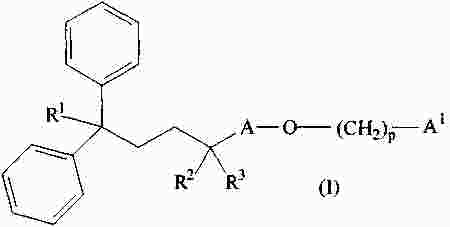
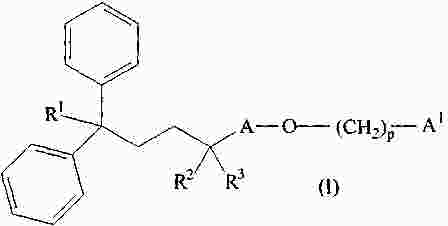
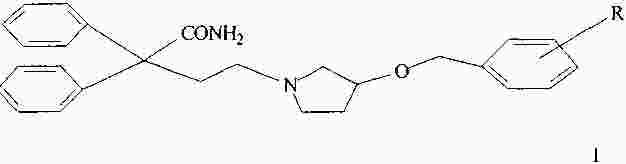
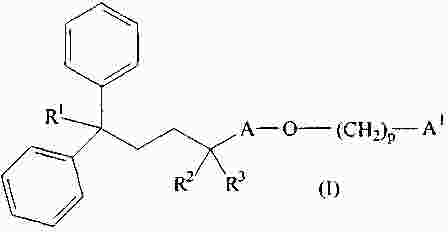
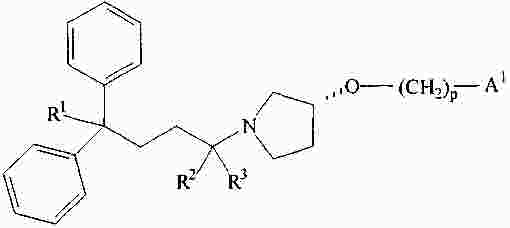
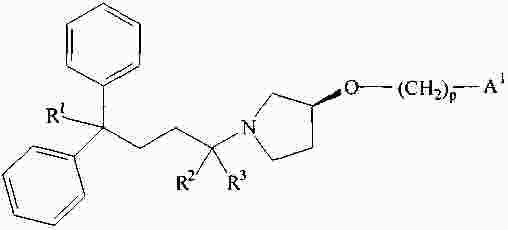
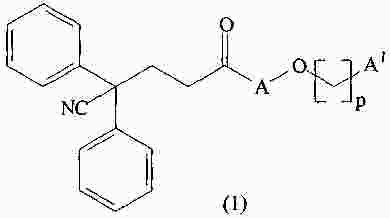
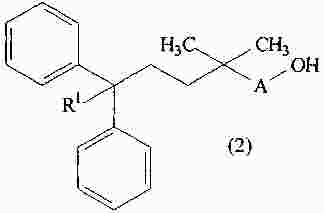
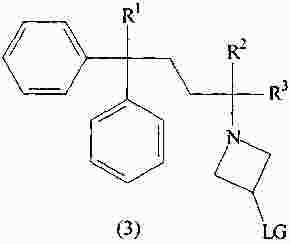
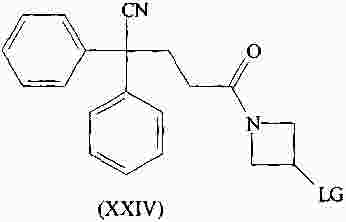
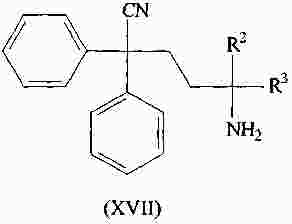
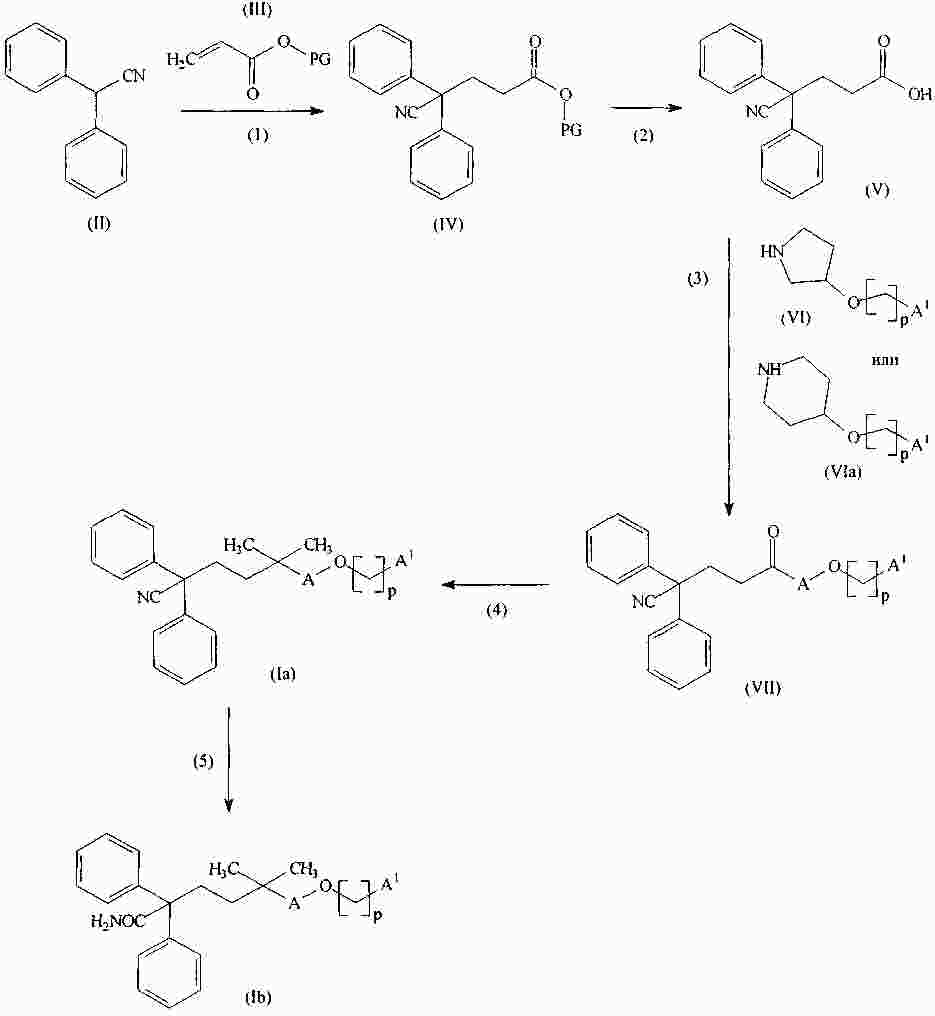
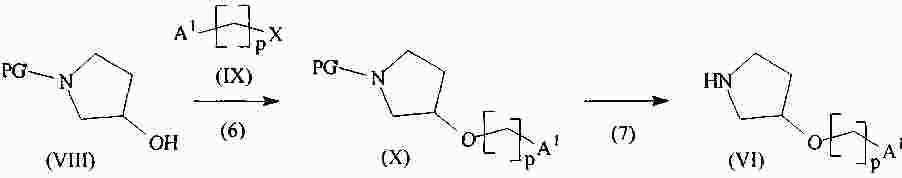
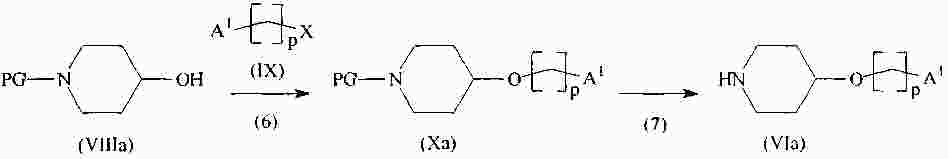
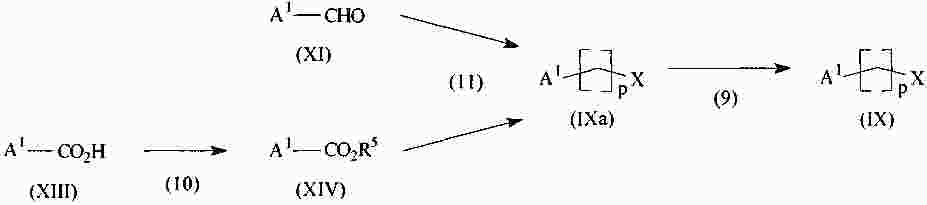
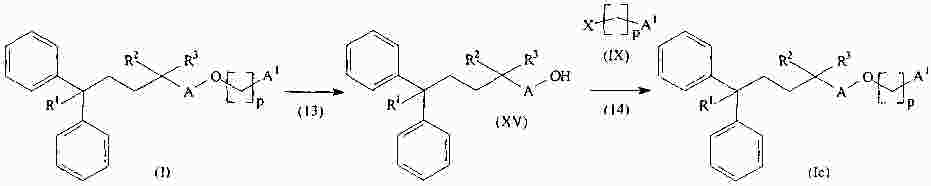
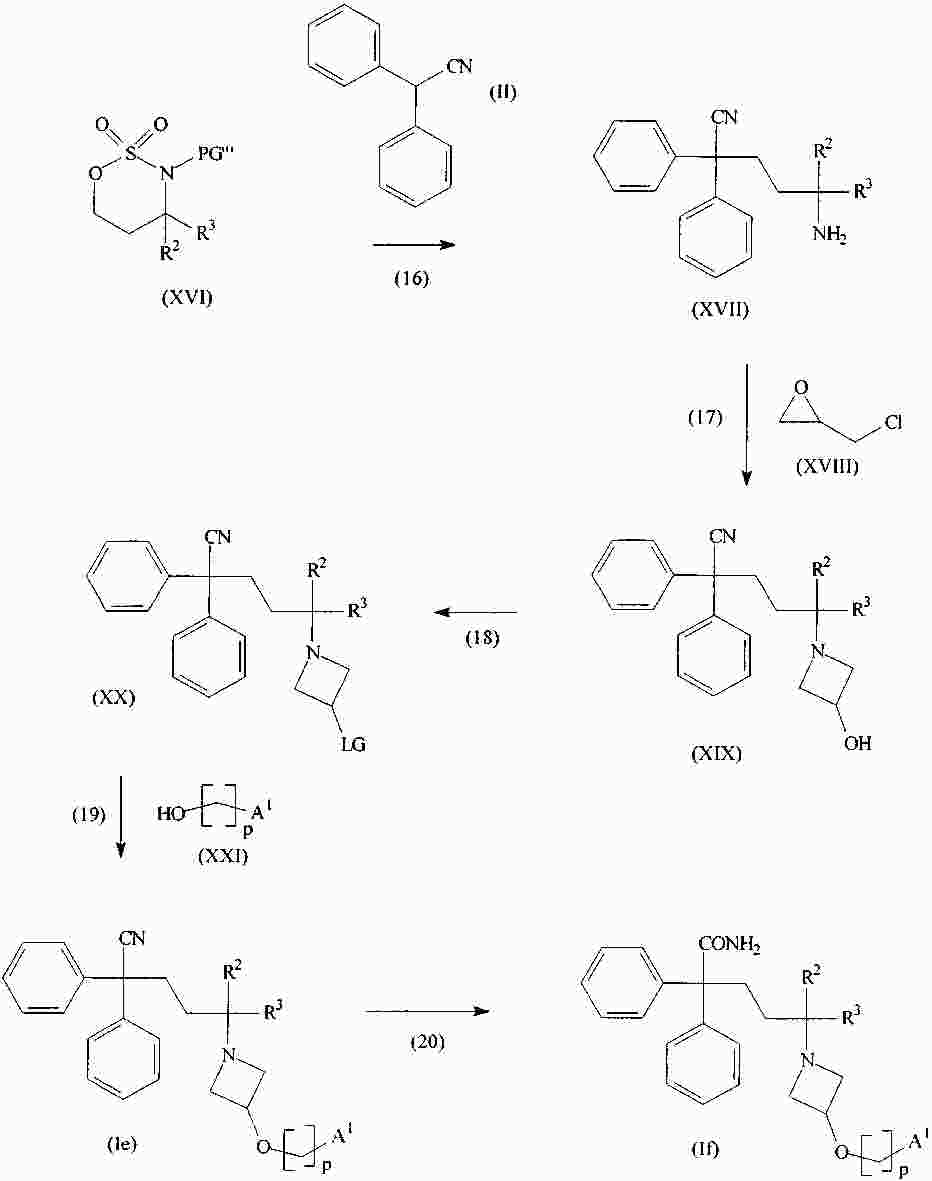
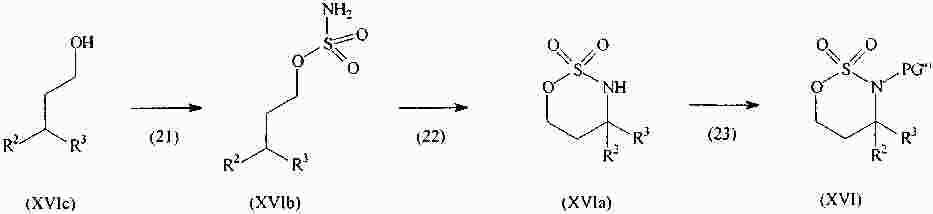
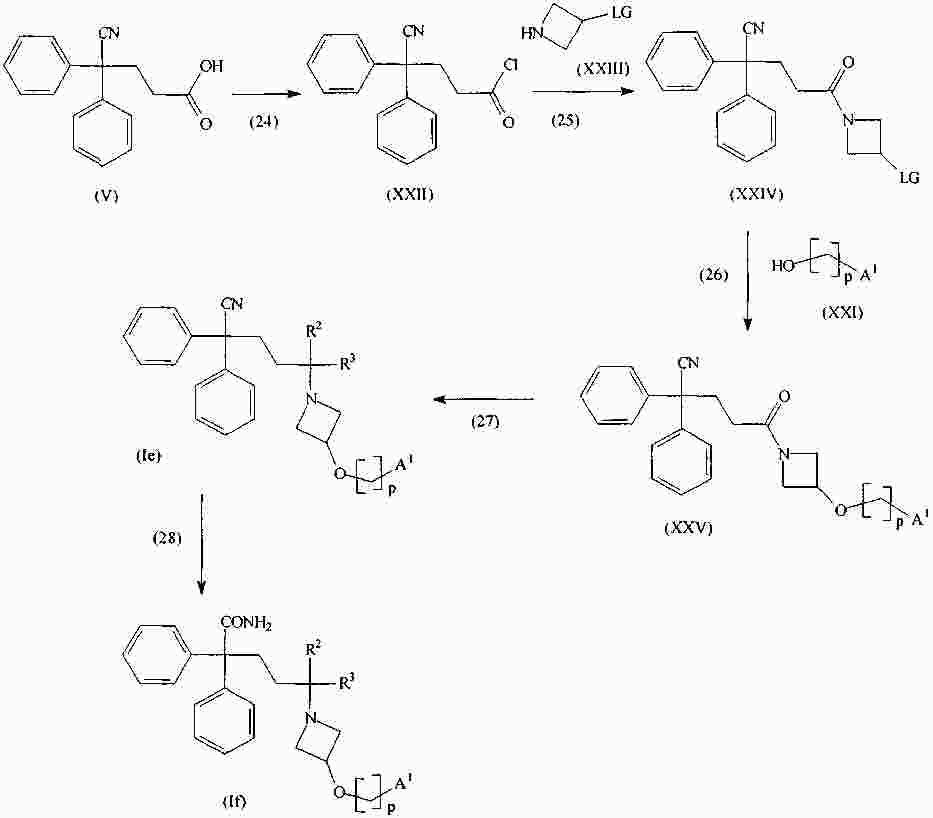
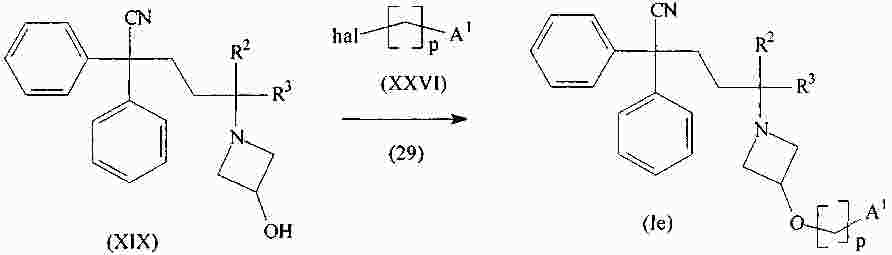
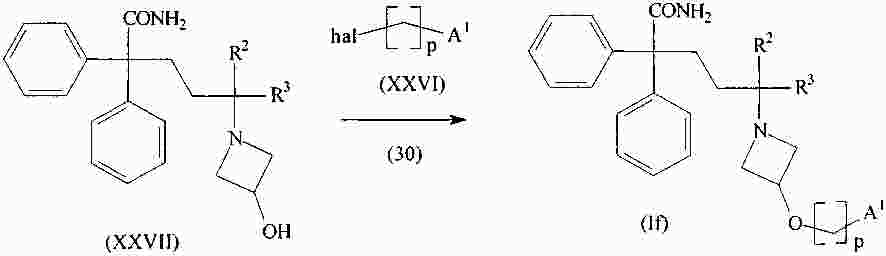
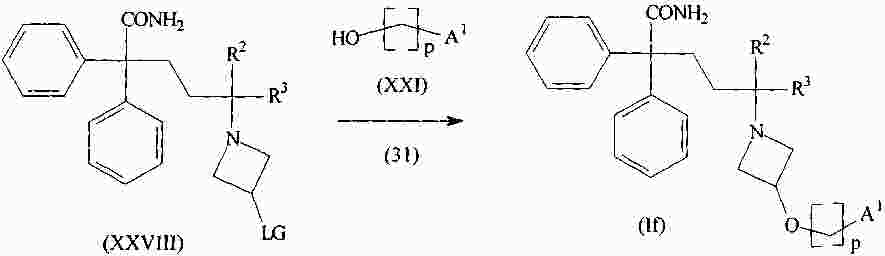
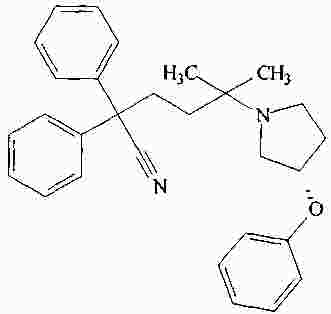
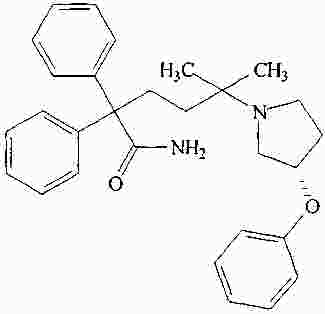
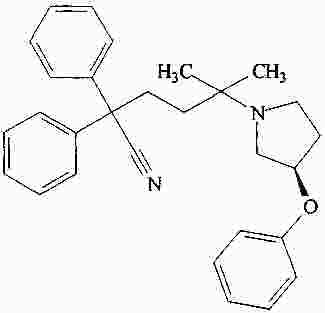
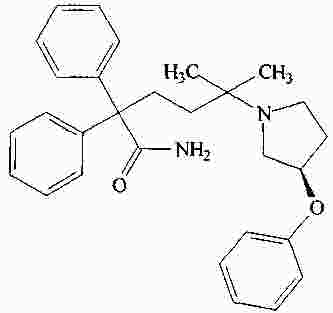
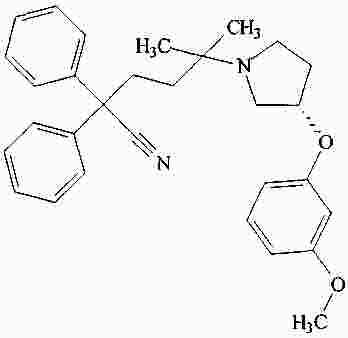
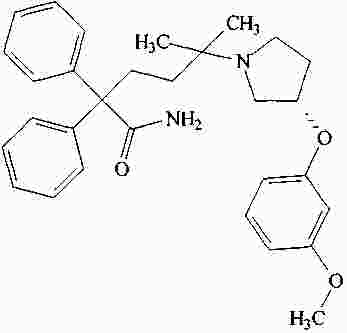
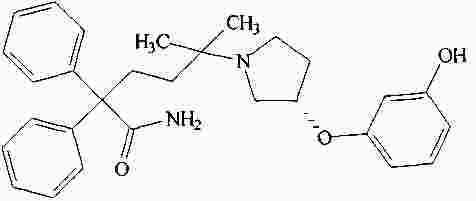
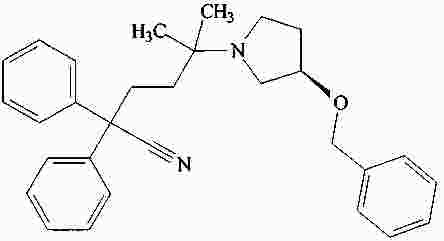
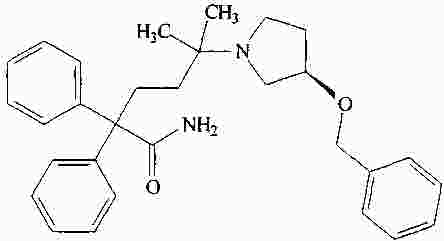
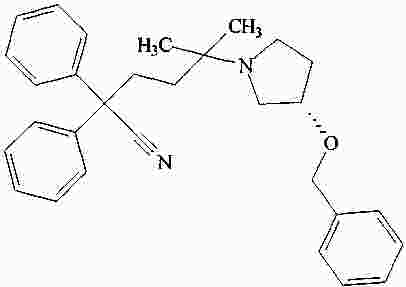
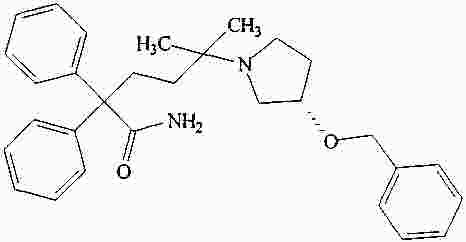
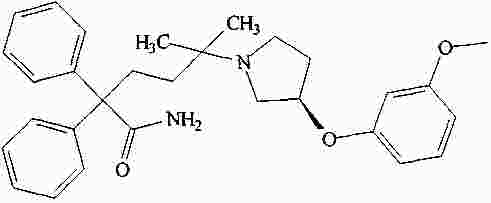
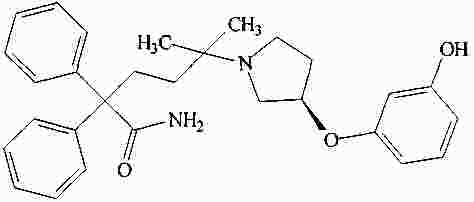
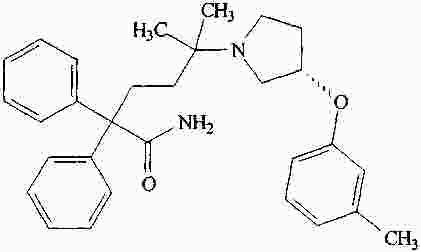
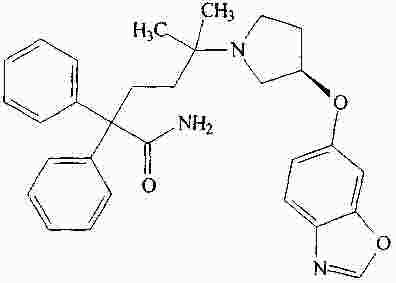
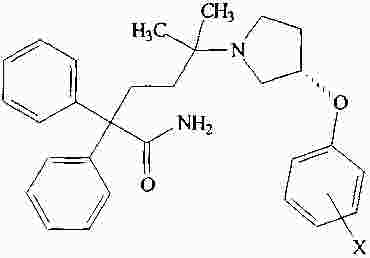
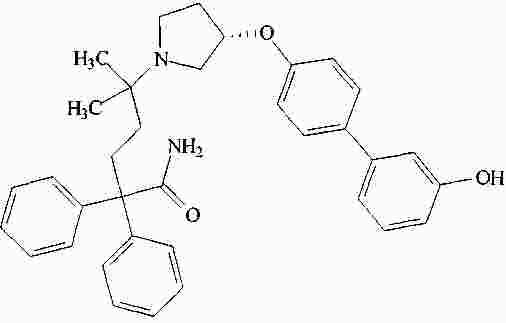
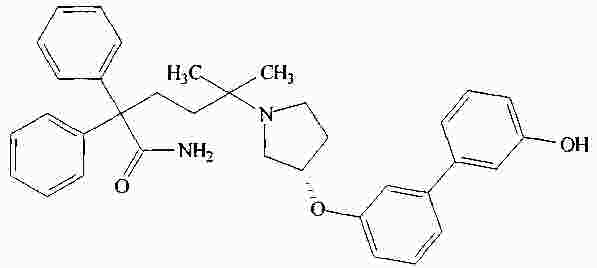
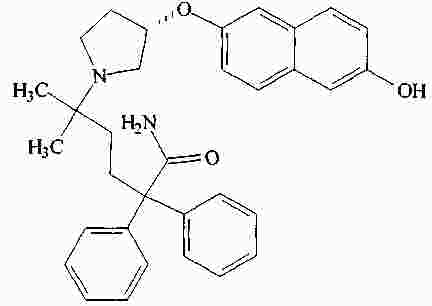
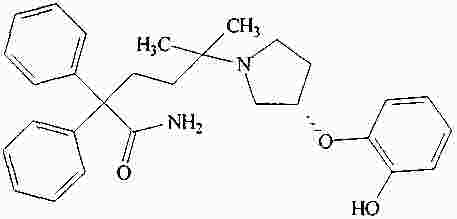
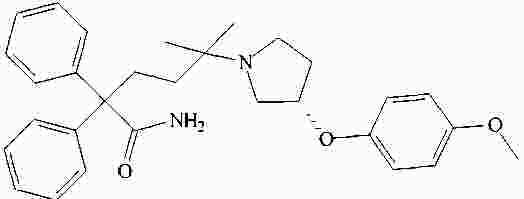
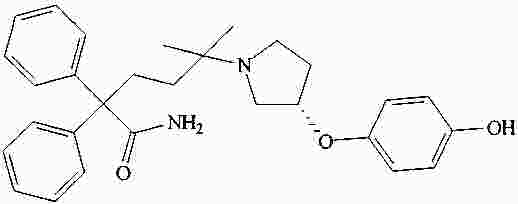
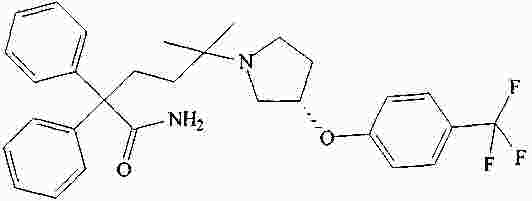
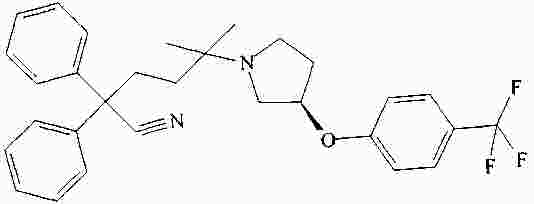
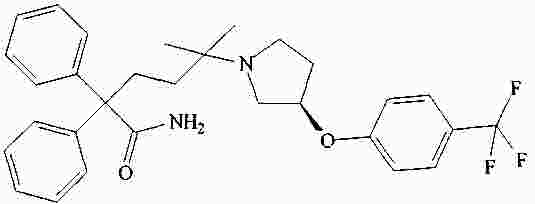
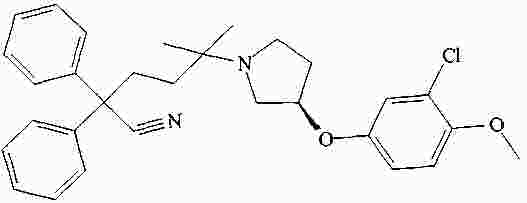
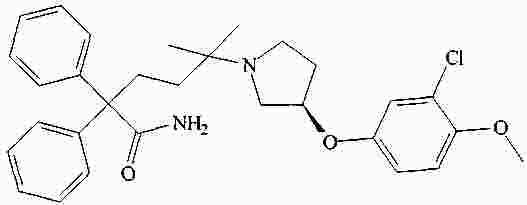
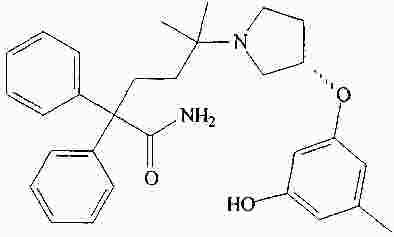
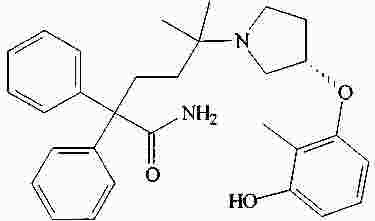
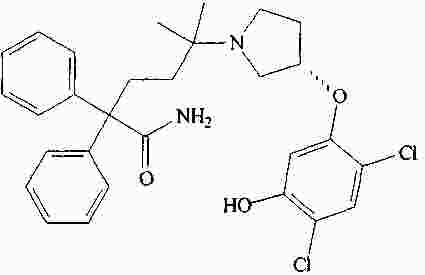
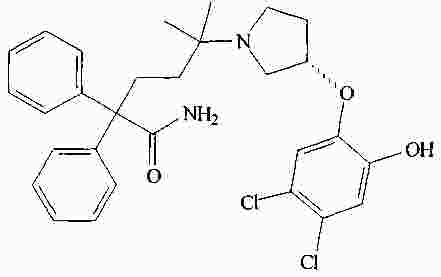
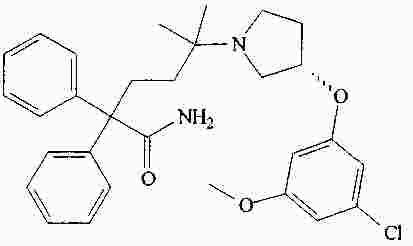
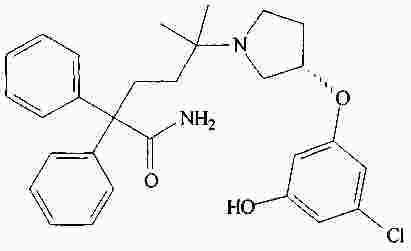
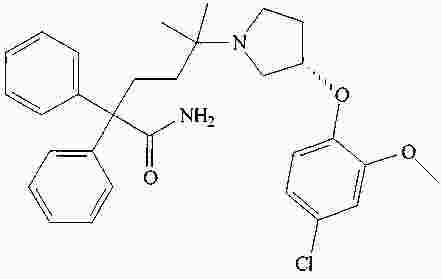
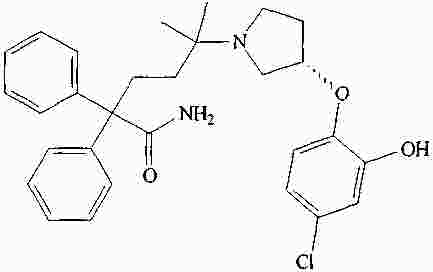
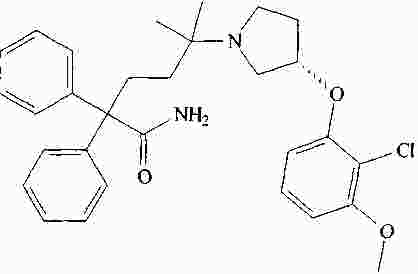
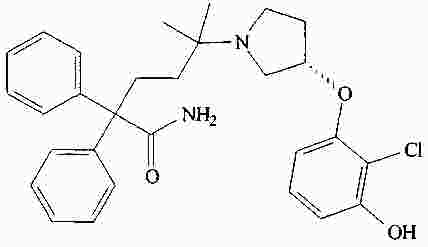
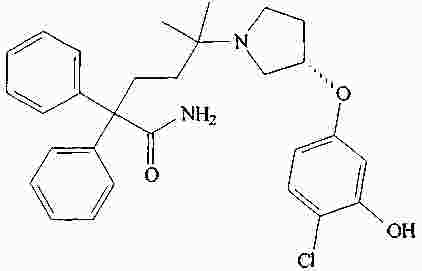
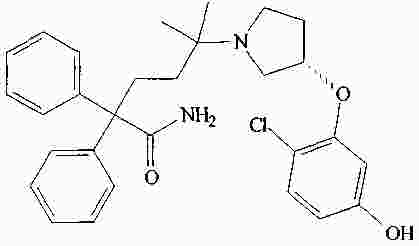
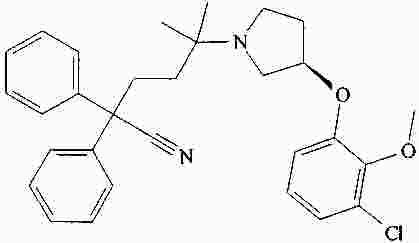
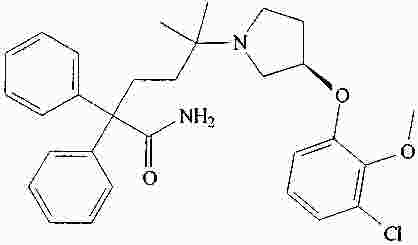
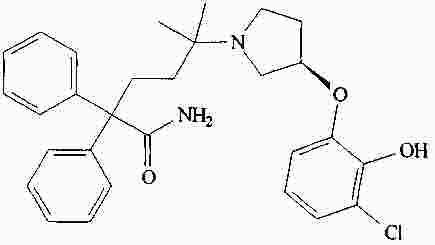
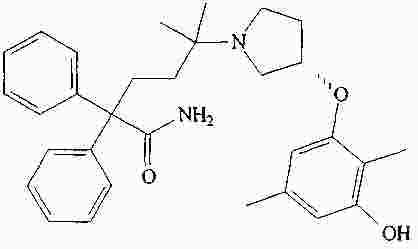
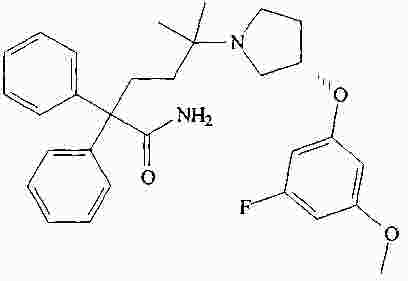
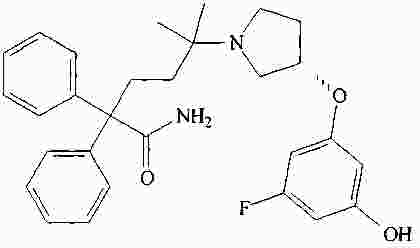
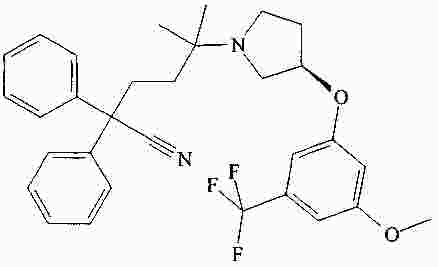
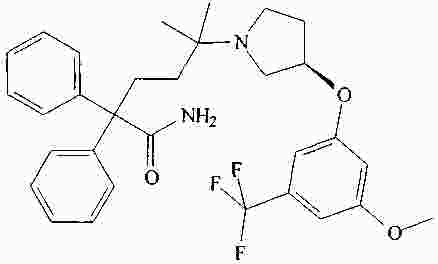
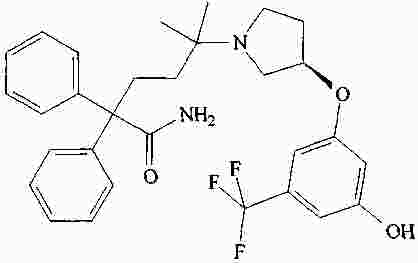
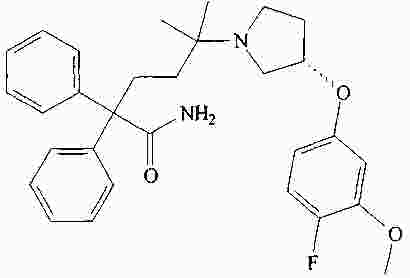
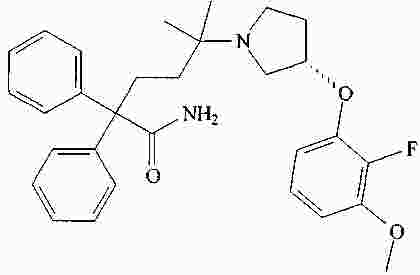
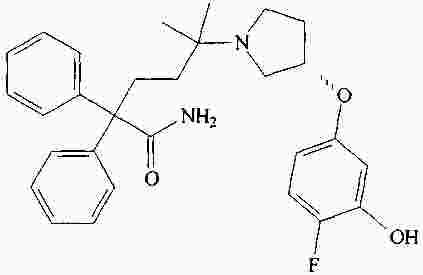
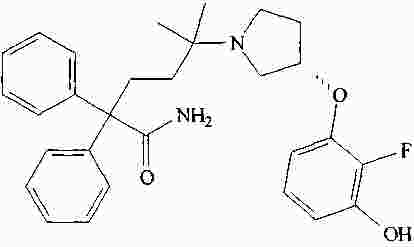
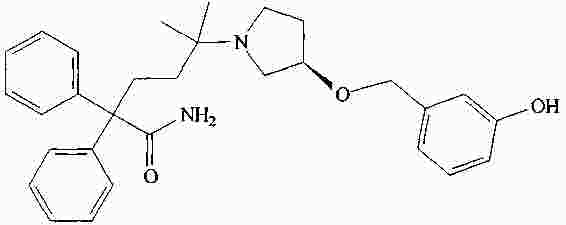
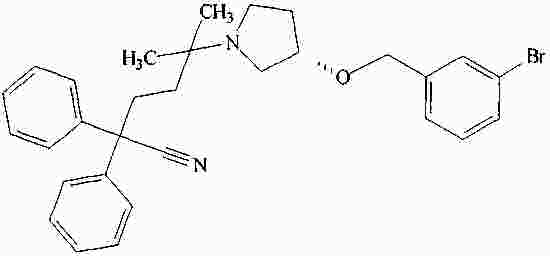
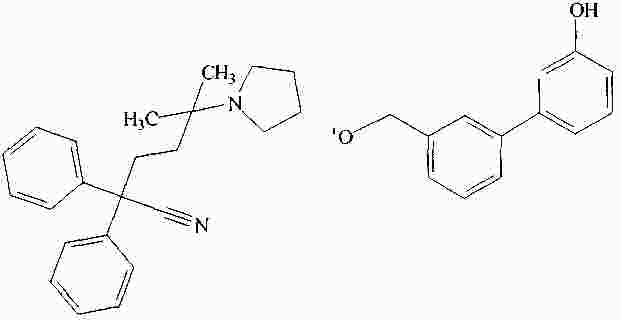
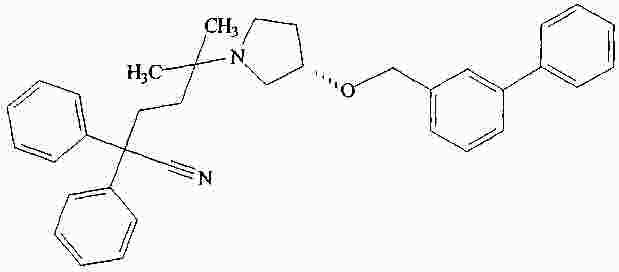
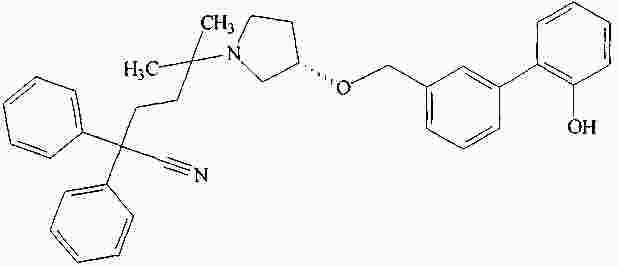
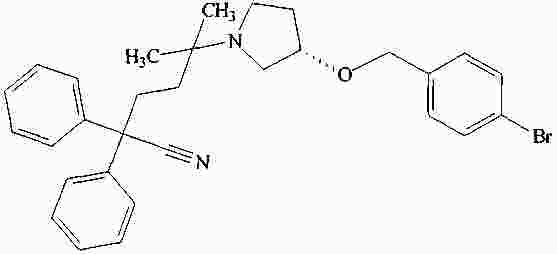
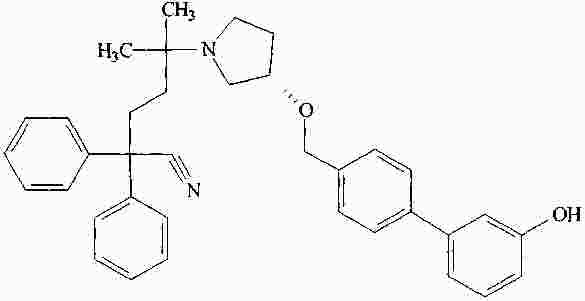
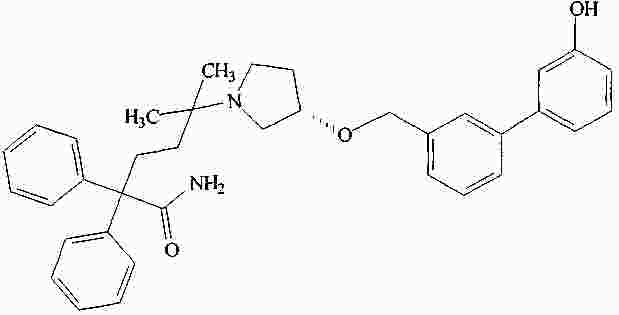
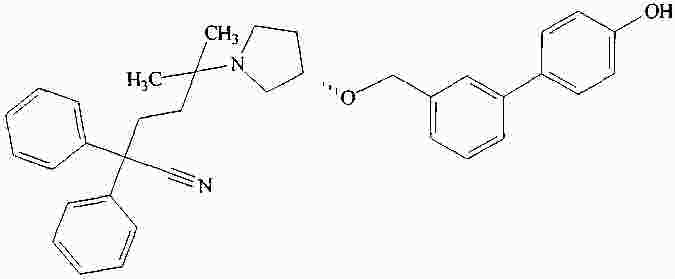
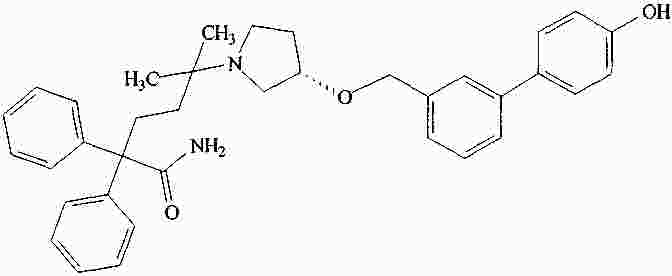
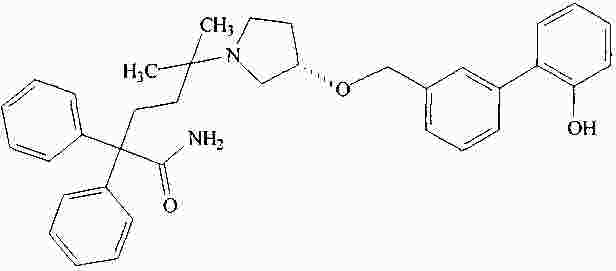
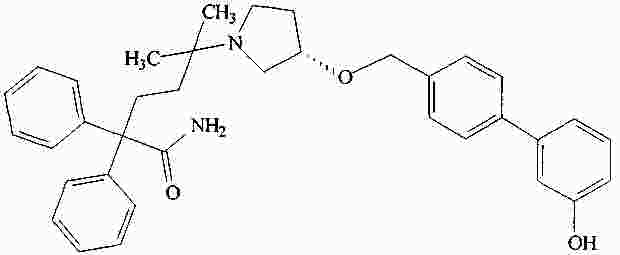
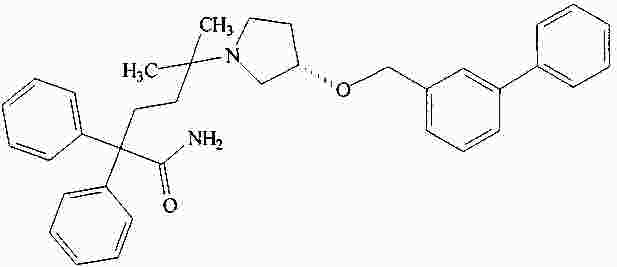
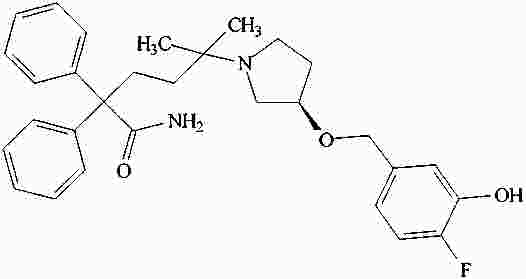
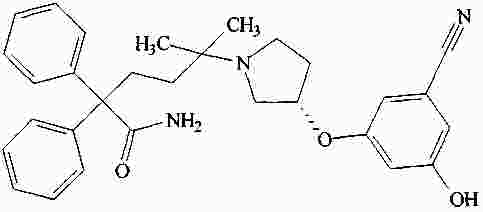
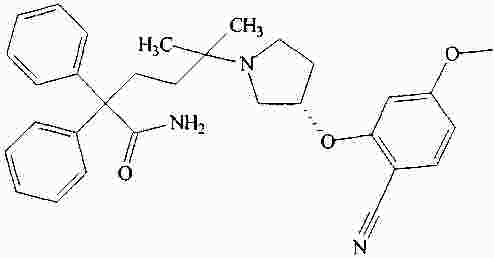
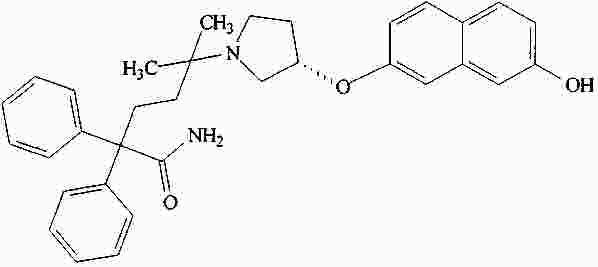
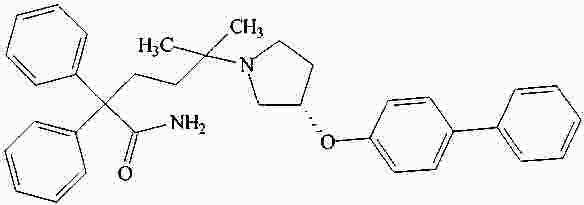
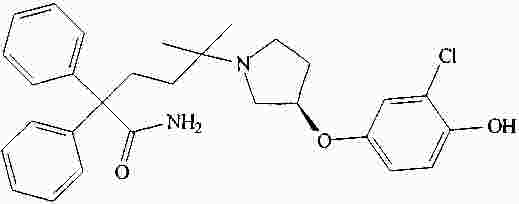
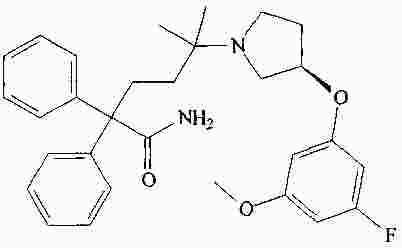
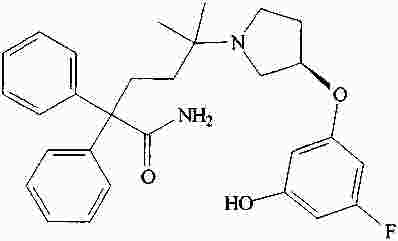
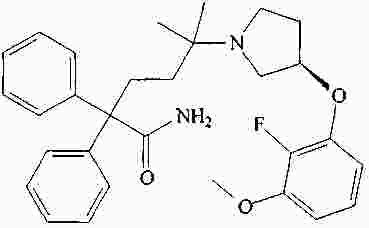
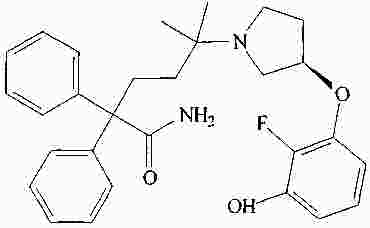
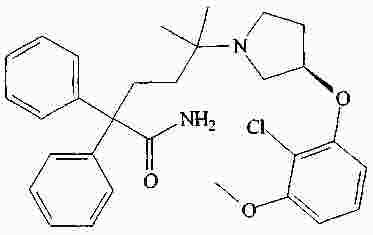
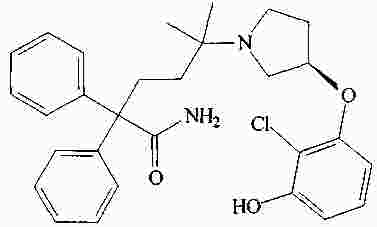
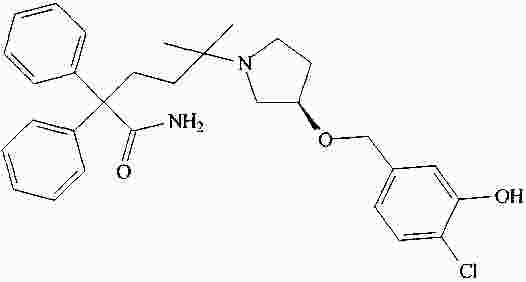
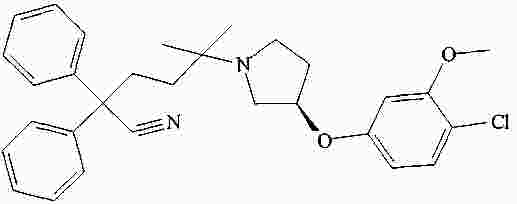
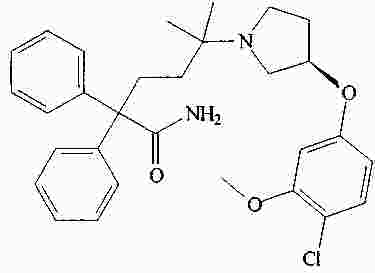
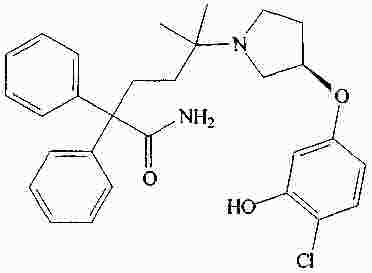
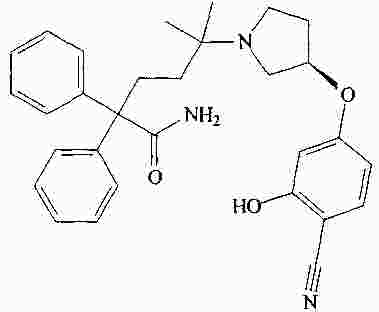
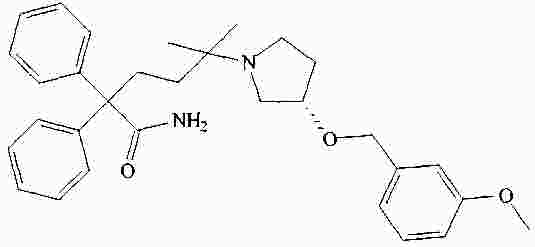
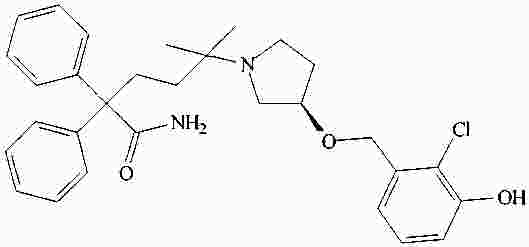
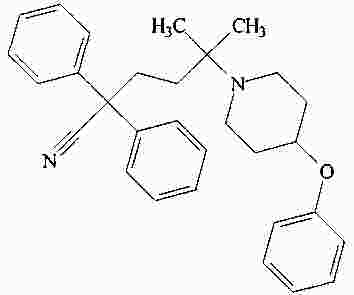
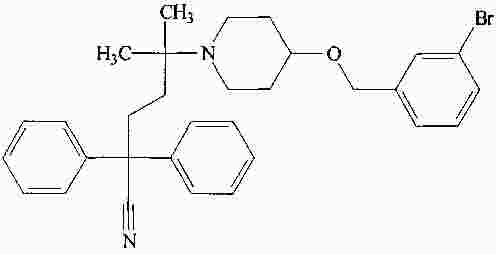
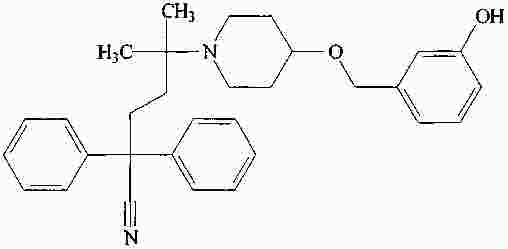
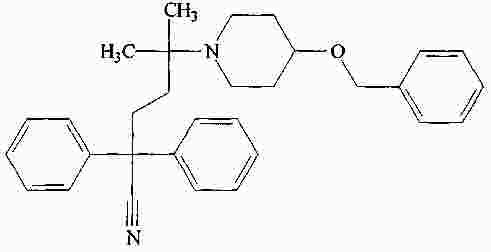
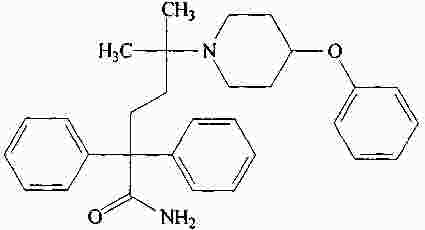
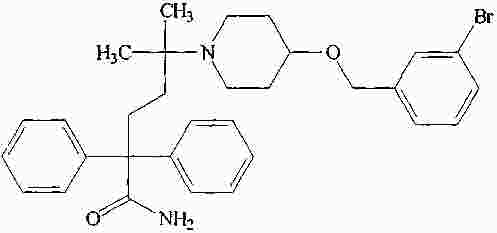
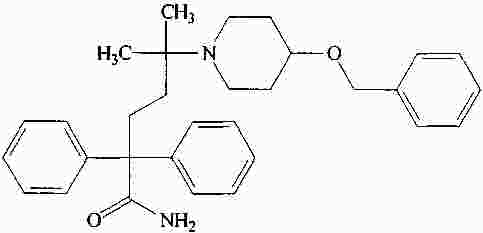
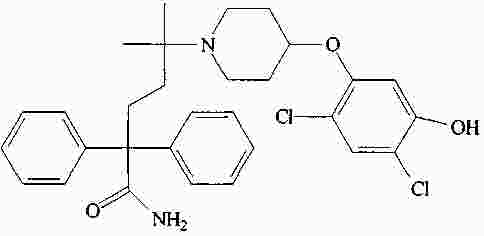
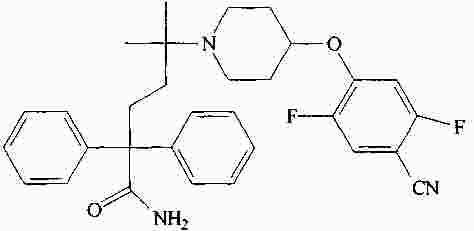
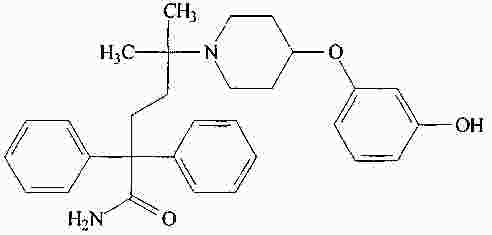
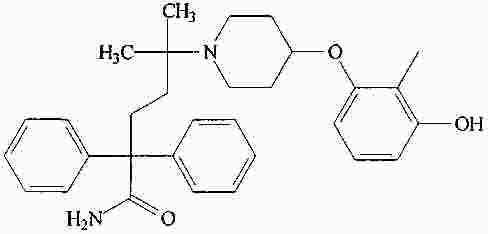
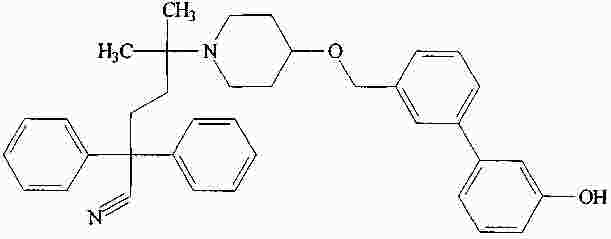
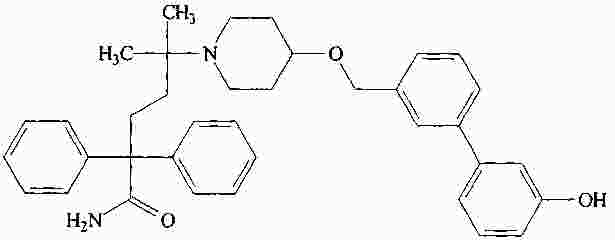
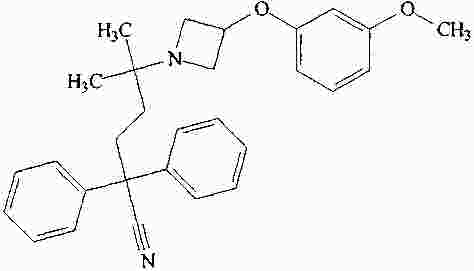
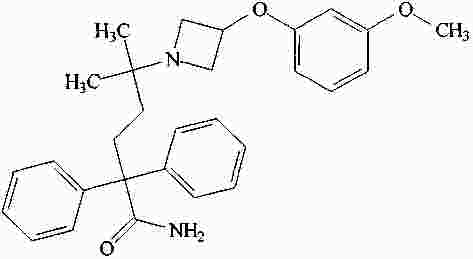
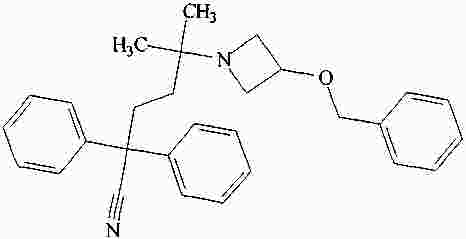
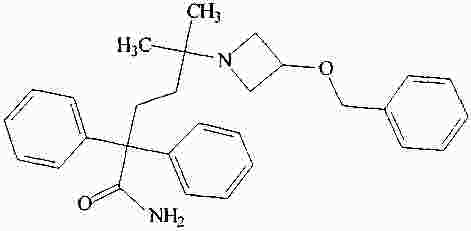
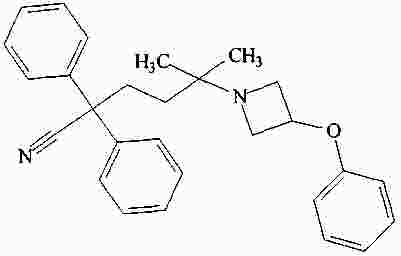
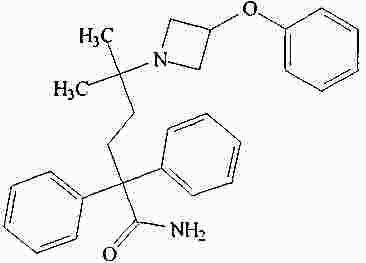
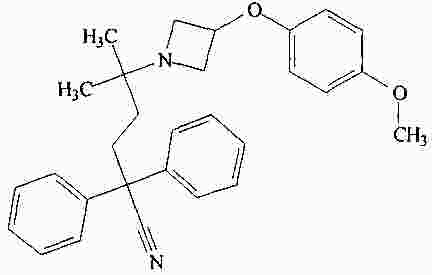
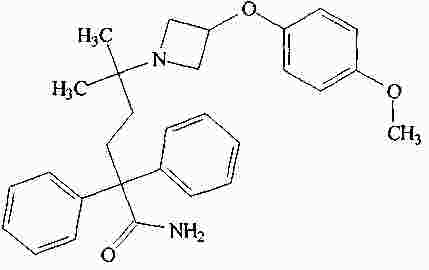
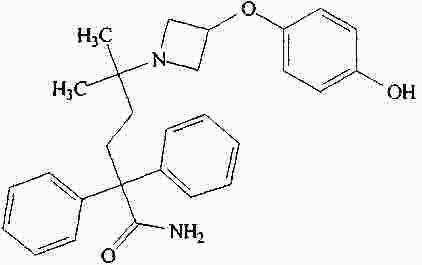
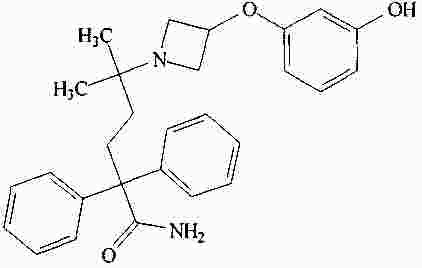
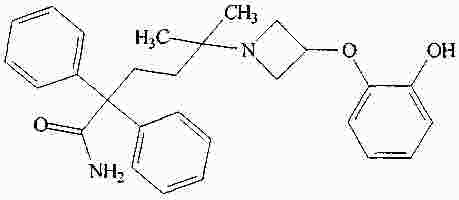
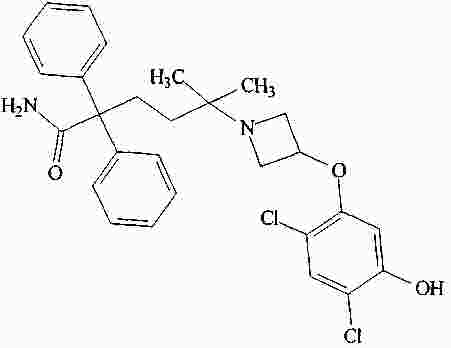
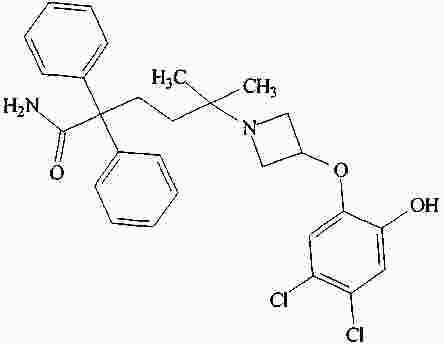
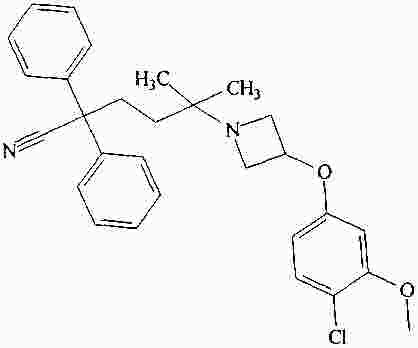
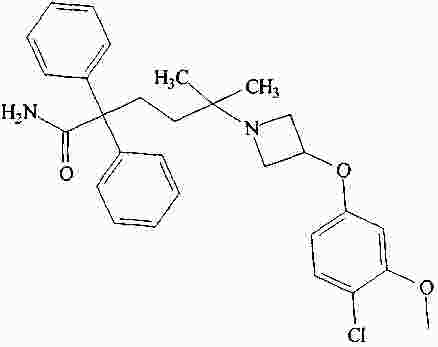
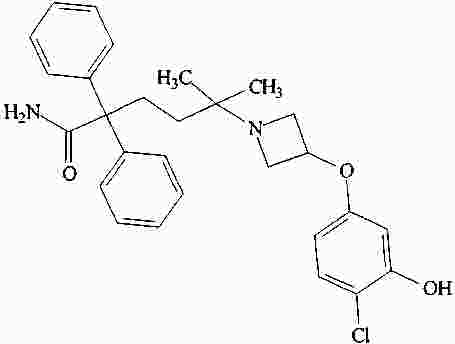
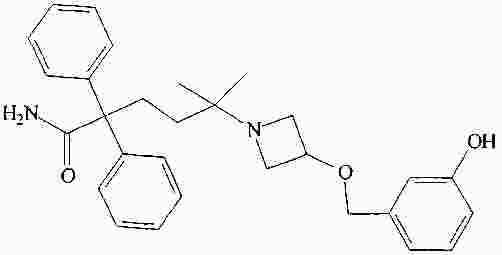
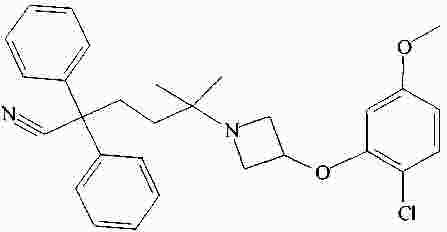
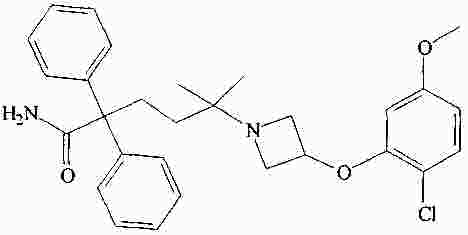
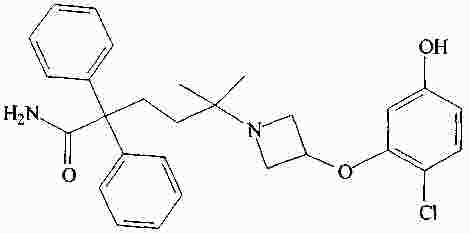
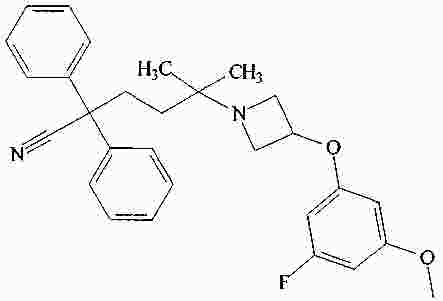
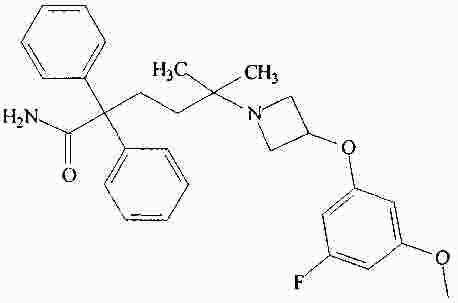
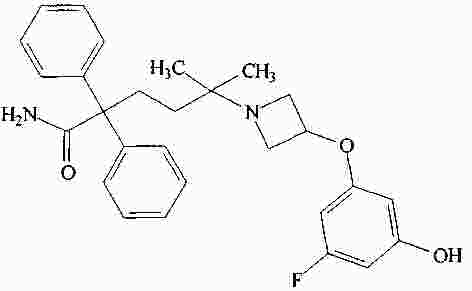
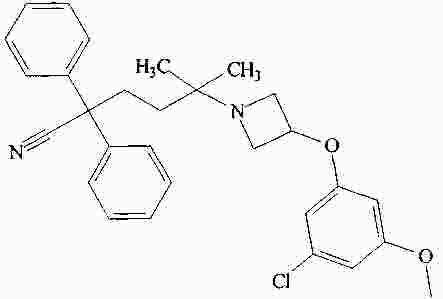
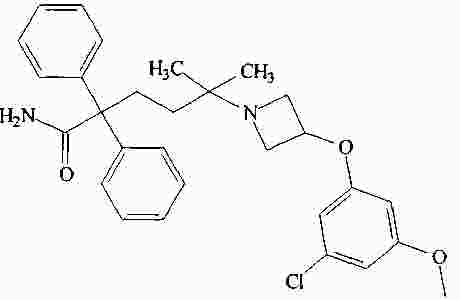
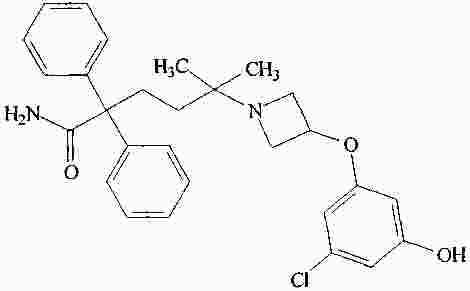
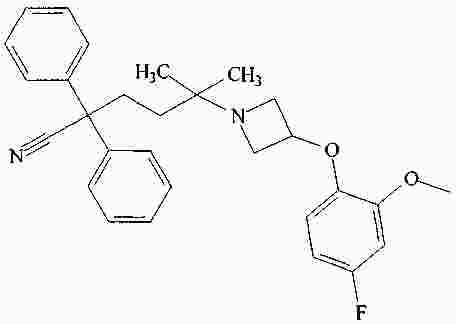
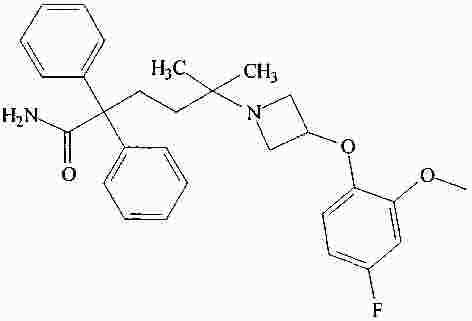
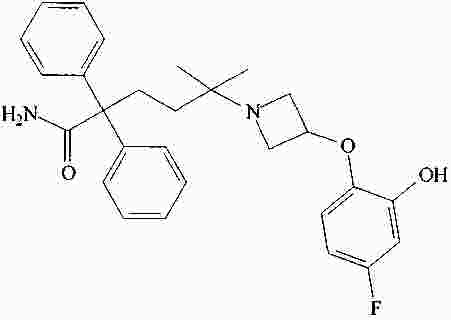
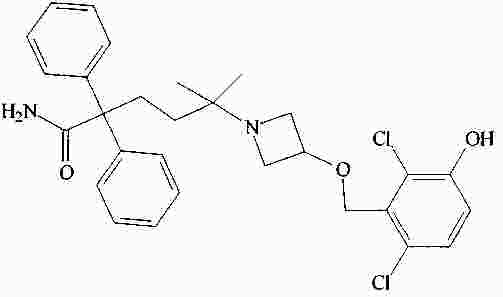
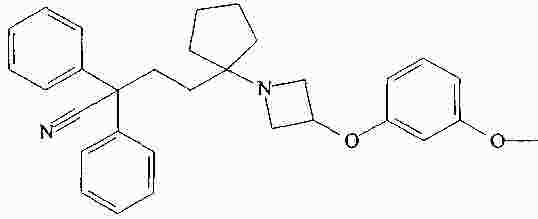
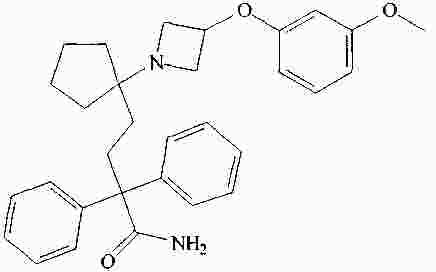
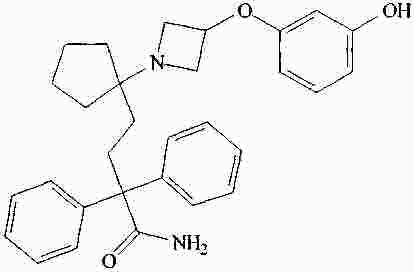
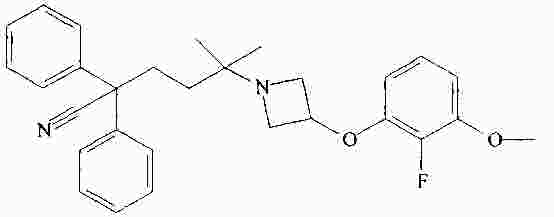
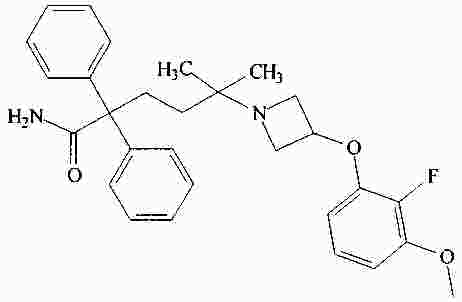
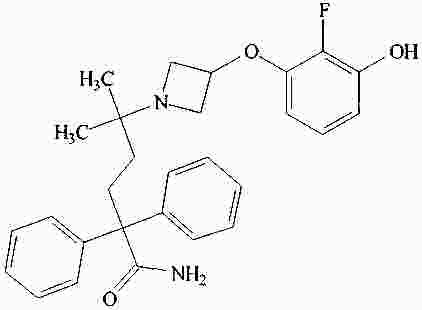
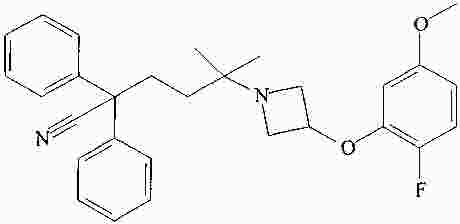
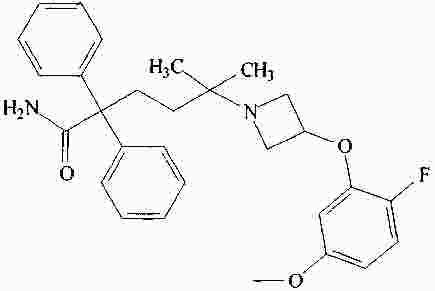
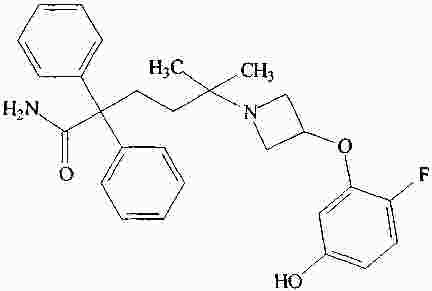
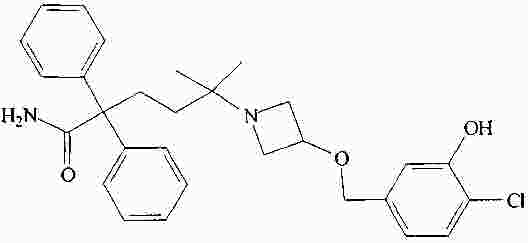
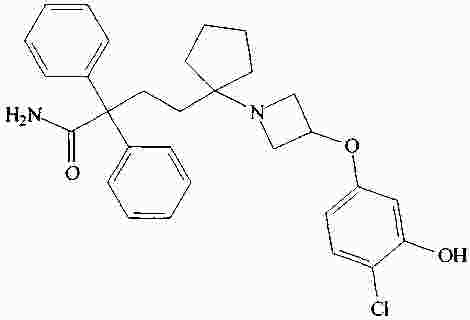
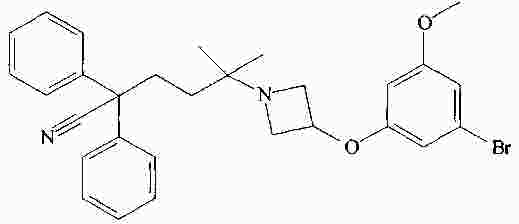
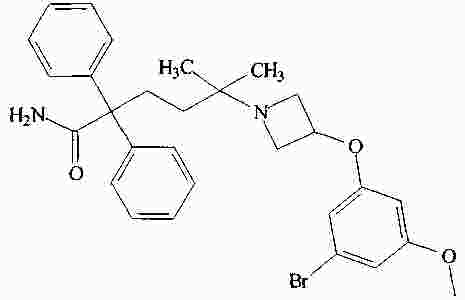
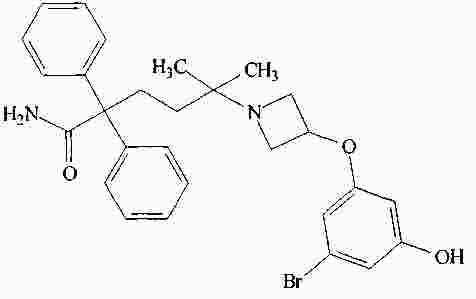
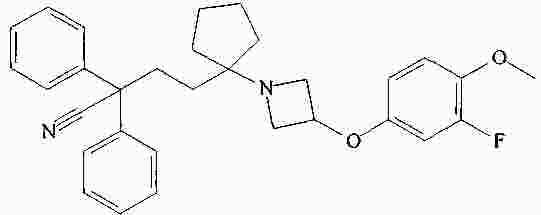
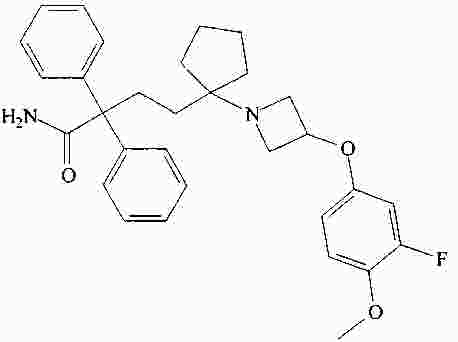
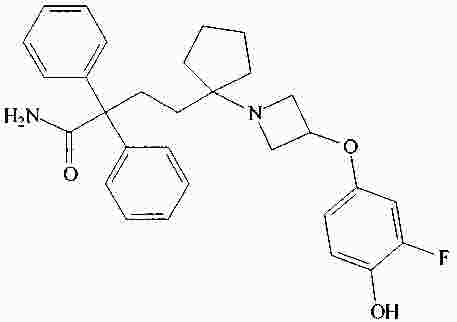
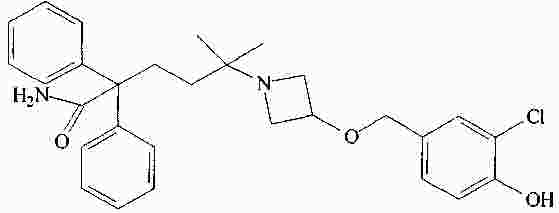
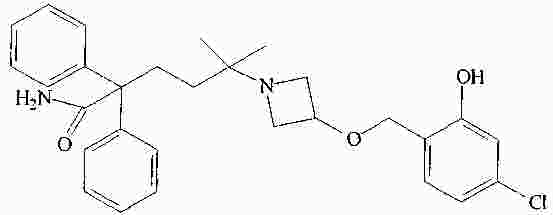
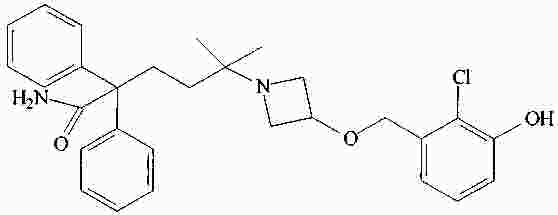
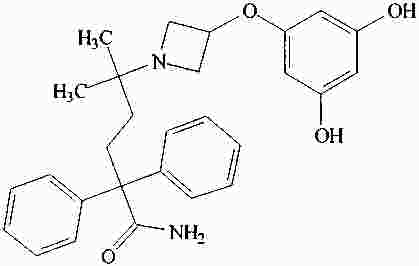
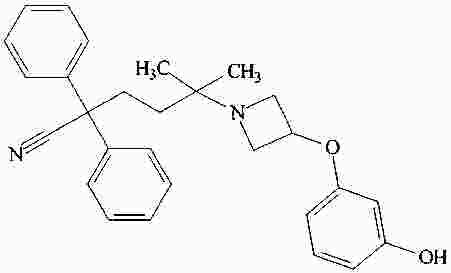
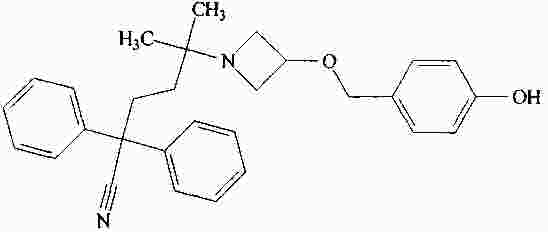
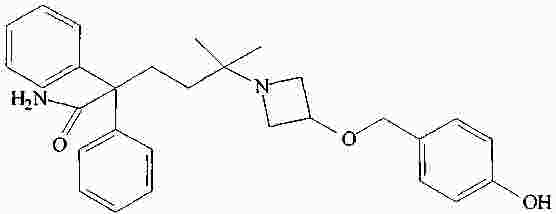
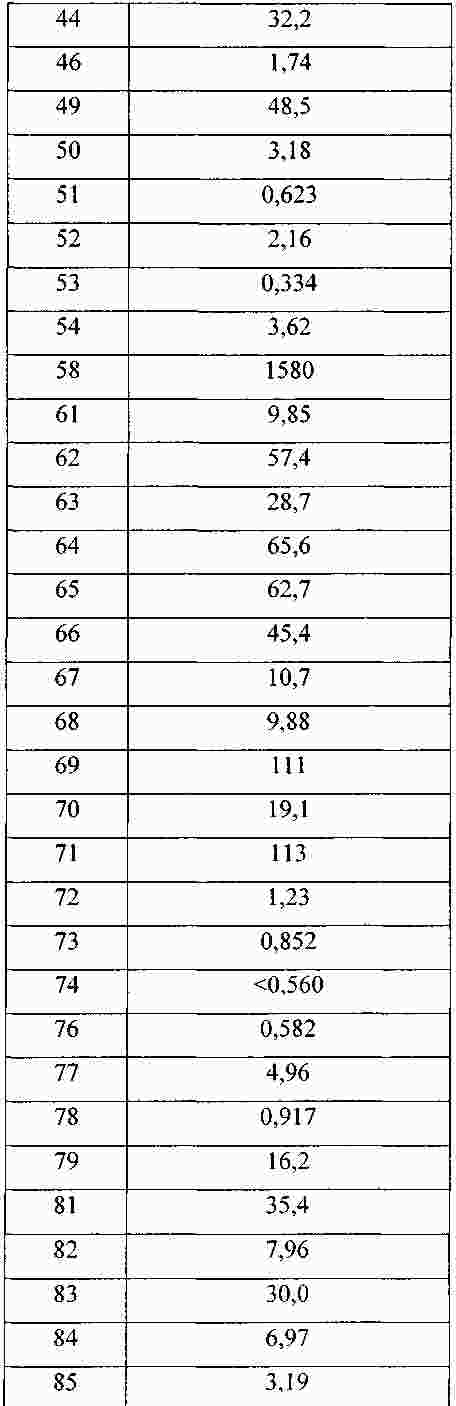
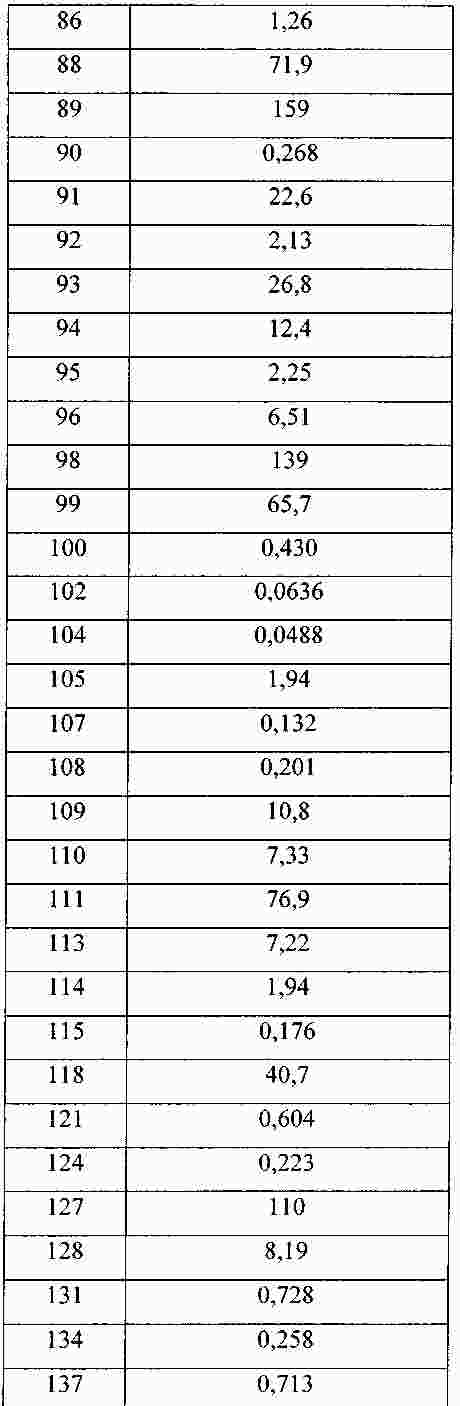
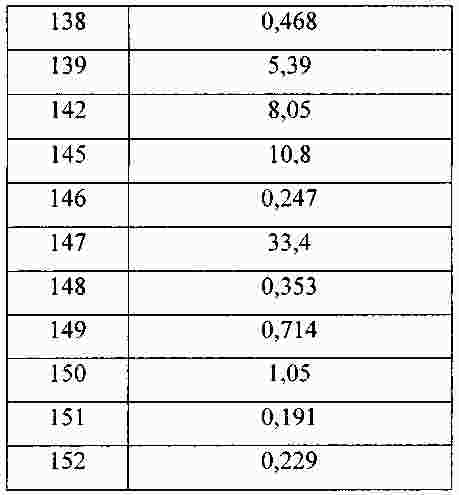
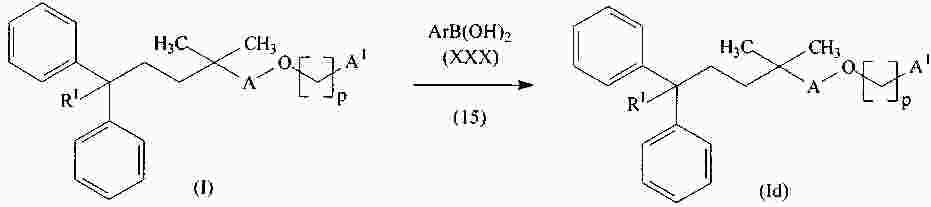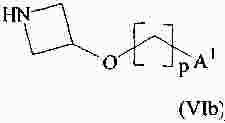|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea000013083b*\id |
|
Термины запроса в документе Реферат Данное изобретение относится к соединениям формулы (I), способам и промежуточным соединениям для их получения, их применению в качестве антагонистов мускариновых рецепторов и к содержащим их фармацевтическим композициям. Формула [0001] Соединение формулы (I) [0002] Соединение по п.1, где R1 представляет собой CONH2. [0003] Соединение по п.1 или 2, где р равно 0. [0004] Соединение по п.1 или 2, где р равно 1. [0005] Соединение по любому из пп.1-4, где А1 представляет собой фенил, возможно замещенный 1, 2 или 3 группами, независимо выбранными из F, Cl, CF3, ОН, ОСН3, OCF3 и H3. [0006] Соединение по любому из пп.1-5, где А1 представляет собой фенил, возможно замещенный 1 или 2 группами, независимо выбранными из F, Cl, CF3, ОН, ОСН3, OCF3 и CH3. [0007] Соединение по любому из пп.1-4, где А1 представляет собой нафтил, возможно замещенный группой ОН. [0008] Соединение по любому из пп.1-4, где А1 выбран из индолила, изоиндолила, хинолила, изохинолила, бензофуранила, изобензофуранила, бензотиенила, изобензотиенила, хиназолила, хиноксалила, фталазинила, бензотиазолила, бензоксазолила, бензизотиазолила, бензизоксазолила, бензимидазолила, индазолила, бензотриазолила, бензоксадиазолила, бензизоксадиазолила, бензотиадиазолила и бензизотиадиазолила. [0009] Соединение по любому из пп.1-4, где А1 представляет собой бензоксазолил. [0010] Соединение по любому из пп.1-9, где R2 и R3 представляют собой метил. [0011] Соединение по любому из пп.1-10, где А представляет собой группу формулы [0012] Соединение по любому из пп.1-10, где А представляет собой группу формулы [0013] Соединение по любому из пп.1-10, где А представляет собой группу формулы [0014] Соединение по п.1, выбранное из [0015] Соединение по п.1, которое представляет собой 5-[3-(3-гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексанамид или его фармацевтически приемлемую соль или сольват. [0016] Фармацевтическая композиция, содержащая, по меньшей мере, эффективное количество соединения формулы (I), как оно описано в любом из пп.1-15, или его фармацевтически приемлемых солей или сольватов. [0017] Соединение формулы (I), как оно описано в любом из пп.1-15, или его фармацевтически приемлемые соли или сольваты для применения в качестве лекарственного средства. [0018] Применение соединения формулы (I), как оно описано в любом из пп.1-15, или его фармацевтически приемлемых солей или сольватов для изготовления лекарственного средства, обладающего активностью антагониста рецептора M3. [0019] Применение соединения формулы (I), как оно описано в любом из пп.1-15, или его фармацевтически приемлемых солей или сольватов для изготовления лекарственного средства для лечения заболеваний, расстройств и состояний, выбранных из группы, состоящей из [0020] Комбинация соединения по любому из пп.1-15 с другим(и) терапевтическим(и) агентом(ами), выбранным(и) из: [0021] Промежуточное соединение формулы [0022] Промежуточное соединение формулы [0023] Промежуточное соединение формулы [0024] Промежуточное соединение формулы [0025] Промежуточное соединение формулы Полный текст патента Данное изобретение относится к соединениям общей формулы (I) где R 1 , R 2 , R 3 , А, А 1 и р имеют указанные ниже значения, и к способам получения таких производных, к промежуточным соединениям для получения таких производных, к композициям, содержащим такие производные, и к применениям таких производных. Холинергические мускариновые рецепторы являются членами надсемейства сопряженных с G-белками рецепторов и дополнительно подразделяются на 5 подтипов от M 1 до M 5 . Подтипы мускариновых рецепторов экспрессируются в организме в разных местах и дифференцированно. Были клонированы гены для всех 5 подтипов, и из них рецепторы M 1 , M 2 и М 3 были со всех сторон фармакологически охарактеризованы в ткани животных и человека. Рецепторы M 1 экспрессируются в мозге (в коре головного мозга и гиппокампе), в железах и в ганглиях симпатических и парасимпатических нервов. Рецепторы М 2 экспрессируются в сердце, заднем мозге, гладкой мышце и в синапсах автономной нервной системы. Рецепторы М 3 экспрессируются в мозге, железах и гладкой мышце. В дыхательных путях стимуляция рецепторов М 3 вызывает сокращение гладкой мышцы дыхательных путей, приводящее к бронхоконстрикции, а стимуляция рецепторов М 3 в слюнных железах повышает секрецию жидкости и слизи, что приводит к повышенному слюноотделению. Очевидно, что рецепторы М 2 , экспрессируемые на гладкой мышце, являются просократительными, а пресинаптические рецепторы М 2 модулируют высвобождение ацетилхолина из парасимпатических нервов. Стимуляция рецепторов М 2 , экспрессируемых в сердце, вызывает брадикардию. Мускариновые антагонисты краткосрочного и длительного действия используются в лечении астмы и COPD (хронической обструктивной болезни легких). Они включают агенты краткосрочного действия Atrovent ® (ипратропия бромид) и Oxivent ® (окситропия бромид) и агент длительного действия Spiriva ® (тиотропия бромид). Эти соединения вызывают бронходилатацию после ингаляционного введения. В дополнение к улучшению спирометрических показателей антимускариновое применение при хронической обструктивной болезни легких (COPD) ассоциируется с улучшением состояния здоровья и качества жизни. Поскольку мускариновые рецепторы широко распространены в организме, значительное системное воздействие мускариновых антагонистов ассоциируется с такими эффектами, как сухость во рту, запор, мидриаз, недержание мочи (все это преимущественно опосредовано блокадой рецепторов М 3 ) и тахикардия (опосредованная блокадой рецепторов М 2 ). Обычно наблюдаемым побочным эффектом после ингаляционного введения терапевтической дозы клинически применяемых в настоящее время неселективных мускариновых антагонистов является сухость во рту, и хотя об этом сообщается только как об умеренной по интенсивности, это не ограничивает дозу вводимого ингаляцией агента. Соответственно, все еще имеется потребность в улучшенных антагонистах рецепторов М 3 , которые могли бы иметь соответствующий фармакологический профиль, например, в отношении эффективности, фармакокинетики или продолжительности действия. В данном контексте настоящее изобретение относится к новым антагонистам рецепторов М 3 . В частности, существует потребность в антагонистах рецепторов М 3 , которые могли бы иметь фармакологический профиль, подходящий для введения ингаляционным путем. В научной литературе описано много соединений, обладающих активностью антагонистов мускариновых рецепторов. К примеру, в ЕР 0948964 А1 раскрыты соединения формулы где R означает атом водорода, атом галогена или низшую алкоксигруппу. Данное изобретение относится к соединению формулы (I) где R 1 представляет собой CN или CONH 2 ; А выбран из или где * и ** представляют собой точки присоединения, причем ** означает связывание с кислородом, R 2 и R 3 представляют собой метил или, когда А представляет собой группу формулы R 2 и R 3 также могут образовывать вместе с атомом углерода, с которым они связаны, циклопентановое кольцо; р равно 0 или 1; А 1 выбран из а) фенила, возможно замещенного 1, 2 или 3 группами, независимо выбранными из галогено, CN, CF 3 , OR 4 , SR 4 , OCF 3 , (С 1 -С 4 )алкила и фенила, возможно замещенного группой ОН; б) нафтила, возможно замещенного 1 или 2 группами, выбранными из галогено, CN, CF 3 , OR 4 , SR 4 , OCF 3 и (С 1 -С 4 )алкила; в) 9- или 10-членной бициклической ароматической гетероциклической группы, содержащей 1, 2 или 3 гетероатома, независимо выбранных из О, S или N, причем указанная гетероциклическая группа возможно замещена 1 или 2 заместителями, независимо выбранными из OR 4 , (С 1 -С 4 )алкила и галогено; R 4 представляет собой Н или (С 1 -С 4 )алкил; или, в соответствующем случае, его фармацевтически приемлемым солям или сольватам. В приведенной выше общей формуле (I) (С 1 -С 4 )алкил означает прямоцепочечную или разветвленную группу, содержащую 1, 2, 3 или 4 атома углерода. Это также применимо, если они несут заместители или являются заместителями других радикалов, например в радикалах О-(С 1 -С 4 )алкил, радикалах S-(С 1 -С 4 )алкил и т.д. Примерами подходящих (С 1 -С 4 )алкильных радикалов являются метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил. Примерами подходящих радикалов О-(С 1 -С 4 )алкил являются метокси, этокси, н-пропилокси, изопропилокси, н-бутилокси, изобутилокси, втор-бутилокси и трет-бутилокси. Примерами 9- или 10-членной бициклической ароматической гетероциклической группы, содержащей 1, 2 или 3 гетероатома, независимо выбранных из О, S или N, являются индолил, изоиндолил, хинолил, изохинолил, бензофуранил, изобензофуранил, бензотиенил, изобензотиенил, хиназолил, хиноксалил, фталазинил, бензотиазолил, бензоксазолил, бензизотиазолил, бензизоксазолил, бензимидазолил, индазолил, бензотриазолил, бензоксадиазолил, бензизоксадиазолил, бензотиадиазолил и бензизотиадиазолил. Предпочтительными 9- или 10-членными бициклическими ароматическими гетероциклическими группами являются бензоксазолил, бензотиазолил, бензофуранил, бензотиенил, изохинолил и хинолил. Особенно предпочтителен бензоксазолил. Галогено означает атом галогена, выбранный из группы, состоящей из фторо, хлоро, бромо и йодо. Предпочтительными галогеногруппами являются фторо и хлоро. В вышеуказанных соединениях формулы (I) и в промежуточных соединениях, полезных для их получения, предпочтительны следующие определения. Предпочтительно R 1 представляет собой CONH 2 . Предпочтительно R 4 представляет собой Н или СН 3 . Предпочтительно А 1 представляет собой фенил, возможно замещенный 1, 2 или 3 группами, независимо выбранными из F, Cl, CF 3 , ОН, ОСН 3 , OCF 3 и СН 3 . Более предпочтительно А 1 представляет собой фенил, возможно замещенный 1 или 2 группами, независимо выбранными из F, Cl, CF 3 , ОН, ОСН 3 , OCF 3 и СН 3 . Предпочтительно А 1 представляет собой нафтил, возможно замещенный ОН. Предпочтительно А 1 представляет собой бензоксазолил. Предпочтительно R 2 и R 3 представляют собой метил. В предпочтительном воплощении р равно 0. В другом предпочтительном воплощении р равно 1. В предпочтительном воплощении соединение формулы (I), где А представляет собой группу формулы имеет указанную ниже (R)-конфигурацию: В предпочтительном воплощении соединение формулы (I), где А представляет собой группу формулы имеет указанную ниже (S)-конфигурацию: Предпочтительными соединениями по изобретению являются 5-метил-5-[(3S)-3-феноксипирролидин-1-ил]-2,2-дифенилгексаннитрил; 5-метил-5-[(3S)-3-феноксипирролидин-1-ил]-2,2-дифенилгексанамид; 5-метил-5-[(3R)-3-феноксипирролидин-1-ил]-2,2-дифенилгексаннитрил; 5-метил-5-[(3R)-3-феноксипирролидин-1-ил]-2,2-дифенилгексанамид; 5-[(3S)-3-(3-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; 5-[(3S)-3-(3-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3S)-3-(3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(бензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; 5-[(3R)-3-(бензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3S)-3-(бензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; 5-[(3S)-3-(бензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(3-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-метил-5-[(3S)-3-(3-метилфенокси)пирролидин-1-ил]-2,2-дифенилгексанамид; 5-[(3R)-3-(1,3-бензоксазол-6-илокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3S)-3-(4-хлорфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3S)-3-(3-бромфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-{(3S)-3-[(3'-гидроксибифенил-4-ил)окси]пирролидин-1-ил}-5-метил-2,2-дифенилгексанамид; 5-{(3S)-3-[(3'-гидроксибифенил-3-ил)окси]пирролидин-1-ил}-5-метил-2,2-дифенилгексанамид; 5-{(3S)-3-[(6-гидрокси-2-нафтил)окси]пирролидин-1-ил}-5-метил-2,2-дифенилгексанамид; 5-[(3S)-3-(2-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3S)-3-(4-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3S)-3-(4-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3S)-3-(4-трифторметилфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(4-трифторметилфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; 5-[(3R)-3-(4-трифторметилфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(3-хлор-4-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; 5-[(3R)-3-(3-хлор-4-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; амид 5-[(3S)-3-(3-гидрокси-5-метилфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3S)-3-(3-гидрокси-2-метилфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3S)-3-(2,4-дихлор-5-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3S)-3-(4,5-дихлор-2-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3S)-3-(3-хлор-5-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3S)-3-(3-хлор-5-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3S)-3-(4-хлор-2-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3S)-3-(4-хлор-2-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3S)-3-(2-хлор-3-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3S)-3-(2-хлор-3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3S)-3-(4-хлор-3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3S)-3-(2-хлор-5-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; 5-[(3R)-3-(3-хлор-2-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; амид 5-[(3R)-3-(3-хлор-2-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3R)-3-(3-хлор-2-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3S)-3-(3-гидрокси-2,5-диметилфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3S)-3-(3-фтор-5-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3S)-3-(3-фтор-5-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; 5-[(3R)-3-(3-метокси-5-трифторметилфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; амид 5-[(3R)-3-(3-метокси-5-трифторметилфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3R)-3-(3-гидрокси-5-трифторметилфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3S)-3-(4-фтор-3-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3S)-3-(2-фтор-3-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3S)-3-(4-фтор-3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3S)-3-(2-фтор-3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3R)-3-(3-гидроксибензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; 5-{(3S)-3-[(3-бромбензил)окси]пирролидин-1-ил}-5-метил-2,2-дифенилгексаннитрил; 5-{(3S)-3-[(3'-гидроксибифенил-3-ил)метокси]пирролидин-1-ил}-5-метил-2,2-дифенилгексаннитрил; 5-[(3S)-3-(бифенил-3-илметокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; 5-{(3S)-3-[(2'-гидроксибифенил-3-ил)метокси]пирролидин-1-ил}-5-метил-2,2-дифенилгексаннитрил; 5-{(3S)-3-[(4-бромбензил)окси]пирролидин-1-ил}-5-метил-2,2-дифенилгексаннитрил; 5-{(3S)-3-[(3'-гидроксибифенил-4-ил)метокси]пирролидин-1-ил}-5-метил-2,2-дифенилгексаннитрил; 5-{(3S)-3-[(3'-гидроксибифенил-3-ил)метокси]пирролидин-1-ил}-5-метил-2,2-дифенилгексанамид; 5-{(3S)-3-[(4'-гидроксибифенил-3-ил)метокси]пирролидин-1-ил}-5-метил-2,2-дифенилгексаннитрил; 5-{(3S)-3-[(4'-гидроксибифенил-3-ил)метокси]пирролидин-1-ил}-5-метил-2,2-дифенилгексанамид; 5-{(3S)-3-[(2'-гидроксибифенил-3-ил)метокси]пирролидин-1-ил}-5-метил-2,2-дифенилгексанамид; 5-{(3S)-3-[(3'-гидроксибифенил-4-ил)метокси]пирролидин-1-ил}-5-метил-2,2-дифенилгексанамид; 5-[(3S)-3-(бифенил-3-илметокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(4-фтор-3-гидроксибензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3S)-3-(3-циано-5-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3S)-3-(2-циано-5-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-{(3S)-3-[(7-гидрокси-2-нафтил)окси]пирролидин-1-ил}-5-метил-2,2-дифенилгексанамид; 5-[(3S)-3-(4-фенилфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(3-хлор-4-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(3-фтор-5-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(3-фтор-5-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(2-фтор-3-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(2-фтор-3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(2-хлор-3-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(2-хлор-3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(4-хлор-3-гидроксибензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(3-метокси-4-хлорфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; 5-[(3R)-3-(3-метокси-4-хлорфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(3-гидрокси-4-хлорфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(3-гидрокси-4-цианофенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3S)-3-(3-метоксибензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(2-хлор-3-гидроксибензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-метил-5-(4-феноксипиперидин-1-ил)-2,2-дифенилгексаннитрил; 5-{4-[(3-бромбензил)окси]пиперидин-1-ил}-5-метил-2,2-дифенилгексаннитрил; 5-{4-[(3-гидроксибензил)окси]пиперидин-1-ил}-5-метил-2,2-дифенилгексаннитрил; 5-[4-(бензилокси)пиперидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; 5-метил-5-(4-феноксипиперидин-1-ил)-2,2-дифенилгексанамид; 5-{4-[(3-бромбензил)окси]пиперидин-1-ил}-5-метил-2,2-дифенилгексанамид; 5-[4-(бензилокси)пиперидин-1-ил]-5-метил-2,2-дифенилгексанамид; амид 5-[4-(2,4-дихлор-5-гидроксифенокси)пиперидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[4-(4-циано-2,5-дифторфенокси)пиперидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; 5-[4-(3-гидроксифенокси)пиперидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[4-(3-гидрокси-2-метилфенокси)пиперидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-{4-[(3'-гидроксибифенил-3-ил)метокси]пиперидин-1-ил}-5-метил-2,2-дифенилгексаннитрил; 5-{4-[(3'-гидроксибифенил-3-ил)метокси]пиперидин-1-ил}-5-метил-2,2-дифенилгексанамид; 5-[3-(3-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; 5-[3-(3-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[3-(бензилокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; 5-[3-(бензилокси)азетидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-метил-5-(3-феноксиазетидин-1-ил)-2,2-дифенилгексаннитрил; 5-метил-5-(3-феноксиазетидин-1-ил)-2,2-дифенилгексанамид; 5-[3-(4-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; 5-[3-(4-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[3-(4-гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[3-(3-гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[3-(2-гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-{3-(2,4-дихлор-5-гидроксифенокси)азетидин-1-ил}-5-метил-2,2-дифенилгексанамид; 5-{3-(4,5-дихлор-2-гидроксифенокси)азетидин-1-ил}-5-метил-2,2-дифенилгексанамид; 5-[3-(4-хлор-3-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; 5-{3-(4-хлор-3-метоксифенокси)азетидин-1-ил}-5-метил-2,2-дифенилгексанамид; 5-{3-(4-хлор-3-гидроксифенокси)азетидин-1-ил}-5-метил-2,2-дифенилгексанамид; амид 5-[3-(3-гидроксибензилокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; 5-[3-(2-хлор-5-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; амид 5-[3-(2-хлор-5-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[3-(2-хлор-5-гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; 5-[3-(3-фтор-5-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; 5-[3-(3-фтор-5-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-{3-(3-фтор-5-гидроксифенокси)азетидин-1-ил}-5-метил-2,2-дифенилгексанамид; 5-[3-(3-хлор-5-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; 5-[3-(3-хлор-5-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-{3-(3-хлор-5-гидроксифенокси)азетидин-1-ил}-5-метил-2,2-дифенилгексанамид; 5-[3-(4-фтор-2-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; 5-[3-(4-фтор-2-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-{3-(4-фтор-2-гидроксифенокси)азетидин-1-ил}-5-метил-2,2-дифенилгексанамид; амид 5-[3-(2,6-дихлор-3-гидроксибензилокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; 4-{1-[3-(3-метоксифенокси)азетидин-1-ил]циклопентил}-2,2-дифенилбутиронитрил; 4-{1-[3-(3-метоксифенокси)азетидин-1-ил]циклопентил}-2,2-дифенилбутирамид; 4-{1-[3-(3-гидроксифенокси)азетидин-1-ил]циклопентил}-2,2-дифенилбутирамид; 5-[3-(2-фтор-3-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; амид 5-[3-(2-фтор-3-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[3-(2-фтор-3-гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; 5-[3-(2-фтор-5-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; амид 5-[3-(2-фтор-5-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[3-(2-фтор-5-гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[3-(4-хлор-3-гидроксибензилокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; 4-{1-[3-(4-хлор-3-гидроксифенокси)азетидин-1-ил]циклопентил}-2,2-дифенилбутирамид; 5-[3-(3-бром-5-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; амид 5-[3-(3-бром-5-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[3-(3-бром-5-гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; 4-{1-[3-(3-фтор-4-метоксифенокси)азетидин-1-ил]циклопентил}-2,2-дифенилбутиронитрил; 4-{1-[3-(3-фтор-4-метоксифенокси)азетидин-1-ил]циклопентил}-2,2-дифенилбутирамид; 4-{1-[3-(3-фтор-4-гидроксифенокси)азетидин-1-ил]циклопентил}-2,2-дифенилбутирамид; амид 5-[3-(3-хлор-4-гидроксибензилокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[3-(4-хлор-2-гидроксибензилокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[3-(2-хлор-3-гидроксибензилокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[3-(3,5-дигидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; 5-[3-(3-гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил; 5-{3-[(4-гидроксибензил)окси]азетидин-1-ил}-5-метил-2,2-дифенилгексаннитрил и амид 5-[3-(4-гидроксибензилокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Более предпочтительными соединениями являются 5-метил-5-[(3S)-3-феноксипирролидин-1-ил]-2,2-дифенилгексанамид; 5-метил-5-[(3R)-3-феноксипирролидин-1-ил]-2,2-дифенилгексанамид; 5-[(3S)-3-(3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; амид 5-[(3S)-3-(3-фтор-5-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[(3S)-3-(2-фтор-3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; 5-[(3R)-3-(2-фтор-3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(2-хлор-3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-метил-5-(4-феноксипиперидин-1-ил)-2,2-дифенилгексанамид; 5-[4-(3-гидроксифенокси)пиперидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-метил-5-(3-феноксиазетидин-1-ил)-2,2-дифенилгексанамид; 5-[3-(3-гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-{3-(4-хлор-3-гидроксифенокси)азетидин-1-ил}-5-метил-2,2-дифенилгексанамид; 5-{3-(3-фтор-5-гидроксифенокси)азетидин-1-ил}-5-метил-2,2-дифенилгексанамид; 5-{3-(3-хлор-5-гидроксифенокси)азетидин-1-ил}-5-метил-2,2-дифенилгексанамид; 4-{1-[3-(3-гидроксифенокси)азетидин-1-ил]циклопентил}-2,2-дифенилбутирамид; амид 5-[3-(2-фтор-3-гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[3-(2-фтор-5-гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты и амид 5-[3-(4-хлор-3-гидроксибензилокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Наиболее предпочтительными соединениями являются 5-[(3S)-3-(3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[(3R)-3-(2-фтор-3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-[3-(3-гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексанамид; 5-{3-(4-хлор-3-гидроксифенокси)азетидин-1-ил}-5-метил-2,2-дифенилгексанамид; 5-{3-(3-фтор-5-гидроксифенокси)азетидин-1-ил}-5-метил-2,2-дифенилгексанамид; 5-{3-(3-хлор-5-гидроксифенокси)азетидин-1-ил}-5-метил-2,2-дифенилгексанамид; 4-{1-[3-(3-гидроксифенокси)азетидин-1-ил]циклопентил}-2,2-дифенилбутирамид; амид 5-[3-(2-фтор-3-гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты; амид 5-[3-(2-фтор-5-гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты и амид 5-[3-(4-хлор-3-гидроксибензилокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Изобретение также относится к способам получения соединений формулы (I), a также к промежуточным соединениям, используемым для их получения. В частности, изобретение относится к указанным ниже промежуточным соединениям где А, р и А 1 такие, как определено в соединениях формулы (I); где А и R 1 такие, как определено в соединениях формулы (I); где R 1 , R 2 и R 3 такие, как определено в соединениях формулы (I), a LG является подходящей уходящей группой, такой как мезилат или тозилат; где LG является подходящей уходящей группой, такой как мезилат или тозилат; и где R 2 и R 3 такие, как определено в соединениях формулы (I). Соединение формулы (I) может быть получено разными способами. Приведенные ниже пути иллюстрируют один из таких способов получения этих соединений. Специалисту должно быть ясно, что для практического применения равным образом могут быть использованы другие способы. Соединения формулы (I), где А представляет собой или могут быть получены согласно описанным ниже путям. где А представляет собой или А 1 и р такие, как определено выше для соединений формулы (I), и PG является подходящей защитной группой для карбоксила, такой как метил или трет-бутил, и обычно является трет-бутилом. Соединение формулы (II) коммерчески доступно. Соединения формулы (III) либо коммерчески доступны, либо их получение известно из литературы. Соединения формулы (IV) могут быть получены из соединений формулы (II) и (III) в результате осуществления стадии (I) способа: соединение (II) обрабатывают соединением (III) в присутствии подходящего основания, такого как гидроксид калия или гидроксид натрия, в подходящем растворителе, таком как метанол, этанол или трет-бутанол, при температуре от 25 °С до повышенной температуры в течение 6-24 ч. Типичные условия включают 1,0 экв. соединения (II), 0,05 экв. гидроксида калия и 1,0 экв. соединения (III) в трет-бутаноле при температуре 25-60 °С в течение до 24 ч включительно. Соединения формулы (V) могут быть получены из соединений формулы (IV) в результате осуществления стадии (2) способа. Удаления защиты с соединения (IV) можно добиться, используя стандартную методологию, которая описана T.W. Greene и P. Wutz в "Protecting Groups in Organic Synthesis". Когда PG является трет-бутилом, типичные условия включают: 1,0 экв. соединения (IV) в присутствии соляной кислоты (4М в диоксане) в дихлорметане при комнатной температуре в течение до 18 ч включительно. Соединение формулы (VI) может быть получено, как указано на схеме 2. Соединения формулы (VIa) либо коммерчески доступны, либо могут быть получены, как представлено на схеме 2а. где А 1 и р такие, как определено выше для соединений формулы (I). В некоторых случаях А 1 может быть защищен подходящей защитной группой. Например, когда А 1 содержит фенол, тогда он может быть защищен подходящей защитной группой для гидроксила. PG' является подходящей защитной группой для амино, такой как трет-бутоксикарбонил (ВОС), бензилоксикарбонил (CBz), и обычно представляет собой BOC. X является подходящей функциональной группой, такой как гидрокси, фторо, бромо, хлоро, йодо, О-мезилат или О-тозилат, и обычно представляет собой гидрокси или бромо. Если PG' представляет собой ВОС, соединения формулы (VIII) и (VIIIa) являются коммерчески доступными соединениями. Соединения общей формулы (IX) либо коммерчески доступны, либо известны в литературе, либо могут быть получены, как показано на схемах 3-5. где А 1 такой, как определено выше для соединений формулы (I), или возможно может быть защищенным. Соединения формулы (IX), где А 1 такой, как определено для соединений формулы (I), р равно 0 и X представляет собой ОН, могут быть получены из соединений формулы (XI) в результате окисления по Байеру-Виллигеру и последующего гидролиза (стадия (8) способа). Типичные условия включают взаимодействие 1,0 экв. соединения (XI) с 3,0 экв. 3-хлорпероксибензойной кислоты в подходящем растворителе, таком как дихлорметан, при комнатной температуре в течение 18 ч и последующую обработку этого продукта подходящим основанием, таким как триэтиламин, в подходящем растворителе, таком как метанол, при комнатной температуре в течение 18 ч. Соединения формулы (IX), где А 1 содержит подходящим образом замещенный фенол, р равно 0 и X представляет собой ОН, могут быть получены из соединений формулы (IXa), где А 1 содержит фенол, р равно 0 и X представляет собой ОН, путем присоединения подходящей защитной группы для фенола, такой как бензил, в результате осуществления стадии (9) способа. Типичные условия включают взаимодействие 1,0 экв. соединения (IXa) с 1,0 экв. бензилбромида и 1,0 экв. подходящего основания, такого как карбонат цезия, в подходящем растворителе, таком как диметилформамид, при 80 °С в течение 30 мин. Альтернативно, соединения формулы (IX), где А 1 содержит подходящим образом замещенный фенол, р равно 0 и X представляет собой ОН, могут быть получены из соединений формулы (IXa), где А 1 содержит подходящим образом замещенный фенол, р равно 0 и X представляет собой ОМе, в результате удаления одной защитной группы, используя условия, описанные T.W. Greene и P. Wutz в "Protecting Groups in Organic Synthesis". Соединения формулы (IX), где А 1 содержит подходящим образом защищенный фенол, р равно 0 и X представляет собой F, могут быть получены из соединений формулы (IXa), где А 1 содержит фенол, р равно 0 и X представляет собой F, путем присоединения подходящей защитной группы для фенола, такой как метил, в результате осуществления стадии (9) способа. Типичные условия включают взаимодействие 1,0 экв. соединения (IXa) с 2,0 экв. метилйодида и 1,0 экв. подходящего основания, такого как карбонат калия, в подходящем растворителе, таком как тетрагидрофуран, при комнатной температуре в течение 3 ч. где А 1 такой, как определено выше для соединений формулы (I), или возможно может быть защищенным. Соединения формулы (IXa), где р равно 1 и X представляет собой ОН, могут быть получены из соединений формулы (XI) путем восстановления альдегида подходящим восстанавливающим агентом, таким как боргидрид натрия, в результате осуществления стадии (11) способа. Типичные условия включают взаимодействие 1,0 экв. соединения (XI) с 1,0 экв. боргидрида натрия в подходящем растворителе, таком как этанол, при комнатной температуре в течение 18 ч. Соединения формулы (XIV), где R 5 представляет собой подходящую защитную группу для кислоты, такую как метил, этил или аллил, могут быть получены из соединений формулы (XIII) путем присоединения подходящей защитной группы, такой как аллил, в результате осуществления стадии (10) способа. Типичные условия включают взаимодействие 1,0 экв. соединения (XIII) с 2,0 экв. аллилбромида и 2,0 экв. подходящего основания, такого как карбонат калия, в подходящем растворителе, таком как диметилформамид, при комнатной температуре в течение 18 ч. Соединения формулы (IXa), где р равно 1 и X представляет собой ОН, могут быть получены из соединений формулы (XIV) путем восстановления сложного эфира подходящим восстанавливающим агентом, таким как алюмогидрид лития, в результате осуществления стадии (11) способа. Типичные условия включают взаимодействие 1,0 экв. соединения (XIV) с 2,0 экв. алюмогидрида лития, в подходящем растворителе, таком как тетрагидрофуран, при температуре от 0 °С до комнатной температуры в течение 5 ч. Соединения формулы (IX), где р равно 1 и X представляет собой галогено, могут быть получены из соединений формулы (IXa), где р равно 1 и X представляет собой ОН, путем галогенирования первичного спирта с использованием подходящего реагента, такого как тионилхлорид, дибромтрифенилфосфоран или йод плюс трифенилфосфин, предпочтительно тионилхлорид или дибромтрифенилфосфоран, в подходящем растворителе, таком как дихлорметан или ацетонитрил, в результате осуществления стадии (12) способа. Когда X представляет собой бромо, типичные условия включают взаимодействие 1,0 экв. соединения (IXa) с 1,0 экв. дибромтрифенилфосфорана в подходящем растворителе, таком как ацетонитрил, при комнатной температуре в течение 18 ч. Когда X представляет собой хлоро, типичные условия включают взаимодействие 1,0 экв. соединения (IXa) с 2,5 экв. тионилхлорида в дихлорметане при комнатной температуре в течение 10 мин. Соединения формулы (X), где р равно 0, могут быть получены из соединений формул (VIII) и (IX), где X представляет собой ОН и р равно 0, в результате реакции Мицунобу между соединениями (VIII) и (IX) в присутствии подходящего фосфина, такого как три-н-бутилфосфин или трифенилфосфин, и подходящего азосоединения, такого как диэтилазодикарбоксилат, диизопропилазодикарбоксилат или ди-трет-бутилазодикарбоксилат, в растворителе, таком как дихлорметан, тетрагидрофуран или N,N-диметилформамид, при температуре 25-115 °С в течение 1-48 ч в результате осуществления стадии (6) способа. Типичные условия включают 1,0 экв. соединения (VIII), 1,0 экв. соединения (IX), 1,0-1,2 экв. трифенилфосфина и 1,0-1,2 экв. диизопропилазодикарбоксилата в тетрагидрофуране при 25 °С в течение до 18 ч включительно. Соединения формулы (Ха), где р равно 0, могут быть получены из соединений формулы (VIIIa) и (IX) в таких же условиях. Соединения формулы (X), где р равно 1, могут быть получены из соединений формул (VIII) и (IX), где X представляет собой галогено и р равно 1, путем обработки соединения (VIII) подходящим сильным основанием, таким как гидрид натрия или трет-бутилат калия, с последующим гашением соединением (IX) в подходящем растворителе, таком как тетрагидрофуран или диметилформамид, при температуре от 0 °С до комнатной температуры в течение 1-18 ч в результате осуществления стадии (6) способа. Типичные условия включают 1,0 экв. соединения (VIII), 1,0 экв. гидрида натрия и 1,0 экв. соединения (IX) в тетрагидрофуране при температуре 0-25 °С в течение до 18 ч включительно. Соединения формулы (Ха), где р равно 1, могут быть получены из соединений формулы (VIIIa) и (IX) в таких же условиях. Соединения формулы (VI) могут быть получены из соединений формулы (X) в результате осуществления стадии (7) способа. Удаления защиты с соединения (X) можно добиться, используя стандартную методологию, которая описана T.W. Greene и P. Wutz в "Protecting Groups in Organic Synthesis". Когда PG' представляет собой ВОС, типичные условия включают 1,0 экв. соединения (X) в присутствии соляной кислоты (4M в диоксане) в дихлорметане при комнатной температуре в течение до 18 ч включительно. Соединения формулы (VIa) можно получить из соединений формулы (Ха) и используя те же условия. Соединения формулы (VII) могут быть получены из соединений формул (V) и (VI) или (VIa) в результате осуществления стадии (3) способа проведением реакции сочетания (V) и (VI) или (VIa) в присутствии подходящего агента сочетания, такого как 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид, N,N'-карбонилдиимидазол, N,N'-дициклогексилкарбодиимид, возможно в присутствии катализатора, такого как 1-гидроксибензотриазола гидрат или 1-гидрокси-7-азабензотриазол, и возможно в присутствии основания, представляющего собой третичный амин, такой как N-метилморфолин, триэтиламин или N,N-диизопропилэтиламин, в подходящем растворителе, таком как N,N-диметилформамид, тетрагидрофуран или диметилсульфоксид, в условиях окружающей среды в течение 1-48 ч. Типичные условия включают 1,0 экв. соединения (V), 1,0 экв. соединения (VI) или (VIa) и 1,0-1,2 экв. 