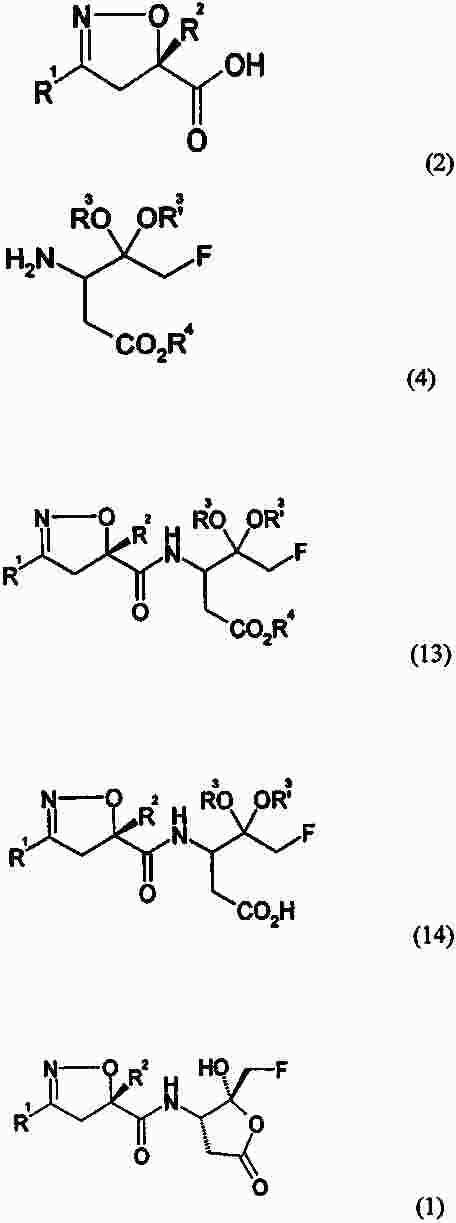
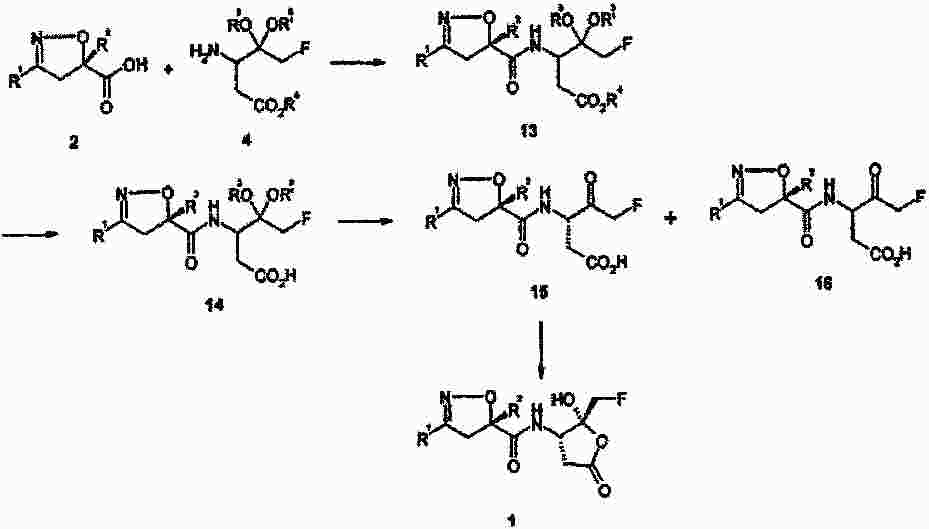
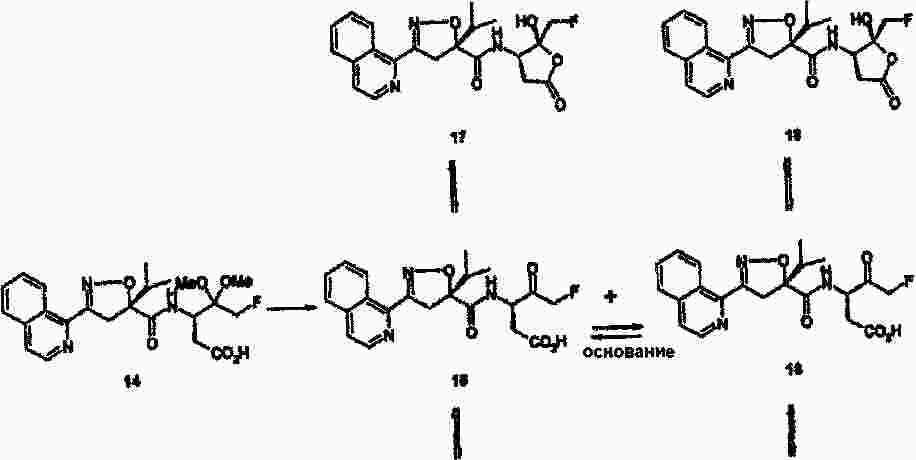
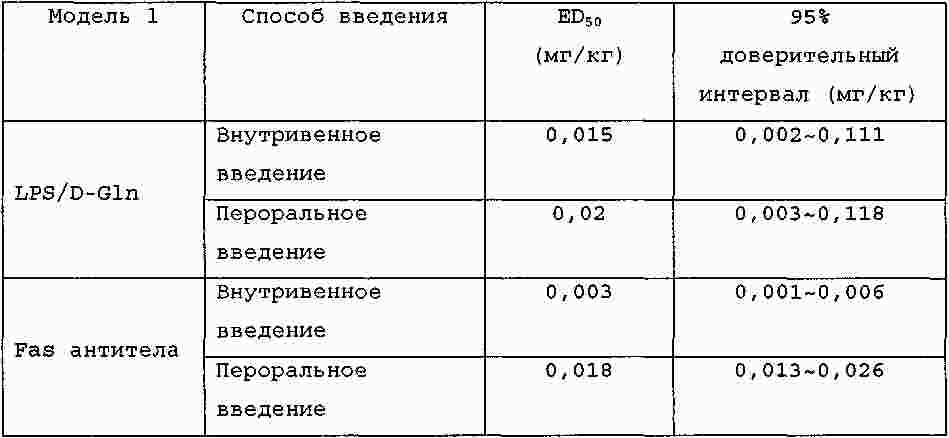
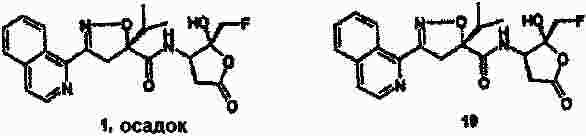|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea000013005b*\id |
|
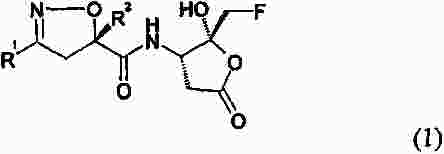
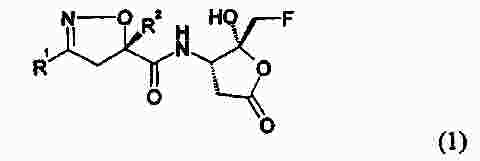
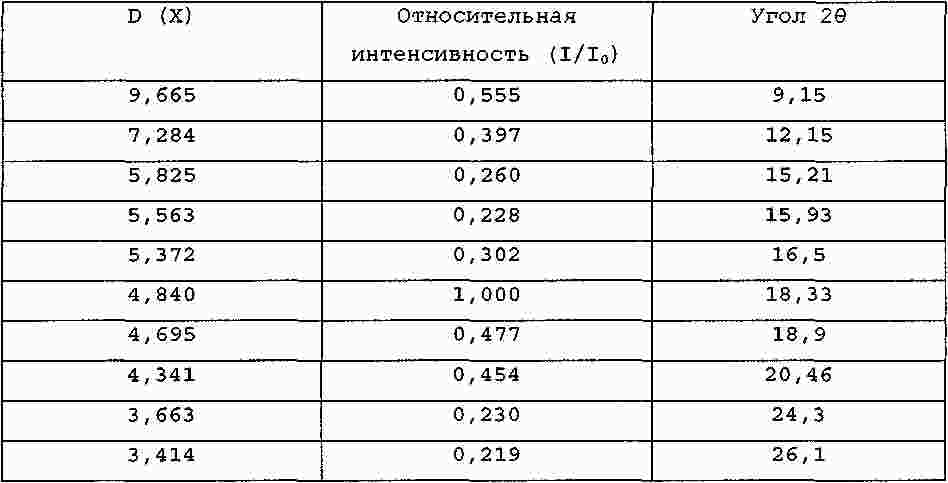
Термины запроса в документе Реферат Настоящее изобретение относится к производному изоксазолина, имеющему циклическую карбоксильную кислотную гемикетальную группу формулы (1) в которой R 1 представляет алкил или арил и R 2 представляет собой алкил, причем алкил относится к линейной или разветвленной алкильной группе, состоящей из 1-8 атомов углерода, или к циклоалкильной группе, состоящей из 3-10 атомов углерода, арил относится к ароматическим группам, гетероароматическим группам и частично восстановленным их производным, где ароматическая группа относится к 5-15-членным ненасыщенным углеводородам в простой или конденсированной циклической форме, и упомянутая гетероароматическая группа относится к ароматическим группам, имеющим 1-5 гетероатомов, выбранных из группы, состоящей из кислорода, серы и азота, предназначенному для применения в качестве ингибитора каспазы, к способу получения его, к фармацевтической композиции на его основе. Формула [0001] Соединение формулы (1) [0002] Соединение по п.1, в котором R1 представляет изохинолинил, хинолинил или нафтил и R2 представляет метил, этил, пропил или бутил. [0003] Соединение по п.2, в котором R1 представляет изохинолинил и R2 представляет изопропил. [0004] Соединение по п.3, в котором соединение представляет собой кристаллическую форму, имеющую нижеследующий спектр рентгенодифракции: [0005] Способ получения соединения формулы (1), включающий [0006] Способ по п.5, в котором R1 представляет изохинолинил, хинолинил или нафтил, R2 представляет метил, этил, пропил или бутил, R3 и R'3, каждый, представляет метил, этил или пропил или R3 и R'3 вместе с атомом кислорода, к которому они присоединены, образуют диоксолан или диоксан и R4 представляет метил, этил, пропил или бутил. [0007] Способ по п.5 или 6, в котором соединение формулы (2) на стадии (а) активируется активирующим реагентом, выбранным из группы, состоящей из оксалилхлорида, триметилацетилхлорида, фосфорилтрихлорида и тионилхлорида. [0008] Способ по п.5 или 6, в котором реакция на стадии (а) осуществляется в присутствии основания, выбранного из группы, состоящей из триэтиламина, три(н-бутил)амина, диизопропилэтиламина, пиридина, 4-диметиламинопиридина и 4-(4-метилпиперидин-1-ил)пиридина. [0009] Способ по п.5 или 6, в котором гидролиз на стадии (b) осуществляется в присутствии основания, выбранного из группы, состоящей из гидроксида лития, гидроксида натрия, гидроксида калия и гидроксида кальция. [0010] Способ по п.5 или 6, в котором снятие защитных групп на стадии (с) осуществляется в присутствии кислоты, выбранной из группы, состоящей из хлористо-водородной кислоты, серной кислоты и трифторуксусной кислоты. [0011] Способ по п.5 или 6, в котором снятие защитных групп на стадии (с) осуществляется в присутствии растворителя, выбранного из дихлорметана или хлороформа. [0012] Способ по п.5 или 6, в котором проведение кристаллизации, обусловленной динамическим переходом, на стадии (d) осуществляется добавлением соединения формулы (1) в качестве затравочного кристалла. [0013] Способ по п.12, в котором проведение кристаллизации, обусловленной динамическим переходом, на стадии (d) осуществляется в присутствии затравочного кристалла и каталитического количества основания. [0014] Способ по п.13, где основанием является амин, выбранный из группы, состоящей из триэтиламина, три(н-бутил)амина, диизопропилэтиламина, диизопропиламина, пиридина, 4-диметиламинопиридина и 4-(4-метилпиперидин-1-ил)пиридина, оптически активного 1-фенилэтиламина и оптически активного 1-нафтилэтиламина. [0015] Способ по п.14, где амин используется в количестве от 0,001 до 1,0 экв. к соединению формулы (14). [0016] Соединение формулы (4) [0017] Соединение по п.16, в котором R3 и R'3, каждый, представляет метил, этил или пропил или R3 и R'3 вместе с атомом кислорода, к которому они присоединены, образуют диоксолан или диоксан и R4 представляет метил, этил, пропил или бутил. [0018] Способ получения соединения формулы (4), включающий: [0019] Способ по п.18, в котором R3 и R'3, каждый, представляет метил, этил или пропил или R3 и R'3 вместе с атомом кислорода, к которому они присоединены, образуют диоксолан или диоксан и R4 представляет метил, этил, пропил или бутил. [0020] Способ по п.18 или 19, в котором реакция введения защитных групп на стадии (а) осуществляется с использованием триметилортоформиата или триэтилортоформиата. [0021] Способ по п.18 или 19, в котором снятие защитных групп на стадии (а) осуществляется в присутствии основания, выбранного из гидроксида натрия, гидроксида калия, карбоната натрия и гидрокарбоната натрия. [0022] Способ по п.18 или 19, в котором реакция на стадии (b) осуществляется с использованием этилхлорформиата или метилхлорформиата. [0023] Способ по п.22, в котором реакция на стадии (b) осуществляется в присутствии основания, выбранного из н-бутиллития, литийдиизопропиламина или литийгексаметилдисилазида. [0024] Способ по п.18 или 19, в котором реакция восстановления на стадии (с) осуществляется с использованием уксусной кислоты и боргидрида натрия. [0025] Способ по п.18 или 19, в котором металлическим катализатором является катализатор палладиевого типа или катализатор типа никеля Ренея. [0026] Способ получения соединения по п.3, включающий преобразование соединений формулы (15) и формулы (16) в соединение, раскрытое в п.3, в присутствии каталитического количества основания вместе с затравочным кристаллом путем селективного осаждения. [0027] Фармацевтическая композиция для лечения воспаления или предотвращения апоптоза, содержащая соединение формулы (1) в соответствии с п.1 и фармацевтически приемлемые носители. [0028] Фармацевтическая композиция для лечения или предотвращения деменции, мозгового инсульта, повреждения головного мозга, обусловленного СПИДом, диабета, язвы желудка, церебрального нарушения из-за вируса гепатита, заболеваний печени из-за вируса гепатита, острого гепатита, скоротечной печеночной недостаточности, цирроза печени, сепсиса, отторжения органа при трансплантации, ревматоидного артрита или сердечного клеточного некроза из-за ишемической болезни сердца. [0029] Фармацевтическая композиция для лечения воспаления или предотвращения апоптоза, содержащая соединение по п.3 и один или несколько фармацевтически приемлемых носителей. [0030] Способ лечения