|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea000013079b*\id |
|
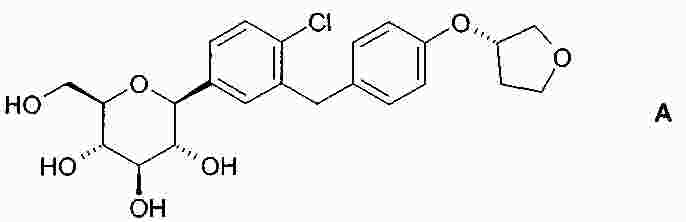
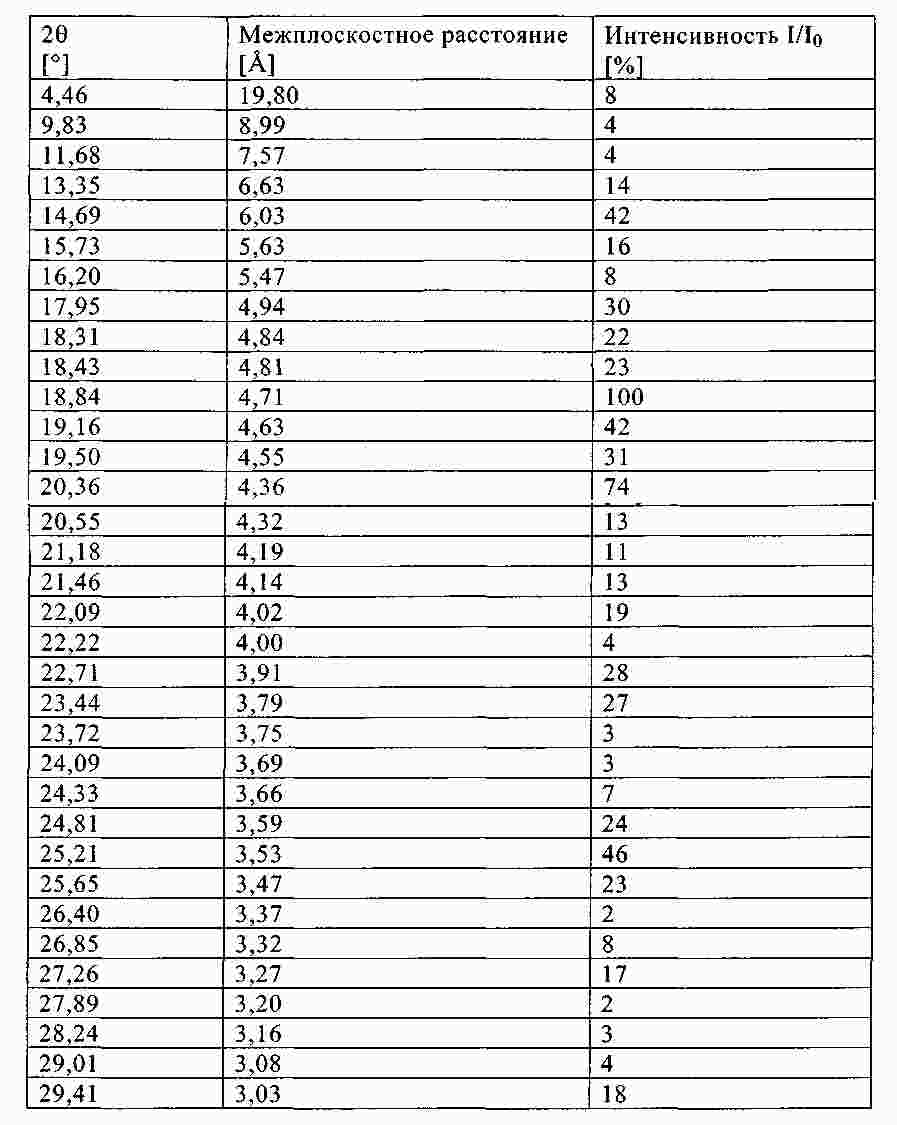
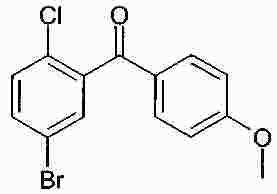
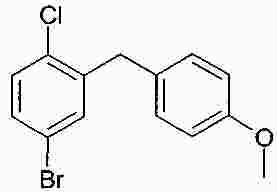
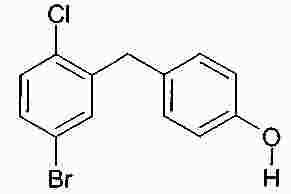
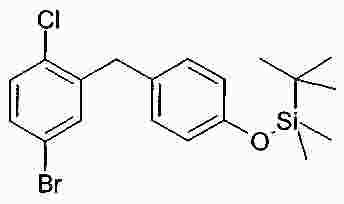
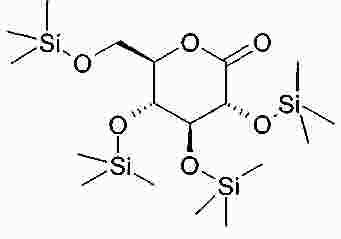
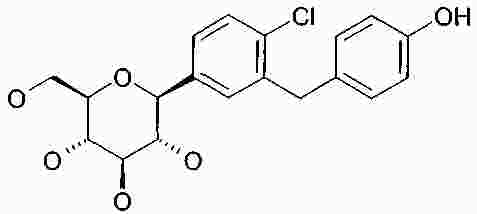
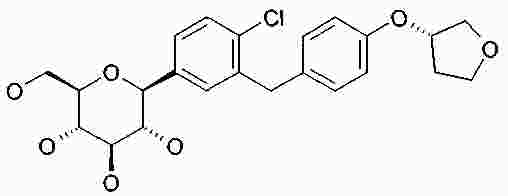
Термины запроса в документе Реферат В изобретении описана кристаллическая форма 1-хлор-4-( β-D-глюкопираноз-1-ил)-2-[4-((S)-тетрагидрофуран-3-илокси)бензил]бензола, способ ее получения, а также ее применение для приготовления лекарственных средств. Формула [0001] Кристаллическая форма 1-хлор-4-( β-D-глюкопираноз-1-ил)-2-[4-((S)-тетрагидрофуран-3-илокси)бензил]бензола, обладающая порошковой рентгенограммой, которая включает пики при 18,84, 20,36 и 25,21 ° 2 θ ( ±0,05 ° 2 θ), и указанная порошковая рентгенограмма получена с использованием излучения CuK α1. [0002] Кристаллическая форма по п.1, порошковая рентгенограмма которой дополнительно включает пики при 14,69, 19,16 и 19,50 ° 2 θ ( ±0,05 ° 2 θ), и указанная порошковая рентгенограмма получена с использованием излучения CuK α1. [0003] 1-Хлор-4-( β-D-глюкопираноз-1-ил)-2-[4-((S)-тетрагидрофуран-3-илокси)бензил]бензол, в котором не менее 50% указанного соединения содержится в виде кристаллической формы по п.1 или 2. [0004] Фармацевтическая композиция, включающая кристаллическую форму по п.1 или 2. [0005] Применение кристаллической формы по п.1 или 2 для получения фармацевтической композиции, которая пригодна для лечения или предупреждения заболеваний или патологических состояний, на которые можно повлиять путем ингибирования натрийзависимого сопереносчика глюкозы SGLT. [0006] Применение кристаллической формы по п.1 или 2 для получения фармацевтической композиции, которая пригодна для лечения или предупреждения метаболических нарушений, предпочтительно метаболического нарушения, выбранного из группы, включающей сахарный диабет типа 1 и 2, осложнения при диабете, метаболический ацидоз или кетоз, реактивную гипогликемию, гиперинсулинемию, нарушение метаболизма глюкозы, резистентность к инсулину, метаболический синдром, дислипидемии различной этиологии, атеросклероз и родственные заболевания, ожирение, высокое артериальное давление, хроническую сердечную недостаточность, отек и гиперурикемию. [0007] Применение кристаллической формы по п.1 или 2 для приготовления фармацевтической композиции, предназначенной для ингибирования натрийзависимого сопереносчика глюкозы SGLT2. [0008] Применение кристаллической формы по п.1 или 2 для приготовления фармацевтической композиции, предназначенной для предупреждения дегенерации бета-клеток панкреатических островков и/или для улучшения и/или восстановления функциональности бета-клеток панкреатических островков. [0009] Способ получения кристаллической формы по п.1 или 2, включающий следующие стадии: Полный текст патента Настоящее изобретение относится к кристаллической форме 1-хлор-4-( β-D-глюкопираноз-1-ил)-2-[4-((S)-тетрагидрофуран-3-илокси)бензил]бензола, к способу ее получения, а также к ее применению для приготовления лекарственных средств. Соединение 1-хлор-4-( β-D-глюкопираноз-1-ил)-2-[4-((S)-тетрагидрофуран-3-илокси)бензил]бензол (далее обозначаемое как "соединение А") описано в заявке WO 2005/092877 и обладает химической структурой, описываемой формулой А Соединения, описанные в настоящем изобретении, оказывают полезное ингибирующее воздействие на натрийзависимый сопереносчик глюкозы SGLT, в особенности SGLT2. Способ получения соединения А, описанный в настоящем изобретении, не приводит к кристаллической форме. Разумеется, наличие определенной фармацевтической активности является главным предварительным условием для того, чтобы фармацевтически активное средство было утверждено для продажи в качестве лекарственного препарата. Однако существует множество дополнительных требований, которым должно соответствовать фармацевтически активное средство. Эти требования относятся к разным параметрам, связанным с природой самого