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида, 1,0-1,2 экв. 1-гидроксибензотриазола гидрата и 1,0-1,2 экв. триэтиламина в дихлорметане при комнатной температуре в течение 18 ч. Соединения формулы (Ia) могут быть получены из соединений формулы (VII) в результате осуществления стадии (4) способа. Соединение (Ia) может быть получено по аналогии со способами Дентона (Denton) и Вуда (Wood) (Synlett, 1999, 1, 55). Соединение (VII) обычно предварительно активируют подходящей кислотой Льюиса, такой как хлорид титана или хлорид циркония, затем обрабатывают избытком подходящего металлоорганического реагента, такого как MeMgCl или MeMgBr, в подходящем растворителе, таком как тетрагидрофуран или диэтиловый эфир, при температуре от -78 до 25 °С в течение 1-18 ч. Типичные условия включают 1,0 экв. соединения (VII), 2 экв. хлорида циркония и 9,0 экв. MeMgCl в тетрагидрофуране при -30 °С в течение 4-8 ч. Соединения формулы (Ib) могут быть получены из соединений формулы (Ia) в результате гидролиза соединения (Ia) избытком гидроксида калия в 3-метил-3-пентаноле при повышенной температуре в течение до 24 ч включительно (стадия (5) способа). Типичные условия включают 1,0 экв. соединения (Ia) и 20 экв. гидроксида калия в 3-метил-3-пентаноле при повышенной температуре в течение до 24 ч включительно. В еще одном воплощении, когда р равно 1, соединения формулы (I) могут быть дополнительно функционализированы с получением соединений формулы (Ic), как показано на схеме 6. где А представляет собой или и R 2 и R 3 представляют собой метил. Соединения формулы (XV) могут быть получены из соединений формулы (I), где р равно 1, в результате осуществления стадии (13) способа. Когда R 1 представляет собой CN, отщепление группы простого бензилового эфира обычно достигается путем обработки соединения (I) избытком хлорида железа(III) в подходящем растворителе, таком как дихлорметан, в условиях окружающей среды в течение 1-8 ч с получением соединений формулы (XV). Когда R 1 представляет собой CONH 2 , в результате обработки соединения (I) газообразным водородом в присутствии подходящего катализатора гидрирования, такого как 20% Pd(OH) 2 или 10% Pd/C, возможно в присутствии подходящей кислоты, такой как соляная кислота, в подходящем растворителе, таком как метанол, этанол или тетрагидрофуран, при повышенной температуре в течение 1-18 ч образуется соединение формулы (XV). Соединения формулы (Ic), где р равно 0, могут быть получены из соединений формулы (XV) и соединений формулы (IX), где р равно 0 и X представляет собой ОН, в результате осуществления стадии (14) способа в условиях, указанных для стадии (6). Соединения формулы (Ic), где р равно 0, могут быть получены из соединений формулы (XV) и соединения формулы (IX), где р равно 0 и X представляет собой F, в результате осуществления стадии (14) способа путем обработки соединения (XV) подходящим сильным основанием, таким как гидрид натрия или трет-бутилат калия, с последующим гашением соединением (IX) в подходящем растворителе, таком как тетрагидрофуран или N,N-диметилформамид, при температуре от 0 °С до повышенной температуры в течение 1-96 ч. Типичные условия включают 1,0 экв. соединения (XV), 1,0-2,0 экв. гидрида натрия и 1,0 экв. соединения (IX) в N,N-диметилформамиде при температурах 0-60 °С в течение 18-96 ч. Соединения формулы (Ic), где р равно 1, могут быть получены из соединений формулы (XV) и соединения формулы (IX), где р равно 1 и X представляет собой Cl, Br, I, О-мезилат или О-тозилат, в результате осуществления стадии (14) способа путем обработки соединения (XV) подходящим сильным основанием, таким как гидрид натрия или трет-бутилат калия, с последующим гашением соединением (IX) в подходящем растворителе, таком как тетрагидрофуран или N,N-диметилформамид, при температуре от 0 °С до повышенной температуры в течение 1-96 ч. Типичные условия включают 1,0 экв. соединения (XV), 1,0-2,0 экв. гидрида натрия и 1,0 экв. соединения (IX) в N,N-диметилформамиде при температуре 0-60 °С в течение 18-96 ч. В еще одном воплощении соединения формулы (I) могут быть также функционализированы с получением соединений формулы (Id), как описано на схеме 7. где А представляет собой или Соединения формулы (Id), где А 1 представляет собой фенил, замещенный фенилом, возможно замещенным ОН, могут быть получены из соединений формулы (I), где А 1 представляет собой фенил, замещенный Cl, Br или I, на стадии (15) способа в результате реакции сочетания Сузуки с соединением (XXX) в подходящем растворителе, таком как 1,4-диоксан или тетрагидрофуран, в присутствии воды, подходящего основания, такого как карбонат натрия или карбонат цезия, и палладиевого катализатора, такого как [1,1'-бис(дифенилфосфино)ферроцен]палладия(II) хлорид или тетракис(трифенилфосфин) палладий(0). Реакции сочетания Сузуки можно проводить, как описано в литературе: Suzuki, A. Pure & Appl. Chem. 1985, 57, 1749 и содержащихся там ссылках; Angew. Chem. Int. Ed. 2002, 41, 4176 и содержащихся там ссылках. Типичные условия включают 1,0 экв. соединения (I), 2,0 экв. соединения (XXX), 2,0 экв. карбоната натрия и 0,05 экв. палладиевого катализатора в тетрагидрофуране и воде при повышенной температуре в течение до 16 ч включительно. Соединение (XVI) либо коммерчески доступно, либо может быть получено способами, описанными в литературе. Соединения формулы (Ic) и (Id), где R 1 представляет собой CONH 2 , также могут быть получены из соединений формулы (Ic) и (Id), где R 1 представляет собой CN, в результате гидролиза избытком гидроксида калия в 3-метил-3-пентаноле при повышенной температуре в течение до 24 ч включительно. Типичные условия включают 1,0 экв. соединения (Ic) или (Id), где R 1 представляет собой CN, и 20 экв. гидроксида калия в 3-метил-3-пентаноле при температуре дефлегмации в течение до 24 ч включительно. Соединения формулы (I), где А представляет собой могут быть получены описанными ниже путями. где LG является подходящей уходящей группой, такой как мезилат или тозилат, и предпочтительно представляет собой мезилат. Соединения формулы (XVI) могут быть получены, как описано в WO 2003037327, стр. 83. PG'" является защитной группой, такой как трет-бутоксикарбонил или бензилоксикарбонил, и предпочтительно представляет собой трет-бутоксикарбонил. Альтернативно соединения формулы (XVI) могут быть получены согласно следующему способу: Соединения формулы (XVIc) коммерчески доступны или известны в литературе. Соединения формулы (XVIb) могут быть получены из соединений формулы (XVIc) путем взаимодействия соединений (XVIc) с хлорсульфонилизоцианатом, муравьиной кислотой и пиридином в подходящем растворителе, таком как дихлорметан, при пониженной температуре в течение 2 ч (стадия (21) способа). Типичные условия включают 1,0 экв. соединения (XVIc), 1,5 экв. хлорсульфонилизоцианата, 1,5 экв. муравьиной кислоты и 1,5 экв. пиридина в дихлорметане при пониженной температуре в течение 2 ч. Соединения формулы (XVIa) могут быть получены из соединений формулы (XVIb) путем взаимодействия соединений (XVIb) с оксидом магния, диацетатом йодбензола и димером ацетата родия в подходящем растворителе, таком как дихлорметан, при комнатной температуре в течение до 24 ч включительно (стадия (22) способа). Типичные условия включают взаимодействие 1,0 экв. соединения (XVIb), 2,3 экв. диоксида магния, 1,1 экв. диацетата йодбензола и 0,02 экв. димера ацетата родия в дихлорметане при комнатной температуре в течение 18 ч. Соединения формулы (XVI) могут быть получены из соединений формулы (XVIa) путем введения подходящей защитной группы, такой как трет-бутоксикарбонил или бензилоксикарбонил, предпочтительно трет-бутоксикарбонил, в условиях, описанных Т.W. Greene и P. Wutz в "Protecting Groups in Organic Synthesis". Типичные условия включают взаимодействие 1,0 экв. соединения (XVIa), 1,2 экв. ди-трет-бутилдикарбоната, 2,0 экв. триэтиламина и 0,2 экв. 4-диметиламинопиридина в дихлорметане при комнатной температуре в течение 3 ч. Соединения формулы (XVII) могут быть получены из соединений формулы (II) и соединений формулы (XVI) в результате осуществления стадии (16) способа. 1) Взаимодействие соединений (II) и (XVI) в присутствии сильного основания, такого как трет-бутилат калия или гидрид натрия, в подходящем растворителе, таком как N,N-диметилформамид или диметилсульфоксид, в условиях окружающей среды или при повышенной температуре в течение до 18 ч включительно. 2) Удаление защитной группы (если она использована) в подходящих условиях, таких как 4н. соляная кислота в диоксане или трифторуксусная кислота, или гидрирование в присутствии каталитического палладия, как описано T.W. Greene и Р. Wutz в "Protecting Groups in Organic Synthesis". Типичные условия включают 1,2 экв. соединения (II), 1,0 экв. соединения (XVI) и 1,2 экв. трет-бутилата калия в N,N-диметилформамиде в условиях окружающей среды в течение до 18 ч включительно с последующей обработкой 4н. соляной кислотой в диоксане. Соединения формулы (XVIII) коммерчески доступны. Соединения формулы (XIX) могут быть получены из соединений формулы (XVII) и (XVIII) в результате осуществления стадии (17) способа, где образование гетероцикла может быть достигнуто путем нуклеофильного присоединения соединения (XVIII) к соединению (XVII) с последующей циклизацией in situ в подходящем растворителе, таком как метанол или этанол, при повышенной температуре в течение до 48 ч включительно. Типичные условия включают 1,0 экв. соединения (XVII) и 1,1 экв. соединения (XVIII) в метаноле при повышенной температуре в течение до 48 ч включительно. Соединения формулы (XX) могут быть получены из соединений формулы (XIX) в результате осуществления стадии (18) способа получения путем введения подходящей уходящей группы (LG), такой как мезилатные или тозилатные группы, путем взаимодействия соединения (XIX) с мезил-хлоридом/ангидридом или тозилхлоридом в присутствии подходящего основания, такого как основание Хюнига, триэтиламин или пиридин, возможно в подходящем растворителе, таком как дихлорметан или диэтиловый эфир, при пониженной температуре в течение 1-2 ч. Типичные условия включают 1,0 экв. соединения (XIX) и 3 экв. мезилхлорида в пиридине при пониженной температуре в течение до 1-2 ч включительно. Соединения общей формулы (XXI) либо коммерчески доступны, либо известны в литературе. Соединения формулы (XXI), содержащие фенольную группу, могут быть защищены путем присоединения подходящей защитной группы для фенола, такой как аллил. Типичные условия включают взаимодействие 1,0 экв. соединения (XXI), содержащего фенол, с 1,0 экв. аллилбромида и 1,0 экв. подходящего основания, такого как карбонат калия, в подходящем растворителе, таком как диметилформамид, при комнатной температуре в течение 18 ч. Альтернативно, соединения формулы (XXI) могут быть получены в результате удаления одной защитной группы бис-защищенного фенола в условиях, описанных Т.W. Greene и P. Wutz в "Protecting Groups in Organic Synthesis". Соединения формулы (Ie) могут быть получены из соединений общей формулы (XX) и (XXI) путем обработки соединения (XXI) подходящим основанием, таким как карбонат цезия или карбонат натрия, с последующим гашением соединением (XX) в подходящем растворителе, таком как N,N-диметилформамид или диметилсульфоксид, при повышенной температуре в течение до 18 ч включительно (стадия (19) способа). Типичные условия включают 1,0 экв. соединения (XX), 3,0 экв. карбоната цезия и 3,0 экв. соединения (XXI) в N,N-диметилформамиде при повышенной температуре в течение до 18 ч включительно. В еще одном воплощении соединения формулы (If) могут быть получены из соединений формулы (Ie) в результате гидролиза соединения (Ie) избытком гидроксида калия в 3-метил-3-пентаноле при повышенной температуре в течение до 24 ч включительно (стадия (20) способа). Типичные условия включают 1,0 экв. соединения (Ie) и 20 экв. гидроксида калия в 3-метил-3-пентаноле при повышенной температуре в течение до 24 ч включительно. Альтернативно, соединения формулы (I), где А представляет собой и R 2 и R 3 представляют собой метил, могут быть получены, как показано на схеме 9. где LG является подходящей уходящей группой, такой как мезилат или тозилат, и предпочтительно представляет собой мезилат. Соединения общей формулы (V) могут быть получены, как описано в WO 97/24325. Соединения формулы (XXII) могут быть получены из соединений общей формулы (V) в результате осуществления стадии (24) способа: карбоновая кислота (V) может быть обработана подходящим хлорирующим агентом, таким как тионилхлорид или оксалилхлорид, в подходящем растворителе, таком как N,N-диметилформамид, ацетонитрил или дихлорметан, в условиях окружающей среды в течение до 8 ч включительно. Типичные условия включают 1,0 экв. соединения (V) и 2 экв. оксалилхлорида в N,N-диметилформамиде при комнатной температуре в течение 2 ч. Соединения формулы (XXIII) могут быть получены, как описано в J. Org. Chem. 1991, 56, 6729-30. Соединения общей формулы (XXIV) могут быть получены из соединений общей формулы (XXII) и (XXIII) в результате осуществления стадии (25) способа: соединение (XXII) подвергают нуклеофильному замещению соединением (XXIII) в присутствии основания, представляющего собой третичный амин, такой как N-метилморфолин, триэтиламин или N,N-диизопропилэтиламин, в подходящем растворителе, таком как N,N-диметилформамид, тетрагидрофуран или дихлорметан, при пониженной температуре в течение 1-8 ч. Типичные условия включают 1 экв. соединения (XXII), 1 экв. соединения (XXIII) и 3 экв. триэтиламина в дихлорметане при пониженной температуре в течение 1 ч. Соединения общей формулы (XXV) могут быть получены из соединений общих формул (XXIV) и (XXI) в результате осуществления стадии (26) способа в условиях, указанных для стадии (19) на схеме 8. Соединения формулы (Ie) могут быть получены из соединений формулы (XXV), используя условия, указанные для стадии (4) схемы 1. Соединения формулы (If) могут быть получены из соединений формулы (Ie), используя условия, указанные для стадии (20) схемы 8. Альтернативно, соединения формулы (I) могут быть получены, как показано на схеме 1, с использованием промежуточного соединения формулы (VIb) которое коммерчески доступно или известно в литературе. Типичные условия для стадии (3) включают 1,0 экв. соединения (V), 1,2 экв. соединения (VIb), 1,0-1,2 экв. 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида, 1,0-1,2 экв. 1-гидроксибензотриазола гидрата и 2,5 экв. N,N-диизопропилэтиламина в дихлорметане при комнатной температуре в течение 18 ч. Альтернативно, соединение формулы (Ie), где р равно 1, может быть получено, как показано на схеме 10. где р равно 1, a Hal представляет собой галогено и обычно представляет собой хлоро или бромо, предпочтительно хлоро. Соединение формулы (XXVI) может быть получено, как показано на схеме 5. Соединения общей формулы (Ie) могут быть получены из соединений общих формул (XIX) и (XXVI) путем обработки соединения (XIX) подходящим сильным основанием, таким как гидрид натрия или трет-бутилат калия, с последующим гашением соединением (XXVI) в подходящем растворителе, таком как тетрагидрофуран, при температуре от 0 °С до комнатной температуры в течение 1-8 ч. Типичные условия включают 1,0 экв. соединения (XIX), 1,2 экв. гидрида натрия и 1,5 экв. соединения (XXVI) в тетрагидрофуране при температурах 0-25 °С в течение до 1-2 ч включительно. Альтернативно, соединения формулы (If) могут быть получены, как показано на схеме 11. где Hal представляет собой галогено и обычно представляет собой хлоро или бромо, предпочтительно хлоро. Соединения формулы (XXVII) получают из соединений формулы (If), где А 1 представляет собой фенил и р равно 1, путем удаления бензильной группы в стандартных условиях гидрирования, которые описаны Т.W. Greene и P. Wutz в "Protecting Groups in Organic Synthesis". Соединения формулы (If) получают из соединений формулы (XXVII) и соединений формулы (XXVI) в условиях, указанных для стадии (29). Альтернативно, соединения формулы (If) могут быть получены, как показано на схеме 12. где LG является подходящей уходящей группой, такой как мезилат или тозилат, и предпочтительно представляет собой мезилат. Соединения формулы (XXVIII) могут быть получены из соединений формулы (XXVII) в результате осуществления стадии (18) способа, как показано на схеме 8. Соединения формулы (If) могут быть получены из соединений формулы (XXVIII) и соединений формулы (XXI) в результате осуществления стадии (19) способа, как показано на схеме 8. Когда А 1 содержит подходящим образом замещенный фенол, с соединений формулы (I) удаляют защиту с получением соответствующего фенола. Подходящие защитные группы (PG") включают метил, бензил, аллил и трет-бутилдиметилсилил (TBDMS). Удаления защиты можно достичь, используя стандартную методологию, которая описана T.W. Greene и P. Wutz в "Protecting Groups in Organic Synthesis". Когда PG" представляет собой метил, типичные условия этой методики включают 1,0 экв. защищенного соединения формулы (I) и 1-4 экв. 1М трибромида бора в дихлорметане в подходящем растворителе, таком как дихлорметан, при температуре окружающей среды в течение 1-18 ч. Когда PG" представляет собой аллил, типичные условия этой методики включают 1,0 экв. защищенного соединения формулы (I) и 20 экв. гидроксида калия в 3-метил-3-пентаноле при температуре дефлегмации в течение 1-24 ч с последующим выделением остатка и обработкой соляной кислотой (4М в диоксане), в воде, при 60 °С в течение 20 мин. Альтернативные условия этой методики включают 1,0 экв. защищенного соединения формулы (I), 6 экв. боргидрида натрия и 0,1 экв. тетракис(трифенилфосфин)палладия(0) в тетрагидрофуране при повышенной температуре в течение 30 мин. Когда PG" представляет собой TBDMS, типичные условия этой методики включают 1,0 экв. защищенного соединения формулы (I) и 10 экв. фторида аммония в метаноле и воде при 50 °С в течение 18-24 ч. Фармацевтически приемлемые соли соединений формулы (I) включают их соли присоединения кислот и соли с основаниями. Подходящие соли присоединения кислот образуются из кислот, которые образуют нетоксичные соли. Примеры включают следующие соли: ацетат, адипат, аспартат, бензоат, безилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камзилат, цитрат, цикламат, эдизилат, эзилат, формиат, фумарат, глюцептат, глюконат, глюкуронат, гексафторфосфат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидройодид/йодид, изетионат, лактат, малат, малеат, малонат, мезилат, метилсульфат, нафтилат, 2-напсилат, никотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, пироглутамат, сахарат, стеарат, сукцинат, таннат, тартрат, тозилат, трифторацетат и ксинафоат. Подходящие соли с основаниями образуются из оснований, которые образуют нетоксичные соли. Примеры включают соли алюминия, аргинина, бензатина, кальция, холина, диэтиламина, диоламина, глицина, лизина, магния, меглумина, оламина, калия, натрия, трометамина и цинка. Также могут быть образованы гемисоли кислот и оснований, например гемисульфатная и гемикальциевая соли. Обзор подходящих солей см. в Stahl and Wermuth, Handbook of Pharmaceutical Salts: Properties, Selection, and Use (Wiley-VCH, 2002). Фармацевтически приемлемые соли соединений формулы (I) могут быть получены одним или более чем одним из трех способов: (1) путем осуществления взаимодействия соединения формулы (I) с желаемой(ым) кислотой или основанием; (2) путем удаления лабильной по отношению к кислоте или основанию защитной группы из подходящего предшественника соединения формулы (I) или в результате раскрытия кольца подходящего циклического предшественника, например лактона или лактама, с использованием желаемой(ого) кислоты или основания; или (3) путем превращения одной соли соединения формулы (I) в другую в результате взаимодействия с соответствующей(им) кислотой или основанием или с помощью подходящей ионообменной колонки. Все три взаимодействия обычно проводят в растворе. Полученную соль можно осадить и собрать фильтрованием или можно выделить выпариванием растворителя. Степень ионизации полученной соли может меняться от полностью ионизированного до почти неионизированного. Соединения по изобретению могут существовать в континууме твердых состояний в диапазоне от полностью аморфного до полностью кристаллического. Термин "аморфный" относится к состоянию, в котором вещество на молекулярном уровне не имеет дальнего порядка и в зависимости от температуры может проявлять физические свойства твердого тела или жидкости. Обычно такие вещества не дают характерных картин дифракции рентгеновских лучей, и хотя они проявляют свойства твердого тела, их формально описывают скорее как жидкость. При нагревании происходит изменение свойств от твердого тела до жидкости, которое характеризуется изменением состояния, обычно второго порядка ("стеклование"). Термин "кристаллический" относится к твердой фазе, в которой вещество на молекулярном уровне имеет регулярную упорядоченную внутреннюю структуру и дает характерную картину дифракции рентгеновских лучей с определенными пиками. Если нагреть такие вещества в достаточной степени, то они также будут демонстрировать свойства жидкости, но изменение состояния с твердого в жидкое характеризуется фазовым переходом, обычно первого порядка ("точка плавления"). Соединения по изобретению также могут существовать в несольватированной и в сольватированной формах. Термин "сольват" использован здесь для описания молекулярного комплекса, содержащего соединение по изобретению и одного или более чем одну молекулу фармацевтически приемлемого растворителя, например этанола. Термин "гидрат" используется, когда указанным растворителем является вода. Одной из принятых в настоящее время систем классификации для органических гидратов является система, которая дает определение гидратов по изолированным центрам, канальных гидратов или гидратов, координированных с ионом металла (см. K.R. Morris, Polymorphism in Pharmaceutical Solids (Ed. H. G. Brittain, Marcel Dekker, 1995)). Гидратами по изолированным центрам являются гидраты, в которых молекулы воды изолированы от непосредственного контакта с каждой другой молекулой воды находящимися между ними органическими молекулами. В канальных гидратах молекулы воды расположены в каналах решетки, где они соседствуют с другими молекулами воды. В гидратах, координированных с ионом металла, молекулы воды связаны с этим ионом металла. Если растворитель или вода связан(а) прочно, то комплекс будет иметь вполне определенную стехиометрию независимо от влажности. Однако если растворитель или вода связан(а) слабо, как в канальных сольватах и гигроскопических соединениях, то содержание воды/растворителя будет зависеть от влажности и условий сушки. В таких случаях нормой будет отсутствие стехиометрии. В объем изобретения также входят многокомпонентные комплексы (отличающиеся от солей и сольватов), где лекарственное средство и по меньшей мере один другой компонент присутствуют в стехиометрических или нестехиометрических количествах. Комплексы этого типа включают клатраты (комплексы включения лекарственное средство-хозяин) и сокристаллы. Последних обычно определяют как кристаллические комплексы нейтральных молекулярных составляющих, которые связаны вместе посредством нековалентных взаимодействий, но которые также могут представлять собой комплекс нейтральной молекулы с солью. Сокристаллы могут быть получены путем кристаллизации из расплава, путем перекристаллизации из растворителей или путем совместного физического измельчения компонентов (см. О. Almarsson and M.J. Zaworotko, Chem. Commun., 17, 1889-1896 (2004)). Общий обзор таких многокомпонентных комплексов см. в Haleblian, J. Pharm. Sci., 64 (8), 1269-1288 (August 1975). В подходящих условиях соединения по изобретению также могут существовать в мезоморфном состоянии (мезофаза или жидкий кристалл). Мезоморфное состояние является промежуточным состоянием между истинным кристаллическим состоянием и истинным жидким состоянием (либо расплав, либо раствор). Мезоморфизм, возникающий в результате изменения температуры, характеризуется как "термотропный", а мезоморфизм, возникающий в результате добавления второго компонента, такого как вода или другой растворитель, характеризуется как "лиотропный". Соединения, способные образовывать лиотропные мезофазы, характеризуются как "амфифильные" и состоят из молекул, которые обладают ионной (например -COO - Na + , -COO - K + или -SO 3 - Na + ) или неионной (например -N - N + (СН 3 ) 3 ) полярной головной группой. Дополнительную информацию см. в N.Н. Hartshorne and A. Stuart, Crystals and the Polarizing Microscope, 4 th Edition (Edward Arnold, 1970). Далее все ссылки на соединения формулы (I) включают ссылки на их соли, сольваты, многокомпонентные комплексы и жидкие кристаллы и на сольваты, многокомпонентные комплексы и жидкие кристаллы их солей. Соединения по изобретению включают соединения формулы (I), которые определены здесь выше, в том числе все их полиморфы и кристаллические формы, их пролекарства и изомеры (включая оптические, геометрические и таутомерные изомеры), которые определены выше, и меченные изотопом соединения формулы (I). Как указано, так называемые "пролекарства" соединений формулы (I) также входят в объем изобретения. Так, некоторые производные соединений формулы (I), которые сами могут иметь незначительную фармакологическую активность или не иметь никакой активности, могут при введении в организм или на тело превращаться в соединения формулы (I), имеющие требуемую активность, например, в результате гидролитического расщепления. Такие производные называются "пролекарствами". Дополнительную информацию по использованию пролекарств можно найти в Pro-drugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T. Higuchi and W. Stella) и Bioreversible Carriers in Drug Design, Pergamon Press, 1987 (Ed. E.B. Roche, American Pharmaceutical Association). Пролекарства по изобретению могут быть получены, например, путем замещения соответствующих функциональных групп, присутствующих в соединениях формулы (I), определенными группировками, известными специалистам в данной области как "прогруппировки", как описано, например, в Н. Bundgaard Design of Prodrugs (Elsevier, 1985). Некоторые примеры пролекарств по изобретению включают: (1) если соединение формулы (I) содержит функциональную группу карбоновой кислоты (-СООН), то его сложный эфир, например соединение, где водород функциональной группы карбоновой кислоты соединения формулы (I) замещен (С 1 -С 8 )алкилом; (2) если соединение формулы (I) содержит спиртовую функциональную группу (-ОН), то его простой эфир, например соединение, где водород спиртовой функциональной группы соединения формулы (I) замещен (C 1 -С 6 )алканоилоксиметилом; и (3) если соединение формулы (I) содержит функциональную первичную или вторичную аминогруппу (-NH 2 или -NHR, где R не представляет собой Н), то его амид, например соединение, где, в зависимости от обстоятельств, один или оба водорода функциональной аминогруппы соединения формулы (I) замещен/замещены (С 1 -С 10 )алканоилом. Дополнительные примеры замещающих групп в соответствии с вышеизложенными примерами и примеры других типов пролекарств могут быть найдены в вышеупомянутых ссылках. Более того, некоторые соединения формулы (I) сами могут действовать как пролекарства других соединений формулы (I). В объем изобретения также входят метаболиты соединений формулы (I), то есть соединения, образовавшиеся in vivo после введения лекарственного средства. Некоторые примеры метаболитов по изобретению включают: (1) если соединение формулы (I) содержит метильную группу, то его гидроксиметильное производное (-СН 3 → -СН 2 ОН); (2) если соединение формулы (I) содержит алкоксигруппу, то его гидроксипроизводное (-OR → -ОН); (3) если соединение формулы (I) содержит третичную аминогруппу, то его производное, представляющее собой вторичный амин (-NR 1 R 2 → -NHR 1 или -NHR 2 ); (4) если соединение формулы (I) содержит вторичную аминогруппу, то его производное, представляющее собой первичный амин (-NHR 1 → -NH 2 ); (5) если соединение формулы (I) содержит фенильную группу, то его фенольное производное (-Ph → -PhOH); и (6) если соединение формулы (I) содержит амидную группу, то его производное, представляющее собой карбоновую кислоту (-CONH 2 → -СООН). Соединения формулы (I), содержащие один или более асимметрических атомов углерода, могут существовать в форме двух или более стереоизомеров. Если соединение формулы (I) содержит алкенильную или алкениленовую группу, то возможны геометрические цис/транс (или Z/E) изомеры. Если структурные изомеры способны взаимопревращаться через низкий энергетический барьер, то может иметь место таутомерная изомерия ("таутомерия"). Она может принимать форму протонной таутомерии в соединениях формулы (I), содержащих, например, имино, кето или оксимную группу, или так называемой валентной таутомерии в соединениях, которые содержат ароматическую группировку. Следовательно, одно соединение может проявлять больше одного типа изомерии. В объем настоящего изобретения входят все стереоизомеры, геометрические изомеры и таутомерные формы соединений формулы I, в том числе соединения, проявляющие больше одного типа изомерии, и смеси одного или более из них. В объем изобретения также входят соли присоединения кислот или соли с основаниями, где противоион является оптически активным, например d-лактат или l-лизин, или рацемическим, например dl-тартрат или dl-аргинин. цис/транс-Изомеры могут быть разделены общепринятыми методами, хорошо известными специалистам в данной области, например хроматографией и фракционной кристаллизацией. Общепринятые методы получения/выделения индивидуальных энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника или разделение рацемата (или рацемата соли или производного) с использованием, например, хиральной жидкостной хроматографии высокого давления (ЖХВД). Альтернативно, рацемат (или рацемический предшественник) может быть подвергнут взаимодействию с подходящим оптически активным соединением, например спиртом или, если соединение формулы (I) содержит кислотную или основную группировку, основанием или кислотой, такими как 1-фенилэтиламин или винная кислота. Полученная диастереомерная смесь может быть разделена хроматографией и/или фракционной кристаллизацией, и один или оба диастереоизомера могут быть превращены в соответствующий чистый(ые) энантиомер(ы) способами, которые хорошо знает специалист. Хиральные соединения по изобретению (и их хиральные предшественники) могут быть получены в обогащенной энантиомером форме с использованием хроматографии, обычно ЖХВД, на асимметрической смоле с подвижной фазой, состоящей из углеводорода, обычно гептана или гексана, содержащего 0-50 об.% изопропанола, обычно 2-20% и 0-5 об.% алкиламина, обычно 0,1% диэтиламина. Концентрирование элюата дает обогащенную смесь. При кристаллизации любого рацемата возможно получение кристаллов двух разных типов. Первый тип представляет собой упомянутое выше рацемическое соединение (истинный рацемат), когда образуется одна гомогенная форма кристалла, содержащего оба энантиомера в эквимолярных количествах. Второй тип представляет собой рацемическую смесь или конгломерат, когда образуются две формы кристалла в эквимолярных количествах, каждая из которых содержит единственный энантиомер. Несмотря на то, что обе кристаллические формы, присутствующие в рацемической смеси, имеют идентичные физические свойства, их физические свойства по сравнению с истинным рацематом могут различаться. Рацемические смеси могут быть разделены общепринятыми методами, известными специалистам в данной области (см., например, Е.L. Eliel and S.H. Wilen, Stereochemistry of Organic Compounds (Wiley, 1994)). Настоящее изобретение охватывает все фармацевтически приемлемые меченные изотопом соединения формулы (I), где один или более атомов заменены атомами, имеющими такой же атомный номер, но атомная масса или массовое число которых отличается от атомной массы или массового числа, преобладающей(его) в природе. Примеры изотопов, подходящих для введения в соединения по изобретению, включают изотопы водорода, такие как 2 Н и 3 Н, углерода, такие как 11 С, 13 С и 14 С, хлора, такие как 36 Cl, фтора, такие как 18 F, йода, такие как 123 I и 125 I, азота, такие как 13 N и 15 N, кислорода, такие как 15 О, 17 О и 18 О, фосфора, такие как 32 Р, и серы, такие как 35 S. Некоторые меченные изотопами соединения формулы I, например соединения, включающие в себя радиоактивный изотоп, полезны в исследованиях по распределению лекарственного средства и/или субстрата в тканях. Радиоактивные изотопы тритий, то есть Н, и углерод-14, то есть 14 С, особенно полезны для этой цели ввиду легкости их введения и наличия готовых средств детектирования. Замещение более тяжелыми изотопами, такими как дейтерий, то есть 2 Н, может дать некоторые терапевтические преимущества, вытекающие из более высокой метаболической стабильности, например увеличение времени полувыведения in vivo и понижение требований по дозировке, и, следовательно, в некоторых случаях может быть предпочтительным. Замещение позитрон-излучающими изотопами, такими как 11 С, 18 F, 15 О и 13 N, может быть полезно в исследованиях методом позитронной эмиссионной томографии (PET) для проверки занятости рецепторов субстратом. В общем случае, меченные изотопами соединения формулы (I) могут быть получены общепринятыми методами, известными специалистам в данной области, или способами, аналогичными описанным с приведенных ниже примерах и подготовительных примерах, с использованием соответствующего меченного изотопом реагента вместо ранее используемого немеченого реагента. Фармацевтически приемлемые сольваты по изобретению включают сольваты, где растворитель для кристаллизации может быть замещенным изотопами, например D 2 O, d 6 -ацетон, d 6 -DMSO (диметилсульфоксид). В объем изобретения также входят промежуточные соединения формулы II, как определено выше, все их соли, сольваты и комплексы и все сольваты и комплексы их солей, как определено выше для соединений формулы I. Изобретение охватывает все полиморфы вышеупомянутых видов веществ и их кристаллические формы. При получении соединений формулы (I) по изобретению для специалиста в данной области остается открытой возможность рутинного выбора формы соединения формулы II, которая обеспечивает наилучшую комбинацию характеристик для этой цели. Такие характеристики включают точку плавления, растворимость, эксплуатационные характеристики и выход промежуточной формы и результирующую легкость, с которой продукт может быть очищен при его выделении. Для выбора наиболее подходящей лекарственной формы и пути введения для лечения предполагаемого показания, должны быть оценены биофармацевтические свойства соединений формулы (I), такие как растворимость и стабильность раствора (во всем диапазоне рН), проницаемость и т.д. Соединения по изобретению, предназначенные для фармацевтического применения, можно вводить в виде кристаллических или аморфных продуктов. Они могут быть получены, например, в виде твердых масс, порошков или пленок такими способами, как осаждение, кристаллизация, сублимационная сушка, распылительная сушка или сушка выпариванием. Для этой цели может быть использована микроволновая или радиочастотная сушка. Их можно вводить одних или в комбинации с одним или более другими соединениями по изобретению, или в комбинации с одним или более другими лекарственными средствами (или в виде любой их комбинации). Как правило, их будут вводить в виде композиции вместе с одним или более фармацевтически приемлемыми эксципиентами. Используемый здесь термин "эксципиент" означает любой ингредиент, отличающийся от соединения(й) по изобретению. Выбор эксципиента будет в значительной степени зависеть от таких факторов, как конкретный способ введения, влияние эксципиента на растворимость и стабильность, и тип лекарственной формы. Специалистам в данной области будут очевидны фармацевтические композиции, подходящие для доставки соединений по настоящему изобретению, и способы их получения. Такие композиции и способы их получения можно найти, например, в Remington's Pharmaceutical Sciences, 19-e издание (Mack Publishing Company, 1995). Соединения по изобретению можно вводить перорально. Пероральное введение может включать проглатывание, при котором соединение поступает в желудочно-кишечный тракт, и/или буккальное, лингвальное или сублингвальное введение, при котором соединение поступает в кровоток непосредственно из ротовой полости. Препараты, подходящие для перорального введения, включают твердые, полутвердые и жидкие системы, например таблетки; мягкие или твердые капсулы, содержащие мульти- и наночастицы, жидкости или порошки; лепешки (в том числе с жидким наполнителем); жевательные резинки; гели; быстрораспадающиеся лекарственные формы; пленки; овули; спреи; и буккальные/мукоадгезивные пластыри. Жидкие препараты включают суспензии, растворы, сиропы и эликсиры. Такие препараты можно использовать в качестве наполнителей в мягких или твердых капсулах (изготовленных, например, из желатина или гидроксипропилметилцеллюлозы), и обычно они содержат носитель, например воду, этанол, полиэтиленгликоль, пропиленгликоль, метилцеллюлозу или подходящее масло, и один или более эмульгирующих агентов и/или суспендирующих агентов. Жидкие композиции могут быть приготовлены также разведением твердого вещества, например из саше, в жидкости. Соединения по изобретению могут быть использованы также в быстрорастворяющихся, быстрораспадающихся лекарственных формах, таких как формы, описанные в Liang and Chen, Expert Opinion in Therapeutic Patents, 11 (6), 981-986 (2001). Для таблетированных лекарственных форм в зависимости от дозы содержание лекарственного средства может составлять от 1 до 80 мас.% от массы лекарственной формы, более типично от 5 до 60 мас.% от массы лекарственной формы. В дополнение к лекарственному средству таблетки обычно содержат разрыхлитель. Примеры разрыхлителей включают крахмальный гликолят натрия, натриевую соль карбоксиметилцеллюлозы, кальциевую соль карбоксиметилцеллюлозы, натриевую соль кроскармелозы, кросповидон, поливинилпирролидон, метилцеллюлозу, микрокристаллическую целлюлозу, замещенную низшим алкилом гидроксипропилцеллюлозу, крахмал, предварительно желатинизированный крахмал и альгинат натрия. Как правило, содержание разрыхлителя будет составлять от 1 до 25 мас.%, предпочтительно от 5 до 20 мас.% от массы лекарственной формы. Для придания когезионных свойств композиции в форме таблетки обычно используют связывающие вещества. Подходящие связывающие вещества включают микрокристаллическую целлюлозу, желатин, сахара, полиэтиленгликоль, природные и синтетические смолы, поливинилпирролидон, предварительно желатинизированный крахмал, гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу. Кроме того, таблетки могут содержать разбавители, такие как лактоза (моногидрат, высушенный распылительной сушкой моногидрат, лактоза безводная и т.п.), маннит, ксилит, декстроза, сахароза, сорбит, микрокристаллическая целлюлоза, крахмал и дигидрат двухосновного фосфата кальция. Таблетки возможно могут содержать также поверхностно-активные вещества, такие как лаурилсульфат натрия и полисорбат 80, и глиданты, такие как диоксид кремния и тальк. Содержание поверхностно-активных веществ, если они присутствуют, может составлять от 0,2 до 5 мас.% от массы таблетки, а содержание глидантов может составлять от 0,2 до 1 мас.% от массы таблетки. Обычно таблетки могут содержать также смазывающие вещества, такие как стеарат магния, стеарат кальция, стеарат цинка, стеарилфумарат натрия и смеси стеарата магния с лаурилсульфатом натрия. Обычно содержание смазывающих веществ составляет от 0,25 до 10 мас.%, предпочтительно от 0,5 до 3 мас.% от массы таблетки. Другие возможные ингредиенты включают антиоксиданты, красители, корригенты, консерванты и агенты, маскирующие вкус. Типичные таблетки содержат вплоть до приблизительно 80% лекарственного средства, от приблизительно 10 до приблизительно 90 мас.% связывающего вещества, от приблизительно 0 до приблизительно 85 мас.% разбавителя, от приблизительно 2 до приблизительно 10 мас.% разрыхлителя и от приблизительно 0,25 до приблизительно 10 мас.