воспаления или предотвращения апоптоза у субъекта, включающий введение указанному субъекту терапевтически эффективного количества соединения формулы (1) по п.1. [0031] Способ по п.30, в котором способ применяется при деменции, мозговом инсульте, повреждении головного мозга, обусловленном СПИДом, диабете, язве желудка, церебральном нарушении из-за вируса гепатита, заболеваниях печени из-за вируса гепатита, остром гепатите, скоротечной печеночной недостаточности, циррозе печени, сепсисе, отторжении органа при трансплантации, ревматоидном артрите или сердечном клеточном некрозе из-за ишемической болезни сердца. [0032] Применение соединения формулы (1) по п.1 для производства лекарственного средства для лечения воспаления или предотвращения апоптоза. [0033] Применение соединения формулы (I) по п.1 для изготовления лекарственного средства для лечения воспаления или предотвращения деменции, мозгового инсульта, повреждения головного мозга, обусловленного СПИДом, диабета, язвы желудка, церебрального нарушения из-за вируса гепатита, заболеваний печени из-за вируса гепатита, острого гепатита, скоротечной печеночной недостаточности, цирроза печени, сепсиса, отторжения органа при трансплантации, ревматоидного артрита или сердечного клеточного некроза из-за ишемической болезни сердца. [0034] Применение соединения по п.3 для производства лекарственного средства для лечения воспаления или предупреждения апоптоза. [0035] Применение соединения по п.3 для изготовления лекарственного средства для лечения деменции, мозгового инсульта, повреждения головного мозга, обусловленного СПИДом, диабета, язвы желудка, церебрального нарушения из-за вируса гепатита, заболеваний печени из-за вируса гепатита, острого гепатита, скоротечной печеночной недостаточности, цирроза печени, сепсиса, отторжения органа при трансплантации, ревматоидного артрита или сердечного клеточного некроза из-за ишемической болезни сердца. [0036] Применение по п.35, где лекарственное средство используется для лечения церебрального нарушения из-за вируса гепатита, заболеваний печени из-за вируса гепатита, острого гепатита, скоротечной печеночной недостаточности, цирроза печени. Полный текст патента Настоящее изобретение относится к производному изоксазолина, имеющему циклическую карбоксильную кислотную гемикетальную группу, предназначенному для применения в качестве ингибитора каспазы, к способу получения его, к фармацевтической композиции на его основе и к их применению. К ингибитору каспазы относится соединение, которое может подавлять активность каспазы, вследствие этого регулирующее воспаление или апоптоз, вызванные действием каспазы. Среди ингибиторов каспазы необратимый ингибитор, как известно, проявляет наиболее эффективную ингибирующую активность, поскольку он необратимо инактивирует фермент, контролирующий апоптоз (Wu J. et al., Methods: A Companion to Methods in Enzymology, 1999, 17, 320). Нижеследующие соединения известны в качестве необратимого ингибитора и имеют общую группу 3-амино-5-фтор-4-оксопентановой кислоты. Так, заболеваниями, которые можно излечить или облегчить с помощью вышеупомянутых соединений, являются ревматоидный артрит, воспалительное заболевание кишечника, реакция трансплантант против хозяина, сепсис, остеоартрит, остеопороз, острая и хроническая миелогенная лейкемия, менингит, сальпингит, септический шок, холангит, колит, энцефалит, эндокардит, гломерулонефрит, гепатит, миокардит, панкреатит, сахарный диабет I типа, рассеянный склероз, болезнь Альцгеймера, болезнь Паркинсона, цирроз печени и др. Ссылки. Деменция: Arch. Neurol., 2003, Mar.; 60(3): 369-76, Caspase gene expression in the brain as a function of the clinical progression of Alzheimer disease. Pompl P.N., Yemul S., Xiang Z., Ho L., Haroutunian V., Purohit D., Mohs R., Pasinetti G.M. Мозговой инсульт: Proc. Natl. Acad. Sci. USA, 2002, Nov., 12; 99(23): 15188-93, Caspase activation and neuroprotection in caspase-3-deficient mice after in vivo cerebral ischemia and in vitro oxygen glucose deprivation. Le D.A., Wu Y., Huang Z., Matsushita K., Plesnila N., Augustinack J.C., Hyman B.T., Yuan J., Kuida K., Flavell R.A., Moskowitz M.A. Повреждение головного мозга, обусловленное СПИД: J. Neurosci., 2002, May 15; 22(10): 4015-24, Caspase cascade in human immunodeficiency virus-associated neurodegeneration. Garden G.A., Budd S.L., Tsai E., Hanson L., Kaul M., D'Emilia D.M., Friedlander R.M., Yuan J., Masliah E., Lipton S.A. Диабет: Diabetes, 2002, Jun.; 51(6): 1938-48, Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Cai L., Li W., Wang G., Guo L., Jiang Y., Kang Y.J. Язва желудка: J. Physiol. Pharmacol., 1998, Dec.; 49(4): 489-500, Role of basic fibroblast growth factor in the suppression of apoptotic caspase-3 during chronic gastric ulcer healing. Slomiany B.L., Piotrowski J., Slomiany A. Церебральные нарушения при гепатите: J. Viral. Hepat., 2003, Mar.; 10(2): 81-6, Cerebral dysfunction in chronic hepatitis С infection. Forton D.M., Taylor-Robinson S.D., Thomas H.C. Скоротечная печеночная недостаточность: Gastroenterology, 2000, Aug.; 119(2): 446-60, Tumor necrosis factor alpha in the pathogenesis of human and murine fulminant hepatic failure. Streetz K., Leifeld L., Grundmann D., Remakers J., Eckert K., Spengler U., Brenner D., Manns M., Trautwein C. Сепсис: Nat. Immunol., 2000, Dec; 1(6): 496-501, Caspase inhibitors improve survival in sepsis: a critical role of the lymphocyte. Hotchkiss R.S., Chang K.C., Swanson P.E., Tinsley K.W., Hui J.J., Klender P., Xanthoudakis S., Roy S., Black C., Grimm E., Aspiotis R., Han Y., Nicholson D.W., Karl I.E. Отторжение органа при трансплантации: Xenotransplantation, 2001, May; 8(2); 115-24, In vivo prevention of cell-mediated xeno-graft rejection via the Fas/FasL-pathway in CrmA-transducted porcine kidney cells. Fujino M., Li X.K., Suda Т., Hashimoto M., Okabe K., Yaginuma H., Mikoshiba K., Guo L., Okuyama T., Enosawa S., Amemiya H., Amano T., Suzuki S. Ревматоидный артрит: Prog. Med. Chem., 2002; 39: 1-72, Caspase inhibitors as anti-inflammatory and antiapoptotic agents. Graczyk P.P. Ишемические заболевания сердца: Am. J. Physiol. Heart Circ Physiol., 2002, Sep.; 283(3): H990-5, Hypoxia-induced cleavage of caspase-3 and DFF45/ICAD in human failed cardiomyocytes. Todor A., Sharov V.G., Tanhehco E.J., Silverman N., Bernabei A., Sabbah H.N. Противовоспаление: J. Immunol., 2003, Mar. 15; 170(6): 3386-91, A broad-spectrum caspase inhibitor attenuates allergic airway inflammation in murine asthma model. Iwata A., Nishio K., Winn R.K., Chi E.Y., Henderson W.R. Jr., Harlan J.M. Цирроз печени: i) J. Pharmacol. Exp. Ther., 2004, Mar.; 308(3): 1191-6, The caspase inhibitor Idn-6556 attenuates