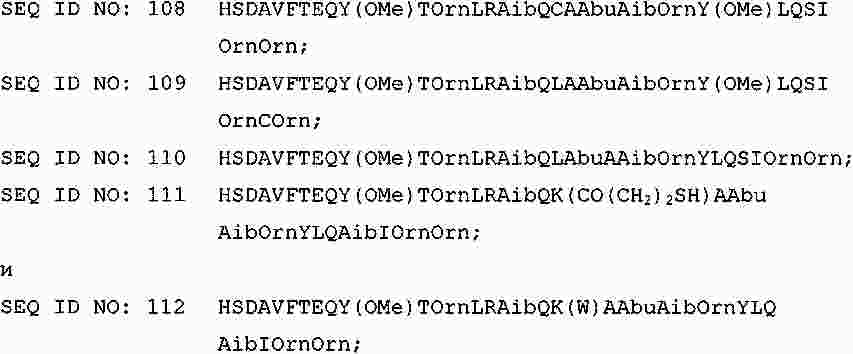
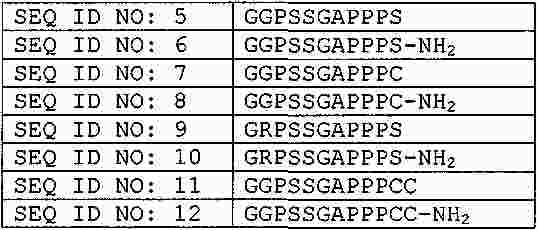
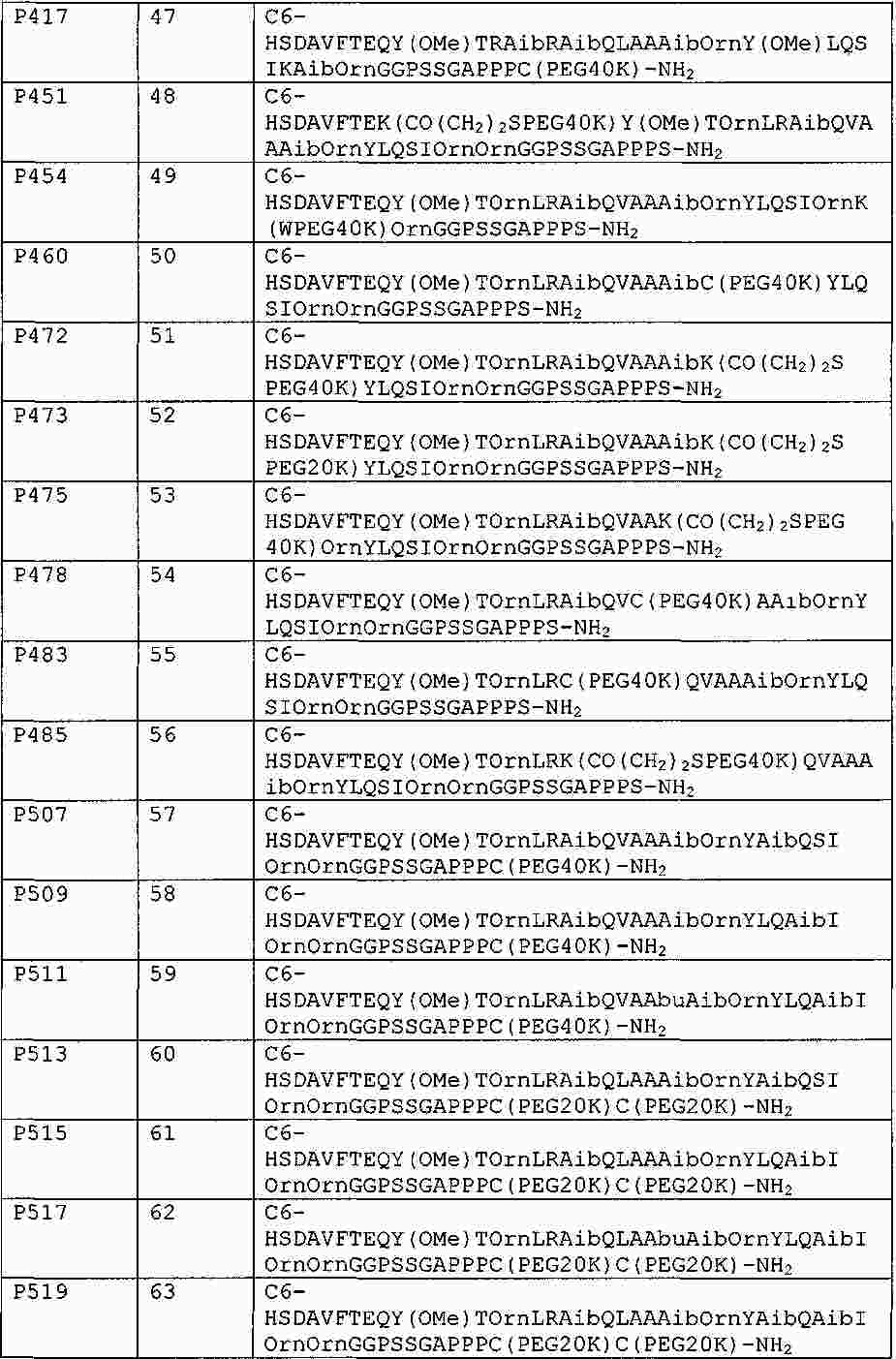
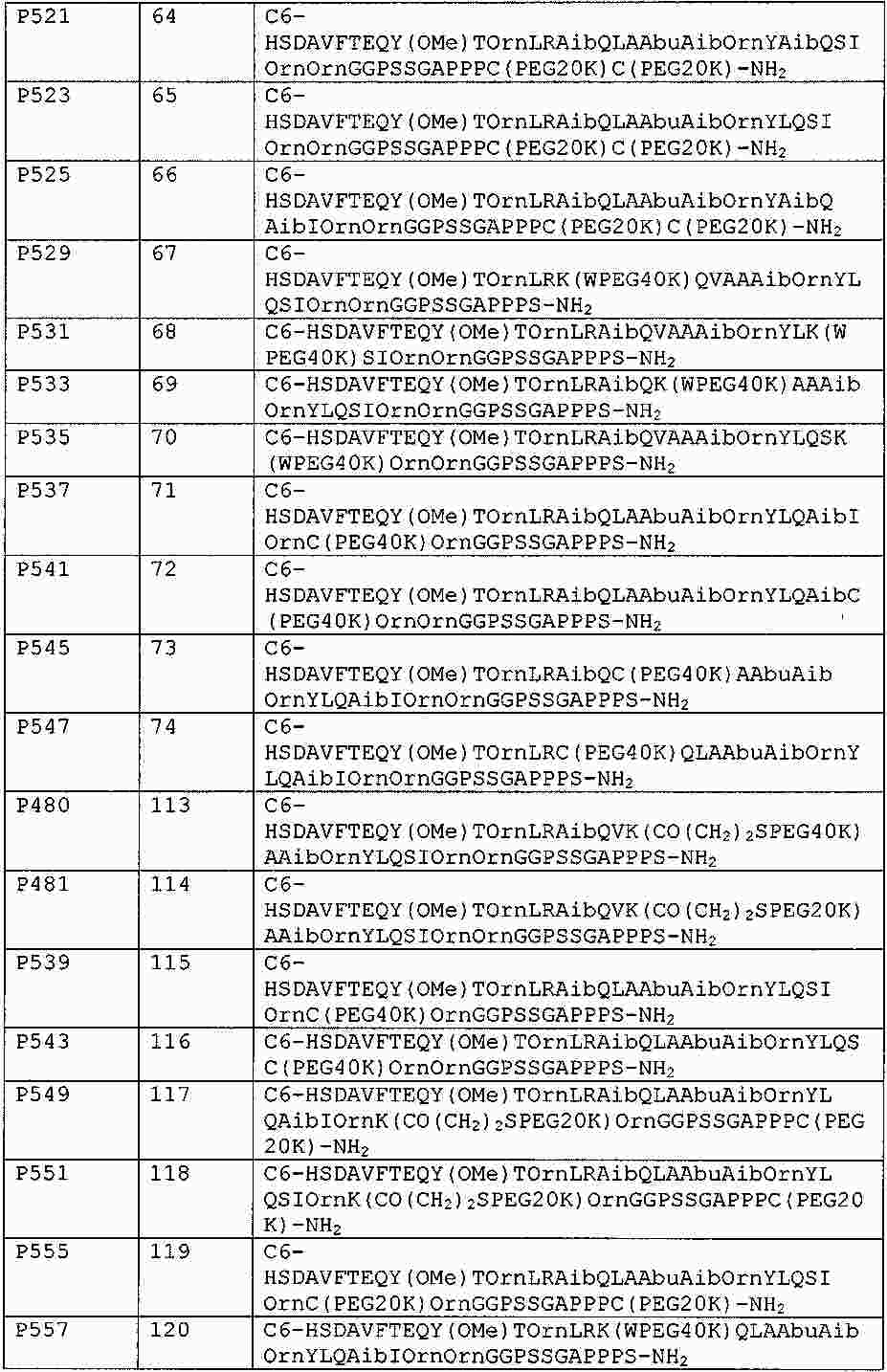
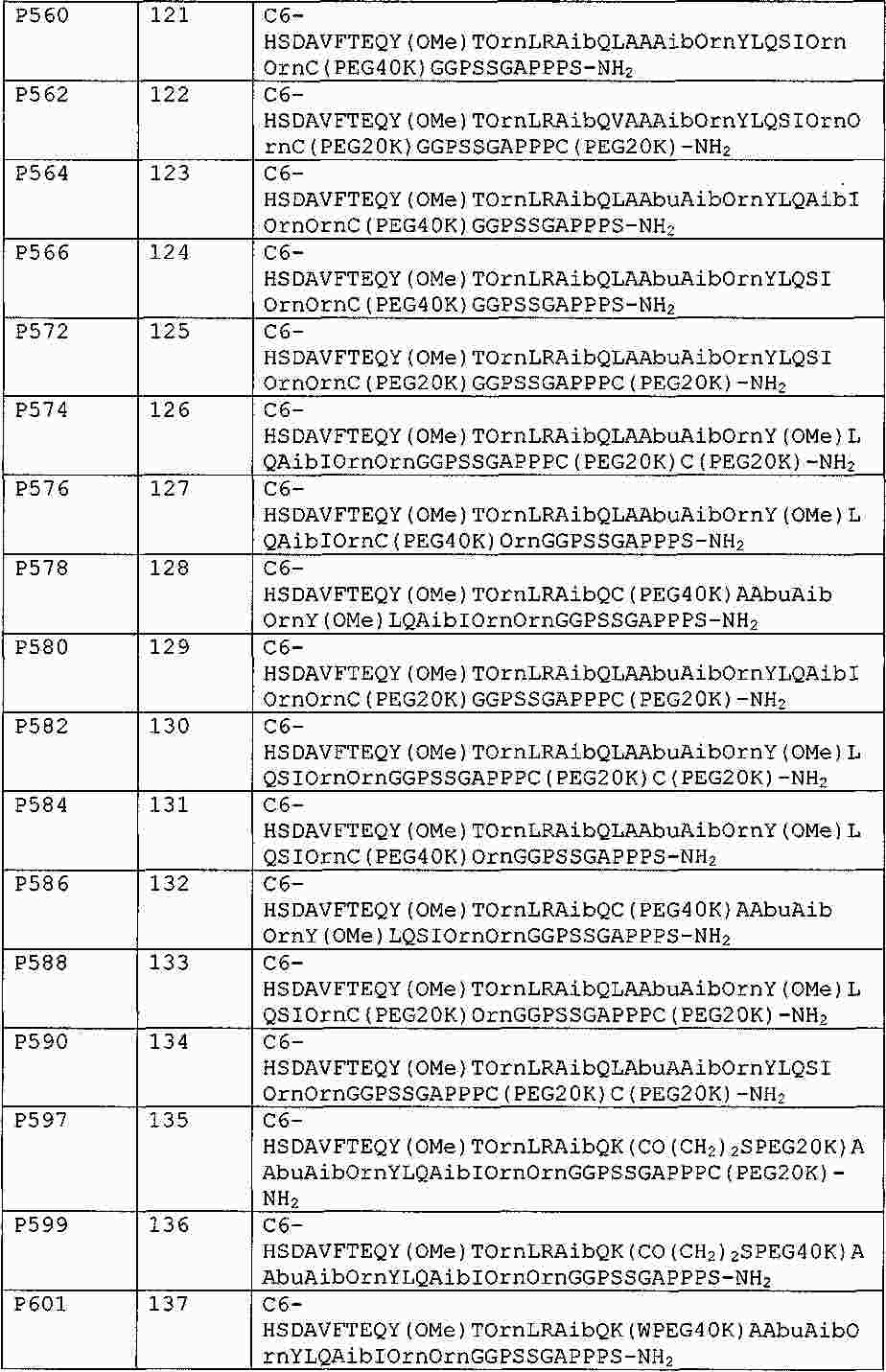
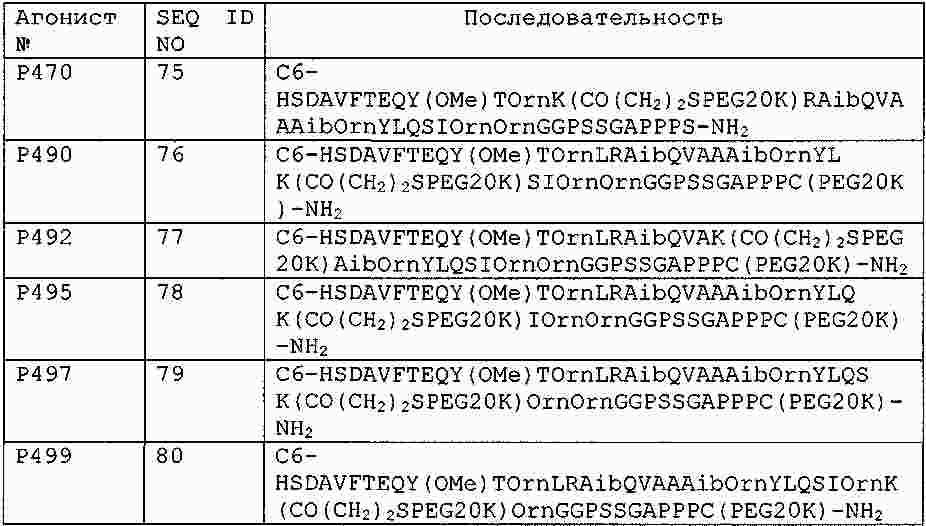
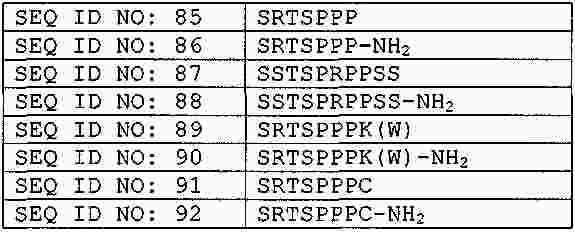