активного средства. Примерами этих параметров являются, но не ограничиваются только ими, стабильность активного средства при различных условиях окружающей среды, его стабильность во время изготовления фармацевтической композиции и стабильность активного средства в готовых лекарственных композициях. Фармацевтически активное вещество, применяющееся для получения фармацевтических композиций, должно быть как можно более чистым и должна быть гарантирована его стабильность при длительном хранении при различных условиях окружающей среды. Это необходимо для того, чтобы исключить использование фармацевтических композиций, которые в дополнение к конкретному активному веществу содержат, например, продукты его разложения. В таких случаях содержание активного средства в лекарственном средстве может быть меньше заданного. Равномерное распределение лекарственного средства в композиции является критически важным фактором, в особенности когда необходимо вводить небольшие дозы лекарственного средства. Для обеспечения равномерного распределения необходимо уменьшить размер частиц активного вещества до подходящих значений, например, путем размола. Поскольку необходимо по возможности уменьшить разложение фармацевтически активного вещества, являющееся побочным эффектом размола (или микронизации), несмотря на жесткие условия, необходимые для его проведения, важно, чтобы активное вещество во время размола обладало высокой стабильностью. Только если активное вещество достаточно стабильно во время размола, можно воспроизводимым образом получать однородные фармацевтической композиции, которые всегда содержат заданное количество активного вещества. Другим затруднением, которое может возникнуть во время размола при получении фармацевтической композиции, является подвод энергии, обусловленный размолом, и давление на поверхности кристаллов. При определенных условиях это может привести к полиморфным превращениям, образованию аморфного вещества или к изменениям кристаллической решетки. Поскольку для обеспечения фармацевтического качества фармацевтической композиции необходимо, чтобы кристаллы активного вещества всегда обладали одинаковой морфологией, с этой точки зрения к стабильности и характеристикам кристаллического активного вещества предъявляются строгие требования. Стабильность фармацевтически активного вещества в фармацевтических композициях также важна для установления срока годности конкретного лекарственного препарата; сроком годности является промежуток времени, в течение которого лекарственный препарат можно вводить без какого-либо риска. Поэтому высокая стабильность лекарственного вещества в указанных выше фармацевтических композициях при различных условиях хранения является дополнительным преимуществом как для пациента, так и для изготовителя. Поглощение влаги уменьшает содержание фармацевтически активного вещества вследствие увеличения массы, обусловленной поступлением воды. Фармацевтические композиции, склонные к поглощению влаги, при хранении необходимо защищать от воздействия влаги, например, путем прибавления подходящих осушающих средств или путем хранения лекарственного средства в среде, которая защищена от поступления влаги. Поэтому предпочтительно, чтобы фармацевтически активное вещество было лишь незначительно гигроскопичным. Кроме того, наличие строго определенной кристаллической формы позволяет выполнить очистку лекарственного вещества путем перекристаллизации. Наряду с указанными выше требованиями в общем случае также следует учитывать, что любое изменение физического состояния фармацевтической композиции, которое может повысить ее физическую и химическую стабильность, обеспечивает значительное преимущество по сравнению с менее стабильными формами того же самого лекарственного средства. В основу настоящего изобретения положена задача получения стабильной кристаллической формы соединения А, которое соответствует указанным выше требованиям, предъявляемым к фармацевтически активным веществам. Первым объектом настоящего изобретения является кристаллическая форма соединения А. Вторым объектом настоящего изобретения является кристаллическая форма соединения