hepatic injuri and fibrosis in the bile duct ligated mouse. Canbay A., Fledstein A., Baskin-Bey E., Bronk F.S. Gores G.J.; ii) Hepatology, 2004, Feb., 39(2): 273-8, Apoptosis: the nexus of liver injury and fibrosis. Canbay A., Friedman S., Gores G.J.; iii) Hepatology, 2003, Nov.; 38(5): 1188-98, Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Canbay A., Feldstein A.E., Higuchi H., Werneburg N., Grambihler A., Bronk S.F., Gores G.J. С другой стороны, для 3-амино-5-фтор-4-оксопентановой кислотной группы ингибитора каспазы такой способ получения, как на нижеследующей схеме реакций 1 (Revesz et al., Tetrahedron Lett., 1994, 35, 9693), известен из уровня техники. Заявители продолжили изучение соединения, которое может использоваться в качестве ингибитора каспазы, и способа его получения. В результате было обнаружено, что производное изоксазолина, имеющее циклическую карбоксильную кислотную гемикетальную группу, в соответствии с настоящим изобретением имеет хорошую активность ингибирования каспазы и может быть получено с высокой степенью чистоты с использованием кристаллизации, обусловленной динамическим переходом, дополняя настоящее изобретение. Таким образом, целью настоящего изобретения является предоставление соединения нижеследующей формулы (1) со структурой изоксазолина и циклическими карбоксильными кислотными гемикетальными группами и нового способа эффективного получения этого соединения. Другой целью настоящего изобретения является предоставление аминопроизводного нижеследующей формулы (4), промежуточного соединения нижеследующей формулы (1) и способа получения его. Другой целью настоящего изобретения является предоставление фармацевтической композиции для лечения воспаления или предотвращения апоптоза, содержащей соединение нижеследующей формулы (1) и фармацевтически приемлемые носители. Другой целью настоящего изобретения является предоставление способа лечения воспаления или предотвращения апоптоза у субъекта, включающего введение терапевтически эффективного количества соединения нижеследующей формулы (1) субъекту. Другой целью настоящего изобретения является применение соединения нижеследующей формулы (1) для приготовления лекарственного средства для лечения воспаления или предотвращения апоптоза. Другой целью настоящего изобретения является предоставление кристаллической формы соединения нижеследующей формулы (1), имеющей хорошую стабильность. Фиг. 1 представляет собой рентгенодифракционный спектр кристаллической формы соединения формулы (1) (R 1 =изохинолинил, R 2 =изопропил) в соответствии с настоящим изобретением. Фиг. 2 представляет собой график, показывающий результаты теста на стабильность кристаллической формы и аморфной формы соединения формулы (1) (R 1 =изохинолинил, R 2 =изопропил) в соответствии с настоящим изобретением. Некоторые важные термины, используемые в настоящем изобретении, могут быть определены как нижеследующие. В формулах и схемах реакций, используемых в настоящем изобретении, алкил относится к линейной или разветвленной алкильной группе, состоящей из 1-8 атомов углерода, или к циклоалкильной группе, состоящей из 3-10 атомов углерода. Также, арил включает все ароматические группы, гетероароматические группы и частично восстановленные их производные. Упомянутая ароматическая группа относится к 5-15-членным ненасыщенным углеводородам в простой или конденсированной циклической форме и упомянутая гетероароматическая группа относится к ароматическим группам, имеющим 1-5 гетероатомов, выбираемых из группы, состоящей из кислорода, серы и азота. Кроме того, один или более водородов упомянутого алкила и упомянутого арила могут быть замещены другими заместителями. Примеры выбираемых заместителей включают ацил, амино, карбоалкокси, карбокси, карбоксиамино, циано, галоген, гидрокси, нитро, тиол, алкил, циклоалкил, алкокси, арилокси, сульфокси, гуанидо и др. Настоящее изобретение относится к производному изоксазолина, имеющему циклическую карбоксильную кислотную гемикетальную группу, нижеприведенной формулы (1), предназначенному для применения в качестве ингибитора каспазы где R 1 представляет алкил или арил и R 2 представляет алкил. Предпочтительно R 1 представляет изохинолинил, хинолинил или нафтил и R 2 представляет метил, этил, пропил или бутил. Более предпочтительно R 1 представляет изохинолинил и R 2 представляет изопропил. Соединение формулы (1), где R 1 представляет изохинолинил и R 2 представляет изопропил, может быть в кристаллической форме, показывающей нижеследующий спектр рентгенодифракции: Также настоящее изобретение относится к способу получения соединения формулы (1), включающему: (a) активацию соединения нижеследующей формулы (2), затем взаимодействие его с соединением нижеследующей формулы (4) с образованием соединения нижеследующей формулы (13); (b) гидролиз соединения нижеследующей формулы (13) с образованием соединения нижеследующей формулы (14); (c) снятие защитных групп соединения нижеследующей формулы (14) и (d) осуществление кристаллизации, обусловленной динамическим переходом где R 1 представляет алкил или арил, R 2 представляет алкил, R 3 и R' 3 , каждый, представляет алкил или R 3 и R' 3 вместе с атомом кислорода, к которому они присоединены, образуют гетероцикл и R 4 представляет алкил. Предпочтительно R 1 представляет изохинолинил, хинолинил или нафтил, R 2 представляет метил, этил, пропил или бутил, R 3 и R' 3 , каждый, представляет метил, этил или пропил или R 3 и R' 3 вместе с атомом кислорода, к которому они присоединены, образуют диоксолан или диоксан и R 4 представляет метил, этил, пропил или бутил. Каждая стадия вышеупомянутого способа получения соединения формулы (1) может быть описана более подробно следующим образом. В качестве активирующего реагента для активации соединения формулы (2) в вышеупомянутой стадии (а) предпочтительно применять один, выбранный из группы, состоящей из оксалилхлорида, триметилацетилхлорида, фосфорилтрихлорида и тионилхлорида. Также предпочтительно, чтобы реакция стадии (а) осуществлялась в присутствии основания, выбранного из группы, состоящей из триэтиламина, три(н-бутил)амина, диизопропилэтиламина, пиридина, 4-диметиламинопиридина и 4-(4-метилпиперидин-1-ил)пиридина, и предпочтительно, чтобы основание использовалось в количестве 1,0-10,0 экв. к соединению формулы (2). Кроме того, предпочтительно, чтобы реакция стадии (а) осуществлялась в одном или более растворителях, выбранных из группы, состоящей из дихлорметана, хлороформа, тетрагидрофурана, диметоксиэтана, диоксана и этилацетата. С другой стороны, предпочтительно, чтобы соединение формулы (4) на стадии (а) использовалось в количестве 1,0-3,0 экв. к соединению формулы (2). Предпочтительно, чтобы гидролиз