% смазывающего вещества. Для формования таблеток таблеточные смеси могут быть подвергнуты прямому прессованию или прессованию с помощью валика. Альтернативно, перед таблетированием таблеточные смеси или порции смесей могут быть подвергнуты влажному гранулированию, сухому гранулированию или гранулированию из расплава, гранулированию в результате застывания расплава или гранулированию экструзией. Конечный препарат может содержать один или более слоев и может быть покрытым оболочкой или может не иметь никакого покрытия и даже может быть инкапсулированным. Технология изготовления таблеток обсуждается в Н. Liebeman and L. Lachman, Pharmaceutical Dosage Forms: Tablets, Vol. 1 (Marcel Dekker, New York, 1980). Принимаемые перорально пленки для применения в медицине или ветеринарии обычно представляют собой гибкие водорастворимые или набухаемые в воде тонкие пленочные лекарственные формы, которые могут быть быстрорастворимыми или мукоадгезивными и обычно содержат соединение формулы I, пленкообразующий полимер, связывающее вещество, растворитель, увлажнитель, пластификатор, стабилизатор или эмульгатор, модификатор вязкости и растворитель. Некоторые компоненты композиции могут выполнять больше чем одну функцию. Соединение формулы (I) может быть водорастворимым или нерастворимым. Водорастворимое соединение обычно составляет от 1 до 80 мас.%, более типично от 20 до 50 мас.% растворенных веществ. Менее растворимые соединения могут составлять большую долю композиции, обычно вплоть до 88 мас.% растворенных веществ. Альтернативно, соединение формулы (I) может быть в форме гранул, состоящих из множества частиц. Пленкообразующий полимер может быть выбран из природных полисахаридов, белков или синтетических гидроколлоидов и обычно присутствует в пределах 0,01-99 мас.%, более типично в пределах 30-80 мас.%. Другие возможные ингредиенты включают антиоксиданты, красители, корригенты и усилители вкуса и запаха, консерванты, средства, стимулирующие слюноотделение, охлаждающие агенты, сорастворители (в том числе масла), смягчители, наполнители, пеногасители, поверхностно-активные вещества и агенты, маскирующие вкус. Пленки по изобретению обычно получают испарительной сушкой тонких водных пленок, нанесенных на легкоснимающуюся подложку или бумагу. Это может быть сделано в сушильном шкафу или в туннельной сушилке, обычно в сушилке, в которой сушка объединена с нанесением покрытия, или сублимационной сушкой или вакуумированием. Могут быть приготовлены твердые композиции для перорального введения с немедленным и/или модифицированным высвобождением. Композиции с модифицированным высвобождением включают отсроченное, замедленное, импульсное, контролируемое, направленное и программируемое высвобождение. Подходящие композиции с модифицированным высвобождением для задач данного изобретения описаны в патенте США № 6106864. Подробные сведения о других подходящих технологиях высвобождения, таких как высокоэнергетические дисперсии и осмотические и покрытые оболочкой частицы, можно найти в Verma et al. (2001), Pharmaceutical Technology On-line, 25(2), 1-14. Применение жевательной резинки для достижения контролируемого высвобождения описано в WO 00/35298. Соединения по изобретению можно вводить также прямо в кровоток, в мышцу или во внутренний орган. Подходящие способы парентерального введения включают внутривенный, внутриартериальный, внутрибрюшинный, интратекальный, интравентрикулярный, интрауретральный, интрастернальный, интракраниальный, внутримышечный, интрасиновиальный и подкожный. Подходящие устройства для парентерального введения включают игольные (в том числе микроигольные) инъекторы, безыгольные инъекторы и технические средства для инфузии. Парентеральные композиции обычно представляют собой водные растворы, которые могут содержать такие эксципиенты, как соли, углеводы и буферные агенты (предпочтительно до рН 3-9), но для некоторых применений более предпочтительно они могут быть приготовлены в виде стерильного неводного раствора или в виде высушенной формы, которую используют совместно с подходящим разбавителем, таким как стерильная апирогенная вода. Приготовление парентеральных композиций в стерильных условиях, например лиофилизацией, легко может быть осуществлено по стандартным фармацевтическим методикам, которые хорошо знают специалисты в данной области. Растворимость соединений формулы (I), используемых в приготовлении парентеральных растворов, можно увеличить, используя соответствующие методы приготовления композиций, такие как введение агентов, повышающих растворимость. Могут быть приготовлены композиции для парентерального введения с немедленным и/или модифицированным высвобождением. Композиции с модифицированным высвобождением включают отсроченное, замедленное, импульсное, контролируемое, направленное и программируемое высвобождение. Так, соединения по изобретению могут быть приготовлены в виде суспензии, в виде твердого вещества, полутвердого вещества или тиксотропной жидкости для введения в качестве имплантированного депо, обеспечивающего модифицированное высвобождение активного соединения. Примеры таких композиций включают покрытые лекарственным средством стенты и полутвердые вещества и суспензии, содержащие загруженные лекарственным средством микросферы из поли(dl-молочной-когликолевой)кислоты (PGLA). Соединения по изобретению можно вводить также местно, (интра)дермально или трансдермально на кожу или слизистую оболочку. Типичные композиции для этой цели включают гели, гидрогели, лосьоны, растворы, кремы, мази, присыпки, повязки, пенки, пленки, кожные пластыри, облатки, имплантаты, губки, нити, бандажи и микроэмульсии. Можно использовать также липосомы. Типичные носители включают спирт, воду, минеральное масло, жидкий вазелин, белый вазелин, глицерин, полиэтиленгликоль и пропиленгликоль. Можно вводить вещества, усиливающие проницаемость (см., например, Finnin and Morgan, J. Pharm. Sci., 88 (10), 955-958 (October 1999)). Другие методы местного введения включают доставку электропорацией, ионтофорезом, фонофорезом, сонофорезом и микроигольной или безыгольной (например, Powderject ™, Bioject ™ и т.д.) инъекцией. Могут быть приготовлены композиции для местного введения с немедленным и/или модифицированным высвобождением. Композиции с модифицированным высвобождением включают отсроченное, замедленное, импульсное, контролируемое, направленное и программируемое высвобождение. Соединения по изобретению можно вводить также интраназально или ингаляцией, обычно в форме сухого порошка (либо соединения одного, в виде смеси, например в виде сухой смеси с лактозой, либо в виде частицы из смеси компонентов, например смеси с фосфолипидами, такими как фосфатидилхолин) из сухого порошкового ингалятора, в виде аэрозольного спрея из находящегося под давлением контейнера, насоса, распылителя, пульверизатора (предпочтительно пульверизатора, использующего электрогидродинамику для продуцирования тонкодисперсного тумана) или небулайзера, с использованием или без использования подходящего пропеллента, такого как 1,1,1,2-тетрафторэтан или 1,1,1,2,3,3,3-гептафторпропан, либо в виде назальных капель. Порошок для интраназального применения может содержать биоадгезивный агент, например хитозан или циклодекстрин. Находящийся под давлением контейнер, насос, распылитель, пульверизатор или небулайзер содержит раствор или суспензию соединения(й) по изобретению, содержащий(ую), например, этанол, водный раствор этанола или подходящий альтернативный агент для диспергирования, солюбилизации или расширительного высвобождения активного начала, пропеллент(ы) в качестве растворителя и возможно поверхностно-активное вещество, такое как триолеат сорбитана, олеиновая кислота или олигомолочная кислота. Лекарственный продукт перед его применением в сухой порошковой или суспензионной лекарственной форме микронизируют до размера, подходящего для доставки ингаляцией (обычно менее 5 мкм). Этого можно достичь любым подходящим способом измельчения, таким как помол в спиральной струйной мельнице, помол в струйной мельнице с кипящим слоем, обработка суперкритической жидкостью с образованием наночастиц, гомогенизация при высоком давлении или распылительная сушка. Могут быть приготовлены капсулы (изготовленные, например, из желатина или гидроксипропилметилцеллюлозы), блистеры и картриджи для использования в ингаляторе или инсуффляторе, которые содержат порошковую смесь соединения по изобретению, подходящей порошковой основы, такой как лактоза или крахмал, и модификатора технологических характеристик, такого как l-лейцин, маннит или стеарат магния. Лактоза может быть безводной или может быть в форме моногидрата, причем последнее предпочтительно. Другие подходящие эксципиенты включают декстран, глюкозу, мальтозу, сорбит, ксилит, фруктозу, сахарозу и трегалозу. Подходящая композиция раствора для использования в пульверизаторе, использующем электрогидродинамику для продуцирования тонкодисперсного тумана, может содержать от 1 мкг до 20 мг соединения по изобретению на срабатывание, и объем срабатывания может варьировать от 1 до 100 мкл. Типичная композиция может содержать соединение формулы I, пропиленгликоль, стерильную воду, этанол и хлорид натрия. Альтернативные растворители, которые могут быть использованы вместо пропиленгликоля, включают глицерин и полиэтиленгликоль. В такие композиции по изобретению, предназначенные для ингаляционного/интраназального введения, могут быть добавлены подходящие корригенты, такие как ментол и левоментол, или подсластители, такие как сахарин или натриевая соль сахарина. Композиции для ингаляционного/интраназального введения с немедленным и/или модифицированным высвобождением могут быть приготовлены с использованием, например, PGLA. Композиции с модифицированным высвобождением включают отсроченное, замедленное, импульсное, контролируемое, направленное и программируемое высвобождение. В случае сухих порошковых ингаляторов и аэрозолей единица дозировки определяется с помощью клапана, который доставляет отмеренное количество. Единицы дозировки согласно изобретению обычно устанавливают для введения отмеренной дозы, или «пшика », содержащей от 0,001 до 10 мг соединения формулы (I). Суммарная суточная доза обычно будет находиться в пределах от 0,001 до 40 мг, которые могут быть введены разовой дозой или чаще разделенными дозами на протяжении суток. Соединения формулы (I) особенно подходят для введения ингаляцией. Соединения по изобретению можно вводить ректально или вагинально, например в форме суппозитория, пессария или клизмы. Масло какао является традиционной суппозиторной основой, однако, если это целесообразно, могут быть использованы различные альтернативные варианты. Могут быть приготовлены композиции для ректального/вагинального введения с немедленным и/или модифицированным высвобождением. Композиции с модифицированным высвобождением включают отсроченное, замедленное, импульсное, контролируемое, направленное и программируемое высвобождение. Соединения по изобретению также можно вводить непосредственно в глаз или ухо, обычно в форме капель микронизированной суспензии или раствора в изотоническом, с отрегулированным значением рН, стерильном физиологическом растворе. Другие композиции, подходящие для введения в глаз и ухо, включают мази, гели, биоразлагаемые (например рассасывающиеся гелевые губки, коллаген) и небиоразлагаемые (например силикон) имплантаты, облатки, линзы и дисперсные или везикулярные системы, такие как ниосомы или липосомы. Такой полимер, как поперечно-сшитая полиакриловая кислота, поливиниловый спирт, гиалуроновая кислота, целлюлозный полимер, например гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза или метилцеллюлоза, или гетерополисахаридный полимер, например гелановая камедь, могут быть инкорпорированы вместе с консервантом, таким как бензалкония хлорид. Такие композиции также можно доставлять ионофорезом. Могут быть приготовлены композиции для введения в глаз/ухо с немедленным и/или модифицированным высвобождением. Композиции с модифицированным высвобождением включают отсроченное, замедленное, импульсное, контролируемое, направленное или программируемое высвобождение. Для использования в любом из вышеупомянутых способов введения соединения по изобретению могут быть объединены с растворимыми макромолекулярными веществами, такими как циклодекстрин и подходящие его производные или полиэтиленгликольсодержащие полимеры, с целью улучшения их растворимости, скорости растворения, маскировки вкуса, биодоступности и/или стабильности. Например, установлено, что комплексы лекарственное средство-циклодекстрин, как правило, полезны для большинства лекарственных форм и путей введения. Могут быть использованы как комплексы включения, так и комплексы невключения. В качестве альтернативы прямому образованию комплекса с лекарственным средством, циклодекстрин может быть использован в качестве вспомогательной добавки, то есть в качестве носителя, разбавителя или солюбилизатора. Наиболее часто используемыми для этих целей являются альфа-, бета- и гамма-циклодекстрины, примеры которых можно найти в международных патентных заявках WO 91/11172, WO 94/02518 и WO 98/55148. Так как может быть желательным введение комбинации активных соединений, например, с целью лечения конкретного заболевания или состояния, в пределах объема настоящего изобретения две фармацевтические композиции или более, по меньшей мере одна из которых содержит соединение по изобретению, могут быть объединены в форме набора, подходящего для совместного введения этих композиций. Так, набор по изобретению включает в себя две отдельные фармацевтические композиции или более, по меньшей мере одна из которых содержит соединение формулы (I) по изобретению, и средства для раздельного хранения указанных композиций, такие как контейнер, секционный флакон или секционный пакет из фольги. Примером такого набора является известная блистерная упаковка, используемая для упаковки таблеток, капсул и т.п. Набор по изобретению особенно подходит для введения разных лекарственных форм, например пероральной и парентеральной, для введения отдельных композиций с разными дозировочными интервалами или для титрования отдельных композиций относительно друг друга. Для облегчения соблюдения схемы приема набор обычно включает в себя инструкции для введения и может быть снабжен так называемой памяткой. Для введения пациентам-людям суммарная суточная доза соединений по изобретению обычно находится в пределах от 0,001 до 5000 мг, разумеется, в зависимости от способа введения. Например, для перорального введения может требоваться суммарная суточная доза от 0,1 до 1000 мг, а требуемая внутривенная доза может составлять только от 0,001 до 100 мг. Суммарную суточную дозу можно вводить разовой дозой или разделенными дозами, и по усмотрению лечащего врача она может выходить за пределы типичного диапазона, указанного выше. Эти дозировки рассчитаны на среднего субъекта-человека, имеющего массу тела приблизительно 60-70 кг. Лечащий врач может без труда определить дозы для субъектов, чья масса тела выходит за пределы этого диапазона, таких как дети и пожилые люди. Во избежание сомнений здесь ссылка на "лечение" включает ссылку на куративное, паллиативное и профилактическое лечение. Соединения формулы (I) способны взаимодействовать с мускариновыми рецепторами и поэтому имеют разнообразное терапевтическое применение, как дополнительно описано ниже, благодаря существенной роли, которую мускариновые рецепторы играют в физиологии всех млекопитающих. Таким образом, данное изобретение относится к применению соединений формулы (I) для изготовления лекарственного средства для лечения или предупреждения заболеваний, расстройств и состояний, в которые вовлечен рецептор М3. Данное изобретение также относится к способу лечения млекопитающего, в том числе человека, антагонистом М3, включающему лечение указанного млекопитающего эффективным количеством соединения формулы (I) или его фармацевтически приемлемой соли, производной формы или композиции. Поэтому в еще одном аспекте настоящее изобретение относится к соединениям формулы (I) или их фармацевтически приемлемым солям, производным формам или композициям для применения в лечении заболеваний, расстройств и состояний, в которые вовлечены мускариновые рецепторы. Примерами таких заболеваний, расстройств и состояний являются воспалительное кишечное заболевание, синдром раздраженного кишечника, дивертикулёз, морская болезнь, язвы желудка, радиологическое исследование кишечника, симптоматическое лечение ВРН (доброкачественной гипертрофии предстательной железы), вызванное NSAID (нестероидными противовоспалительными лекарственными средствами) изъязвление желудка, недержание мочи (включая безотлагательное, частое, позывное недержание, сверхактивный мочевой пузырь, никтурию и симптомы нижних мочевыводящих путей), циклоплегия, мидриаз, болезнь Паркинсона. Более конкретно настоящее изобретение также относится к соединениям формулы (I) или их фармацевтически приемлемым солям, производным формам или композициям для применения в лечении заболеваний, расстройств и состояний, выбранных из группы, состоящей из хронического или острого бронхостеноза, хронического бронхита, обструкции малых дыхательных путей и эмфиземы, обструктивных или воспалительных заболеваний дыхательных путей любого типа, этиологии или патогенеза, в частности обструктивного или воспалительного заболевания дыхательных путей, которое выбрано из группы, состоящей из хронической эозинофильной пневмонии, хронической обструктивной болезни легких (COPD), COPD, которая включает в себя хронический бронхит, легочной эмфиземы или диспноэ, ассоциированной или не ассоциированной с COPD, COPD, которая характеризуется необратимой, прогрессирующей обструкцией дыхательных путей, респираторного дистресс-синдрома взрослых (ARDS), обострения гиперреактивности дыхательных путей после другой лекарственной терапии и заболевания дыхательных путей, которое ассоциировано с легочной гипертензией, бронхита любого типа, этиологии или патогенеза, в частности бронхита, который выбран из группы, состоящей из острого бронхита, острого ларинготрахеального бронхита, арахидного бронхита, катарального бронхита, крупозного бронхита, сухого бронхита, инфекционного астматического бронхита, продуктивного бронхита, стафилококкового или стрептококкового бронхита и везикулярного бронхита, астмы любого типа, этиологии или патогенеза, в частности астмы, которая выбрана из группы, состоящей из атопической астмы, неатопической астмы, аллергической астмы, атопической бронхиальной IgE-опосредованной астмы, бронхиальной астмы, эссенциальной астмы, истинной астмы, эндогенной астмы, вызванной патофизиологическими нарушениями, экзогенной астмы, вызванной факторами окружающей среды, эссенциальной астмы неизвестного или инаппарантного происхождения, неатопической астмы, бронхиальной астмы, эмфизематозной астмы, астмы, индуцированной физической нагрузкой, астмы, индуцированной аллергеном, астмы, индуцированной холодом, профессиональной астмы, инфекционной астмы, вызванной бактериальной, грибковой, протозойной или вирусной инфекцией, неаллергической астмы, астмы в начальной стадии, синдрома стерторозного дыхания у детей и бронхиолита, острого поражения легких, бронхоэктаза любого типа, этиологии или патогенеза, в частности бронхоэктаза, который выбран из группы, состоящей из цилиндрического бронхоэктаза, мешотчатого бронхоэктаза, веретенообразного бронхоэктаза, капиллярного бронхоэктаза, кистозного бронхоэктаза, сухого бронхоэктаза и фолликулярного бронхоэктаза. Более конкретно, настоящее изобретение также относится к соединениям формулы (I) или их фармацевтически приемлемым солям, производным формам или композициям для применения в лечении COPD или астмы. Примеры других подходящих терапевтических агентов, которые можно использовать в комбинации с соединением(ями) формулы (I) или их фармацевтически приемлемыми солями, производными формами или композициями, включают, но никоим образом ими не ограничиваются: (а) ингибиторы 5-липоксигеназы (5-LO) или антагонисты 5-липоксигеназа-активирующего белка (FLAP), (б) антагонисты лейкотриенов (LTRA), в том числе антагонисты LTB 4 , LTC 4 , LTD 4 и LTE 4 , (в) антагонисты гистаминового рецептора, в том числе H1- и Н3-антагонисты, (г) сосудосуживающие симпатомиметические агенты, являющиеся агонистами α 1 - и α 2 -адренорецепторов, для использования в качестве противозастойного средства, (д) β 2 -агонисты кратковременного или длительного действия, (е) ингибиторы PDE (фосфодиэстеразы), например ингибиторы PDE3, PDE4 и PDE5, (ж) теофиллин, (з) кромогликат натрия, (и) ингибиторы СОХ (циклооксигеназы), как неселективные, так и селективные ингибиторы СОХ-1 или СОХ-2 (NSAID (нестероидные противовоспалительные лекарственные средства)), (к) пероральные и ингалируемые глюкокортикостероиды, (л) моноклональные антитела, активные против эндогенных воспалительных объектов, (м) агенты против фактора некроза опухоли (анти-TNF- α), (н) ингибиторы молекул адгезии, в том числе антагониста VLA-4 (очень позднего антигена-4), (о) антагонисты кинин-B 1 - и В 2 -рецепторов, (п) иммуносупрессивные агенты, (р) ингибиторы матриксных металлопротеаз (ММР), (с) антагонисты тахикининовых NK 1 -, NK 2 - и NK 3 -рецепторов, (т) ингибиторы эластазы, (у) агонисты аденозинового А2а-рецептора, (ф) ингибиторы урокиназы, (х) соединения, воздействующие на дофаминовые рецепторы, например D2-агонисты, (ц) модуляторы NF κB-пути, например ингибиторы IKK, (ш) модуляторы цитокиновых сигнальных путей, например р38 МАР-киназные или syk-киназные, (щ) агенты, которые можно классифицировать как муколитические или противокашлевые средства, (э) антибиотики, (ю) ингибиторы HDAC (гистондеацетилазы), (я) ингибиторы PI3-киназы и (аа) CXCR-2-антагонисты. Согласно настоящему изобретению предпочтительной является комбинация соединений формулы (I) с Н3-антагонистами, β 2 -агонистами, ингибиторами PDE4, стероидами, особенно глюкокортикостероидами, агонистами аденозинового А2а-рецептора, модуляторами цитокиновых сигнальных путей, например р38 МАР-киназных или syk-киназных, или лейкотриеновыми антагонистами (LTRA), включая антагонисты LTB 4 , LTC 4 , LTD 4 и LTE 4 . Согласно настоящему изобретению более предпочтительной является комбинация соединений формулы (I) с глюкокортикостероидами, в частности с ингалируемыми глюкокорткостероидами со сниженными системными побочными эффектами, включая преднизон, преднизолон, флунизолид, триамцинолона ацетонид, беклометазона дипропионат, будесонид, флутиказона пропионат, циклесонид и мометазона фуроат, или β 2 -агонистами, включая, в частности салбутамол, тербуталин, бамбутерол, фенотерол, салметерол, формотерол, тулобутерол и их соли. Приведенные ниже примеры иллюстрируют получение соединений формулы (I). Подготовительный пример 1. трет-Бутил-4-циано-4,4-дифенилбутаноат. Суспензию дифенилацетонитрила (38,6 г; 200 ммоль) в трет-бутаноле (200 мл) нагревали до 60 °С в течение 30 мин. Полученный раствор охлаждали до 50 °С и добавляли раствор гидроксида калия (0,6 г; 10,69 ммоль) в метаноле (2 мл). Затем по каплям добавляли трет-бутилакрилат (30 мл; 200 ммоль), и смесь перемешивали при 50 °С в течение 2 ч и при комнатной температуре в течение 18 ч. Добавляли дополнительное количество гидроксида калия (0,6 г; 10,69 ммоль), и смесь вновь нагревали до 50 °С в течение 3 ч. Затем реакционную смесь концентрировали в вакууме, и остаток разбавляли диэтиловым эфиром (300 мл), промывали водой (200 мл), сушили над сульфатом натрия и концентрировали в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества с выходом 90% (57,95 г). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.41 (s, 9H), 2.29 (t, 2H), 2.72 (t, 2H), 7.29-7.33 (m, 1Н), 7.36-7.42 (m, 9H); ХИАД-МСНР (масс-спектрометрии низкого разрешения с химической ионизацией при атмосферном давлении в) m/z 322 [М+Н] + . Подготовительный пример 2. 4-Циано-4,4-дифенилбутановая кислота. Смесь продукта подготовительного примера 1 (57,5 г; 179,13 ммоль) в соляной кислоте (4н. в диоксане, 500 мл) перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь концентрировали в вакууме и остаток обрабатывали теплым диизопропиловым эфиром (150 мл), затем охлаждали до комнатной температуры. Полученное твердое вещество отфильтровывали, промывая диизопропиловым эфиром (2 ×30 мл) и сушили под вакуумом с получением указанного в заголовке соединения в виде кристаллического белого твердого вещества с выходом 77% (36,45 г). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 2.35 (t, 2Н), 2.76 (t, 2H), 7.30-7.44 (m, 10Н); ХИАД-МСНР m/z 266 [М+Н] + . Подготовительный пример 3. трет-Бутил-(3S)-3-феноксипирролидин-1-карбоксилат. Диизопропилазодикарбоксилат (5,7 мл; 29,38 ммоль) добавляли к охлажденному во льду раствору (R)-(-)-N-boc-3-пирролидинола (5 г; 26,71 ммоль), фенола (2,51 г; 26,71 ммоль) и трифенилфосфина (7,71 г; 29,38 ммоль) в тетрагидрофуране (70 мл), и эту смесь перемешивали при комнатной температуре в течение 18 ч. Затем реакционную смесь концентрировали в вакууме и остаток дважды растирали с диэтиловым эфиром и фильтровали. Фильтрат промывали 1н. раствором гидроксида натрия (20 мл), сушили над сульфатом натрия и концентрировали в вакууме. После очистки остатка колоночной хроматографией на силикагеле с элюированием смесью пентан:этилацетат от 90:10 до 83:17 получили указанное в заголовке соединение в виде бесцветного масла с выходом 75% (5,27 г). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.45 (m, 9H), 2.10-2.16 (m, 2H), 3.40-3.59 (m, 4H), 4.95-4.97 (m, 1H), 6.88-6.95 (m, 3Н), 7.24-7.28 (m, 2H). Подготовительный пример 4. трет-Бутил-(3S)-3-(3-метоксифенокси)пирролидин-1-карбоксилат. Указанное в заголовке соединение получили из (R)-(-)-N-boc-3-пирролидинола и 3-метоксифенола способом, который описан для подготовительного примера 3, в виде смолы с выходом 81%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.45 (m, 9H), 2.14 (bs, 2H), 3.40-3.58 (m, 4H), 3.75 (s, 3Н), 4.95 (m, 1Н), 6.45-6.54 (m, 3Н), 7.14-7.18 (m, 1H). Подготовительный пример 5. трет-Бутил-(3S)-3-(бензилокси)пирролидин-1-карбоксилат. Гидрид натрия (60% дисперсия в минеральном масле; 2,13 г; 53,41 ммоль) порциями добавляли к охлажденному во льду раствору (S)-(-)-N-boc-3-пирролидинола (10 г; 53,41 ммоль) в тетрагидрофуране (100 мл) и эту смесь перемешивали при 0 °С в течение 1 ч. Добавляли бензилбромид (6,4 мл; 53,41 ммоль) и тетрагидрофуран (50 мл) и смесь перемешивали в течение 6 ч, давая температуре подняться до 25 °С. Затем реакционную смесь медленно разбавляли водой (50 мл), концентрировали в вакууме и водный остаток экстрагировали этилацетатом (3 ×70 мл). Объединенный органический раствор промывали рассолом (30 мл), сушили над сульфатом натрия и концентрировали в вакууме до получения оранжевого масла. Это масло очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 100:0:0 до 90:10:1. Соответствующие фракции упаривали при пониженном давлении и остаток дополнительно очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат 66:33, с получением указанного в заголовке соединения в виде бесцветного масла с выходом 74% (10,93 г). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.45 (s, 9Н), 1.91-2.00 (m, 1H), 2.02-2.08 (m, 1H), 3.35-3.44 (m, 4H), 4.13-4.17 (m, 1H), 4.51-4.52 (m, 2H), 7.24-7.29 (m, 1H), 7.30-7.32 (m, 4H); ХИАД-МСНР m/z 278 [M+H] + . Подготовительный пример 6. трет-Бутил-(3R)-3-(бензилокси)пирролидин-1-карбоксилат. Указанное в заголовке соединение получили из (R)-(-)-N-boc-3-пирролидинола и бензилбромида способом, который описан для подготовительного примера 5. Сырое соединение очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 95:5:0,5, с получением целевого продукты с выходом 97%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.46 (s, 3Н), 1.92-2.02 (m, 1H), 2.03-2.10 (m, 1H), 3.35-3.48 (m, 4H), 4.14-4.19 (m, 1H), 4.49-4.57 (m, 2H), 7.24-7.33 (m, 5H). Подготовительный пример 7. Гидрохлорид (3S)-3-феноксипирролидина. Смесь продукта подготовительного примера 3 (5,25 г; 19,96 ммоль) в соляной кислоте (4н. в диоксане, 50 мл) перемешивали при комнатной температуре в течение 2 ч. Затем реакционную смесь концентрировали в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества с выходом 100%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 2.29-2.33 (m, 2H), 3.42-3.56 (m, 4H), 5.18-5.21 (m, 1Н), 6.95-7.01 (m, 3Н), 7.29-7.32 (m, 2H); ХИАД-МСНР m/z 164 [М+Н] + . Подготовительный пример 8. (3S)-3-(3-Метоксифенокси)пирролидин. Смесь продукта подготовительного примера 4 (3,19 г; 10,8 ммоль) в соляной кислоте (4н. в диоксане, 27 мл) перемешивали при комнатной температуре в течение 3 ч. Затем реакционную смесь концентрировали в вакууме и остаток очищали колоночной хроматографией с использованием картриджа Isolute ® SCX-2, элюируя метанолом, затем 1М аммиаком в метаноле, с получением указанного в заголовке соединения в виде желтого масла с выходом 85% (1,77 г). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.91-1.98 (m, 1H), 2.02-2.11 (m, 1H), 2.85-2.91 (m, 1H), 3.02-3.08 (m, 3H), 3.75 (s, 3H), 4.85-4.89 (m, 1H), 6.43-6.51 (m, 3H), 7.12-7.16 (m, 1H); ХИАД-МСНР m/z 194 [M+H] + . Подготовительный пример 9. Гидрохлорид (3S)-3-(бензилокси)пирролидина. Указанное в заголовке соединение получили из продукта подготовительного примера 5 способом, который описан для подготовительного примера 7, в виде твердого вещества с выходом 100%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 2.02-2.12 (m, 1H), 2.22-2.29 (m, 1H), 3.26-3.46 (m, 4H), 4.35-4.37 (m, 1H), 4.55 (s, 2H), 7.25-7.37 (m, 5H); ХИАД-МСНР m/z 178 [M+H] + . Подготовительный пример 10. Гидрохлорид (3R)-3-(бензилокси)пирролидина. Указанное в заголовке соединение получили из продукта подготовительного примера 6 способом, который описан для подготовительного примера 7, в виде твердого вещества с выходом 100%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.96-2.05 (m, 1H), 2.20-2.15 (m, 1H), 3.49-3.32 (m, 4H), 4.23-4.25 (m, 1H), 4.44-4.54 (m, 2H), 7.26-7.36 (m, 5H), 9.74-9.88 (m, 2H); ХИАД-МСНР m/z 178 [M+H] + . Подготовительный пример 11. 5-Оксо-5-[(3S)-3-феноксипирролидин-1-ил]-2,2-дифенилпентаннитрил. Смесь продуктов подготовительных примеров 2 (2,40 г; 9,05 ммоль) и 7 (1,99 г; 9,96 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (1,55 г; 9,96 ммоль), 1-гидроксибензотриазола гидрата (1,35 г; 9,96 ммоль) и триэтиламина (1,38 мл, 9,96 ммоль) в дихлорметане (40 мл) перемешивали при комнатной температуре в течение 18 ч. Затем реакционную смесь разбавляли дихлорметаном (100 мл), промывали 1М соляной кислотой (70 мл), 1М раствором гидроксида натрия (30 мл) и рассолом (30 мл), сушили над сульфатом натрия и концентрировали в вакууме. Остаток подвергали перекристаллизации из горячего этанола с получением указанного в заголовке соединения в виде кристаллического твердого вещества с выходом 76% (2,83 г). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 2.07-2.21 (m, 2H), 2.33-2.38 (m, 1H), 2.43-2.47 (m, 1Н), 2.69-2.84 (m, 2H), 3.44-3.55 (m, 2H), 3.58-3.67 (m, 2H), 4.96-5.04 (m, 1H), 6.89 (d, 2H), 6.93-6.96 (m, 1H), 7.24-7.46 (m, 12H); ХИАД-МСНР m/z 412 [M+H] + . Подготовительный пример 12. 5-Оксо-5-[(3R)-3-феноксипирролидин-1-ил]-2,2-дифенилпентаннитрил. Указанное в заголовке соединение получили из (3R)-3-феноксипирролидина (WO 2005/061457) и продукта подготовительного примера 2 способом, который описан для подготовительного примера 11, с выходом 41%. Подготовительный пример 13. 5-[(3S)-3-(3-Метоксифенокси)пирролидин-1-ил]-5-оксо-2,2-дифенилпентаннитрил. Указанное в заголовке соединение получили из продуктов подготовительных примеров 2 и 8 способом, который описан для подготовительного примера 11. Сырое соединение очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 98:2:0,2 до 95:5:0,5, с получением целевого продукта в виде бесцветного масла с количественным выходом. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 2.03-2.20 (m, 2H), 2.32-2.37 (m, 1H), 2.43-2.47 (m, 1H), 2.66-2.84 (m, 2H), 3.44-3.67 (m, 4H), 3.74 (s, 3H), 4.94-5.00 (m, 1H), 6.44-6.55 (m, 3H), 7.13-7.19 (m, 1H), 7.28-7.46 (m, 10Н); ХИАД-МСНР m/z 412 [M+H] + . Подготовительный пример 14. 5-[(3S)-3-(Бензилокси)пирролидин-1-ил]-5-оксо-2,2-дифенилпентаннитрил. Указанное в заголовке соединение получили из продуктов подготовительного примера 2 и 9 способом, который описан для подготовительного примера 11, в виде бледно-оранжевого твердого вещества с выходом 70%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.86-2.15 (m, 2H), 2.32-2.43 (m, 2H), 2.75-2.82 (m, 2Н), 3.35-3.62 (m, 4Н), 4.14-4.21 (m, 1Н), 4.45-4.56 (m, 2H), 7.22-7.44 (m, 15H); ХИАД-МСНР m/z 412 [M+H] + . Подготовительный пример 15. 5-[(3R)-3-(Бензилокси)пирролидин-1-ил]-5-оксо-2,2-дифенилпентаннитрил. N,N'-Карбонилдиимидазол (27 г; 162 ммоль) добавляли к раствору продукта подготовительного примера 2 (36 г; 135 ммоль) в тетрагидрофуране (600 мл), и эту смесь перемешивали в течение 3 ч при комнатной температуре. Добавляли раствор продукта подготовительного примера 10 (31 г; 141,75 ммоль) в тетрагидрофуране (300 мл) и смесь перемешивали при комнатной температуре в течение 18 ч. Затем реакционную смесь фильтровали, промывая тетрагидрофураном, и фильтрат концентрировали в вакууме. Остаток распределяли между водой (200 мл) и этилацетатом (600 мл) и органический слой отделяли, промывали водой, 2н. соляной кислотой (2 ×100 мл) и рассолом, сушили над сульфатом магния и концентрировали в вакууме. Растирание остатка с диэтиловым эфиром затем дало указанное в заголовке соединение с выходом 83% (48,2 г). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.87-2.15 (m, 2H), 2.32-2.44 (m, 2H), 2.74-2.82 (m, 2Н), 3.35-3.61 (m, 4Н), 4.14-4.22 (m, 1Н), 4.46-4.57 (m, 2H), 7.23-7.44 (m, 15H); ХИАД-MCHP m/z 425 [М+Н] + . Подготовительный пример 16. 5-[(3S)-3-Гидроксипирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Хлорид железа(III) (1,57 г; 9,70 ммоль) добавляли к раствору продукта примера 11 (385 мг; 0,84 ммоль) в дихлорметане (10 мл) и эту смесь перемешивали при комнатной температуре в течение 3 ч. Реакцию гасили добавлением 2М соляной кислоты (6 мл), фильтровали через Arbocel ® и фильтрат подщелачивали раствором аммиака 0,88 (20 мл). Слои фильтрата разделяли и водный раствор экстрагировали дихлорметаном (2 ×25 мл). Объединенный органический раствор сушили над сульфатом натрия, концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат:метанол:аммиак 0,88 от 95:5:0,5 до 85:15:1,5, с получением указанного в заголовке соединения в виде коричневой смолы с выходом 60% (415 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.99 (s, 6H), 1.23-1.27 (m, 2H), 1.56-1.64 (m, 1H), 1.89-1.98 (m, 1Н), 2.40-2.44 (m, 3Н), 2.47-2.52 (m, 1H), 2.61-2.67 (m, 1H), 2.74-2.78 (m, 1H), 4.17-4.22 (m, 1H), 7.22-7.26 (m, 2H), 7.29-7.33 (m, 4H), 7.37 (d, 4H); ХИАД-МСНР m/z 367 [M+H] + . Подготовительный пример 17. 5-[(3R)-3-Гидроксипирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. 1н. Соляную кислоту (10,95 мл; 10,95 ммоль) и 20% Pd(OH) 2 (1 г) добавляли к раствору продукта примера 9 (5 г; 10,95 ммоль) в этаноле (250 мл) и эту смесь перемешивали при 50 °С под давлением газообразного водорода 50 фунт/кв.дюйм (345 кПа) в течение 4 ч. Затем реакционную смесь фильтровали через Arbocel ® и фильтрат концентрировали в вакууме, получив белую пену. Пену перерастворяли в этаноле (250 мл), добавляли 20% Pd(ОН) 2 (1 г) и смесь перемешивали при 50 °С под давлением газообразного водорода 50 фунт/кв.дюйм (345 кПа) в течение 24 ч. Затем реакционную смесь фильтровали через Arbocel ®, промывая этанолом, и фильтрат концентрировали в вакууме. Остаток суспендировали в аммиаке 0,88, экстрагировали этилацетатом (3 ×50 мл) и объединенный органический раствор промывали рассолом (50 мл), сушили над сульфатом натрия и концентрировали в вакууме с получением указанного в заголовке соединения в виде белой пены с выходом 90% (3,63 г). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.99 (s, 6H), 1.23-1.27 (m, 2H), 1.56-1.63 (m, 1H), 1.89-1.96 (m, 1H), 2.39-2.44 (m, 3H), 2.46-2.52 (m, 1H), 2.61-2.67 (m, 1H), 2.74-2.78 (m, 1H), 4.17-4.22 (m, 1H), 7.22-7.26 (m, 2H), 7.29-7.33 (m, 4H), 7.36-7.39 (m, 4H); ХИАД-MCHP m/z 367 [M+H] + . Подготовительный пример 18. 5-[(3S)-3-Гидроксипирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Хлорид железа(III) (1,33 г; 8,22 ммоль) добавляли к раствору продукта примера 10 (1,2 г; 2,74 ммоль) в дихлорметане (25 мл) и эту смесь перемешивали при комнатной температуре в течение 3 ч. Растворитель удаляли в вакууме и остаток распределяли между 2М соляной кислотой (водн.) (20 мл) и диэтиловым эфиром (30 мл). Водный слой отделяли и подщелачивали до рН 14 твердым гидроксидом натрия. Полученный коричневый осадок собирали фильтрованием и очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 95:5:0,5 до 85:15:1,5, с получением указанного в заголовке соединения в виде бесцветной смолы с выходом 50% (475 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.11 (s, 6H), 1.52-1.56 (m, 2H), 1.69-1.76 (m, 1H), 1.95-2.04 (m, 1H), 2.53-2.57 (m, 2H), 2.62-2.65 (m, 1H), 2.69-2.76 (m, 1H), 2.83-2.95 (m, 2H), 4.27-4.31 (m, 1H), 7.29-7.33 (m, 2H), 7.36-7.45 (m, 8H); ХИАД-МСНР m/z 349 [M+H] + . Подготовительный пример 19. 5-{(3S)-3-[(6-{[трет-Бутил(диметил)силил]окси}-2-нафтил)окси] пирролидин-1-ил}-5-метил-2,2-дифенилгексанамид. 1,1'-Азобис(N,N'-диметилформамид) (95 мг; 0,553 ммоль) добавляли к охлажденному во льду раствору трифенилфосфина (145 мг; 0,553 ммоль), 6-(трет-бутилдиметилсилилокси)-2-нафтола [(102 мг; 0,372 ммоль) ЕР 625510, р. 13] и продукта подготовительного примера 17 (150 мг; 0,41 ммоль) в тетрагидрофуране (5 мл) и эту смесь нагревали при 60 °С в течение 18 ч. Добавляли дополнительное количество трифенилфосфина (145 мг; 0,553 ммоль) и 1,1'-азобис(N,N'-диметилформамида) (95 мг; 0,553 ммоль) и нагревание продолжали в течение следующих 18 ч. Смесь концентрировали в вакууме и остаток очищали с использованием картриджа Isolute ® SCX-2, элюируя метанолом, затем 0,25М аммиаком в метаноле. Основные фракции концентрировали в вакууме и затем очищали с использованием силикагелевого картриджа RediSep ®, элюируя смесью дихлорметан:метанол:аммиак 0,88 (от 100:0:0 до 92:8:0,8), с получением указанного в заголовке соединения с выходом 20% (46 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.24 (s, 6H), 0.95 (s, 3Н), 1.04 (s, 9H), 1.06 (s, 3Н) 1.15-1.29 (m, 2Н), 1.88-1.99 (m, 1Н), 2.11-2.22 (m, 1H), 2.30-2.52 (m, 2H), 2.54-2.62 (m, 1H), 2.64-2.69 (m, 1H), 2.72-2.82 (m, 1H), 2.82-2.90 (m, 1H), 4.83-4.89 (m, 1H), 6.96-7.06 (m, 2H), 7.12-7.34 (m, 12H), 7.53-7.57 (m, 1H), 7.60-7.65 (m, 1H); ХИАД-МСНР m/z 623 [M+H] + . Подготовительный пример 20. 5-[(3R)-3-Гидроксипирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Хлорид железа(III) (13,3 г; 82,192 ммоль) добавляли к раствору продукта примера 8 (9 г; 20,548 ммоль) в дихлорметане (200 мл) и эту смесь перемешивали при комнатной температуре в течение 1 ч. Реакцию гасили добавлением 2М соляной кислоты (150 мл) и перемешивали в течение 30 мин. Органический слой отделяли, а водный слой повторно экстрагировали дополнительными 100 мл дихлорметана. Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме. Очистка колоночной хроматографией на силикагеле с элюированием смесью дихлорметан:метанол:аммиак 0,88 от 95:5:0,5 до 90:1:1,0 дала указанное в заголовке соединение в виде бледно-коричневой пены с выходом 89% (6,37 г). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.18 (s, 6Н), 1.58-1.62 (m, 2Н), 1.76-1.84 (m, 1H), 1.96-2.05 (m, 1H), 2.55-2.59 (m, 2H), 2.74-2.81 (m, 1H), 2.87-2.93 (m, 1H), 2.98-3.06 (m, 2H), 4.32-4.36 (m, 1H), 7.30-7.34 (m, 2H), 7.37-7.46 (m, 8H); ХИАД-МСНР m/z 349 [M+H] + . Подготовительный пример 21. 2-Хлор-3-метоксифенол. 3-Хлорпероксибензойную кислоту (760 мг; 4,396 ммоль) порциями добавляли к раствору 2-хлор-3-метоксибензальдегида (500 мг; 2,931 ммоль) в дихлорметане (12 мл) и эту смесь перемешивали в течение 3 ч при комнатной температуре. Добавляли дополнительно 3-хлорпероксибензойную кислоту (760 мг; 4,396 ммоль) и смесь оставляли перемешиваться в течение 18 ч. Раствор разбавляли 12 мл дихлорметана и промывали насыщенным раствором сульфита натрия (15 мл) и насыщенным раствором гидрокарбоната натрия (15 мл). Органический слой сушили над сульфатом натрия и концентрировали в вакууме до желтой смолы. Остаток растворяли в метаноле (12 мл), добавляли триэтиламин (0,05 мл) и смесь перемешивали в течение 18 ч при комнатной температуре. Раствор концентрировали в вакууме, растворяли в диэтиловом эфире (20 мл) и экстрагировали 1н. гидроксидом натрия (20 мл). Водный слой подкисляли до рН 1 добавлением по каплям 2н. соляной кислоты и экстрагировали диэтиловым эфиром (2 ×25 мл). Эти объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме с получением указанного в заголовке соединения в виде коричневой смолы с выходом 70% (325 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 3.83 (s, 3Н), 6.52-6.54 (d, 2Н), 7.01-7.06 (t, 1H). Подготовительный пример 22. 1-Хлор-3-фтор-2-метоксибензол. Метилйодид (850 мкл; 13,646 ммоль) и карбонат калия (943 мг; 6,824 ммоль) добавляли к 2-хлор-6-фторфенолу (1,0 г; 6,824 ммоль) в тетрагидрофуране (10 мл) и эту смесь перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь распределяли между диэтиловым эфиром (50 мл) и водой (50 мл). Органическую фазу экстрагировали и затем промывали водой (2 ×20 мл), затем сушили над сульфатом натрия и концентрировали в вакууме с получением указанного в заголовке соединения в виде бесцветной жидкости с выходом 94% (1,03 г). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 3.90-3.91 (s, 3Н), 7.01-7.12 (m, 2H), 7.18-7.21 (m, 1Н). Подготовительный пример 23. 3-Фтор-5-метоксифенол. Трибромид бора (1M в дихлорметане; 9 мл; 89,985 ммоль) по каплям добавляли к охлажденному во льду раствору 3,5-диметоксифторбензола (3 мл; 22,496 ммоль) в дихлорметане (20 мл) и эту смесь перемешивали при увеличении температуры от 0 °С до комнатной температуры в течение 4 ч. Раствор охлаждали до 0 °С, добавляли дополнительное количество трибромида бора (4 мл; 44,992 ммоль) и перемешивание продолжали, нагревая до комнатной температуры в течение дополнительных 18 ч. Реакцию гасили раствором аммиака 0,88 и перемешивали при комнатной температуре в течение 90 мин. Органический слой отделяли и экстрагировали 2н. гидроксидом натрия (30 мл), который затем подкисляли до рН 1 добавлением по каплям концентрированной соляной кислоты. Водный слой затем повторно экстрагировали дихлорметаном (3 ×15 мл), объединенный органический раствор сушили над сульфатом натрия и концентрировали в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества с выходом 54% (1,72 г). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 3.72 (s, 3Н), 6.07-6.15 (m, 3Н). Подготовительный пример 24. 1-Фтор-3-метокси-5-трифторметилбензол. Указанное в заголовке соединение получили из 3-фтор-5-трифторметилфенола способом, который описан для подготовительного примера 22, в виде бесцветного масла с выходом 90%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 3.84 (s, 3Н), 6.92-6.97 (m, 2H), 7.00 (s, 1H). Подготовительный пример 25. 4-Фтор-3-метоксифенол. Указанное в заголовке соединение получили из 4-фтор-3-метоксибензальдегида способом, который описан для подготовительного примера 21, в виде коричневого масла с выходом 55%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 3.79 (s, 3Н), 6.23-6.27 (m, 1H), 6.47-6.50 (dd, 1H), 6.81-6.86 (m, 1H). Подготовительный пример 26. 2-Фтор-3-метоксифенол. Указанное в заголовке соединение получили из 2-фтор-3-метоксибензальдегида способом, который описан для подготовительного примера 21, в виде коричневого масла с выходом 33%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 3.82 (s, 3Н), 6.47-6.55 (m, 2H), 6.82-6.87 (t, 1H). Подготовительный пример 27. 1-Аллилокси-3-бромметилбензол. К раствору (3-аллилоксифенил)метанола (Tetrahedron, 2000, 56(13), 1873-1882) (1,07 г; 6,49 ммоль) в THF (тетрагидрофуран) (7 мл) при 3 °С добавляли четырехбромистый углерод (2,69 г; 8,11 ммоль), затем трифенилфосфин (2,13 г; 8,11 ммоль) в THF (2 мл). Реакционную смесь перемешивали при 5 °С в течение 1 ч. Реакционную смесь фильтровали и концентрировали в вакууме. Остаток промывали пентаном с получением желтого твердого вещества, которое очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат от 100:0 до 95:5, с получением указанного в заголовке соединения в виде бледно-желтого масла с выходом 24% (350 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 4.47 (s, 2H), 4.51-4.60 (m, 2H), 5.26-5.35 (m, 1H), 5.37-5.47 (m, 1H), 5.99-6.11 (m, 1H), 6.82-6.90 (m, 1H), 6.92-7.01 (m, 2H), 7.21-7.30 (m, 1H). Подготовительный пример 28. 5-[(3R)-3-(3-Аллилоксибензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Продукт подготовительного примера 20 (179 мг; 0,514 ммоль) в диметилформамиде (3 мл) по каплям добавляли к охлажденному во льду раствору гидрида натрия (60% дисперсия в минеральном масле; 31 мг; 0,770 ммоль) в диметилформамиде (1 мл). После перемешивания в течение 1 ч добавляли продукт подготовительного примера 27 (175 мг; 0,514 ммоль) в диметилформамиде (1 мл) и эту смесь оставляли нагреваться до комнатной температуры в течение 18 ч. Затем реакционную смесь вновь охлаждали до 0 °С и добавляли дополнительное количество гидрида натрия (60% дисперсия в минеральном масле; 31 мг; 0,770 ммоль) при перемешивании при комнатной температуре в течение еще 3 ч. Раствор гасили добавлением по каплям воды, концентрировали в вакууме и распределяли между этилацетатом (10 мл) и водой (10 мл). Органический слой экстрагировали и промывали водой (10 мл), затем сушили над сульфатом натрия и концентрировали в вакууме. Очистка колоночной хроматографией на силикагеле с элюированием смесью дихлорметан:метанол:аммиак 0,88 (97:3:0,3) дала указанное в заголовке соединение в виде бледно-желтой смолы с выходом 43% (108 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.03 (s, 3Н), 1.05 (s, 3Н), 1.44-1.51 (m, 2H), 1.78-1.85 (m, 1Н), 1.93-2.01 (m, 1H), 2.50-2.55 (m, 3H), 2.64-2.78 (m, 3H), 4.03-4.08 (m, 1H), 4.43 (s, 2H), 4.50-4.55 (m, 2H), 5.20-5.24 (m, 1H), 5.34-5.41 (m, 1H), 5.98-6.09 (m, 1H), 6.79-6.84 (m, 1H), 6.87-6.94 (m, 2H), 7.18-7.21 (t, 1H), 7.26-7.43 (m, 10Н); ЭРИ-МСНР (масс-спектрометрия низкого разрешения с электрораспылительной ионизацией) m/z 495 [М+Н] + . Подготовительный пример 29. Амид 5-метил-2,2-дифенил-5-[(3R)-3-(3-пропенилоксибензилокси) пирролидин-1-ил]гексановой кислоты. Указанное в заголовке соединение получили из продукта подготовительного примера 28 способом, который описан в примере 2, с добавлением дополнительного количества гидроксида калия (2 экв.) через 20 ч и продолжением нагревания в течение дополнительных 4 ч, в виде желтой смолы с выходом 88%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.99 (s, 3Н), 1.01 (s, 3Н), 1.23-1.28 (m, 2H), 1.67-1.69 (d, 3Н), 1.74-1.82 (m, 1H), 1.88-1.95 (m, 1Н), 2.40-2.44 (m, 2H), 2.47-2.53 (m, 1H), 2.55-2.74 (m, 3H), 3.99-4.04 (m, 1H), 4.42 (s, 2H), 4.85-4.87 (m, 1H), 6.40-6.42 (d, 1H), 6.88-6.90 (d, 1H), 6.96-6.99 (m, 2H), 7.21-7.38 (m, 11Н); ЭРИ-МСНР m/z 513 [M+H] + . Подготовительный пример 30. Аллиловый эфир 3-аллилокси-4-фторбензойной кислоты. Аллилбромид (3,04 мл; 35,2 ммоль) по каплям добавляли к суспензии 4-фтор-3-гидроксибензойной кислоты (2,5 г; 16,0 ммоль) и карбоната калия (4,43 г; 32,03 ммоль) в диметилформамиде (50 мл) при комнатной температуре и эту смесь перемешивали в течение 18 ч. Растворитель удаляли в вакууме и остаток распределяли между диэтиловым эфиром (30 мл) и водой (30 мл). Водный слой отделяли и экстрагировали дополнительно диэтиловым эфиром (2 ×20 мл). Объединенные органические слои промывали водой (3 ×10 мл), сушили над сульфатом натрия и концентрировали в вакууме с получением указанного в заголовке соединения в виде бесцветной жидкости с выходом 100% (3,82 г). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 4.65-4.66 (d, 2H), 4.80-4.82 (d, 2H), 5.28-5.48 (m, 4Н), 5.98-6.12 (m, 2Н), 7.10-7.14 (dd, 1Н), 7.65-7.69 (m, 2H); ХИАД-МСНР m/z 237 [М+Н] + . Подготовительный пример 31. (3-Аллилокси-4-фторфенил)метанол. Раствор продукта подготовительного примера 30 (2,0 г; 8,47 ммоль) в тетрагидрофуране (30 мл) по каплям добавляли в течение 20 мин к раствору алюмогидрида лития в тетрагидрофуране (1М; 16,9 мл; 16,9 ммоль) при 0 °С в атмосфере азота и этот раствор оставляли нагреваться до комнатной температуры в течение 5 ч. Смесь охлаждали до 0 °С и гасили последовательным добавлением по каплям воды (1 мл), водного раствора гидроксида натрия (2М, 2 мл) и воды (3 мл) и смесь перемешивали при комнатной температуре в течение 18 ч. Смесь фильтровали через Celite ® и фильтровальную подушку промывали этилацетатом (3 ×20 мл). Фильтрат отделяли и органический слой промывали рассолом (10 мл), сушили над сульфатом натрия и концентрировали в вакууме с получением указанного в заголовке соединения в виде бесцветной жидкости с выходом 100% (1,75 г). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 4.61-4.63 (m, 4H), 5.29-5.32 (d, 1H), 5.41-5.45 (d, 1H), 6.02-6.11 (m, 1H), 6.85-6.89 (m, 1H), 7.00-7.07 (m, 2H); ХИАД-МСНР m/z 165 [M-OH] + . Подготовительный пример 32. 2-Аллилокси-4-бромметил-1-фторбензол. Дибромтрифенилфосфоран (2,3 г; 5,43 ммоль) добавляли к раствору продукта подготовительного примера 31 (900 мг; 4,94 ммоль) в ацетонитриле (40 мл) при комнатной температуре и раствор перемешивали в течение 18 ч. Растворитель удаляли в вакууме, и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат (80:20), с получением указанного в заголовке соединения в виде бесцветной жидкости с выходом 31% (380 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 4.44 (s, 2Н), 4.61-4.63 (m, 2Н), 5.30-5.34 (m, 1H), 5.41-5.47 (m, 1H), 6.02-6.11 (m, 1H), 6.91-6.94 (m, 1H), 6.99-7.05 (m, 2H); ХИАД-МСНР m/z 165 [M-Br] + . Подготовительный пример 33. 5-[(3R)-3-(3-Аллилокси-4-фторбензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Гидрид натрия (62 мг; 1,55 ммоль) порциями добавляли к охлажденному во льду раствору продукта подготовительного примера 20 (270 мг; 0,775 ммоль) в диметилформамиде (3 мл) в атмосфере азота. После перемешивания в течение 1,5 ч добавляли раствор продукта подготовительного примера 32 (380 мг; 1,55 ммоль) в диметилформамиде (2 мл), смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 18 ч. Растворитель удаляли в вакууме, и остаток распределяли между этилацетатом (10 мл) и насыщенным раствором гидрокарбоната натрия (5 мл). Водный слой отделяли и экстрагировали дополнительно этилацетатом (10 мл). Объединенные органические слои промывали водой (5 мл), рассолом (5 мл), сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 (от 99:1:0,1 до 98:2:0,2 и до 95:5:0,5), с получением указанного в заголовке соединения в виде желтого масла с выходом 38% (150 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.03 (s, 3Н), 1.06 (s, 3Н), 1.44-1.53 (m, 2H), 1.78-1.86 (m, 1Н), 1.93-2.02 (m, 1H), 2.50-2.57 (m, 3H), 2.67-2.79 (m, 3H), 4.03-4.07 (m, 1H), 4.41 (s, 2H), 4.56-4.58 (m, 2H), 5.22-5.26 (m, 1H), 5.36-5.41 (m, 1H), 5.99-6.08 (m, 1H), 6.85-6.88 (m, 1H), 6.98-7.06 (m, 2H), 7.28-7.43 (m, 10Н); ЭРИ-МСНР m/z 513 [M+H] + . Подготовительный пример 34. 5-[(3R)-3-(4-Фтор-3-пропенилоксибензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта подготовительного примера 33 способом, который описан в примере 2, в виде бесцветного масла с выходом 100%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.02 (s, 3Н), 1.04 (s, 3Н), 1.23-1.32 (m, 2H), 1.69-1.71 (d, 3Н), 1.77-1.86 (m, 1Н), 1.89-1.97 (m, 1H), 2.40-2.82 (m, 6H), 4.02-4.07 (m, 1H), 4.39 (s, 2H), 4.88-4.95 (m, 1H), 6.37-6.40 (m, 1H), 6.95-6.99 (m, 1H), 7.04-7.11 (m, 2H), 7.22-7.38 (m, 10Н); ЭРИ-МСНР m/z 531 [M+H] + . Подготовительный пример 35. 3-Бензилокси-5-гидроксибензонитрил. Карбонат цезия (2,41 г; 7,4 ммоль) добавляли к раствору 3,5-дигидроксибензонитрила (1,0 г; 7,4 ммоль) в диметилформамиде (5 мл) при комнатной температуре и эту смесь перемешивали в течение 10 мин. По каплям добавляли бензилбромид (0,880 мл; 7,4 ммоль) и смесь нагревали при 80 °С в течение 30 мин. Растворитель удаляли в вакууме, остаток обрабатывали водой (10 мл), подкисляли водной соляной кислотой (2М) и экстрагировали этилацетатом (3 ×25 мл). Объединенные органические слои промывали водой (2 ×10 мл), рассолом (10 мл), сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией, элюируя смесью этилацетат/пентан (от 20:80 до 50:50), с получением указанного в заголовке соединения в виде белого твердого вещества с выходом 26% (445 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 5.07 (s, 2H), 6.67-6.70 (m, 2H), 6.80 (d, 1H), 7.29-7.43 (m, 5H); ЭРИ-МСНР m/z 224 [М] - . Подготовительный пример 36. 5-[(3S)-3-(3-Бензилокси-5-цианофенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Диизопропилазодикарбоксилат (215 мкл; 1,09 ммоль) по каплям добавляли к охлажденному во льду раствору трифенилфосфина (286 мг; 1,09 ммоль), продукта подготовительного примера 35 (369 мг; 1,64 ммоль) и продукта подготовительного примера 17 (200 мг; 0,546 ммоль) в тетрагидрофуране (10 мл) и эту смесь перемешивали при комнатной температуре в течение 18 ч. Смесь концентрировали в вакууме и очищали с использованием картриджа Isolute ® SCX-2, элюируя метанолом, затем 1М аммиаком метаноле. Основные фракции концентрировали в вакууме и дополнительно очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 (от 98:2:0,2 до 97:3:0,3), с получением указанного в заголовке соединения в виде оранжевой пены с выходом 25% (80 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.98 (s, 3Н), 1.02 (s, 3Н), 1.20-1.28 (m, 3H), 1.81-1.87 (m, 1Н), 2.07-2.16 (m, 1H), 2.34-2.83 (m, 5H), 4.74-4.79 (m, 1H), 5.10 (s, 2H), 6.72-6.74 (m, 2H), 6.93 (s, 1H), 7.20-7.43 (m, 15H); ЭРИ-МСНР m/z 574 [M+H] + . Подготовительный пример 37. 5-{(3S)-3-[(7-{[трет-Бутил(диметил)силил]окси}-2-нафтил)окси]пирролидин-1-ил}-5-метил-2,2-дифенилгексанамид. Раствор 1,1'-азобис(N,N'-диметилформамида) (109 мг; 0,630 ммоль) в тетрагидрофуране (2 мл) по каплям добавляли к охлажденному во льду раствору трифенилфосфина (165 мг; 0,630 ммоль), 7-[[диметил(1,1-диметилэтил)силил]окси]нафталин-2-ола [(115 мг; 0,420 ммоль), Journal of Medicinal Chemistry, 1993, Vol. 36, № 22, p. 3316] и продукта подготовительного примера 17 (171 мг; 0,467 ммоль) в тетрагидрофуране (3 мл) и эту смесь перемешивали при комнатной температуре в течение 72 ч, затем при 60 °С в течение 18 ч. Смесь концентрировали в вакууме и остаток очищали с использованием картриджа Isolute ® SCX-2, элюируя метанолом, затем 0,5М аммиаком в метаноле. Основные фракции концентрировали в вакууме и дополнительно очищали с использованием силикагелевого картриджа RediSep ®, элюируя смесью дихлорметан:метанол:аммиак 0,88 (от 100:0:0 до 92:8:0,8), с получением указанного в заголовке соединения в виде бесцветной смолы с выходом 34% (90 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.25 (s, 6H), 0.95-1.10 (m, 15H), 1.19-1.33 (m, 2H), 1.95 (m, 1H), 2.18 (m, 1H), 2.34-2.52 (m, 2H), 2.56-2.75 (m, 2H), 2.76-2.86 (m, 1H), 2.87-2.94 (m, 1H), 4.90 (m, 1H), 6.83-6.96 (m, 3H), 7.12 (m, 1H), 7.13-7.36 (m, 10H), 7.58-7.66 (m, 2H); ХИАД-МСНР m/z 623 [M+H] + . Подготовительный пример 38. Аллиловый эфир 3-метокси-4-хлорбензойной кислоты. Карбонат калия (4,44 г; 32,156 ммоль) и аллилбромид (2,78 мл; 32,156 ммоль) добавляли к перемешиваемому раствору 4-хлор-3-метоксибензойной кислоты (3 г; 16,078 ммоль) в N,N-диметилформамиде (30 мл) и оставляли перемешиваться при комнатной температуре в течение 18 ч. Реакционную смесь распределяли между диэтиловым эфиром (200 мл) и водой (150 мл), органическую фазу экстрагировали и дополнительно промывали водой (150 мл), сушили над сульфатом натрия и концентрировали в вакууме с получением указанного в заголовке соединения в виде оранжевого масла с выходом 98% (3,57 г). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 3.92 (s, 3H), 4.79-4.82 (d, 2H), 5.26-5.29 (d, 1H), 5.37-5.42 (d, 1H), 6.00-6.10 (m, 1H), 7.44-7.46 (d, 1H), 7.56-7.59 (dd, 1H), 7.62-7.63 (d, 1H); ЭРИ-МСНР m/z 227 [M+H] + . Подготовительный пример 39. 3-Гидрокси-4-хлорбензойная кислота. Трибромид бора (1М в дихлорметане; 31 мл; 31,504 ммоль) добавляли к охлажденному во льду раствору продукта подготовительного примера 38 (3,56 г; 15,752 ммоль) в дихлорметане (30 мл) и эту смесь перемешивали при увеличении температуры от 0 °С до комнатной температуры в течение 18 ч. Реакцию гасили раствором аммиака 0,88 и перемешивали при комнатной температуре в течение 90 мин. Реакционную смесь подкисляли до рН 1 добавлением по каплям 2н. соляной кислоты (водн.) и экстрагировали диэтиловым эфиром (2 ×50 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества с выходом 90% (2,45 г). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 7.36-7.38 (d, 1H), 7.44-7.47 (dd, 1H), 7.54-7.55 (d, 1H); ХИАД-МСНР m/z 171 [M-H] - . Подготовительный пример 40. Аллиловый эфир 3-аллилокси-4-хлорбензойной кислоты. Карбонат калия (4,9 г; 35,057 ммоль) и аллилбромид (3,07 мл; 35,507 ммоль) добавляли к перемешиваемому раствору продукта подготовительного примера 39 (2,45 г; 14,203 ммоль) в N,N-диметилформамиде (30 мл) и оставляли перемешиваться при комнатной температуре в течение 18 ч. Реакционную смесь распределяли между диэтиловым эфиром (70 мл) и водой (70 мл), органическую фазу затем экстрагировали и дополнительно промывали водой (50 мл), сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат от 100:0 до 90:10, с получением указанного в заголовке соединения в виде бледно-оранжевого масла с выходом 74% (2,65 г). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 4.66-4.68 (d, 2H), 4.79-4.81 (d, 2H), 5.25-5.31 (t, 2H), 5.36-5.49 (q, 2Н), 6.00-6.12 (m, 2Н), 7.45-7.47 (d, 1H), 7.56-7.59 (dd, 1H), 7.62 (d, 1H). Подготовительный пример 41. (3-Аллилокси-4-хлорфенил)метанол. Раствор продукта подготовительного примера 40 (2,6 г; 10,30 ммоль) в тетрагидрофуране (40 мл) по каплям добавляли к перемешиваемому раствору алюмогидрида лития (1M в тетрагидрофуране, 21 мл; 20,60 ммоль) в течение 30 мин при комнатной температуре и оставляли перемешиваться в течение 18 ч. Реакцию гасили добавлением по каплям воды (1 мл), 2М гидроксида натрия (2 мл) и воды (3 мл) и оставляли перемешиваться в течение 3 ч. Затем смесь фильтровали, промывая диэтиловым эфиром (2 ×20 мл) и водой (10 мл). Органическую фазу экстрагировали, сушили над сульфатом натрия и концентрировали в вакууме с получением указанного в заголовке соединения в виде бесцветного масла с выходом 69% (1,4 г). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 4.50 (s, 2H), 4.60-4.62 (d, 2H), 5.24-5.28 (d, 1H), 5.42-5.48 (d, 1H), 6.03-6.12 (m, 1H), 6.87-6.90 (d, 1H), 7.05 (s, 1H), 7.29-7.31 (d, 1H); ЭРИ-MCHP m/z 198 [M+H] + . Подготовительный пример 42. 2-Аллилокси-4-бромметил-1-хлорбензол. Дибромтрифенилфосфоран (3,27 г; 7,758 ммоль) добавляли к перемешиваемому раствору продукта подготовительного примера 41 (1,4 г; 7,053 ммоль) в ацетонитриле (50 мл) и оставляли перемешиваться при комнатной температуре в течение 18 ч. Добавляли дополнительное количество дибромтрифенилфосфорана (3,27 г; 7,758 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение следующих 6 ч. Раствор затем концентрировали в вакууме и подвергали перекристаллизации из горячего этилацетата (25 мл) и диэтилового эфира (25 мл). Твердое вещество отфильтровывали от раствора и фильтрат концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат от 100:0 до 80:20, с получением указанного в заголовке соединения в виде бесцветного масла с выходом 48% (890 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 4.52 (s, 2Н), 4.61-4.63 (d, 2Н), 5.25-5.29 (d, 1H), 5.42-5.48 (d, 1H), 6.02-6.11 (m, 1H), 6.95-6.98 (d, 1H), 7.10 (d, 1H), 7.30-7.32 (d, 1H). Подготовительный пример 43. 5-[(3R)-3-(3-Аллилокси-4-хлорбензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Раствор продукта подготовительного примера 20 (180 мг; 0,515 ммоль) в N,N-диметилформамиде (3 мл) добавляли к охлажденному во льду раствору гидрида натрия (60% дисперсия в минеральном масле; 41 мг; 1,032 ммоль) в N,N-диметилформамиде (2 мл) и эту смесь перемешивали при 0 °С в течение 30 мин. Добавляли продукт подготовительного примера 42 (270 мг; 1,032 ммоль) и смесь перемешивали в течение 18 ч при увеличении температуры от 0 °С до комнатной температуры. Реакционную смесь охлаждали во льду, добавляли дополнительно гидрид натрия (41 мг; 1,032 ммоль) и смесь перемешивали в течение следующих 3 ч при комнатной температуре. Затем реакционную смесь гасили водой (3 мл), концентрировали в вакууме и водный остаток распределяли между этилацетатом (30 мл) и водой (20 мл). Органический слой отделяли и промывали водой (2 ×20 мл), затем концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 (95:5:0,5), с получением указанного в заголовке соединения в виде коричневой смолы с выходом 62% (170 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.02 (s, 3Н), 1.05 (s, 3Н), 1.43-1.52 (m, 2H), 1.78-1.86 (m, 1Н), 1.93-2.02 (m, 1H), 2.49-2.56 (m, 3H), 2.65-2.79 (m, 3H), 4.03-4.08 (m, 1H), 4.43 (s, 2H), 4.56-4.58 (d, 2H), 5.22-5.26 (d, 1H), 5.39-5.45 (d, 1H), 5.99-6.08 (m, 1H), 6.85-6.88 (dd, 1H), 7.01-7.02 (d, 1H), 7.26-7.42 (m, 11Н); ХИАД-МСНР m/z 529 [M+H] + . Подготовительный пример 44. 5-[(3R)-3-(4-Хлор-3-пропенилоксибензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Гидроксид калия (340 мг; 6,055 ммоль) добавляли к раствору продукта подготовительного примера 43 (160 мг; 0,303 ммоль) в 3-метил-3-пентаноле (7 мл) и эту смесь нагревали при температуре дефлегмации в течение 24 ч. Затем реакционную смесь охлаждали до комнатной температуры, концентрировали в вакууме и остаток распределяли между этилацетатом (25 мл) и водой (25 мл). Водный слой отделяли, экстрагировали дополнительно этилацетатом (25 мл) и объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме с получением указанного в заголовке соединения в виде желтой смолы с выходом 97% (161 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.97 (s, 3Н), 0.99 (s, 3Н), 1.21-1.27 (m, 2H), 1.70-1.72 (d, 3Н), 1.74-1.80 (m, 1H), 1.87-1.93 (m, 1H), 2.39-2.44 (m, 2H), 2.45-2.50 (m, 1H), 2.53-2.57 (dd, 1H), 2.59-2.65 (q, 1H), 2.68-2.72 (m, 1H), 3.98-4.02 (m, 1H), 4.39 (s, 2H), 4.93-5.00 (m, 1H), 6.38-6.40 (m, 1H), 6.94-6.97 (dd, 1H), 7.04 (d, 1H), 7.20-7.39 (m, 11H); ХИАД-МСНР m/z 547 [M+H] + . Подготовительный пример 45. 5-[(3R)-3-(3-Бензилокси-4-цианофенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Гидрид натрия (60% дисперсия в минеральном масле; 26 мг; 0,656 ммоль) добавляли к охлажденному во льду раствору продукта подготовительного примера 17 (200 мг; 0,546 ммоль) в N,N-диметилформамиде (5 мл) и эту смесь перемешивали в течение 60 мин. Добавляли 2-бензилокси-4-фтор-бензонитрил (136 мг; 0,601 ммоль) и смесь перемешивали в течение 18 ч при комнатной температуре. Реакцию гасили водой (3 мл), концентрировали в вакууме и водный остаток распределяли между этилацетатом (20 мл) и водой (15 мл). Водный слой отделяли и экстрагировали дополнительно этилацетатом (2 ×10 мл). Объединенные органические слои концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 (95:5:0,5), с получением указанного в заголовке соединения в виде белой пены с выходом 33% (105 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.95 (s, 3Н), 1.02 (s, 3Н), 1.19-1.25 (m, 2H), 1.79-1.88 (m, 1Н), 2.07-2.16 (m, 1H), 2.33-2.49 (m, 2H), 2.51-2.58 (m, 2H), 2.67-2.74 (m, 1H), 2.79-2.83 (m, 1H), 4.80-4.84 (m, 1H), 5.20 (s, 2H), 6.49-6.52 (dd, 1H), 6.57-6.58 (d, 1H), 7.20-7.38 (m, 13H), 7.44-7.46 (m, 2H), 7.48-7.50 (d, 1H); ХИАД-МСНР m/z 574 [M+H] + . Подготовительный пример 46. (3-Аллилокси-2-хлорфенил)метанол. Боргидрид натрия (185 мг; 4,883 ммоль) добавляли к раствору продукта подготовительного примера 99 (800 мг; 4,07 ммоль) в этаноле (30 мл) и эту смесь перемешивали при комнатной температуре в течение 18 ч. Смесь гасили добавлением воды (30 мл) с последующим добавлением по каплям ледяной уксусной кислоты до прекращения выделения газа. Смесь затем экстрагировали диэтиловым эфиром (2 ×50 мл), объединенные органические слои промывали насыщенным водным раствором гидрокарбоната натрия (40 мл), сушили над сульфатом натрия и концентрировали в вакууме с получением указанного в заголовке соединения в виде бесцветной смолы с выходом 100% (805 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 4.60-4.62 (m, 2H), 4.66-4.69 (d, 2H), 5.24-5.28 (d, 1H), 5.43-5.47 (d, 1H), 6.01-6.12 (m, 1H), 6.82-6.84 (d, 1H), 7.09-7.12 (m, 1H), 7.22-7.26 (m, 1H); ЭРИ-МСНР m/z 198 [M+H] + . Подготовительный пример 47. 3-Аллилокси-2-хлорбензилбромид. Дибромтрифенилфосфоран (1,87 г; 4,431 ммоль) добавляли к раствору продукта подготовительного примера 46 (800 мг; 4,028 ммоль) в ацетонитриле (30 мл) и эту смесь оставляли перемешиваться при комнатной температуре в течение 18 ч. Добавляли дополнительное количество дибромтрифенилфосфорана (1,87 г; 4,431 ммоль) и смесь перемешивали при комнатной температуре в течение следующих 6 ч. Раствор концентрировали в вакууме и остаток подвергали перекристаллизации из горячего этилацетата (15 мл) и диэтилового эфира (15 мл). Твердое вещество отфильтровывали из раствора и фильтрат концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат от 100:0 до 90:10, с получением указанного в заголовке соединения в виде бесцветного масла с выходом 27% (285 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 4.61-4.63 (m, 2H), 4.65 (s, 2H), 5.26-5.29 (d, 1H), 5.43-5.49 (d, 1H), 6.00-6.11 (m, 1H), 7.01-7.03 (d, 1H), 7.08-7.10 (d, 1H), 7.20-7.24 (t, 1H); ХИАД-МСНР m/z 262 [M+H] + . Подготовительный пример 48. 5-[(3R)-3-(3-Аллилокси-2-хлорбензилокси)пирролщщн-1-ил]-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из продукта подготовительного примера 47 и продукта подготовительного примера 20 способом, который описан для подготовительного примера 43, в виде коричневой смолы с выходом 46%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.03 (s, 3Н), 1.05 (s, 3Н), 1.45-1.51 (m, 2H), 1.81-1.90 (m, 1Н), 1.95-2.04 (m, 1H), 2.50-2.58 (m, 3H), 2.65-2.79 (m, 3H), 4.07-4.12 (m, 1H), 4.49-4.54 (m, 2H), 4.59-4.61 (m, 2H), 5.24-5.27 (d, 1H), 5.41-5.47 (d, 1H), 6.02-6.11 (m, 1H), 6.96-6.98 (d, 1H), 7.06-7.08 (d, 1H), 7.17-7.42 (m, 11Н); ХИАД-МСНР m/z 529 [M+H] + . Подготовительный пример 49. 5-[(3R)-3-(2-Хлор-3-пропенилоксибензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта подготовительного примера 48 способом, который описан для подготовительного примера 44, в виде желтой смолы с выходом 100%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.98 (s, 3Н), 1.01 (s, 3Н), 1.23-1.27 (m, 2H), 1.71-1.73 (d, 3Н), 1.78-1.85 (m, 1Н), 1.90-1.97 (m, 1H), 2.40-2.44 (m, 2H), 2.46-2.58 (m, 2H), 2.61-2.67 (q, 1H), 2.71-2.75 (m, 1H), 4.03-4.09 (m, 1H), 4.52-4.53 (d, 2H), 4.93-5.00 (m, 1H), 6.40-6.42 (m, 1H), 7.00-7.02 (d, 1H), 7.11-7.40 (m, 12H); ХИАД-МСНР m/z 547 [M+H] + . Подготовительный пример 50. 5-Оксо-5-(4-феноксипиперидин-1-ил)-2,2-дифенилпентаннитрил. 1-Гидроксибензотриазола гидрат (5,67 г; 42 ммоль) и триэтиламин (14,88 мл; 107 ммоль) добавляли к 4-циано-4,4-дифенилбутановой кислоте (WO 97/24325) (10,39 г; 39 ммоль) и гидрохлориду 4-фенокси-пиперидина (6,32 г; 36 ммоль) в N,N-диметилформамиде (150 мл). Смесь перемешивали в течение 10 мин. Затем добавляли 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (8,20 г; 42 ммоль) и смесь перемешивали при комнатной температуре в течение 18 ч. Затем реакционную смесь концентрировали в вакууме и остаток распределяли между этилацетатом (100 мл) и водой (100 мл). Органический слой отделяли, промывали водой (100 мл) и рассолом (30 мл), сушили над сульфатом магния и концентрировали в вакууме. Остаток растирали с диэтиловым эфиром, отфильтровывали, промывали диэтиловым эфиром и очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат от 75:25 до 50:50, с получением указанного в заголовке соединения в виде белого твердого вещества с выходом 95% (14,4 г). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.74-1.90 (m, 4H), 2.45-2.49 (m, 2H), 2.79-2.83 (m, 2Н), 3.27-3.33 (m, 1Н), 3.54-3.61 (m, 1H), 3.62-3.74 (m, 2H), 4.49-4.53 (m, 1H), 6.87-6.91 (m, 2H), 6.93-6.98 (m, 1H), 7.26-7.43 (m, 12H); ХИАД-МСНР m/z 426 [M+H] + . Подготовительный пример 51. трет-Бутил-4-[(3-бромбензил)окси]пиперидин-1-карбоксилат. Гидрид натрия (60% дисперсия в минеральном масле; 3,31 г; 83 ммоль) добавляли к охлажденному во льду раствору 1-трет-бутоксикарбонил-4-гидроксипиперидина (16,64 г; 83 ммоль) в тетрагидрофуране (200 мл) и эту смесь перемешивали при 0 °С в течение 30 мин. Добавляли 3-бромбензилбромид (20,66 г; 83 ммоль) и смесь перемешивали при комнатной температуре в течение 18 ч. Затем реакционную смесь гасили водой (50 мл) и концентрировали в вакууме. Водный остаток экстрагировали этилацетатом (2 ×100 мл), объединенные органические фазы промывали рассолом (100 мл), сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат от 90:10 до 80:20, с получением указанного в заголовке соединения в виде бесцветного масла с выходом 61% (18,64 г). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.45 (s, 9H), 1.54-1.62 (m, 2H), 1.81-1.88 (m, 2H), 3.08-3.14 (m, 2Н), 3.52-3.58 (m, 1H), 3.73-3.79 (m, 2H), 4.51 (s, 2H), 7.18-7.26 (m, 2H), 7.38-7.41 (m, 1H), 7.49 (m, 1H); ХИАД-МСНР m/z 372 [M+H] + . Подготовительный пример 52. 4-[(3-Бромбензил)окси]пиперидин. Соляную кислоту (4М в диоксане, 340 мл) добавляли к раствору продукта подготовительного примера 51 (18,64 г; 50 ммоль) в диоксане (50 мл) и эту смесь перемешивали при комнатной температуре в течение 2,5 ч. Реакционную смесь концентрировали в вакууме, остаток растворяли в 2М водной соляной кислоте (200 мл) и промывали диэтиловым эфиром (2 ×100 мл). рН водного слоя затем доводили до значения 10 добавлением 2М водного раствора гидроксида натрия и экстрагировали диэтиловым эфиром (3 ×200 мл). Объединенные органические фазы сушили над сульфатом натрия и концентрировали в вакууме с получением указанного в заголовке соединения в виде бесцветного масла с выходом 98% (13,39 г). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.45-1.55 (m, 2H), 1.92-1.97 (m, 2H), 2.58-2.64 (m, 2Н), 3.07-3.13 (m, 2Н), 3.42-3.49 (m, 1Н), 4.51 (s, 2H), 7.17-7.27 (m, 2H), 7.37-7.40 (m, 1H), 7.50 (m, 1H); ХИАД-МСНР m/z 270 [M+H] + . Подготовительный пример 53. 5-{4-[(3-Бромбензил)окси]пиперидин-1-ил}-5-оксо-2,2-дифенилпентаннитрил. Указанное в заголовке соединение получили из 4-циано-4,4-дифенилбутановой кислоты (WO 9724325) и продукта подготовительного примера 52 способом, который описан для подготовительного примера 50, с выходом 95%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.63 (m, 2H), 1.82 (m, 2H), 2.46 (m, 2H), 2.80 (m, 2Н), 3.17 (m, 1Н), 3.35 (m, 1H), 3.51-3.64 (m, 2H), 3.88 (m, 1H), 4.44-4.53 (m, 2H), 7.17-7.42 (m, 14H); ХИАД-МСНР m/z 519 [M+H] + . Подготовительный пример 54. Амид 5-(4-гидроксипиперидин-1-ил)-5-метил-2,2-дифенилгексановой кислоты. 1М водный раствор HCl (9,43 мл; 9,43 ммоль) добавляли к перемешиваемому раствору продукта примера 91 в этаноле (250 мл). Добавляли 20% гидроксид палладия(II) на угле (1 г) и полученную смесь перемешивали в атмосфере газообразного водорода (50 фунт/кв.дюйм (345 кПа)) при 50 °С в течение 18 ч. Реакционную смесь фильтровали через арбоцел и затем концентрировали в вакууме. Остаток растворяли в воде (100 мл), рН раствора доводили до значения 12 добавлением 2М водного раствора гидроксида натрия и экстрагировали этилацетатом (3 ×100 мл). Объединенные органические слои сушили над сульфатом магния и концентрировали в вакууме с получением белой пены с выходом 95% (3,39 г). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.97 (s, 6H), 1.23-1.27 (m, 2H), 1.39-1.48 (m, 2H), 1.74-1.78 (m, 2H), 2.06-2.11 (m, 2H), 2.40-2.44 (m, 2H), 2.65-2.68 (m, 2H), 3.47 (m, 1H), 7.23-7.39 (m, 10Н); ЭРИ-МСНР m/z 381 [М+Н] + . Подготовительный пример 55. 5-Амино-5-метил-2,2-дифенилгексаннитрил. трет-Бутилат калия (203 мг; 1,81 ммоль) и 2,2-диоксид трет-бутил-4,4-диметил-1,2,3-оксатиазинан-3-карбоксилата [(400 мг; 1,51 ммоль), WO 2003037327, с. 83] добавляли к раствору дифенилацетонитрила (349 мг; 1,81 ммоль) в N,N-диметилформамиде (5 мл) и эту смесь перемешивали в течение 18 ч при комнатной температуре. Затем реакционную смесь концентрировали в вакууме и остаток обрабатывали соляной кислотой (4M в диоксане, 10 мл) и нагревали при 40 °С в течение 2,5 ч. Реакционную смесь концентрировали в вакууме и остаток подщелачивали насыщенным раствором гидрокарбоната натрия и экстрагировали этилацетатом (2 ×30 мл). Объединенный органический раствор сушили над сульфатом магния, концентрировали в вакууме, и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 (90:10:1), с получением указанного в заголовке соединения в виде бесцветного масла с выходом 77% (324 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.17 (m, 6H), 1.48-1.57 (m, 2H), 2.20-2.40 (brs, 2H), 2.42-2.53 (m, 2Н), 7.22-7.43 (m, 10Н); ХИАД-МСНР m/z 279 [М+Н] + . Подготовительный пример 56. 5-(3-Гидроксиазетидин-1-ил)-5-метил-2,2-дифенилгексаннитрил. Смесь (+/-)-эпихлоргидрина (1,47 мл; 18,76 ммоль) и продукта подготовительного примера 55 (4,74 г; 17 ммоль) в метаноле (50 мл) нагревали при 60 °С в течение 48 ч. Затем реакционную смесь концентрировали в вакууме и остаток распределяли между этилацетатом (50 мл) и раствором гидрокарбоната натрия (30 мл). Водный слой отделяли и экстрагировали этилацетатом (2 ×50 мл). Объединенный органический раствор сушили над сульфатом магния, концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 100:0:0 до 95:5:0,5, с получением указанного в заголовке соединения в виде бледно-желтого масла с выходом 50% (2,86 г). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.93 (s, 6H), 1.29-1.39 (m, 2H), 2.38-2.50 (m, 2H), 2.90-3.00 (m, 2H), 3.29-3.39 (m, 2H), 4.29-4.39 (m, 1H), 7.24-7.45 (m, 10Н); ХИАД-МСНР m/z 335 [М+Н] + . Подготовительный пример 57. 1-(4-Циано-1,1-диметил-4,4-дифенилбутил)азетидин-3-ил-метансульфонат. Метансульфонилхлорид (3,3 мл; 43 ммоль) добавляли к раствору продукта подготовительного примера 56 (4,82 г; 14,4 ммоль) в пиридине (50 мл), охлажденному до -15 °С. Смесь перемешивали в течение 2 ч, давали температуре подняться до 0 °С, затем концентрировали в вакууме. Остаток распределяли между этилацетатом (100 мл) и раствором гидрокарбоната натрия (100 мл) и органический слой отделяли, сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат/метанол/аммиак 0,88 (90/10/1) 2:1, с получением указанного в заголовке соединения в виде желтого масла с выходом 81% (4,80 г). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.30-1.41 (m, 2H), 2.42-2.55 (m, 2H), 2.98 (s, 3Н), 3.25-3.37 (m, 2H), 3.44-3.56 (m, 2H), 5.00-5.06 (m, 1H), 7.23-7.44 (m, 10Н); ХИАД-МСНР m/z 413 [М+Н] + . Подготовительный пример 58. Гидрохлорид азетидин-3-ил-метансульфоната. Смесь 1-(дифенилметил)азетидин-3-ил-метансульфоната (WO 9725322, с. 64), (20 г; 63 ммоль) и хлорэтилхлорформиата (10 мл; 95 ммоль) в дихлорметане (100 мл) нагревали при температуре дефлегмации в течение 2,5 ч. Затем реакционную смесь концентрировали в вакууме, остаток перерастворяли в метаноле (100 мл) и нагревали при температуре дефлегмации в течение следующих 2,5 ч. Смесь затем охлаждали до комнатной температуры и концентрировали в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества с количественным выходом (9,6 г). 1 Н-ЯМР (400 МГц, DMSO-d 6 ) δ: 3.28 (s, 3Н), 4.00-4.15 (m, 2H), 4.31 (m, 2Н), 5.28-5.38 (m, 1Н); ХИАД-МСНР m/z 152 [М+Н] + . Подготовительный пример 59. 4-Циано-4,4-дифенилбутаноилхлорид. N,N-Диметилформамид (1 каплю) добавляли к суспензии 4-циано-4,4-дифенилбутановой кислоты [(7,8 г; 29 ммоль), WO 97/24325] и оксалилхлорида (5,2 мл; 60 ммоль) в дихлорметане (40 мл) и эту смесь перемешивали при комнатной температуре в течение 2 ч. Затем реакционную смесь концентрировали в вакууме и остаток подвергали азеотропной перегонке с толуолом (3 ×50 мл) с получением сырого указанного в заголовке соединения. Это вещество использовали в подготовительном примере 60 без дополнительной очистки. Подготовительный пример 60. 1-(4-Циано-4,4-дифенилбутаноил)азетидин-3-ил-метансульфонат. Триэтиламин (12,3 мл; 87 ммоль) и раствор продукта подготовительного примера 59 (8,23 г; 29 ммоль) в дихлорметане по каплям добавляли к раствору подготовительного примера 58 (5,53 г; 29 ммоль) в дихлорметане (50 мл), охлаждали до -78 °С и смесь перемешивали при этой температуре в течение 1 ч. Реакцию гасили 2н. соляной кислотой (50 мл), органический слой отделяли, сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат:пентан от 50:50 до 100:0, с получением указанного в заголовке соединения в виде желтого масла с выходом 97% (11,4 г). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 2.17-2.29 (m, 2H), 2.71-2.80 (m, 2H), 3.05 (s, 3Н), 4.03-4.20 (m, 2Н), 4.26-4.38 (m, 2Н), 5.18-5.22 (m, 1H), 7.24-7.45 (m, 10Н); ХИАД-МСНР m/z 399 [М+Н] + . Подготовительный пример 61. 5-{3-[2-(Бензилокси)фенокси]азетидин-1-ил}-5-оксо-2,2-дифенилпентаннитрил. Указанное в заголовке соединение получили из продукта подготовительного примера 60 и 2-(бензилокси)фенола способом, который описан в примере 99, в виде желтого масла с выходом 77%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 2.18-2.24 (m, 2H), 2.75-2.80 (m, 2H), 4.02-4.10 (m, 2Н), 4.23-4.33 (m, 2Н), 4.82-4.91 (m, 1Н), 5.08 (s, 2H), 6.62-6.65 (m, 1H), 6.84-6.99 (m, 3Н), 7.24-7.43 (m, 15H); ХИАД-МСНР m/z 503 [М+Н] + . Подготовительный пример 62. 5-{3-[2-(Бензилокси)фенокси]азетидин-1-ил}-5-метил-2,2-дифенилгексаннитрил. Раствор продукта подготовительного примера 61 (700 мг; 1,39 ммоль) в тетрагидрофуране (10 мл) охлаждали до -35 °С. Добавляли хлорид циркония (650 г; 2,78 ммоль) и эту реакционную смесь перемешивали при -35 °С в течение 1 ч. Затем по каплям добавляли хлорид метилмагния (3М в тетрагидрофуране; 4,2 мл; 12,6 ммоль) и смесь перемешивали в течение 3 ч, поддерживая температуру ниже -20 °С. Реакцию гасили 1н. раствором гидроксида натрия (10 мл) и смесь фильтровали через Arbocel ®, промывая этилацетатом. Органический слой фильтрата отделяли, промывали рассолом (20 мл), сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат от 85:15 до 50:50, с получением указанного в заголовке соединения в виде желтого масла с выходом 10% (69 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.98 (s, 6H), 1.35-1.42 (m, 2H), 2.45-2.58 (m, 2H), 3.20-3.32 (m, 2Н), 3.40-3.55 (m, 2Н), 4.68-4.78 (m, 1Н), 5.13 (s, 2H), 6.64-6.72 (d, 1H), 6.85-6.99 (m, 3H), 7.24-7.48 (m, 15H); ЭРИ-МСНР m/z 517 [M+H] + . Подготовительный пример 63. 5-{3-[2-(Бензилокси)фенокси]азетидин-1-ил}-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта подготовительного примера 62 способом, который описан в примере 100, в виде бесцветной смолы с выходом 55%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.90 (s, 6H), 1.15-1.22 (m, 2H), 2.42-2.52 (m, 2H), 3.18-3.32 (m, 2Н), 3.40-3.58 (m, 2Н), 4.65-4.78 (m, 1Н), 5.13 (s, 2H), 5.40-5.60 (m, 2H), 6.62-6.70 (m, 1H), 6.82-6.97 (m, 3H), 7.23-7.44 (m, 15H); ХИАД-МСНР m/z 535 [M+H] + . Подготовительный пример 64. Гидрохлорид 3-феноксиазетидина. 10% Pd/C (2,5 г) добавляли к раствору 1-(дифенилметил)-3-феноксиазетидина (27,7 г; 88 ммоль) в этаноле (100 мл) и уксусной кислоте (100 мл) и эту смесь перемешивали при комнатной температуре под давлением водорода 50 фунт/кв.дюйм (345 кПа) в течение 24 ч. Затем реакционную смесь фильтровали через Arbocel ® и фильтрат концентрировали в вакууме. Остаток растворяли в диэтиловом эфире (200 мл), охлаждали до 0 °С и обрабатывали соляной кислотой (1M в диэтиловом эфире, 120 мл). Затем растворитель упаривали при пониженном давлении, остаток подвергали азеотропной перегонке с толуолом и растирали с этилацетатом с получением указанного в заголовке соединения в виде белого твердого вещества с выходом 86% (13,99 г). ХИАД-МСНР m/z 160 [М+Н] + . Подготовительный пример 65. 5-Оксо-5-(3-феноксиазетидин-1-ил)-2,2-дифенилпентаннитрил. Смесь продукта подготовительного примера 64 (1,13 г; 7,6 ммоль), 4-циано-4,4-дифенилбутановой кислоты [(2,4 г; 9,12 ммоль), WO 97/24325], 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (1,76 г; 9,12 ммоль), 1-гидроксибензотриазола гидрата (1,30 г; 9,12 ммоль) и N,N-диизопропилэтиламина (5,3 мл; 19 ммоль) в дихлорметане (50 мл) перемешивали при комнатной температуре в течение 18 ч. Затем реакционную смесь разбавляли этилацетатом (50 мл), промывали 2М соляной кислотой (30 мл) и раствором гидрокарбоната натрия (30 мл), сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат от 100:0 до 0:100, с получением указанного в заголовке соединения в виде бесцветного масла с выходом 75% (2,28 г). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 2.20-2.25 (m, 2H), 2.75-2.82 (m, 2H), 3.97-4.08 (m, 2Н), 4.28-4.40 (m, 2Н), 4.85-4.90 (m, 1Н), 6.65-6.74 (m, 2H), 6.98-7.04 (m, 1H), 7.24-7.43 (m, 12H); ХИАД-МСНР m/z 397 [M+H] + . Подготовительный пример 66. трет-Бутил[4-(йодметил)фенокси]диметилсилан. Трифенилфосфин (1,32 г; 5,03 ммоль), имидазол (370 мг; 5,47 ммоль) и йод (1,16 г; 4,61 ммоль) добавляли к охлажденному во льду раствору 4-(трет-бутилдиметилсилилокси)бензилового спирта [(1 г; 4,19 ммоль), Tet. Lett. (2004), 45, 9617] в тетрагидрофуране (50 мл) и эту смесь перемешивали при 0 °С в течение 10 мин и комнатной температуре в течение 30 мин. Затем реакционную смесь концентрировали в вакууме и остаток распределяли между этилацетатом и водой. Органический слой отделяли, сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя пентаном, с получением указанного в заголовке соединения с выходом 41% (600 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.00 (s, 6H), 0.78 (s, 9H), 4.26 (s, 2H), 6.72-6.98 (m, 2Н), 7.22-7.28 (m, 2Н). Подготовительный пример 67. 5-Аллилокси-2,4-дихлорфенол. 4,6-Дихлоррезорцин (3,80 г; 21 ммоль), аллилбромид (1,82 мл; 21 ммоль) и карбонат калия (2,24 г; 21 ммоль) объединяли в DMF (диметилформамид) и перемешивали при комнатной температуре в течение 18 ч. DMF удаляли в вакууме, остаток подкисляли 2М соляной кислотой (50 мл) и экстрагировали этилацетатом (2 ×50 мл). Объединенный органический раствор сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат от 9:1 до 4:1, с получением указанного в заголовке соединения в виде желтого масла с выходом 27% (1,24 г). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 4.55-4.60 (m, 2H), 5.33-5.52 (m, 2H), 5.95-6.10 (m, 1H), 6.62 (s, 1H), 7.3 (s, 1H); ЭРИ-МСНР m/z 217 [M-H] + . Подготовительный пример 68. 5-[3-(5-Аллилокси-2,4-дихлорфенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из продуктов подготовительного примера 67 и подготовительного примера 57 способом, который описан в примере 99, в виде бесцветной смолы с выходом 93%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.98 (s, 6H), 1.31-1.40 (m, 2H), 2.41-2.55 (m, 2H), 3.20-3.25 (m, 2H), 3.48-3.58 (m, 2H), 4.55-4.58 (m, 2H), 4.62-4.75 (m, 1H), 5.32-5.38 (m, 1H), 5.40-5.48 (m, 1H), 5.95-6.10 (m, 1H), 6.28 (s, 1H), 7-25-7.45 (m, 11Н); ХИАД-МСНР m/z 535 [M+H] + . Подготовительный пример 69. 