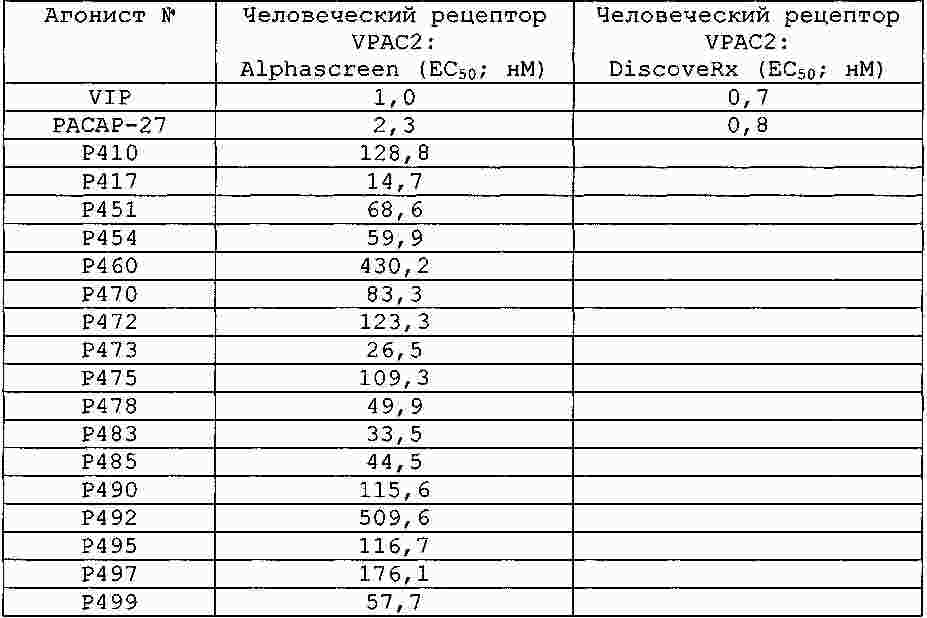
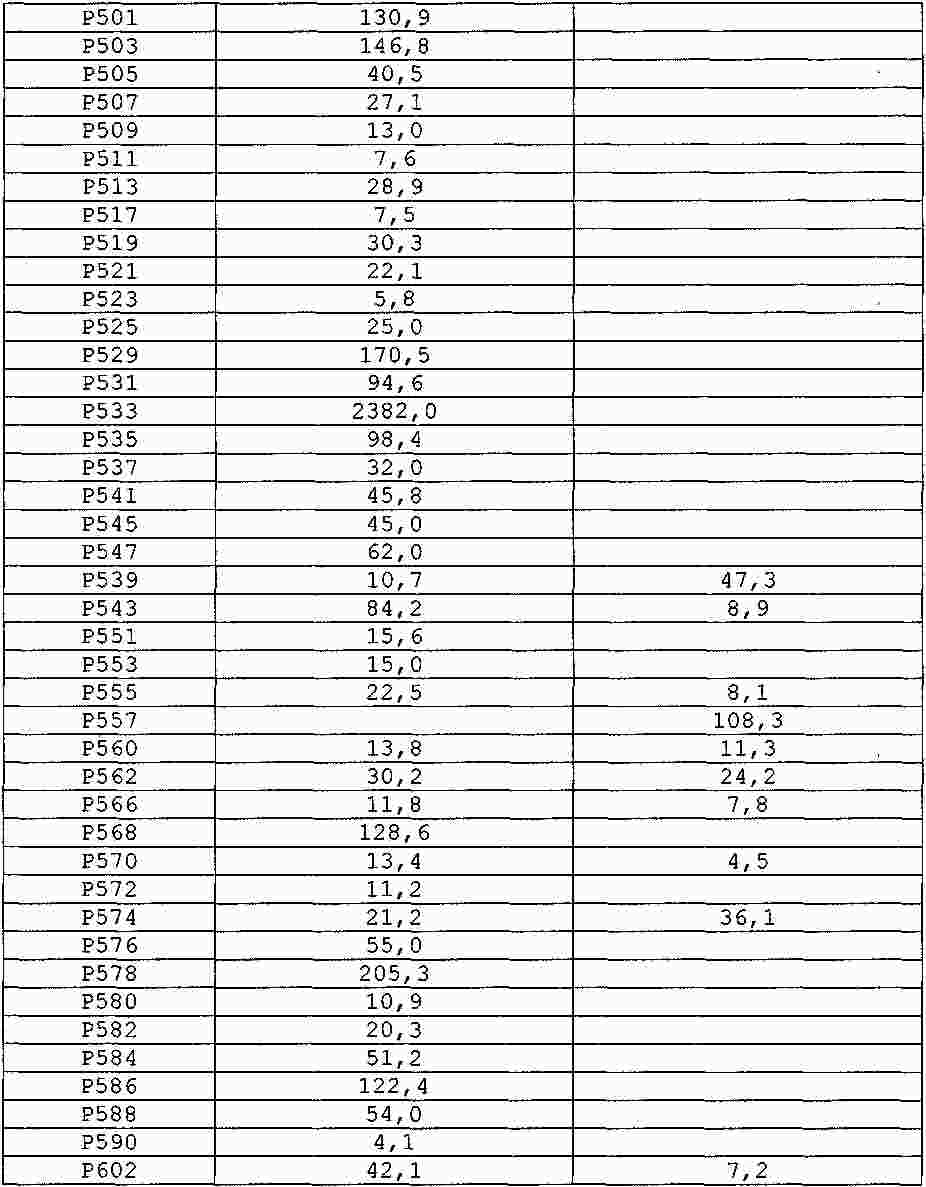
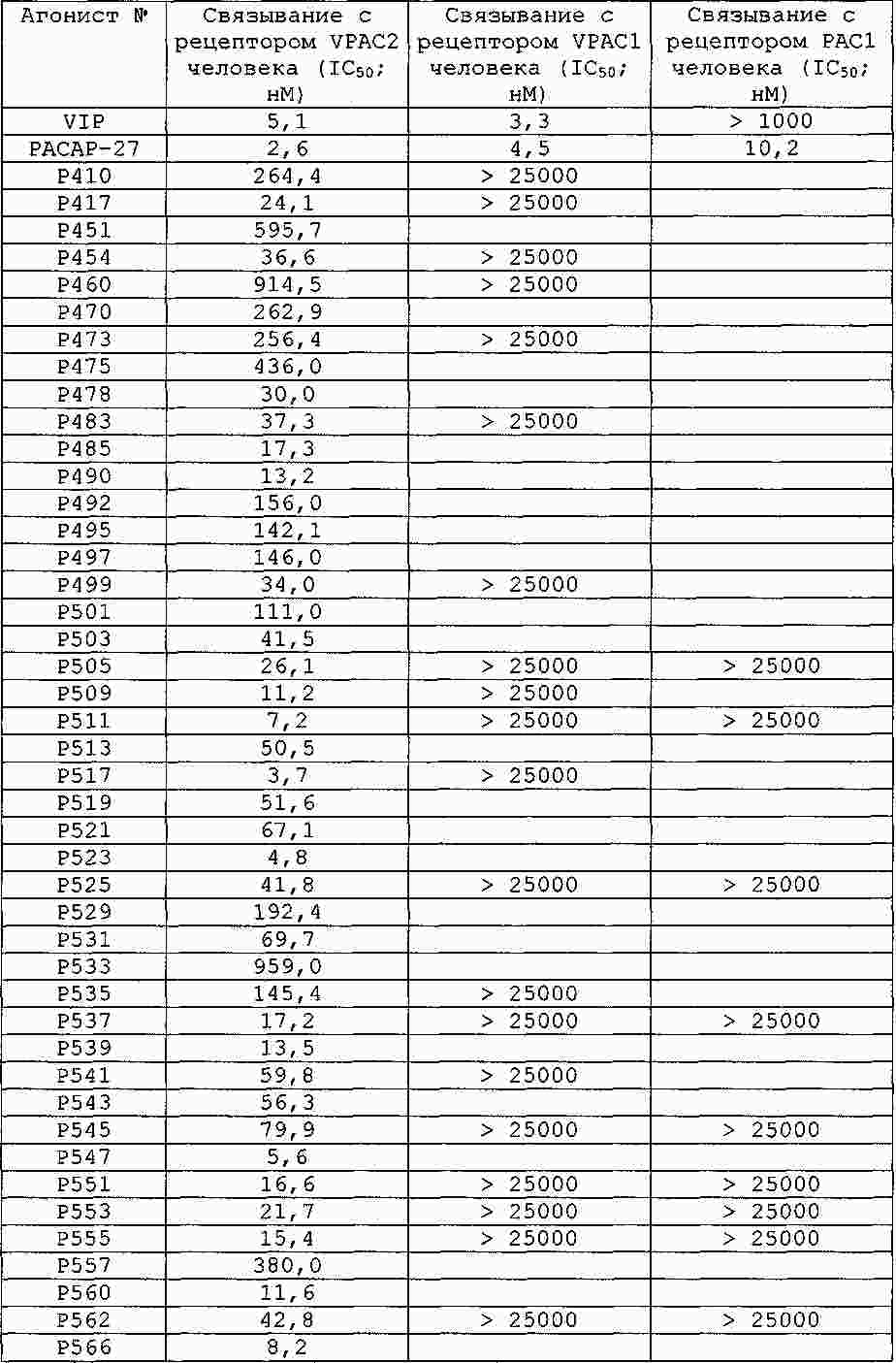
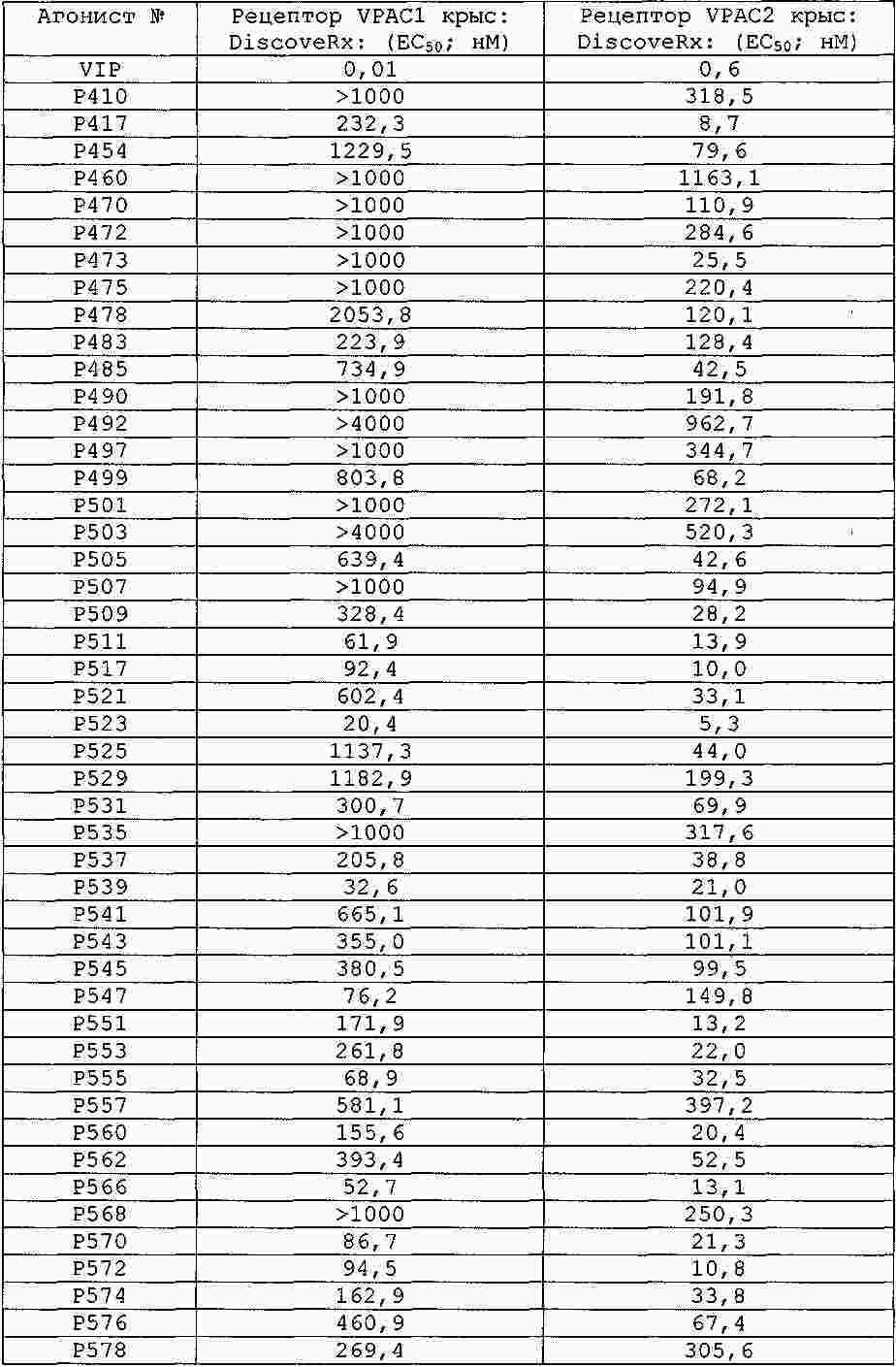
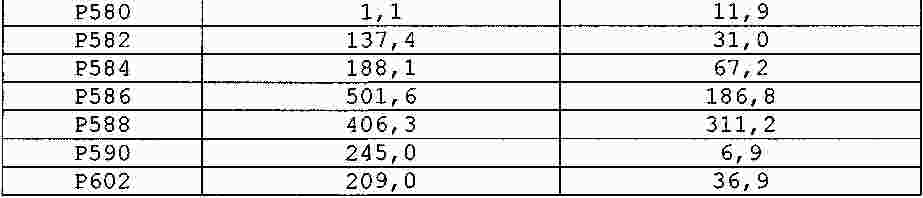
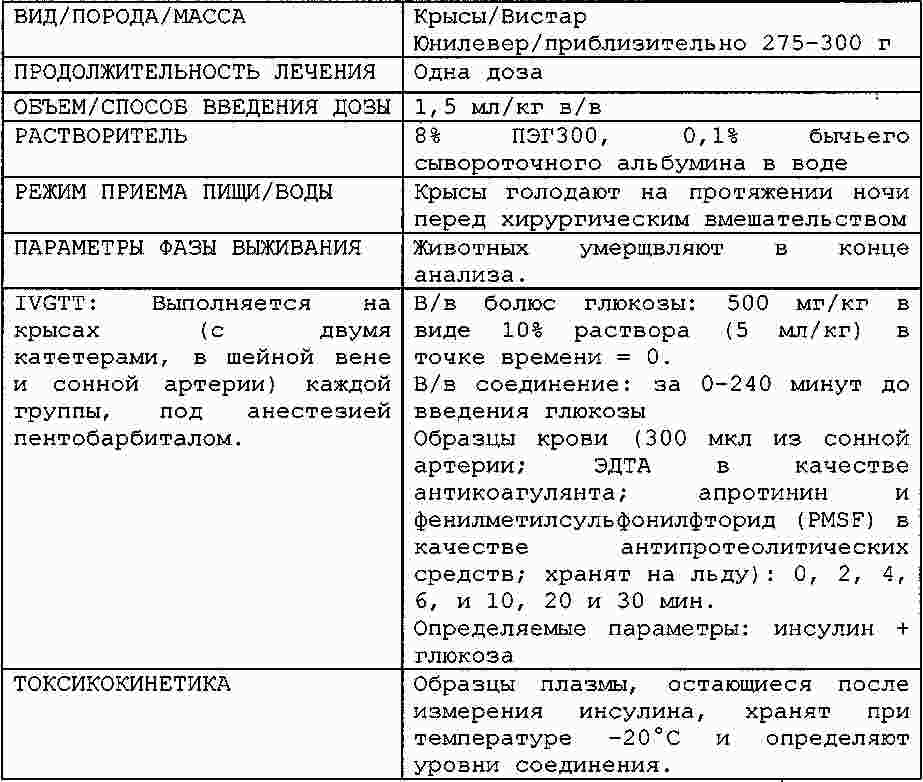
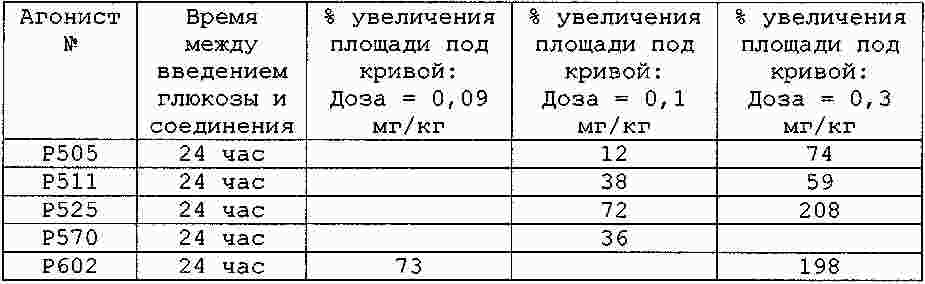