на стадии (b) осуществлялся в присутствии основания, выбранного из группы, состоящей из гидроксида лития (предпочтительно безводного или кристалломоногидрата), гидроксида натрия, гидроксида калия и гидроксида кальция, и также предпочтительно, чтобы основание использовалось в количестве 0,1-10,0 экв. к соединению формулы (13). Кроме того, предпочтительно, чтобы реакция стадии (b) осуществлялась в одном или более растворителях, выбранных из группы, состоящей из метанола, этанола, н-пропанола, изопропанола, тетрагидрофурана, диметоксиэтана, диоксана и дихлорметана, или в смешанном растворителе из растворителя, выбранного из вышеупомянутой группы, и воды. Предпочтительно, чтобы снятие защитных групп на стадии (с) осуществлялось в присутствии кислоты, выбранной из группы, состоящей из хлористо-водородной кислоты, серной кислоты и трифторуксусной кислоты, и предпочтительно, чтобы кислота использовалась в количестве 0,1-20,0 экв. к соединению формулы (14). Также предпочтительно, чтобы снятие защитных групп на стадии (с) осуществлялось в присутствии или в отсутствие растворителя, выбранного из дихлорметана или хлороформа. Реакция кристаллизации, обусловленная динамическим переходом, стадии (d) может осуществляться добавлением соединения формулы (1) в качестве затравочного кристалла или осуществляться в присутствии затравочного кристалла и каталитического количества основания, где основанием является предпочтительно амин, выбранный из группы, состоящей из триэтиламина, три(н-бутил)амина, диизопропилэтиламина, диизопропиламина, пиридина, 4-диметиламинопиридина и 4-(4-метилпиперидин-1-ил)пиридина, оптически активного 1-фенилэтиламина и оптически активного 1-нафтилэтиламина. На стадии (d) предпочтительно использовать вышеупомянутый амин в количестве 0,001-1,0 экв. к соединению формулы (14) и более предпочтительно использовать от 0,03 до 0,5 экв. Если количество использованного амина слишком мало, скорость реакции становится медленнее, и если количество слишком велико, выход соединения формулы (1) понижается. Кроме того, предпочтительно, чтобы реакция кристаллизации, обусловленная динамическим переходом, стадии (d) осуществлялась в одном или более растворителях, выбранных из группы, состоящей из толуола, бензола, дихлорбензола, тетрагидрофурана, диметоксиэтана, диоксана, этилацетата, дихлорметана, ацетонитрила, метил-трет-бутилового эфира и диэтилового эфира. Ниже способ получения соединения формулы (1) в соответствии с настоящим изобретением будет объяснен более подробно со ссылкой на схему реакций 2. Производное изоксазолина формулы (2), имеющее высокую оптическую активность, получается в соответствии со способом, раскрытым в PCT/KR2004/002060 заявке, поданной 17 августа 2004 г. настоящим заявителем, и затем соединяется с соединением формулы (4) для получения соединения формулы (13). Затем соединение формулы (13) гидролизуется по эфирной группе с получением соединения формулы (14) и осуществляется реакция снятия защитной группы кетальной группы соединения формулы (14) с получением смеси соединений формулы (15) и формулы (16), которые эффективно трансформируются в соединение формулы (1) селективной динамической кристаллизацией. В частности, если смесь соединений формулы (15) и формулы (16) растворяется в органическом растворителе и в раствор добавляется затравочный кристалл соединения формулы (1), только соединение формулы (15) в смеси трансформируется в соединение формулы (1), выделяемое в твердом виде. где R 1 представляет алкил или арил, R 2 представляет алкил, R 3 и R 3 , каждый, представляет алкил или R 3 и R' 3 вместе с атомом кислорода, к которому они присоединены, образуют гетероцикл и R 4 представляет алкил. Также смесь соединений формулы (15) и формулы (16) обрабатывается каталитическим количеством основания вместе с добавлением затравочного кристалла, и соединение формулы (15), и соединение формулы (16) трансформируются в соединение формулы (1), приводя к получению соединения формулы (1) с высоким выходом (схема реакций 3). Соединение формулы (15) находится в равновесии с соединением формулы (16) благодаря присутствию основания в растворе. Также соединение формулы (15) находится в равновесии с соединениями формулы (17) и формулы (1), и соединение формулы (16) находится в равновесии с соединениями формулы (18) и формулы (19). Среди них соединение формулы (1), имеющее хорошие кристаллизационные свойства, селективно осаждается, и, таким образом, равновесие всех соединений смещается в сторону соединения формулы (1), селективно давая только соединение формулы (1) с высоким выходом из смеси соединений формулы (15) и формулы (16). Также настоящее изобретение относится к соединению нижеследующей формулы (4), промежуточному соединению, используемому для получения соединения формулы (1): где R 3 и R' 3 , каждый, представляет алкил или R 3 и R' 3 вместе с атомом кислорода, к которому они присоединены, образуют гетероцикл и R 4 представляет алкил. Предпочтительно R 3 и R' 3 , каждый, представляет метил, этил или пропил или R 3 и R' 3 вместе с атомом кислорода, к которому они присоединены, образуют диоксолан или диоксан и R 4 представляет метил, этил, пропил или бутил. Также настоящее изобретение относится к способу получения соединения формулу (4), включающему: (a) введение защитных групп и снятие защитных групп соединения нижеследующей формулы (9) с получением соединения нижеследующей формулы (10); (b) проведение реакции углерод-углеродного связывания соединения формулы (10) с получением соединения нижеследующей формулы (11); (c) взаимодействие соединения формулы (11) с бензиламином и восстановление его с получением соединения нижеследующей формулы (12) и (d) гидрирование соединения формулы (12) где R 3 и R' 3 , каждый, представляет алкил или R 3 и R' 3 вместе с атомом кислорода, к которому они присоединены, образуют гетероцикл и R 4 представляет алкил. Предпочтительно R 3 и R' 3 , каждый, представляет метил, этил или пропил или R 3 и R' 3 вместе с атомом кислорода, к которому они присоединены, образуют диоксолан или диоксан и R 4 представляет метил, этил, пропил или бутил. Вышеупомянутый способ получения соединения формулы (4) более конкретно будет описан ниже. На стадии (а) предпочтительно, чтобы введение защитных групп в соединение формулы (9) осуществлялось с использованием триметилортоформиата или триэтилортоформиата, и реакция снятия защитных групп осуществлялась в присутствии основания, выбранного из гидроксида натрия, гидроксида калия, карбоната натрия и гидрокарбоната натрия. Кроме того, предпочтительно, чтобы основание в вышеупомянутой реакции снятия защитных групп применялось в количестве 1,0-2,0 экв. к соединению формулы (9). Также предпочтительно, чтобы