А, обладающая порошковой рентгенограммой, которая включает пики при 18,84, 20,36 и 25,21 ° 2 θ ( ±0,05 ° 2 θ), и указанная порошковая рентгенограмма получена с использованием излучения CuK α 1 . Третьим объектом настоящего изобретения является соединение А, в котором не менее 50% указанного соединения содержится в виде кристаллической формы, определенной выше и ниже в настоящем изобретении. Вследствие фармацевтической эффективности соединения А четвертым объектом настоящего изобретения является фармацевтическая композиция или лекарственное средство, включающее кристаллическую форму, определенную выше и ниже в настоящем изобретении. Пятым объектом настоящего изобретения является применение кристаллической формы, определенной выше или ниже в настоящем изобретении, для получения фармацевтической композиции, которая пригодна для лечения или предупреждения заболеваний или патологических состояний, на которые можно повлиять путем ингибирования натрийзависимого сопереносчика глюкозы SGLT, предпочтительно SGLT2. Шестым объектом настоящего изобретения является применение кристаллической формы, определенной выше или ниже в настоящем изобретении, для получения фармацевтической композиции, которая пригодна для лечения или предупреждения метаболических нарушений. Седьмым объектом настоящего изобретения является применение кристаллической формы, определенной выше или ниже в настоящем изобретении, для приготовления фармацевтической композиции, предназначенной для ингибирования натрийзависимого сопереносчика глюкозы SGLT2. Восьмым объектом настоящего изобретения является применение кристаллической формы, определенной выше или ниже в настоящем изобретении, для приготовления фармацевтической композиции для предупреждения дегенерации бета-клеток панкреатических островков и/или для улучшения и/или восстановления функциональности бета-клеток панкреатических островков. Девятым объектом настоящего изобретения является применение кристаллической формы, определенной выше или ниже в настоящем изобретении, для приготовления фармацевтической композиции для предупреждения, замедления, задерживания или лечения заболеваний или патологических состояний, обусловленных аномальным накоплением жира в печени, у нуждающегося в нем пациента. Десятым объектом настоящего изобретения является способ получения кристаллической формы, определенной выше или ниже в настоящем изобретении, указанный способ включает следующие стадии: (a) растворение соединения А в растворителе или смеси растворителей с получением насыщенного или почти насыщенного раствора; (b) выдерживание раствора, предпочтительно с охлаждением, для осаждения кристаллической формы и вследствие этого - для образования суспензии; (c) выделение осадка из суспензии и (d) сушку осадка до удаления избытка указанного растворителя или смеси растворителей. Другие объекты настоящего изобретения станут понятными для специалиста в данной области техники из последующего подробного описания настоящего изобретения и примеров. На фиг. 1 приведена порошковая рентгенограмма кристаллической формы. На фиг. 2 приведены результаты термического анализа и определения температуры плавления кристаллической формы с помощью ДСК. Согласно изобретению неожиданно было установлено, что существует кристаллическая форма соединения А, которая соответствует важным требованиям, указанным выше в настоящем изобретении. В соответствии с этим настоящее изобретение относится к а кристаллической форме соединения А. Эту кристаллическую форму можно идентифицировать по ее характеристической порошковой рентгенограмме (ПРРГ). Кристаллическая форма характеризуется порошковой рентгенограммой, которая включает пики при 18,84, 20,36 и 25,21 ° 2 θ ( ±0,05 ° 2 θ), и указанная порошковая рентгенограмма получена с использованием излучения CuK α 1 . Предпочтительно, если указанная порошковая рентгенограмма включает пики при 14,69, 18,84, 19,16, 19,50, 20,36 и 25,21 ° 2 θ ( ±0,05 ° 2 θ), и указанная порошковая рентгенограмма получена с использованием излучения CuK α 1 . Более предпочтительно, если кристаллическая форма характеризуется порошковой рентгенограммой, полученной с