|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea000013082b*\id |
|
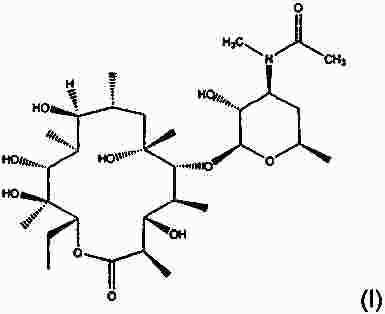
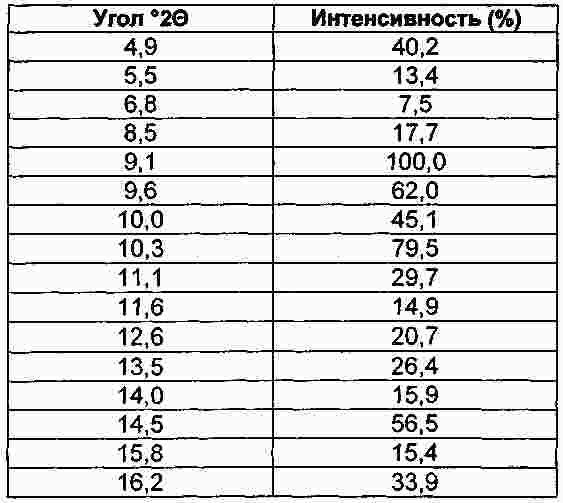
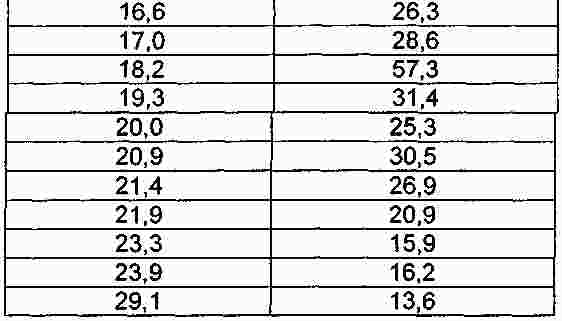
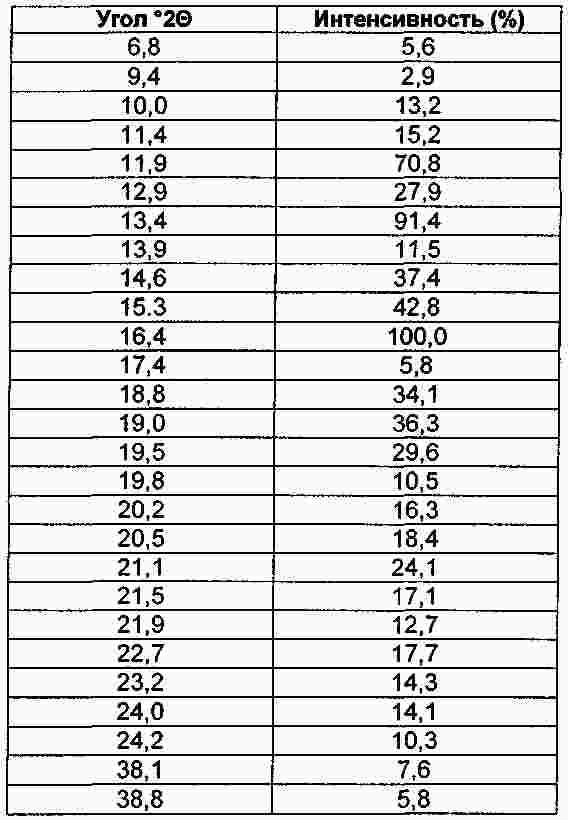
Термины запроса в документе Реферат Настоящее изобретение относится к макролидным соединениям, обладающим противовоспалительной активностью, и, в частности, оно относится к новым устойчивым кристаллическим формам макролидного производного с противовоспалительной активностью, способу получения таких форм, фармацевтическим композициям, содержащим их в качестве активного ингредиента, и к применению кристаллических форм для лечения воспалительных заболеваний. Формула [0001] Кристаллическая форма I соединения формулы (I) [0002] Кристаллическая форма по п.1, характеризующаяся порошковой рентгенограммой, в основном соответствующей порошковой рентгенограмме, проиллюстрированной на фиг. 1. [0003] Кристаллическая форма по одному из предшествующих пунктов, характеризующаяся кривой дифференциальной сканирующей калориметрии, показывающей начало плавления при 163-174 °С. [0004] Кристаллическая форма по п.3, характеризующаяся кривой дифференциальной сканирующей калориметрии, показывающей начало плавления при 163-168 °С. [0005] Кристаллическая форма II соединения формулы I, характеризующаяся порошковой рентгенограммой, содержащей значения угла 2 θ, составляющие приблизительно 11,9; приблизительно 13,4; приблизительно 13,9; приблизительно 14,6; приблизительно 15,3; приблизительно 16,4; приблизительно 17,4; приблизительно 18,8; приблизительно 19,0; приблизительно 19,5; приблизительно 21,1; приблизительно 22,7. [0006] Кристаллическая форма по п.5, характеризующаяся порошковой рентгенограммой, в основном соответствующей порошковой рентгенограмме, проиллюстрированной на фиг. 2. [0007] Кристаллическая форма по п.5 или 6, характеризующаяся кривой дифференциальной сканирующей калориметрии, показывающей начало плавления при 218-226 °С. [0008] Кристаллическая форма по п.7, характеризующаяся кривой дифференциальной сканирующей калориметрии, показывающей начало плавления при 218-223 °С. [0009] Способ получения кристаллической формы I, включающий следующие этапы:добавление метил-трет-бутилового эфира к раствору соединения формулы I в этилацетате, охлаждение полученной реакционной смеси, фильтрование и сушка кристаллизованного продукта. [0010] Способ получения кристаллической формы II, включающий преобразование из кристаллической формы I в кристаллическую форму II путем нагрева водной суспензии кристаллической формы I. [0011] Способ по п.10, в котором суспензию нагревают до температуры в пределах от 30 до 35 °С. [0012] Способ получения кристаллической формы II, включающей кристаллизацию соединения формулы I их метилэтилкетона. [0013] Способ по п.12, дополнительно включающий затравливание раствора кристаллической формой II. [0014] Применение кристаллической формы I в качестве лекарственного средства для лечения воспалительных заболеваний. [0015] Применение кристаллической формы II в качестве лекарственного средства для лечения воспалительных заболеваний. [0016] Фармацевтическая композиция, применяемая при лечении воспалительных заболеваний, включающая терапевтически эффективное количество кристаллической формы I в качестве активного ингредиента в смеси с фармацевтически приемлемым носителем. [0017] Фармацевтическая композиция, применяемая при лечении воспалительных заболеваний, включающая терапевтически эффективное количество кристаллической формы II в качестве активного ингредиента в смеси с фармацевтически приемлемым носителем. [0018] Фармацевтическая композиция по п.16 или 17, являющаяся приемлемой при лечении респираторных заболеваний. [0019] Фармацевтическая