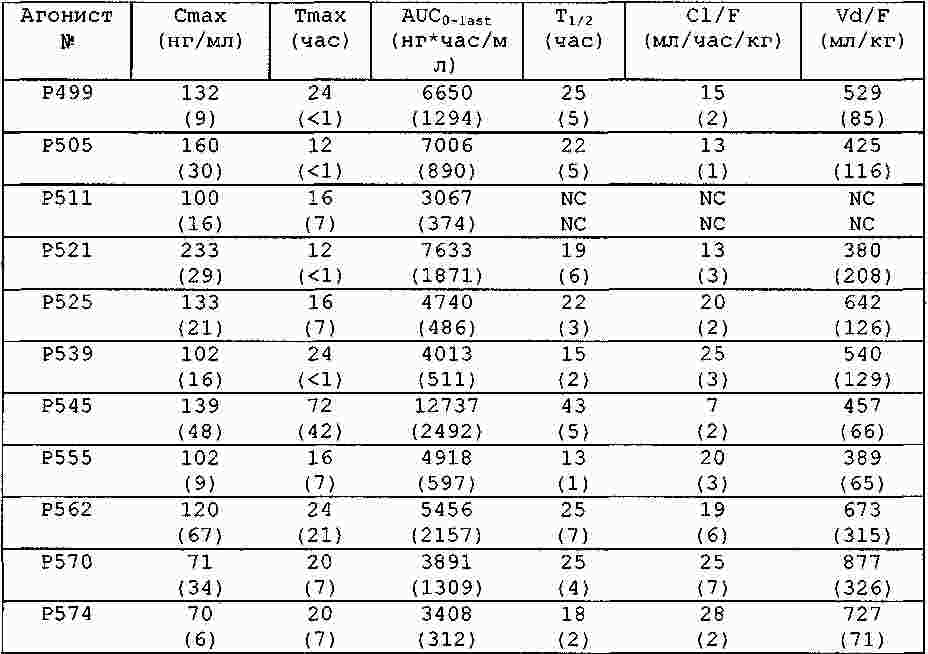
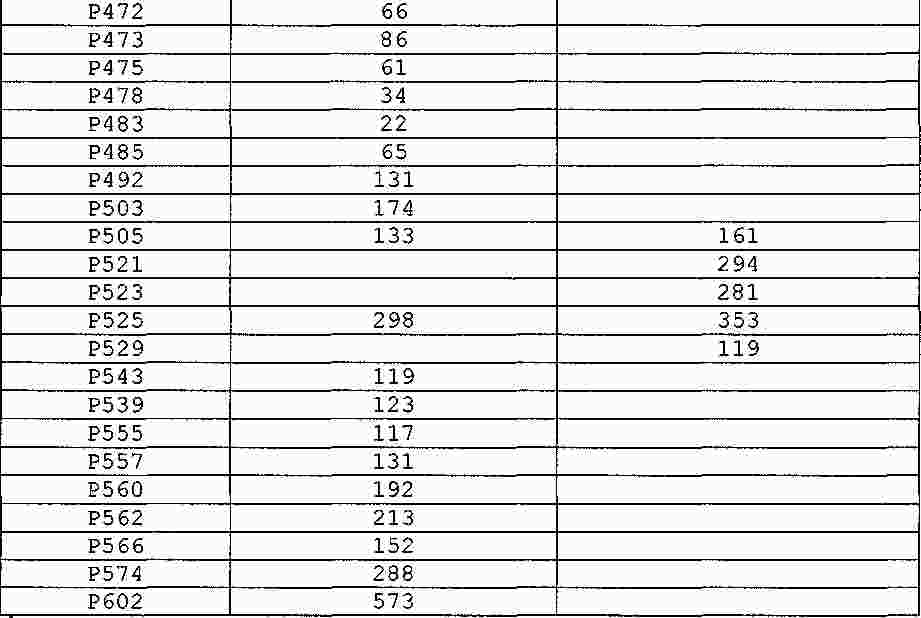
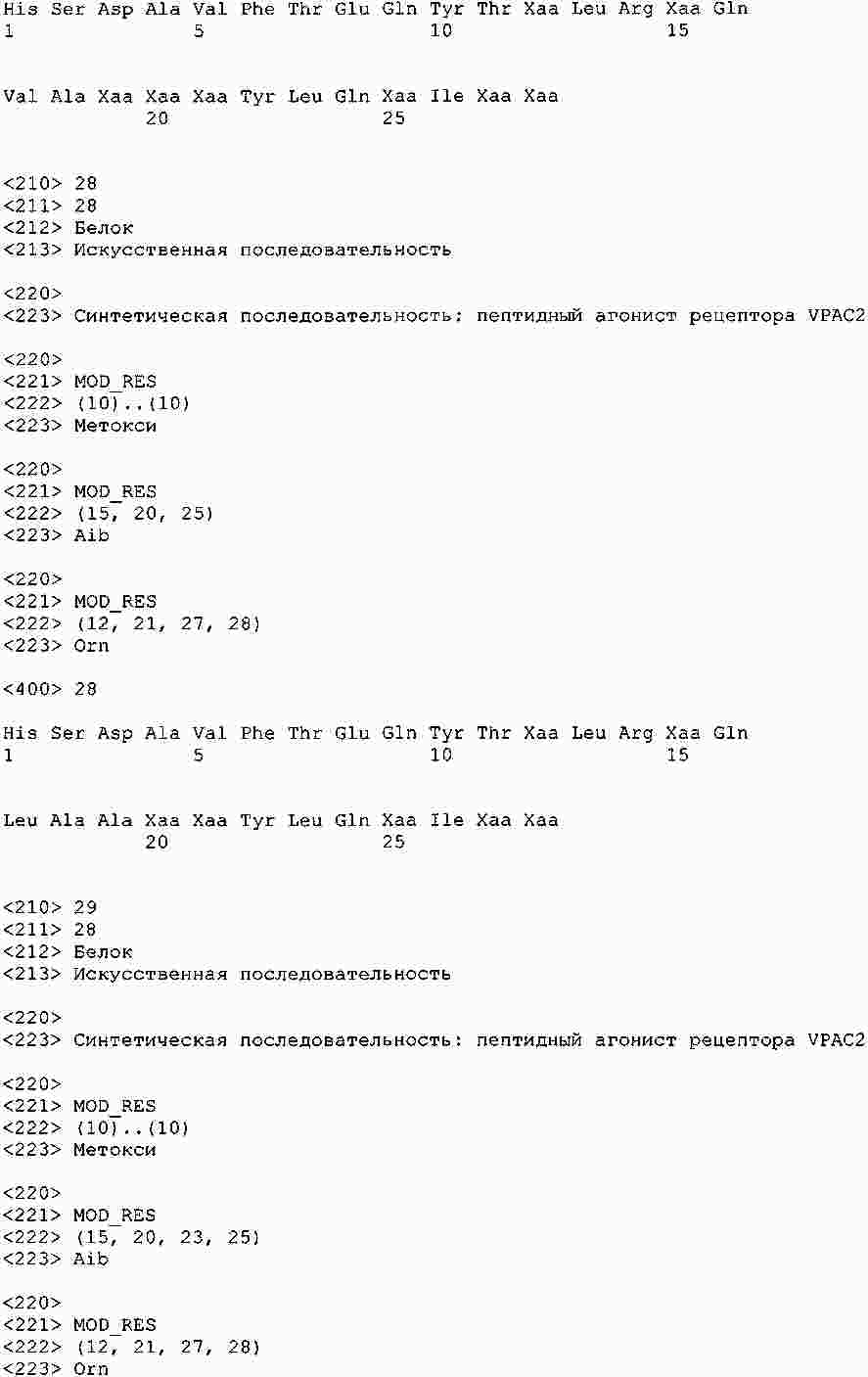
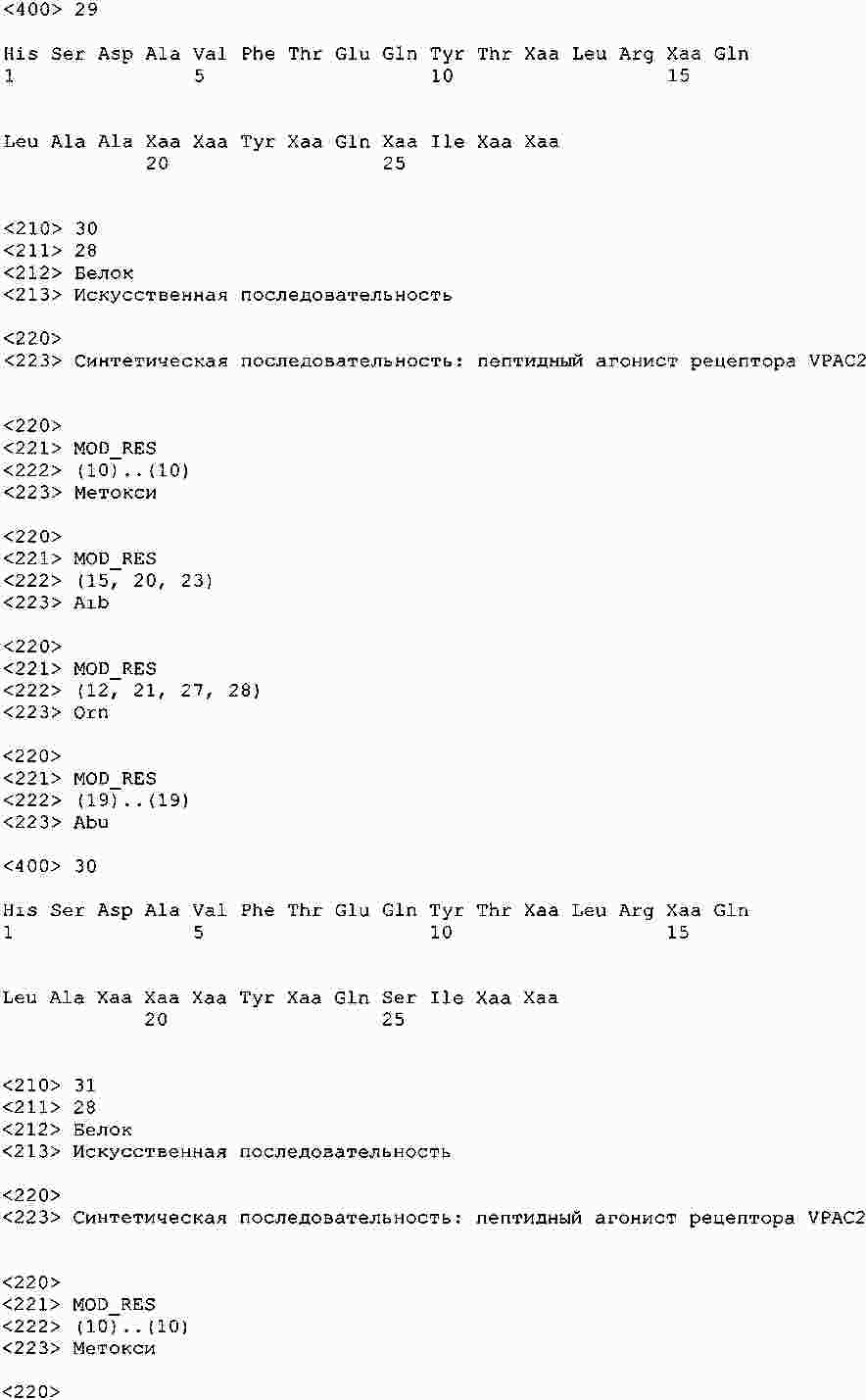
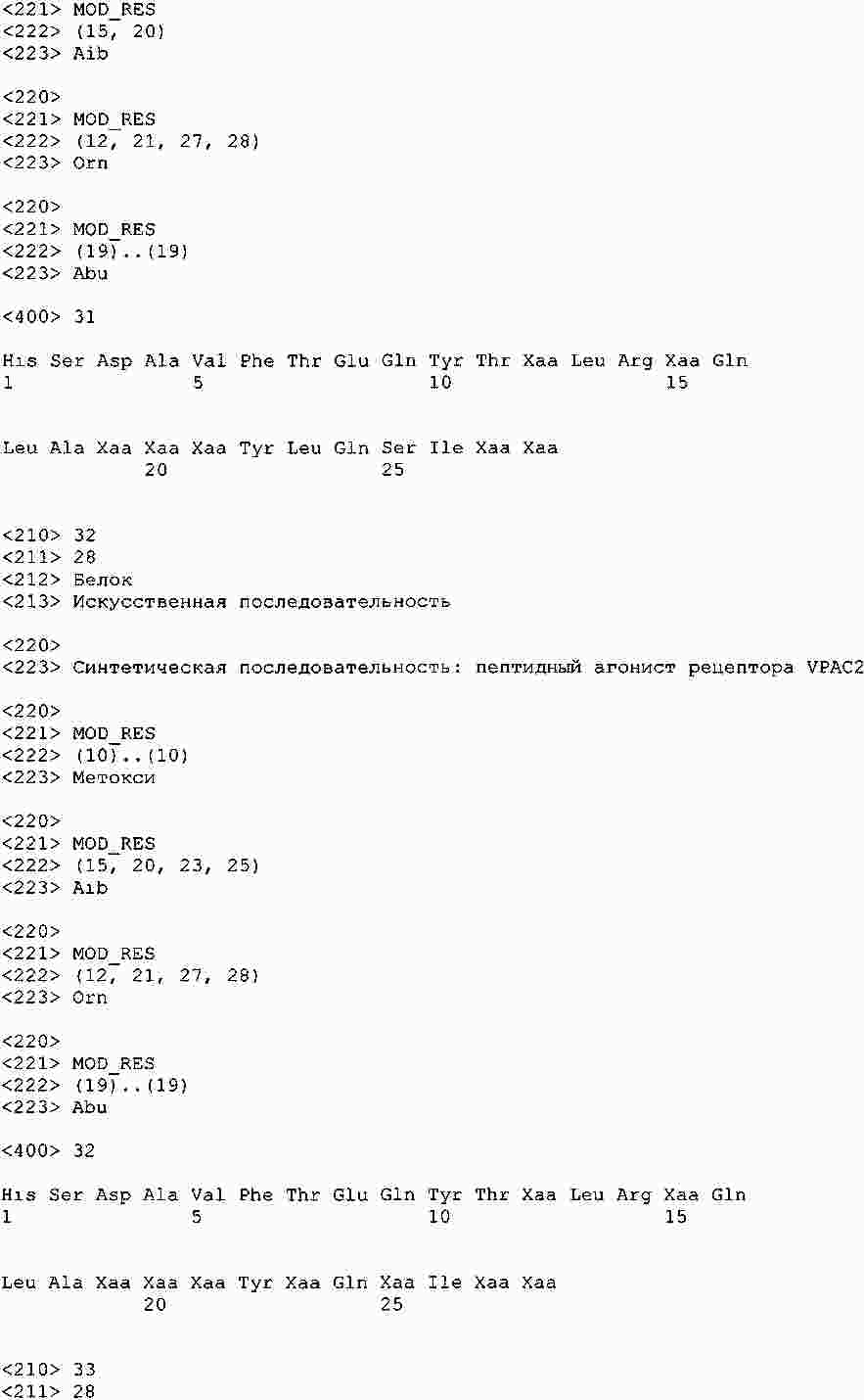
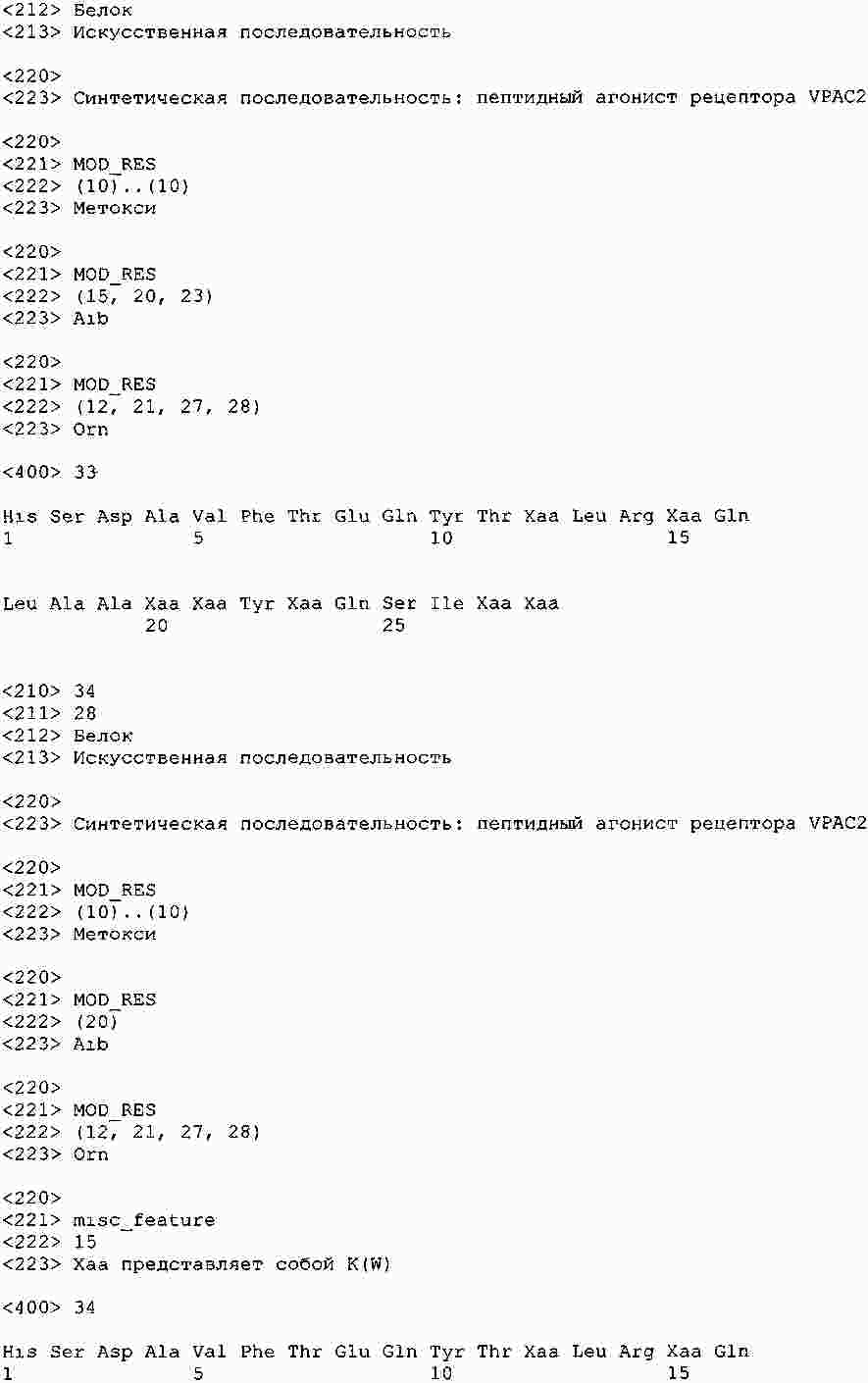
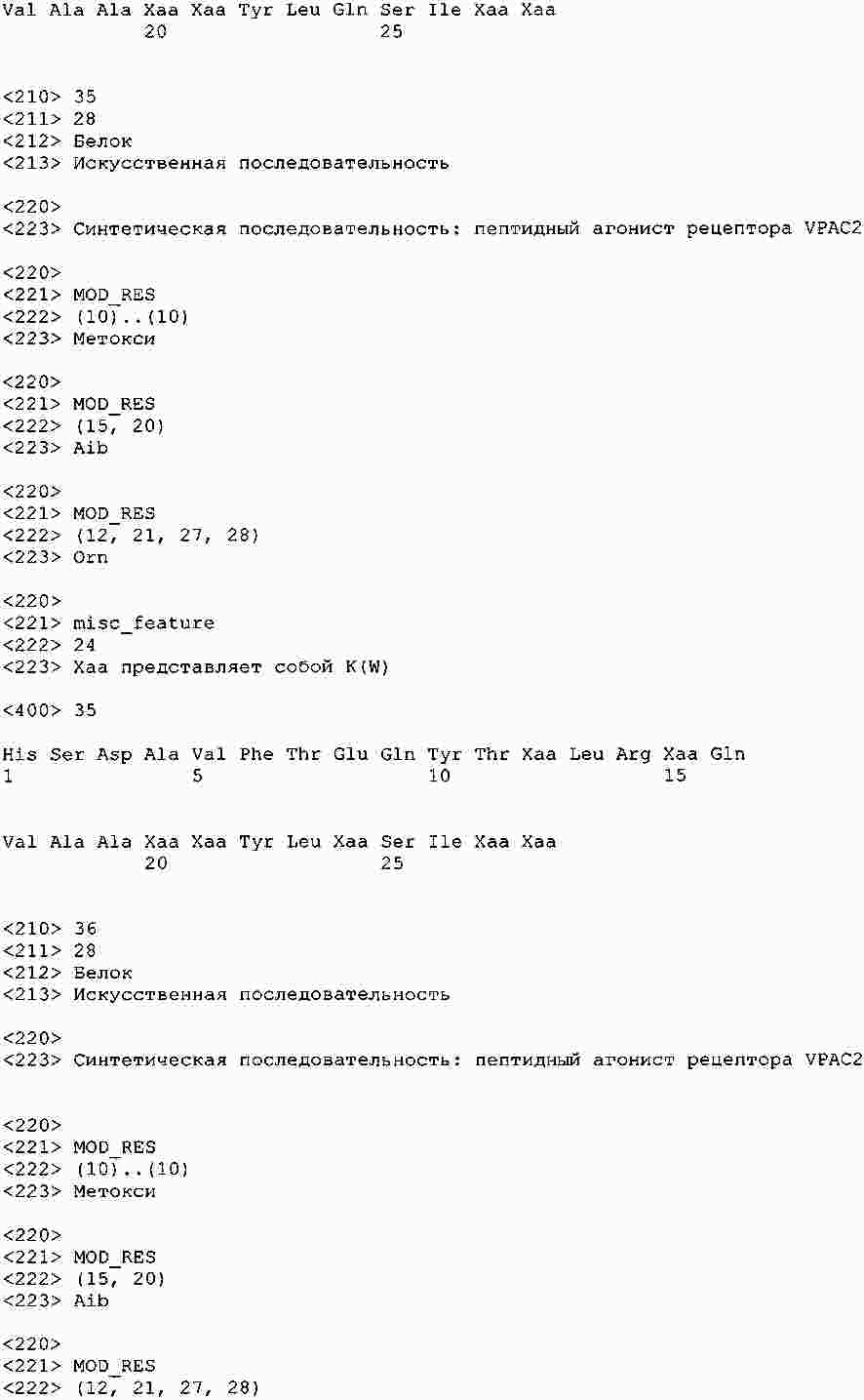