2-Аллилокси-4,5-дихлорфенол. Указанное в заголовке соединение получили из 4,5-дихлоркатехина способом, который описан для подготовительного примера 67, в виде розового твердого вещества с выходом 62%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 4.55-4.60 (m, 2Н), 5.35-5.45 (m, 2Н), 5.81 (1Н, s), 5.95-6.10 (m, 1H), 6.90 (s, 1H), 7.03 (s, 1H); ХИАД-МСНР m/z 217 [M-H] + . Подготовительный пример 70. 5-[3-(2-Аллилокси-4,5-дихлорфенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из продуктов подготовительного примера 69 и подготовительного примера 57 способом, который описан в примере 99, в виде бесцветной смолы с выходом 75%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.98 (s, 6H), 1.31-1.40 (m, 2H), 2.41-2.55 (m, 2H), 3.15-3.25 (m, 2H), 3.36-3.50 (m, 2H), 4.50-4.58 (m, 2H), 4.60-4.66 (m, 1H), 5.28-5.42 (m, 2H), 5.95-6.10 (m, 1H), 6.65 (s, 1H), 6.95 (s, 1H), 7,25-7.45 (m, 10Н); ХИАД-МСНР m/z 535 [M+H] + . Подготовительный пример 71. 1-Аллилокси-3-бромметилбензол. К раствору (3-аллилоксифенил)метанола (Tetrahedron (2000), 56(13), 1873-1882) (1,07 г; 6,49 ммоль) в THF (7 мл) при 3 °С добавляли четырехбромистый углерод (2,69 г; 8,11 ммоль), затем трифенилфосфин (2,13 г; 8,11 ммоль) в THF (2 мл). Реакционную смесь перемешивали при 5 °С в течение 1 ч. Реакционную смесь фильтровали и концентрировали в вакууме. Остаток промывали пентаном и полученное желтое твердое вещество очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат от 100:0 до 95:5, с получением указанного в заголовке соединения в виде бледно-желтого масла с выходом 24% (350 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 4.47 (s, 2H), 4.51-4.60 (m, 2H), 5.26-5.35 (m, 1H), 5.37-5.47 (m, 1H), 5.99-6.11 (m, 1H), 6.82-6.90 (m, 1H), 6.92-7.01 (m, 2H), 7.21-7.30 (m, 1H). Подготовительный пример 72. 5-[3-(3-Аллилоксибензилокси)азетидин-1-ил]-5-метил-2,2-дифенил-гексаннитрил. Гидрид натрия (60% дисперсия в минеральном масле; 24 мг; 0,596 ммоль) порциями добавляли к охлажденному во льду раствору продукта подготовительного примера 56 (166 мг; 0,496 ммоль) в N,N-диметилформамиде (2 мл) и эту смесь перемешивали при 0 °С в течение 15 мин. Добавляли продукт подготовительного примера 71 (169 мг; 0,746 ммоль) и смесь перемешивали в течение 0,5 ч при 0 °С. Затем реакционную смесь гасили 2н. соляной кислотой (2 мл), затем подщелачивали насыщенным раствором гидрокарбоната натрия. Полученную смесь затем распределяли между этилацетатом (50 мл) и водой (10 мл). Водный слой отделяли и экстрагировали этилацетатом (1 ×50 мл). Объединенный органический раствор сушили над сульфатом магния, концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 100:0:0 до 98:2:0,2, с получением указанного в заголовке соединения в виде бесцветной смолы с выходом 54% (130 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.93 (s, 6H), 1.26-1.38 (m, 2H), 2.38-2.50 (m, 2H), 2.96-3.10 (m, 2Н), 3.18-3.36 (m, 2Н), 4.06-4.18 (m, 1H), 4.39 (s, 2H), 4.52-4.58 (m, 2H), 5.25-5.33 (m, 1H), 5.37-5.47 (m, 1H), 6.00-6.11 (m, 1H), 6.81-6.93 (m, 3H), 7.20-7.45 (m, 11H); ХИАД-МСНР m/z 481 [M+H] + . Подготовительный пример 73. Амид 5-метил-2,2-дифенил-5-[3-(3-пропенилоксибензилокси)азетидин-1-ил]гексановой кислоты. Гидроксид калия (303 мг; 5,42 ммоль) добавляли к раствору соединения подготовительного примера 72 (130 мг; 0,271 ммоль) в 3-метил-3-пентаноле (5 мл) и эту смесь нагревали при температуре дефлегмации в течение 24 ч. Затем реакционную смесь охлаждали до комнатной температуры, концентрировали в вакууме и остаток распределяли между этилацетатом (50 мл) и водой (10 мл). Водный слой отделяли, экстрагировали этилацетатом (2 ×30 мл) и объединенный органический раствор сушили над сульфатом магния и концентрировали в вакууме с получением бесцветной смолы с выходом 96% (130 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.87 (s, 6H), 1.07-1.19 (m, 2H), 1.64-1.76 (m, 3Н), 2.37-2.47 (m, 2H), 2.94-3.10 (m, 2H), 3.20-3.40 (m, 2H), 4.08-4.20 (m, 1H), 4.35 (s, 2H), 4.83-4.93 (m, 1H), 5.35-5.65 (d, 2H), 6.34-6.40 (d, 1H), 6.88-6.98 (m, 3H), 7.20-7.40 (m, 11H); ХИАД-МСНР m/z 499 [M+H] + . Подготовительный пример 74. 3-Аллилокси-2,6-дихлорбензальдегид. 2,6-Дихлор-3-гидроксибензальдегид (960 мг; 5,03 ммоль) (Synthesis, 2004, 12, 2062), аллилбромид (431 мкл; 5,03 ммоль) и карбонат калия (563 мг; 10,06 ммоль) объединяли в DMF (5 мл) и перемешивали при комнатной температуре в течение 18 ч. DMF удаляли в вакууме и остаток распределяли между диэтиловым эфиром (50 мл) и водой (30 мл). Слои разделяли и водный слой экстрагировали диэтиловым эфиром (2 ×30 мл). Объединенный органический раствор сушили над сульфатом магния и концентрировали в вакууме с получением указанного в заголовке соединения в виде желтого твердого вещества, которое использовали без дополнительной очистки в подготовительном примере 75. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 4.62-4.65 (m, 2Н), 5.35-5.38 (m, 1H), 5.45-5.52 (m, 1H), 6.00-6.13 (m, 1H), 7.00-7.04 (m, 1H), 7.27-7.35 (m, 1H), 10.46 (s, 1H); ХИАД-МСНР m/z 232 [M+H] + . Подготовительный пример 75. (3-Аллилокси-2,6-дихлорфенил)метанол. Продукт подготовительного примера 74 (приблизительно 5,03 ммоль) растворяли в этаноле (30 мл) и добавляли боргидрид натрия (284 мг; 7,79 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Реакционную смесь разбавляли водой (30 мл) и по каплям добавляли ледяную уксусную кислоту до тех пор, пока не прекращалось выделение газа. Смесь экстрагировали диэтиловым эфиром (2 ×50 мл). Объединенные органические экстракты промывали рассолом, сушили над сульфатом магния и концентрировали в вакууме с получением указанного в заголовке соединения в виде бесцветного твердого вещества, которое использовали без дополнительной очистки в подготовительном примере 76. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 4.54-4.64 (m, 2H), 4.98 (s, 2H), 5.33-5.38 (m, 1H), 5.43-5.52 (m, 1H), 5.99-6.11 (m, 1H), 6.83-6.88 (m, 1H), 7.24-7.28 (m, 1H). Подготовительный пример 76. 1-Аллилокси-2,4-дихлор-3-хлорметилбензол. Продукт подготовительного примера 75 (400 мг; 1,72 ммоль) растворяли в дихлорметане (20 мл) и в течение 1 мин добавляли тионилхлорид (312 мкл; 4,29 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Реакцию гасили водой (2 ×10 мл). Органический слой сушили над сульфатом магния и концентрировали в вакууме с получением указанного в заголовке соединения в виде желтого твердого вещества с выходом 91% (390 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 4.54-4.64 (m, 2Н), 4.88 (s, 2Н), 5.28-5.37 (m, 1H), 5.42-5.50 (m, 1H), 5.99-6.11 (m, 1H), 6.83-6.91 (m, 1H), 7.24-7.33 (m, 1H). Подготовительный пример 77. 5-[3-(3-Аллилокси-2,6-дихлорбензилокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Гидрид натрия (60% дисперсия в минеральном масле; 25 мг; 0,629 ммоль) порциями добавляли к охлажденному во льду раствору продукта подготовительного примера 56 (140 мг; 0,419 ммоль) в N,N-диметилформамиде (2 мл) и эту смесь перемешивали при 0 °С в течение 30 мин. Добавляли продукт подготовительного примера 76 (137 мг; 0,546 ммоль) в DMF (1 мл) и смесь перемешивали в течение 18 ч при комнатной температуре. Затем реакционную смесь гасили насыщенным раствором гидрокарбоната натрия (10 мл) и полученную смесь экстрагировали диэтиловым эфиром (3 ×50 мл). Объединенный органический раствор сушили над сульфатом магния, концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 100:0:0 до 98:2:0,2, с получением указанного в заголовке соединения в виде бесцветной смолы с выходом 87% (200 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.28-1.38 (m, 2H), 2.40-2.48 (m, 2H), 3.00-3.10 (m, 2Н), 3.20-3.38 (m, 2Н), 4.15-4.25 (m, 1Н), 4.58-4.62 (m, 2H), 4.68 (s, 2H), 5.26-5.35 (m, 1H), 5.42-5.48 (m, 1H), 6.00-6.08 (m, 1H), 6.82-6.85 (m, 1H), 7.21-7.44 (m, 11H); ХИАД-МСНР m/z 549 [M+H] + . Подготовительный пример 78. Амид 5-(3-{2,6-дихлор-3-[((Е)-пропенил)окси]бензилокси}азетидин-1-ил)-5-метил-2,2-дифенилгексановой кислоты. Гидроксид калия (400 мг; 7,14 ммоль) добавляли к раствору соединения подготовительного примера 77 (200 мг; 0,364 ммоль) в 3-метил-3-пентаноле (3 мл) и эту смесь нагревали при 120 °С в течение 24 ч. Затем реакционную смесь охлаждали до комнатной температуры, концентрировали в вакууме и остаток распределяли между этилацетатом (50 мл) и водой (30 мл). Водный слой отделяли, экстрагировали этилацетатом (2 ×50 мл), объединенный органический раствор сушили над сульфатом магния и концентрировали в вакууме. В результате перекристаллизации из диизопропилового эфира получили указанное в заголовке соединение в виде бесцветного твердого вещества с выходом 73% (150 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.90 (s, 6H), 1.12-1.20 (m, 2H), 1.72-1.78 (m, 3Н), 2.40-2.52 (m, 2Н), 3.02-3.15 (m, 2Н), 3.24-3.40 (m, 2Н), 4.15-4.25 (m, 1H), 4.68 (s, 2H), 4.98-5.07 (m, 1Н), 5.35-5.63 (br m, 2Н), 6.25-6.28 (m, 1Н), 6.92-6.95 (m, 1Н), 7.21-7.38 (m, 11Н); ЭРИ-МСНР m/z 567 [М+Н] + . Подготовительный пример 79. 2-Циклопентилэтиловый эфир сульфаминовой кислоты. К хлорсульфонилизоцианату (275 мл; 3,15 моль) при 0 °С по каплям добавляли муравьиную кислоту (119 мл; 3,15 моль). Полученное твердое вещество оставляли перемешиваться при 0 °С в течение следующих 20 мин. Остаток разбавляли дихлорметаном (875 мл) и нагревали до комнатной температуры в течение 1 ч. Реакционную смесь охлаждали в бане лед/соль и добавляли раствор 2-циклопентилэтанола (240 г; 2,1 моль) в пиридине (255 мл; 3,15 моль) и дихлорметане (2,1 л), поддерживая температуру ниже 7 °С. Через 2 ч реакционную смесь концентрировали в вакууме, разбавляли этилацетатом, промывали насыщенным раствором бикарбоната натрия и рассолом. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением бесцветного масла. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью гептан:этилацетат от 90:10 до 50:50, с получением указанного в заголовке соединения в виде бесцветного масла с выходом 95% (276 г). Подготовительный пример 80. 7,7-Диоксид 8-окса-7-тиа-6-аза-спиро[4.5]декана. К продукту подготовительного примера 79 (276 г; 1,43 моль) в дихлорметане (7,1 л) при комнатной температуре добавляли оксид магния (132,5 г; 3,29 моль), диацетат йодбензола (507 г; 1,57 моль) и димер ацетата родия (12,6 г; 0,028 моль). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Остаток фильтровали через набивку целлита и растворитель удаляли в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью гептан:этилацетат от 90:10 до 50:50, с получением указанного в заголовке соединения в виде белого кристаллического твердого вещество с выходом 75% (205 г). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.63-1.78 (m, 4H), 1.79-2.00 (m, 4H), 2.00-2.11 (m, 2Н), 4.64-4.67 (m, 2H), 4.71 (s, 1H); ХИАД-МСНР m/z 214 [M+Na] + . Подготовительный пример 81. трет-Бутиловый эфир 7,7-диоксо-8-окса-7 λ*6*-тиа-6-аза-спиро[4.5]декан-6-карбоновой кислоты. Продукт подготовительного примера 80 (1,0 г; 5,23 ммоль), ди-трет-бутилдикарбонат (1,36 г; 6,24 ммоль), триэтиламин (1,06 г; 10,5 ммоль) и 4-диметиламинпиридин (126 мг; 1,03 ммоль) объединяли при комнатной температуре в дихлорметане (50 мл). Через 3 ч реакционную смесь промывали хлоридом аммония (50 мл; насыщенный водный раствор), органический раствор сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат от 100:0 до 80:20, с получением указанного в заголовке соединения в виде бесцветного масла с выходом 46% (700 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.48 (s, 9H), 1.48-1.66 (m, 2H), 1.82-1.96 (m, 4H), 2.17-2.33 (m, 4H), 4.57-4.63 (m, 2H). Подготовительный пример 82. 4-(1-Аминоциклопентил)-2,2-дифенилбутиронитрил. Указанное в заголовке соединение получили из продукта подготовительного примера 81, используя способ, аналогичный описанному для подготовительного примера 55, с выходом 67%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.43-1.58 (m, 4H), 1.58-1.67 (m, 4H), 1.72-1.80 (m, 2Н), 2.49-2.60 (m, 2H), 7.25-7.46 (m, 10Н); ХИАД-МСНР m/z 305 [М+Н] + . Подготовительный пример 83. 4-[1-(3-Гидроксиазетидин-1-ил)циклопентил]-2,2-дифенилбутиронитрил. Указанное в заголовке соединение получили из продукта подготовительного примера 82, используя способ, аналогичный описанному для подготовительного примера 56, с выходом 52%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.25-1.38 (m, 2H), 1.39-1.64 (m, 6H), 1.65-1.78 (m, 2Н), 2.42-2.55 (m, 2H), 2.86-2.97 (m, 2H), 3.36-3.40 (m, 2H), 4.29-4.40 (m, 1H), 7.25-7.48 (m, 10Н); ХИАД-МСНР m/z 361 [М+Н] + . Подготовительный пример 84. 1-[1-(3-Циано-3,3-дифенилпропил)циклопентил]азетидин-3-иловый эфир метансульфоновой кислоты. Указанное в заголовке соединение получили из продукта подготовительного примера 83, используя способ, аналогичный описанному для подготовительного примера 57, с выходом 59%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.25-1.67 (m, 10Н), 2.42-2.52 (m, 2H), 3.00 (s, 3Н), 3.15-3.26 (m, 2H), 3.40-3.55 (m, 2H), 4.95-5.05 (m, 1H), 7.27-7.45 (m, 10Н); ХИАД-МСНР m/z 439 [М+Н] + . Подготовительный пример 85. 5-[3-(3-Аллилокси-4-хлорбензилокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из продукта подготовительного примера 56 и продукта подготовительного примера 42, используя способ, аналогичный способу, описанному в примере 101, с выходом 83%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.92 (s, 6H), 1.32-1.38 (m, 2Н), 2.40-2.48 (m, 2H), 2.97-3.06 (m, 2Н), 3.23-3.32 (m, 2Н), 4.05-4.16 (m, 1Н), 4.36 (s, 2H), 4.58-4.65 (m, 2H), 5.28-5.35 (m, 1H), 5.43-5.63 (m, 1H), 6.02-6.16 (m, 1H), 6.80-6.85 (m, 1H), 6.92 (s, 1H), 7.23-7.42 (m, 11H); ХИАД-МСНР m/z 515 [M+H] + Подготовительный пример 86. Амид 5-(3-{4-хлор-3-[((Е)-пропенил)окси]бензилокси}азетидин-1-ил)-5-метил-2,2-дифенилгексановой кислоты. Указанное в заголовке соединение получили из продукта подготовительного примера 85, используя способ, аналогичный описанному для подготовительного примера 73, с выходом 75%. ХИАД-МСНР m/z 533 [М+Н] + . Подготовительный пример 87. 4-{1-[3-(3-Аллилокси-4-хлорфенокси)азетидин-1-ил]циклопентил}-2,2-дифенилбутиронитрил. Указанное в заголовке соединение получили из продукта подготовительного примера 84 и 3-аллилокси-4-хлорфенола (ЕР 78099), используя способ, аналогичный способу, описанному в примере 99, с выходом 82%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.25-1.40 (m, 2H), 1.40-1.65 (m, 6H), 1.66-1.77 (m, 2Н), 2.46-2.58 (m, 2H), 3.07-3.16 (m, 2H), 3.46-3.58 (m, 2H), 4.55-4.59 (m, 2H), 4.60-4.73 (m, 1Н), 5.26-5.35 (m, 1H), 5.43-5.53 (m, 1H), 6.00-6.13 (m, 1H), 6.20-6.28 (m, 1H), 6.42 (s, 1H), 7.19-7.23 (m, 1H), 7.23-7.46 (m, 10Н); ХИАД-МСНР m/z 527 [M+H] + . Подготовительный пример 88. 4-[1-(3-{4-Хлор-3-[((Е)-пропенил)окси]фенокси}азетидин-1-ил)циклопентил]-2,2-дифенилбутирамид. Указанное в заголовке соединение получили из продукта подготовительного примера 87, используя способ, аналогичный описанному для подготовительного примера 73, с выходом 49%. ХИАД-МСНР m/z 545 [М+Н] + . Подготовительный пример 89. 3-Бром-5-метоксифенол. 1-Бром-3,5-диметилоксибензол (3,0 г; 13,8 ммоль) растворяли в дихлорметане (45 мл) и этот раствор охлаждали до -78 °С. Добавляли трибромид бора (1M в дихлорметане; 41 мл; 41 ммоль) и раствор постепенно нагревали до комнатной температуры в течение 18 ч. После охлаждения до -78 °С реакцию гасили водой (100 мл). Органические слои разделяли и промывали раствором тиосульфата натрия, потом водой, затем сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат от 100:0 до 80:20, с получением указанного в заголовке соединения с выходом 16% (470 мг). 1 Н-ЯМР (400 МГц, MeOD) δ: 3.76 (s, 3Н), 6.28 (s, 1H), 6.53-6.57 (m, 2H). Подготовительный пример 90. 4-Аллилокси-3-хлорбензальдегид. Указанное в заголовке соединение получили из 3-хлор-4-гидроксибензальдегида, используя способ, аналогичный описанному для подготовительного примера 74, с выходом 96%. Это вещество использовали без дополнительной очистки в подготовительном примере 91. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 4.68-4.76 (m, 2H), 5.35-5.39 (m, 1H), 5.44-5.56 (m, 1H), 6.02-6.15 (m, 1H), 7.01-7.05 (m, 1H), 7.76-7.79 (m, 1H), 7.93 (s, 1H), 9.86 (s, 1H); ХИАД-МСНР m/z 197 [M+H] + . Подготовительный пример 91. (4-Аллилокси-3-хлорфенил)метанол. Указанное в заголовке соединение получили из продукта подготовительного примера 90, используя способ, аналогичный описанному для подготовительного примера 75, с выходом 100%. Это вещество использовали без дополнительной очистки в подготовительном примере 92. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 4.61-4.65 (m, 4H), 5.28-5.36 (m, 1H), 5.44-5.52 (m, 1H), 6.02-6.13 (m, 1H), 6.88-6.94 (m, 1H), 7.17-7.20 (m, 1H), 7.40 (s, 1H). Подготовительный пример 92. 1-Аллилокси-2-хлор-4-хлорметилбензол. Указанное в заголовке соединение получили из продукта подготовительного примера 91, используя способ, аналогичный описанному для подготовительного примера 76, с выходом 70%. Это вещество использовали без дополнительной очистки в подготовительном примере 94. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 4.52 (s, 2H), 4.60-4.63 (m, 2H), 5.28-5.35 (m, 1H), 5.43-5.54 (m, 1H), 6.01-6.12 (m, 1H), 6.86-6.94 (m, 1H), 7.20-7.24 (m, 1H), 7.42 (s, 1H). Подготовительный пример 93. Амид 5-(3-гидроксиазетидин-1-ил)-5-метил-2,2-дифенилгексановой кислоты. Продукт примера 102 (5,2 г; 12,2 ммоль), формиат аммония (4,25 г; 92 ммоль) и гидроксид палладия 20 мас.% Pd на угле (1,7 г) объединяли в этаноле (150 мл) и перемешивали при температуре дефлегмации в течение 1 ч. После охлаждения остаток фильтровали через Arbocel ®, промывая МеОН (50 мл). Фильтрат концентрировали в вакууме. Остаток разбавляли этилацетатом (100 мл) и промывали раствором гидрокарбоната натрия (30 мл; насыщ. водн.). Органический экстракт сушили над сульфатом магния и концентрировали в вакууме. Указанное в заголовке соединение выделили в виде бесцветной пены с выходом 97% (4,2 г). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.94 (s, 6H), 1.08-1.18 (m, 2H), 2.35-2.45 (m, 2H), 2.85-2.95 (m, 2Н), 3.26-3.35 (m, 2Н), 4.25-4.35 (m, 1H), 5.56-5.80 (br m, 2H), 7.15-7.40 (m, 10Н); ЭРИ-МСНР m/z 353 [M+H] + . Подготовительный пример 94. Амид 5-[3-(4-аллилокси-3-хлорбензилокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Продукт подготовительного примера 93 (300 мг; 0,85 ммоль) растворяли в DMF при 0 °С (5 мл) и добавляли гидрид натрия (60% дисперсия в масле, 62 мг; 1,6 ммоль). Через 30 мин добавляли раствор продукта подготовительного примера 92 (210 мг; 1,15 ммоль) в DMF (1 мл). Через 30 мин реакцию гасили 2н. HCl (20 мл), подщелачивали насыщенным гидрокарбонатом натрия и продукт экстрагировали этилацетатом (20 мл). Органический экстракт сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 100:0:0 до 90:10:1, с получением указанного в заголовке соединения в виде бесцветного масла с выходом 22% (100 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.92 (s, 6H), 1.11-1.20 (m, 2H), 2.39-2.47 (m, 2H), 2.98-3.08 (m, 2Н), 3.23-3.39 (m, 2Н), 4.08-4.17 (m, 1H), 4.28 (s, 2H), 4.58-4.63 (m, 2H), 5.27-5.35 (m, 1H), 5.45-5.50 (m, 1H), 5.40-5.60 (br m, 2H), 6.01-6.13 (m, 1H), 6.85-6.92 (m, 1H), 7.07-7.15 (m, 1H), 7.20-7.37 (m, 11Н); ЭРИ-МСНР m/z 533 [M+H] + . Подготовительный пример 95. 2-Аллилокси-4-хлорбензальдегид. Указанное в заголовке соединение получили из 4-хлор-2-гидроксибензальдегида, используя способ, аналогичный описанному для подготовительного примера 74, с выходом 85%. Это вещество использовали без дополнительной очистки в подготовительном примере 96. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 4.62-4.66 (m, 2H), 5.35-5.39 (m, 1H), 5.40-5.46 (m, 1H), 6.02-6.13 (m, 1H), 6.92-6.96 (m, 1H), 7.45-7.51 (m, 1H), 7.80 (s, 1H), 10.45 (s, 1H); ХИАД-МСНР m/z 197 [M+H] + . Подготовительный пример 96. (2-Аллилокси-4-хлорфенил)метанол. Указанное в заголовке соединение получили из продукта подготовительного примера 95, используя способ, аналогичный описанному для подготовительного примера 75, с выходом 100%. Это вещество использовали без дополнительной очистки в подготовительном примере 97. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 4.55-4.60 (m, 2Н), 4.67 (s, 2Н), 5.28-5.35 (m, 1H), 5.38-5.45 (m, 1H), 5.98-6.08 (m, 1H), 6.76-6.83 (m, 1H), 7.17-7.21 (m, 1H), 7.33 (s, 1H). Подготовительный пример 97. 2-Аллилокси-4-хлор-1-хлорметилбензол. Указанное в заголовке соединение получили из продукта подготовительного примера 96, используя способ, аналогичный описанному для подготовительного примера 76, с выходом 77%. Это вещество использовали без дополнительной очистки в подготовительном примере 98. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 4.57-4.60 (m, 2Н), 4.62 (s, 2Н), 5.28-5.35 (m, 1H), 5.40-5.47 (m, 1H), 6.00-6.09 (m, 1H), 6.78-6.83 (m, 1H), 7.20-7.25 (m, 1H), 7.37 (s, 1H). Подготовительный пример 98. Амид 5-[3-(2-аллилокси-4-хлорбензилокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Указанное в заголовке соединение получили из продуктов подготовительного примера 97 и 93, используя способ, аналогичный описанному для подготовительного примера 94, с выходом 18%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.92 (s, 6H), 1.10-1.98 (m, 2H), 2.38-2.45 (m, 2H), 2.98-3.08 (m, 2Н), 3.28-3.39 (m, 2Н), 4.10-4.20 (m, 1Н), 4.40 (s, 2H), 4.47-4.55 (m, 2H), 5.23-5.32 (m, 1H), 5.35-5.41 (m, 1H), 5.53-5.86 (br m, 2H), 5.97-6.08 (m, 1H), 6.73-6.78 (m, 1H), 7.14-7.20 (m, 1H), 7.20-7.39 (m, 11H); ЭРИ-МСНР m/z 533 [M+H] + . Подготовительный пример 99. 3-Аллилокси-2-хлорбензальдегид. Указанное в заголовке соединение получили из 2-хлор-3-гидроксибензальдегида, используя способ, аналогичный описанному для подготовительного примера 74, с выходом 100%. Это вещество использовали без дополнительной очистки в подготовительном примере 100. Подготовительный пример 100. (3-Аллилокси-2-хлорфенил)метанол. Указанное в заголовке соединение получили из продукта подготовительного примера 99, используя способ, аналогичный описанному для подготовительного примера 31, с выходом 92%. Это вещество использовали без дополнительной очистки в подготовительном примере 101. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 4.62-4.66 (m, 2H), 4.78 (s, 2H), 5.32-5.37 (m, 1H), 5.46-5.52 (m, 1H), 6.02-6.15 (m, 1H), 6.86-6.94 (m, 1H), 7.07-7.14 (m, 1H), 7.20-7.28 (m, 1H). Подготовительный пример 101. 1-Аллилокси-2-хлор-3-хлорметилбензол. Продукт подготовительного примера 100 (740 мг; 3,73 ммоль) растворяли в дихлорметане (20 мл) и добавляли тионилхлорид (678 мкл; 9,32 ммоль) в течение 1 мин. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Добавляли дополнительно 600 мкл тионилхлорида, и реакционную смесь перемешивали в течение 1 ч. Реакцию гасили водой (10 мл). Органический слой промывали насыщенным раствором гидрокарбоната натрия (20 мл) и водой (10 мл), сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат от 100:0 до 99:1, с получением указанного в заголовке соединения в виде бесцветного масла с выходом 21% (168 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 4.59-4.68 (m, 2H), 4.75 (s, 2H), 5.28-5.37 (m, 1H), 5.44-5.55 (m, 1H), 6.00-6.15 (m, 1H), 6.86-6.95 (m, 1H), 7.06-7.13 (m, 1H), 7.17-7.26 (m, 1H). Подготовительный пример 102. 5-[3-(3-Аллилокси-2-хлорбензилокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из продуктов подготовительных примеров 101 и 56, используя способ, аналогичный способу, описанному в примере 101, с выходом 69%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.29-1.38 (m, 2H), 2.40-2.52 (m, 2H), 3.02-3.15 (m, 2Н), 3.24-3.39 (m, 2Н), 4.15-4.22 (m, 1H), 4.53 (s, 2H), 4.60-4.64 (m, 2H), 5.28-5.35 (m, 1H), 5.45-5.50 (m, 1H), 6.03-6.15 (m, 1H), 6.86-6.90 (m, 1H), 7.05-7.08 (m, 1H), 7.16-7.23 (m, 1H), 7.23-7.45 (m, 10Н); ХИАД-МСНР m/z 515 [M+H] + . Подготовительный пример 103. Амид 5-(3-{2-хлор-3-[((Е)-пропенил)окси]бензилокси}азетидин-1-ил)-5-метил-2,2-дифенилгексановой кислоты. Указанное в заголовке соединение получили из продукта подготовительного примера 102, используя способ, аналогичный описанному для подготовительного примера 78, с выходом 61%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.19-1.20 (m, 2H), 1.72-1.78 (m, 3Н), 2.40-2.48 (m, 2H), 3.02-3.15 (m, 2H), 3.20-3.42 (m, 2H), 4.13-4.25 (m, 1H), 4.53 (s, 2H), 4.95-5.03 (m, 1Н), 5.32-5.60 (br m, 2H), 6.30-6.45 (m, 1H), 6.93-6.97 (m, 1H), 7.12-7.40 (m, 12H); ЭРИ-МСНР m/z 533 [M+H] + . Подготовительный пример 104. 1-(4-Карбамоил-1,1-диметил-4,4-дифенилбутил)азетидин-3-иловый эфир метансульфоновой кислоты. Метансульфонилхлорид (102 мкл; 1,33 ммоль) по каплям добавляли к раствору продукта подготовительного примера 93 (156 мг; 0,44 ммоль) в пиридине (5 мл), охлажденному до -20 °С. Смесь постепенно нагревали до 5 °С в течение 2 ч. Добавляли насыщенный раствор гидрокарбоната натрия (10 мл) и реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Остаток экстрагировали этилацетатом (3 ×30 мл) и объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат/метанол/аммиак 0,88 (90/10/1) от 8:1 до 1:2, с получением указанного в заголовке соединения в виде бесцветного масла с выходом 74% (142 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.88 (s, 6H), 1.08-1.15 (m, 2H), 2.38-2.45 (m, 2H), 2.98 (s, 3Н), 3.07-3.22 (m, 2Н), 3.36-3.52 (m, 2Н), 4.95-5.00 (m, 1Н), 5.42-5.53 (br m, 1H), 5.71-5.80 (br m, 1H), 7.23-7.38 (m, 10Н); ЭРИ-МСНР m/z 431 [M+H] + . Подготовительный пример 105. 5-[3-(3-Бензилоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенил-гексаннитрил. Указанное в заголовке соединение получили из продукта подготовительного примера 57 и 3-(бензилокси)фенола способом, который описан в примере 99, в виде желтого масла с выходом 95%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.35-1.42 (m, 2H), 2.40-2.52 (m, 2H), 3.09-3.20 (m, 2Н), 3.40-3.52 (m, 2Н), 4.61-4.72 (m, 1Н), 5.06 (s, 2H), 6.36-6.40 (m, 2H), 6.57-6.62 (m, 1H), 7.10-7.18 (m, 1H), 7.15-7.47 (m, 15H); ХИАД-МСНР m/z 517 [M+H] + . Подготовительный пример 106. (4-Аллилоксифенил)метанол. Указанное в заголовке соединение получили из 4-гидроксибензилового спирта, используя способ, аналогичный описанному для подготовительного примера 74, с выходом 57%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 4.52-4.94 (m, 2H), 4.63 (s, 2H), 5.28-5.35 (m, 1H), 5.38-5.45 (m, 1H), 6.00-6.12 (m, 1H), 6.86-6.94 (m, 2H), 7.24-7.33 (m, 2H). Подготовительный пример 107. 1-Аллилокси-4-хлорметилбензол. Указанное в заголовке соединение получили из продукта подготовительного примера 106, используя способ, аналогичный описанному для подготовительного примера 76, с выходом 43%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 4.53-4.58 (m, 4H), 5.25-5.32 (m, 1H), 5.36-5.43 (m, 1H), 6.00-6.12 (m, 1H), 6.86-6.94 (m, 2H), 7.28-7.35 (m, 2H). Подготовительный пример 108. Амид 5-[3-(4-аллилоксибензилокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Указанное в заголовке соединение получили из продуктов подготовительных примеров 107 и 93, используя способ, аналогичный описанному для подготовительного примера 94, с выходом 25%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.24-1.28 (m, 2H), 2.35-2.45 (m, 2H), 2.94-3.00 (m, 2Н), 3.20-3.28 (m, 2Н), 4.05-4.15 (m, 1Н), 4.35 (s, 2H), 4.52-4.55 (m, 2H), 5.25-5.28 (m, 1H), 5.36-5.43 (m, 1H), 5.40-5.60 (br m, 2H), 6.00, 6.12 (m, 1H), 6.84-6.89 (m, 2H), 7.20-7.38 (m, 12H); ХИАД-МСНР m/z 499 [M+H] + . Пример 1. 5-Метил-5-[(3S)-3-феноксипирролидин-1-ил]-2,2-дифенилгексаннитрил. Раствор продукта подготовительного примера 11 (3,31 г; 8,07 ммоль) в тетрагидрофуране (90 мл) охлаждали до -20 °С. Добавляли хлорид циркония (3,76 г; 16,15 ммоль) и реакционную смесь перемешивали при -20 °С в течение 1 ч. Затем по каплям добавляли хлорид метилмагния (3М в тетрагидрофуране, 24 мл; 72 ммоль), и смесь перемешивали в течение 2 ч при температуре ниже -10 °С. Реакцию гасили 1М водным раствором гидроксида натрия (25 мл) и затем фильтровали через Celite ®, промывая этилацетатом (2 ×50 мл). Фильтрат промывали рассолом (70 мл), концентрировали в вакууме и остаток подвергали перекристаллизации из смеси гексан/этилацетат с получением указанного в заголовке соединения в виде бледно-оранжевого кристаллического твердого вещества с выходом 59% (2 г). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.99 (s, 3Н), 1.04 (s, 3Н), 1.23-1.27 (m, 2H), 1.85-1.93 (m, 1H), 2.07-2.16 (m, 1H), 2.40-2.45 (m, 2H), 2.58-2.67 (m, 2H), 2.72-2.78 (m, 1H), 2.87-2.91 (m, 1H), 4.75-4.79 (m, 1H), 6.80 (d, 2H), 6.88-6.92 (m, 1H), 7.21-7.36 (m, 12H); ХИАД-МСНР m/z 425 [M+H] + . Пример 2. 5-Метил-5-[(3S)-3-феноксипирролидин-1-ил]-2,2-дифенилгексанамид. Гидроксид калия (5,10 г; 91,98 ммоль) добавляли к раствору продукта примера 1 (1,95 г; 4,60 ммоль) в 3-метил-3-пентаноле (40 мл) и эту смесь нагревали при температуре дефлегмации в течение 24 ч. Затем реакционную смесь охлаждали до комнатной температуры, концентрировали в вакууме и остаток распределяли между этилацетатом (70 мл) и водой (40 мл). Водный слой отделяли, экстрагировали этилацетатом (50 мл) и объединенный органический раствор сушили над сульфатом натрия и концентрировали в вакууме. Остаток затем подвергали перекристаллизации из смеси гексан/этилацетат и сушили под вакуумом в течение 18 ч с получением указанного в заголовке соединения в виде белого твердого вещества с выходом 82% (1,66 г). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.97 (s, 3Н), 1.02 (s, 3Н), 1.19-1.33 (m, 2H), 1.82-1.91 (m, 1Н), 2.02-2.17 (m, 1H), 2.37-2.47 (m, 2H), 2.48-2.64 (m, 2H), 2.65-2.75 (m, 1H), 2.81-2.89 (m, 1H), 4.75 (m, 1H), 6.76-6.83 (m, 2H), 6.86-6.92 (m, 1H), 7.17-7.38 (m, 12H); ХИАД-МСНР m/z 425 [M+H] + . Пример 3. 5-Метил-5-[(3R)-3-феноксипирролидин-1-ил]-2,2-дифенилгексаннитрил. Раствор продукта подготовительного примера 12 (0,84 г; 2,05 ммоль) в тетрагидрофуране (15 мл) охлаждали до -10 °С. Добавляли хлорид титана(IV) (0,23 мл; 2,05 ммоль), и реакционную смесь перемешивали при -10 °С в течение 15 мин. Затем по каплям добавляли бромид метилмагния (3М, в диэтиловом эфире; 4,1 мл; 12,3 ммоль) и смесь перемешивали в течение 10 мин при температурах ниже -5 °С и при комнатной температуре в течение 18 ч. Реакционную смесь медленно гасили водой (4 мл), разбавляли этилацетатом (20 мл) и затем декантировали. Оставшееся твердое вещество экстрагировали этилацетатом (3 ×20 мл) и объединенный органический раствор сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат:гексан (60:40), с получением указанного в заголовке соединения с выходом 54% (0,47 г). ХИАД-МСНР m/z 425 [М+Н] + . Пример 4. 5-Метил-5-[(3R)-3-феноксипирролидин-1-ил]-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта примера 3 способом, который описан в примере 2, с выходом 62%. ХИАД-МСНР m/z 443 [М+Н] + . Пример 5. 5-[(3S)-3-(3-Метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из продукта подготовительного примера 13 способом, который описан в примере 1. Сырое соединение очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 97,5:2,5:0,25 до 95:5:0,5, с получением целевого продукта в виде коричневого масла с выходом 78%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.01 (s, 3Н), 1.07 (s, 3Н), 1.43-1.47 (m, 2H), 1.86-1.93 (m, 1H), 2.08-2.19 (m, 1Н), 2.47-2.59 (m, 3Н), 2.65-2.69 (m, 1H), 2.73-2.86 (m, 2H), 3.73 (s, 3H), 4.74-4.79 (m, 1H), 6.38-6.43 (m, 2H), 6.47-6.50 (m, 1H), 7.11-7.15 (m, 1H), 7.24-7.42 (m, 10Н); ХИАД-МСНР m/z 456 [M+H] + . Пример 6. 5-[(3S)-3-(3-Метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта примера 5 способом, который описан в примере 2, в виде бледно-желтой смолы с выходом 96%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.03 (s, 3Н), 1.08 (s, 3Н), 1.24-1.30 (m, 2H), 1.89-1.98 (m, 1Н), 2.08-2.16 (m, 1H), 2.40-2.46 (m, 2H), 2.65-2.76 (m, 2H), 2.79-2.88 (m, 1H), 2.91-2.98 (m, 1H), 3.74 (s, 3H), 4.77-4.82 (m, 1H), 6.37-6.42 (m, 2H), 6.51 (dd, 1H), 7.12-7.16 (m, 1H), 7.22-7.36 (m, 10Н); ХИАД-МСНР m/z 474 [M+H] + . Пример 7. 5-[(3S)-3-(3-Гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Трибромид бора (1М в дихлорметане; 20,7 мл; 20,7 ммоль) добавляли к охлажденному во льду раствору продукта примера 6 (2,45 г; 5,18 ммоль) в дихлорметане (25 мл) и эту смесь перемешивали при 0 °С в течение 20 мин. Затем реакционную смесь гасили раствором аммиака 0,88 и перемешивали при комнатной температуре в течение 20 мин. Реакционную смесь экстрагировали дихлорметаном (3 ×25 мл) и объединенный органический раствор сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат/метанол/аммиак 0,88 (90:10:1) от 50:50 до 33:66, с получением указанного в заголовке соединения в виде белой пены с выходом 60% (1,42 г). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.06 (s, 3Н), 1.11 (s, 3Н), 1.30-1.34 (m, 2H), 1.94-2.01 (m, 1Н), 2.08-2.17 (m, 1H), 2.42-2.46 (m, 2H), 2.77-2.93 (m, 3H), 2.99-3.05 (m, 1H), 4.79 (m, 1H), 6.30 (d, 2H), 6.37-6.40 (d, 1H), 7.02-7.06 (m, 1H), 7.23-7.36 (m, 10Н); ХИАД-MCHP m/z 459 [M+H] + . Пример 8. 5-[(3R)-3-(Бензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Раствор продукта подготовительного примера 15 (40 г; 93 ммоль) в тетрагидрофуране (1 л) охлаждали до -30 °С. Добавляли хлорид циркония (44 г; 186 ммоль) и реакционную смесь перемешивали при -30 °С в течение 1 ч. Затем по каплям добавляли хлорид метилмагния (3М в тетрагидрофуране, 300 мл; 900 ммоль) и смесь перемешивали в течение 2 ч при температуре ниже -10 °С. Реакцию гасили 1М водным раствором гидроксида натрия (300 мл) и смесь затем декантировали. Оставшееся твердое вещество экстрагировали этилацетатом (2 ×500 мл) и объединенный органический раствор упаривали при пониженном давлении. Остаток затем растворяли в дихлорметане (1 л), промывали водой (200 мл) и концентрировали в вакууме. Сырое вещество очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол (97,5:2,5) и остаток подвергали азеотропной отгонке последовательно с пентаном (2 ×250 мл), диэтиловым эфиром (2 ×250 мл) и пентаном (2 ×250 мл) с получением указанного в заголовке соединения в виде твердого вещества. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.02 (s, 3Н), 1.04 (s, 3Н), 1.42-1.54 (m, 2H), 1.78-1.86 (m, 1Н), 1.93-2.02 (m, 1H), 2.47-2.60 (m, 3H), 2.63-2.77 (m, 3H), 4.04-4.08 (m, 1H), 4.43-4.49 (s, 2H), 7.23-7.43 (m, 15H); ХИАД-МСНР m/z 439 [M+H] + . Пример 9. 5-[(3R)-3-(Бензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта примера 8 способом, который описан в примере 2, в виде твердого вещества с выходом 89%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.99-1.01 (m, 6H), 1.24-1.28 (m, 2H), 1.75-1.82 (m, 1Н), 1.88-1.97 (m, 1H), 2.40-2.44 (m, 2H), 2.49-2.68 (m, 2H), 2.71-2.76 (m, 1H), 4.00-4.05 (m, 1H), 4.39-4.46 (m, 2H), 7.22-7.38 (m, 15H); ХИАД-МСНР m/z 425 [M+H] + . Пример 10. 5-[(3S)-3-(Бензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из продукта подготовительного примера 14 способом, который описан в примере 1. Сырое соединение очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 99:1:0,1 до 92:8:0,8, с получением целевого продукта в виде коричневого масла с выходом 76%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.02 (s, 3Н), 1.04 (s, 3Н), 1.41-1.55 (m, 2H), 1.77-1.85 (m, 1Н), 1.93-2.01 (m, 1Н), 2.50-2.55 (m, 3Н), 2.63-2.77 (m, 3Н), 4.03-4.08 (m, 1H), 4.45 (s, 2H), 7.22-7.42 (m, 15H); ХИАД-МСНР m/z 439 [M+H] + . Пример 11. 5-[(3S)-3-(Бензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта примера 10 способом, который описан в примере 2. Сырое соединение очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат:метанол:аммиак 0,88 (98:2:0,2), с получением целевого продукта в виде бесцветной смолы с выходом 78%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.98 (s, 3Н), 1.00 (s, 3Н), 1.23-1.27 (m, 2H), 1.73-1.80 (m, 1Н), 1.87-1.96 (m, 1H), 2.40-2.65 (m, 5H), 2.69-2.73 (m, 1H), 3.98-4.03 (m, 1H), 4.42 (m, 2H), 7.22-7.32 (m, 11H), 7.35-7.38 (m, 4H); ХИАД-МСНР m/z 458 [M+H] + . Пример 12. 5-[(3R)-3-(3-Метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Раствор трифенилфосфина (272 мг; 1,04 ммоль) в тетрагидрофуране (3 мл) и диизопропилазодикарбоксилат (0,20 мл; 1,04 ммоль) добавляли к раствору продукта подготовительного примера 16 (190 мг; 0,52 ммоль) в тетрагидрофуране (2 мл) и эту смесь перемешивали при комнатной температуре в течение 15 мин. Добавляли раствор 3-метоксифенола (129 мг; 1,04 ммоль) в тетрагидрофуране (2 мл) и смесь перемешивали при комнатной температуре в течение 2,5 ч. Затем реакционную смесь концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол от 95:5 до 90:10, с получением указанного в заголовке соединения в виде бесцветного масла с выходом 7% (20 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.05 (s, 3Н), 1.10 (s, 3Н), 1.18-1.37 (m, 2H), 1.92-2.00 (m, 1Н), 2.10-2.16 (m, 1H), 2.40-2.45 (m, 2H), 2.72-2.95 (m, 3H), 2.97-3.05 (m, 1H), 3.74 (s, 3H), 4.80-4.14 (m, 1H), 6.38-6.40 (m, 2H), 6.51 (d, 1H), 7.12-7.16 (m, 2H), 7.22-7.37 (m, 9H); ХИАД-МСНР m/z 472 [М+Н] + . Пример 13. 5-[(3R)-3-(3-Гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Трибромид бора (1M в дихлорметане; 0,17 мл; 169 ммоль) добавляли к раствору продукта примера 12 (20 мг; 42 ммоль) в дихлорметане (2 мл) и эту смесь перемешивали при комнатной температуре. Ход реакции контролировали ТСХ-анализом (тонкослойной хроматографией) и добавляли порции трибромида бора (1M в дихлорметане; 0,17 мл; 42 ммоль) с регулярными интервалами до тех пор, пока все исходное вещество не было потреблено. Через 8 суток реакцию гасили раствором аммиака 0,88, перемешивали при комнатной температуре в течение 1 ч, затем экстрагировали дихлорметаном (3 ×5 мл). Объединенный органический раствор сушили над сульфатом натрия, концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 95:5:0,5 до 92:8:0,8. Соответствующие фракции упаривали при пониженном давлении и остаток растворяли в этаноле. Добавляли формиат аммония (12 мг; 0,19 ммоль) и 20% Pd(OH) 2 /C (2 мг) и смесь нагревали при температуре дефлегмации в течение 4 ч. Затем реакционную смесь охлаждали до комнатной температуры, фильтровали через Arbocel ® и фильтрат концентрировали в вакууме. Фильтрат распределяли между этилацетатом (8 мл) и водным раствором аммиака (2 мл), водный слой отделяли и дополнительно экстрагировали этилацетатом (2 ×3 мл). Объединенный органический раствор сушили над сульфатом натрия, концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 (95:5:0,5), с получением указанного в заголовке соединения в виде бесцветной смолы с выходом 68% (5,8 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.99 (s, 3Н), 1.05 (s, 3Н), 1.23-1.28 (m, 2H), 1.85-1.90 (m, 1Н), 2.06-2.12 (m, 1H), 2.40-2.45 (m, 2H), 2.57-2.69 (m, 2H), 2.72-2.79 (m, 1H), 2.85-2.90 (m, 1H), 4.70-4.73 (m, 1H), 6.26-6.30 (m, 2H), 6.36 (d, 1H), 7.00-7.04 (m, 1H), 7.21-7.25 (m, 2H), 7.27-7.31 (m, 4H), 7.33-7.36 (m, 4H); ЭРИ-МСНР m/z 459 [M+H] + . Пример 14. 5-Метил-5-[(3S)-3-(3-метилфенокси)пирролидин-1-ил]-2,2-дифенилгексанамид. Диизопропилазодикарбоксилат (65 мкл; 0,34 ммоль) и продукт подготовительного примера 17 (62 мг; 0,17 ммоль) добавляли к раствору трифенилфосфина (89 мг; 0,34 ммоль) в тетрагидрофуране (5 мл) и эту смесь перемешивали при комнатной температуре в течение 15 мин. Добавляли раствор 3-метилфенола (27 мг; 0,25 ммоль) в тетрагидрофуране (1 мл) и смесь перемешивали при комнатной температуре в течение 2 ч. Затем реакционную смесь концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол от 100:0 до 93:7, с получением указанного в заголовке соединения в виде бесцветной смолы с выходом 43% (33 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.97 (s, 3Н), 1.03 (s, 3Н), 1.21-1.26 (m, 2H), 1.82-1.90 (m, 1Н), 2.05-2.13 (m, 1H), 2.27 (s, 3H), 2.40-2.45 (m, 2H), 2.51-2.61 (m, 2H), 2.68-2.74 (m, 1H), 2.81-2.85 (m, 1H), 4.71-4.75 (m, 1H), 6.57-6.62 (m, 2H), 6.72 (d, 1H), 7.08-7.12 (m, 1H), 7.35-7.20 (m, 10Н); ХИАД-МСНР m/z 457 [M+H] + . Пример 15. 5-[(3R)-3-(1,3-Бензоксазол-6-илокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Диэтилазодикарбоксилат (125 мкл; 0,74 ммоль) и раствор 6-бензоксазолола [(100 мг; 0,74 ммоль), US 613027, с. 56] в тетрагидрофуране (2 мл) добавляли к охлажденному во льду раствору трифенилфосфина (195 мг; 0,74 ммоль) в тетрагидрофуране (2 мл) и эту смесь перемешивали при 0 °С в течение 10 мин и при комнатной температуре в течение 90 мин. Затем добавляли раствор продукта подготовительного примера 16 (238 мг; 0,65 ммоль) в тетрагидрофуране (2 мл) и смесь перемешивали при комнатной температуре в течение 18 ч. Затем реакционную смесь распределяли между этилацетатом и разбавленным раствором карбоната натрия, и водный слой отделяли и экстрагировали этилацетатом ( ×2). Объединенный органический раствор сушили над сульфатом магния, концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол (90:10), с получением указанного в заголовке соединения с выходом 29% (90 мг). ХИАД-МСНР m/z 484 [М+Н] + . Примеры 16 и 17. Указанные ниже соединения показанной ниже общей формулы получили способом, который описан в примере 15, с использованием продукта подготовительного примера 17 и коммерчески доступных фенолов. Протекание реакций контролировали ТСХ-анализом и реакционные смеси перемешивали при комнатной температуре в течение 18-96 ч. Пример 18. 