использованием излучения CuK α 1 , которая включает пики при 2 θ ( ±0,05 ° 2 θ), приведенные в таблице. Еще более предпочтительно, если кристаллическая форма характеризуется порошковой рентгенограммой, полученной с использованием излучения CuK α 1 , которая включает пики при 2 θ ( ±0,05 ° 2 θ), приведенные на фиг. 1. Кроме того, кристаллическая форма соединения А характеризуется температурой плавления, равной примерно 149 ±3 °С (определена с помощью ДСК; за нее принята температура, соответствующая наступлению перехода; скорость нагрева 10 К/мин). Полученная с помощью ДСК зависимость приведена на фиг. 2. Порошковые рентгенограммы в настоящем изобретении снимали с помощью дифрактометра STOE-STADI Р в режиме пропускания, снабженном детектором положения (OED) и анодом из Cu в качестве источника рентгеновского излучения (излучение CuK α 1 , λ=1,54056 Å, 40 кВ, 40 мА). В приведенной выше таблице значения "2 θ [ °]" означают угол дифракции в градусах и значения "d [ Å]" означают указанные межплоскостные расстояния в Å. Интенсивности, указанные на фиг. 1, приведены в импульсах в секунду. Для учета экспериментальной погрешности описанные выше значения 2 θ следует считать точными с отклонениями ±0,05 ° 2 θ. Это означает, что при решении вопроса о том, является ли данный образец кристаллов соединения А кристаллической формой, предлагаемой в настоящем изобретении, значение 2 θ, полученное экспериментально для образца, следует считать совпадающим с характеристическим значением, указанным выше, если оно отличается от характеристического значения не более чем на ±0,05 ° 2 θ. Температуру плавления определяют с помощью ДСК (дифференциальная сканирующая калориметрия) с использованием прибора ДСК 821 (Mettler Toledo). Другим объектом настоящего изобретения является способ получения кристаллической формы соединения А, определенный выше и ниже в настоящем изобретении, указанный способ включает следующие стадии: (a) растворение соединения А в растворителе или смеси растворителей с получением насыщенного или почти насыщенного раствора; (b) выдерживание раствора для осаждения кристаллической формы из раствора; (c) извлечение осадка из раствора и (d) сушку осадка до удаления избытка указанного растворителя или смеси растворителей. Термины "насыщенный" или "почти насыщенный" означают исходное соединение А, использующееся на стадии (а). Например, раствор, который насыщен исходным соединением А, может быть перенасыщенным его кристаллической формой. Подходящие растворители предпочтительно выбраны из группы, включающей C 1 -C 4 -алканолы, воду, этилацетат, ацетонитрил, ацетон, диэтиловый эфир и смесь одного или большего количества этих растворителей. Более предпочтительные растворители выбраны из группы, включающей метанол, этанол, изопропанол, этилацетат, диэтиловый эфир, ацетон, воду и смесь одного или большего количества этих растворителей, предпочтительно смеси одного или большего количества указанных органических растворителей с водой. Особенно предпочтительные растворители выбраны из группы, включающей этилацетат, этанол, изопропанол и смеси этанола и/или изопропанола с водой. В случае использования смеси воды и одного или большего количества C 1 -C 4 -алканолов, предпочтительно метанола, этанола и/или изопропанола, наиболее предпочтительно этанола предпочтительное объемное отношение вода:алканол находится в диапазоне примерно от 1:4 до 4:1; более предпочтительно примерно от 1:2 до 2:1; еще более предпочтительно примерно от 2:3 до 3:2. Стадию (а) предпочтительно проводить примерно при температуре (примерно 20 °С) или при повышенной температуре, примерно до температуры кипения растворителя или смеси растворителей. Для снижения растворимости соединения А в растворителе на стадии (а) и/или на стадии (b) можно прибавить один или большее количество антирастворителей или нерастворителей, предпочтительно во время стадии (а) или в начале стадии (b). Примером подходящего антирастворителя или нерастворителя является вода. Количество антирастворителя или нерастворителя или их смеси предпочтительно выбирать так, чтобы получить перенасыщенный или почти перенасыщенный раствор. На