реакция введения защитных групп на стадии (а) осуществлялась в растворителе: метаноле или этаноле, и реакция снятия защитных групп осуществлялась в одном или более растворителях, выбранных из группы, состоящей из метанола, этанола, дихлорметана, хлороформа и воды. Также предпочтительно, чтобы реакция на стадии (b) осуществлялась с использованием этилхлорформиата или метилхлорформиата в присутствии основания, выбранного из н-бутиллития, литийдиизопропиламина или литийгексаметилдисилазида, и основание применяется в количестве 0,5-3,0 экв. к соединению формулы (10), и упомянутые этилхлорформиат или метилхлорформиат применяются в количестве 0,5-3,0 экв. к соединению формулы (10). Также предпочтительно, чтобы реакция на стадии (b) осуществлялась в растворителе, выбранном из группы, состоящей из тетрагидрофурана, этилового эфира и метил-трет-бутилового эфира. Предпочтительно, чтобы реакция восстановления на стадии (с) осуществлялась с использованием уксусной кислоты и боргидрида натрия, и упомянутый боргидрид натрия применялся в количестве 1,0-5,0 экв. к соединению формулы (11), и упомянутая уксусная кислота применялась в количестве 1,0-20,0 экв. к соединению формулы (11). Также предпочтительно, чтобы упомянутый бензиламин на стадии (с) применялся в количестве 1,0-10,0 экв. к соединению формулы (11). Кроме того, предпочтительно, чтобы реакция на стадии (с) осуществлялась в присутствии или в отсутствие растворителя, выбранного из этилацетата, тетрагидрофурана, этилового эфира и метил-трет-бутилового эфира, и может осуществляться как однореакторная реакция, если это требуется. Предпочтительно, чтобы реакция на стадии (d) осуществлялась в присутствии металлического катализатора, более предпочтительно катализатора палладиевого типа или катализатора типа никеля Ренея. Конкретно, катализатор палладиевого типа, имеющий от 1 до 20 мас.% палладия (Pd), или катализатор типа никеля Ренея, имеющий 1 мас.% и более никеля Ренея, нанесен на носитель, выбранный из группы, состоящей из углерода, оксида кремния и оксида алюминия, может применяться в количестве от 0,01 до 10 мас.% по металлическому компоненту от соединения формулы (12). Также предпочтительно, чтобы реакция на стадии (d) осуществлялась в одном или более растворителях, выбранных из группы, состоящей из метанола, этанола, н-пропанола, изопропанола, тетрагидрофурана, диметоксиэтана, диоксана, этилацетата и дихлорметана. Кроме того, предпочтительно, чтобы реакция гидрирования на стадии (d) осуществлялась при температуре от 0 до 50 °С и под давлением водорода от 1 до 100 атм. Заявители разработали новый способ получения соединения формулы (4), промежуточного соединения формулы (1), с более высоким выходом, как проиллюстрировано на нижеследующей схеме реакций 4. где R 3 и R' 3 , каждый, представляет алкил или R 3 и R' 3 вместе с атомом кислорода, к которому они присоединены, образуют гетероцикл и R 4 представляет алкил. Также настоящее изобретение относится к фармацевтической композиции для лечения воспаления или предотвращения апоптоза, содержащей соединение вышеуказанной формулы (1) и фармацевтически приемлемые носители, конкретно к фармацевтической композиции для лечения деменции, мозгового инсульта, повреждения головного мозга, обусловленного СПИД, диабета, язвы желудка, церебрального нарушения из-за вируса гепатита, заболеваний печени из-за вируса гепатита, острого гепатита, скоротечной печеночной недостаточности, цирроза печени, сепсиса, отторжения органа при трансплантации, ревматоидного артрита или сердечного клеточного некроза из-за ишемической болезни сердца. Также настоящее изобретение относится к способу лечения воспаления или предотвращения апоптоза у субъекта, включающему введение указанному объекту терапевтически эффективного количества соединения вышеуказанной формулы (1), конкретно к способу лечения деменции, мозгового инсульта, повреждения головного мозга, обусловленного СПИД, диабета, язвы желудка, церебрального нарушения из-за вируса гепатита, заболеваний печени из-за вируса гепатита, острого гепатита, скоротечной печеночной недостаточности, цирроза печени, сепсиса, отторжения органа при трансплантации, ревматоидного артрита или сердечного клеточного некроза из-за ишемической болезни сердца. Также настоящее изобретение относится к применению соединения вышеуказанной формулы (1) для приготовления лекарственного средства для лечения воспаления или предотвращения апоптоза, конкретно лекарственного средства для лечения деменции, мозгового инсульта, повреждения головного мозга, обусловленного СПИД, диабета, язвы желудка, церебрального нарушения из-за вируса гепатита, заболеваний печени из-за вируса гепатита, острого гепатита, скоротечной печеночной недостаточности, цирроза печени, сепсиса, отторжения органа при трансплантации, ревматоидного артрита или сердечного клеточного некроза из-за ишемической болезни сердца. Соединение формулы (1) в соответствии с настоящим изобретением может быть составлено в различные фармацевтические формы для целей введения. При приготовлении фармацевтической композиции в соответствии с настоящим изобретением эффективное количество соединения формулы (1) смешивается с фармацевтически приемлемым носителем, который можно выбрать из широкого разнообразия форм в зависимости от приготавливаемой рецептуры. Соединение формулы (1) может быть приготовлено в виде парентеральной инъекции или введения через кожу, или лекарственного препарата перорального введения, в зависимости от целей применения. И особо успешным является приготовление композиции в виде стандартной лекарственной формы для облегчения введения и равномерности дозировки. Для лекарственного препарата перорального введения могут применяться любые обычные фармацевтические наполнители. Например, вода, гликоли, масла, спирты и им подобные могут применяться для жидких лекарственных препаратов перорального введения, таких как суспензии, сиропы, эликсиры и растворы, или крахмалы, сахара, каолин, скользящие вещества, связующие вещества, дезинтегрирующие агенты и им подобные могут применяться для твердых лекарственных препаратов, таких как порошки, пилюли, капсулы и таблетки. Благодаря легкости введения таблетки и капсулы являются наиболее успешными стандартными лекарственными формами. Также желательно для таблеток и пилюль быть приготовленными в виде лекарственных препаратов с энтеросолюбильным покрытием. Для парентеральных лекарственных препаратов стерильная вода обычно используется в качестве носителя, хотя могут применяться другие ингредиенты, такие как растворяющие добавки. Инъекционные формы, например стерилизованные растворы или маслосодержащие суспензии для инъекций, могут быть приготовлены в соответствии с