5-{(3S)-3-[(3'-Гидроксибифенил-4-ил)окси]пирролидин-1-ил}-5-метил-2,2-дифенилгексанамид. Диизопропилазодикарбоксилат (0,21 мл; 1,09 ммоль) и продукт подготовительного примера 17 (218 мг; 1,09 ммоль) добавляли к раствору трифенилфосфина (286 мг; 1,09 ммоль) в тетрагидрофуране (5 мл) и эту смесь перемешивали при комнатной температуре в течение 15 мин. Добавляли раствор 3-метокси-1,1'-бифенил-4-ола [(200 мг; 0,55 ммоль) Bioinorganic and Medicinal Chemistry, 2003, 11, 2347] в тетрагидрофуране (2 мл) и смесь перемешивали при комнатной температуре в течение 4 ч. Затем реакционную смесь концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол от 100:0 до 93:7. Соответствующие фракции упаривали при пониженном давлении и остаток затем очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат/метанол/аммиак 0,88 (90:10:1) от 100:0 до 50:50. Соответствующие фракции концентрировали в вакууме и остаток растворяли в дихлорметане (2 мл). Добавляли трибромид бора (1М в дихлорметане; 0,58 мл; 0,58 ммоль) и смесь перемешивали при комнатной температуре в течение 90 мин. Затем реакционную смесь гасили добавлением по каплям раствора аммиака 0,88 и смесь перемешивали при комнатной температуре в течение следующих 30 мин. Затем реакционную смесь экстрагировали дихлорметаном (3 ×20 мл) и объединенный органический раствор концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат/метанол/аммиак 0,88 (90:10:1) от 100:0 до 50:50, с получением указанного в заголовке соединения в виде пены с выходом 5% (14 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.96 (s, 3Н), 1.04 (s, 3Н), 1.21-1.26 (m, 2H), 1.86-1.93 (m, 1Н), 2.10-2.15 (m, 1H), 2.39-2.46 (m, 2H), 2.52-2.57 (m, 1H), 2.62 (dd, 1H), 2.70-2.76 (m, 1H), 2.83 (dd, 1H), 4.73-4.79 (m, 1H), 6.70-6.72 (dd, 1H), 6.84-6.87 (m, 2H), 6.97-7.02 (m, 2H), 7.18-7.57 (m, 13H); ХИАД-МСНР m/z 535 [M+H] + . Пример 19. 5-{(3S)-3-[(3'-Гидроксибифенил-3-ил)окси]пирролидин-1-ил}-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта подготовительного примера 17 и 3-метокси-1,1 -бифенил-3-ола [WO 2003 006437, с. 45] способом, который описан в примере 18, в виде бесцветной смолы с выходом 16%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.98 (s, 3Н), 1.03 (s, 3Н), 1.21-1.25 (m, 2H), 1.87-1.94 (m, 1H), 2.09-2.18 (m, 1Н), 2.40-2.45 (m, 2H), 2.53-2.58 (m, 1H), 2.61-2.64 (dd, 1H), 2.71-2.76 (m, 1H), 2.82-2.87 (dd, 1H), 4.80-4.83 (m, 1H), 6.76-6.76 (m, 2H), 6.97-7.04 (m, 3H), 7.12 (d, 1H), 7.16-7.35 (m, 12H); ХИАД-МСНР m/z 535 [M+H] + . Пример 20. 5-{(3S)-3-[(6-Гидрокси-2-нафтил)окси]пирролидин-1-ил}-5-метил-2,2-дифенилгексанамид. Фторид аммония (27 мг; 0,738 ммоль) добавляли к раствору продукта подготовительного примера 19 (46 мг; 0,0738 ммоль) в метаноле (3 мл) и воде (0,3 мл) и эту смесь перемешивали при комнатной температуре в течение 18 ч. Смесь концентрировали в вакууме и остаток очищали с использованием силикагелевого картриджа RediSep ®, элюируя смесью дихлорметан:метанол:аммиак 0,88 (от 100:0:0 до 92:8:0,8). Соответствующие фракции концентрировали в вакууме и остаток растворяли в диэтиловом эфире (30 мл), промывали водой (2 ×10 мл), сушили над сульфатом магния и концентрировали в вакууме с получением указанного в заголовке соединения в виде беловатой пены с выходом 35% (14 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.97 (s, 3Н), 1.06 (s, 3Н), 1.17-1.26 (m, 2H), 1.88-1.98 (m, 1H), 2.09-2.25 (m, 1Н), 2.33-2.51 (m, 2H), 2.54-2.62 (m, 1H), 2.63-2.68 (m, 1H), 2.71-2.80 (m, 1H), 2.81-2.91 (m, 1H), 4.80-4.95 (m, 1H), 6.89-7.00 (m, 2H), 7.00-7.06 (m, 2H), 7.10-7.26 (m, 6H), 7.26-7.35 (m, 4H), 7.47-7.54 (m, 1H), 7.56-7.62 (m, 1H); ХИАД-МСНР m/z 509 [M+H] + . Пример 21. 5-[(3S)-3-(2-Гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Диизопропилазодикарбоксилат (0,22 мл; 1,12 ммоль) добавляли к охлажденному во льду раствору продукта подготовительного примера 17 (205 мг; 0,56 ммоль), трифенилфосфина (293 мг; 1,12 ммоль) и 2-гидроксифенола (616 мг; 5,59 ммоль) в тетрагидрофуране (4 мл) и раствор перемешивали с охлаждением во льду в течение 2 ч. Добавляли трифенилфосфин (293 мг; 1,12 ммоль) и диизопропилазодикарбоксилат (0,22 мл; 1,12 ммоль) и полученный раствор перемешивали при комнатной температуре в течение 16 ч. Добавляли диизопропилазодикарбоксилат (0,22 мл; 1,12 ммоль) и раствор перемешивали при комнатной температуре в течение 4 ч. ТСХ-анализ показал полное завершение реакции. Затем реакционную смесь концентрировали в вакууме и остаток очищали с использованием картриджа Isolute ® SCX-2, элюируя метанолом, затем 1М аммиаком в метаноле. Основные фракции упаривали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:(этилацетат:метанол:аммиак 0,88, 90:10:1) от 1:0 до 1:1, с получением указанного в заголовке соединения в виде коричневой смолы с выходом 38% (98 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.02 (s, 3Н), 1.06 (s, 3Н), 1.25-1.29 (m, 2H), 1.87-1.94 (m, 1Н), 2.03-2.11 (m, 1H), 2.41-2.45 (m, 2H), 2.51-2.57 (m, 1H), 2.66-2.70 (dd, 1H), 2.75-2.84 (m, 2H), 4.80-4.85 (m, 1H), 6.71-6.84 (m, 4H), 7.21-7.37 (m, 10Н); ХИАД-МСНР m/z 459 [M+H] + , 458 [M-1] - . Пример 22. 5-[(3S)-3-(4-Метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Диизопропилазодикарбоксилат (0,21 мл; 1,09 ммоль) добавляли к раствору продукта подготовительного примера 17 (200 мг; 0,546 ммоль), трифенилфосфина (286 мг; 1,09 ммоль) и 4-метоксифенола (135 мг; 1,09 ммоль) в тетрагидрофуране (3 мл) и раствор перемешивали при комнатной температуре в течение 16 ч. Затем реакционную смесь концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат от 100:0 до 50:50. Содержащие продукт фракции упаривали при пониженном давлении и остаток затем очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат:метанол:аммиак 0,88 от 100:0:0 до 95:5:0,5, с получением указанного в заголовке соединения в виде белой пены с выходом 14% (36 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.96 (s, 3Н), 1.02 (s, 3Н), 1.19-1.25 (m, 2H), 1.81-1.91 (m, 1H), 2.00-2.11 (m, 1H), 2.35-2.60 (m, 4H), 2.66-2.72 (m, 1H), 2.77-2.81 (m, 1H), 3.73 (s, 3H), 4.64-4.69 (m, 1H), 6.72-6.81 (m, 4H), 7.20-7.39 (m, 10Н); ХИАД-МСНР m/z 473 [M+H] + . Пример 23. 5-[(3S)-3-(4-Гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Трибромид бора (1М в дихлорметане; 0,76 мл; 0,761 ммоль) добавляли к раствору продукта примера 22 (36 мг; 0,0761 ммоль) в дихлорметане (3 мл) и эту смесь перемешивали при комнатной температуре в течение 1 ч. ТСХ-анализ показал полное завершение реакции. Реакцию гасили добавлением по каплям аммиака 0,88 (2 мл) и реакционную смесь перемешивали в течение 30 мин, при этом выделялся газ. Реакционную смесь расслаивали и водный слой экстрагировали дихлорметаном (2 ×5 мл). Объединенные органические фракции концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя начиная с этилацетата до смеси 95:5:0,5 этилацетат:метанол:аммиак 0,88, с получением указанного в заголовке соединения в виде белой пены с выходом 46% (16 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.97 (s, 3Н), 1.03 (s, 3Н), 1.20-1.25 (m, 2H), 1.81-1.89 (m, 1Н), 2.01-2.09 (m, 1Н), 2.39-2.44 (m, 2Н), 2.51-2.61 (m, 2Н), 2.67-2.73 (m, 1Н), 2.77-2.81 (m, 1H), 4.62-4.66 (m, 1H), 6.63-6.69 (m, 4H), 7.21-7.37 (m, 10Н); ХИАД-МСНР m/z 458 [M-1] - . Пример 24. 5-[(3S)-3-(4-Трифторметилфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Диизопропилазодикарбоксилат (0,11 мл; 0,546 ммоль) добавляли к раствору продукта подготовительного примера 17 (100 мг; 0,273 ммоль), трифенилфосфина (143 мг; 0,546 ммоль) и 4-трифторметилфенола (88 мг; 0,546 ммоль) в тетрагидрофуране (3 мл) и раствор перемешивали при комнатной температуре в течение 4 ч. Затем реакционную смесь концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:(этилацетат:метанол:аммиак 0,88, 90:10:1) от 1:0 до 1:1, с получением указанного в заголовке соединения в виде белой пены с выходом 34% (48 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.97 (s, 3Н), 1.03 (s, 3Н), 1.21-1.26 (m, 2H), 1.84-1.92 (m, 1H), 2.11-2.20 (m, 1Н), 2.35-2.50 (m, 2H), 2.52-2.62 (m, 2H), 2.69-2.75 (m, 1H), 2.85-2.89 (m, 1H), 4.81-4.86 (m, 1H), 6.93-6.96 (d, 2H), 7.20-7.35 (m, 10H), 7.52-7.54 (d, 2H); ХИАД-МСНР m/z 511 [M+H] + , 510 [M-1] - . Пример 25. 5-[(3R)-3-(4-Трифторметилфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Гидрид натрия (60% дисперсия в минеральном масле; 23 мг; 0,574 ммоль) добавляли к охлажденному во льду раствору продукта подготовительного примера 20 (100 мг; 0,287 ммоль) в N,N-диметилформамиде (1,5 мл) и эту смесь перемешивали при 0 °С в течение 30 мин. Добавляли 4-фторбензотрифторид (71 мг; 0,431 ммоль) в N,N-диметилформамиде (0,5 мл), и смесь перемешивали в течение 16 ч, давая температуре подняться до 25 °С. Раствор перемешивали при 50 °С в течение 24 ч и затем перемешивали при 25 °С в течение 48 ч. Добавляли гидрид натрия (60% дисперсия в минеральном масле; 23 мг; 0,574 ммоль) и раствор перемешивали в течение 1,25 ч при 50 °С. Затем реакционную смесь охлаждали до 25 °С и гасили водой (8 мл), концентрировали в вакууме и водный остаток распределяли между этилацетатом (10 мл) и водой (10 мл). Водный слой отделяли и экстрагировали этилацетатом (2 ×15 мл). Объединенный органический раствор промывали рассолом (5 мл), концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат от 3:1 до 0:1, с получением указанного в заголовке соединения в виде бесцветной смолы с выходом 70% (10 мг). 1 H-ЯМР (400 МГц, CD 3 OD) δ: 1.02 (s, 3Н), 1.07 (s, 3Н), 1.44-1.48 (m, 2H), 1.88-1.95 (m, 1Н), 2.16-2.25 (m, 1H), 2.49-2.53 (m, 2H), 2.56-2.62 (m, 1H), 2.67-2.70 (m, 1H), 2.75-2.80 (q, 1H), 2.89-2.93 (dd, 1H), 4.85-4.89 (m, 1H), 6.96-6.98 (d, 2H), 7.42-7.45 (m, 10H), 7.53-7.55 (d, 2H); ХИАД-МСНР m/z 493 [M+H] + . Пример 26. 5-[(3R)-3-(4-Трифторметилфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Гидроксид калия (23 мг; 0,406 ммоль) добавляли к раствору соединения примера 25 (10 мг; 0,0203 ммоль) в 3-метил-3-пентаноле (2 мл) и эту смесь нагревали при температуре дефлегмации в течение 16 ч. Раствор охлаждали до 25 °С, добавляли гидроксид калия (23 мг; 0,406 ммоль) и раствор нагревали при температуре дефлегмации в течение дополнительных 24 ч. Затем реакционную смесь охлаждали до комнатной температуры, концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат:метанол:аммиак 0,88 (95:5:0,5), с получением указанного в заголовке соединения в виде бесцветной смолы с выходом 68% (7 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.97 (s, 3Н), 1.03 (s, 3Н), 1.20-1.25 (m, 2H), 1.84-1.91 (m, 1Н), 2.10-2.20 (m, 1H), 2.34-2.49 (m, 2H), 2.52-2.61 (m, 2H), 2.69-2.74 (m, 1H), 2.84-2.88 (dd, 1H), 4.81-4.85 (m, 1H), 6.94-6.96 (d, 2H), 7.20-7.35 (m, 10H), 7.53-7.55 (d, 2H); ХИАД-МСНР m/z 511 [M+H] + , 510 [M-1] - . Пример 27. 5-[(3R)-3-(3-Хлор-4-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Гидрид натрия (60% дисперсия в минеральном масле; 31 мг; 0,776 ммоль) добавляли к охлажденному во льду раствору продукта подготовительного примера 20 (135 мг; 0,388 ммоль) в N,N-диметилформамиде (4 мл) и эту смесь перемешивали при 0 °С в течение 15 мин. Добавляли 2-хлор-4-фторанизол (93 мг; 0,582 ммоль) в N,N-диметилформамиде (1 мл) и смесь перемешивали в течение 16 ч при 50 °С. Раствор охлаждали до 25 °С, добавляли гидрид натрия (60% дисперсия в минеральном масле; 62 мг; 1,55 ммоль) и раствор перемешивали в течение 16 ч при 80 °С. Затем реакционную смесь охлаждали до 25 °С и гасили водой (3 мл), концентрировали в вакууме и водный остаток распределяли между этилацетатом (20 мл) и водой (5 мл). Водный слой отделяли и экстрагировали этилацетатом (2 ×20 мл). Объединенный органический раствор промывали рассолом (10 мл), концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:(этилацетат:метанол:аммиак 0,88, 90:10:1) от 3:1 до 1:1. Содержащие продукт фракции упаривали при пониженном давлении и остаток дополнительно очищали с использованием картриджа Isolute ® SCX-2, элюируя метанолом, затем 1М аммиаком в метаноле. Основные фракции упаривали с получением указанного в заголовке соединения в виде желтой смолы с выходом 32% (61 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.03 (s, 3Н), 1.08 (s, 3Н), 1.28-1.31 (m, 2H), 1.76-1.80 (m, 1Н), 1.86-1.93 (m, 1H), 2.09-2.18 (m, 1H), 2.49-2.63 (m, 2H), 2.67-2.70 (m, 1H), 2.76-2.87 (m, 2H), 3.80 (s, 3H), 4.71-4.75 (m, 1H), 6.74-6.77 (dd, 1H), 6.89-6.95 (dd, 1H), 7.24-7.46 (m, 10H), 8.45-8.59 (m, 1H); ХИАД-МСНР m/z 489 [M+H] + . Пример 28. 5-[(3R)-3-(3-Хлор-4-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Гидроксид калия (140 мг; 2,49 ммоль) добавляли к раствору продукта примера 27 (61 мг; 0,125 ммоль) в 3-метил-3-пентаноле (5 мл) и эту смесь нагревали при температуре дефлегмации в течение 16 ч. Раствор охлаждали до комнатной температуры, концентрировали в вакууме и остаток распределяли между дихлорметаном (10 мл) и водой (5 мл), водный слой экстрагировали дихлорметаном (3 ×10 мл). Объединенные органические слои концентрировали при пониженном давлении и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат:метанол:аммиак 0,88 (90:10:1). Содержащие продукт фракции упаривали при пониженном давлении с получением указанного в заголовке соединения в виде бесцветной смолы с выходом 33% (21 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.03 (s, 3Н), 1.09 (s, 3Н), 1.26-1.30 (m, 2H), 1.88-1.96 (m, 1Н), 2.05-2.14 (m, 1H), 2.35-2.49 (m, 2H), 2.70-2.75 (m, 2H), 2.82-2.88 (m, 1H), 2.91-2.96 (m, 1H), 3.81 (s, 3H), 4.72-4.76 (m, 1H), 6.73-6.76 (dd, 1H), 6.88-6.89 (d, 1H), 6.94-6.96 (d, 1H), 7.24-7.36 (m, 10Н); ХИАД-МСНР m/z 507 [M+H] + , 505 [M-1] - . Пример 29. Амид 5-[(3S)-3-(3-гидрокси-5-метилфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Диизопропилазодикарбоксилат (212 мкл; 1,093 ммоль) добавляли тремя порциями к охлажденному во льду раствору трифенилфосфина (287 мг; 1,093 ммоль), 5-метилрезорцина (678 мг; 5,464 ммоль) и продукта подготовительного примера 17 (200 мг; 0,546 ммоль) в тетрагидрофуране (8 мл) и эту смесь перемешивали в течение 2 ч, при этом температура повышалась от 0 °С до комнатной температуры. Реакционную смесь концентрировали в вакууме и остаток очищали с использованием картриджа Isolute ® SCX-2, элюируя метанолом, затем 1М аммиаком в метаноле. Основные фракции концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат:метанол:аммиак 0,88 от 98:2:0,2 до 94:6:0,6, с получением указанного в заголовке соединения в виде белой пены с выходом 27% (70 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.00 (s, 3Н), 1.06 (s, 3Н), 1.24-1.27 (m, 2H), 1.87-1.93 (m, 1H), 2.05-2.13 (m, 1H), 2.19 (s, 3H), 2.39-2.45 (m, 2H), 2.61-2.69 (m, 2H), 2.75-2.81 (m, 1H), 2.85-2.90 (m, 1H), 4.69-4.73 (m, 1H), 6.07 (s, 1H), 6.13 (s, 1H), 6.21 (s, 1H), 7.21-7.35 (m, 10Н); ЭРИ-МСНР m/z 473 [M+H] + . Пример 30. Амид 5-[(3S)-3-(3-гидрокси-2-метилфенокси)пирролидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. Указанное в заголовке соединение получили из продукта подготовительного примера 17 и 2-метилрезорцина способом, который описан в примере 29, в виде не совсем белой пены с выходом 68%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.99 (s, 3Н), 1.04 (s, 3Н), 1.23-1.28 (m, 2H), 1.86-1.93 (m, 1Н), 1.98 (s, 3Н), 2.02-2.11 (m, 1H), 2.41-2.45 (m, 2H), 2.57-2.67 (m, 2H), 2.71-2.78 (m, 1H), 2.88-2.92 (m, 1H), 4.69-4.72 (m, 1H), 6.26-6.28 (d, 1H), 6.38-6.40 (d, 1H), 6.87-6.91 (t, 1H), 7.21-7.24 (m, 2H), 7.27-7.30 (t, 4H), 7.34-7.36 (m, 4H); ЭРИ-МСНР m/z 473 [M+H] + . Пример 31. Амид 5-[(3R)-3-(2,4-дихлор-5-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Указанное в заголовке соединение получили из продукта подготовительного примера 17 и 4,6-дихлоррезорцина способом, который описан в примере 29, с добавлением дополнительного количества трифенилфосфина (2 экв.) и диизопропилазодикарбоксилата (2 экв.) и через 18 ч, и через 24 ч, и с последующим перемешиванием в течение еще 24 ч, в виде не совсем белой пены с выходом 21%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.99 (s, 3Н), 1.06 (s, 3Н), 1.24-1.28 (m, 2H), 1.89-1.96 (m, 1Н), 2.05-2.14 (m, 1H), 2.38-2.49 (m, 2H), 2.59-2.70 (m, 2H), 2.75-2.81 (m, 1H), 2.89-2.93 (m, 1H), 4.71-4.74 (m, 1H), 6.48 (s, 1H), 7.21-7.24 (m, 3H), 7.26-7.31 (t, 4H), 7.34-7.37 (m, 4H); ХИАД-МСНР m/z 527 [M+H] + . Пример 32. Амид 5-[(3S)-3-(4,5-дихлор-2-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Указанное в заголовке соединение получили из продукта подготовительного примера 17 и 4,5-дихлоркатехина способом, который описан в примере 29, с добавлением дополнительного количества трифенилфосфина (2 экв.) и диизопропилазодикарбоксилата (2 экв.) и через 18 ч, и через 24 ч, и с последующим перемешиванием в течение еще 24 ч, в виде не совсем белой пены с выходом 18%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.03 (s, 3Н), 1.09 (s, 3Н), 1.19-1.31 (m, 2H), 1.90-1.97 (m, 1H), 2.05-2.14 (m, 1H), 2.34-2.49 (m, 2H), 2.61-2.67 (m, 1H), 2.69-2.73 (m, 1H) 2.76-2.79 (m, 1H), 2.86-2.92 (m, 1H), 4.78-4.83 (m, 1H), 6.85 (s, 1H), 6.94 (s, 1H), 7.21-7.40 (m, 10Н); ЭРИ-МСНР m/z 527 [M+H] + . Пример 33. Амид 5-[(3S)-3-(3-хлор-5-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. Диизопропилазодикарбоксилат (160 мкл; 0,820 ммоль) добавляли тремя порциями к охлажденному во льду раствору трифенилфосфина (215 мг; 0,820 ммоль), 3-хлор-5-метоксифенола (325 мг; 2,049 ммоль) и продукта подготовительного примера 17 (150 мг; 0,410 ммоль) в тетрагидрофуране (8 мл) и эту смесь перемешивали в течение 18 ч, при этом температура повышалась от 0 °С до комнатной температуры. Реакционную смесь концентрировали в вакууме и очищали с использованием картриджа Isolute ® SCX-2, элюируя метанолом, затем 2М аммиаком в метаноле. Основные фракции концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 98:2:0,2 до 94:6:0,6, с получением указанного в заголовке соединения в виде зеленой смолы с выходом 97% (200 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.97 (s, 3Н), 1.03 (s, 3Н), 1.20-1.25 (m, 2H), 1.81-1.89 (m, 1Н), 2.06-2.14 (m, 1H), 2.35-2.49 (m, 2H), 2.51-2.60 (m, 2H), 2.67-2.73 (m, 1H), 2.79-2.83 (m, 1H), 3.75 (s, 3H), 4.70-4.74 (m, 1H), 6.29-6.30 (t, 1H), 6.41-6.42 (t, 1H), 6.51-6.52 (t, 1H), 7.20-7.24 (m, 2H), 7.26-7.30 (t, 4H), 7.33-7.35 (m, 4H); ЭРИ-МСНР m/z 507 [M+H] + . Пример 34. Амид 5-[(3S)-3-(3-хлор-5-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. Трибромид бора (1М в дихлорметане; 1,97 мл; 1,976 ммоль) добавляли к охлажденному во льду раствору продукта примера 33 (200 мг; 0,395 ммоль) в дихлорметане (10 мл) и эту смесь перемешивали при увеличении температуры от 0 °С до комнатной температуры в течение 18 ч. Реакцию гасили раствором аммиака 0,88 и перемешивали при комнатной температуре в течение 90 мин. Реакционную смесь экстрагировали дихлорметаном (3 ×10 мл) и объединенный органический раствор сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 (95:5:0,5), с получением указанного в заголовке соединения в виде белой пены с выходом 45% (88 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.01 (s, 3Н), 1.06 (s, 3Н), 1.24-1.28 (m, 2H), 1.85-1.93 (m, 1Н), 2.06-2.15 (m, 1H), 2.38-2.46 (m, 2H), 2.61-2.69 (m, 2H), 2.74-2.81 (m, 1H), 2.87-2.92 (m, 1H), 4.70-4.74 (m, 1H), 6.18-6.19 (t, 1H), 6.30-6.31 (t, 1H), 6.38-6.39 (t, 1H), 7.22-7.26 (m, 2H), 7.28-7.31 (t, 4H), 7.33-7.36 (m, 4H); ЭРИ-МСНР m/z 493 [M+H] + . Пример 35. Амид 5-[(3S)-3-(4-хлор-2-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. Указанное в заголовке соединение получили из продукта подготовительного примера 17 и 4-хлор-2-метоксифенола способом, который описан в примере 33, с перемешиванием в течение 2 ч и очисткой на картридже Isolute ® SCX, в виде бесцветной смолы с выходом 100%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.96 (s, 3Н), 1.02 (s, 3Н), 1.21-1.25 (m, 2H), 1.83-1.90 (m, 1Н), 2.02-2.11 (m, 1H), 2.42-2.46 (m, 2H), 2.48-2.54 (m, 1H), 2.62-2.66 (dd, 1H), 2.69-2.80 (m, 2H), 3.73 (s, 3H), 4.68-4.73 (m, 1H), 6.75-6.77 (d, 1H), 6.82-6.85 (dd, 1H), 6.91-6.92 (d, 1H), 7.20-7.23 (m, 2H), 7.26-7.29 (t, 4H), 7.33-7.37 (m, 4H); ЭРИ-МСНР m/z 473 [M+H] + . Пример 36. Амид 5-[(3S)-3-(4-хлор-2-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. Указанное в заголовке соединение получили из продукта примера 35 способом, который описан в примере 34, с перемешиванием в течение 3 ч и очисткой колоночной хроматографией на силикагеле, элюируя смесью этилацетат:метанол:аммиак от 98:2:0,2 до 94:6:0,6, в виде белой пены с выходом 72%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.96 (s, 3Н), 1.02 (s, 3Н), 1.21-1.25 (m, 2H), 1.83-1.90 (m, 1Н), 2.02-2.11 (m, 1H), 2.42-2.46 (m, 2H), 2.48-2.54 (m, 1H), 2.62-2.66 (dd, 1H), 2.69-2.80 (m, 2H), 4.68-4.73 (m, 1H), 6.75-6.77 (d, 1H), 6.82-6.85 (dd, 1H), 6.91-6.92 (d, 1H), 7.20-7.23 (m, 2H), 7.26-7.29 (t, 4H), 7.33-7.37 (m, 4H); ЭРИ-МСНР m/z 473 [M+H] + . Пример 37. Амид 5-[(3S)-3-(2-хлор-3-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. Указанное в заголовке соединение получили из продукта подготовительного примера 17 и продукта подготовительного примера 21 способом, который описан в примере 33, с очисткой на картридже Isolute ® SCX, в виде белой пены с выходом 100%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.99 (s, 3Н), 1.04 (s, 3Н), 1.23-1.28 (m, 2H), 1.88-1.95 (m, 1Н), 2.04-2.13 (m, 1Н), 2.42-2.46 (m, 2H), 2.56-2.63 (m, 1H), 2.65-2.70 (m, 1H), 2.72-2.79 (m, 1H), 2.88-2.93 (m, 1H), 3.85 (s, 3H), 4.79-4.81 (m, 1H), 6.54-6.57 (dd, 1H), 6.67-6.69 (dd, 1H), 7.14-7.18 (t, 1H) 7.20-7.24 (m, 2H), 7.27-7.30 (t, 4H), 7.33-7.37 (m, 4H); ЭРИ-МСНР m/z 507 [M+H] + . Пример 38. Амид 5-[(3S)-3-(2-хлор-3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Указанное в заголовке соединение получили из продукта примера 37 способом, который описан в примере 34, в виде белой пены с выходом 37%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.08 (s, 3Н), 1.12 (s, 3Н), 1.31-1.36 (m, 2H), 1.97-2.05 (m, 1Н), 2.07-2.16 (m, 1H), 2.44-2.48 (m, 2H), 2.77-2.96 (m, 3H), 3.05-3.10 (q, 1H), 4.82-4.87 (m, 1H), 6.43-6.45 (dd, 1H), 6.54-6.57 (dd, 1H), 7.01-7.05 (t, 1H), 7.22-7.26 (m, 2H), 7.29-7.33 (t, 4H), 7.36-7.39 (m, 4H); ЭРИ-МСНР m/z 493 [M+H] + . Примеры 39 и 40. Указанные ниже соединения получили из продукта подготовительного примера 17 и 2-хлоррезорцина способом, который описан в примере 33, с добавлением дополнительного количества трифенилфосфина (2 экв.) и диизопропилазодикарбоксилата (2 экв.) через 18 ч и последующим перемешиванием в течение дополнительных 3 ч. Сырое вещество очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат:метанол:аммиак 0,88 от 98:2:0,2 до 94:6:0,6, с получением смеси региоизомеров, которые разделяли ЖХВД на "кислотной" колонке Luna С8(2), элюируя смесью ацетонитрил:вода:диэтиламин (1:1:0,05), с получением указанных в заголовке соединений в виде белых твердых веществ с выходом 12 и 1% соответственно. Пример 39. Амид 5-[(3S)-3-(4-хлор-3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.98 (s, 3Н), 1.04 (s, 3Н), 1.23-1.27 (q, 2H), 1.87-1.95 (m, 1Н), 2.05-2.13 (m, 1H), 2.41-2.47 (m, 2H), 2.54-2.60 (m, 1H), 2.62-2.66 (dd, 1H), 2.71-2.77 (q, 1H), 2.86-2.90 (q, 1H), 4.69-4.74 (m, 1H), 6.32-6.36 (m, 2H), 7.07-7.09 (d, 1H), 7.20-7.24 (m 2H), 7.27-7.30 (t, 4H), 7.35-7.37 (m, 4H); ЭРИ-МСНР m/z 493 [M+H] + . Пример 40. Амид 5-[(3S)-3-(2-хлор-5-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.99 (s, 3Н), 1.04 (s, 3Н), 1.21-1.26 (m, 2H), 1.82-1.90 (m, 1Н), 2.05-2.15 (m, 1H), 2.35-2.46 (m, 2H), 2.53-2.62 (m, 2H), 2.69-2.75 (q, 1H), 2.81-2.86 (q, 1H), 4.66-4.71 (m, 1H), 6.27-6.30 (dd, 1H), 6.39-6.40 (d, 1H), 7.09-7.11 (d, 1H), 7.21-7.36 (m, 10Н); ЭРИ-МСНР m/z 493 [M+H] + . Пример 41. 5-[(3R)-3-(3-Хлор-2-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Продукт подготовительного примера 20 (150 мг; 0,431 ммоль) в диметилформамиде (3 мл) по каплям добавляли к охлажденному во льду раствору гидрида натрия (60% дисперсия в минеральном масле; 52 мг; 1,293 ммоль) в диметилформамиде (1 мл). После перемешивания в течение 1 ч добавляли продукт подготовительного примера 22 (103 мг; 0,646 ммоль) в диметилформамиде (1 мл), и смесь нагревали до 60 °С в течение 96 ч. Раствор концентрировали в вакууме и распределяли между этилацетатом (10 мл) и водой (10 мл). Органический слой экстрагировали и вновь промывали водой (10 мл), затем сушили над сульфатом натрия и концентрировали в вакууме с получением указанного в заголовке соединения в виде коричневого масла с выходом 74% (156 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.03 (s, 3Н), 1.08 (s, 3Н), 1.44-1.49 (m, 2H), 1.90-1.97 (m, 1Н), 2.12-2.21 (m, 1H), 2.50-2.62 (m, 3Н), 2.74-2.83 (m, 2H), 2.87-2.91 (m, 1H), 3.74 (s, 3H), 4.81-4.84 (m, 1H), 6.83-6.85 (dd, 1H), 6.93-7.00 (m, 2H), 7.25-7.42 (m, 10H); ЭРИ-МСНР m/z 489 [M+H] + . Пример 42. Амид 5-[(3R)-3-(3-хлор-2-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. Указанное в заголовке соединение получили из продукта примера 41 способом, который описан в примере 2. Сырое соединение очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 (95:5:0,5), с получением белой пены с выходом 50%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.00 (s, 3Н), 1.05 (s, 3Н), 1.23-1.27 (q, 2H), 1.88-1.96 (m, 1Н), 2.07-2.16 (m, 1H), 2.41-2.46 (m, 2H), 2.56-2.61 (m, 1H), 2.64-2.77 (m, 2H), 2.86-2.92 (m, 1H), 3.75 (s, 3H), 4.77-4.82 (m, 1H), 6.80-6.83 (dd, 1H), 6.96-6.99 (m, 2H), 7.21-7.25 (m, 2H), 7.27-7.31 (t, 4H), 7.34-7.36 (m, 4H); ЭРИ-МСНР m/z 507 [M+H] + . Пример 43. Амид 5-[(3R)-3-(3-хлор-2-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. Указанное в заголовке соединение получили из продукта примера 42 способом, который описан в примере 34, в виде белой пены с выходом 65%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.05 (s, 3Н), 1.09 (s, 3Н), 1.28-1.32 (m, 2H), 1.89-1.98 (m, 1Н), 2.03-2.13 (m, 1H), 2.42-2.46 (m, 2H), 2.59-2.67 (m, 1H), 2.70-2.75 (m, 1H), 2.81-2.93 (m, 2H), 4.84-4.88 (m, 1H), 6.66-6.70 (t, 1H), 6.77-6.79 (d, 1H), 6.88-6.91 (d, 1H), 7.21-7.25 (m, 2H), 7.28-7.32 (t, 4H), 7.35-7.37 (m, 4H); ЭРИ-МСНР m/z 493 [M+H] + . Пример 44. Амид 5-[(3S)-3-(3-гидрокси-2,5-диметилфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Указанное в заголовке соединение получили из продукта подготовительного примера 17 и 2,5-диметилрезорцина способом, который описан в примере 29, с добавлением дополнительного количества трифенилфосфина (2 экв.) и диизопропилазодикарбоксилата (2 экв.) через 18 ч и последующим перемешиванием в течение 1 ч, в виде бледно-коричневой пены с выходом 58%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.99 (s, 3Н), 1.04 (s, 3Н), 1.24-1.28 (m, 2H), 1.85-1.92 (m, 1Н), 1.93 (s, 3Н), 2.01-2.10 (m, 1H), 2.20 (s, 3H), 2.41-2.45 (m, 2H), 2.56-2.66 (m, 2H), 2.70-2.78 (m, 1H), 2.86-2.93 (m, 1H), 4.67-4.873 (m, 1H), 6.11 (s, 1H), 6.24 (s, 1H), 7.20-7.24 (m, 2H), 7.26-7.30 (t, 4H), 7.33-7.37 (m, 4H); ХИАД-МСНР m/z 487 [M+H] + . Пример 45. Амид 5-[(3S)-3-(3-фтор-5-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. Указанное в заголовке соединение получили из продукта подготовительного примера 17 и продукта подготовительного примера 23 способом, который описан в примере 33, в виде бесцветной смолы с выходом 100%. ЭРИ-МСНР m/z 491 [М+Н] + . Пример 46. Амид 5-[(3S)-3-(3-фтор-5-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. Указанное в заголовке соединение получили из продукта примера 45 способом, который описан в примере 34, с добавлением дополнительно трибромида бора (1M в дихлорметане, 4 экв.) через 3 ч и последующим перемешиванием в течение 2 ч. Сырое вещество очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат:метанол:аммиак 0,88 от 98:2:0,2 до 94:6:0,6, с получением белой пены с выходом 35%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.99 (s, 3Н), 1.04 (s, 3Н), 1.21-1.26 (m, 2H), 1.82-1.90 (m, 1Н), 2.05-2.14 (m, 1H), 2.39-2.45 (m, 2H), 2.54-2.63 (m, 2H), 2.69-2.75 (m, 1H), 2.82-2.87 (q, 1H), 4.66-4.71 (m, 1H), 6.02-6.11 (m, 3H), 7.21-7.26 (m, 2H), 7.27-7.31 (t, 4H), 7.33-7.36 (m, 4H); ХИАД-МСНР m/z 477 [M+H] + . Пример 47. 5-[(3R)-3-(3-Метокси-5-трифторметилфенокси)пирролидин-1-ил]-5-метил-2,2-дифенил-гексаннитрил. Указанное в заголовке соединение получили из продукта подготовительного примера 20 и продукта подготовительного примера 24 способом, который описан в примере 41. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 (95:5:0,5), с получением бледно-коричневой смолы с выходом 32%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.03 (s, 3Н), 1.08 (s, 3Н), 1.44-1.48 (m, 2H), 1.87-1.90 (m, 1Н), 2.14-2.22 (m, 1H), 2.49-2.54 (m, 2H), 2.56-2.62 (m, 1H), 2.67-2.70 (d, 1H), 2.75-2.81 (q, 1H), 2.87-2.91 (m, 1H), 3.80 (s, 3H), 4.82-4.85 (m, 1H), 6.61 (s, 1H), 6.67 (s, 1H), 6.74 (s, 1H), 7.24-7.42 (m, 10Н); ХИАД-МСНР m/z 523 [M+H] + . Пример 48. Амид 5-[(3R)-3-(3-метокси-5-трифторметилфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Указанное в заголовке соединение получили из продукта примера 47 способом, который описан в примере 2, в виде бесцветной смолы с выходом 83%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.98 (s, 3Н), 1.02 (s, 3Н), 1.20-1.25 (m, 2H), 1.82-1.89 (m, 1Н), 2.08-2.17 (m, 1H), 2.39-2.46 (m, 2H), 2.49-2.59 (m, 2H), 2.66-2.72 (q, 1H), 2.81-2.86 (m, 1H), 3.80 (s, 3H), 4.76-4.81 (m, 1H), 6.58-6.60 (q, 1H), 6.64 (s, 1H), 6.74 (s, 1H), 7.18-7.36 (m, 10Н); ХИАД-МСНР m/z 541 [M+H] + . Пример 49. Амид 5-[(3R)-3-(3-гидрокси-5-трифторметилфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Указанное в заголовке соединение получили из продукта примера 48 способом, который описан в примере 34, в виде бесцветной смолы с выходом 40%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.99 (s, 3Н), 1.04 (s, 3Н), 1.22-1.27 (m, 2H), 1.84-1.91 (m, 1Н), 2.08-2.16 (m, 1H), 2.38-2.47 (m, 2H), 2.54-2.63 (m, 2H), 2.69-2.75 (q, 1H), 2.85-2.89 (m, 1H), 4.73-4.78 (m, 1H), 6.45-6.47 (t, 1H), 6.52-6.53 (s, 1H), 6.60-6.61 (s, 1H), 7.21-7.24 (m, 2H), 7.27-7.30 (t, 4H), 7.34-7.36 (m, 4H); ХИАД-МСНР m/z 527 [M+H] + . Пример 50. Амид 5-[(3S)-3-(4-фтор-3-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. Указанное в заголовке соединение получили из продукта подготовительного примера 17 и продукта подготовительного примера 25 способом, который описан в примере 33, с добавлением дополнительно трифенилфосфина (2 экв.) и диизопропилазодикарбоксилата (2 экв.) и через 16 ч, и через 17 ч с последующим перемешиванием в течение дополнительно 1 ч и очисткой на картридже Isolute ® SCX, в виде бледно-коричневой пены с выходом 86%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.98 (s, 3Н), 1.03 (s, 3Н), 1.21-1.26 (m, 2Н), 1.82-1.89 (m, 1Н), 2.04-2.13 (m, 1H), 2.37-2.47 (m, 2H), 2.52-2.62 (m, 2H), 2.68-2.74 (q, 1H), 2.81-2.85 (m, 1H), 3.79 (s, 3H), 4.69-4.73 (m, 1H), 6.28-6.32 (m, 1H), 6.52-6.56 (dd, 1H), 6.90-6.95 (dd, 1H), 7.20-7.24 (m, 2H), 7.26-7.30 (t, 4H), 7.33-7.36 (m, 4H); ХИАД-МСНР m/z 491 [M+H] + . Пример 51. Амид 5-[(3S)-3-(2-фтор-3-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. Указанное в заголовке соединение получили из продукта подготовительного примера 17 и продукта подготовительного примера 26 способом, который описан в примере 33, с добавлением дополнительно трифенилфосфина (2 экв.) и диизопропилазодикарбоксилата (2 экв.) и через 16 ч, и через 17 ч с последующим перемешиванием в течение дополнительно 1 ч и очисткой на картридже Isolute ® SCX, в виде бледно-желтой пены с выходом 86%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.98 (s, 3Н), 1.02 (s, 3Н), 1.22-1.26 (m, 2H), 1.85-1.93 (m, 1H), 2.04-2.12 (m, 1H), 2.40-2.45 (m, 2H), 2.51-2.56 (m, 1H), 2.62-2.65 (m, 1H), 2.68-2.74 (q, 1H), 2.82-2.86 (m, 1H), 3.83 (s, 3H), 4.74-4.78 (m, 1H), 6.53-6.57 (t, 1H), 6.66-6.70 (t, 1H), 6.93-6.98 (td, 1H), 7.20-7.24 (m, 2H), 7.27-7.30 (t, 4H), 7.33-7.36 (m, 4H); ХИАД-МСНР m/z 491 [М+Н] + . Пример 52. Амид 5-[(3S)-3-(4-фтор-3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. Указанное в заголовке соединение получили из продукта примера 50 способом, который описан в примере 34, с перемешиванием в течение 3 ч и очисткой колоночной хроматографией на силикагеле, элюируя смесью этилацетат:метанол:аммиак 0,88 от 98:2:0,2 до 94:6:0,6, в виде белой пены с выходом 52%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.99 (s, 3Н), 1.04 (s, 3Н), 1.22-1.27 (m, 2H), 1.82-1.90 (m, 1Н), 2.03-2.12 (m, 1H), 2.38-2.46 (m, 2H), 2.55-2.64 (m, 2H), 2.70-2.76 (q, 1H), 2.83-2.87 (m, 1H), 4.6-4.69 (m, 1H), 6.19-6.23 (dt, 1H), 6.38-6.40 (dd, 1H), 6.85-6.90 (dd, 1H), 7.21-7.25 (m, 2H), 7.27-7.31 (t, 4H), 7.33-7.36 (m, 4H); ХИАД-МСНР m/z 477 [M+H] + . Пример 53. Амид 5-[(3S)-3-(2-фтор-3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. Указанное в заголовке соединение получили из продукта примера 51 способом, который описан в примере 34, с перемешиванием в течение 3 ч и очисткой колоночной хроматографией на силикагеле, элюируя смесью этилацетат:метанол:аммиак от 98:2:0,2 до 94:6:0,6, в виде белой пены с выходом 26%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.00 (s, 3Н), 1.04 (s, 3Н), 1.24-1.28 (m, 2H), 1.88-1.95 (m, 1H), 2.04-2.13 (m, 1Н), 2.41-2.47 (m, 2H), 2.55-2.63 (m, 1H), 2.66-2.71 (m, 1H), 2.72-2.78 (q, 1H), 2.86-2.91 (m, 1H), 4.73-4.77 (m, 1H), 6.38-6.42 (t, 1H), 6.48-6.53 (t, 1H), 6.80-6.85 (td, 1H), 7.21-7.25 (m, 2H), 7.27-7.31 (t, 4H), 7.34-7.37 (m, 4H); ЭРИ-МСНР m/z 477 [M+H] + . Пример 54. Амид 5-[(3R)-3-(3-гидроксибензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. 4М Соляную кислоту в диоксане (3 мл) и воде (0,3 мл) добавляли к продукту подготовительного примера 29 (95 мг; 0,186 ммоль), и полученный раствор перемешивали при 60 °С в течение 20 мин. Реакционную смесь охлаждали до комнатной температуры и распределяли между этилацетатом (20 мл) и насыщенным раствором гидрокарбоната натрия (10 мл). Водный слой экстрагировали и промывали этилацетатом (2 ×10 мл), объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией, элюируя смесью этилацетат:метанол:аммиак 0,88 от 97:3:0,2 до 95:5:0,5, с получением указанного в заголовке соединения в виде бесцветной смолы с выходом 51% (44 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.99 (s, 3Н), 1.00 (s, 3Н), 1.24-1.28 (m, 2H), 1.74-1.81 (m, 1Н), 1.87-1.94 (m, 1H), 2.40-2.44 (m, 2H), 2.49-2.58 (m, 2H), 2.61-2.67 (m, 1H), 2.71-2.76 (m, 1H), 3.98-4.03 (m, 1H), 4.36 (s, 2H), 6.67-6.69 (d, 1H), 6.74-6.76 (m, 2H), 7.09-7.13 (t, 1H), 7.22-7.26 (m, 2H), 7.29-7.32 (t, 4H), 7.35-7.38 (m, 4H); ЭРИ-МСНР m/z 473 [M+H] + . Пример 55. 5-{(3S)-3-[(3-Бромбензил)окси]пирролидин-1-ил}-5-метил-2,2-дифенилгексаннитрил. Гидрид натрия (60% дисперсия в минеральном масле; 150 мг; 3,74 ммоль) порциями добавляли к охлажденному во льду раствору продукта подготовительного примера 18 (1,3 г; 3,74 ммоль) в N,N-диметилформамиде (20 мл) и эту смесь перемешивали при 0 °С в течение 1 ч. Добавляли 3-бромбензилбромид (935 мг; 3,74 ммоль) и смесь перемешивали в течение 4 ч, давая температуре подняться до 25 °С. Затем реакционную смесь гасили водой, концентрировали в вакууме и водный остаток распределяли между этилацетатом (50 мл) и водой (30 мл). Водный слой отделяли и экстрагировали этилацетатом (4 ×30 мл). Объединенный органический раствор сушили над сульфатом натрия, концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат от 50:50 до 100:0, с получением указанного в заголовке соединения в виде бледно-коричневого масла с выходом 70%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.02 (s, 3Н), 1.04 (s, 3Н), 1.41-1.54 (m, 2H), 1.77-1.85 (m, 1Н), 1.93-2.02 (m, 1H), 2.49-2.54 (m, 3H), 2.62-2.76 (m, 3H), 4.02-4.07 (m, 1H), 4.43 (m, 2H), 7.18-7.50 (m, 14H); ХИАД-МСНР m/z 518 [M+H] + . Пример 56. 5-{(3S)-3-[(3'-Гидроксибифенил-3-ил)метокси]пирролидин-1-ил}-5-метил-2,2-дифенилгексаннитрил. [1,1'-Бис(дифенилфосфино)ферроцен]палладий(II)хлорид (16 мг; 19 мкмоль) добавляли к раствору продукта примера 55 (205 мг; 0,38 ммоль), 3-гидроксифенилбороновой кислоты (106 мг; 0,77 ммоль) и карбоната натрия (81 мг; 0,77 ммоль) в тетрагидрофуране (5 мл) и воде (1 мл) и эту смесь нагревали при температуре дефлегмации в течение 16 ч. Охлажденную затем реакционную смесь концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат:метанол от 98:2 до 96:4, с получением указанного в заголовке соединения с выходом 25% (51 мг). 1 Н-ЯМР (CD 3 OD, 400 МГц) δ: 1.04 (s, 3H), 1.06 (s, 3H), 1.46-1.52 (m, 2H), 1.82-1.90 (m, 1Н), 1.96-2.03 (m, 1Н), 2.50-2.55 (m, 3Н), 2.67-2.80 (m, 3Н), 4.08-4.13 (m, 1H), 4.52 (s, 2Н), 6.76 (dd, 1Н), 7.01 (m, 1Н), 7.05 (d, 1Н), 7.20-7.24 (m, 1Н), 7.27-7.42 (m, 12Н), 7.48 (d, 1H), 7.54 (s, 1H); ЭРИ-МСНР m/z 529 [М-Н] - . Пример 57. 5-[(3S)-3-(Бифенил-3-илметокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из продукта примера 55 и бензолбороновой кислоты способом, который описан в примере 56, в виде зеленой смолы с выходом 54%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.11 (s, 3Н), 1.13 (s, 3Н), 1.51-1.58 (m, 2H), 1.94-2.03 (m, 2H), 2.52-2.56 (m, 2H), 2.70-2.77 (m, 1H), 2.82-2.93 (m, 3Н), 4.08-4.13 & 4.13-4.18 (2 ×m, 1H), 4.45 & 4.54 (2 ×m, 2H), 7.26-7.43 (m, 19H); ХИАД-МСНР m/z 516 [M+H] + . Пример 58. 5-{(3S)-3-[(2'-Гидроксибифенил-3-ил)метокси]пирролидин-1-ил}-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из продукта примера 55 и 2-гидроксифенилбороновой кислоты способом, который описан в примере 56. Сырое соединение очищали колоночной хроматографией на силикагеле, элюируя этилацетатом, затем смесью дихлорметан:метанол (95:5), с получением целевого продукта в виде бледно-коричневой пены с выходом 61%. 1 Н-ЯМР (CD 3 OD, 400 МГц) δ: 1.03 (s, 3H), 1.07 (s, 3H), 1.47-1.53 (m, 2Н), 1.83-1.90 (m, 1Н), 1.94-2.03 (m, 1Н), 2.50-2.55 (m, 2Н), 2.57-2.64 (m, 1H), 2.69-2.83 (m, 3Н), 4.09-4.14 (m, 1Н), 4.51 (s, 2Н), 6.85-6.89 (m, 2Н), 7.14 (m, 1Н), 7.20-7.46 (m, 14Н), 7.51 (s, 1H); ЭРИ-МСНР m/z 529 [М-Н] - . Пример 59. 5-{(3S)-3-[(4-Бромбензил)окси]пирролидин-1-ил}-5-метил-2,2-дифенилгексаннитрил. Гидрид натрия (60% дисперсия в минеральном масле; 35 мг; 0,88 ммоль) порциями добавляли к охлажденному во льду раствору продукта подготовительного примера 18 (205 мг; 0,59 ммоль) в N,N-диметилформамиде (5 мл) и эту смесь перемешивали в течение 1 ч. Добавляли 4-бромбензилбромид (220 мг; 0,88 ммоль) и смесь перемешивали в течение 3 ч, давая температуре подняться до 25 °С. Затем реакционную смесь вновь охлаждали до 0 °С, добавляли дополнительно гидрид натрия (60% дисперсия в минеральном масле; 220 мг; 0,88 ммоль) и смесь перемешивали при комнатной температуре. Через 18 ч смесь вновь охлаждали до 0 °С и добавляли гидрид натрия (60% дисперсия в минеральном масле; 293 мг; 1,17 ммоль). После перемешивания в течение 1 ч добавляли дополнительно 4-бромбензилбромид (220 мг; 0,88 ммоль) и смесь перемешивали в течение 3 ч при комнатной температуре. Затем реакционную смесь гасили водой, концентрировали в вакууме, и водный остаток распределяли между этилацетатом (50 мл) и водой (30 мл). Органический слой отделяли, сушили над сульфатом натрия, концентрировали в вакууме, и остаток очищали с использованием картриджа Isolute ® SCX-2, элюируя метанолом, затем 0,5М аммиаком в метаноле. Основные фракции упаривали при пониженном давлении и остаток дополнительно очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 (95:5:5), с получением указанного в заголовке соединения в виде бледно-оранжевой смолы. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.02 (s, 3Н), 1.05 (s, 3Н), 1.40-1.53 (m, 2H), 1.77-1.85 (m, 1Н), 1.93-2.02 (m, 1H), 2.50-2.55 (m, 3H), 2.64-2.78 (m, 3H), 4.03-4.08 (m, 1H), 4.42 (s, 2H), 7.23 (d, 2H), 7.28-7.44 (m, 12H); ХИАД-МСНР m/z 519 [M+H] + . Пример 60. 5-{(3S)-3-[(3'-Гидроксибифенил-4-ил)метокси]пирролидин-1-ил}-5-метил-2,2-дифенилгексаннитрил. 1,1'-Бис[(дифенилфосфино)ферроцен]палладий(II)хлорид (12 мг; 14 мкмоль) добавляли к раствору продукта примера 59 (150 мг; 0,29 ммоль), 3-гидроксифенилбороновой кислоты (80 мг; 0,58 ммоль) и карбоната натрия (62 мг; 0,58 ммоль) в тетрагидрофуране (5,5 мл) и воде (1 мл) и эту смесь нагревали при температуре дефлегмации в течение 18 ч. Добавляли дополнительно 1,1'-бис[(дифенилфосфино)ферроцен]палладий(II)хлорид (12 мг; 14 мкмоль) и смесь нагревали при температуре дефлегмации в течение 6 ч, затем охлаждали до комнатной температуры. Затем реакционную смесь концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 100:0:0 до 95:5:0,5 с получением указанного в заголовке соединения в виде бледно-коричневой пены с выходом 47% (73 мг). ЭРИ-МСНР m/z 531 [М+Н] + . Пример 61. 5-{(3S)-3-[(3'-Гидроксибифенил-3-ил)метокси]пирролидин-1-ил}-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта примера 56 способом, который описан в примере 2, в виде бесцветной смолы с выходом 17%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.22 (s, 3Н), 1.24 (s, 3Н), 1.42-1.47 (m, 2H), 1.96-2.06 (m, 1Н), 2.11-2.19 (m, 1H), 2.42-2.47 (m, 2H), 3.14-3.23 (m, 4H), 4.23-4.27 (m, 1H), 4.56 (m, 2H), 6.76-6.79 (dd, 1H), 7.02 (m, 1H), 7.06 (d, 1H), 7.23-7.41 (m, 13H), 7.49-7.51 (d, 1H), 7.54 (s, 1H); ЭРИ-МСНР m/z 549 [M+H] + . Пример 62. 5-{(3S)-3-[(4'-Гидроксибифенил-3-ил)метокси]пирролидин-1-ил}-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из продукта примера 55 и 4-гидроксифенилбороновой кислоты способом, который описан в примере 56. Сырое соединение очищали колоночной хроматографией на силикагеле, элюируя этилацетатом. Соответствующие фракции упаривали при пониженном давлении и остаток очищали далее колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 98:2:0,2 до 95:5:0,5, с получением целевого соединения в виде бледно-коричневой смолы с выходом 78%. 1 Н-ЯМР (CD 3 OD, 400 МГц) δ: 1.03 (s, 3H), 1.06 (s, 3H), 1.46-1.52 (m, 2H), 1.80-1.88 (m, 1Н), 1.93-2.02 (m, 1Н), 2.49-2.60 (m, 3Н), 2.66-2.80 (m, 3Н), 4.04-4.09 (m, 1H), 4.44 (s, 2Н), 6.80 (d, 2Н), 7.26-7.43 (m, 15Н), 7.50 (s, 1H); ЭРИ-МСНР m/z 529 [М-Н] - . Пример 63. 