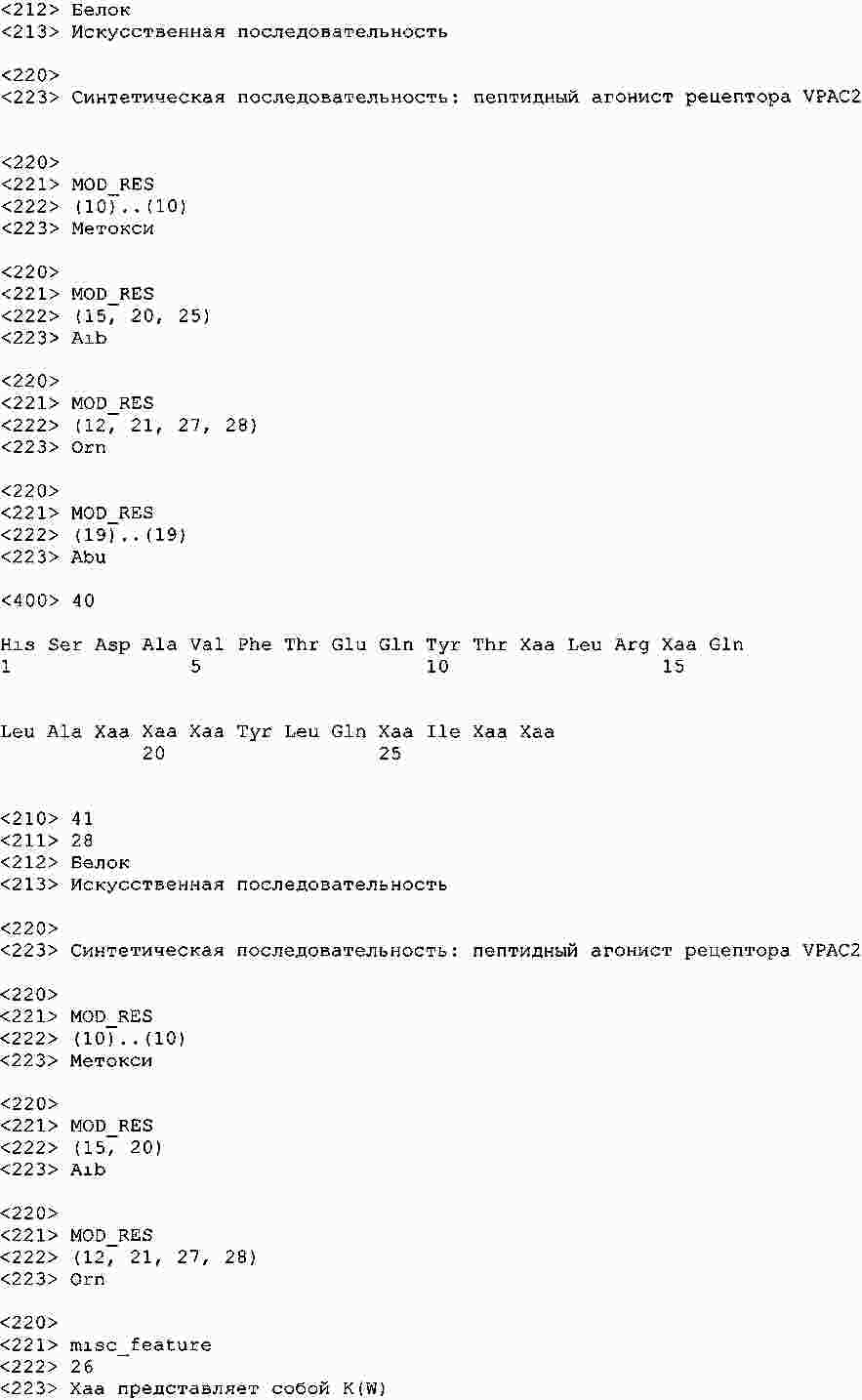
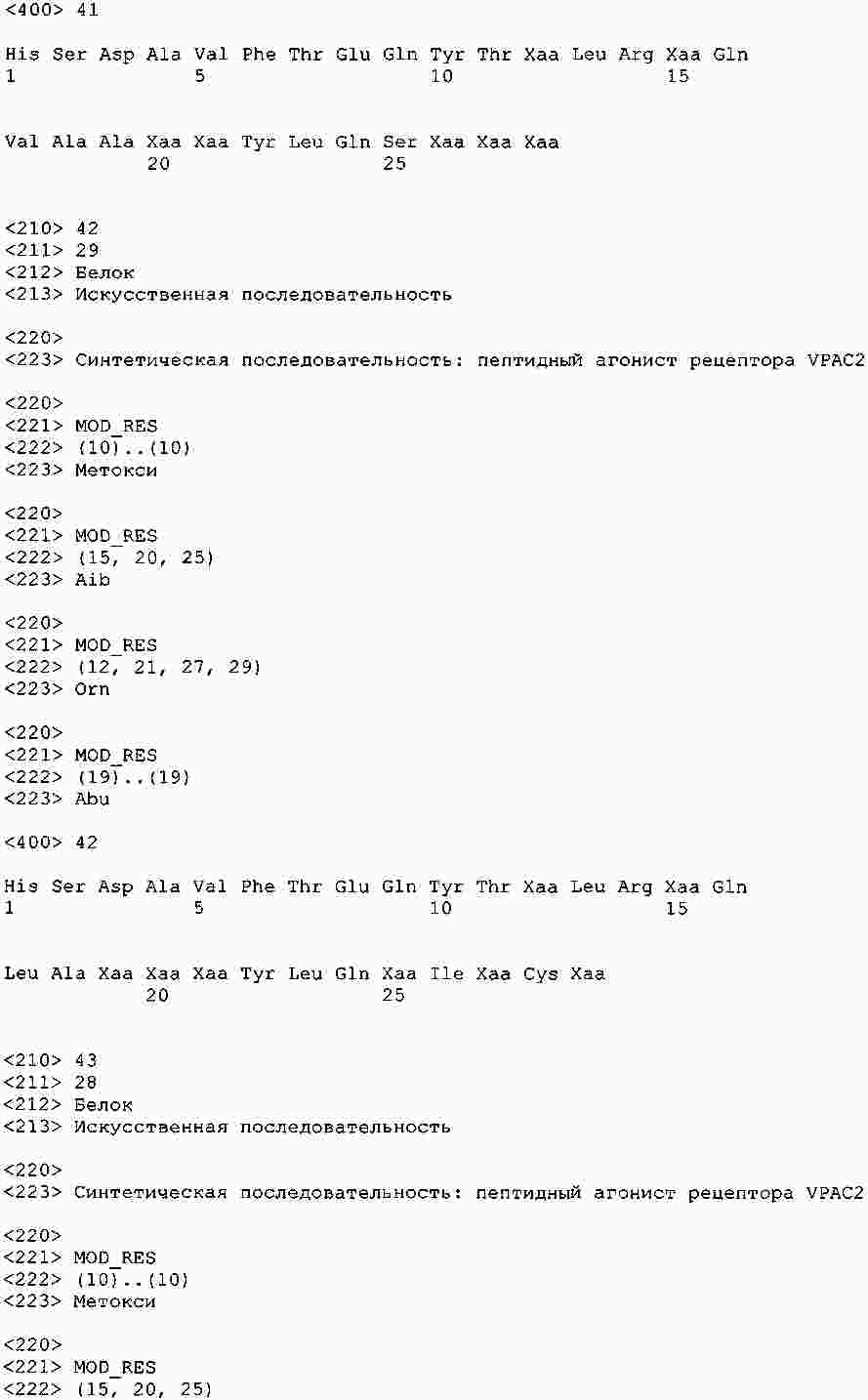
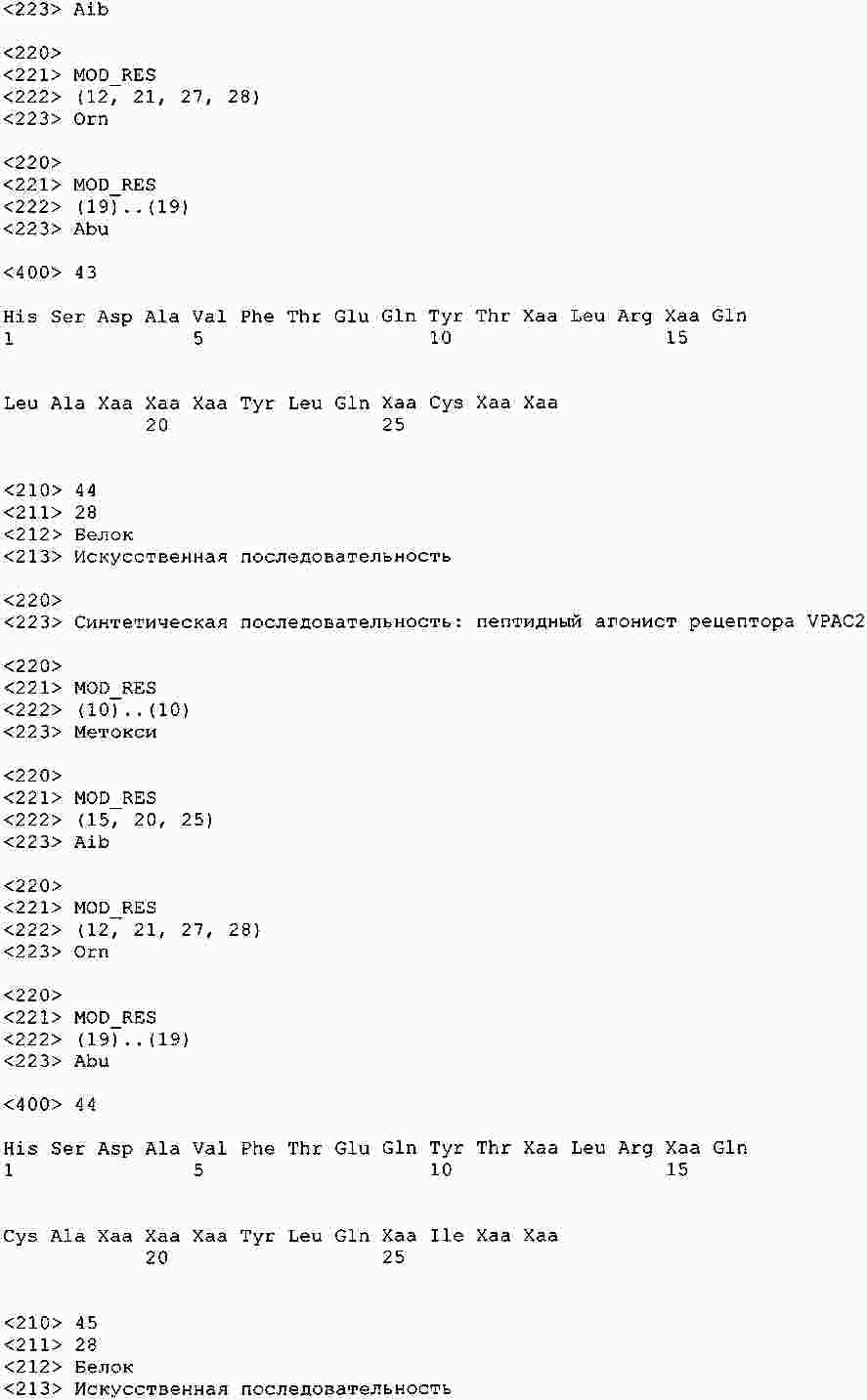
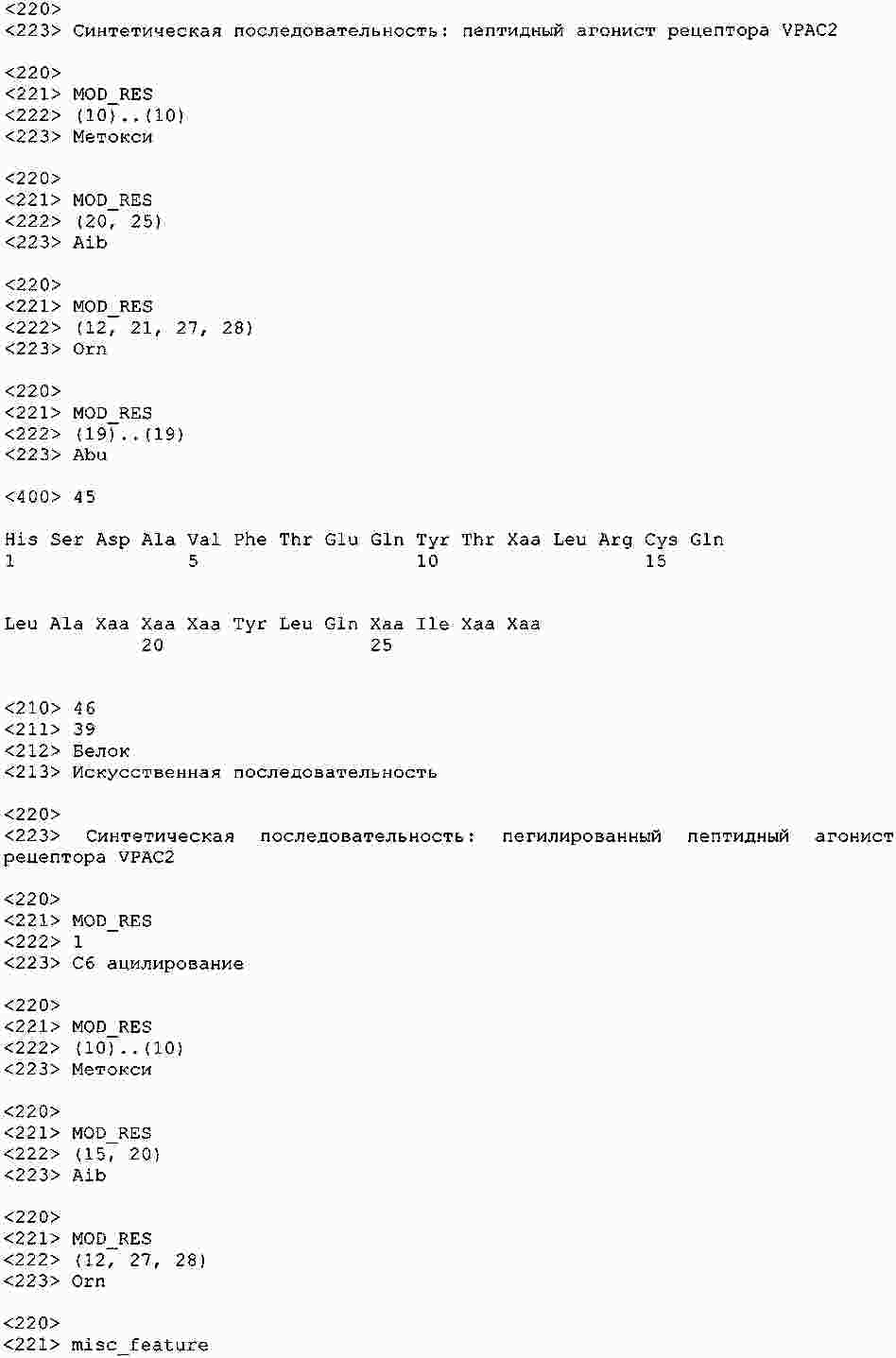
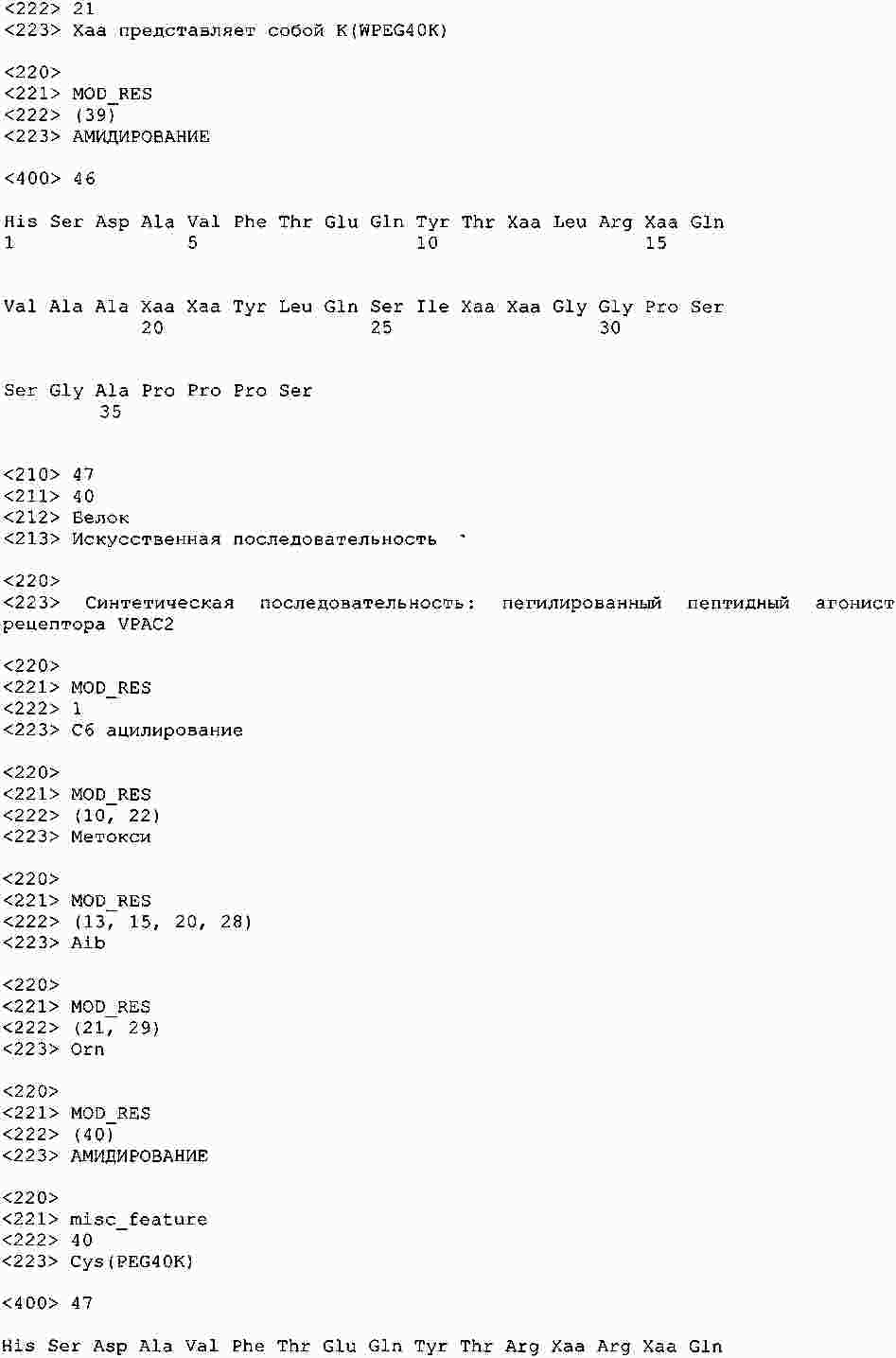
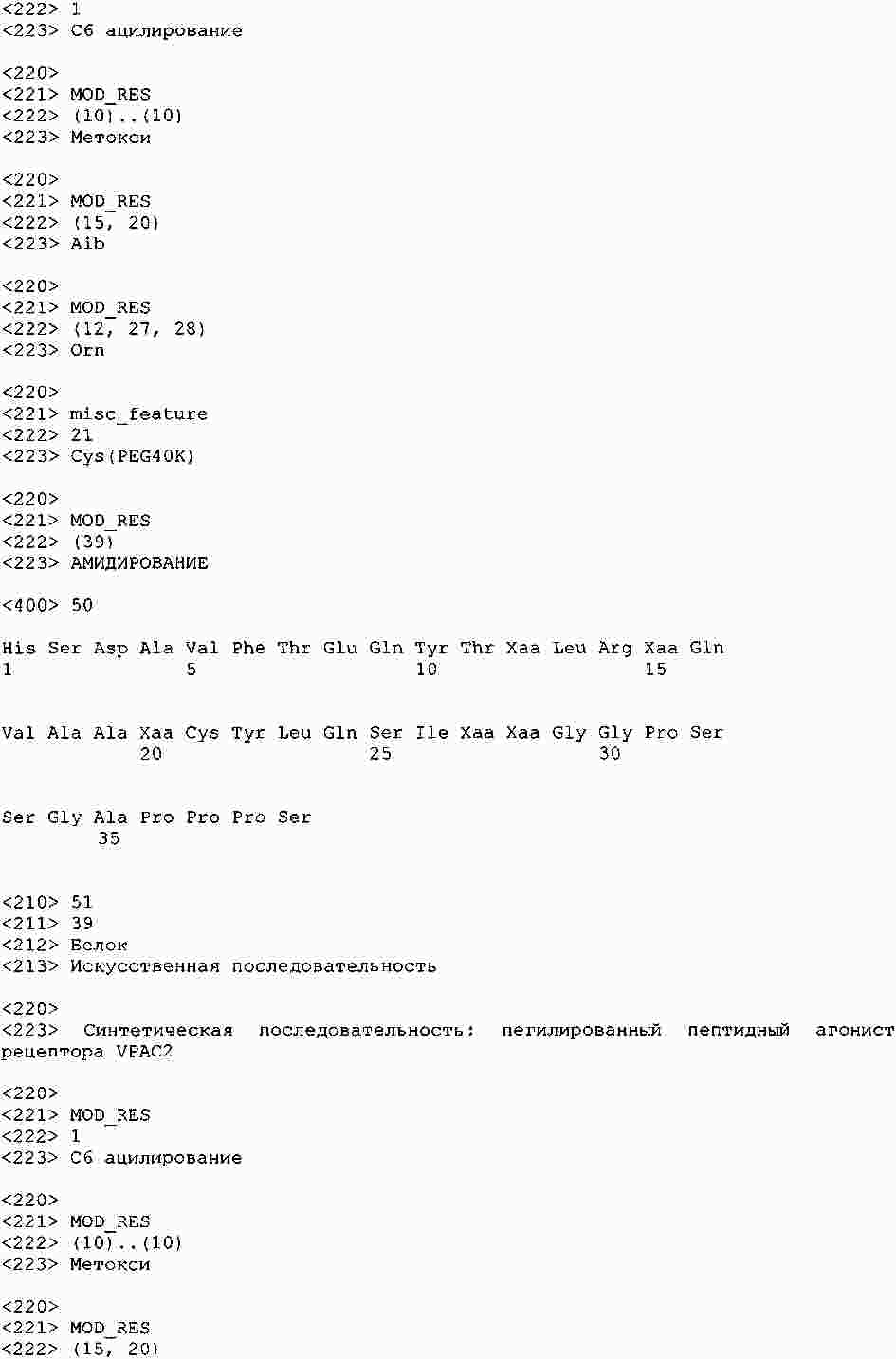
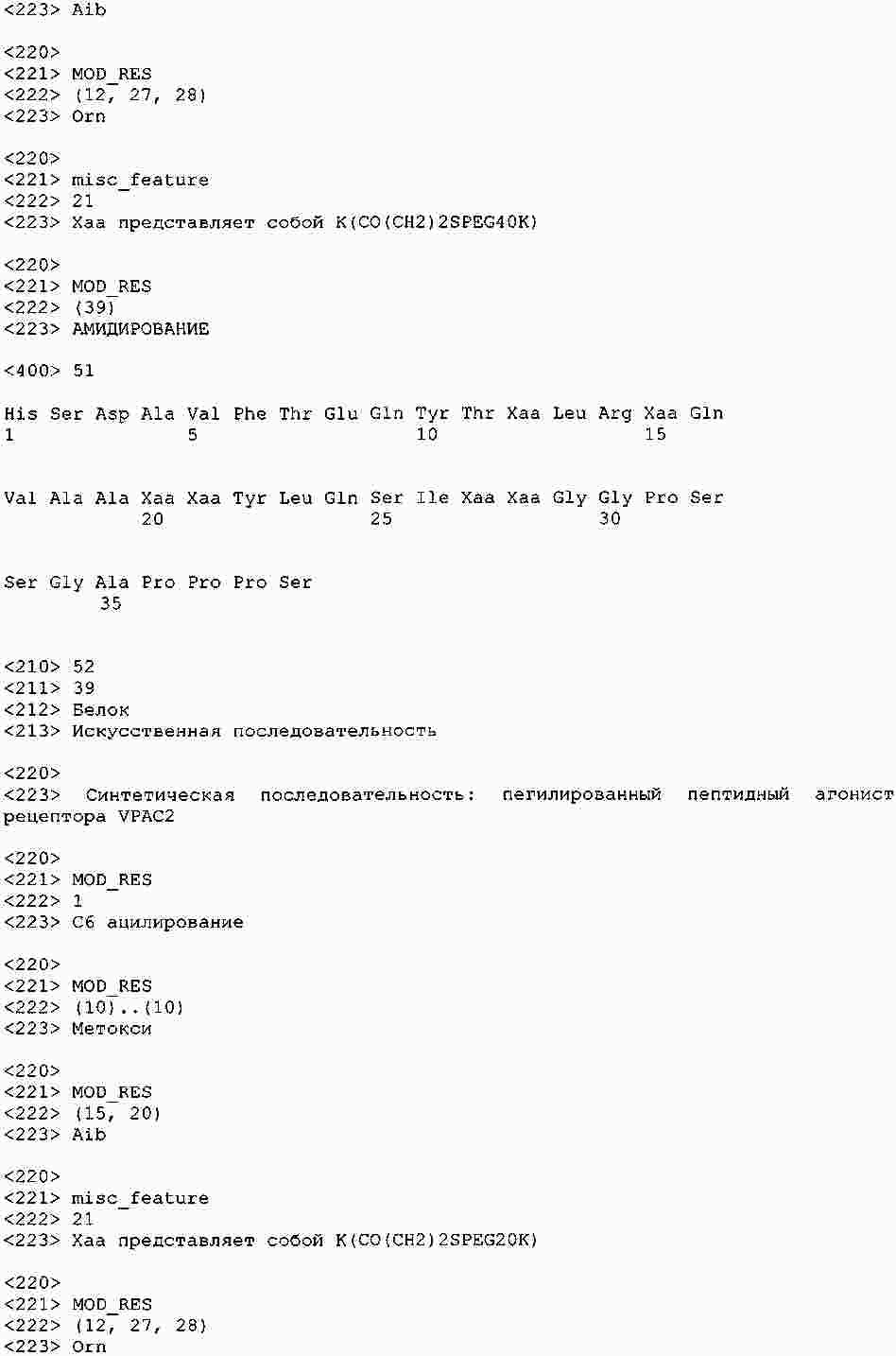