стадии (b) раствор выдерживают в течение периода времени, достаточного для получения осадка. Температура раствора на стадии (b) является примерно такой же или ниже, чем на стадии (а). Во время выдерживания температуру раствора, содержащего соединение А, предпочтительно понизить, предпочтительно до температуры в диапазоне от 20 до 0 °С или даже более низкой. Стадию (b) можно проводить с перемешиванием или без него. Как известно специалисту в данной области техники, путем изменения периода времени и разности температур на стадии (b) можно менять размеры, форму и качество получаемых кристаллов. Кроме того, кристаллизацию можно инициировать по методикам, известным в данной области техники, например, путем царапания или растирания. В (почти) насыщенный раствор необязательно можно внести затравочные кристаллы. На стадии (с) растворитель (растворители) можно удалить из осадка по известным методикам, таким как, например, фильтрование, фильтрование с отсасыванием, декантация или центрифугирование. На стадии (d) избыток растворителя (растворителей) удаляют из осадка по методикам, известным специалисту в данной области техники, таким как, например, снижение парциального давления растворителя (растворителей), предпочтительно в вакууме, и/или путем нагревания до температуры выше примерно 20 °С, предпочтительно в температурном диапазоне ниже 80 °С, еще более предпочтительно ниже 50 °С. Соединение А можно синтезировать по методикам, в частности и/или в общем виде описанным или цитированным в заявке WO 2005/092877. Кроме того, биологические характеристики соединения А можно исследовать так, как это описано в заявке WO 2005/092877, которая во всей своей полноте включена в настоящее изобретение в качестве ссылки. Кристаллическую форму, предлагаемую в настоящем изобретении, предпочтительно использовать, как лекарственное активное вещество в основном в чистой форме, т. е. в основном не содержащей других кристаллических форм соединения А. Тем не менее, настоящее изобретение также включает кристаллическую форму, определенную в настоящем изобретении, в смеси с другой кристаллической формой или формами. Если активное лекарственное вещество должно быть смесью кристаллических форм, то предпочтительно, если это вещество включает не менее 50% кристаллической формы, описанной в настоящем изобретении. Вследствие своей способности ингибировать активность SGLT кристаллическая форма, предлагаемая в настоящем изобретении, пригодна для приготовления фармацевтических композиций, предназначенных для лечения и/или предупредительного лечения всех тех патологических состояний или заболеваний, на которые можно повлиять путем ингибирования активности SGLT, предпочтительно активности SGLT-2. Поэтому кристаллическая форма является особенно подходящей для приготовления фармацевтических композиций, предназначенных для предупреждения или лечения заболеваний, предпочтительно метаболических нарушений или патологических состояний, таких как сахарный диабет типа 1 и 2, осложнения при диабете (таких как, ретинопатия, нефтопатия или невропатии, диабетическая стопа, язвы, макроангиопатии), метаболический ацидоз или кетоз, реактивная гипогликемия, гиперинсулинемия, нарушение метаболизма глюкозы, резистентность к инсулину, метаболический синдром, дислипидемии различной этиологии, атеросклероз и родственные заболеваний, ожирение, высокое артериальное давление, хроническая сердечная недостаточность, отек и гиперурикемия. Кристаллическая форма также является подходящей для приготовления фармацевтических композиций, предназначенных для предупреждения дегенерации бета-клеток, такой как, например, апоптоз или некроз бета-клеток панкреатических островков. Кристаллическая форма также является подходящей для приготовления фармацевтических композиций, предназначенных для улучшения или восстановления функциональности клеток поджелудочной железы, а также для увеличения количества и размера бета-клеток панкреатических островков. Кристаллическую форму, предлагаемую в настоящем изобретении, также можно использовать для приготовления фармацевтических композиций, применимых в качестве диуретиков или гипотензивных