композиция по п.18, являющаяся приемлемой при лечении желудочно-кишечных заболеваний. Полный текст патента Настоящее изобретение относится к макролидным соединениям, обладающим противовоспалительной активностью, и, в частности, настоящее изобретение относится к новым устойчивым кристаллическим формам производного макролида с противовоспалительной активностью, к способам получения таких форм, к фармацевтическим композициям, содержащим их в качестве активного ингредиента, и к применению указанных кристаллических форм для лечения воспалительных заболеваний. Известно, что многие антибиотики, в частности класс основанных на эритромицине макролидов, имеющих 14 кольцевых атомов, обладают противовоспалительными свойствами дополнительно к их антибактериальной активности [Clin. Immunother., (1996), 6, 454-464]. Эритромицин является природным макролидом (The Merck Index, XIII Edition, No. 3714, p. 654), который нашел широкое применение при лечении инфекционных заболеваний, вызванных грамположительными бактериями, рядом грамотрицательных бактерий и микоплазмами. В последнее время научное сообщество обратило свой интерес к противовоспалительным и иммуномодуляторным свойствам эритромицина и его производных [Journal of Antimicrobial Chemotherapy, (1998), 41, Suppl. В, 37-46]. Указанная активность в полной мере описана как при проведении клинических исследований, так и при проведении экспериментов in vivo и in vitro. Тем не менее, тот факт, что известные макролидные соединения обладают высокой антибактериальной активностью, не позволяет использовать их в более широком масштабе при длительном лечении воспалительных процессов, не вызванных патогенными микроорганизмами, так как это может привести к быстрому развитию устойчивых штаммов. Вышеуказанная техническая проблема была успешно решена в международной заявке на патент WO 2004/013153 (WO' 153) на имя того же самого заявителя, в которой приведено описание макролидных производных, обладающих противовоспалительной активностью и лишенных антибиотической активности. В частности, соединение (9S)-3-дескладинозил-3'-десметил-3'-ацетил-9-деоксо-9-дегидроэритромицин А (именуемое в дальнейшем «соединение »), имеющее формулу получено в твердой аморфной форме. Способность вещества кристаллизоваться с более чем одной кристаллической структурой известно как полиморфизм, и конкретная кристаллическая форма называется полиморфом. В заявке на патент WO' 153 не раскрывается или не предлагается возможное существование кристаллических полиморфных форм соединения. В данной области техники известно, что различные твердые формы одного и того же активного соединения могут проявлять различные свойства, такие как растворимость, скорость растворения и (или) устойчивость при хранении, которые могут привести к различной степени эффективности. Кроме того, различные физические свойства твердых форм по отношению к кристаллическому или аморфному состоянию могут оказать существенное влияние на возможность производства соединения химическими и фармацевтическими методами, в частности при его изготовлении и применении в промышленном масштабе. Например, важным является обеспечение создания лекарственных веществ в форме, обладающей максимальной степенью чистоты. Обычно в процессе работы и составления формул трудности вызывают аморфные вещества, а не кристаллические формы, и они нередко создают проблемы в отношении устойчивости и чистоты. Таким образом, специалистам в данной области техники желательно получить соединение в основном в кристаллической устойчивой форме, которое может быть легко выделено и, в частности, доведено до состояния, приемлемого для применения в качестве лекарственного средства. В патентной заявке WO' 153 предлагается эффективный способ для получения соединения в