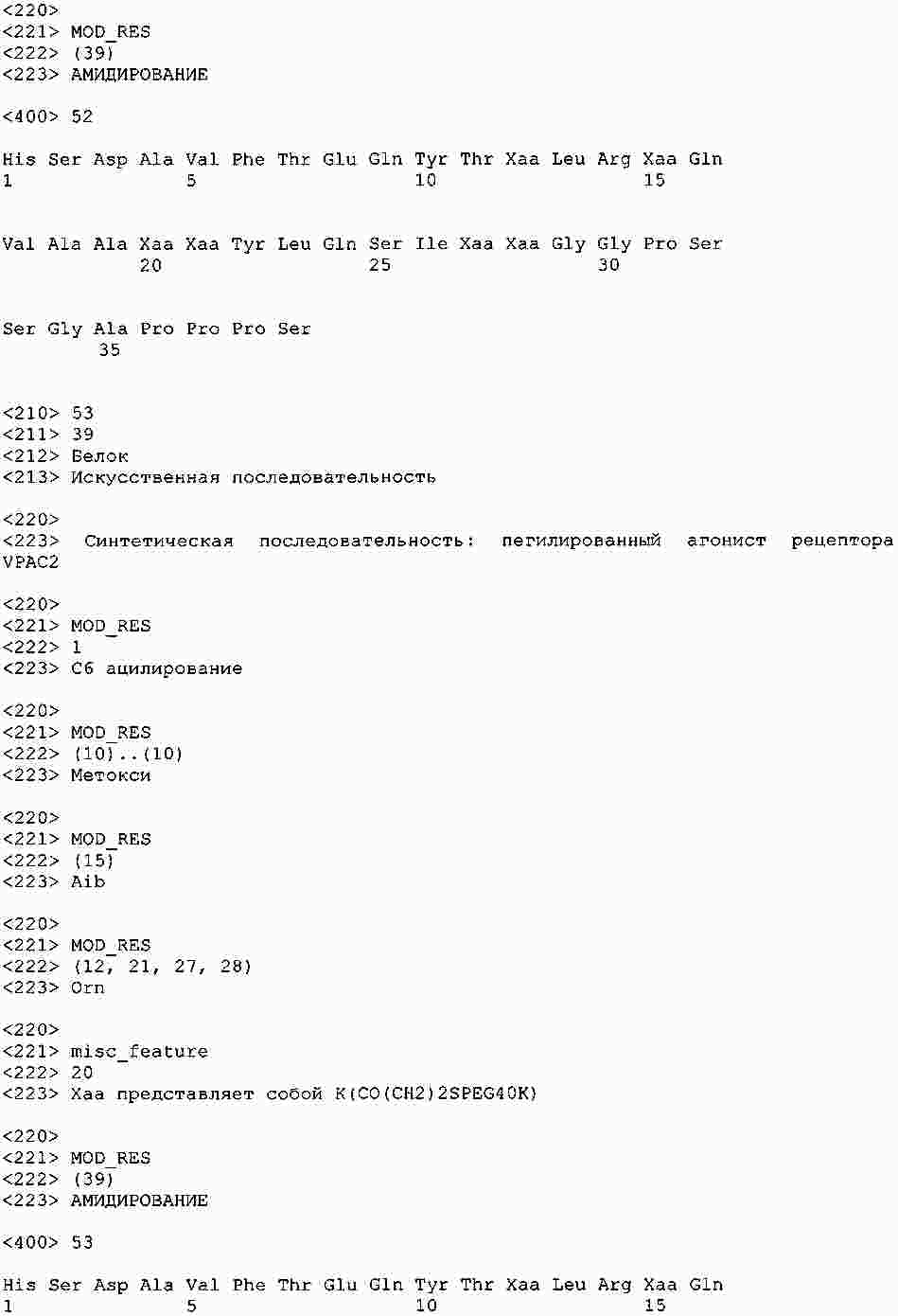
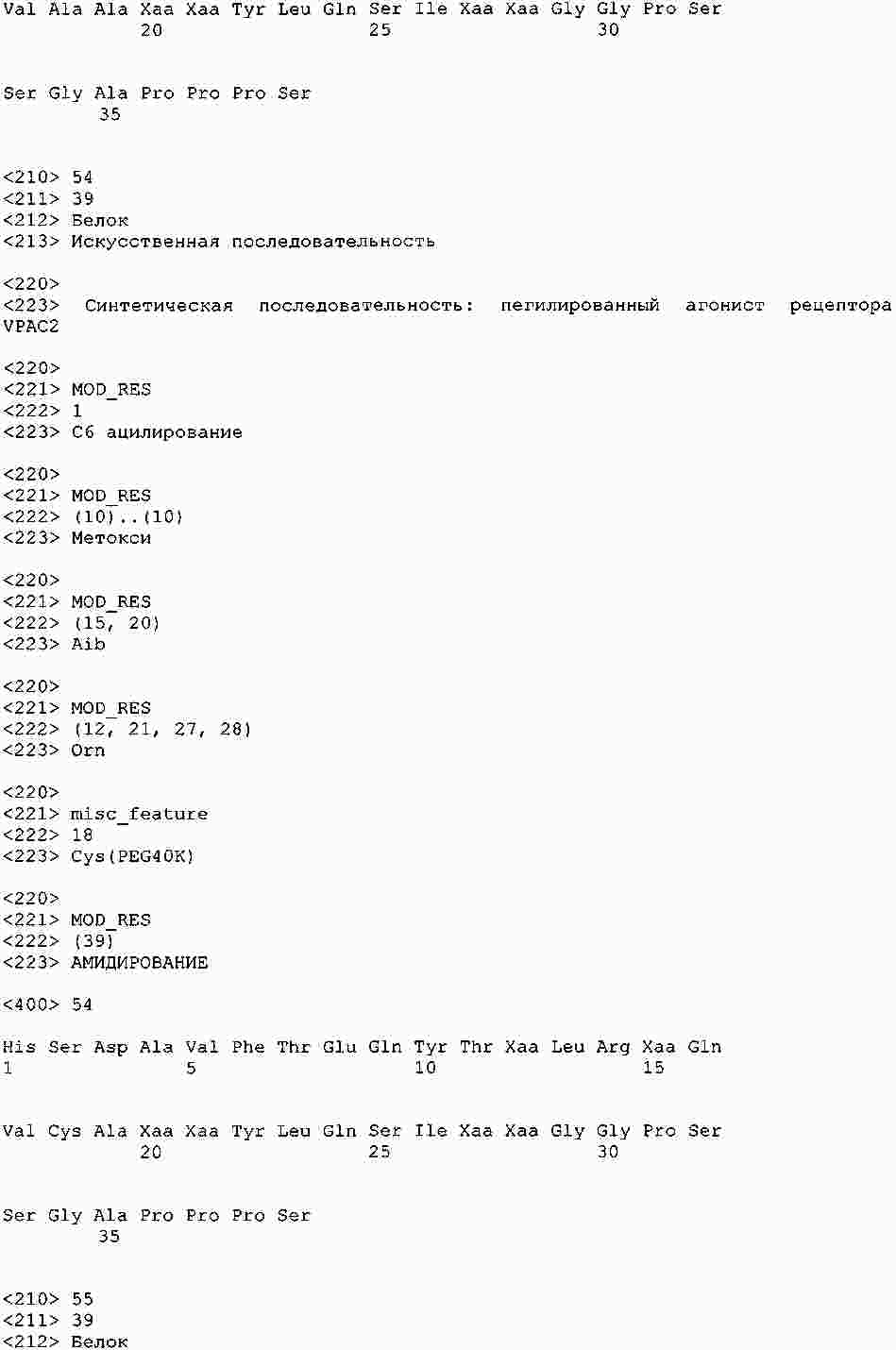
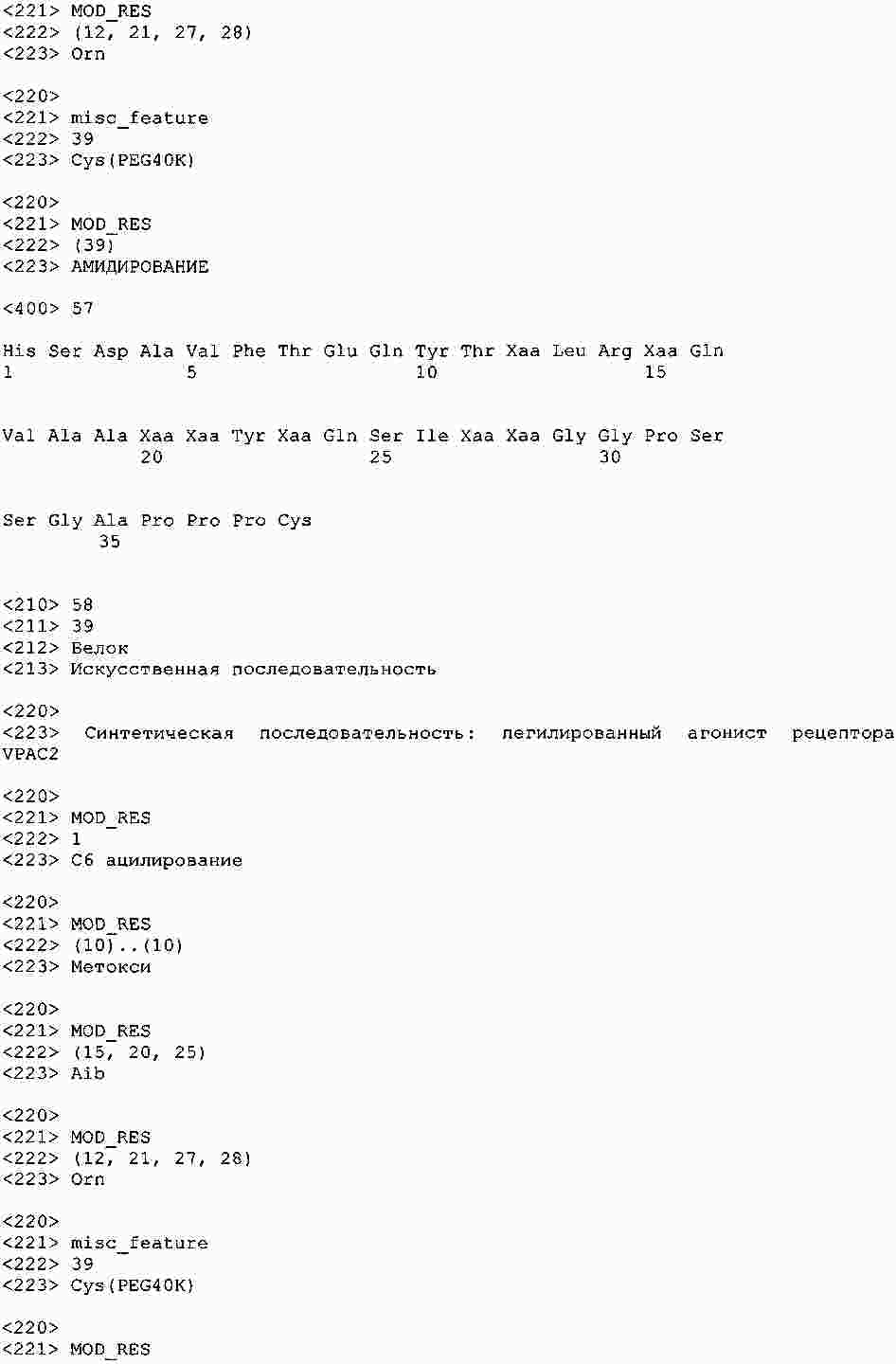
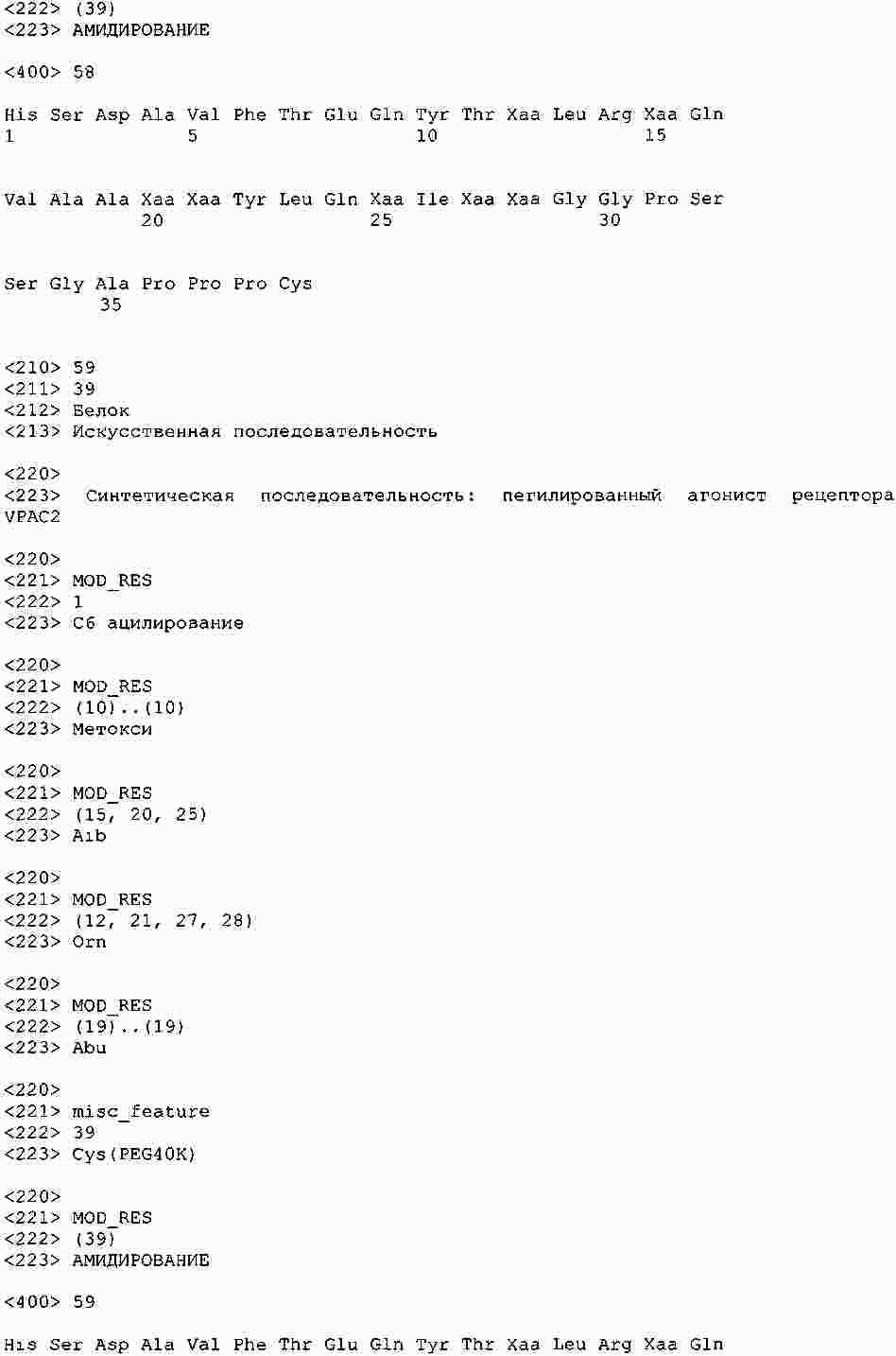
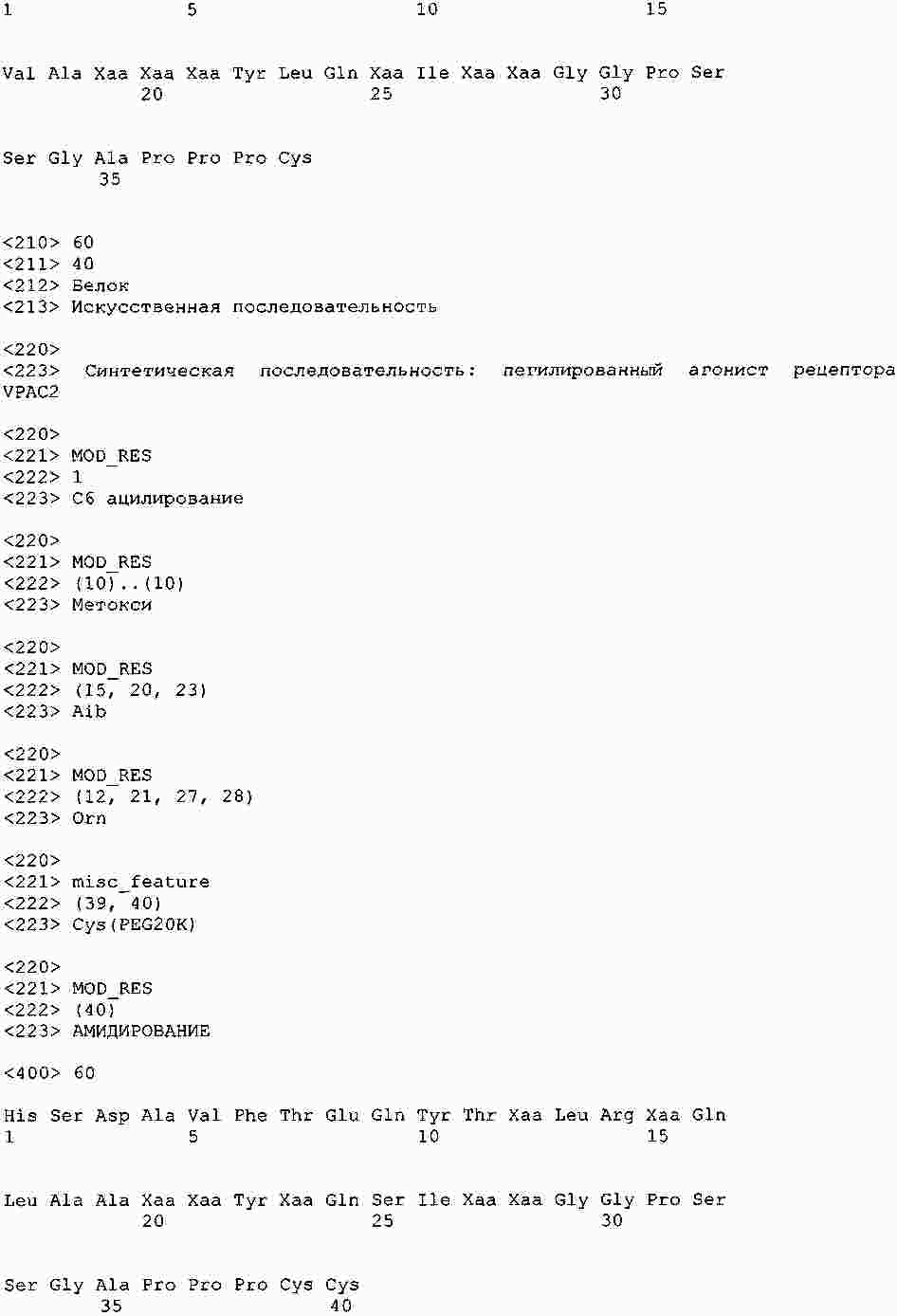
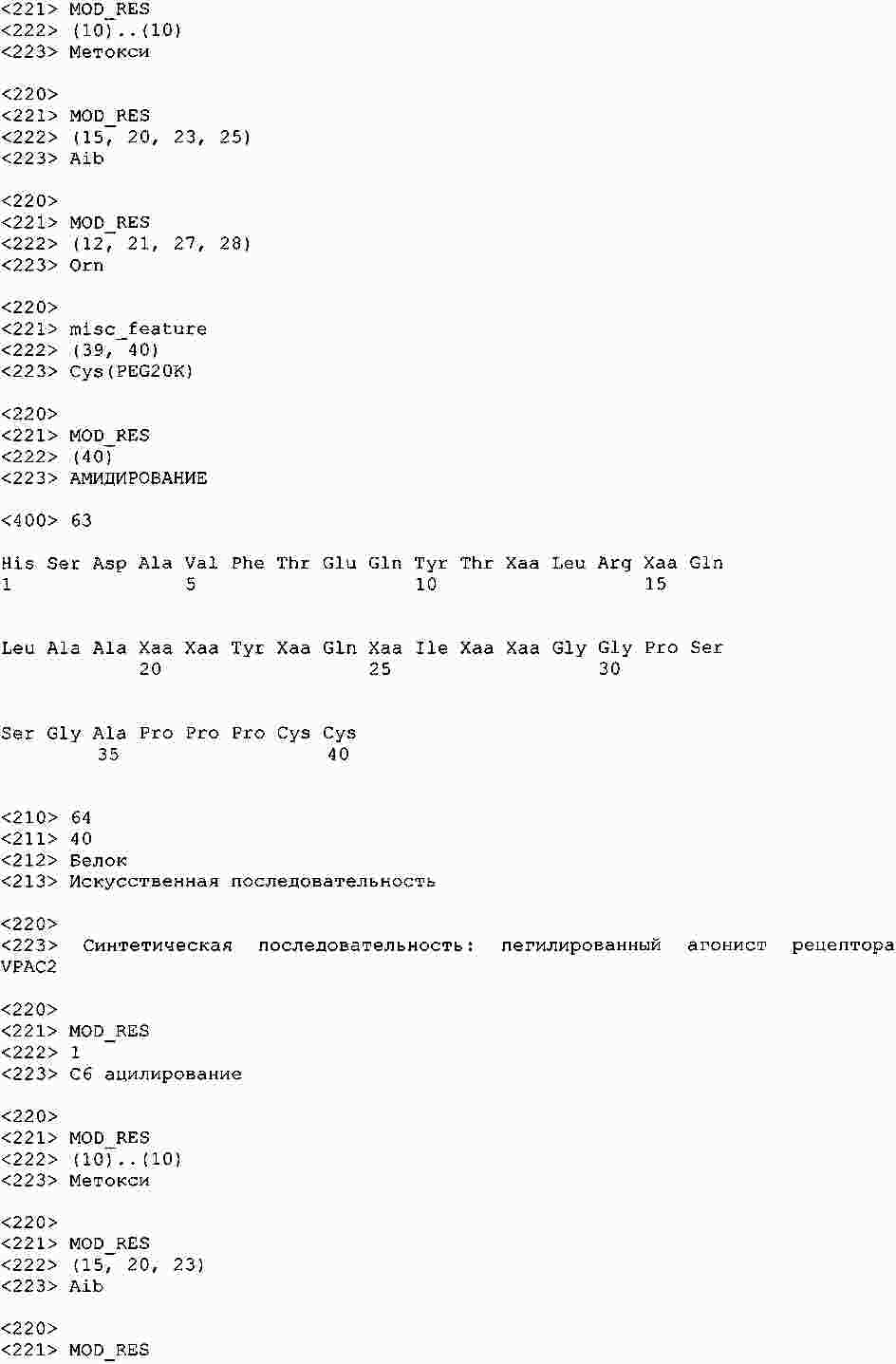
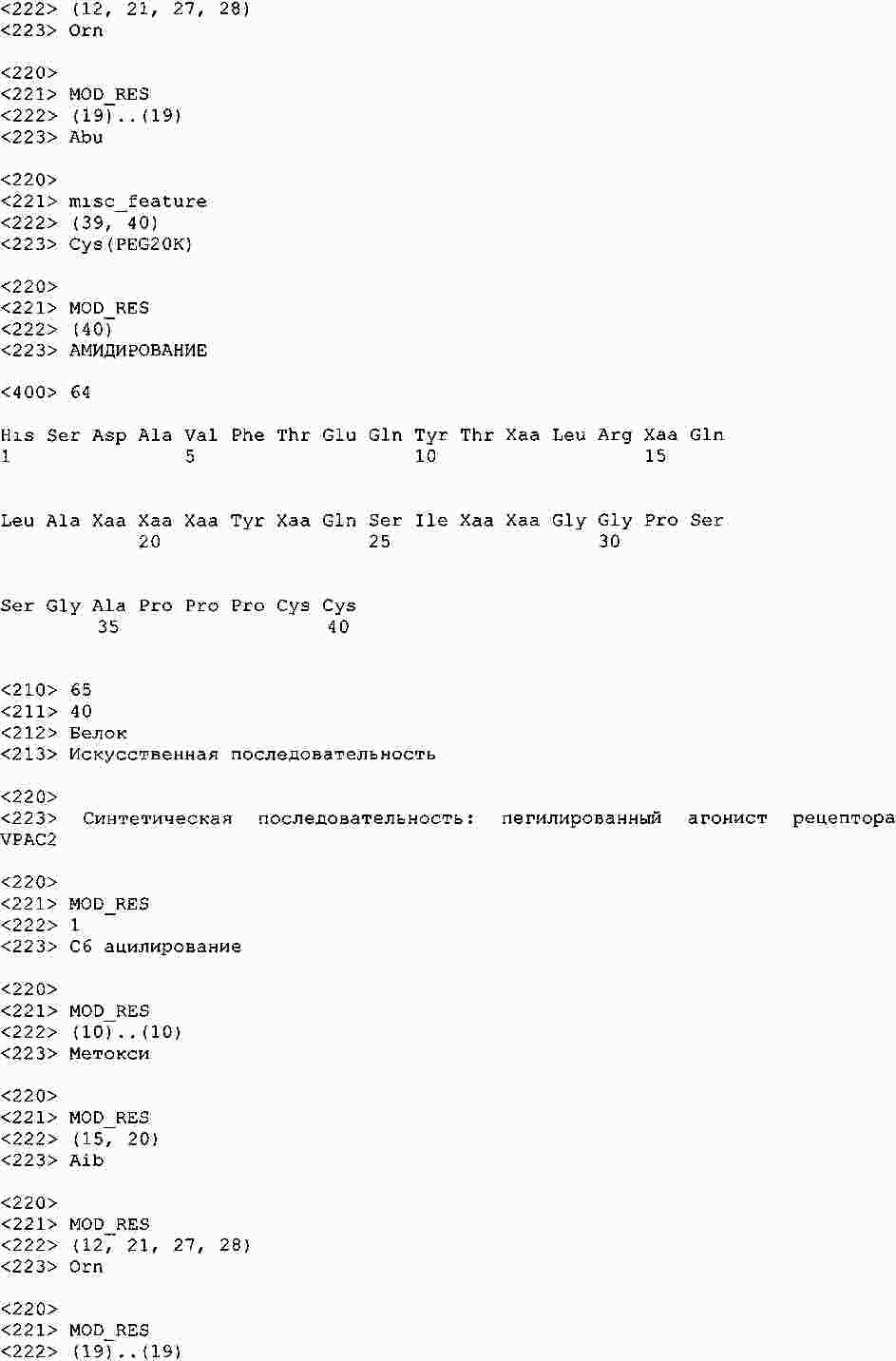
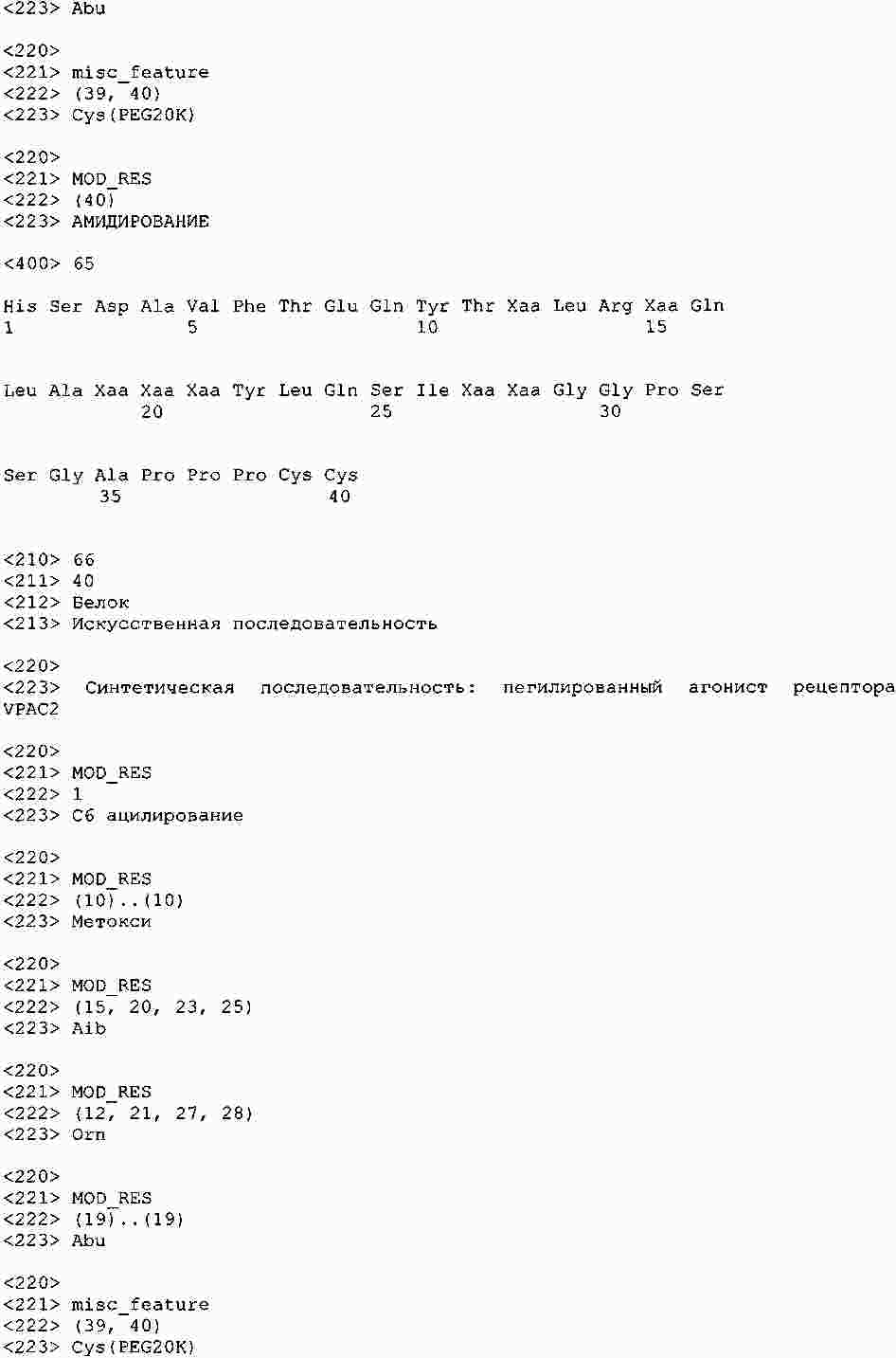