5-{(3S)-3-[(4'-Гидроксибифенил-3-ил)метокси]пирролидин-1-ил}-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта примера 62 способом, который описан в примере 2, с выходом 6%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.00 (s, 3Н), 1.02 (s, 3Н), 1.25-1.29 (m, 2H), 1.78-1.87 (m, 1H), 1.88-1.99 (m, 1H), 2.40-2.44 (m, 2H), 2.52-2.59 (m, 1H), 2.60-2.71 (m, 2H), 2.72-2.78 (m, 1H), 4.03-4.08 (m, 1H), 4.48 (s, 2H), 6.84 (d, 2H), 7.20-7.38 (m, 12H), 7.41-7.45 (m, 3H), 7.48 (s, 1H); ЭРИ-МСНР m/z 549 [M+H] + . Пример 64. 5-{(3S)-3-[(2'-Гидроксибифенил-3-ил)метокси]пирролидин-1-ил}-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта примера 58 способом, который описан в примере 2. Неочищенный продукт очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 90:10:1 до 80:20:2, с получением целевого продукта в виде белого твердого вещества с выходом 21%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.98 (s, 3Н), 1.00 (s, 3Н), 1.23-1.27 (m, 2H), 1.75-1.84 (m, 1H), 1.88-1.98 (m, 1Н), 2.39-2.44 (m, 2H), 2.47-2.66 (m, 3Н), 2.71-2.75 (m, 1H), 4.02-4.08 (m, 1H), 4.47 (s, 2H), 6.86-6.89 (m, 2H), 7.12-7.16 (m, 1H), 7.20-7.37 (m, 13H), 7.44 (d, 1H), 7.49 (s, 1H); ЭРИ-МСНР m/z 549 [M+H] + . Пример 65. 5-{(3S)-3-[(3'-Гидроксибифенил-4-ил)метокси]пирролидин-1-ил}-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта примера 60 способом, который описан в примере 2. Неочищенный продукт очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 95:5:0,5 до 90:10:1, с получением целевого продукта в виде белого твердого вещества с выходом 28%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.03 (s, 3Н), 1.05 (s, 3Н), 1.27-1.31 (m, 1H), 1.52-1.56 (m, 1H), 1.81-1.89 (m, 1H), 1.90-2.00 (m, 1H), 2.34-2.38 (m, 1H), 2.41-2.45 (m, 2H), 2.59-2.64 (m, 1H), 2.67-2.76 (m, 1H), 2.79-2.83 (m, 1H), 4.04-4.10 (m, 1H), 4.47 (m, 2H), 6.75 (d, 1H), 7.01 (m, 1H), 7.05 (d, 1H), 7.18-7.38 (m, 11H), 7.43 (d, 2H), 7.53 (d, 2H); ЭРИ-МСНР m/z 549 [M+H] + . Пример 66. 5-[(3S)-3-(Бифенил-3-илметокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта примера 57 способом, который описан в примере 2, в виде коричневой смолы с выходом 23%. 1 H-ЯМР (400 МГц, CD 3 OD) δ: 1.18 (s, 3Н), 1.20 (s, 3Н), 1.38-1.44 (m, 2H), 1.97-2.04 (m, 1Н), 2.05-2.12 (m, 1H), 2.41-2.46 (m, 2H), 3.03-3.14 (m, 4H), 4.21-4.24 (m, 1H), 4.56 (m, 2H), 7.23-7.44 (m, 16H), 7.50-7.55 (m, 1H), 7.57-7.60 (m, 2H); ХИАД-МСНР m/z 533 [M+H] + . Пример 67. 5-[(3R)-3-(4-Фтор-3-гидроксибензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Соляную кислоту в диоксане (4М, 4 мл) и воде (0,5 мл) добавляли к продукту подготовительного примера 34 (150 мг; 0,248 ммоль) и полученный раствор кипятили с обратным холодильником в течение 45 мин. Растворитель удаляли в вакууме и остаток распределяли между этилацетатом (10 мл) и насыщенным раствором гидрокарбоната натрия (3 мл). Водный слой отделяли и экстрагировали дополнительно этилацетатом (2 ×10 мл). Объединенные органические слои промывали водой (5 мл), рассолом (5 мл), сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией, элюируя смесью дихлорметан:метанол:аммиак 0,88 (от 95:5:0,5 до 93:7:0,7 и до 90:10:1), с получением указанного в заголовке соединения в виде беловатой пены с выходом 44% (54 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.05 (s, 3Н), 1.06 (s, 3Н), 1.28-1.33 (m, 2H), 1.81-1.97 (m, 2H), 2.41-2.45 (m, 2H), 2.63-2.86 (m, 4H), 4.02-4.07 (m, 1H), 4.34 (s, 2H), 6.70-6.74 (m, 1H), 6.87-6.90 (dd, 1H), 6.94-6.99 (dd, 1H), 7.23-7.38 (m, 10Н); ЭРИ-МСНР m/z 491 [M+H] + . Пример 68. 5-[(3S)-3-(3-Циано-5-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Формиат аммония (33 мг; 0,523 ммоль) и 20% Pd(OH) 2 /C (3 мг) добавляли к раствору продукта подготовительного примера 36 (30 мг; 0,0523 ммоль) в этаноле (2 мл) и эту смесь кипятили с обратным холодильником в течение 15 мин. Затем реакционную смесь фильтровали через Arbocel ® и фильтрат концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 (95:5:0,5), с получением указанного в заголовке соединения в виде желтой пены с выходом 87% (22 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.00 (s, 3Н), 1.04 (s, 3Н), 1.23-1.28 (m, 2H), 1.84-1.93 (m, 1Н), 2.08-2.17 (m, 1H), 2.36-2.50 (m, 2H), 2.56-2.63 (m, 2H), 2.71-2.77 (q, 1H), 2.85-2.89 (m, 1H), 4.73-4.79 (m, 1H), 6.53-6.54 (d, 1H), 6.60-6.61 (d, 1H), 6.66-6.67 (d, 1H), 7.22-7.36 (m, 10Н); ЭРИ-МСНР m/z 484 [M+H] + . Пример 69. 5-[(3S)-3-(2-Циано-5-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта подготовительного примера 17 и 2-гидрокси-4-метоксибензонитрила способом, который описан для подготовительного примера 36, в виде оранжевой пены с выходом 36%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.00 (s, 3Н), 1.04 (s, 3Н), 1.21-1.29 (m, 2H), 1.89-1.98 (m, 1Н), 2.10-2.20 (m, 1H), 2.35-2.51 (m, 2H), 2.57-2.68 (m, 2H), 2.73-2.83 (m, 1H), 2.91-2.99 (m, 1H), 3.86 (s, 3H), 4.88-4.91 (m, 1H), 6.51 (d, 1H), 6.61-6.64 (dd, 1H), 7.20-7.39 (m, 10H), 7.48-7.50 (d, 1H); ЭРИ-МСНР m/z 498 [M+H] + . Пример 70. 5-{(3S)-3-[(7-Гидрокси-2-нафтил)окси]пирролидин-1-ил}-5-метил-2,2-дифенилгексанамид. Фторид аммония (53 мг; 1,44 ммоль) добавляли к раствору продукта подготовительного примера 37 (90 мг; 0,144 ммоль) в метаноле (3 мл) и воде (0,3 мл) и эту смесь нагревали при 50 °С в течение 18 ч. Смесь концентрировали в вакууме и остаток очищали с использованием силикагелевого картриджа RediSep ®, элюируя смесью дихлорметан:метанол:аммиак 0,88 (от 100:0:0 до 90:10:1). Соответствующие фракции концентрировали в вакууме и остаток затем очищали с использованием силикагелевого картриджа RediSep ®, элюируя смесью дихлорметан:метанол:аммиак 0,88 (от 100:0:0 до 93:7:0,7), с получением указанного в заголовке соединения в виде бесцветной смолы с выходом 6% (4,5 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.96 (s, 3Н), 1.06 (s, 3Н), 1.19-1.30 (m, 2H), 1.88-1.98 (m, 1Н), 2.11-2.23 (m, 1H), 2.34-2.52 (m, 2H), 2.54-2.64 (m, 1H), 2.64-2.68 (m, 1H), 2.72-2.82 (m, 1H), 2.85-2.91 (m, 1H), 4.82-4.89 (m, 1H), 6.77-6.83 (m, 1H), 6.85-6.87 (m, 1H), 6.88-6.91 (m, 1H), 6.98-7.01 (m, 1H), 7.15-7.35 (m, 10H), 7.54-7.62 (m, 2H); ХИАД-MCHP m/z 509 [M+H] + . Пример 71. 5-[(3S)-3-(4-Фенилфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Раствор трифенилфосфина (143 мг; 0,546 ммоль) в тетрагидрофуране (1 мл) и диизопропилазодикарбоксилат (0,11 мл; 0,546 ммоль) добавляли к раствору продукта подготовительного примера 17 (100 мг; 0,273 ммоль) в тетрагидрофуране (1 мл) и эту смесь перемешивали при комнатной температуре в течение 15 мин. Добавляли 4-фенилфенол (93 мг; 0,546 ммоль) и смесь перемешивали при комнатной температуре в течение 18 ч. Добавляли дополнительно трифенилфосфин (143 мг; 0,546 ммоль) и диизопропилазодикарбоксилат (0,11 мл; 0,546 ммоль) и раствор перемешивали при комнатной температуре в течение 72 ч. Смесь концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол (от 100:0 до 95:5), с получением указанного в заголовке соединения в виде бесцветного масла с выходом 28% (40 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.99 (s, 3Н), 1.07 (s, 3Н), 1.23-1.26 (m, 2H), 1.90-1.98 (m, 1H), 2.10-2.19 (m, 1H), 2.39-2.46 (m, 2H), 2.58-2.68 (m, 2H), 2.75-2.89 (m, 2H), 4.80-4.84 (m, 1H), 6.88-6.90 (d, 2H), 7.20-7.41 (m, 13H), 7.49-7.56 (dd, 4H); ХИАД-МСНР m/z 519 [M+H] + . Пример 72. 5-[(3R)-3-(3-Хлор-4-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Трибромид бора (1М в дихлорметане; 0,16 мл; 0,158 ммоль) добавляли к раствору продукта примера 28 (20 мг; 0,039 ммоль) в дихлорметане (3 мл) и эту смесь перемешивали при комнатной температуре в течение 72 ч. Реакцию гасили, добавляя по каплям аммиак 0,88 (5 мл) и реакционную смесь перемешивали в течение 24 ч. Водный слой отделяли и экстрагировали дихлорметаном (3 ×20 мл). Объединенные органические слои концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат:метанол:аммиак 0,88 (от 100:0:0 до 95:5:0,5), с получением бесцветной смолы. Затем эту смолу очищали ЖХВД с использованием неподвижной фазы Curosil PFP-Acid (150 ×21,2), элюируя смесью 0,1% муравьиной кислоты(водн.):(ацетонитрил+0,1% муравьиной кислоты) от 95:5 до 0:100. Соответствующие фракции концентрировали в вакууме и остаток распределяли между дихлорметаном (15 мл) и водой (5 мл), водный слой отделяли и экстрагировали дополнительно дихлорметаном (2 ×15 мл). Объединенные органические слои промывали раствором гидроксида натрия (1М, 5 мл), рассолом (10 мл) и концентрировали в вакууме. Затем остаток очищали препаративной тонкослойной хроматографией, элюируя смесью дихлорметан:метанол:аммиак 0,88 (90:10:1). Чистый продукт вымывали из силикагеля, используя смесь дихлорметан:метанол:аммиак 0,88 (95:5:0,5), фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения в виде бесцветной смолы с выходом 16% (3 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.18 (s, 3Н), 1.22 (s, 3Н), 1.28-1.30 (m, 2H), 1.39-1.44 (m, 2Н), 2.08-2.14 (m, 2Н), 2.42-2.47 (m, 2Н), 3.04-3.27 (m, 2Н), 4.82-4.87 (m, 1Н), 6.66-6.70 (m, 1Н), 6.82-6.87 (m, 2Н), 7.25-7.38 (m, 10Н); ХИАД-МСНР m/z 493 [М+Н] + 491. Пример 73. 5-[(3R)-3-(3-Фтор-5-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Диизопропилазодикарбоксилат (160 мкл; 0,822 ммоль) добавляли тремя порциями к охлажденному во льду раствору трифенилфосфина (215 мг; 0,822 ммоль), продукта подготовительного примера 23 (175 мг; 1,232 ммоль) и продукта подготовительного примера 16 (150 мг; 0,411 ммоль) в тетрагидрофуране (7 мл) и эту смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали в вакууме и остаток очищали с использованием картриджа Isolute ® SCX-2, элюируя метанолом, затем 2М аммиаком в метаноле. Основные фракции концентрировали в вакууме с получением указанного в заголовке соединения в виде бледно-коричневой пены с выходом 75% (150 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.98 (s, 3Н), 1.03 (s, 3Н), 1.21-1.26 (m, 2H), 1.81-1.89 (m, 1H), 2.06-2.15 (m, 1H), 2.38-2.46 (m, 2H), 2.50-2.61 (m, 2H), 2.67-2.72 (m, 1H), 2.78-2.85 (m, 1H), 3.74 (s, 3H), 4.69-4.74 (m, 1H), 6.14-6.19 (m, 2H), 6.24-6.28 (m, 1H), 7.21-7.36 (m, 10Н); ЭРИ-МСНР m/z 491 [M+H] + . Пример 74. 5-[(3R)-3-(3-Фтор-5-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Трибромид бора (1М в дихлорметане; 1,2 мл; 1,224 ммоль) добавляли к охлажденному во льду раствору продукта примера 73 (150 мг; 0,306 ммоль) в дихлорметане (4 мл) и эту смесь перемешивали при комнатной температуре в течение 18 ч. Реакцию гасили раствором аммиака 0,88 и перемешивали при комнатной температуре в течение 2 ч. рН реакционной смеси подводили до значения 8 добавлением по каплям 2н. соляной кислоты (водн.) и экстрагировали дихлорметаном (3 ×10 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 (95:5:0,5), с получением указанного в заголовке соединения в виде бледно-коричневой пены с выходом 49% (71 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.01 (s, 3Н), 1.05 (s, 3Н), 1.24-1.28 (m, 2H), 1.85-1.92 (m, 1Н), 2.07-2.15 (m, 1Н), 2.40-2.45 (m, 2Н), 2.59-2.67 (m, 2Н), 2.73-2.79 (q, 1H), 2.87-2.91 (m, 1Н), 4.68-4.72 (m, 1Н), 6.03-6.12 (m, 3Н), 7.21-7.26 (m, 2Н), 7.28-7.32 (m, 4Н), 7.33-7.37 (m, 4Н); ЭРИ-МСНР m/z 477 [М+Н] + , 475 [М-Н] - . Пример 75. 5-[(3R)-3-(2-Фтор-3-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Диизопропилазодикарбоксилат (160 мкл; 0,822 ммоль) добавляли тремя порциями к охлажденному во льду раствору трифенилфосфина (215 мг; 0,822 ммоль), продукта подготовительного примера 26 (160 мг; 1,127 ммоль) и продукта подготовительного примера 16 (150 мг; 0,411 ммоль) в тетрагидрофуране (7 мл) и эту смесь перемешивали при увеличении температуры от 0 °С до комнатной температуры в течение 16 ч. Добавляли дополнительно трифенилфосфин (215 мг; 0,822 ммоль) и диизопропилазодикарбоксилат (160 мкл; 0,822 ммоль) и смесь перемешивали в течение следующих 48 ч. Реакционную смесь концентрировали в вакууме и остаток очищали с использованием картриджа Isolute ® SCX-2, элюируя метанолом, затем 2М аммиаком в метаноле. Основные фракции концентрировали в вакууме с получением указанного в заголовке соединения в виде бледно-коричневой пены с выходом 75% (145 мг). ЭРИ-МСНР m/z 491 [М+Н] + . Пример 76. 5-[(3R)-3-(2-Фтор-3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Трибромид бора (1М в дихлорметане; 1,1 мл; 1,224 ммоль) добавляли к охлажденному во льду раствору продукта примера 75 (140 мг; 0,286 ммоль) в дихлорметане (4 мл) и эту смесь перемешивали при комнатной температуре в течение 2 ч. Реакцию гасили раствором аммиака 0,88 и перемешивали при комнатной температуре в течение 16 ч. рН реакционной смеси подводили до значения 8 добавлением по каплям 2н. соляной кислоты (водн.) и экстрагировали дихлорметаном (2 ×10 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 (95:5:0,5), с получением указанного в заголовке соединения в виде бледно-желтой пены с выходом 15% (20 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.00 (s, 3Н), 1.04 (s, 3Н), 1.25-1.28 (m, 2H), 1.85-1.93 (m, 1Н), 2.06-2.12 (m, 1H), 2.42-2.46 (m, 2H), 2.54-2.60 (m, 1H), 2.65-2.75 (m, 2H), 2.85-2.90 (q, 1H), 4.74-4.78 (m, 1H), 6.38-6.42 (t, 1H), 6.49-6.53 (t, 1H), 6.80-6.85 (t, 1H), 7.22-7.25 (m, 2H), 7.28-7.32 (m, 4H), 7.35-7.38 (m, 4H); ЭРИ-МСНР m/z 477 [M+H] + , 475 [M-H] - . Пример 77. 5-[(3R)-3-(2-Хлор-3-метоксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Диизопропилазодикарбоксилат (118 мкл; 0,606 ммоль) добавляли тремя порциями к охлажденному во льду раствору трифенилфосфина (160 мг; 0,606 ммоль), продукта подготовительного примера 21 (120 мг; 0,757 ммоль) и продукта подготовительного примера 16 (111 мг; 0,411 ммоль) в тетрагидрофуране (7 мл) и эту смесь перемешивали при увеличении температуры от 0 °С до комнатной температуры в течение 3 ч. Добавляли дополнительно трифенилфосфин (160 мг; 0,606 ммоль) и диизопропилазодикарбоксилат (118 мкл; 0,606 ммоль) и смесь перемешивали в течение следующих 16 ч. Реакционную смесь концентрировали в вакууме и остаток очищали с использованием картриджа Isolute ® SCX-2, элюируя метанолом, затем 2М аммиаком в метаноле. Основные фракции концентрировали в вакууме с получением указанного в заголовке соединения в виде бледно-коричневой пены с выходом 82% (124 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.00 (s, 3Н), 1.05 (s, 3Н), 1.24-1.29 (m, 2Н), 1.85-1.97 (m, 1Н), 2.05-2.15 (m, 1H), 2.43-2.47 (m, 2H), 2.58-2.64 (m, 1H), 2.66-2.72 (m, 1H), 2.73-2.80 (m, 1H), 2.89-2.95 (m, 1H), 3.85 (s, 3H), 4.79-4.81 (m, 1H), 6.55-6.57 (d, 1H), 6.67-6.70 (d, 1H), 7.15-7.19 (t, 1H), 7.21-7.24 (m, 2H), 7.27-7.31 (m, 4H), 7.34-7.38 (m, 4H); ЭРИ-МСНР m/z 507 [M+H] + . Пример 78. 5-[(3R)-3-(2-Хлор-3-гидроксифенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта примера 77 способом, который описан в примере 74, в виде бледно-оранжевой пены с выходом 36%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.03 (s, 3Н), 1.07 (s, 3Н), 1.27-1.32 (m, 2H), 1.92-1.99 (m, 1Н), 2.06-2.14 (m, 1H), 2.43-2.47 (m, 2H), 2.65-2.71 (m, 1H), 2.74-2.78 (d, 1H), 2.80-2.86 (q, 1H), 2.96-3.00 (m, 1H), 4.79-4.82 (m, 1H), 6.41-6.43 (dd, 1H), 6.53-6.55 (dd, 1H), 7.00-7.04 (t, 1H), 7.21-7.25 (m, 2H), 7.28-7.32 (m, 4H), 7.36-7.39 (m, 4H); ЭРИ-МСНР m/z 493 [M+H] + , 491 [M-H] - . Пример 79. 5-[(3R)-3-(4-Хлор-3-гидроксибензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Соляную кислоту (4М в диоксане, 4 мл) и воду (0,5 мл) добавляли к продукту подготовительного примера 44 (161 мг; 0,295 ммоль) и эту смесь нагревали при температуре дефлегмации в течение 30 мин. Затем реакционную смесь охлаждали до комнатной температуры и распределяли между этилацетатом (20 мл) и насыщенным водным раствором гидрокарбоната натрия (20 мл). Водный слой отделяли, экстрагировали дополнительно этилацетатом (20 мл) и объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 95:5:0,5 до 90:10:1, с получением указанного в заголовке соединения в виде бледно-коричневой пены с выходом 19% (29 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.01 (s, 3Н), 1.02 (s, 3Н), 1.24-1.28 (m, 2H), 1.75-1.82 (m, 1Н), 1.87-1.94 (m, 1H), 2.40-2.44 (m, 2H), 2.51-2.60 (m, 2H), 2.63-2.69 (q, 1H), 2.73-2.77 (q, 1H), 3.98-4.03 (m, 1H), 4.34 (s, 2H), 6.70-6.73 (dd, 1H), 6.87-6.88 (d, 1H), 7.18-7.21 (d, 1H), 7.22-7.26 (m, 2H), 7.29-7.33 (m, 4H), 7.35-7.38 (m, 4H); ХИАД-МСНР m/z 505 [M-H] - . Пример 80. 5-[(3R)-3-(3-Метокси-4-хлорфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Гидрид натрия (60% дисперсия в минеральном масле; 60 мг; 1,466 ммоль) добавляли к охлажденному во льду раствору продукта подготовительного примера 20 (170 мг; 0,489 ммоль) в N,N-диметилформамиде (5 мл) и эту смесь перемешивали при 0 °С в течение 30 мин. Добавляли 2-хлор-5-фторанизол (73 мкл; 0,586 ммоль) и смесь перемешивали в течение 18 ч при комнатной температуре. Смесь затем нагревали при 60 °С в течение 6 ч. Смесь охлаждали до комнатной температуры, добавляли дополнительно гидрид натрия (60% дисперсия в минеральном масле; 60 мг; 1,466 ммоль) и нагревание продолжали при 60 °С в течение 18 ч. Раствор охлаждали до 0 °С, добавляли дополнительно гидрид натрия (60% дисперсия в минеральном масле; 100 мг; 2,443 ммоль) и раствор перемешивали в течение 30 мин. Добавляли дополнительно 2-хлор-5-фторанизол (122 мкл; 0,977 ммоль) и смесь нагревали при 80 °С в течение 18 ч. Затем реакционную смесь охлаждали до комнатной температуры, гасили водой (3 мл), концентрировали в вакууме и водный остаток распределяли между этилацетатом (10 мл) и водой (5 мл). Водный слой отделяли и дополнительно экстрагировали этилацетатом (2 ×10 мл). Объединенные органические слои промывали рассолом (10 мл), сушили над сульфатом натрия, концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат:метанол:аммиак 0,88 (99:1:0,1), с получением указанного в заголовке соединения в виде бесцветной смолы с выходом 34% (80 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.03 (s, 3Н), 1.08 (s, 3Н), 1.44-1.49 (m, 2Н), 1.88-1.95 (m, 1Н), 2.12-2.21 (m, 1H), 2.49-2.54 (m, 2H), 2.56-2.62 (m, 1H), 2.68-2.70 (d, 1H), 2.76-2.82 (m, 1H), 2.85-2.89 (m, 1H), 3.80 (s, 3H), 4.79-4.81 (m, 1H), 6.39-6.42 (d, 1H), 6.54 (s, 1H), 7.16-7.20 (d, 1H), 7.26-7.42 (m, 10Н); ХИАД-МСНР m/z 489 [M+H] + . Пример 81. 5-[(3R)-3-(3-Метокси-4-хлорфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Гидроксид калия (185 мг; 3,275 ммоль) добавляли к раствору продукта примера 80 (80 мг; 0,163 ммоль) в 3-метил-3-пентаноле (3 мл) и эту смесь нагревали при температуре дефлегмации в течение 24 ч. Добавляли дополнительно гидроксид калия (93 мг; 1,638 ммоль) и смесь нагревали при температуре дефлегмации в течение следующих 5 ч. Затем реакционную смесь охлаждали до комнатной температуры, концентрировали в вакууме, и остаток распределяли между этилацетатом (15 мл) и водой (10 мл). Водный слой отделяли, дополнительно экстрагировали этилацетатом (15 мл) и объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 97:3:0,3 до 94:6:0,6, с получением указанного в заголовке соединения в виде белой пены с выходом 64% (53 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.98 (s, 3Н), 1.05 (s, 3Н), 1.20-1.26 (m, 2H), 1.84-1.93 (m, 1Н), 2.07-2.16 (m, 1H), 2.34-2.49 (m, 2H), 2.53-2.65 (m, 2H), 2.70-2.77 (m, 1H), 2.81-2.88 (m, 1H), 3.80 (s, 3H), 4.74-4.79 (m, 1H), 6.36-6.38 (dd, 1H), 6.51-6.52 (d, 1H), 7.18-7.20 (d, 1H), 7.21-7.35 (m, 10Н); ХИАД-МСНР m/z 507 [M+H] + . Пример 82. 5-[(3R)-3-(3-Гидрокси-4-хлорфенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Трибромид бора (1М в дихлорметане; 0,4 мл; 0,4 ммоль) добавляли к охлажденному во льду раствору продукта примера 81 (50 мг; 0,099 ммоль) в дихлорметане (2 мл) и эту смесь перемешивали при комнатной температуре в течение 18 ч. Добавляли дополнительно трибромид бора (1М в дихлорметане; 0,2 мл; 0,2 ммоль) и смесь перемешивали при комнатной температуре в течение еще 3 ч. Реакцию гасили раствором аммиака 0,88 и перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь подкисляли до рН 6 добавлением по каплям 2н. соляной кислоты (водн.) и экстрагировали дихлорметаном (2 ×10 мл). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 97:3:0,3 до 94:6:0,6, с получением указанного в заголовке соединения в виде белой пены с выходом 70% (34 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.00 (s, 3Н), 1.06 (s, 3Н), 1.24-1.28 (m, 2H), 1.83-1.93 (m, 1Н), 2.06-2.15 (m, 1H), 2.35-2.47 (m, 2H), 2.60-2.68 (m, 2H), 2.74-2.81 (m, 1H), 2.87-2.91 (m, 1H), 4.68-4.73 (m, 1H), 6.28-6.31 (dd, 1H), 6.40-6.41 (d, 1H), 7.10-7.12 (d, 1H), 7.22-7.35 (m, 10Н); ЭРИ-МСНР m/z 493 [M+H] + , 491 [M-H] - . Пример 83. 5-[(3R)-3-(3-Гидрокси-4-цианофенокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Формиат аммония (44 мг; 0,698 ммоль) и гидроксид палладия (20% на угле, 8 мг) добавляли к раствору продукта подготовительного примера 45 (40 мг; 0,070 ммоль) в этаноле (2 мл) и эту смесь перемешивали при температуре дефлегмации в течение 20 мин. Реакционную смесь охлаждали, катализатор удаляли фильтрованием через Arbocel ® и фильтрат концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 95:5:0,5 до 90:10:1, с получением указанного в заголовке соединения в виде белой пены с выходом 55% (18 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.00 (s, 3Н), 1.06 (s, 3Н), 1.24-1.28 (m, 2H), 1.87-1.94 (m, 1Н), 2.09-2.18 (m, 1H), 2.35-2.49 (m, 2H), 2.61-2.71 (m, 2H), 2.75-2.82 (q, 1H), 2.91-2.95 (q, 1H), 4.76-4.79 (m, 1H), 6.32-6.35 (m, 2H), 7.21-7.36 (m, 11Н); ХИАД-МСНР m/z 484 [M+H] + . Пример 84. 5-[(3S)-3-(3-Метоксибензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Гидрид натрия (60% дисперсия в минеральном масле; 26 мг; 0,638 ммоль) добавляли к охлажденному во льду раствору продукта подготовительного примера 18 (185 мг; 0,532 ммоль) в N,N-диметилформамиде (5 мл) и эту смесь перемешивали при 0 °С в течение 60 мин. Добавляли 3-метоксибензилбромид (128 мг; 0,638 ммоль) и смесь перемешивали в течение 3 ч при комнатной температуре. Добавляли дополнительно 3-метоксибензилбромид (160 мг; 0,797 ммоль) и смесь перемешивали в течение следующих 2 ч. Реакцию гасили водой (3 мл), реакционную смесь концентрировали в вакууме и водный остаток распределяли между этилацетатом (20 мл) и водой (10 мл). Водный слой отделяли и дополнительно экстрагировали этилацетатом (2 ×10 мл). Объединенные органические слои концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя дихлорметаном, с получением бесцветного масла (180 мг). Гидроксид калия (430 мг; 7,692 ммоль) добавляли к раствору этого бесцветного масла (180 мг; 0,385 ммоль) в 3-метил-3-пентаноле (5 мл) и эту смесь нагревали при температуре дефлегмации в течение 24 ч. Реакционную смесь охлаждали до комнатной температуры, концентрировали в вакууме и остаток распределяли между этилацетатом (20 мл) и водой (10 мл). Водный слой отделяли, дополнительно экстрагировали этилацетатом (10 мл), объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме. Остаток очищали колоночной хроматографией, элюируя сначала пентаном, затем смесью дихлорметан:метанол:аммиак 0,88 от 95:5:0,5 до 90:10:1, с получением указанного в заголовке соединения в виде белой пены с выходом 16% (40 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.01 (s, 3Н), 1.03 (s, 3Н), 1.26-1.30 (m, 2H), 1.77-1.85 (m, 1Н), 1.88-1.95 (m, 1H), 2.40-2.45 (m, 2H), 2.53-2.72 (m, 3H), 2.75-2.79 (m, 1H), 3.76 (s, 3H), 4.00-4.05 (m, 1H), 4.41 (s, 2H), 6.80-6.83 (d, 1H), 6.86-6.88 (m, 2H), 7.16-7.37 (m, 11H); ЭРИ-МСНР m/z 487 [M+H] + . Пример 85. 5-[(3R)-3-(2-Хлор-3-гидроксибензилокси)пирролидин-1-ил]-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта подготовительного примера 49 способом, который описан в примере 79, в виде белой пены с выходом 34%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.11 (s, 3H), 1.13 (s, 3Н), 1.34-1.38 (m, 2H), 1.90-2.02 (m, 2H), 2.41-2.45 (m, 2H), 2.83-3.02 (m, 4H), 4.14-4.18 (m, 1H), 4.49-4.57 (m, 2H), 6.84-6.87 (dd, 1H), 6.91-6.93 (d, 1H), 7.06-7.10 (t, 1H), 7.24-7.27 (m, 2H), 7.30-7.38 (m, 8H); ХИАД-МСНР m/z 505 [M-H] - . Пример 86. 5-Метил-5-(4-феноксипиперидин-1-ил)-2,2-дифенилгексаннитрил. Раствор продукта подготовительного примера 50 (9,03 г; 21 ммоль) в тетрагидрофуране (270 мл) охлаждали до -20 °С. Добавляли тетрахлорид циркония (9,91 г; 43 ммоль) и реакционную смесь перемешивали при -20 °С в течение 1 ч. Затем по каплям добавляли хлорид метилмагния (3М в тетрагидрофуране, 63,8 мл; 191 ммоль) и смесь перемешивали в течение 1 ч при температуре ниже -10 °С. Реакцию гасили этанолом (20 мл), реакционную смесь концентрировали в вакууме и остаток распределяли между 2н. раствором гидроксида натрия (200 мл) и этилацетатом (250 мл). Водный слой отделяли и экстрагировали этилацетатом (2 ×200 мл), а объединенный органический раствор сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат от 75:25 до 67:33. Соответствующие фракции упаривали при пониженном давлении и остаток дополнительно очищали с использованием картриджа Isolute ® SCX-2, элюируя сначала метанолом, затем 1М аммиаком в метаноле, с получением указанного в заголовке соединения в виде желтой смолы с выходом 41% (3,83 г). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.04 (s, 6H), 1.54-1.58 (m, 2H), 1.71-1.80 (m, 2H), 1.99-2.02 (m, 2H), 2.25 (m, 2H), 2.53-2.57 (m, 2H), 2.70-2.75 (m, 2H), 4.22-4.28 (m, 1H), 6.92-6.96 (m, 3Н), 7.28-7.32 (m, 4H), 7.35-7.39 (m, 4H), 7.45-7.47 (m, 4H); ХИАД-МСНР m/z 439 [М+Н] + . Пример 87. 5-{4-[(3-Бромбензил)окси]пиперидин-1-ил}-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из продукта подготовительного примера 53 способом, который описан в примере 86, в виде желтой смолы с выходом 33%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.98 (s, 6H), 1.46-1.60 (m, 4H), 1.88-1.91 (m, 2Н), 2.04-2.09 (m, 2H), 2.46-2.50 (m, 2H), 2.65-2.68 (m, 2H), 3.28-3.34 (m, 1H), 4.50 (s, 2H), 7.18-7.42 (m, 13H), 7.51 (s, 1H); ЭРИ-МСНР m/z 533 [M+H] + . Пример 88. 5-{4-[(3-Гидроксибензил)окси]пиперидин-1-ил}-5-метил-2,2-дифенилгексаннитрил. н-Бутиллитий (2,5М в гексанах; 0,18 мл; 0,45 ммоль) по каплям добавляли к раствору продукта примера 87 (200 мг; 0,38 ммоль) в тетрагидрофуране (5 мл), охлажденному до -78 °С, и эту смесь перемешивали в течение 15 мин. Добавляли триметилборат (0,13 мл; 1,13 ммоль) и смесь перемешивали при -78 °С в течение 30 мин и при комнатной температуре в течение 2 ч. Добавляли N-оксид 4-метилморфолина (132 мг; 1,13 ммоль) и смесь нагревали при температуре дефлегмации в течение 4 ч и перемешивали при комнатной температуре в течение 18 ч. Затем реакционную смесь распределяли между этилацетатом (30 мл) и водой (30 мл), органический слой отделяли и промывали водой (20 мл). Органический раствор сушили над сульфатом натрия, концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 100:0:0 до 90:10:1. Соответствующие фракции упаривали при пониженном давлении и остаток очищали далее препаративной ТСХ, элюируя смесью дихлорметан:метанол:аммиак 0,88 (90:10:1), с получением указанного в заголовке соединения в виде смолы с выходом 3% (5 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.02 (s, 6Н), 1.46-1.58 (m, 4H), 1.86-1.90 (m, 2Н), 2.13-2.19 (m, 2H), 2.49-2.53 (m, 2H), 2.70-2.75 (m, 2H), 3.33-3.39 (m, 1H), 4.45 (s, 2H), 6.67-6.70 (m, 1H), 6.77-6.79 (m, 2H), 7.12 (m, 1H), 7.28-7.43 (m, 10Н); ЭРИ-МСНР m/z 469 [M+H] + . Пример 89. 5-[4-(Бензилокси)пиперидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. н-Бутиллитий (2,5М в гексанах; 0,23 мл; 0,56 ммоль) по каплям добавляли к раствору продукта примера 87 (200 мг; 0,38 ммоль) в тетрагидрофуране (8 мл), охлажденному до -78 °С, и эту смесь перемешивали в течение 30 мин. Затем через раствор пропускали углекислый газ при перемешивании при -78 °С в течение 3,5 ч, после чего смесь оставляли нагреваться до комнатной температуры. Реакционную смесь концентрировали в вакууме и остаток распределяли между этилацетатом (30 мл) и водой (20 мл). Органический слой отделяли, промывали водой, сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 100:0:0 до 98:2:0,2, с получением указанного в заголовке соединения как побочного продукта в виде не совсем белого твердого вещества с выходом 68% (116 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.00 (s, 6H), 1.45-1.49 (m, 2H), 1.50-1.58 (m, 2H), 1.86-1.90 (m, 2H), 2.11-2.17 (m, 2H), 2.48-2.53 (m, 2H), 2.69-2.72 (m, 2H), 3.33-3.39 (m, 1Н), 4.51 (s, 2H), 7.29-7.42 (m, 15H); ЭРИ-МСНР m/z 453 [M+H] + . Пример 90. 5-Метил-5-(4-феноксипиперидин-1-ил)-2,2-дифенилгексанамид. Гидроксид калия (512 мг; 9,12 ммоль) добавляли к раствору продукта примера 86 (200 мг; 0,46 ммоль) в 3-метил-3-пентаноле (4 мл) и эту смесь нагревали при температуре дефлегмации в течение 20 ч. Затем реакционную смесь охлаждали до комнатной температуры, концентрировали в вакууме и остаток распределяли между этилацетатом (20 мл) и водой (20 мл). Органический слой отделяли, сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 100:0 до 90:10:1, с получением указанного в заголовке соединения в виде бесцветной стекловидной массы с выходом 93% (193 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.99 (s, 6H), 1.25-1.29 (m, 2H), 1.60-1.68 (m, 2H), 1.89-1.93 (m, 2H), 2.20-2.25 (m, 2H), 2.42-2.46 (m, 2H), 2.64-2.68 (m, 2H), 4.20-4.27 (m, 1H), 6.85-6.89 (m, 3Н), 7.20-7.26 (m, 4H), 7.30-7.34 (m, 4H), 7.37-7.40 (m, 4H); ЭРИ-МСНР m/z 457 [М+Н] + . Пример 91. 5-{4-[(3-Бромбензил)окси]пиперидин-1-ил}-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта примера 87 способом, который описан в примере 90, в виде бесцветной смолы с выходом 99%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.97 (s, 6Н), 1.23-1.27 (m, 2Н), 1.48-1.57 (m, 2Н), 1.82-1.87 (m, 2Н), 2.07-2.12 (m, 2Н), 2.40-2.44 (m, 2Н), 2.62-2.66 (m, 2Н), 3.29-3.37 (m, 1Н), 4.48 (s, 2Н), 7.21-7.42 (m, 13Н), 7.49 (s, 1Н); ЭРИ-МСНР m/z 551 [М+Н] + . Пример 92. 5-[4-(Бензилокси)пиперидин-1-ил]-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта примера 89 способом, который описан в примере 90, в виде бесцветной стекловидной массы с выходом 79%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.01 (s, 6Н), 1.26-1.30 (m, 2Н), 1.57-1.59 (m, 2Н), 1.86-1.89 (m, 2H), 2.21 (m, 2H), 2.40-2.45 (m, 2H), 2.72 (m, 2H), 3.38 (m, 1H), 4.50 (s, 2H), 7.23-7.39 (m, 15H); ХИАД-МСНР m/z 471 [M+H] + . Пример 93. Амид 5-[4-(2,4-дихлор-5-гидроксифенокси)пиперидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Диизопропилазодикарбоксилат (0,05 мл; 0,26 ммоль) по каплям добавляли к смеси продукта подготовительного примера 54 (50 мг; 0,13 ммоль), 4,6-дихлорбензол-1,3-диола (47 мг; 0,26 ммоль) и трифенилфосфина (69 мг; 0,26 ммоль) в тетрагидрофуране (1 мл) и эту смесь оставляли перемешиваться в течение 7 суток при комнатной температуре. Растворитель концентрировали в вакууме и остаток очищали с использованием картриджа Isolute ® SCX-2, элюируя метанолом, затем 1М аммиаком в метаноле. Соответствующие фракции объединяли, упаривали при пониженном давлении и остаток дополнительно очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол от 98:2 до 95:5, с получением указанного в заголовке соединения в виде бесцветной стекловидной массы с выходом 8% (6 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.04 (s, 6H), 1.28-1.32 (m, 2H), 1.71-1.78 (m, 2H), 1.86-1.92 (m, 2H), 2.36-2.46 (m, 4H), 2.69-2.75 (m, 2H), 4.29 (m, 1H), 6.57 (s, 1H), 7.21-7.39 (m, 11Н); ЭРИ-МСНР m/z 541 [M+H] + . Пример 94. Амид 5-[4-(4-циано-2,5-дифторфенокси)пиперидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Гидрид натрия (60% дисперсия в минеральном масле; 12 мг; 0,29 ммоль) добавляли к раствору продукта подготовительного примера 54 (100 мг; 0,26 ммоль) в тетрагидрофуране (0,5 мл) при комнатной температуре, перемешивали в течение 20 мин, после чего охлаждали до -70 °С. Смесь добавляли к раствору 2,4,5-трифторбензонитрила (41 мг; 0,26 ммоль) в тетрагидрофуране (0,5 мл) при -70 °С и перемешивали в течение 3 ч. Затем реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение еще 18 ч. Реакцию гасили водой (5 капель) и растворитель выпаривали в вакууме. Остаток распределяли между этилацетатом (40 мл) и водой (20 мл), органический слой отделяли и промывали рассолом (20 мл). Органическую фазу сушили над сульфатом магния, концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 100:0:0 до 90:10:1. Соответствующие фракции упаривали при пониженном давлении с получением указанного в заголовке соединения в виде бесцветной стекловидной массы с выходом 40% (54 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 0.99 (s, 6H), 1.25-1.29 (m, 2H), 1.66-1.75 (m, 2H), 1.92-1.97 (m, 2H) 2.25-2.30 (m, 2H) 2.42-2.46 (m, 2H), 4.43-4.49 (m, 1H), 7.13-7.52 (m, 12H); ХИАД-МСНР m/z 518 [M+H] + . Пример 95. 5-[4-(3-Гидроксифенокси)пиперидин-1-ил]-5-метил-2,2-дифенилгексанамид. Диизопропилазодикарбоксилат (0,23 мл; 1,20 ммоль) по каплям добавляли к охлажденному на льду раствору продукта подготовительного примера 54 (226 мг; 0,594 ммоль), резорцина (196 мг; 1,71 ммоль) и трифенилфосфина (312 мг; 1,19 ммоль) в тетрагидрофуране (2 мл) и эту смесь перемешивали при комнатной температуре в течение 12 ч. Растворитель удаляли при пониженном давлении и остаток очищали с использованием картриджа Isolute ® SCX-2, элюируя метанолом, затем 1М аммиаком в метаноле. Основные фракции концентрировали в вакууме и дополнительно очищали хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 98:2:0,2 до 95:5:0,5. Соответствующие фракции концентрировали в вакууме и половину полученного остатка (55 мг) очищали далее препаративной тонкослойной хроматографией, используя покрытую диоксидом кремния пластинку и элюируя смесью дихлорметан:метанол:аммиак 0,88 (80:20:2), с получением указанного в заголовке соединения в виде смолы с выходом 14%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.05 (s, 6H), 1.30-1.34 (m, 2H), 1.66-1.74 (m, 2H), 1.91-1.96 (m, 2H), 2.34-2.47 (m, 4H), 2.70-2.79 (m, 2H), 4.21-4.27 (m, 1H), 6.32-6.38 (m, 3Н), 7.00-7.05 (m, 1H), 7.23-7.40 (m, 10Н); ХИАД-МСНР m/z 473 [M+H] + . Пример 96. 5-[4-(3-Гидрокси-2-метилфенокси)пиперидин-1-ил]-5-метил-2,2-дифенилгексанамид. Диизопропилазодикарбоксилат (0,1 мл; 0,53 ммоль) по каплям добавляли к раствору продукта подготовительного примера 54 (100 мг; 0,263 ммоль), 2-метилрезорцина (130 мг; 1,05 ммоль) и трифенилфосфина (139 мг; 0,53 ммоль) в тетрагидрофуране (2 мл) и эту смесь перемешивали при комнатной температуре в течение 12 ч. Растворитель удаляли при пониженном давлении и очищали с использованием картриджа Isolute ® SCX-2, элюируя метанолом, затем 1М аммиаком в метаноле. Основные фракции концентрировали в вакууме и дополнительно очищали хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 98:2:0,2 до 95:5:0,5. Соответствующие фракции концентрировали в вакууме и остаток очищали далее, используя колонку Phenomenex Curosil PFP (размеры 21,2 ×150 мм) на препаративной ЖХВД-системе Agilent 1100. Использовали две подвижные фазы: 0,1% (об./об.) муравьиную кислоту (водн.) (А) и 0,1% (об./об.) муравьиную кислоту в ацетонитриле (В), элюируя с градиентом 20-80% В в течение 18 мин при скорости потока 18 мл/мин. Пики детектировали, используя УФ-детектор при 225 нМ, и соответствующие фракции концентрировали в вакууме с получением указанного в заголовке соединения в виде смолы с выходом 9%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.06 (s, 6H), 1.28-1.34 (m, 2H), 1.71-1.81 (m, 2H), 1.89-1.94 (m, 2Н), 2.02 (s, 3Н), 2.37-2.46 (m, 4Н), 2.68-2.77 (m, 2Н), 4.24-4.31 (m, 1H), 6.37-6.41 (m, 2H), 6.86-6.90 (m, 1H), 7.23-7.40 (m, 10Н); ХИАД-МСНР m/z 487 [M+H] + . Пример 97. 5-{4-[(3'-Гидроксибифенил-3-ил)метокси]пиперидин-1-ил}-5-метил-2,2-дифенилгексаннитрил. 1,1'-Бис(дифенилфосфино)ферроцендихлорпалладий(II) (8 мг; 0,09 ммоль) добавляли к суспензии продукта примера 87 (100 мг; 0,19 ммоль), 3-гидроксифенилбороновой кислоты (52 мг; 0,038 ммоль) и карбоната натрия (40 мг; 0,038 ммоль) в тетрагидрофуране (5 мл) и воде (1 мл) и эту смесь нагревали при 60 °С в течение 12 ч. Суспензию оставляли охлаждаться до комнатной температуры и затем упаривали досуха при пониженном давлении. Остаток очищали хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 99:1:0 до 98:2:0,2, с получением указанного в заголовке соединения в виде смолы с выходом 72%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.09 (s, 6H), 1.52-1.56 (m, 2H), 1.61-1.68 (m, 2Н), 1.93-1.96 (m, 2H), 2.24-2.42 (m, 2H), 2.51-2.55 (m, 2H), 2.81-2.84 (m, 2H), 3.45-3.49 (m, 1Н), 4.58 (s, 2H), 6.75-6.78 (m, 1H), 7.02-7.07 (m, 2H), 7.21-7.43 (m, 13H), 7.47-7.50 (m, 1H), 7.54 (s, 1H); ЭРИ-МСНР m/z 545 [M+H] + . Пример 98. 5-{4-[(3'-Гидроксибифенил-3-ил)метокси]пиперидин-1-ил}-5-метил-2,2-дифенилгексанамид. Суспензию продукта примера 97 (70 мг; 0,13 ммоль) и порошкообразного гидроксида калия (144 мг; 2,57 ммоль) в 3-метил-4-пентаноле (4 мл) нагревали при температуре дефлегмации в течение 36 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры и растворитель удаляли при пониженном давлении. Остаток распределяли между этилацетатом (30 мл) и водой (20 мл). Органический слой отделяли, сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 95:5:0,5 до 90:10:1. Соответствующие фракции концентрировали в вакууме и остаток дополнительно очищали хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 98:2:0,2 до 95:5:0,5, с получением указанного в заголовке соединения в виде смолы с выходом 21%. 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.05 (s, 6H), 1.22-1.35 (m, 2H), 1.62-1.68 (m, 2H), 1.85-1.93 (m, 2H), 2.35-2.45 (m, 4H), 2.78-2.82 (m, 2H), 3.43-3.49 (m, 1H), 4.57 (s, 2H), 6.75-6.78 (m, 1Н), 7.02-7.07 (m, 2Н), 7.21-7.40 (m, 13Н), 7.47-7.50 (m, 1Н), 7.54 (s, 1H); ХИАД-МСНР m/z 563 [М+Н] + . Пример 99. 5-[3-(3-Метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Смесь продукта подготовительного примера 57 (500 мг; 1,21 ммоль), карбоната цезия (1,18 г; 3,64 ммоль) и 3-метоксифенола (0,41 мл; 3,64 ммоль) в N,N-диметилформамиде (10 мл) перемешивали при 80 °С в течение 18 ч. Затем реакционную смесь концентрировали в вакууме и остаток распределяли между диэтиловым эфиром (50 мл) и водой (20 мл). Водный слой отделяли, экстрагировали диэтиловым эфиром (2 ×30 мл), объединенный органический раствор сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат от 90:10 до 75:25, затем смесью дихлорметан:метанол:аммиак 0,88 от 100:0:0 до 95:5:0,5, с получением указанного в заголовке соединения в виде бесцветной смолы с выходом 71% (380 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.90-1.03 (m, 6H), 1.31-1.44 (m, 2H), 2.41-2.56 (m, 2Н), 3.07-3.24 (m, 2H), 3.42-3.54 (m, 2H), 3.77 (s, 3Н), 4.63-4.74 (m, 1H), 6.28-6.38 (m, 2H), 6.48-6.55 (m, 1H), 7.26-7.49 (m, 11H); ХИАД-МСНР m/z 441 [M+H] + . Пример 100. 5-[3-(3-Метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексанамид. Гидроксид калия (215 мг; 3,83 ммоль) добавляли к раствору продукта примера 99 (85 мг; 0,19 ммоль) в 3-метил-3-пентаноле (5 мл) и смесь нагревали при температуре дефлегмации в течение 24 ч. Затем реакционную смесь охлаждали до комнатной температуры, концентрировали в вакууме и остаток распределяли между этилацетатом (20 мл) и водой (5 мл). Водный слой отделяли, экстрагировали этилацетатом (20 мл) и объединенный органический раствор сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 100:0:0 до 93:7:0,7, с получением указанного в заголовке соединения в виде бесцветной смолы с выходом 74% (65 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.92 (s, 6H), 1.12-1.23 (m, 2H), 2.40-2.50 (m, 2H), 3.10-3.25 (m, 2H), 3.44-3.58 (m, 2H), 3.78 (s, 3Н), 4.62-4.72 (m, 1H), 6.30-6.38 (m, 2H), 6.47-6.54 (m, 1H), 7.10-7.18 (m, 1H), 7.22-7.45 (m, 10Н); ЭРИ-МСНР m/z 459 [M+H] + . Пример 101. 5-[3-(Бензилокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Гидрид натрия (60% дисперсия в минеральном масле; 24 мг; 0,60 ммоль) добавляли к охлажденному во льду раствору продукта подготовительного примера 56 (166 мг; 0,50 ммоль) в N,N-диметилформамиде (5 мл) и эту смесь перемешивали в течение 30 мин, давая температуре подняться до 25 °С. Затем реакционную смесь вновь охлаждали до 0 °С, добавляли бензилбромид (89 мкл; 0,75 ммоль), и смесь перемешивали при 0 °С в течение 30 мин. Реакцию гасили 2М соляной кислотой (2 мл), подщелачивали до рН 8 насыщенным раствором гидрокарбоната натрия и экстрагировали этилацетатом (3 ×50 мл). Объединенный органический раствор сушили над сульфатом магния, концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 100:0:0 до 97:3:0,3, с получением указанного в заголовке соединения в виде бесцветного масла с выходом 65% (137 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.91 (s, 6Н), 0.90-0.97 (m, 2Н), 2.40-2.48 (m, 2Н), 2.97-3.06 (m, 2Н), 3.21-3.30 (m, 2Н), 4.07-4.17 (m, 1H), 4.41 (s, 2Н), 7.26-7.46 (m, 15Н); ХИАД-МСНР m/z 425 [М+Н] + . Пример 102. 5-[3-(Бензилокси)азетидин-1-ил]-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта примера 101 способом, который описан в примере 100, в виде бесцветной смолы с выходом 77%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.89 (s, 6H), 1.05-1.13 (m, 2H), 2.32-2.41 (m, 2H), 2.95-3.05 (m, 2H), 3.15-3.25 (m, 2H), 4.02-4.13 (m, 1H), 4.39 (s, 2H), 7.21-7.42 (m, 15H); ХИАД-МСНР m/z 443 [М+Н] + . Пример 103. 5-Метил-5-(3-феноксиазетидин-1-ил)-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из продукта подготовительного примера 65 способом, который описан для подготовительного примера 62, в виде бесцветного масла с выходом 38%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.34-1.41 (m, 2H), 2.43-2.54 (m, 2H), 3.15-3.22 (m, 2H), 3.45-3.55 (m, 2H), 4.65-4.75 (m, 1H), 6.73-6.78 (m, 2H), 6.90-6.98 (m, 1H), 7.22-7.45 (m, 12H); ХИАД-МСНР m/z 411 [M+H] + . Пример 104. 