аморфной форме, начиная с эритромицина. Было обнаружено, что путем создания приемлемых условий реакции и применения приемлемых растворителей соединение может быть выделено из реакционной смеси в устойчивых твердых кристаллических формах с высоким выходом и высокой степенью чистоты. Кристалличность полиморфных форм была подтверждена путем рентгенографического измерения порошкообразного образца. Таим образом, целью настоящего изобретения является создание кристаллической устойчивой формы соединения, которая в настоящем описании имеет название кристаллическая форма I. Кристаллическая форма I позволяет получить порошковую рентгенограмму, в основном соответствующую порошковой рентгенограмме, проиллюстрированной на фиг. 1. Значения угла 2 θ дифракционных пиков, а также их соответствующая относительная интенсивность приведены в табл. 1. Кристаллическая форма I соединения характеризуется созданием порошковой рентгенограммы, содержащей значения угла 2 θ, составляющие приблизительно 4,9; приблизительно 8,5; приблизительно 9,1; приблизительно 9,6; приблизительно 10,3; приблизительно 11,1; приблизительно 14,5; приблизительно 17,0; приблизительно 18,2; приблизительно 19,3. Кристаллическая форма I дополнительно характеризуется образованием кривой дифференциальной сканирующей калориметрии, задающей начало плавления, находящееся в пределах 163-174 °С (начало: 168,2 °С; пик: 174,8 °С). Кристаллическая форма I предпочтительно образует кривую дифференциальной сканирующей калориметрии, задающей начало плавления, находящееся в пределах 163-168 °С. Кристаллическая форма I наиболее полно соответствует вышеупомянутым требованиям по обработке и устойчивости, чем известная твердая аморфная форма, полученная в соответствии с описанием в заявке на патент WO' 153. Также было обнаружено, что соединение может быть получено в более устойчивой кристаллической форме. Таким образом, дополнительной целью настоящего изобретения является создание кристаллической устойчивой формы соединения, которая в настоящем описании имеет название кристаллическая форма II. Кристаллическая форма II может образовывать порошковую рентгенограмму, в основном соответствующую порошковой рентгенограмме на фиг. 2. Значения угла 2 θ дифракционных пиков, а также их соответствующая относительная интенсивность приведены в табл. 2. Кристаллическая форма II соединения характеризуется созданием порошковой рентгенограммы, содержащей значения угла 2 θ, составляющие приблизительно 11,9; приблизительно 13,4; приблизительно 13,9; приблизительно 14,6; приблизительно 15,3; приблизительно 16,4; приблизительно 17,4; приблизительно 18,8; приблизительно 19,0; приблизительно 19,5; приблизительно 21,1; приблизительно 22,7. Форма II дополнительно характеризуется образованием кривой дифференциальной сканирующей калориметрии, задающей начало плавления, находящееся в пределах 218-226 °С (начало: 222,7 °С; пик: 225,0 °С). Кристаллическая форма II предпочтительно образует кривую дифференциальной сканирующей калориметрии, задающей начало плавления, находящееся в пределах 218-223 °С. Кристаллическая форма II характеризуется термодинамической активностью, тем самым проявляя предпочтительные свойства, заключающиеся в том, что она является термодинамически более устойчивой, чем кристаллическая форма I, в особенности в биологических жидкостях. Кристаллическую форму II выделяют из реакционной смеси с помощью технологических процессов, обычно используемых в промышленном масштабе, с высоким выходом и высокой степенью чистоты, и благодаря ее термодинамической устойчивости она является исключительно приемлемой для применения в качестве лекарственного средства. В соответствии с настоящим изобретением дополнительно предусматривается способ получения кристаллической формы