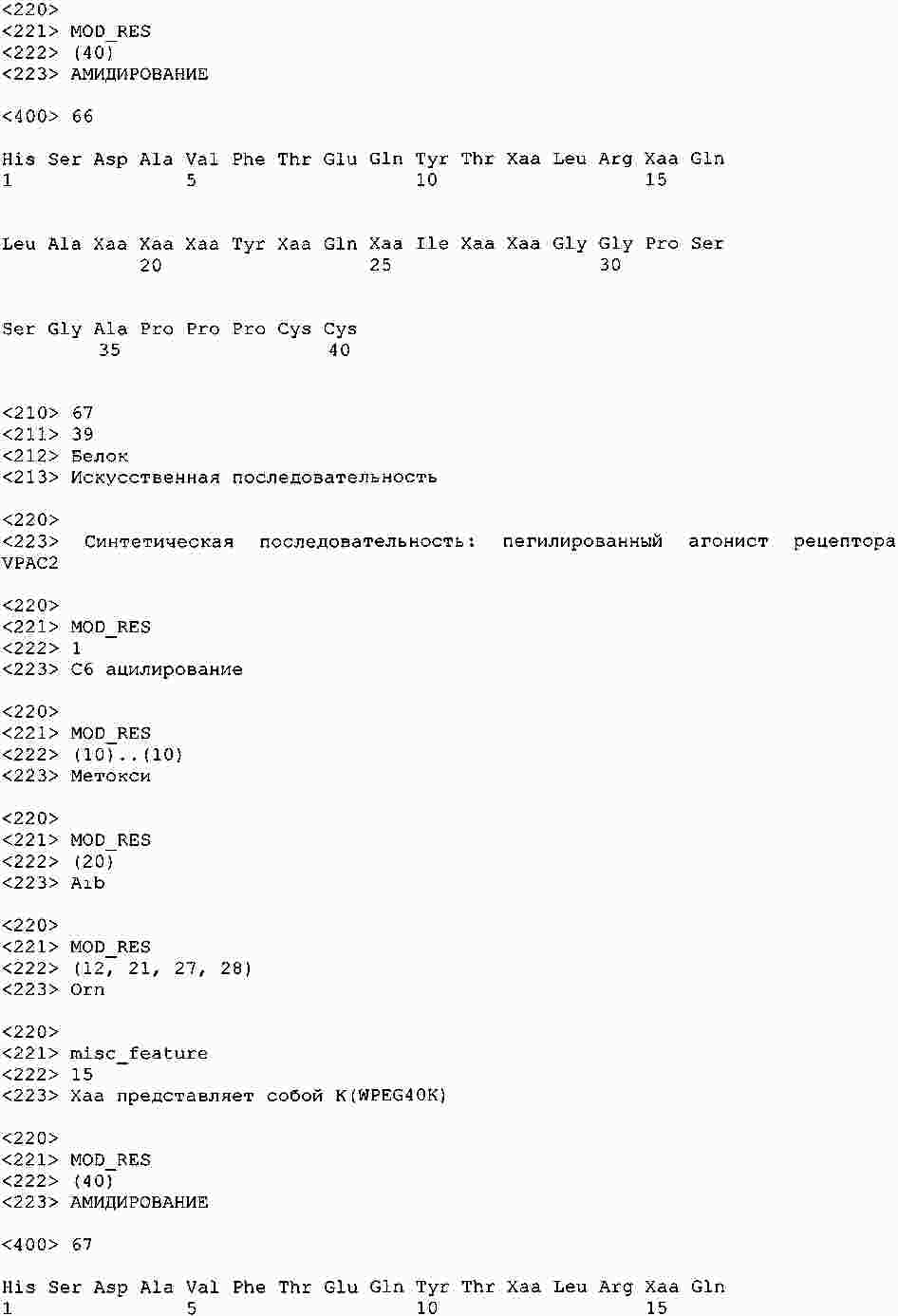
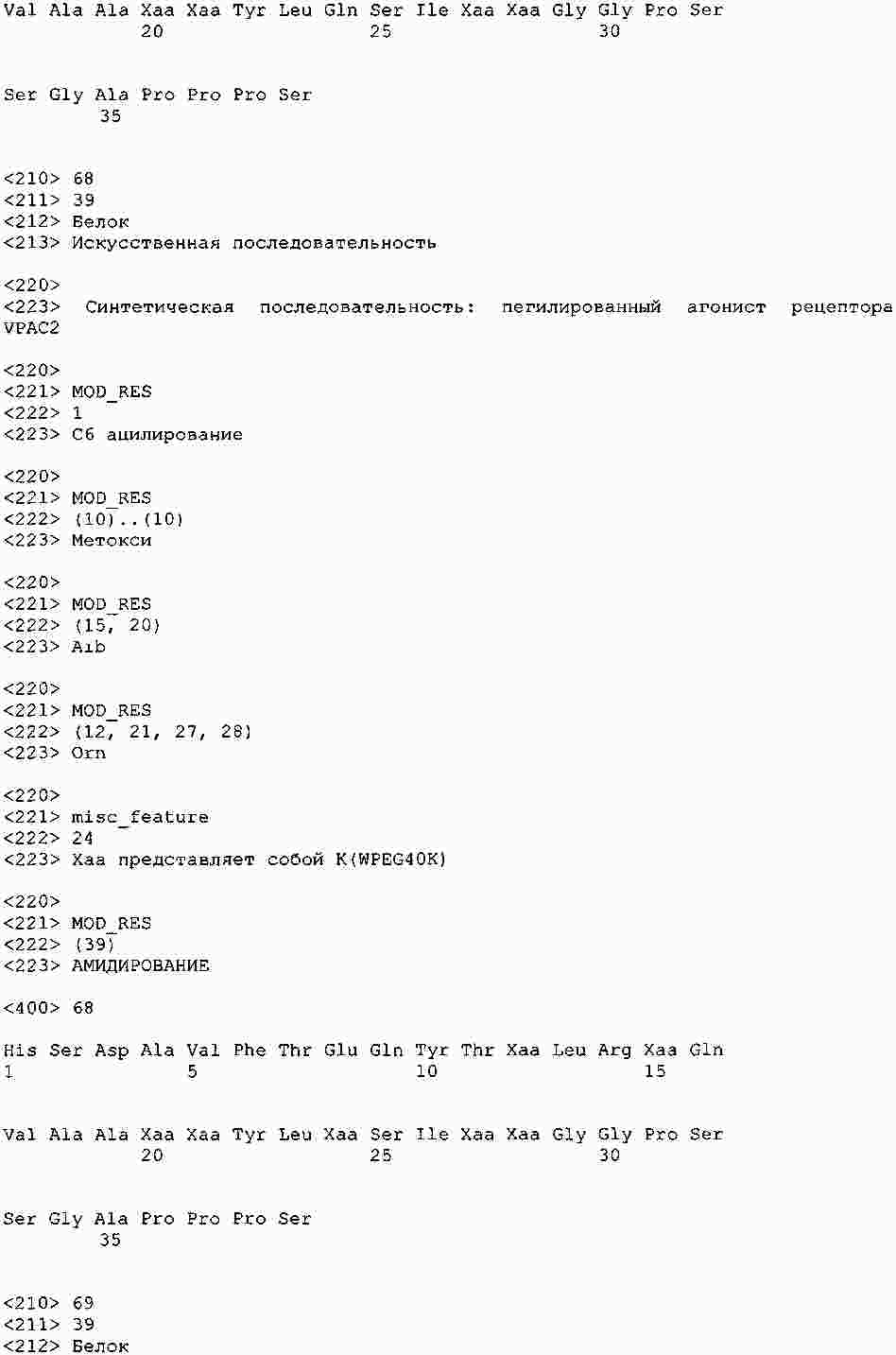
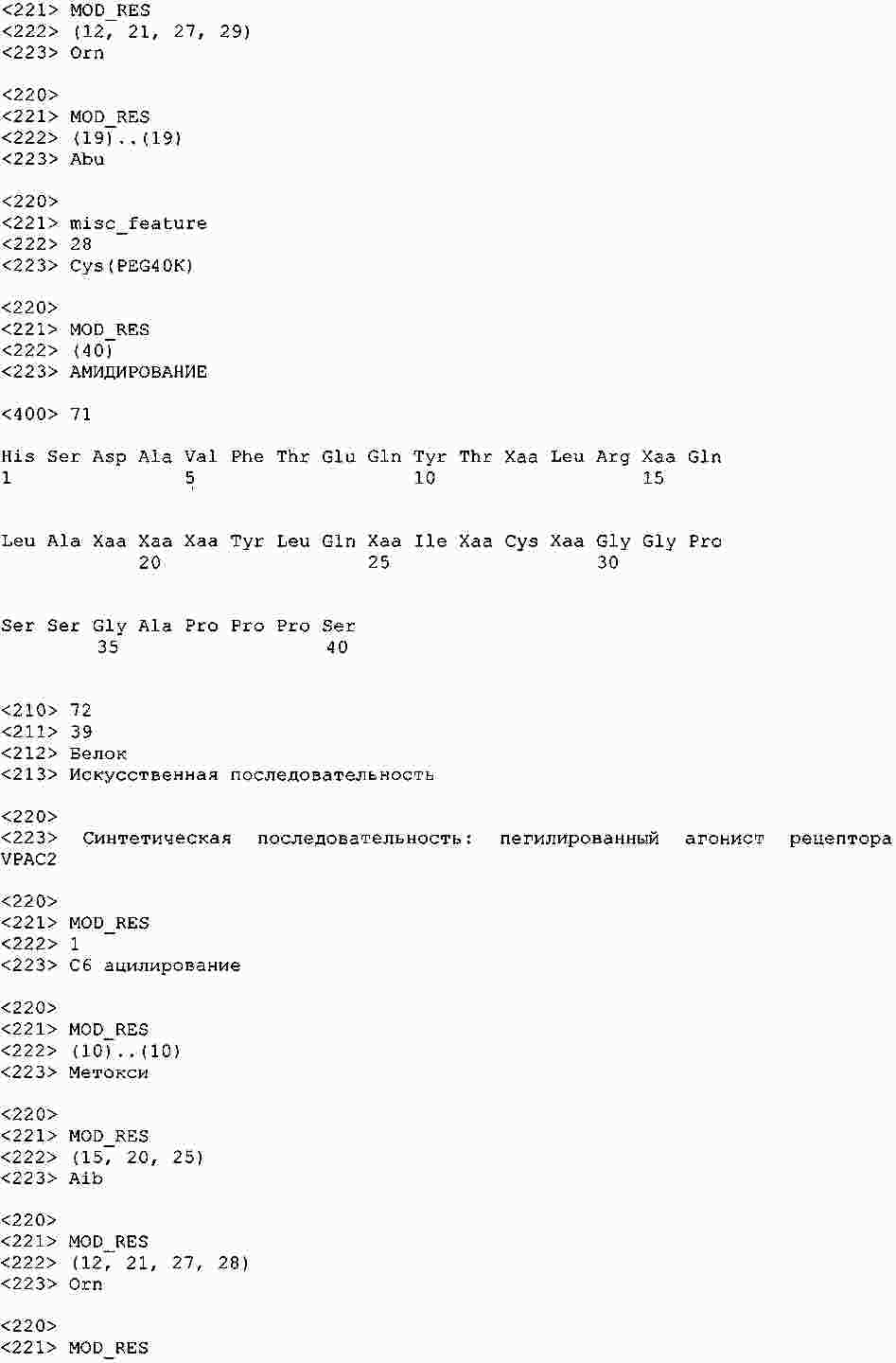
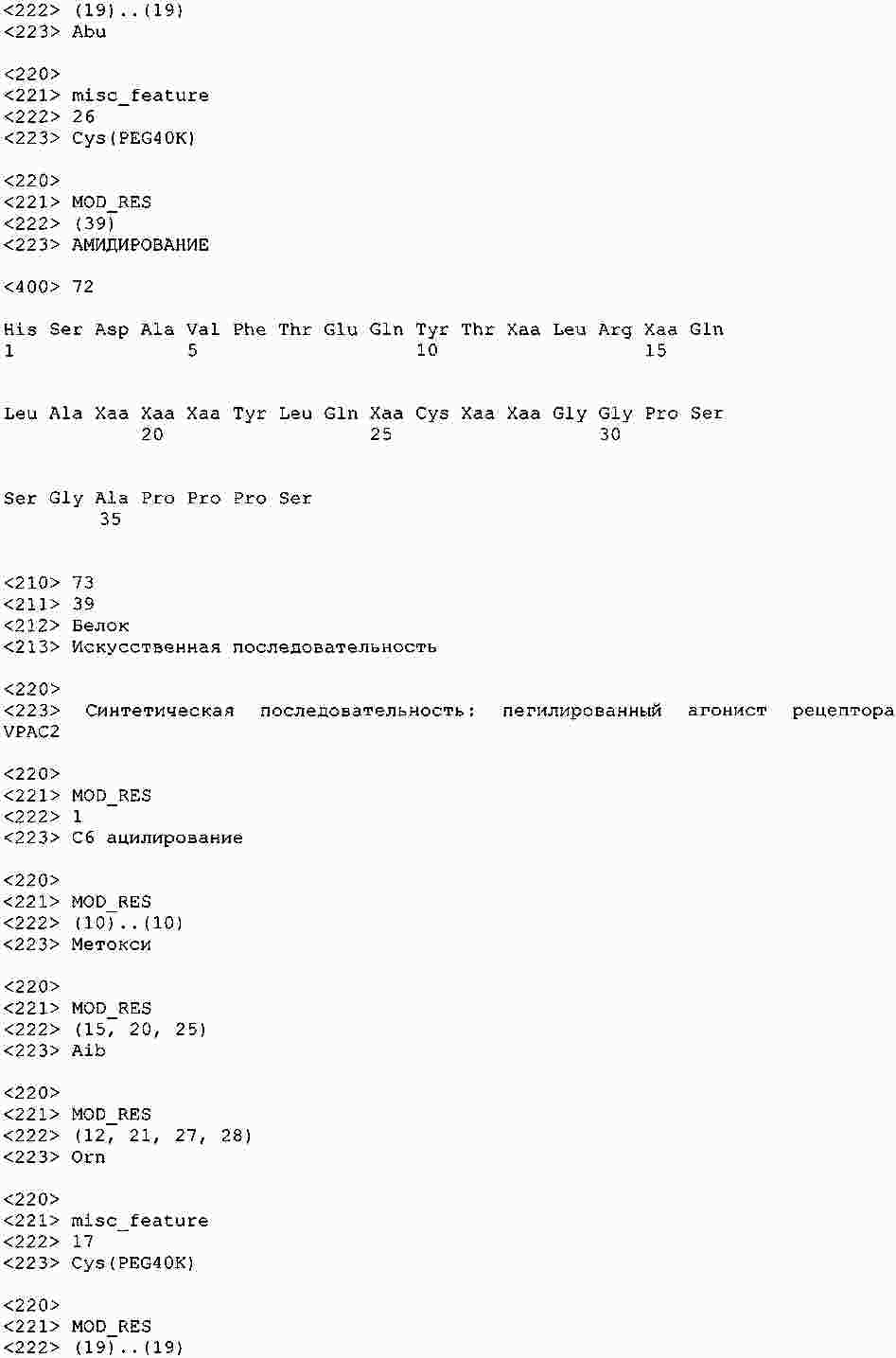
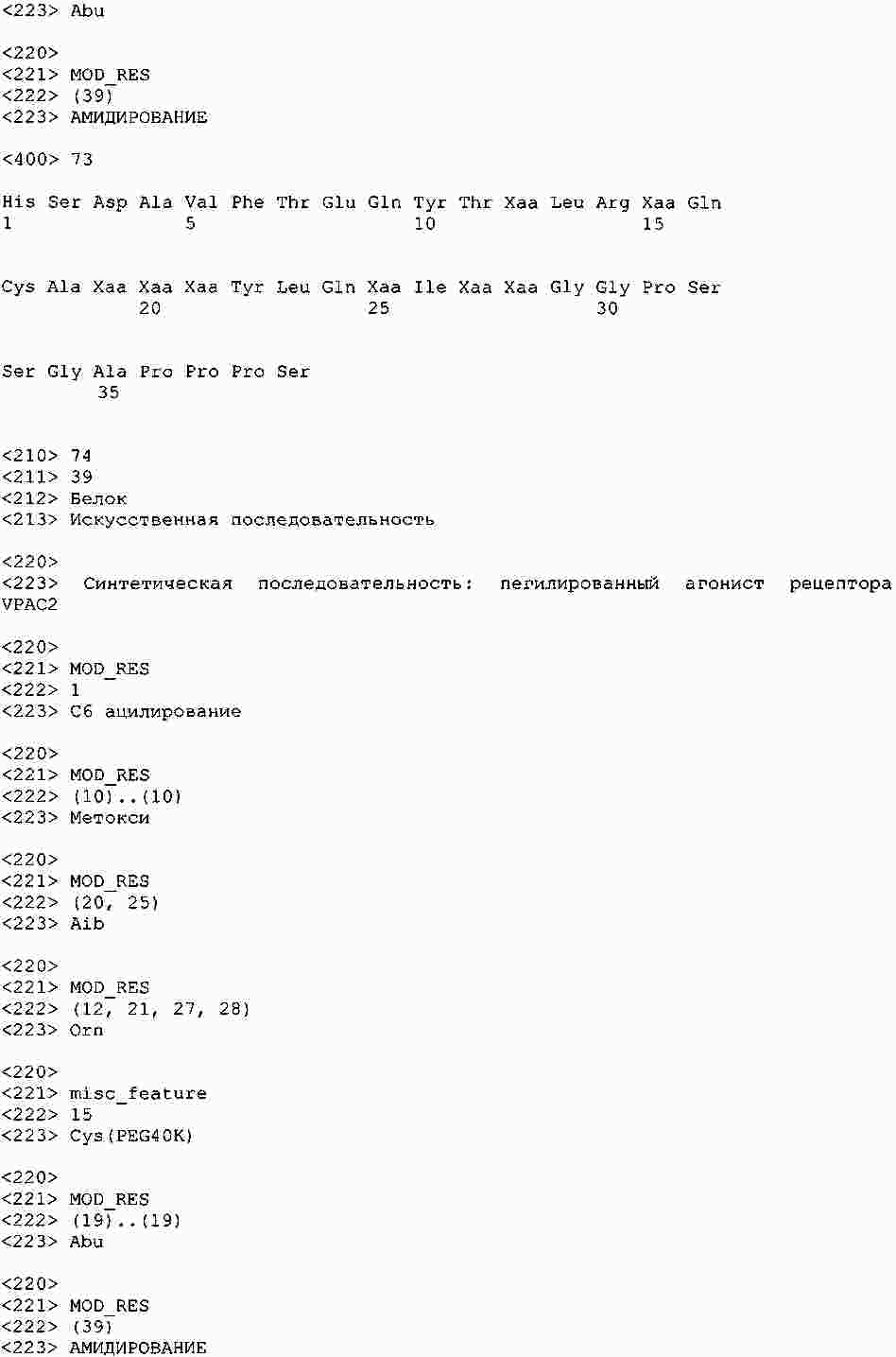
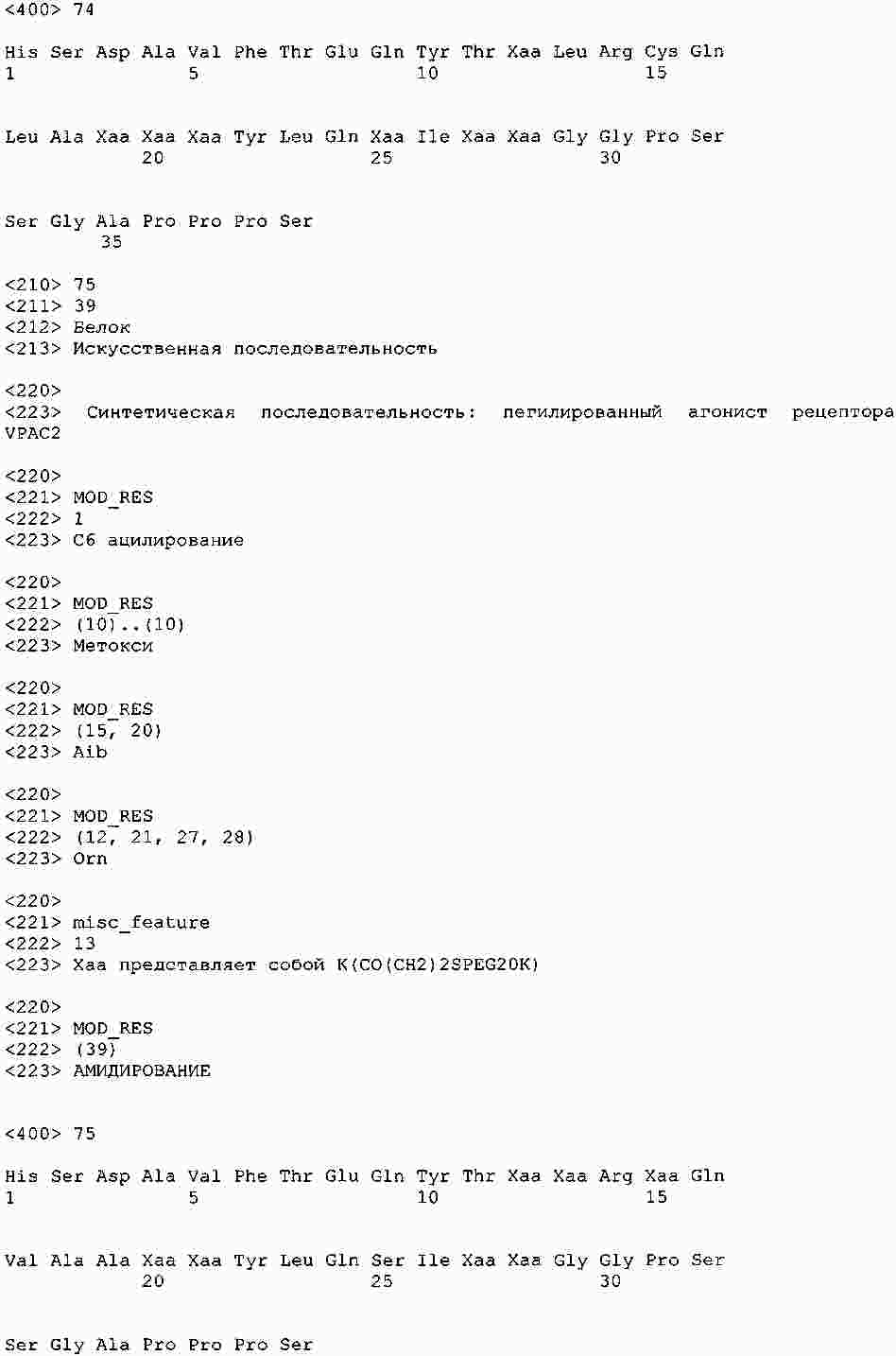
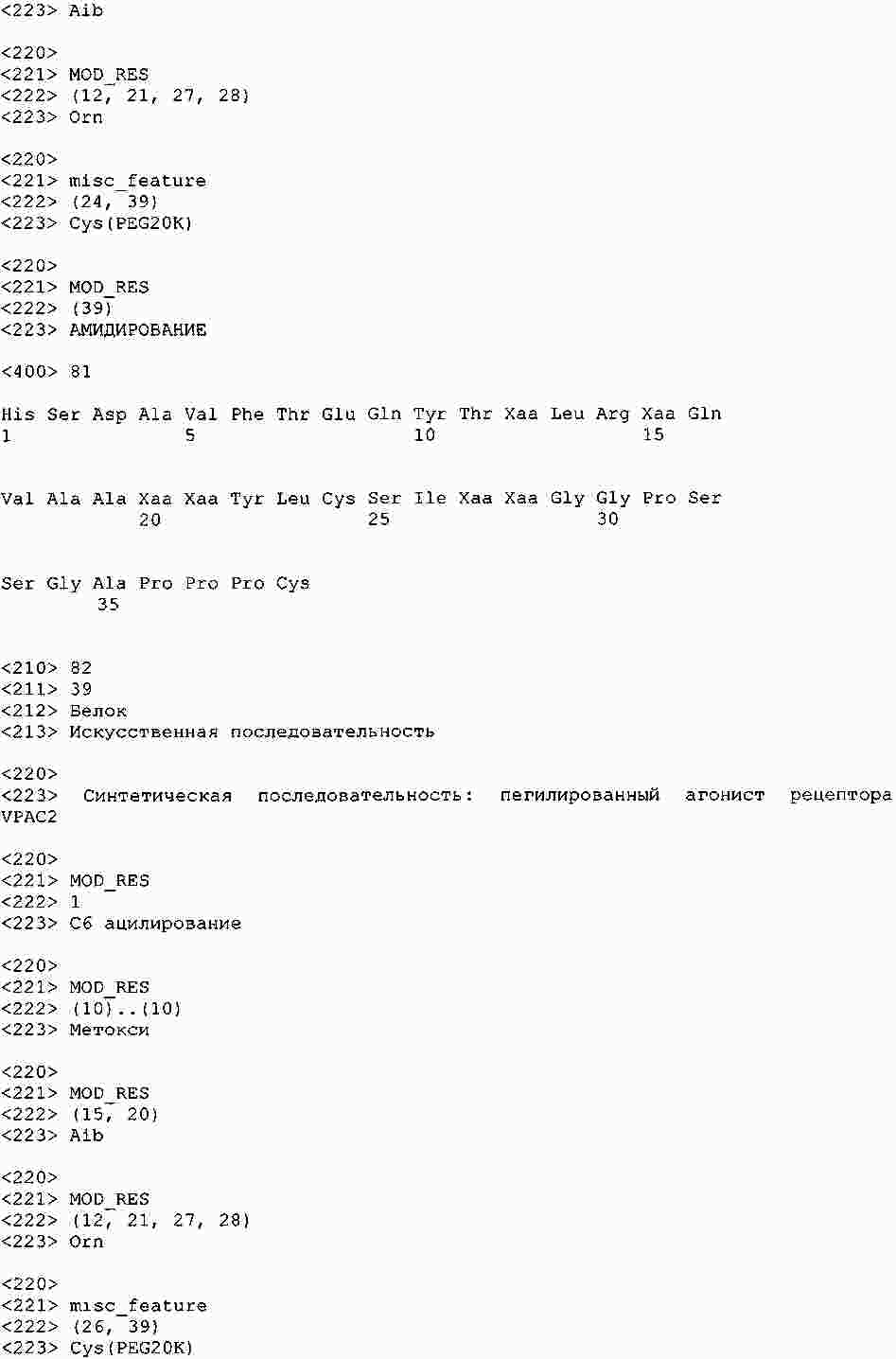