известной методикой с использованием подходящего диспергирующего агента, увлажняющего агента или суспендирующего агента. Растворители, которые могут применяться для приготовления инъекций, включают воду, раствор Рингера и изотонический NaCl раствор, и стерилизованное фиксирующее масло можно также применять, что удобно в качестве растворителя или суспендирующей среды. Какое-либо нестимулирующее фиксирующее масло, включая моно-, диглицерид, может применяться для этих целей. Жирная кислота, такая как олеиновая кислота, также может применяться для инъекций. В лекарственном препарате для введения через кожу носитель может включать способствующий проникновению агент и/или подходящий увлажняющий агент, необязательно совмещенный с подходящими добавками, обладающими незначительной способностью раздражения кожи. Упомянутые добавки могут облегчать введение через кожу и/или могут облегчать приготовление желаемой композиции. Эти лекарственные препараты для введения через кожу вводятся посредством различных способов, например как трансдермальный пластырь местного применения или как мазь. Когда соединение формулы (1) применяется в клинических целях, его предпочтительно вводить конкретному пациенту в количестве, выбираемом из интервала от 0,1 до 100 мг/кг массы тела в день. Общая дневная доза может быть введена однократно или за несколько раз. Однако специальная доза введения индивидуальному пациенту может меняться в соответствии со спецификой применяемого соединения, массой тела, полом, гигиеническим состоянием или диетой конкретного пациента, временем или способом введения, скоростью выведения из организма, соотношением агента с другими компонентами при смешении, тяжестью излечиваемой болезни и др. В дальнейшем настоящее изобретение будет описано более подробно со ссылкой на нижеследующие примеры, но не предназначенные никоим образом для ограничения объема изобретения. Пример получения 1. 1-Фтор-4-триметилсиланил-3-бутин-2-он (9). 49,1 г (499 ммоль) триметилсилилацетилена растворяли в 250 мл безводного тетрагидрофурана и внутреннюю температуру понижали приблизительно до -55 °С, и затем 210 мл (525 ммоль) 2,5 М n-BuLi в н-гексане добавляли за приблизительно 25 мин с поддержанием внутренней температуры ниже -30 °С. После перемешивания в течение приблизительно 40 мин к реакционной смеси добавляли 52,9 г (499 ммоль) этилфторацетата за 5 мин с поддержанием внутренней температуры ниже -25 °С и затем добавляли 74,4 г (524 ммоль) BF 3 -OEt за 15 мин с поддержанием внутренней температуры от -55 до -65 °С. После окончания добавления реакционную смесь перемешивали при 20 °С 2 ч и добавляли 250 мл 10% водного раствора хлорида аммония для окончания реакции. Органический слой отделяли и водный слой экстрагировали 200 мл этилацетата. Объединенную органическую фазу промывали 250 мл солевого раствора и концентрировали при пониженном давлении. Остаток перегоняли в вакууме при 10 мбар и 68 °С с получением соединения формулы (9) (67,3 г, 85%) в виде бесцветного масла. 1 H-ЯМР (500 МГц, CDCl 3 ): 4,90 (д, J=47,1 Гц, 2Н), 0,26 (с, 9Н). 13 С-ЯМР (125 МГц, CDCl 3 ): 181,0 (д, J=21,5 Гц), 104,0, 98,1, 84,8 (д, J=187 Гц). Пример получения 2. 4-Фтор-3,3-диметокси-1-бутин (10, R 3 , R' 3 =метил). 33,6 г (316 ммоль) триметилортоформиата и 6,0 г (31,5 ммоль) п-TsOH-H 2 O вместе с 50,0 г (316 ммоль) соединения формулы (9), полученного в примере получения 1, помещали в 260 мл метанола и перемешивали при температуре кипения с обратным холодильником (внутренняя температура 60-64 °С) в течение 6 ч. Реакционную смесь концентрировали при пониженном давлении, удаляя около 130 мл растворителя, и разбавляли 260 мл метиленхлорида. Добавляли 130 мл 10% водного раствора гидрокарбоната натрия и слои разделяли, и водный слой экстрагировали с использованием 130 мл метиленхлорида. Объединенный органический слой концентрировали при пониженном давлении с получением 4-фтор-3,3-диметокси-1-триметилсилилбутина (59,0 г, 92%) в качестве промежуточного соединения соединения-предшественника для целевого соединения (10). Это соединение использовали в следующей реакции без дополнительной очистки. 1 Н-ЯМР (500 МГц, CDCl 3 ): 4,38 (д, J=47,1 Гц, 2Н), 3,40 (с, 6Н), 0,20 (с, 9Н). 59,0 г (289 ммоль) 4-фтор-3,3-диметокси-1-триметилсилилбутина соединения-предшественника для целевого соединения (10), полученного выше, растворяли в 280 мл метиленхлорида, обрабатывали 59 мг (0,183 ммоль) тетра-н-бутиламмонийбромида и 347 мл (347 ммоль) 1 N водного раствора гидроксида натрия и перемешивали около 2 ч. Органический слой отделяли и водный слой экстрагировали 110 мл метиленхлорида. Объединенный органический слой промывали 110 мл солевого раствора и концентрировали при пониженном давлении с получением целевого соединения (10, R 3 , R' 3 =метил; 40,9 г, количественный выход). Это соединение использовали в следующей реакции без дополнительной очистки. 1 Н-ЯМР (500 МГц, CDCl 3 ): 4,42 (д, J=47,1 Гц, 2Н), 3,42 (с, 6Н), 2,64 (с, 1Н). 13 С-ЯМР (125 МГц, CDCl 3 ): 96,1 (д, J=20,3 Гц), 82,9 (д, J=180 Гц), 77,5, 75,5, 51,0. Пример получения 3. Этил 5-фтор-4,4-диметокси-2-пентиноат (11, R 3 , R' 3 =метил, R 4 =этил). Раствор 40,9 г (405 ммоль) диизопропиламина в 270 мл тетрагидрофурана охлаждали до 0 °С и 112 г (405 ммоль) 2,5 М n-BuLi в н-гексане добавляли в течение приблизительно 1 ч с поддерживанием внутренней температуры ниже 14 °С. Реакционную смесь перемешивали при 0 °С около 30 мин и температуру понижали до -78 °С. Раствор 41,0 г (311 ммоль) соединения, полученного в вышеприведенном примере получения 2 (10, R 3 , R' 3 =метил), растворенного в 160 мл тетрагидрофурана, добавляли к реакционной смеси за приблизительно 2 ч с поддерживанием внутренней температуры ниже -40 °С, затем 60,4 г (557 ммоль) этилхлорформиата добавляли за приблизительно 1 ч с поддерживанием внутренней температуры ниже -40 °С и далее реакционную смесь перемешивали при 0 °С около 2 ч. 250 мл 10% водного раствора хлорида аммония добавляли к реакционной смеси для завершения реакции и органический слой отделяли. Водный слой экстрагировали 100 мл этилацетата и объединенный органический слой промывали 100 мл солевого раствора и концентрировали при пониженном давлении с получением неочищенного целевого соединения (11, R 3 , R' 3 =метил, R 4 =этил; 95,0 г, рассчитанный выход 70%). Это соединение использовали в следующей реакции без дополнительной очистки. 1 H-ЯМР (500 МГц, CDCl 3 ): 4,45 (д, J=46,5 Гц, 2Н), 4,25 (кв., J=7,1 Гц, 2Н), 3,43 (с, 6Н), 1,31 (т, J=7,3 Гц, 3Н). Пример получения 4. Этил 3-(бензиламино)-5-фтор-4,4-диметоксипентаноат (12, R 3 , R' 3 =метил, R 4 =этил). 