: 5,9 °, 13,4 °, 16,3 °, 17,6 °, 23,9 °. Аналитически вычислено для C 22 H 30 N 6 OS ∙2С 4 Н 4 О 4 ∙2Н 2 О: С, 51,86%; Н, 6,09%; N, 12,10%; найдено: С, 51,80%; Н, 5,84%; N, 12,10%. Пример 17. 2 экв. Фумарат 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидин. (1) Соединение (1,50 г), полученное в ссылочном примере 1, растворяли в этаноле (20 мл). При комнатной температуре добавляли раствор фумаровой кислоты (814 мг) в этаноле (25 мл) и смесь перемешивали при комнатной температуре в течение 1 ч, затем с охлаждением льдом в течение 1 ч. 1/3 Объема реакционного растворителя упаривали. Осадок собирали фильтрованием и высушивали под пониженным давлением с получением твердого вещества (1,77 г, выход 77%). (2) Твердое вещество (200 мг), полученное в (1), суспендировали в ацетонитриле (5 мл), суспензию нагревали до кипения с обратным холодильником в течение 4 ч и позволили охладиться до комнатной температуры. Осадок собирали фильтрованием с получением указанного в заголовке соединения в виде кристаллов (141 мг, выход 71%). Поскольку кристалл обнаруживал два эндотермических пика на DSC, кристалл был отнесен как являющийся смесью двух кристаллических форм (форма А, форма В), или как претерпевающий превращение из формы А в форму В действием тепла. DSC: 128-(135 или 142) °С. XRD: 3,1 °, 15,2 °, 17,4 °, 23,4 °, 25,5 °. Аналитически вычислено для C 22 H 30 N 6 OS ∙2С 4 Н 4 О 4 : С, 54,70; Н, 5,81; N, 12,76; найдено: С, 54,40; Н, 5,88; N, 12,63. 1 H-ЯМР (ДМСО-d 6 ): δ 1,50-1,78 (1Н, м), 2,14 (3Н, м), 2,37-3,90 (16Н, м), 4,10-4,72 (3Н, м), 5,79 (1Н, с), 6,57 (4Н, с), 7,27 (1Н, т, J=7,2 Гц), 7,46 (2Н, т, J=8,1 Гц), 7,74 (2Н, д, J=7,7 Гц). (3) Твердое вещество (200 мг), полученное в (1), растворяли в воде (2 мл) и раствор перемешивали при комнатной температуре. Осадок собирали фильтрованием с получением указанного в заголовке соединения в виде кристаллов (47,5 мг, выход 24%). Кристалл имел порошковую рентгенограмму, отличную от той, что была получена для кристалла в (2). XRD: 9,4 °, 17,8 °, 19,6 °, 21,0 °, 23,5 °, 24,3 °. Пример 18. 1,6 экв. Сульфат 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина. Соединение (2,00 г), полученное в ссылочном примере 1, растворяли в THF (40 мл). Водный раствор серной кислоты (14,5 мл, 0,5 моль/л) добавляли при комнатной температуре и смесь перемешивали в течение 0,5 ч. Осадок собирали фильтрованием и высушивали под пониженным давлением с получением указанного в заголовке соединения в виде твердого вещества (2,57 г, выход 94%). Аналитически вычислено для C 22 H 30 N 6 OS ∙1,6H 2 O 4 S ∙2Н 2 О: С, 42,65; Н, 6,05; N, 13,57; найдено: С, 42,56; Н, 5,67; N, 13,44. Пример 19. 2 экв. L-тартрат 3-{(2S,4S)-4-[4-(3-метил-1-фенил-1Н-пиразол-5-ил)пиперазин-1-ил]пирролидин-2-илкарбонил}тиазолидина. (1) Соединение (1,17 г), полученное в ссылочном примере 1, растворяли в ИПС (30 мл). Добавляли L-винную кислоту (823 мг) при комнатной температуре и смесь перемешивали в течение 3 ч. Осадок собирали фильтрованием и высушивали под пониженным давлением с получением кристаллов (1,55 г, выход 78%). 2) Продукт (254 мг), упомянутый выше, суспендировали в этилацетате (10 мл), суспензию нагревали с обратным холодильником в течение 1,5 ч и позволили охладиться до комнатной температуры. Осадок собирали фильтрованием и высушивали под пониженным давлением с получением указанного в заголовке соединения в виде кристаллов (250 мг, выход 98%). 