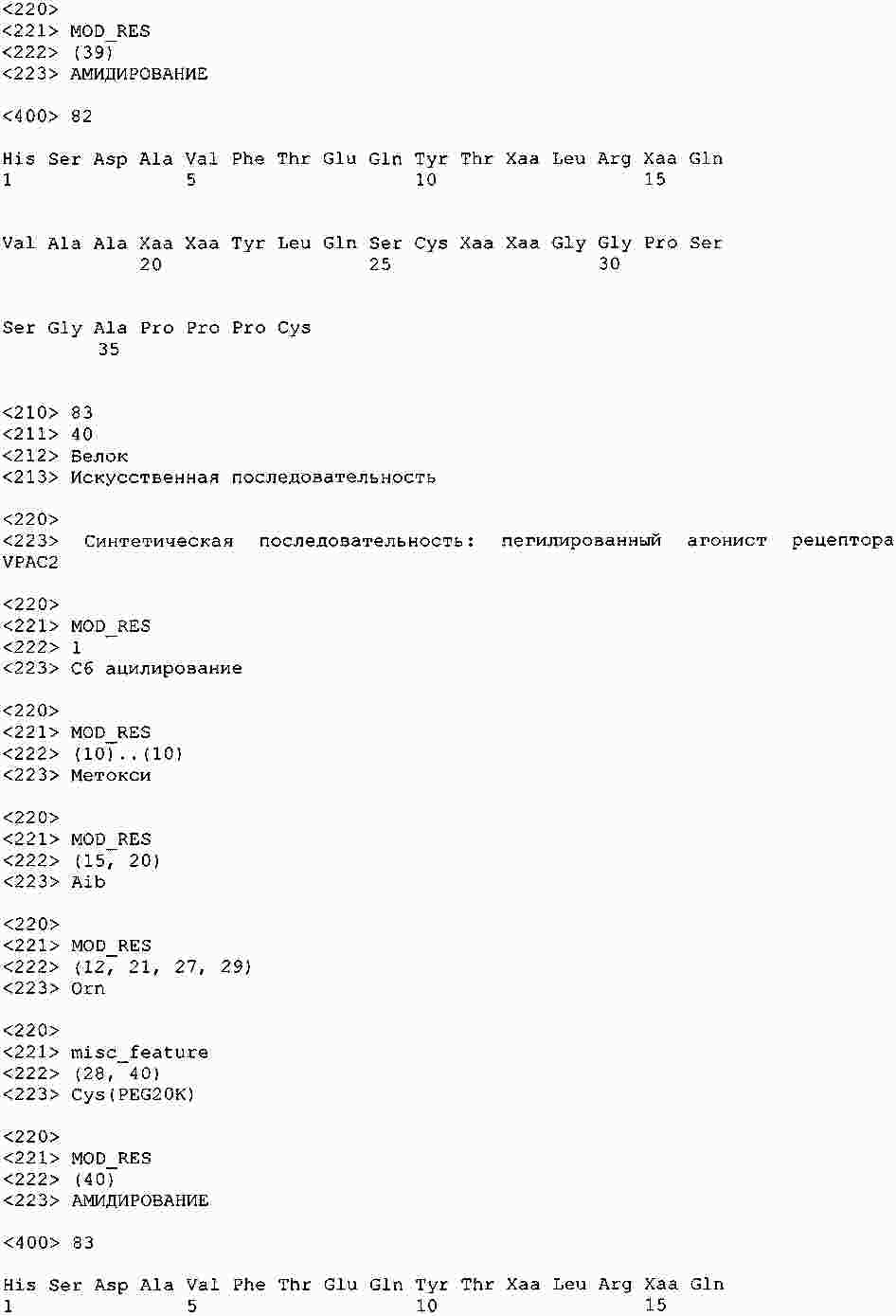
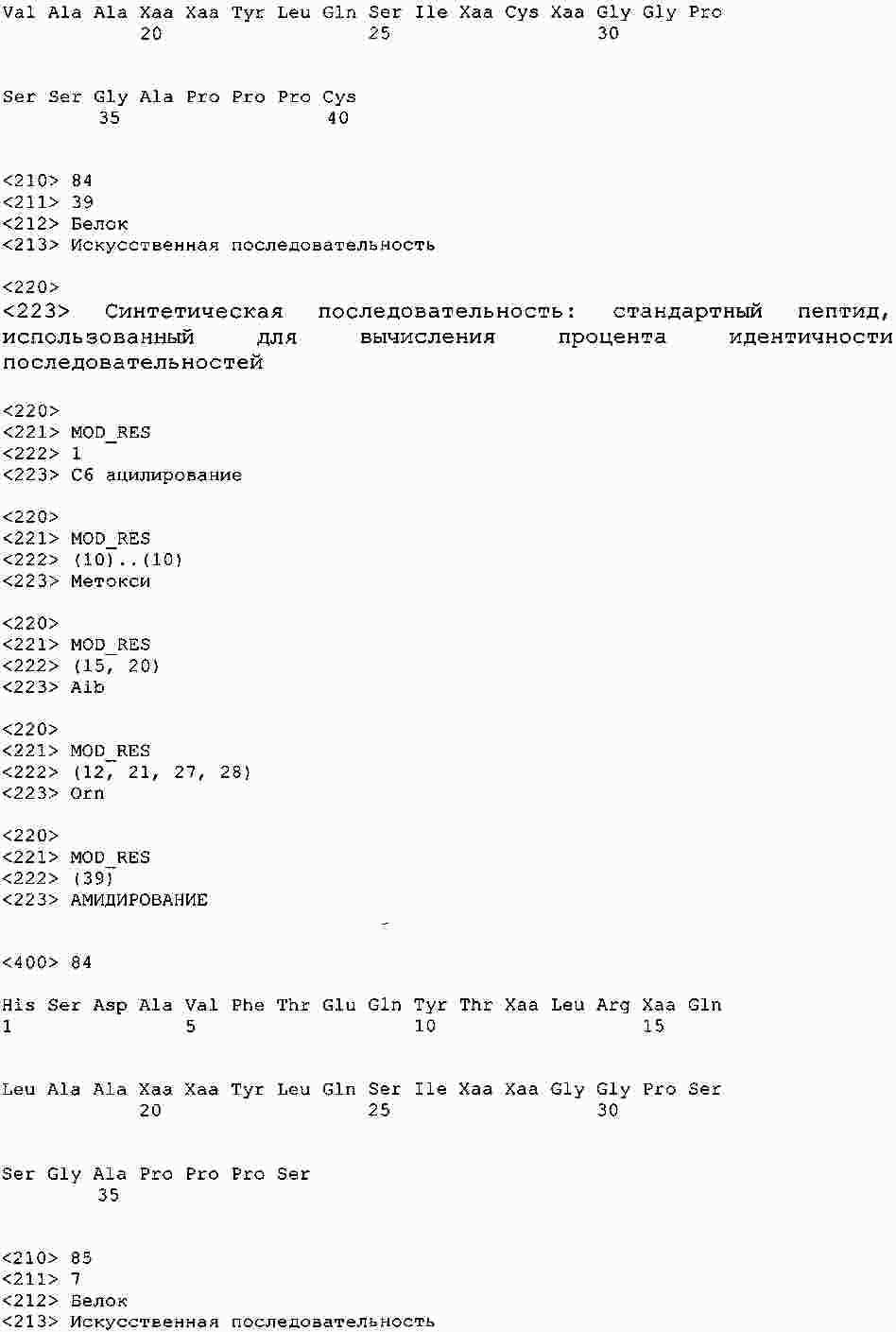
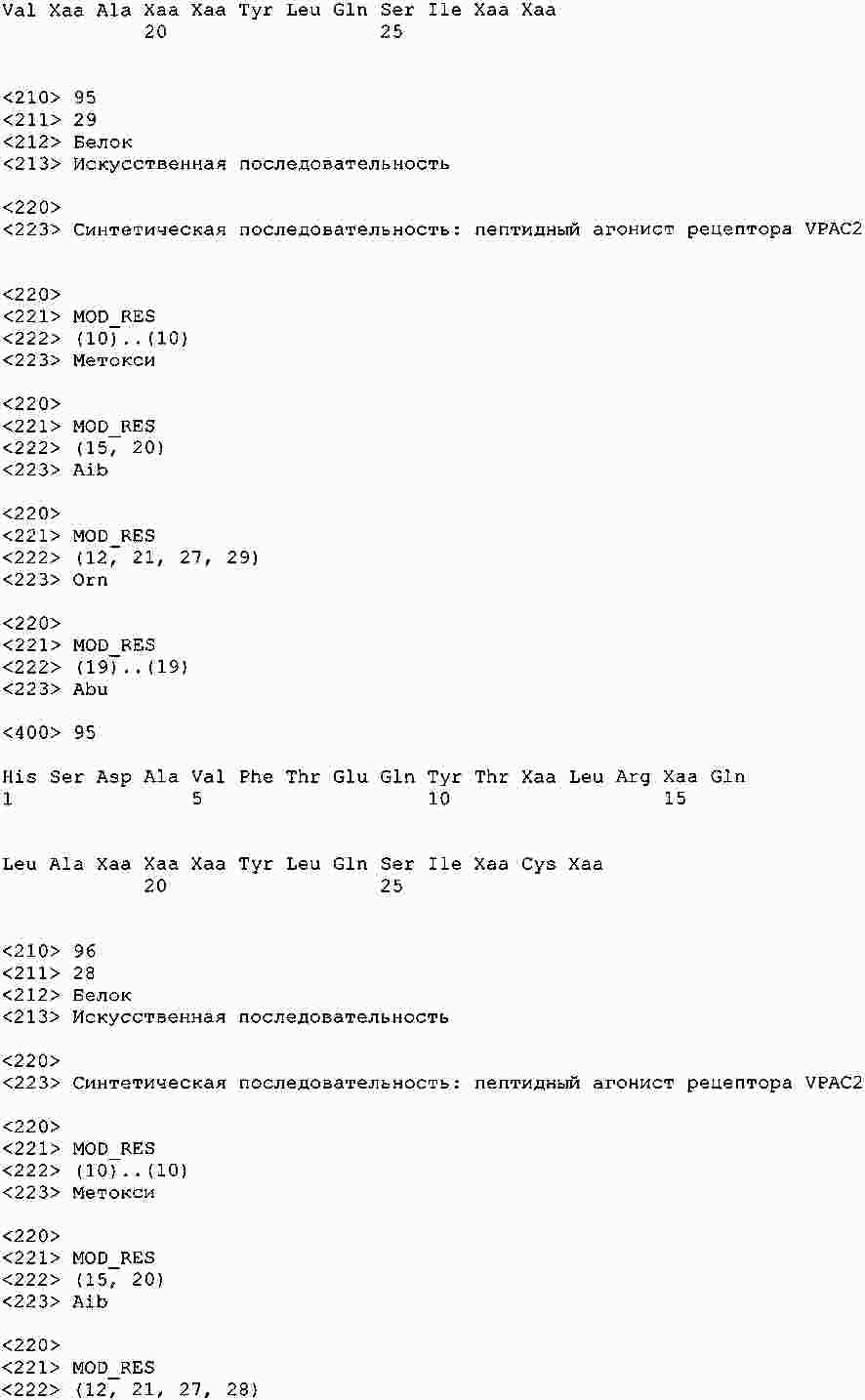
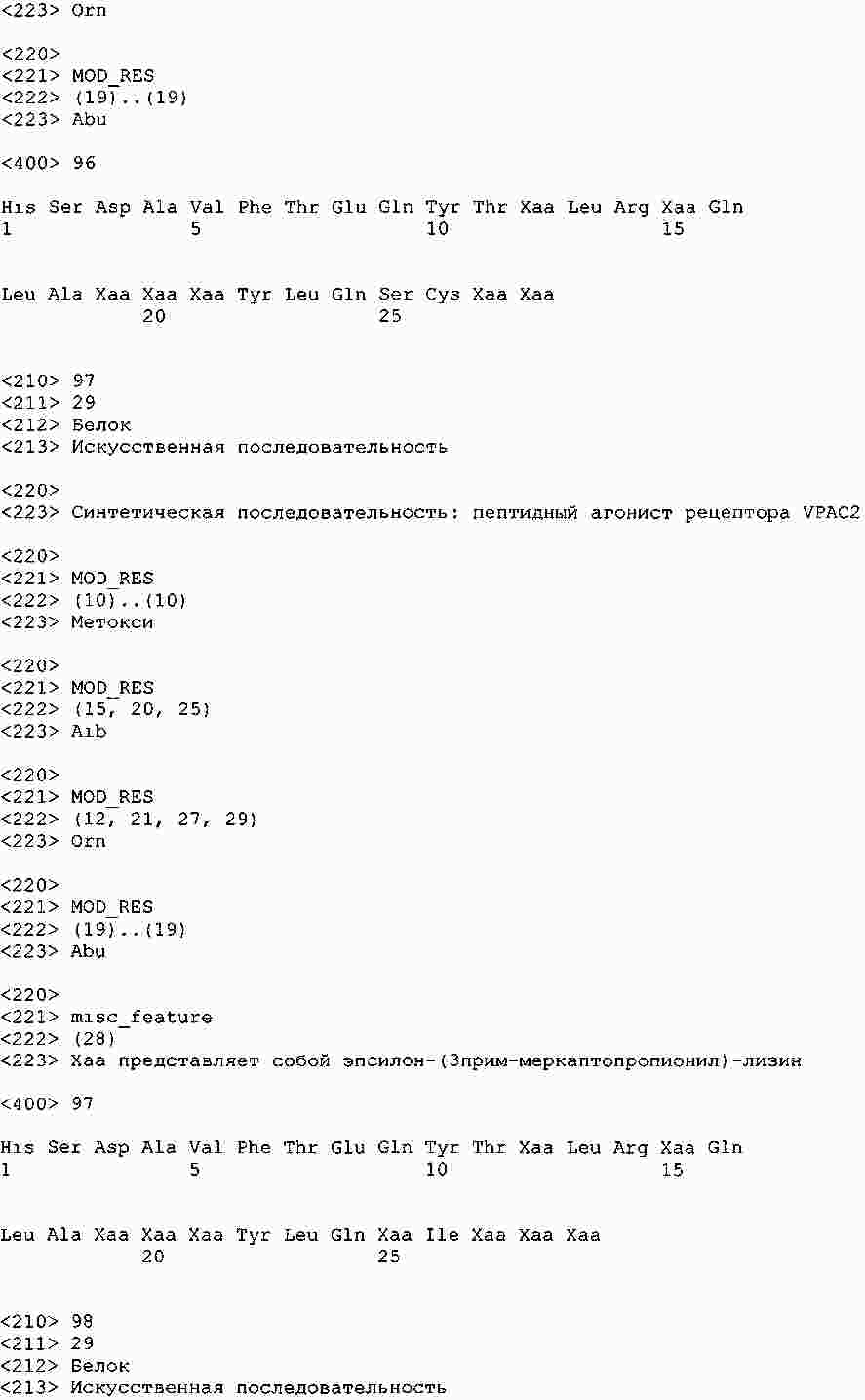
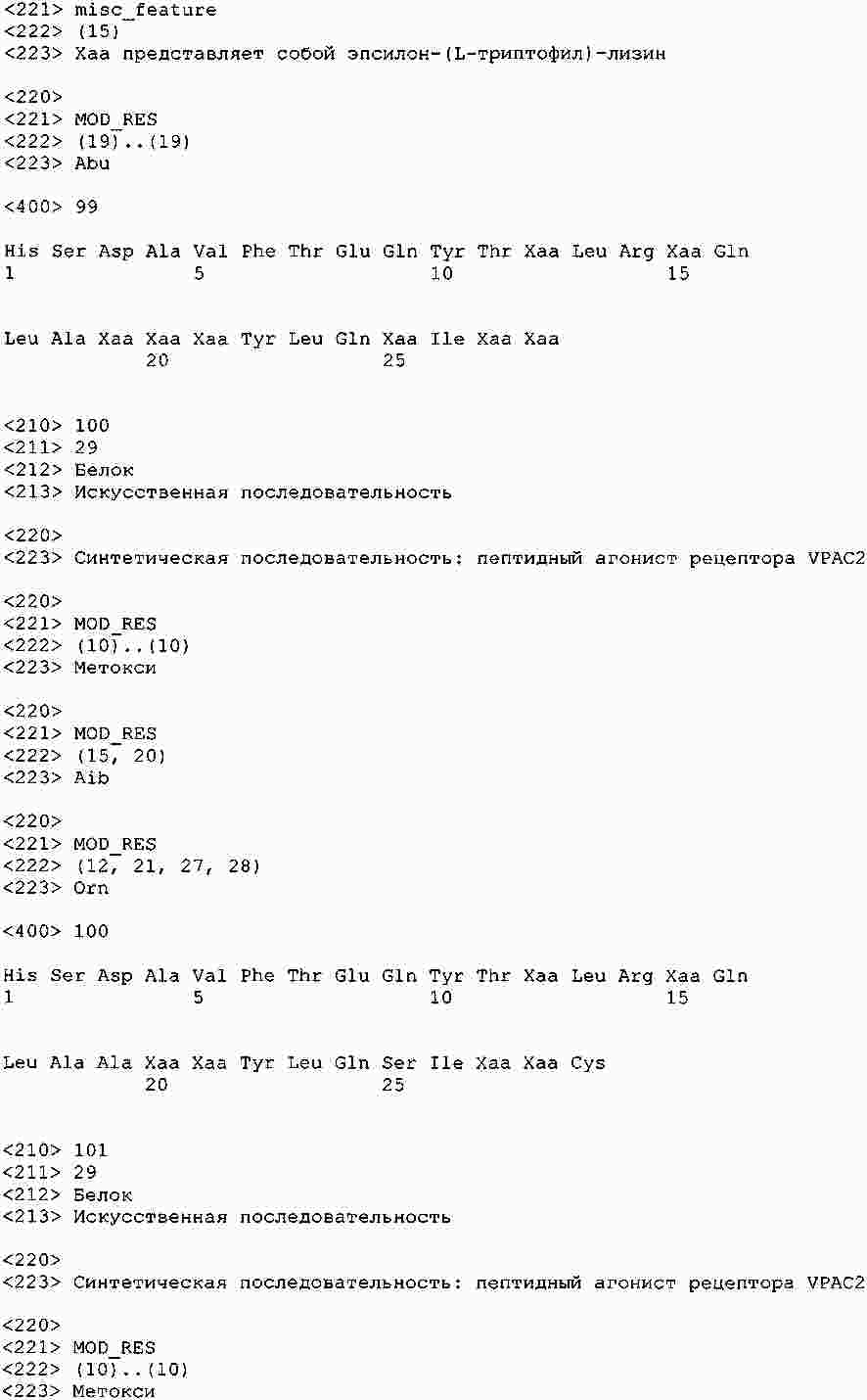
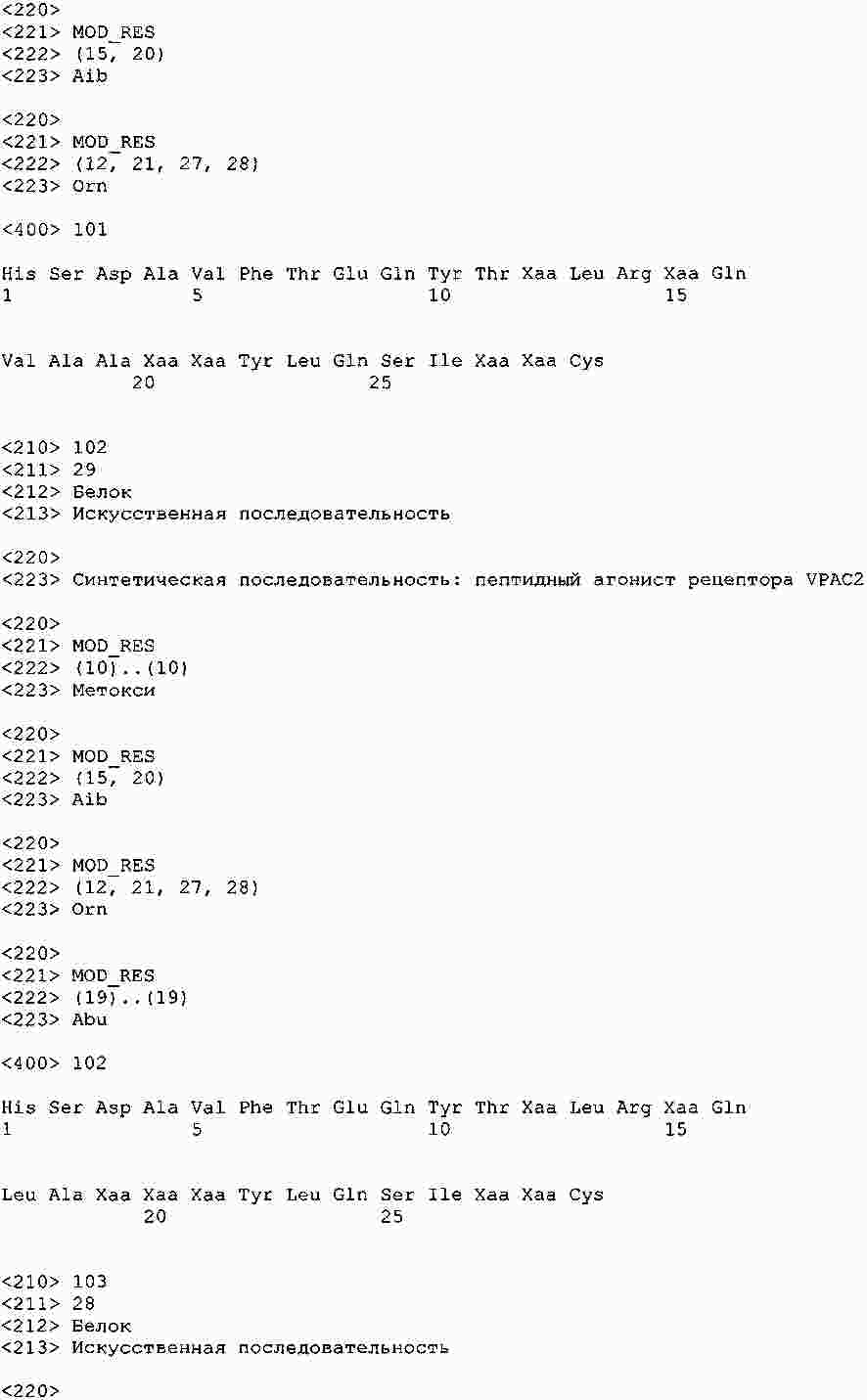
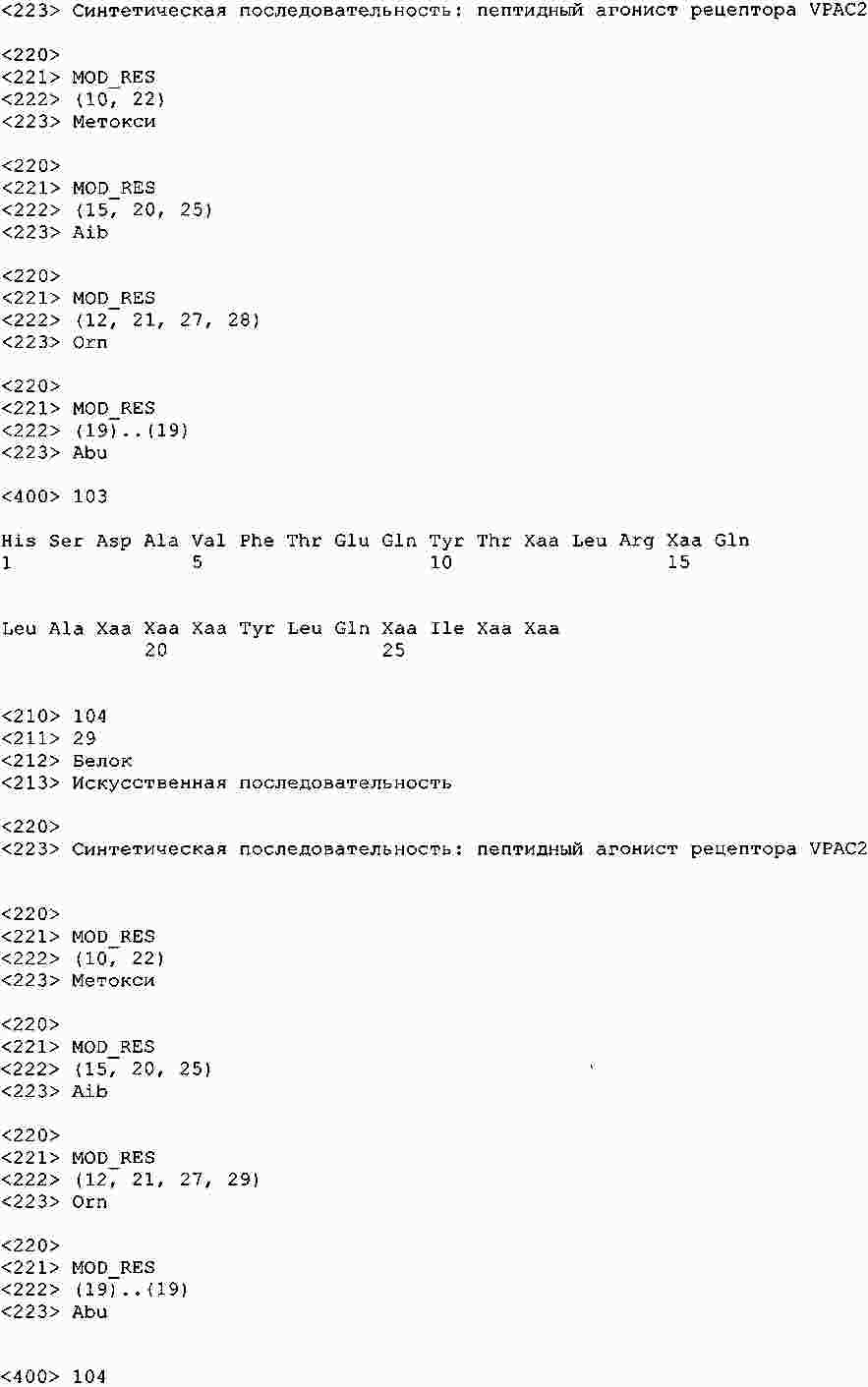