I путем выделения продукта с применением процесса осаждения антирастворителем. В заявке на патент WO' 153 при втором способе синтезирования изготовление соединения проводили, начиная с эритромицина А, путем восстановления кето-группы в позиции 9, монодеметилирования атома азота в положении 3', гидролиза кладинозы и ацетилирования полученного амина. В конце этапа ацетилирования сухой остаток соединения растворяли в этилацетате и удаление примесей проводили промывкой разбавленным раствором лимонной кислоты. Полученную таким образом органическую фазу фильтровали и растворитель отгоняли. Для выделения требуемой кристаллической формы I добавляли метил-трет-бутиловый эфир в качестве антирастворителя к раствору соединения в этилацетате. Таким образом, дополнительной целью настоящего изобретения является создание способа получения кристаллической формы I, включающего следующие этапы: добавление метил-трет-бутилового эфира к раствору соединения в этилацетате, охлаждение полученной реакционной смеси, фильтрование и сушка кристаллизованного продукта. Например, получение кристаллической формы I осуществляли путем добавления метил-трет-бутилового эфира в качестве антирастворителя к раствору, полученному путем растворения в этилацетате сухого остатка соединения, полученного в соответствии с процессом, описанным в заявке на патент WO' 153. В альтернативном случае смесь растворитель/антирастворитель, содержащую соединение, т.е. смесь, содержащую этилацетат/метил-трет-бутиловый эфир, получали путем добавления метил-трет-бутилового эфира к раствору соединения, полученному непосредственно на этапе ацетилирования. Другими словами, раствор соединения, являющийся предпочтительным в качестве исходного материала в осаждении антирастворителем, является реакционной смесью, полученной непосредственно в процессе этапа ацетилирования. Предпочтительно, чтобы весовое соотношение между этилацетатом и метил-трет-бутиловым эфиром составляло приблизительно 1:1. Предпочтительно, чтобы антирастворитель добавляли при температуре приблизительно 50 °С. Предпочтительно, чтобы раствор этилацетата имел концентрацию продукта по весу приблизительно 30-40%. Предпочтительно, чтобы смесь охлаждали в течение 3 ч в соответствии с процессом, предусматривающим понижение температуры на 0,08 °С/мин до приблизительно 10 °С. Практический пример осуществления способа настоящего изобретения включает отгонку раствора этилацетата до тех пор, пока не будет достигнута концентрация по весу в пределах 30-40% при постоянном медленном перемешивании при комнатной температуре. Продукт начинает осаждаться и суспензию нагревают до приблизительно 50 °С, далее добавляют метил-трет-бутиловый эфир и спустя приблизительно 3 ч реакционную смесь охлаждают до приблизительно 10 °С. Твердый осадок фильтруют, промывают смесью этилацетата-метил-трет-бутилового эфира в отношении 1/1 и высушивают в вакууме. Продукт выделяют в виде кристаллической формы I. В соответствии с настоящим изобретением дополнительно предусматривается создание способа преобразования кристаллической формы I в более устойчивую кристаллическую форму II. Таким образом, дополнительной целью настоящего изобретения является создание способа получения кристаллической формы II, включающего преобразование кристаллической формы I в кристаллическую форму II путем нагрева водной суспензии кристаллической формы Form I. Предпочтительно, чтобы весовое соотношение вода-субстрат составляло приблизительно 15:1 с целью повышения реологических свойств суспензии и исключения недостатков, которые могут возникнуть в процессе перемешивания и фильтрования. Предпочтительно, чтобы суспензию нагревали до температуры в пределах от 30 до 35 °С. Практический пример осуществления способа настоящего изобретения