88 г (431 ммоль) неочищенного соединения, полученного в вышеприведенном примере получения 3 (11, R 3 , R' 3 =метил, R 4 =этил), растворяли в 430 мл метил-трет-бутилового эфира (МТВЕ) и температуру понижали до 0 °С. 31,4 г (293 ммоль) бензиламина добавляли к реакционной смеси, перемешивали при 20 °С в течение приблизительно 1 ч и разбавляли 450 мл метил-трет-бутилового эфира. Снова температуру реакционной смеси понижали до 0 °С, 33 г (873 ммоль) NaBH 4 добавляли к реакционной смеси и затем 259 г (4320 ммоль) уксусной кислоты добавляли за приблизительно 30 мин. Реакционную смесь выдерживали при 0 °С и 880 мл (2640 ммоль) 3 N водного раствора гидроксида натрия медленно добавляли за приблизительно 2 ч. Органический слой отделяли и отделенный органический слой промывали 880 мл 10% водного раствора хлорида аммония и затем 880 мл 1 N водного раствора хлористо-водородной кислоты добавляли. Водный слой отделяли, промывали 400 мл метил-трет-бутилового эфира и подщелачивали, используя 246 мл 10 N водного раствора гидроксида натрия, и экстрагировали 700 мл × 2 метил-трет-бутилового эфира. Объединенный органический слой промывали 400 мл солевого раствора, концентрировали при пониженном давлении с получением целевого соединения [12, R 3 , R' 3 =метил, R 4 =этил; 60,0 г, 44%, и 65% соединения формулы (10)]. Это соединение использовали в следующей реакции без дополнительной очистки. 1 Н-ЯМР (400 МГц, CDCl 3 ): 7,35-7,21 (м, 5Н), 4,53 (2дд, J=46,8, 10,4 Гц, 2Н), 4,13 (кв., J=7,2 Гц, 2Н), 3,80 (2д, J=12,8 Гц, 2Н), 3,53 (дд, J=8,4, 4,0 Гц, 1Н), 3,30 (с, 3Н), 3,22 (с, 3Н), 2,79 (дд, J=15,6, 3,6 Гц, 1Н), 2,40 (ддд, J=15,6, 8,0, 1,6 Гц, 1Н), 1,25 (т, J=7,2 Гц, 3Н). Пример 1. Этил 3-амино-5-фтор-4,4-диметоксипентаноат (4, R 3 , R' 3 =метил, R 4 =этил). 18,3 г (58,5 ммоль) соединения, полученного в вышеприведенном примере получения 4 (12, R 3 , R' 3 =метил, R 4 =этил), растворяли в 180 мл этанола и снятие бензильной защиты осуществляли с использованием катализатора 5% палладия-на-активированном угле (5% Pd/C) при давлении водорода 50 фунт/кв.дюйм в течение приблизительно 4 ч. Реакционную смесь фильтровали через 5,0 г слоя целита и промывали 90 мл этанола и фильтрат концентрировали при пониженном давлении с получением целевого соединения (4, 12,8 г, 98%). Это соединение использовали на следующей стадии без какой-либо очистки. 1 Н-ЯМР (500 МГц, CDCl 3 ): 4,53 (2дд, J=46,5, 10,4 Гц, 2Н), 4,14 (кв., J=7,3 Гц, 2Н), 3,57 (дд, J=11,0, 1,9 Гц, 1H), 3,29 (д, J=11,7 Гц, 6Н), 2,73 (дд, J=16,5, 2,5 Гц, 1Н), 2,36 (ддд, J=16,5, 10,4, 2,5 Гц, 1Н), 1,25 (т, J=7,3 Гц, 3Н). Пример получения 5. Этиловый эфир 5-фтор-3-[((R)-5-изопропил-3-(1-изохинолинил)-4,5-дигидроизоксазол-5-карбонил)амино]-4,4-диметоксипентановой кислоты (13, R 1 =1-изохинолинил, R 2 =изопропил, R 3 , R' 3 =метил, R 4 =этил). 15,5 г (54,5 ммоль) (5R)-5-изопропил-3-(1-изохинолинил)-4,5-дигидро-5-изоксазол карбоновой кислоты (2, R 1 =1-изохинолинил, R 2 =изопропил) растворяли в 150 мл метиленхлорида, температуру понижали до 0 °С и затем добавляли к указанному раствору 7,1 мл (81,7 ммоль) оксалилхлорида и 0,2 мл (2,6 ммоль) DMF с поддерживанием внутренней температуры ниже 12 °С. Реакционную смесь перемешивали при 20 °С около 2 ч и концентрировали при пониженном давлении. Реакционную смесь растворяли в 150 мл метиленхлорида, температуру понижали до 0 °С, триэтиламин добавляли и раствор 12,8 г (57,4 ммоль) соединения, полученного в примере 1 (4, R 3 , R' 3 =метил, R 4 =этил), растворенного в 30 мл метиленхлорида, медленно добавляли за 20 мин. Реакционную смесь перемешивали при 25 °С в течение 1,5 ч, смешанный раствор 120 мл 10% водного раствора гидрокарбоната натрия и 60 мл 1 N водного раствора гидроксида натрия добавляли для окончания реакции. Органический слой отделяли и водный слой экстрагировали 150 мл × 3 метиленхлорида. Объединенный органический слой концентрировали при пониженном давлении с получением целевого соединения (13, R 1 =1-изохинолинил, R 2 =изопропил, R 3 , R' 3 =метил, R 4 =этил; 30,1 г, количественный выход). Это соединение использовали на следующей стадии без какой-либо очистки. 1 Н-ЯМР (500 МГц, CDCl 3 ): 9,12 (кв., 1Н), 8,53 (м, 1Н), 7,85-7,25 (м, 4Н), 4,80 (м, 1Н), 4,54-4,34 (м, 2Н), 4,14 (кв., J=7,4 Гц, 2Н), 3,99 (2д, J=18,4 Гц, 1Н), 3,81 (м, 1Н), 3,78 (2д, J=8,6 Гц, 1Н), 3,33 (д, 3Н), 3,20 (д, 3Н), 2,75 (м, 3Н), 2,53 (м, 1Н), 2,39 (гептет, J=6,7 Гц, 1Н), 1,27 (т, J=7,4 Гц, 1,5Н), 1,07 (м, 6Н), 0,97 (т, J=7,4 Гц, 1,5Н). Пример получения 6. 5-Фтор-3-[((R)-5-изопропил-3-(1-изохинолинил)-4,5-дигидроизоксазол-5-карбонил)амино]-4,4-диметоксипентановая кислота (14, R 1 =1-изохинолинил, R 2 =изопропил, R 3 , R' 3 =метил). 30,1 г (61,6 ммоль) соединения, полученного в вышеприведенном примере получения 5 (13, R 1 =1-изохинолинил, R 2 =изопропил, R 3 , R' 3 =метил, R 4 =этил), вместе с 7,76 г (185 ммоль) моногидрата гидроксида лития растворяли в смешанном растворителе: 168 мл тетрагидрофурана и 42 мл воды, и перемешивали приблизительно при 40 °С 4 ч. Реакционную смесь концентрировали при пониженном давлении для удаления тетрагидрофурана в растворителе, 180 мл 1 N водного раствора гидроксида натрия добавляли и смесь промывали 120 мл × 2 толуола. Водный слой подкисляли 66 мл 6 N водным раствором хлористо-водородной кислоты и экстрагировали 150 мл × 3 метиленхлорида, и объединенный органический слой концентрировали при пониженном давлении с получением целевого соединения (14, R 1 =1-изохинолинил, R 2 =изопропил, R 3 , R' 3 =метил; 25,4 г, 89%). Это соединение использовали на следующей стадии без какой-либо очистки. 1 Н-ЯМР (400 МГц, CDCl 3 ): 9,10-8,92 (м, 1Н), 8,52 (м, 1Н), 7,86-7,13 (м, 4Н), 4,77 (м, 1Н), 4,54-4,34 (м, 2Н), 3,95 (2д, J=8,0 Гц, 1Н), 3,75 (2д, J=18,4 Гц, 1Н), 3,35-3,16 (2д, 6Н), 2,78 (2дд, J=16,0, 4,4 Гц, 1Н), 2,54 (м, 1Н), 2,39 (м, 1Н), 2,35 (с, 1Н), 1,06 (м, 6Н). Пример 2. (4S,5S)-5-фторметил-5-гидрокси-4-({[(5R)-5-изопропил-3-(1-изохинолинил)-4,5-дигидро-5-изоксазолил]карбонил}амино)-2-дигидрофуранон (1, R 1 =1-изохинолинил, R 2 =изопропил). 17,0 г (36,9 ммоль) соединения, полученного в вышеприведенном примере получения 6 (14, R 1 =1-изохинолинил, R 2 =изопропил, R 3 , R' 3 =метил) и 6,6 мл (110 ммоль) уксусной кислоты растворяли в 123 мл (738 ммоль) 6 N водного раствора хлористо-водородной кислоты и перемешивали около 4 ч. Внутреннюю температуру реакционной смеси доводили до 0 °С и 150 мл этилацетата добавляли. 220 мл (660 ммоль) 3 N водного раствора гидроксида натрия медленно добавляли для доведения рН приблизительно до 3. Органический слой отделяли и водный слой экстрагировали 150 мл × 2 этилацетата. Объединенную органическую фазу промывали 100 мл солевого раствора и концентрировали при пониженном давлении. Остаток разбавляли 50 мл толуола и снова концентрировали при пониженном давлении с получением смеси соединений формулы (15) и формулы (16) (R 1 =1-изохинолинил, R 2 =изопропил) (15,4 г, количественный выход, химическая чистота: 87,0%). 