5-Метил-5-(3-феноксиазетидин-1-ил)-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта примера 103 способом, который описан в примере 100, в виде белого твердого вещества с выходом 88%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.91 (s, 6H), 1.12-1.20 (m, 2H), 2.42-2.48 (m, 2H), 3.12-3.18 (m, 2Н), 3.45-3.55 (m, 2Н), 4.62-4.73 (m, 1Н), 5.55 (brs, 2H), 6.75-6.78 (m, 2H), 6.92-6.96 (m, 1H), 7.20-7.41 (m, 12H); ХИАД-МСНР m/z 429 [M+H] + . Пример 105. 5-[3-(4-Метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из продукта подготовительного примера 57 и 4-метоксифенола способом, который описан в примере 99, в виде бесцветной смолы с выходом 55%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.98 (s, 6H), 1.33-1.45 (m, 2Н), 2.45-2.55 (m, 2H), 3.14-3.24 (m, 2H), 3.42-3.55 (m, 2H), 3.78 (s, 3Н), 4.58-4.68 (m, 1H), 6.66-6.74 (m, 2H), 6.76-6.85 (m, 2H), 7.25-7.47 (m, 10Н); ХИАД-МСНР m/z 441 [М+Н] + . Пример 106. 5-[3-(4-Метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексанамид. Гидроксид калия (135 мг; 2,41 ммоль) добавляли к раствору продукта примера 105 (53 мг; 0,12 ммоль) в 3-метил-3-пентаноле (5 мл) и эту смесь нагревали при температуре дефлегмации в течение 18 ч. Затем реакционную смесь охлаждали до комнатной температуры, концентрировали в вакууме и остаток распределяли между этилацетатом (20 мл) и водой (5 мл). Водный слой отделяли, экстрагировали этилацетатом (2 ×20 мл) и объединенный органический раствор сушили над сульфатом магния и концентрировали в вакууме с получением бесцветного масла с выходом 96% (53 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.92 (s, 6H), 1.12-1.23 (m, 2H), 2.40-2.50 (m, 2H), 3.10-3.25 (m, 2H), 3.44-3.58 (m, 2H), 3.78 (s, 3Н), 4.62-4.72 (m, 1H), 6.30-6.38 (m, 2H), 6.47-6.54 (m, 1H), 7.10-7.18 (m, 1H), 7.22-7.45 (m, 10Н); ЭРИ-МСНР m/z 459 [M+H] + . Пример 107. 5-[3-(4-Гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексанамид. Продукт примера 106 (53 мг; 0,16 ммоль) растворяли в дихлорметане (5 мл) и этот раствор охлаждали до 0 °С. Добавляли трибромид бора (1M в дихлорметане; 0,52 мл; 0,52 ммоль) и раствор перемешивали при 0 °С в течение 35 мин. Добавляли дополнительно трибромид бора (1М в дихлорметане; 0,52 мл; 0,52 ммоль) и перемешивание продолжали при 0 °С в течение 30 мин. Затем реакционную смесь гасили насыщенным раствором гидрокарбоната натрия (20 мл) и перемешивали при комнатной температуре в течение 18 ч. Водный слой отделяли, экстрагировали этилацетатом (25 мл) и объединенный органический раствор сушили над сульфатом магния и концентрировали в вакууме с получением смолы. Эту смолу перерастворяли в дихлорметане (5 мл) и раствор охлаждали до -10 °С. Добавляли трибромид бора (1М в дихлорметане; 0,52 мл; 0,52 ммоль) и смесь перемешивали при -10 °С в течение 1 ч. Затем реакционную смесь гасили насыщенным раствором гидрокарбоната натрия (20 мл) и органический слой отделяли и экстрагировали этилацетатом (20 мл). Объединенный органический раствор сушили над сульфатом магния, концентрировали в вакууме и остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат/метанол/аммиак 0,88 (90/10/1) от 75:25 до 50:50, с получением указанного в заголовке соединения в виде бесцветной пены с выходом 27% (14 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.92 (s, 6H), 1.13-1.26 (m, 2H), 2.40-2.50 (m, 2H), 3.08-3.24 (m, 2H), 3.42-3.58 (m, 2Н), 4.52-4.63 (m, 1H), 5.50-5.68 (brs, 2H), 6.53-6.62 (m, 2H), 6.67-6.78 (m, 2H), 7.21-7.42 (m, 10Н); ХИАД-МСНР m/z 445 [М+Н] + . Пример 108. 5-[3-(3-Гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексанамид. Трибромид бора (1М в дихлорметане; 1,75 мл; 1,75 ммоль) добавляли к охлажденному во льду раствору продукта примера 100 (200 мг; 0,44 ммоль) в дихлорметане (5 мл) и эту смесь перемешивали при 0 °С в течение 1 ч. Добавляли дополнительно трибромид бора (1М в дихлорметане; 0,5 мл; 0,5 ммоль) и смесь перемешивали при 0 °С в течение 30 мин. Затем реакционную смесь гасили 1М раствором гидроксида натрия (5 мл), разбавляли дихлорметаном (20 мл) и перемешивали при комнатной температуре в течение 40 мин. Водный слой отделяли, экстрагировали этилацетатом (2 ×25 мл) и объединенный органический раствор сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат/метанол/аммиак 0,88 (90/10/1) от 75:25 до 50:50, с получением указанного в заголовке соединения в виде бесцветной пены с выходом 91% (176 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.10 (s, 6H), 1.22-1.34 (m, 2H), 2.42-2.55 (m, 2H), 3.28-3.40 (m, 2H), 3.65-3.88 (m, 2H), 4.70-4.80 (m, 1H), 5.55-5.70 (brs, 2H), 6.23-6.36 (m, 2H), 6.45-6.53 (m, 1H), 7.03-7.12 (m, 1H), 7.19-7.39 (m, 10Н); ЭРИ-МСНР m/z 445 [M+H] + . Пример 109. 5-[3-(2-Гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексанамид. Формиат аммония (25 мг; 0,4 ммоль) добавляли к смеси продукта подготовительного примера 63 (35 мг; 65 мкмоль) и 20% Pd(OH) 2 /C (10 мг) в этаноле (10 мл), и эту смесь нагревали при температуре дефлегмации в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры, добавляли дополнительно формиат аммония (25 мг; 0,4 ммоль) и 20% Pd(OH) 2 /C (10 мг) и смесь вновь нагревали при температуре дефлегмации в течение 2 ч. Реакционную смесь фильтровали через Arbocel ®, промывая метанолом, и фильтрат концентрировали в вакууме. Остаток разбавляли насыщенным раствором гидрокарбоната натрия, экстрагировали этилацетатом (2 ×20 мл) и объединенный органический раствор сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат:метанол (95:5), с получением указанного в заголовке соединения в виде бесцветного масла с количественным выходом (30 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.02 (s, 6H), 1.22-1.32 (m, 2H), 2.31-2.49 (m, 2H), 2.57-2.72 (m, 2Н), 3.98-4.04 (m, 1Н), 4.15-4.24 (m, 2H), 5.40-5.70 (br m, 2H), 6.77-6.86 (m, 4H), 7.22-7.38 (m, 10Н); ХИАД-МСНР m/z 445 [M+H] + . Пример 110. 5-{3-(2,4-Дихлор-5-гидроксифенокси)азетидин-1-ил}-5-метил-2,2-дифенилгексанамид. Гидроксид калия (523 мг; 9,33 ммоль) добавляли к раствору продукта подготовительного примера 68 (250 мг; 0,46 ммоль) в 3-метил-3-пентаноле (5 мл) и эту смесь нагревали при температуре дефлегмации в течение 18 ч. Затем реакционную смесь охлаждали до комнатной температуры, концентрировали в вакууме и остаток распределяли между этилацетатом (20 мл) и водой (20 мл). Водный слой отделяли и экстрагировали этилацетатом (20 мл) и объединенный органический раствор сушили над сульфатом магния и концентрировали в вакууме. Сырое вещество обрабатывали 4М соляной кислотой в диоксане (10 мл; 40 ммоль) и раствор перемешивали при 60 °С в течение 30 мин. Диоксан удаляли в вакууме и остаток подщелачивали аммиаком 0,88. Водный слой экстрагировали этилацетатом (2 ×20 мл). Объединенный органический раствор сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол от 100:1 до 10:1, с получением указанного в заголовке соединения в виде бесцветного масла с выходом 61% (147 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.98 (s, 6Н), 1.18-1.25 (m, 2H), 2.41-2.55 (m, 2H), 3.32-3.38 (m, 2H), 3.75-3.82 (m, 2H), 4.68-4.75 (m, 1H), 5.65-5.75 (m, 1H), 5.95-6.05 (m, 1H), 6.43 (s, 1H), 7-20-7.40 (m, 11H); ХИАД-МСНР m/z 513 [M+H] + . Пример 111. 5-{3-(4,5-Дихлор-2-гидроксифенокси)азетидин-1-ил}-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта подготовительного примера 70 способом, который описан в примере 110, в виде бесцветного масла с выходом 53%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.02 (s, 6H), 1.22-1.30 (m, 2H), 2.35-2.50 (m, 2H), 2.60-2.75 (m, 2Н), 3.95-4.01 (m, 1Н), 4.10-4.25 (m, 2H), 5.40-5.60 (m, 2H), 6.92-6.98 (m, 2H), 7-20-7.40 (m, 10Н); ЭРИ-МСНР m/z 513 [М+Н] + . Пример 112. 5-[3-(4-Хлор-3-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из 4-хлор-3-метоксифенола (ЕР230379, р. 52) и продукта подготовительного примера 57 способом, который описан в примере 99, в виде бесцветного масла с выходом 85%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.35-1.40 (m, 2H), 2.45-2.53 (m, 2H), 3.16-3.20 (m, 2Н), 3.45-3.52 (m, 2H), 3.86 (s, 3Н), 4.62-4.70 (m, 1H), 6.20-6.25 (m, 1H), 6.40-6.43 (m, 1H), 7-18-7.45 (m, 11H); ХИАД-МСНР m/z 475 [M+H] + . Пример 113. 5-{3-(4-Хлор-3-метоксифенокси)азетидин-1-ил}-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта примера 112 способом, который описан в примере 100, в виде бесцветного масла с выходом 91%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.85 (s, 6H), 1.08-1.22 (m, 2H), 2.38-2.50 (m, 2H), 3.15-3.20 (m, 2H), 3.40-3.55 (m, 2H), 3.85 (s, 3Н), 4.60-4.72 (m, 1H), 5.30-5.55 (m, 2H), 6.20-6.23 (m, 1H), 6.38-6.41 (m, 1H), 7-18-7.40 (m, 11H); ХИАД-МСНР m/z 493 [M+H] + . Пример 114. 5-{3-(4-Хлор-3-гидроксифенокси)азетидин-1-ил}-5-метил-2,2-дифенилгексанамид. Трибромид бора (1М в дихлорметане; 1,75 мл; 1,75 ммоль) добавляли к охлажденному во льду раствору продукта примера 113 (180 мг; 0,36 ммоль) в дихлорметане (5 мл) и эту смесь перемешивали при 0 °С в течение 1,5 ч. Затем реакционную смесь гасили аммиаком 0,88 (30 мл) и раствор перемешивали при комнатной температуре в течение 18 ч. Органический слой отделяли, сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя от дихлорметана до смеси дихлорметан/метанол/аммиак 0,88 (97:3:0,3), с получением указанного в заголовке соединения в виде бесцветной пены с выходом 93% (270 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.85 (s, 6Н), 1.08-1.22 (m, 2H), 2.40-2.50 (m, 2H), 3.10-3.20 (m, 2H), 3.50-3.58 (m, 2H), 4.58-4.66 (m, 1H), 5.45-5.55 (m, 1H), 5.75-5.90 (m, 1H), 6.25-6.32 (m, 1H), 6.38-6.40 (m, 1H), 7-15-7.40 (m, 11Н); ХИАД-МСНР m/z 479 [M+H] + . Пример 115. Амид 5-[3-(3-гидроксибензилокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Продукт подготовительного примера 73 (65 мг; 0,131 ммоль) обрабатывали 4М соляной кислотой в диоксане (2 мл; 8 ммоль), добавляли воду (0,2 мл) и раствор перемешивали при 85 °С в течение 30 мин. Диоксан удаляли в вакууме и остаток распределяли между этилацетатом (25 мл) и насыщенным раствором гидрокарбоната натрия (20 мл). Водный слой экстрагировали этилацетатом (20 мл). Объединенный органический раствор сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат/метанол/аммиак 0,88 (90/10/1) от 75:25 до 50:50, с получением указанного в заголовке соединения в виде бесцветной пены с выходом 70% (42 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.90 (s, 6H), 1.16-1.25 (m, 2H), 2.38-2.48 (m, 2H), 3.02-3.12 (m, 2H), 3.28-3.40 (m, 2H), 4.06-4.17 (m, 1H), 4.31 (s, 2H), 5.58-5.74 (br s, 2H), 6.72-6.83 (m, 3Н), 7,12-7.20 (m, 1Н), 7.21-7.40 (m, 10Н); ХИАД-МСНР m/z 459 [M+H] + . Пример 116. 5-[3-(2-Хлор-5-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из продукта подготовительного примера 57 и 2-хлор-5-метоксифенола способом, который описан в примере 99, в виде коричневой смолы с выходом 55%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.96 (s, 6H), 1.31-1.44(m, 2H), 2.40-2.57 (m, 2H), 3.14-3.35 (m, 2H), 3.40-3.60 (m, 2H), 3.77 (s, 3Н), 4.64-4.79 (m, 1H), 6.20-6.25 (m, 1H), 6.39-6.47 (m, 1H), 7.20-7.48 (m, 11Н); ХИАД-МСНР m/z 475 [M+H] + . Пример 117. Амид 5-[3-(2-хлор-5-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Указанное в заголовке соединение получили из продукта примера 116 способом, который описан в примере 100, в виде бесцветной смолы с выходом 61%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.92 (s, 6H), 1.11-1.20 (m, 2H), 2.40-2.48 (m, 2H), 3.15-3.24 (m, 2H), 3.42-3.55 (m, 2H), 3.76 (s, 3Н), 4.64-4.72 (m, 1H), 5.48-5.75 (m, 2H), 6.20-6.25 (m, 1H), 6.38-6.45 (m, 1H), 7.20-7.41 (m, 11Н); ХИАД-МСНР m/z 493 [M+H] + . Пример 118. Амид 5-[3-(2-хлор-5-гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Трибромид бора (1М в дихлорметане; 1,42 мл; 1,42 ммоль) добавляли к охлажденному во льду раствору продукта примера 117 (70 мг; 0,142 ммоль) в дихлорметане (5 мл) и эту смесь перемешивали при 0 °С в течение 1 ч. Затем реакционную смесь гасили аммиаком 0,88 (20 мл) и раствор перемешивали при комнатной температуре в течение 18 ч. Органический слой отделяли. Водный слой экстрагировали дихлорметаном (20 мл). Объединенный органический раствор промывали насыщенным раствором гидрокарбоната натрия (10 мл), сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат/метанол/аммиак 0,88 (90/10/1) от 85:15 до 50:50. Полученную смолу растворяли в метаноле (5 мл) и добавляли 1н. соляную кислоту в диэтиловом эфире (0,5 мл). Раствор упаривали и твердое вещество подвергали перекристаллизации из смеси этилацетат/метанол, с получением гидрохлоридной соли указанного в заголовке соединения в виде пурпурного твердого вещества с выходом 30% (22 мг). 1 Н-ЯМР (400 МГц, CD 3 OD) δ: 1.28 (s, 6H), 1.33-1.44 (m, 2Н), 2.40-2.48 (m, 2H), 4.02-4.30 (m, 2Н), 4.33-4.62 (m, 2Н), 4.95-5.06 (m, 1Н), 6.20-6.28 (m, 1H), 6.44-6.49 (m, 1H), 7.15-7.20 (m, 1H), 7.24-7.41 (m, 10Н); ХИАД-МСНР m/z 479 [M+H] + . Пример 119. 5-[3-(3-Фтор-5-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из 3-фтор-5-метоксифенола (WO 2005037763, с. 95) и продукта подготовительного примера 57 способом, который описан в примере 99, в виде бесцветного масла с выходом 90%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.35-1.40 (m, 2H), 2.45-2.53 (m, 2H), 3.06-3.15 (m, 2H), 3.40-3.48 (m, 2H), 3.75 (s, 3Н), 4.58-4.64 (m, 1H), 6.05-6.12 (m, 2H), 6.20-6.26 (m, 1H), 7.22-7.45 (m, 10Н); ХИАД-МСНР m/z 459 [M+H] + . Пример 120. 5-[3-(3-Фтор-5-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта примера 119 способом, который описан в примере 100, в виде бесцветного масла с выходом 90%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.42-1.55 (m, 2H), 2.40-2.48 (m, 2H), 3.06-3.15 (m, 2H), 3.40-3.45 (m, 2H), 3.78 (s, 3Н), 4.58-4.66 (m, 1H), 5.40-5.55 (m, 2H), 6.03-6.12 (m, 2H), 6.20-6.25 (m, 1H), 7.22-7.45 (m, 10H). Пример 121. 5-{3-(3-Фтор-5-гидроксифенокси)азетидин-1-ил}-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта примера 120 способом, который описан в примере 108, в виде бесцветного масла с выходом 25%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.20-1.26 (m, 2H), 2.40-2.46 (m, 2H), 3.20-3.28 (m, 2Н), 3.60-3.65 (m, 2Н), 4.64-4.70 (m, 1H), 5.50-5.60 (m, 1H), 6.00 (s, 1H), 6.02-6.06 (m, 1H), 6.18-6.23 (m, 1H), 6.25-6.35 (га, 1Н), 7.20-7.35 (m, 10Н); ХИАД-МСНР m/z 463 [M+H] + . Пример 122. 5-[3-(3-Хлор-5-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из 3-хлор-5-метоксифенола и продукта подготовительного примера 57 способом, который описан в примере 99, в виде бесцветного масла с выходом 75%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.35-1.40 (m, 2H), 2.45-2.53 (m, 2H), 3.06-3.15 (m, 2Н), 3.40-3.48 (m, 2Н), 3.78 (s, 3Н), 4.58-4.66 (m, 1Н), 6.20 (s, 2H), 6.35 (s, 1Н), 6.50 (s, 1H), 7.22-7.45 (m, 10Н); ХИАД-МСНР m/z 475 [M+H] + . Пример 123. 5-[3-(3-Хлор-5-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта примера 122 способом, который описан в примере 100, в виде бесцветного масла с выходом 90%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.15-1.20 (m, 2H), 2.38-2.45 (m, 2H), 3.02-3.10 (m, 2H), 3.37-3.43 (m, 2H), 3.75 (s, 3Н), 4.55-4.63 (m, 1H), 5.40-5.65 (m, 2H), 6.18 (s, 1H), 6.30 (s, 1H), 6.48 (s, 1H), 7.20-7.40 (m, 10Н); ХИАД-МСНР m/z 493 [M+H] + . Пример 124. 5-{3-(3-Хлор-5-гидроксифенокси)азетидин-1-ил}-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта примера 123 способом, который описан в примере 114, в виде бесцветного масла с выходом 92%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.90 (s, 6H), 1.08-1.18 (m, 2H), 2.35-2.45 (m, 2H), 3.06-3.15 (m, 2Н), 3.40-3.46 (m, 2Н), 4.55-4.60 (m, 1Н), 5.45 (brs, 2H), 6.15 (s, 1H), 6.25 (s, 1H), 6.40 (s, 1H), 7.20-7.40 (m, 10H). Пример 125. 5-[3-(4-Фтор-2-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из 4-фтор-2-метоксифенола и продукта подготовительного примера 57 способом, который описан в примере 99, в виде бесцветного масла с выходом 72%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.35-1.40 (m, 2H), 2.45-2.53 (m, 2H), 3.20-3.25 (m, 2H), 3.42-3.48 (m, 2H), 3.83 (s, 3Н), 4.58-4.66 (m, 1H), 6.53-6.65 (m, 3H), 7.22-7.45 (m, 10Н); ХИАД-МСНР m/z 459 [М+Н] + . Пример 126. 5-[3-(4-Фтор-2-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта примера 125 способом, который описан в примере 100, в виде бесцветного масла с выходом 87%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ 0.95 (s, 6H), 1.10-1.18 (m, 2H), 2.40-2.48 (m, 2H), 3.15-3.20 (m, 2H), 3.40-3.46 (m, 2H), 3.80 (s, 3Н), 4.58-4.66 (m, 1H), 5.50-5.75 (m, 2H), 6.48-6.65 (m, 3Н), 7.22-7.40 (m, 10Н); ХИАД-МСНР m/z 477 [M+H] + . Пример 127. 5-{3-(4-Фтор-2-гидроксифенокси)азетидин-1-ил}-5-метил-2,2-дифенилгексанамид. Указанное в заголовке соединение получили из продукта примера 126 способом, который описан в примере 114, в виде бесцветного масла с выходом 32%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.98 (s, 6H), 1.20-1.28 (m, 2H), 2.35-2.48 (m, 2H), 2.56-2.68 (m, 2Н), 3.95-3.42 (m, 1Н), 4.05-4.18 (m, 1H), 4.18-4.25 (m, 1H), 5.40-5.65 (m, 2H), 6.45-6.53 (m, 1H), 6.55-6.60 (m, 1H), 6.72-6.78 (m, 1H), 7.20-7.35 (m, 10Н); ХИАД-MCHP m/z 463 [M+H] + . Пример 128. Амид 5-[3-(2,6-дихлор-3-гидроксибензилокси)азетидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. Продукт подготовительного примера 78 (150 мг; 0,265 ммоль) обрабатывали 4М соляной кислотой в диоксане (5 мл; 20 ммоль) и водой (0,5 мл) и раствор перемешивали при 70 °С в течение 25 мин. Диоксан удаляли в вакууме и остаток распределяли между этилацетатом (30 мл) и насыщенным раствором гидрокарбоната натрия (20 мл). Водный слой экстрагировали этилацетатом (20 мл). Объединенный органический раствор сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:этилацетат/метанол/аммиак 0,88 (90/10/1) от 5:1 до 1:3, с получением указанного в заголовке соединения с выходом 52% (73 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.22 (s, 6H), 1.44-1.53 (m, 2H), 2.60-2.68 (m, 2H), 3.60-3.68 (m, 2Н), 4.26-4.37 (m, 2Н), 4.57-4.64 (m, 1Н), 4.70 (s, 2H), 5.48-5.55 (br m, 1H), 5.78-5.84 (br m, 1H), 7.06-7.12 (m, 1H), 7.18-7.23 (m, 1H), 7.23-7.38 (m, 10Н); ХИАД-MCHP m/z 527 [M+H] + . Пример 129. 4-{1-[3-(3-Метоксифенокси)азетидин-1-ил]циклопентил}-2,2-дифенилбутиронитрил. Указанное в заголовке соединение получили из продукта подготовительного примера 84 и 3-метоксифенола, используя способ, аналогичный описанному в примере 99, с выходом 73%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.25-1.38 (m, 2H), 1.40-1.65 (m, 6H), 1.66-1.76 (m, 2Н), 2.46-2.57 (m, 2H), 3.06-3.16 (m, 2H), 3.48-3.57 (m, 2H), 3.78 (s, 3H), 4.64-4.72 (m, 1Н), 6.34 (s, 1H), 6.33-6.37 (m, 1H), 6.48-6.53 (m, 1H), 7.13-7.19 (m, 1H), 7.27-7.46 (m, 10Н); ХИАД-МСНР m/z 467 [M+H] + . Пример 130. 4-{1-[3-(3-Метоксифенокси)азетидин-1-ил]циклопентил}-2,2-дифенилбутирамид. Указанное в заголовке соединение получили из продукта примера 129, используя способ, аналогичный способу, описанному в примере 106, с выходом 48%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.22-1.70 (m, 10Н), 2.46-2.53 (m, 2H), 3.07-3.15 (m, 2Н), 3.47-3.59 (m, 2Н), 3.77 (s, 3Н), 4.63-4.73 (m, 1H), 5.44-5.60 (br m, 2H), 6.33 (s, 1H), 6.33-6.36 (m, 1H), 6.48-6.53 (m, 1H), 7.11-7.18 (m, 1H), 7.20-7.40 (m, 10Н); ХИАД-МСНР m/z 485 [M+H] + . Пример 131. 4-{1-[3-(3-Гидроксифенокси)азетидин-1-ил]циклопентил}-2,2-дифенилбутирамид. Указанное в заголовке соединение получили из продукта примера 130, используя способ, аналогичный способу, описанному в примере 108, с выходом 53%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.31-1.47 (m, 6H), 1.48-1.60 (m, 2H), 1.63-1.77 (m, 2Н), 2.45-2.54 (m, 2H), 3.22-3.28 (m, 2H), 3.73-3.80 (m, 2H), 4.75-4.83 (m, 1H), 5.40-5.55 (br m, 1H), 6.24 (s, 1H), 6.36-6.39 (m, 1H), 6.48-6.55 (m, 1H), 6.56-6.67 (br m, 1H), 7.08-7.16 (m, 1H), 7.17-7.38 (m, 10Н); ХИАД-МСНР m/z 471 [M+H] + . Пример 132. 5-[3-(2-Фтор-3-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из продукта подготовительного примера 57 и 2-фтор-3-метоксифенола (J. Comb. Chem. 2002, 4, 329), используя способ, аналогичный способу, описанному в примере 99, с выходом 73%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.34-1.40 (m, 2H), 2.44-2.53 (m, 2H), 3.20-3.27 (m, 2H), 3.43-3.52 (m, 2H), 3.88 (s, 3Н), 4.65-4.74 (m, 1H), 6.34-6.39 (m, 1H), 6.58-6.64 (m, 1H), 6.88-6.95 (m, 1H), 7.25-7.47 (m, 10Н); ХИАД-МСНР m/z 459 [М+Н] + . Пример 133. Амид 5-[3-(2-фтор-3-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Указанное в заголовке соединение получили из продукта примера 132 способом, который описан в примере 100, в виде бесцветного масла с выходом 72%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.17-1.25 (m, 2H), 2.40-2.48 (m, 2H), 3.20-3.28 (m, 2Н), 3.50-3.60 (m, 2Н), 4.65-4.74 (m, 1Н), 5.46-5.75 (br m, 2H), 6.20-6.27 (m, 1H), 6.56-6.63 (m, 1H), 6.80-6.86 (m, 1H), 7.25-7.40 (m, 10Н); ХИАД-МСНР m/z 477 [M+H] + . Пример 134. Амид 5-[3-(2-фтор-3-гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. Трибромид бора (1М в дихлорметане; 1,5 мл; 1,5 ммоль) добавляли к охлажденному во льду раствору продукта примера 133 (45 мг; 0,094 ммоль) в дихлорметане (5 мл) и эту смесь перемешивали при 0 °С в течение 45 мин. Реакционную смесь нагревали до комнатной температуры. Через 15 мин добавляли дополнительно 1,5 мл трибромида бора. Через 20 мин реакцию гасили аммиаком 0,88 (20 мл) и раствор перемешивали при комнатной температуре в течение 18 ч. Органический слой отделяли и промывали насыщенным раствором гидрокарбоната натрия (10 мл), сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 100:0:0 до 98:2:0,2, с получением указанного в заголовке соединения с выходом 100% (44 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.17-1.26 (m, 2H), 2.43-2.49 (m, 2H), 3.20-3.28 (m, 2Н), 3.45-3.60 (m, 2Н), 4.65-4.74 (m, 1Н), 5.49-5.76 (br m, 2H), 6.20-6.27 (m, 1H), 6.57-6.62 (m, 1H), 6.80-6.85 (m, 1H), 7.23-7.38 (m, 10Н); ХИАД-МСНР m/z 463 [M+H] + . Пример 135. 5-[3-(2-Фтор-5-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из продукта подготовительного примера 57 и 2-фтор-5-метоксифенола (J. Can. Chem. 1988, 66, 1479), используя способ, аналогичный способу, описанному в примере 99, с выходом 68%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.36-1.42 (m, 2H), 2.44-2.53 (m, 2H), 3.20-3.27 (m, 2H), 3.43-3.52 (m, 2H), 3.77 (s, 3Н), 4.65-4.74 (m, 1H), 6.27-6.34 (m, 1H), 6.38-6.42 (m, 1H), 6.95-7.02 (m, 1H), 7.25-7.47 (m, 10Н); ХИАД-МСНР m/z 459 [M+H] + . Пример 136. Амид 5-[3-(2-фтор-5-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Указанное в заголовке соединение получили из продукта примера 135 способом, который описан в примере 100, в виде бесцветного масла с выходом 96%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.92 (s, 6H), 1.12-1.20 (m, 2H), 2.40-2.48 (m, 2H), 3.16-3.21 (m, 2H), 3.43-3.50 (m, 2H), 3.76 (s, 3Н), 4.60-4.72 (m, 1H), 5.46-5.60 (br m, 1H), 5.65-5.75 (br m, 1H), 6.25-6.33 (m, 1Н), 6.34-6.40 (m, 1Н), 6.91-6.99 (m, 1H), 7.22-7.40 (m, 10Н); ХИАД-МСНР m/z 477 [М+Н] + . Пример 137. Амид 5-[3-(2-фтор-5-гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Указанное в заголовке соединение получили из продукта примера 136 способом, который описан в примере 118, в виде бесцветного масла с выходом 51%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.04 (s, 6H), 1.22-1.33 (m, 2H), 2.46-2.57 (m, 2H), 3.43-3.55 (m, 2H), 3.92-4.00 (m, 2H), 4.75-4.86 (m, 1H), 5.72-5.82 (br m, 1H), 6.35-6.48 (m, 3Н), 6.83-6.92 (m, 1H), 7.20-7.38 (m, 10Н); ХИАД-МСНР m/z 463 [M+H] + . Пример 138. Амид 5-[3-(4-хлор-3-гидроксибензилокси)азетидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. Указанное в заголовке соединение получили из продукта подготовительного примера 86 способом, который описан в примере 115, в виде бесцветного масла с выходом 37%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.88 (s, 6H), 1.15-1.20 (m, 2Н), 2.38-2.45 (m, 2H), 3.00-3.08 (m, 2Н), 3.26-3.36 (m, 2Н), 4.03-4.15 (m, 1Н), 4.27 (s, 2H), 5.55-5.80 (br m, 2H), 6.73-6.78 (m, 1H), 6.95 (s, 1H), 7.20-7.38 (m, 11H); ХИАД-МСНР m/z 493 [M+H] + . Пример 139. 4-{1-[3-(4-Хлор-3-гидроксифенокси)азетидин-1-ил]циклопентил}-2,2-дифенил-бутирамид. Указанное в заголовке соединение получили из продукта подготовительного примера 88 способом, который описан в примере 115, в виде бесцветного масла с выходом 20%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.22-1.60 (m, 8H), 1.60-1.73 (m, 2H), 2.43-2.55 (m, 2Н), 3.14-3.20 (m, 2Н), 3.60-3.68 (m, 2Н), 4.62-4.73 (m, 1H), 5.48-5.62 (br m, 1H), 6.11-6.25 (br m, 1H), 6.26-6.35 (m, 1H), 6.36-6.40 (m, 1H), 7.15-7.38 (m, 11Н); ХИАД-МСНР m/z 505 [M+H] + . Пример 140. 5-[3-(3-Бром-5-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из продукта подготовительного примера 57 и продукта подготовительного примера 89, используя способ, аналогичный способу, описанному в примере 99, с выходом 90%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.33-1.40 (m, 2Н), 2.42-2.53 (m, 2H), 3.06-3.18 (m, 2Н), 3.38-3.50 (m, 2Н), 3.75 (s, 3Н), 4.60-4.66 (m, 1Н), 6.25 (s, 1H), 6.48 (s, 1H), 6.67 (s, 1H), 7.24-7.46 (m, 10Н); ЭРИ-МСНР m/z 519 [M+H] + . Пример 141. Амид 5-[3-(3-бром-5-метоксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Указанное в заголовке соединение получили из продукта примера 140 способом, который описан в примере 100, в виде бесцветного масла с выходом 80%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.92 (s, 6Н), 1.12-1.18 (m, 2Н), 2.38-2.46 (m, 2Н), 3.05-3.13 (m, 2Н), 3.37-3.44 (m, 2Н), 3.75 (s, 3Н), 4.56-4.63 (m, 1Н), 5.45-5.58 (br m, 1H), 5.95-6.08 (br m, 1H), 6.23 (s, 1H), 6.48 (s, 1H), 6.65 (s, 1H), 7.20-7.40 (m, 10Н); ЭРИ-МСНР m/z 539 [М+Н] + . Пример 142. Амид 5-[3-(3-бром-5-гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. Указанное в заголовке соединение получили из продукта примера 141 способом, который описан в примере 114, в виде бесцветной пены с выходом 27%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.18-1.25 (m, 2H), 2.38-2.46 (m, 2H), 3.19-3.26 (m, 2Н), 3.55-3.63 (m, 2Н), 4.60-4.66 (m, 1H), 5.45-5.58 (br m, 1H), 6.13 (s, 1H), 6.20-6.35 (br m, 1H), 6.48 (s, 1H), 6.65 (s, 1H), 7.20-7.35 (m, 10Н); ЭРИ-МСНР m/z 525 [M+H] + . Пример 143. 4-{1-[3-(3-Фтор-4-метоксифенокси)азетидин-1-ил]циклопентил}-2,2-дифенилбутиронитрил. Указанное в заголовке соединение получили из продукта подготовительного примера 84 и 3-фтор-4-метоксифенола (J. Het. Chem., 1989, 26, 1547), используя способ, аналогичный способу, описанному в примере 99, с выходом 75%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.25-1.39 (m, 2H), 1.40-1.65 (m, 6H), 1.65-1.77 (m, 2Н), 2.47-2.58 (m, 2H), 3.12-3.22 (m, 2H), 3.46-3.57 (m, 2H), 3.77 (s, 3H), 4.66-4.74 (m, 1Н), 6.27-6.33 (m, 1H), 6.35-6.43 (m, 1H), 6.94-7.03 (m, 1H), 7.24-7.48 (m, 10Н); ЭРИ-МСНР m/z 485 [M+H] + . Пример 144. 4-{1-[3-(3-Фтор-4-метоксифенокси)азетидин-1-ил]циклопентил}-2,2-дифенилбутирамид. Указанное в заголовке соединение получили из продукта примера 143 способом, который описан в примере 100, в виде бесцветной пены с выходом 65%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.20-1.37 (m, 4H), 1.39-1.58 (m, 4H), 1.58-1.67 (m, 2Н), 2.40-2.53 (m, 2H), 3.06-3.15 (m, 2H), 3.46-3.53 (m, 2H), 3.75 (s, 3H), 4.60-4.71 (m, 1Н), 5.50-5.67 (br m, 1H), 6.17-6.38 (m, 3H), 6.88-6.98 (m, 1H), 7.18-7.40 (m, 10H); ХИАД-МСНР m/z 503 [M+H] + . Пример 145. 4-{1-[3-(3-Фтор-4-гидроксифенокси)азетидин-1-ил]циклопентил}-2,2-дифенилбутирамид. Указанное в заголовке соединение получили из продукта примера 144, используя способ, аналогичный способу, описанному в примере 114, в виде бесцветной пены с выходом 25%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 1.31-1.47 (m, 6H), 1.48-1.62 (m, 2H), 1.64-1.78 (m, 2Н), 2.43-2.54 (m, 2H), 3.27-3.37 (m, 2H), 3.76-3.85 (m, 2H), 4.75-4.84 (m, 1H), 5.44-5.56 (br m, 1H), 6.22-6.28 (m, 1H), 6.38-6.44 (m, 1H), 6.84-7.00 (m, 2H), 7.16-7.34 (m, 10H). Пример 146. Амид 5-[3-(3-хлор-4-гидроксибензилокси)азетидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. Продукт подготовительного примера 94 (100 мг; 0,19 ммоль) растворяли в тетрагидрофуране (10 мл) и добавляли боргидрид натрия (47 мг; 1,2 ммоль) и тетракис(трифенилфосфин)палладий(0) (22 мг; 0,02 ммоль). После перемешивания при 45 °С в течение 30 мин реакционную смесь охлаждали до комнатной температуры и гасили несколькими каплями ледяной уксусной кислоты. Реакционную смесь подщелачивали насыщенным раствором гидрокарбоната натрия. Органические слои экстрагировали этилацетатом (20 мл), органический раствор сушили над сульфатом магния и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью дихлорметан:метанол:аммиак 0,88 от 100:0:0 до 95:5:0,5, с получением указанного в заголовке соединения с выходом 38% (35 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.90 (s, 6H), 1.12-1.20 (m, 2H), 2.38-2.45 (m, 2H), 2.98-3.07 (m, 2Н), 3.28-3.39 (m, 2Н), 4.06-4.15 (m, 1H), 4.26 (s, 2H), 5.53-5.65 (br m, 2H), 6.88-6.95 (m, 1H), 7.02-7.06 (m, 1H), 7.20-7.37 (m, 11Н); ХИАД-МСНР m/z 493 [M+H] + . Пример 147. Амид 5-[3-(4-хлор-2-гидроксибензилокси)азетидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. Указанное в заголовке соединение получили из продукта подготовительного примера 98, используя способ, аналогичный способу, описанному в примере 146, с выходом 19%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.92 (s, 6H), 1.12-1.20 (m, 2H), 2.40-2.46 (m, 2H), 3.04-3.15 (m, 2Н), 3.35-3.43 (m, 2Н), 4.13-4.20 (m, 1Н), 4.47 (s, 2H), 5.56-5.74 (br m, 2H), 6.75-6.80 (m, 1H), 7.03 (s, 1H), 7.08-7.14 (m, 1H), 7.23-7.38 (m, 10Н); ЭРИ-МСНР m/z 493 [M+H] + . Пример 148. Амид 5-[3-(2-хлор-3-гидроксибензилокси)азетидин-1-ил]-5-метил-2,2-дифенил-гексановой кислоты. Указанное в заголовке соединение получили из продукта подготовительного примера 103, используя способ, аналогичный способу, описанному в примере 128, с выходом 25%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.12-1.24 (m, 2H), 2.38-2.45 (m, 2H), 3.02-3.12 (m, 2Н), 3.28-3.39 (m, 2Н), 4.13-4.20 (m, 1Н), 4.45 (s, 2H), 5.37-5.60 (br m, 2H), 6.94-6.99 (m, 2H), 7.12-7.18 (m, 1H), 7.20-7.40 (m, 10Н); ХИАД-МСНР m/z 493 [M+H] + . Пример 149. Амид 5-[3-(3,5-дигидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. 1,3,5-Тригидроксибензола дигидрат (10 г; 79 ммоль) помещали в круглодонную колбу, оснащенную ловушкой Дина-Старка, и дегидратировали, используя 23 мл толуола. После удаления воды и концентрирования в вакууме выделили безводный фенол в виде белого твердого вещества. Смесь карбоната цезия (107 мг; 0,33 ммоль) и безводного 1,3,5-тригидроксибензола (125 мг; 0,99 ммоль) в N,N-диметилформамиде (3 мл) перемешивали при 80 °С в течение 10 мин. Добавляли продукт подготовительного примера 104 (142 мг; 0,33 ммоль) и реакционную смесь перемешивали при 80 °С в течение 10 мин. Сырое вещество распределяли между этилацетатом (20 мл) и водой (30 мл), органический слой отделяли и водный слой повторно экстрагировали этилацетатом (2 ×20 мл). Объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью пентан:(90:10:1, этилацетат:метанол:аммиак 0,88) от 100:1 до 30:70, с получением указанного в заголовке соединения в виде бесцветной смолы с выходом 5% (7 мг). 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.20-1.28 (m, 2H), 2.35-2.48 (m, 2H), 3.15-3.24 (m, 2Н), 3.52-3.60 (m, 2Н), 4.48-4.58 (m, 1Н), 5.62-5.65 (br s, 1H), 5.78 (s, 2H), 6.06 (s, 1H), 6.35-6.40 (br s, 1H), 7.15-7.28 (m, 10Н); ХИАД-МСНР m/z 461 [M+H] + . Пример 150. 5-{3-(3-Гидроксифенокси)азетидин-1-ил]-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из продукта подготовительного примера 105 способом, который описан в примере 109, в виде бесцветного масла с выходом 73%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.98 (s, 6H), 1.35-1.40 (m, 2H), 2.42-2.48 (m, 2H), 3.16-3.22 (m, 2Н), 3.45-3.57 (m, 2Н), 4.64-4.72 (m, 1H), 6.25 (s, 1H), 6.30-6.35 (m, 1H), 6.42-6.48 (m, 1H), 7.05-7.15 (m, 1H), 7.25-7.42 (m, 10Н); ХИАД-МСНР m/z 427 [M+H] + . Пример 151. 5-(3-[(4-Гидроксибензил)окси]азетидин-1-ил}-5-метил-2,2-дифенилгексаннитрил. Указанное в заголовке соединение получили из продуктов подготовительных примеров 56 и 66 способом, который описан в примере 101, с выходом 15%. 1 Н-ЯМР (400 МГц, CDCl 3 ) δ: 0.95 (s, 6H), 1.32-1.40 (m, 2Н), 2.40-2.48 (m, 2H), 3.04-3.08 (m, 2Н), 3.26-3.35 (m, 2Н), 4.05-4.15 (m, 1Н), 4.32 (s, 2H), 6.72-6.76 (m, 2H), 7.10-7.15 (m, 2H), 7.18-7.46 (m, 10Н). Пример 152. Амид 5-[3-(4-гидроксибензилокси)азетидин-1-ил]-5-метил-2,2-дифенилгексановой кислоты. Указанное в заголовке соединение получили из продукта подготовительного примера 108, используя способ, аналогичный способу, описанному в примере 146, с выходом 53%. 1 Н-ЯМР (400 МГц, MeOD) δ: 0.95 (s, 6H), 1.23-1.28 (m, 2H), 2.35-2.45 (m, 2H), 2.96-3.05 (m, 2Н), 3.22-3.28 (m, 2Н), 4.01-4.18 (m, 1Н), 4.30 (s, 2H), 5.55-5.65 (br s, 2H), 6.70-6.75 (m, 2H), 7.05-7.12 (m, 2H), 7.20-7.35 (m, 10Н); ЭРИ-МСНР m/z 459 [M+H] + . Анализ эффективности. М 3 -эффективность определяли на CHO-K1-клетках, трансфицированных геном NFAT-бета-лактамазы (NFAT (nuclear factor of activated T-cells, ядерный фактор активации Т-клеток)). Клетки СНО (яичник китайского хомячка), рекомбинантно экспрессирующие человеческий мускариновый рецептор M 3 (hM 3 ), трансфицировали плазмидой NFAT_ β-Lac_Zeo. Клетки выращивали на среде DMEM (среда Игла, модифицированная по Дульбекко) с Glutamax-1, дополненной 25 мМ HEPES (N-2-гидроксиэтил-пиперазин-N-2-этансульфоновая кислота; Life Technologies 32430-027), содержащей 10% FCS (фетальная сыворотка теленка; Sigma F-7524), 1 нМ пируват натрия (Sigma S-8636), NEAA (не являющиеся незаменимыми аминокислоты; Invitrogen 11140-035) и 200 мкг/мл зеоцина (Invitrogen R250-01). Протокол анализа hM 3 с использованием β-Lac. Клетки собирали для анализа по достижении ими 80-90% конфлюэнтности, используя не содержащий ферментов клеточный диссоциирующий раствор (Life technologies 13151-014), инкубируемый с данными клетки в течение 5 мин при 37 °С в атмосфере, содержащей 5% CO 2 . Отделенные клетки собирали в нагретые ростовые среды и центрифугировали при 2000 об./мин в течение 10 мин, промывали в PBS (забуференный фосфатом физиологический раствор; Life Technologies 14190-094) и снова центрифугировали, как описано непосредственно выше. Клетки ресуспендировали в концентрации 2 ×10 5 клеток/мл в ростовой среде (состав описан выше). По 20 мкл этой клеточной суспензии добавляли в каждую лунку 384-луночного черного планшета с прозрачным дном (Greiner Bio One 781091-PFI). В качестве буфера для анализа использовали PBS, дополненный 0,05% Pluronic F-127 (Sigma 9003-11-6) и 2,5% DMSO. Сигнализацию мускаринового рецептора M 3 стимулировали, используя 80 нМ карбамилхолин (Aldrich N240-9), инкубируемый с клетками в течение 4 ч при 37 °С/5% СО 2 , и регистрировали в конце инкубационного периода, используя планшет-ридер Tecan SpectraFluor+( λ(длина волны) - возбуждение 405 нМ, эмиссия 450 нМ и 503 нМ). Тестируемые антагонисты рецептора M 3 добавляли в анализ в начале 4-часового инкубационного периода и измеряли активность соединений как зависящее от концентрации ингибирование индуцируемого карбамилхолином сигнала. Строили кривые ингибирования и рассчитывали величины IC 50 , используя 4-параметрическое сигмоидное выравнивание, и переводили в величины Ki, используя поправку Ченга-Пруссофа и величину K D для карбамилхолина в этом анализе. Таким образом, было обнаружено, что карбоксамидные производные формулы (I) по настоящему изобретению, которые были протестированы в описанным выше анализе, демонстрируют антагонистическую активностью в отношении рецептора M 3 , которая приведена ниже в таблице. Анализ на трахее морской свинки. Самцов морских свинок Dunkin-Hartley массой 350-450 г умерщвляют, поднимая концентрацию СО 2 и затем вскрывая полую вену. Трахеи иссекают из гортани до точки входа в полость грудной клетки и затем помещают в свежий оксигенированный модифицированный буферный раствор Кребса (буферный раствор Кребса, содержащий 10 мМ пропранолола, 10 мкМ гуанетидина и 3 мкМ индометацина) при комнатной температуре. Трахеи вскрывают, разрезая по хрящу напротив трахеальной мышцы. Нарезают полоски шириной приблизительно по 3-5 хрящевых колец. На одном конце полоски к хрящу присоединяют хлопковую нить для прикрепления к датчику силы, а на другом конце делают петлю из хлопковой нити для закрепления ткани в ванне для органов. Полоски помещают в ванны для органов емкостью 5 мл, заполненные теплым (37 °С) аэрированным модифицированным буферным раствором Кребса. Устанавливают скорость потока насоса 1,0 мл/мин и ткани постоянно промывают. Ткани помещают в условия начального натяжения 1000 мг. Ткани подвергают повторному натяжению через 15 и 30 мин, затем оставляют уравновешиваться в течение следующих 30-45 мин. Ткани подвергают стимуляции электрическим полем (EFS), используя следующие параметры: испытания в течение 10 с каждые 2 мин, длительность импульса 0,1 мс, 10 Гц и 10-30 В. Напряжение повышают на 5 В каждые 10 мин в пределах установленного диапазона до тех пор, пока не будет наблюдаться максимальный сократительный ответ для каждой ткани. Именно это максимальное напряжение для каждой ткани далее используют на протяжении остальной части эксперимента. После установления равновесия к EFS в течение 20 мин насос останавливают и через 15 мин производят контрольные считывания в течение периода времени 8-10 мин (4-5 ответов). Затем к каждой ткани добавляют соединение в виде болюсной дозы, составляющей 30 ×Ki (определенной для человеческого рецептора M 3 , экспрессированного в СНО-клетках, в анализе по связыванию с фильтрованием), и оставляют инкубироваться в течение 2 ч. Далее ткани отмывают от соединения, используя быстрое промывание модифицированным буферным раствором Кребса в течение 1 мин, и скорость потока вновь устанавливают 1 мл/мин на оставшуюся часть эксперимента. В конце эксперимента в ткани вводят гистамин (1 мкМ) для определения жизнеспособности. Считывания, производимые во время эксперимента, автоматически собирают с использованием программного обеспечения Notocord ®. Необработанные данные переводят в процент ответа, учитывая измерения ингибирования EFS-ответа. После начала промывки регистрируют промежутки времени, необходимые для того, чтобы ткань восстановилась на 25% от индуцированного ингибирования, и их используют в качестве меры продолжительности действия соединения. Жизнеспособность ткани ограничивает продолжительность эксперимента 16 ч промывки после применения соединения. В типичном случае для установления продолжительности действия соединения тестируют при n=2-5. Альтернативно может быть использован также следующий анализ на трахее морской свинки. У самцов морских свинок Dunkin-Hartley (массой 350-450 г) извлекали трахею и после удаления прикрепленной соединительной ткани делали разрез по всему хрящу напротив трахеальной мышцы и готовили трахеальные полоски шириной по 3-5 хрящевых колец. Трахеальные полоски закрепляли между датчиком изометрического натяжения и фиксирующим ткань крючком, располагая мышцу в горизонтальной плоскости в ваннах для тканей емкостью 5 мл с начальным натяжением 1 г, и погружали в нагретый (37 °С) аэрируемый (95% О 2 /5% СО 2 ) раствор Кребса, содержащий 3 мкМ индометацина и 10 мкМ гуанетидина. Ткани располагали между параллельными электродами из платиновой проволоки (зазор приблизительно 1 см). Используя перистальтические насосы, поддерживали постоянный (1 мл/мин) ток свежего раствора Кребса (указанного выше состава) через ванны для тканей. Ткани оставляли уравновешиваться в течение часа с повторным натяжением до 1 г через 15 мин и 30 мин от начала периода уравновешивания. По окончании уравновешивания ткани подвергали стимуляции электрическим полем (EFS), используя следующие параметры: 10 В, 10 Гц, длительность импульса 0,1 мс с испытаниями в течение 10 с каждые 2 мин. Для каждой ткани получали кривую ответа на напряжение в диапазоне 10-30 В (поддерживая все остальные параметры стимуляции постоянными) для определения максимальной стимуляции. С использованием этих параметров стимуляции EFS-ответы были на 100% нервноопосредоваными и на 100% холинергическими, что подтверждалось блокадой 1 мкМ тетродотоксином или 1 мкМ атропином. Затем ткани многократно стимулировали с 2-минутными интервалами до воспроизводимости. Перистальтический насос останавливали за 20 мин до добавления исследуемого соединения и в течение последних 10 мин регистрировали среднее судорожное сокращение как контрольный ответ. В ванны для тканей добавляли исследуемое соединение, при этом к каждой ткани добавляли соединение в одной концентрации и оставляли уравновешиваться в течение 2 ч. Через 2 ч после добавления регистрировали ингибирование EFS-ответа и получали кривые IC 50 , используя диапазон концентраций соединения для трахеальных полосок от одного и того же животного. Затем ткани быстро промывали и вновь устанавливали перфузию раствором Кребса (1 мл/мин). Ткани стимулировали в течение следующих 16 ч и регистрировали восстановление EFS-ответа. По окончании 16 ч в ванны добавляли 10 мкМ гистамин для подтверждения жизнеспособности ткани. Максимальную концентрацию (тестируемую концентрацию, дающую ответ более 70%, но менее 100% ингибирования) антагониста идентифицировали из кривой IC 50 и в тканях, получавших эту концентрацию, рассчитывали время, необходимое для 25% восстановления от индуцируемого ингибирования (Т 25 ). Обычно для установления продолжительности действия соединения тестируют при n=2-5. |