включает этапы нагревания суспензии кристаллической формы I в воде до температуры приблизительно 30-35 °С при постоянном помешивании в течение приблизительно одних суток при одной и той же температуре, охлаждения до комнатной температуры, фильтрования реакционной смеси, промывки водой и сушки в вакууме. Продукт выделяют в виде кристаллической формы II. В соответствии с настоящим изобретением дополнительно предусматривается создание способа изготовления термодинамически устойчивой кристаллической формы II путем исключения выделения кинетической кристаллической формы I и последующего преобразования из нее формы II. Было обнаружено, что имеется возможность вызвать непосредственное осаждение термодинамической кристаллической формы II путем кристаллизации из метилэтилкетона. Таким образом, дополнительной целью настоящего изобретения является создание способа получения кристаллической формы II, включающего кристаллизацию соединения из метилэтилкетона. Например, получение кристаллической формы II осуществляют путем кристаллизации раствора, полученного в результате растворения в метилэтилкетоне сухого остатка соединения, полученного в соответствии с процессом, описанным в заявке на патент WO' 153. В альтернативном случае раствор соединения в метилэтилкетоне, являющийся предпочтительным в качестве исходного продукта при кристаллизации в соответствии с настоящим изобретением, является реакционной смесью, полученной непосредственно в процессе этапа ацетилирования при замене растворителя этилацетата на метилэтилкетон в соответствии с известными способами. Предпочтительно, чтобы раствор соединения в метилэтилкетоне включал остаточное количество этилацетата менее 15 вес.%. Предпочтительно, чтобы раствор соединения в метилэтилкетоне концентрировали до тех пор, пока не будет достигнута концентрация продукта в пределах 25-50 вес.%. Более предпочтительно, чтобы раствор концентрировали до тех пор, пока не будет достигнута концентрация продукта приблизительно 36 вес.%. Применение кристаллической формы II в качестве затравки может явиться эффективным для ускорения осаждения продукта. Предпочтительно, чтобы раствор затравливали при комнатной температуре до процесса отгонки до тех пор, пока не будет достигнута концентрация раствора, приемлемая для осаждения кристаллической формы II. Предпочтительно, чтобы при достижении приемлемой концентрации и начальном осаждении продукта температуру полученной суспензии поддерживали на уровне приблизительно 75 °С в течение нескольких часов. Предпочтительно, чтобы смесь охлаждали до приблизительно 0 °С в течение 5 ч и поддерживали такую температуру в течение приблизительно 1 ч. Практический пример осуществления процесса настоящего изобретения включает растворение соединения, полученного в твердой аморфной форме в соответствии с описанием в заявке на патент WO' 153, в метилэтилкетоне. Раствор отгоняли до тех пор, пока не была достигнута концентрация продукта приблизительно 36 вес.%. После охлаждения до приблизительно 75 °С реакционную смесь продолжали перемешивать в течение нескольких часов. Затем ее охлаждали до 0 °С в течение 5 ч и температуру поддерживали на этом уровне в течение 1 ч. Смесь фильтровали и промывали метилэтилкетоном. Продукт выделяли в виде кристаллической формы II. Другой пример практического примера осуществления способа настоящего изобретения включает замену растворителя с этилацетата на метилэтилкетон в реакционной смеси, поступающей с процесса этапа ацетилирования. После фильтрования и промывки метилэтилкетоном раствор затравливали кристаллической формой II, и смесь отгоняли до тех пор, пока не была достигнута концентрация продукта приблизительно 36 вес.