1 H-ЯМР (ДМСО-d 6 ): δ 1,50-1,69 (1Н, м), 2,14 (3Н, м), 2,40-3,90 (16Н, м), 4,08 (2Н, с), 4,30-4,70 (3Н, м), 5,79 (1Н, с), 7,27 (1Н, т, J=7,3 Гц), 7,46 (2Н, м), 7,73 (2Н, д, J=7,8 Гц). Экспериментальный пример 1. Измерение порошковой рентгенограммы. Порошковую рентгенограмму соединения, обозначенного в заголовке примеров 4 и 5, измеряли в следующих условиях измерения: аппарат: XRD-6000, производимый Shimadzu Corporation; антикатод: Cu; монохроматор: Graphite; напряжение на трубке: 40 кВ; ток на трубке: 40 мА; отклоняющая щель: 1 °; принимающая щель: 0,15 мм; щель разброса: 1 °; диапазон сканирования: 2-40 ° (2 θ); вращение образца: 60 об./мин. Результаты измерения порошковой рентгенограммы соединения, обозначенного в заголовке примера 4, показаны на фиг. 1. Результаты измерения порошковой рентгенограммы кристаллической формы А соединения, обозначенного в заголовке примера 5, показаны на фиг. 2. Результаты измерения порошковой рентгенограммы кристаллической формы В соединения, обозначенного в заголовке примера 5, показаны на фиг. 3. Результаты измерения порошковой рентгенограммы кристаллической формы С соединения, обозначенного в заголовке примера 5, показаны на фиг. 4. Экспериментальный пример 2. Измерение гигроскопичности. Гигроскопичность соединения, указанного в заголовке примера 3, измеряли в следующих условиях, с применением влагоадсорбционного измеряющего аппарата: аппарат: MB-300G, производимый VTI; температура измерения: 25 °С; диапазон измерения: 0-95% RH; Результаты измерения гигроскопичности показаны на фиг. 5. Соединение, обозначенное в заголовке примера 3, подвергали измерению адсорбции влаги с применением аппарата для измерения адсорбции влаги под пониженным давлением. В результате, соединение, как было найдено, имеет уменьшенное содержание воды, соответствующее 1,8 экв. гидрату при 50% RH, и является практически полностью сухим при 0% RH. Экспериментальный пример 3. Измерение растворимости. (1) Измерение растворимости в воде. Как способ измерения применяли визуальное наблюдение, что допускает удобное приблизительное исследование растворимости с небольшим количеством образца. Температура в течение измерения составляла 37 °С. Около 3 мг соединения, указанного в заголовке примера 3, помещали в бутылку для образца с навинчивающейся крышкой, добавляли тестовый раствор (0,15 мл) и крышку на ней плотно закручивали. Образец облучали ультразвуком в течение 1 мин, что приводило к дисперсии, которую помещали в термостатирующую водную баню трясущего типа, удерживаемую при 37 °С, и трясли в течение 1 ч. Затем растворение подтверждали визуальным наблюдением. В результате растворимость в воде соединения, указанного в заголовке примера 3, составляла не менее чем 20 мг/мл при 37 °С. (2) Измерение растворимости при рН 9-13. С применением в качестве тестового раствора смешанного раствора из 0,2 моль/л NaOH и 0,1 моль/л NaCl анализировали методом жидкостной хроматографии (HPLC) (n=3) растворимость соединения, обозначенного в заголовке примера 3, имеющего рН 9-13 при комнатной температуре. В результате растворимость составила 6,4-8,4 мг/мл. Из указанных данных был сделан вывод, что растворимость соединения, обозначенного в заголовке примера 3, составляет около 7 мг/мл. Соль соединения I или ее сольват и их новые полиморфные кристаллы имеют одно или несколько свойств, выбранных из улучшенной стабильности, улучшенной гигроскопичности (расплывание в адсорбированной влаге), быстрого выделения из растворителя и простого способа получения, которые обеспечивают использование соединения I в качестве фармацевтического продукта. Настоящая заявка основана на опубликованной в Японии заявке № 2005-041851, содержание которой включено в настоящее описание посредством ссылки. |