средств и подходящих для предупреждения и лечения острой сердечной недостаточности. Путем введения кристаллической формы, предлагаемой в настоящем изобретении, можно уменьшить или подавить аномальное накопление жира в печени. Поэтому другим объектом настоящего изобретения является способ предупреждения, замедления, задерживания или лечения заболеваний или патологических состояний, обусловленных аномальным накоплением жира в печени, у нуждающегося в нем пациента, характеризующийся тем, что вводят фармацевтическую композицию, предлагаемую в настоящем изобретении. Заболевания или патологические состояния, которые обусловлены аномальным накоплением жира в печени, предпочтительно выбраны из группы, включающей генерализованную жировую инфильтрацию печени, неалкогольную жировую инфильтрацию печени (НАИП), неалкогольный стеатогепатит (НАСГ), вызванную перееданием жировую инфильтрацию печени, диабетическую жировую инфильтрацию печени, алкогольную жировую инфильтрацию печени и токсическую жировую инфильтрацию печени. В частности, кристаллическая форма, предлагаемая в настоящем изобретении, применима для приготовления фармацевтических композиций, предназначенных для предупреждения или лечения диабета, предпочтительно сахарного диабета типа 1 и 2, и/или осложнений при диабете. Кроме того, кристаллическая форма, предлагаемая в настоящем изобретении, является особенно подходящей для предупреждения или лечения избыточной массы, ожирения (включая ожирение класса I, класса II и/или класса III), висцерального ожирения и/или абдоминального ожирения. Дозы, необходимые для обеспечения соответствующей активности с целью лечения или предупреждения, обычно зависят от пациента, характера и тяжести заболевания или патологического состояния и методики и частоты введения и определяются лечащим врачом. Целесообразная доза может составлять от 1 до 100 мг, предпочтительно - от 1 до 30 мг при внутривенном введении и от 1 до 1000 мг, предпочтительно от 1 до 100 мг при пероральном введении и в каждом случае средство вводят от 1 до 4 раз в сутки. Для этого фармацевтические композиции, предлагаемые в настоящем изобретении, предпочтительно включают кристаллическую форму совместно с одним или большим количеством обычных инертных наполнителей и/или разбавителей. Такие фармацевтические композиции можно приготовить в виде обычных галеновых препаратов, таких как таблетки без покрытия или с покрытием, капсулы, порошки, суспензии или суппозитории. Приведенные ниже примеры синтеза предназначены для иллюстрации методики получения соединения А и его кристаллической формы. Их следует понимать только, как возможные методики, приведенные в качестве примера, а не для ограничения настоящего изобретения их описанием. Получение исходных соединений. Пример I. (5-Бром-2-хлорфенил)-(4-метоксифенил)метанон 38,3 мл оксалилхлорида и 0,8 мл диметилформамида прибавляют к смеси 100 г 5-бром-2-хлорбензойной кислоты с 500 мл дихлорметана. Реакционную смесь перемешивают в течение 14 ч, затем фильтруют и с помощью роторного испарителя отделяют от всех летучих компонентов. Остаток растворяют в 150 мл дихлорметана, раствор охлаждают до -5 °С и прибавляют 46,5 г анизола. Затем порциями прибавляют 51,5 г трихлорида алюминия, так чтобы температура не превышала 5 °С. Раствор перемешивают в течение еще 1 ч при температуре от 1 до 5 °С и затем выливают на измельченный лед. Органическую фазу отделяют и водную фазу еще трижды экстрагируют дихлорметаном. Объединенные органические фазы промывают 1 М водным раствором хлористо-водородной кислоты, дважды 1 М водным раствором гидроксида натрия и рассолом. Затем органическую фазу сушат, растворитель удаляют и остаток перекристаллизовывают из этанола. Выход: 86,3 г (64% от теоретического). Масс-спектр (ESI + ): m/z=325/327/329 (Br+Cl) [M+H] + . Пример II. 4-Бром-1-хлор-2-(4-метоксибензил)бензол Раствор 86,2 г (5-бром-2-хлорфенил)-(4-метоксифенил)метанона и 101,5 мл триэтилсилана в 75 мл дихлорметана и 150 мл ацетонитрила охлаждают до 10 °С. Затем при перемешивании прибавляют 50,8 мл эфирата трифторида бора, так чтобы температура не превышала 20 °С. Раствор перемешивают