%. После охлаждения до приблизительно 75 °С реакционную смесь выдерживали в течение нескольких часов при постоянном перемешивании. Затем смесь охлаждали до 0 °С в течение 5 ч, и указанную температуру поддерживали в течение приблизительно 1 ч. Смесь фильтровали и промывали метилэтилкетоном. Продукт выделяли в виде кристаллической формы II. Специалистам в данной области техники должно быть очевидно, что процесс кристаллизации с помощью метилэтилкетона является также приемлемым для получения кристаллической формы II исходя из кристаллической формы I. Соединения в соответствии с настоящим изобретением представляют собой противовоспалительные макролиды, не обладающие антибиотической активностью и, таким образом, являющиеся эффективными при лечении и профилактике воспалительных заболеваний. Таким образом, дополнительной целью настоящего изобретения является применение соединения в качестве лекарственного средства в его кристаллической форме I. Дополнительной целью настоящего изобретения является применение соединения в качестве лекарственного средства в его кристаллической форме II. Соединения в соответствии с настоящим изобретением, предназначенные для терапевтического или профилактического применения в вышеуказанных патологиях, предпочтительно используют в фармацевтических композициях, приемлемых для перорального, ректального, сублингвального, парентерального, топикального, трансдермального и ингаляторного приема. Таким образом, дополнительной целью настоящего изобретения является фармацевтическая композиция, содержащая терапевтически эффективное количество соединения в кристаллической форме I в качестве активного ингредиента в смеси с фармацевтически приемлемым носителем. Другой целью настоящего изобретения является фармацевтическая композиция, содержащая терапевтически эффективное количество соединения в кристаллической форме II в качестве активного ингредиента в смеси с фармацевтически приемлемым носителем. Фармацевтические композиции в соответствии с настоящим изобретением могут быть жидкими, приемлемыми для перорального и (или) парентерального приема, такими как, например, капли, сиропы, растворы, готовые к применению инъецируемые растворы, либо приготовленные путем разбавления лиофилизированного препарата, и твердыми или полутвердыми, такими как таблетки, капсулы, грануляты, порошки, пилюли, вагинальные суппозитории, суппозитории, кремы, притирания, гели, мази; либо дистиллированными растворами, суспензиями, эмульсиями и иными формами, приемлемыми для ингаляторного или трансдермального приема. В зависимости от типа композиции кроме терапевтически эффективного количества соединений в соответствии с настоящим изобретением они могут содержать определенные твердые или жидкие наполнители или разбавители для фармацевтического применения и произвольно дополнительные добавки, обычно используемые при приготовлении фармацевтических композиций, такие как загустители, связующие вещества, смазывающие вещества, дезинтеграторы, ароматизирующие вещества и пигменты. Получение фармацевтических композиций в соответствии с настоящим изобретением может быть осуществлено в соответствии с известными способами. Изобретение проиллюстрировано со ссылкой на прилагаемые рисунки, описанные ниже. Фиг. 1 - порошковая рентгенограмма кристаллической формы I. Фиг. 2 - порошковая рентгенограмма кристаллической формы II. Фиг. 3 - термограмма дифференциальной сканирующей калориметрии кристаллической формы I. Фиг. 4 - термограмма дифференциальной сканирующей калориметрии кристаллической формы II. Термограммы дифференциальной сканирующей калориметрии были определены на анализаторе ТА Q100 (10 °С/мин; алюминиевая чашка). Начало расплавления определяют как точку, при которой происходит значительное изменение исходного состояния, и производили измерение указанной точки. Специалистам в данной области