1 Н-ЯМР (500 МГц, ДМСО-d 6 ): 8,99 (м, 1Н), 8,65 (м, 1Н), 8,19-7,78 (м, 4Н), 5,15 (м, 1,5Н), 4,77 (м, 1Н), 4,42 (м, 0,5Н), 3,91 (2д, J=17,6 Гц, 1Н), 3,74 (м, 1Н), 2,99 (м, 0,2Н), 2,82 (м, 1Н), 2,63 (м, 0,8Н), 2,33 (м, 1Н), 0,97 (м, 6Н). К 146 мл толуола добавляли 14,6 г (35,2 ммоль) смеси соединений формулы (15) и формулы (16) (R 1 =1-изохинолинил, R 2 =изопропил) (химическая чистота: 87,0%) и смесь нагревали до 100 °С до полного растворения. Затем добавляли 14 мг затравочного кристалла целевого соединения (1, R 1 =1-изохинолинил, R 2 =изопропил), температуру медленно понижали до 20 °С и реакционную смесь перемешивали с получением твердого вещества. 0,25 мл (1,8 ммоль) диизопропиламина добавляли и перемешивали при 20 °С около 2 недель, подтверждая, что соотношение между соединением формулы (15) и соединением формулы (16) (R 1 =1-изохинолинил, R 2 =изопропил) составляет 92,8:7,2 по данным ВЭЖХ. Реакционную смесь концентрировали при пониженном давлении для удаления толуола, 88 мл этилацетата добавляли и смесь нагревали до 65 °С до полного растворения ее. Затем 88 мл нормального гексана добавляли и температуру медленно понижали и перемешивали приблизительно при 20 °С 2 дня. Полученное в результате твердое вещество фильтровали и промывали смешанным раствором 15 мл этилацетата и 15 мл нормального гексана. После сушки твердого вещества с азотом было получено твердое целевое вещество белого цвета (1, R 1 =1-изохинолинил, R 2 =изопропил) с выходом 54,7% из расчета от соединения формулы (2) (8,0 г, химическая чистота 98,6%). Данные ЯМР в твердой фазе для кристаллической формы были получены с применением VACP MAS (кросс-поляризация с переменной амплитудой и вращение образца под магическим углом) при скорости вращения 9 кГц. 1 H-ЯМР (CDCl 3 ): 9,02 (ушир.с, 1Н), 8,54 (д, J=5,5 Гц, 1Н), 7,85 (д, J=7,95 Гц, 1Н), 7,70 (м, 3Н), 7,60 (ушир.с, 1Н), 4,86 (ушир.с, 1Н), 4,2-5,2 (ушир.с, 1Н), 4,05 (ушир., J=19,0 Гц, 1Н), 3,78 (ушир. J=19,0 Гц, 1Н), 2,7-3,1 (ушир.м, 2Н), 2,40 (м, 1Н), 1,08 (дд, J=6,7, 4,9 Гц, 6Н). 13 С-ЯМР (CDCl 3 ): 173,8, 172,4, 160,2, 147,6, 141,7, 136,8, 130,7, 129,0, 127,4, 127,3, 126,8, 122,9, 92,3, 82,7 (д, J=215 Гц), 48,9, (ушир.), 44,6, 34,4, 33,9, 17,7, 16,3. 13 С-ЯМР (твердое вещество): 176,4, 171,8, 160,3, 150,2, 139,5, 137,5, 132,3 (2С), 127,7 (3С), 123,0, 104,3, 94,1, 86,4, 48,8, 42,9, 32,7 (2С), 19,6, 15,4. Масс (ESI) (ионизация электрораспылением) 416,14 (М+1). [ α] D 25 =+3,2 (с=1,0, ацетонитрил). Экспериментальный пример 1. Тест на стабильность. Как показано на фиг. 2, в качестве результата теста на стабильность аморфной формы и кристаллической формы соединения формулы (1) (R 1 =1-изохинолинил, R 2 =изопропил) наблюдали, что 50% аморфной формы было разрушено через 28 дней в неблагоприятных условиях (60 °С), но количественное содержание кристаллической формы нисколько не понизилось при тех же условиях (60 °С) даже спустя 28 дней. Таким образом, следует понимать, что кристаллическая форма имеет стабильность, лучшую по сравнению с аморфной формой, достаточную для применения в композиции ингибитора или терапевтического агента. Экспериментальный пример 2. Лечебный эффект на LPS-индуцированный острый гепатит у мышей. Стадия 1. Подготовка пробы крови. Самцов мышей Balb/c (6 недель, Charles River Laboratory, Osaka, Japan) выращивали при 22 °C, при 55% относительной влажности, в режиме чередования ночь/день через 12 ч. Кормом и водой снабжали без ограничения. LPS (липополисахарид) и D-галактозамин растворяли в концентрациях 0,4 мг/мл и 280 мг/мл, соответственно, в физиологическом растворе, не содержащем пирогенов, и смешивали в соотношении 1:1. Раствор вводили как инъекцию мышам в количестве 5 мл/кг. Сразу после инъекции LPS и D-галактозамина среду, в которой тестируемое соединение растворено (смесь, состоящая из PEG400:этанол:Tween 80, 15:7,5:2,5, разбавленная 1/5 физиологическим раствором) или только среду вводили как инъекцию тестируемому животному. Пробы крови отбирали из сердец мышей спустя 8 ч после инъекции лекарственного средства. Стадия 2. Анализ аминотрансферазной активности плазмы крови. ALT активность плазмы крови полученных на стадии 1 проб крови измеряли с использованием набора реактивов для анализа ALT активности (Asan pharmaceutical company) в соответствии с регламентированной процедурой производителя. Наблюдали, что введение LPS и D-галактозамина быстро повышает ALT активность плазмы крови и тестируемый материал дозозависимо подавляет такое повышение ферментативной активности. Основываясь на этих результатах, величину ED 50 для каждого тестируемого материала подсчитывали, используя программное обеспечение Prism (GraphPad Co.) Экспериментальный пример 3. Лечебный эффект на индуцированный Fas антителами острый гепатит у мышей. Самцов мышей Balb/c (6 недель, Charles River Laboratory, Osaka, Japan) выращивали при 22 °C, при 55% относительной влажности, в режиме чередования ночь/день через 12 ч. Кормом и водой снабжали без ограничения. Fas антитела растворяли в концентрации 30 мг/мл в физиологическом растворе, не содержащем пирогенов, и раствор вводили как инъекцию мышам в количестве 5 мл/кг. Сразу после инъекции Fas антител среду, в которой растворено тестируемое соединение (смесь, состоящая из PEG4 00:этанол:Tween 80, 15:7,5:2,5, разбавленная 1/5 физиологическим раствором), или только среду вводили как инъекцию тестируемому животному. Пробы крови отбирали из сердец мышей спустя 8 ч после инъекции лекарственного средства. Величину ED 50 для отобранной пробы крови подсчитывали, используя вышеуказанный способ тестирования. Нижеследующая таблица показывает фармакологический эффект результатов теста на модели острого гепатита в соответствии со способом введения соединения формулы (1), полученного в вышеприведенных примерах 2 и 3. Промышленное применение. Производное изоксазолина, имеющее циклическую карбоксильную кислотную гемикетальную группу, формулы (1) в соответствии с настоящим изобретением имеет превосходную активность ингибирования каспазы и превосходную стабильность. Кроме того, способ в соответствии с настоящим изобретением приводит к получению только одного диастереоизомера высокой степени чистоты в результате использования кристаллизации, обусловленной динамическим переходом. Далее, если аминопроизводное, имеющее кетальную группу, в соответствии с настоящим изобретением применяется как промежуточное соединение, специалист в данной области техники может легко получить производное изоксазолина, имеющее циклическую карбоксильную кислотную гемикетальную группу, без дополнительной очистки. |