в течение 14 ч при температуре окружающей среды, а затем прибавляют еще 9 мл триэтилсилана и 4,4 мл эфирата трифторида бора. Раствор перемешивают в течение еще 3 ч при температуре от 45 до 50 °С и затем охлаждают до температуры окружающей среды. Прибавляют раствор 28 г гидроксида калия в 70 мл воды и полученную смесь перемешивают в течение 2 ч. Затем органическую фазу отделяют и водную фазу еще трижды экстрагируют диизопропиловым эфиром. Объединенные органические фазы дважды промывают 2 М водным раствором гидроксида калия и один раз рассолом и затем сушат над сульфатом натрия. После удаления растворителя остаток промывают этанолом, повторно отделяют и сушат при 60 °С. Выход: 50,0 г (61% от теоретического). Масс-спектр (ESI + ): m/z=310/312/314 (Br+Cl) [M+H] + . Пример III. 4-(5-Бром-2-хлорбензил)фенол Раствор 14,8 г 4-бром-1-хлор-2-(4-метоксибензил)бензола в 150 мл дихлорметана охлаждают в бане со льдом. Затем прибавляют 50 мл 1 М раствора трибромида бора в дихлорметане, и раствор перемешивают в течение 2 ч при температуре окружающей среды. Затем раствор повторно охлаждают в бане со льдом и по каплям прибавляют насыщенный водный раствор карбоната калия. При температуре окружающей среды значение рН смеси доводят до 1 с помощью 1 М водного раствора хлористо-водородной кислоты, органическую фазу отделяют и водную фазу еще трижды экстрагируют этилацетатом. Объединенные органические фазы сушат над сульфатом натрия и растворитель полностью удаляют. Выход: 13,9 г (98% от теоретического). Масс-спектр (ESI - ): m/z=295/297/299 (Br+Cl) [M-H] - . Пример IV. [4-(5-Бром-2-хлорбензил)фенокси]-трет-бутилдиметилсилан Раствор 13,9 г 4-(5-бром-2-хлорбензил)фенола в 140 мл дихлорметана охлаждают в бане со льдом. Затем прибавляют 7,54 г трет-бутилдиметилсилилхлорида в 20 мл дихлорметана, а после этого 9,8 мл триэтиламина и 0,5 г 4-диметиламинопиридина. Раствор перемешивают в течение 16 ч при температуре окружающей среды и затем разбавляют с помощью 100 мл дихлорметана. Органическую фазу дважды промывают 1 М водным раствором хлористо-водородной кислоты и один раз водным раствором гидрокарбоната натрия и затем сушат над сульфатом натрия. После удаления растворителя остаток фильтруют через силикагель (циклогексан/этилацетат 100:1). Выход: 16,8 г (87% от теоретического). Масс-спектр (EI): m/z=410/412/414 (Br+Cl) [M] + . Пример V. 2,3,4,6-Тетракис-О-(триметилсилил)-D-глюкопиранон Раствор 20 г D-глюконо-1,5-лактона и 98,5 мл N-метилморфолина в 200 мл тетрагидрофурана охлаждают до -5 °С. Затем по каплям прибавляют 85 мл триметилсилилхлорида, так чтобы температура не превышала 5 °С. Затем раствор перемешивают в течение 1 ч при температуре окружающей среды, в течение 5 ч при 35 °С и повторно в течение 14 ч при температуре окружающей среды. После прибавления 300 мл толуола раствор охлаждают в бане со льдом и прибавляют 500 мл воды, так чтобы температура не превышала 10 °С. Затем органическую фазу отделяют и по одному разу промывают водным раствором дигидрофосфата натрия, водой и рассолом. Растворитель удаляют, остаток растворяют в 250 мл толуола и растворитель повторно полностью удаляют. Выход: 52,5 г (чистота примерно 90%). Масс-спектр (ESI + ): m/z=467 [М+Н] + . Пример VI. 1-Хлор-4-( β-D-глюкопираноз-1-ил)-2-(4-гидроксибензил)бензол Раствор 4,0 г [4-(5-бром-2-хлорбензил)фенокси]-трет-бутилдиметилсилана в 42 мл сухого диэтилового эфира охлаждают до -80 °С в атмосфере аргона. К охлажденному раствору по каплям медленно прибавляют 11,6 мл 1,7 М раствора трет-бутиллития в пентане и затем перемешивают в течение 30 мин при -80 °С. Затем этот раствор с помощью шприца, который охлаждают твердым диоксидом углерода, по каплям прибавляют к раствору 4,78 г 2,3,4,6-тетракис-О-(триметилсилил)-D-глюкопиранона в 38 мл диэтилового эфира, охлажденного до -80 °С. Полученный раствор перемешивают в течение 3 ч при -78 °С. Затем прибавляют раствор 1,1 мл метансульфоновой кислоты в 35 мл метанола и раствор перемешивают в течение 16 ч при температуре окружающей среды. Затем раствор нейтрализуют гидрокарбонатом натрия, прибавляют этилацетат и метанол удаляют вместе с эфиром. К оставшемуся