техники должно быть очевидно, что степень чистоты соединения, вес образца, скорость нагрева и размер частиц оказывают влияние на точное значение точки плавления. Рентгенограммы кристаллической формы I и II в соответствии с настоящим изобретением измеряли на рентгеновском дифрактометре Philips X'Pert (геометрия Брэгга-Брентано) с источником радиации Cu K альфа-1. Положение и интенсивность максимумов измеряли с помощью программы X' Pert Philips Analitycal. Относительная интенсивность максимумов порошковой рентгенограммы может варьироваться в зависимости от способа получения образца, процедуры установки и конкретного используемого прибора. Специалистам в данной области техники должно быть очевидно, что несмотря на то что описание изобретения приведено в сочетании с его предпочтительными примерами осуществления, могут быть выполнены иные примеры осуществления изобретения в пределах существа изобретения. В целях более полной иллюстрации настоящего изобретения ниже приведены следующие примеры. Пример 1. Получение (9S)-3-дескладинозил-3'-десметил-3'-ацетил-9-деоксо-9-дегидроэритромицина А кристаллической формы I. Соединение (26,0 г) в твердой аморфной форме, полученное в процессе на этапе ацетилирования в соответствии с описанием в заявке на патент WO' 153, растворили в этилацетате (39,0 г). Смесь перемешивали в течение 2 ч при комнатной температуре, начиналось осаждение продукта, и полученную суспензию нагревали до приблизительно 50 °С. Затем в течение 30 мин добавляли метил-трет-бутиловый эфир (39,0 г), и суспензию перемешивали в течение 2 ч при 50 °С, далее охлаждали в течение 8 ч до 10 °С, и указанную температуру поддерживали в течение 2 ч при постоянном перемешивании. Полученное твердое вещество выделяли путем промывки фильтрованием смесью этилацетата-метил-трет-бутилового эфира (1:1) (3 ×5 мл) и затем высушивали в вакууме при 45 °С в течение 16 ч до получения кристаллической формы I (14,4 г) в виде белого твердого вещества. Пример 2. Преобразование кристаллической формы I в кристаллическую форму II. Суспензию кристаллической формы I (55,4 г, 0,098 моль) в деминерализованной воде (898,5 г) нагревали до 30-35 °С и температуру суспензии поддерживали в течение как минимум 24 ч при постоянном перемешивании. Преобразование считалось завершенным, когда в профиле DSC наблюдался единичный пик, и начальная температура составляла > 219,0 °С. Далее суспензию охлаждали до 25 °С в течение 30 мин и температуру суспензии поддерживали на уровне комнатной температуры в течение 1 ч. Затем твердое вещество отфильтровывали на тигеле Гуча и панель промывали деминерализованной водой (60,0 г). Твердое вещество высушивали при 50 °С в вакууме до получения кристаллической формы II (52,4 г, титр 96,9%, выход 92,7%). Пример 3. Получение (9S)-3-дескладинозил-3'-десметил-3'-ацетил-9-деоксо-9-дегидроэритромицина А кристаллической формы II. Соединение (126,7 г), полученное в соответствии с процессом, описанным в заявке на патент WO' 153, растворяли в метилэтилкетоне (1586,0 г). Полученный в результате этого раствор перегоняли при нормальном давлении до концентрации приблизительно 15-17% в весовом отношении, затем затравливали и дополнительно перегоняли до достижения окончательной концентрации приблизительно 25-26% в весовом отношении. Полученную суспензию перемешивали при 75 °С в течение 5 ч, охлаждали до 0 °С в течение 7,5 ч (приблизительно 10 °С/ч), и указанную суспензию выдерживали при этой температуре дополнительно в течение 2 ч при постоянном перемешивании. Полученное твердое вещество выделяли путем промывки фильтрованием охлажденным (0 °С) метилэтилкетоном (2 ×56 г) и высушивали в вакууме при 50 °С в течение 16 ч до получения кристаллической формы Form II (110,2 г) в виде белого твердого вещества. |