раствору прибавляют водный раствор гидрокарбоната натрия и полученную смесь четырежды экстрагируют этилацетатом. Органические фазы сушат над сульфатом натрия и выпаривают. Остаток растворяют в 30 мл ацетонитрила и 30 мл дихлорметана и раствор охлаждают до -10 °С. После прибавления 4,4 мл триэтилсилана по каплям прибавляют 2,6 мл эфирата трифторида бора, так чтобы температура не превышала -5 °С. После завершения прибавления раствор перемешивают в течение еще 5 ч при температуре от -5 до -10 °С и затем реакцию останавливают путем прибавления водного раствора гидрокарбоната натрия. Органическую фазу отделяют и водную фазу четырежды экстрагируют этилацетатом. Объединенные органические фазы сушат над сульфатом натрия, растворитель удаляют и остаток очищают с помощью хроматографии на силикагеле (дихлорметан/метанол 1:0-> 3:1). Получают продукт в виде смеси состава примерно 6:1 β/ α, которую можно превратить в чистый β-аномер путем полного ацетилирования гидроксигрупп уксусным ангидридом и пиридином в дихлорметане и перекристаллизации продукта из этанола. Полученный таким образом продукт превращают в исходное соединение путем дезацетилирования в метаноле 4 М водным раствором гидроксида калия. Выход: 1,6 г (46% от теоретического). Масс-спектр (ESI + ): m/z=398/400 (Cl) [M+H] + . Получение соединения А. 1-Хлор-4-( β-D-глюкопираноз-1-ил)-2-[4-((S)-тетрагидрофуран-3-илокси)бензил]бензол 0,19 г (R)-3-(4-Метилфенилсульфонилокси)тетрагидрофуран прибавляют к смеси 0,20 г 1-хлор-4-( β-D-глюкопираноз-1-ил)-2-(4-гидроксибензил)бензола и 0,29 г карбоната цезия в 2,5 мл диметилформамида. Смесь перемешивают при 75 °С в течение 4 ч, а затем прибавляют еще 0,29 г карбоната цезия и 0,19 г (R)-3-(4-метилфенилсульфонилокси)тетрагидрофурана. После еще 14 ч перемешивания при 75 °С смесь охлаждают до температуры окружающей среды и прибавляют рассол. Полученную смесь экстрагируют этилацетатом, объединенные органические экстракты сушат над сульфатом натрия и растворитель удаляют. Остаток очищают с помощью хроматографии на силикагеле (дихлорметан/метанол 1:0-> 5:1). Выход: 0,12 г (49% от теоретического). Масс-спектр (ESI + ): m/z=451/453 (Cl) [M+H] + . Получение кристаллической формы. Вариант 1. 30 мг 1-хлор-4-( β-D-глюкопираноз-1-ил)-2-[4-((S)-тетрагидрофуран-3-илокси)бензил]бензола (полученного, как описано выше) растворяют в 0,8 мл этилацетата (содержащего 0,5-3% воды) при нагревании примерно до 50 °С. Раствору дают медленно охладиться (примерно от 1 до 3 ч) примерно до 20 °С. Через 48 ч кристаллическую форму выделяют в виде белых кристаллов путем фильтрования. Избыток растворителя удаляют путем выдерживания кристаллов при повышенной температуре (от 40 до 50 °С) в течение примерно от 3 до 4 ч при пониженном давлении. Вариант 2. 1 г 1-хлор-4-( β-D-глюкопираноз-1-ил)-2-[4-((S)-тетрагидрофуран-3-илокси)бензил]бензола растворяют в 5 мл смеси вода/этанол (объемное отношение 2:3) при нагревании примерно до 50 °С. Прибавляют 8 мл воды и раствору дают охладиться примерно до 20 °С в течение от 1 до 3 ч. Через 16 ч кристаллическую форму выделяют в виде белых кристаллов путем фильтрования. Избыток растворителя удаляют путем выдерживания кристаллов при повышенной температуре (от 40 до 50 °С) в течение примерно от 4 до 6 ч. Вариант 3. 1 г 1-хлор-4-( β-D-глюкопираноз-1-ил)-2-[4-((S)-тетрагидрофуран-3-илокси)бензил]бензола растворяют в 11 мл изопропанола при нагревании примерно до 50 °С. Раствору дают охладиться примерно до 20 °С в течение от 1 до 3 ч. Через 16 ч кристаллическую форму выделяют в виде белых кристаллов путем фильтрования. Остаточный растворитель удаляют путем выдерживания кристаллов при повышенной температуре (от 40 до 50 °С) в течение примерно от 4 до 6 ч. Вариант 4. 8,9 г 1-хлор-4-( β-D-глюкопираноз-1-ил)-2-[4-((S)-тетрагидрофуран-3-илокси)бензил]бензола растворяют в 60 мл смеси вода/этанол (объемное отношение 2:3) при нагревании примерно до 50 °С. Раствору дают охладиться примерно до 20 °С в течение 3 ч и кристаллическое соединение выделяют путем фильтрования. Выделенное твердое вещество сушат при 40 °С в течение 16 ч и получают примерно 6 г кристаллической формы. |