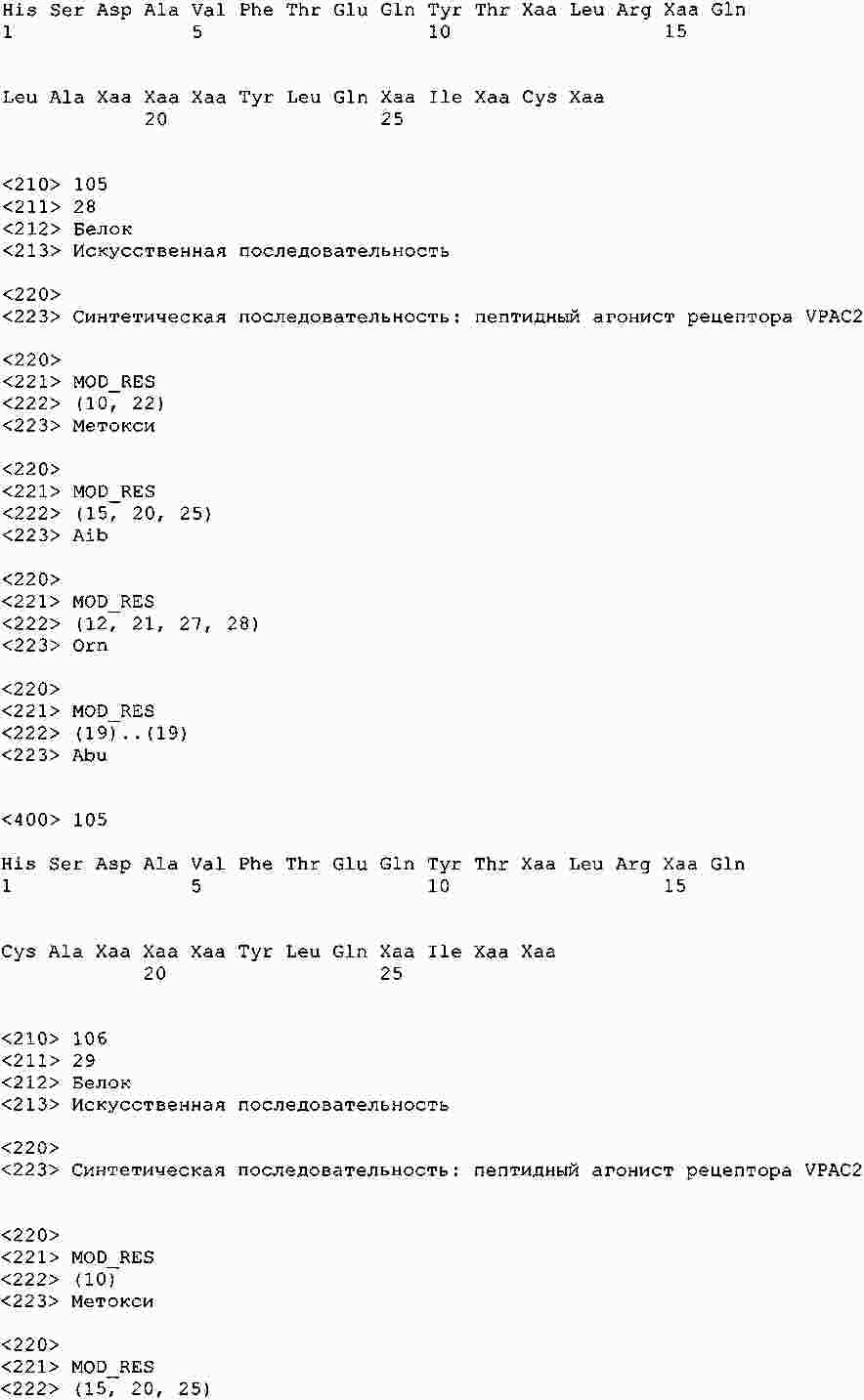
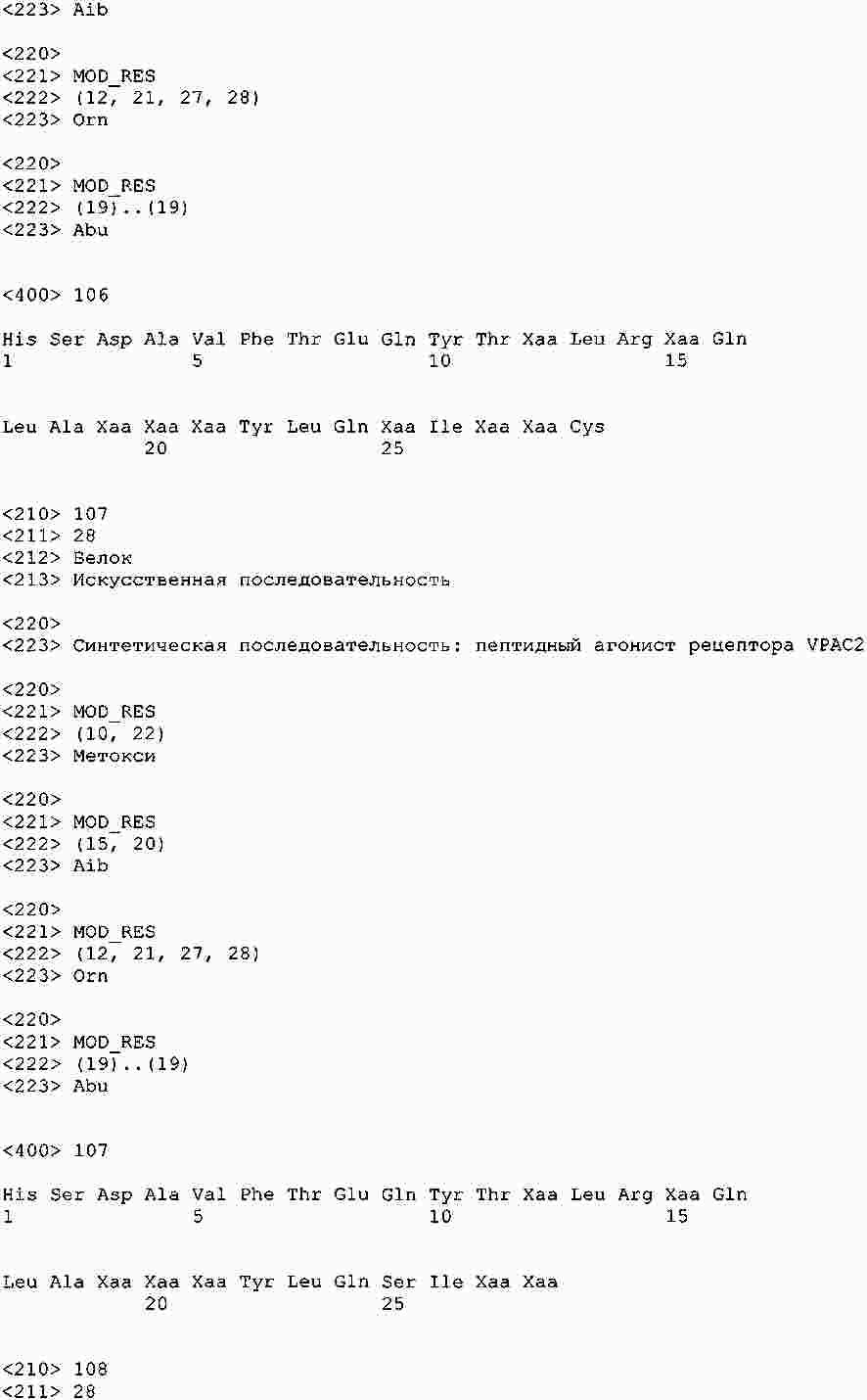
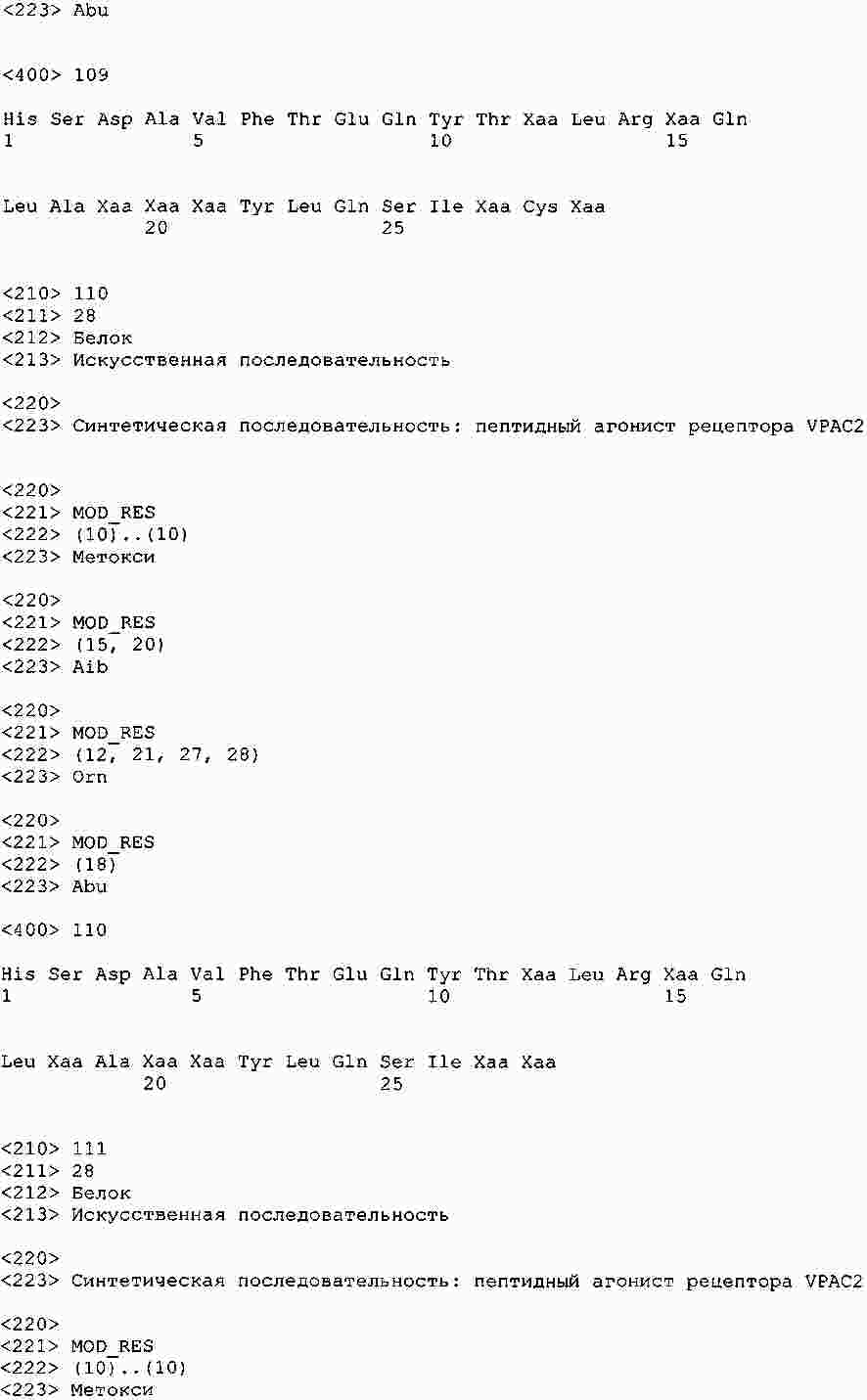
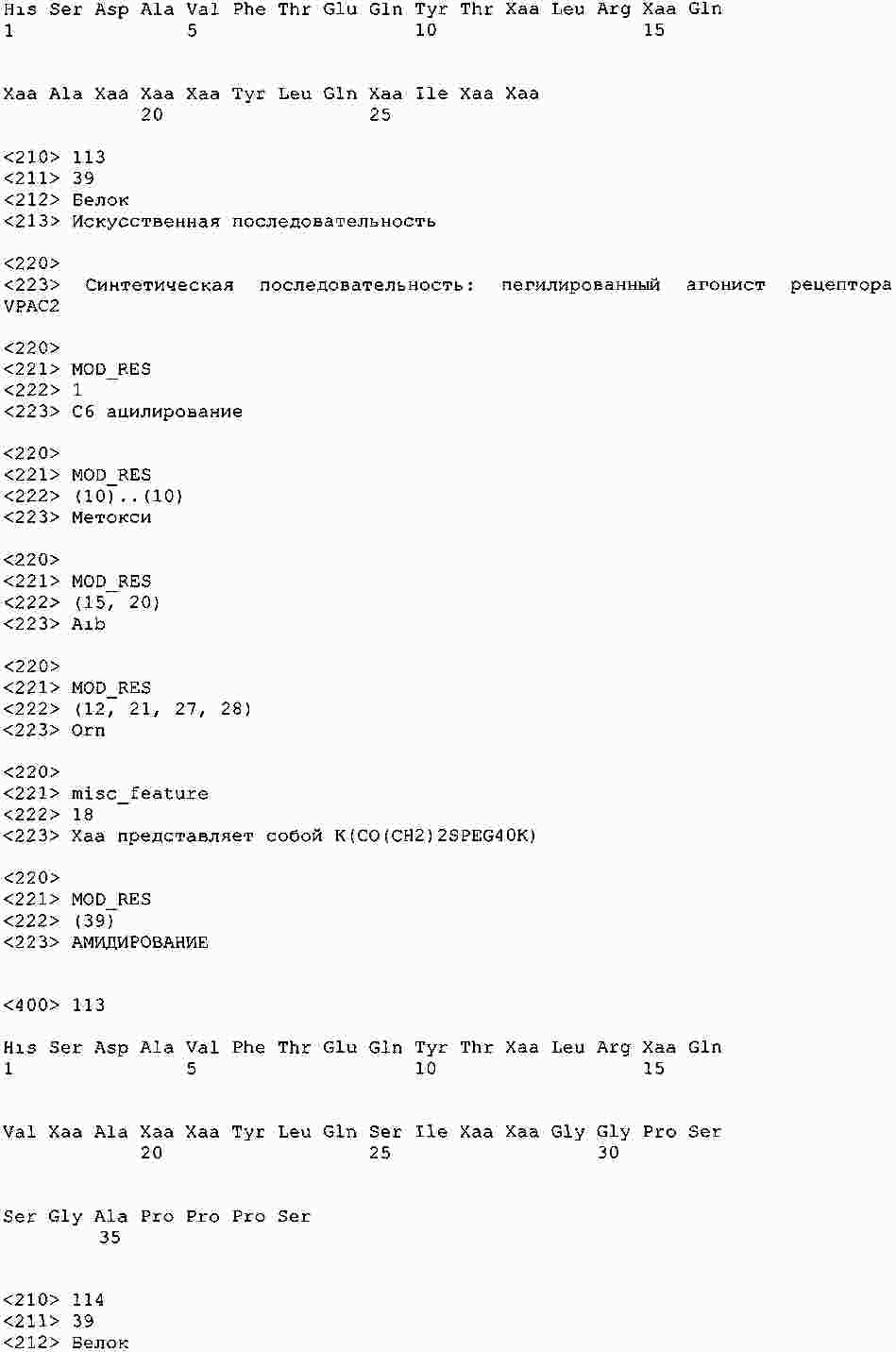
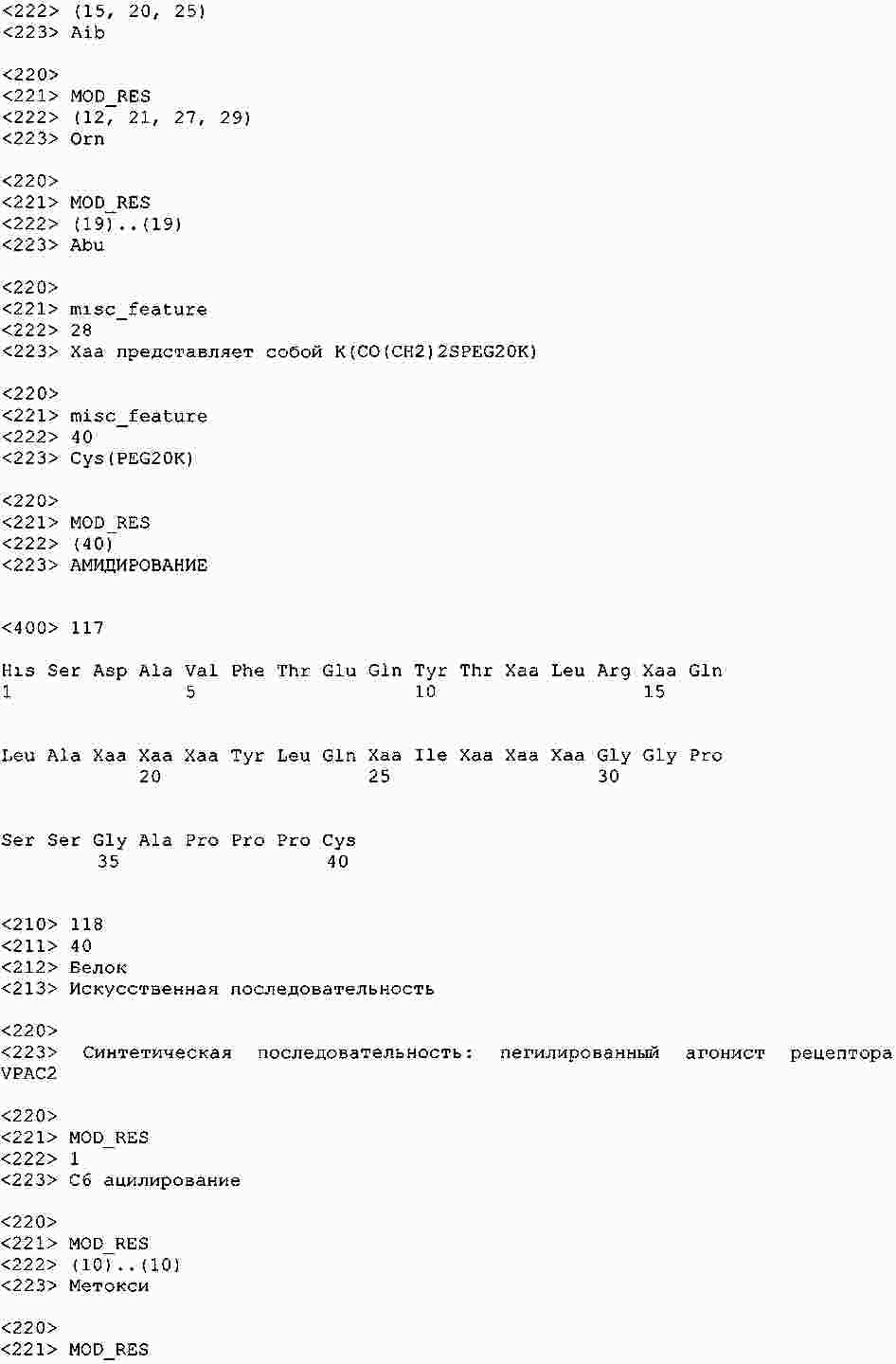
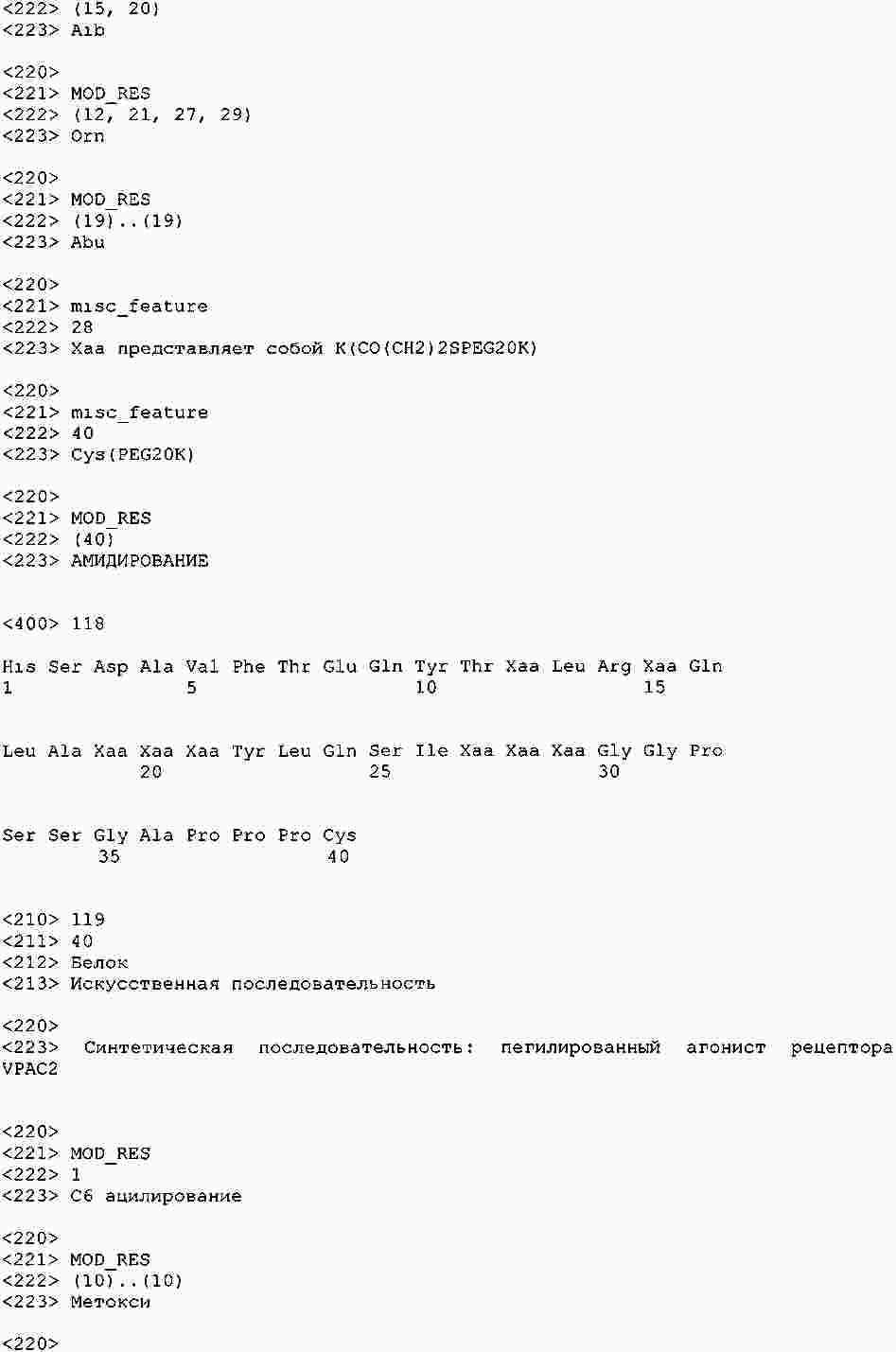
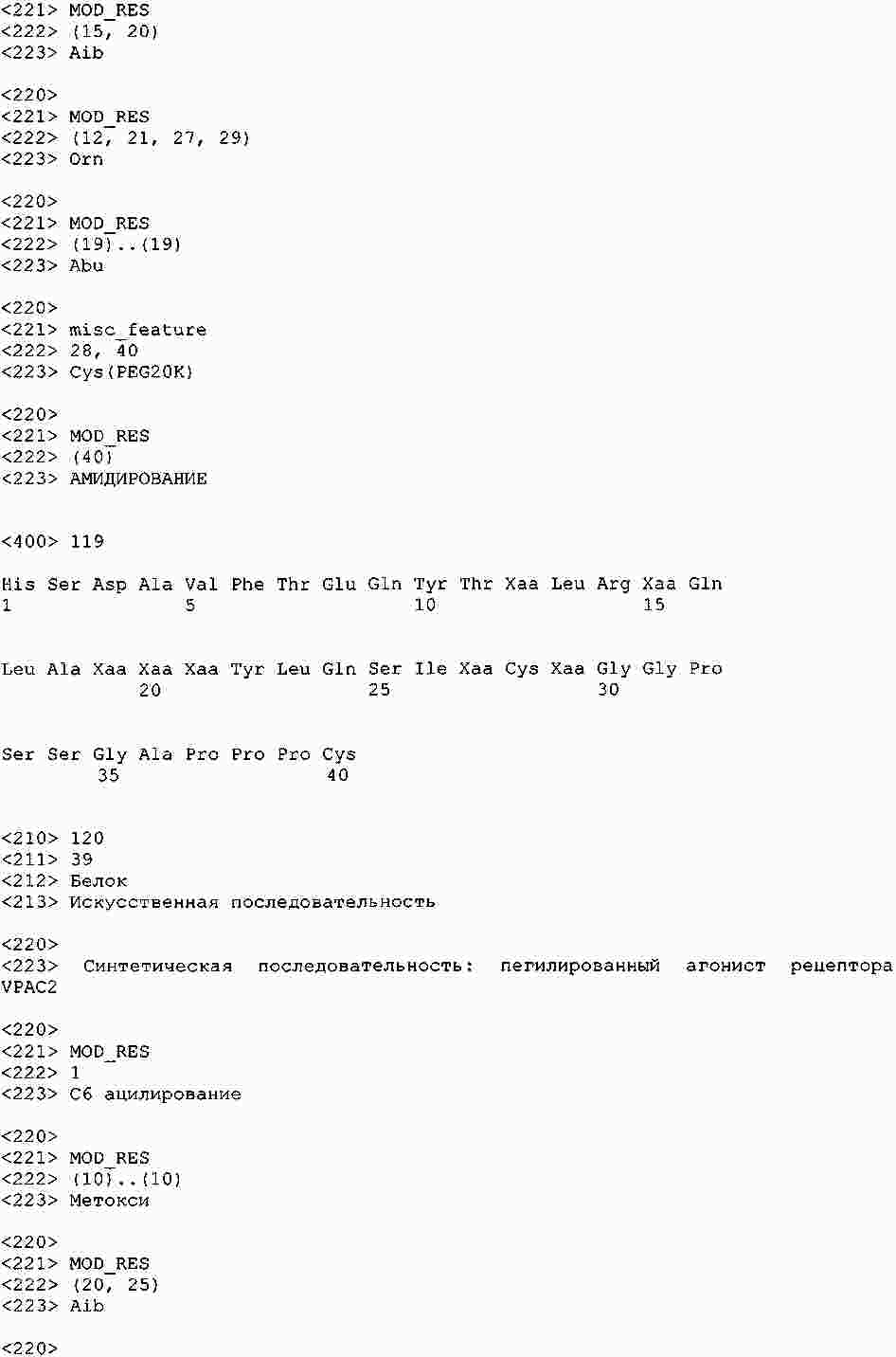
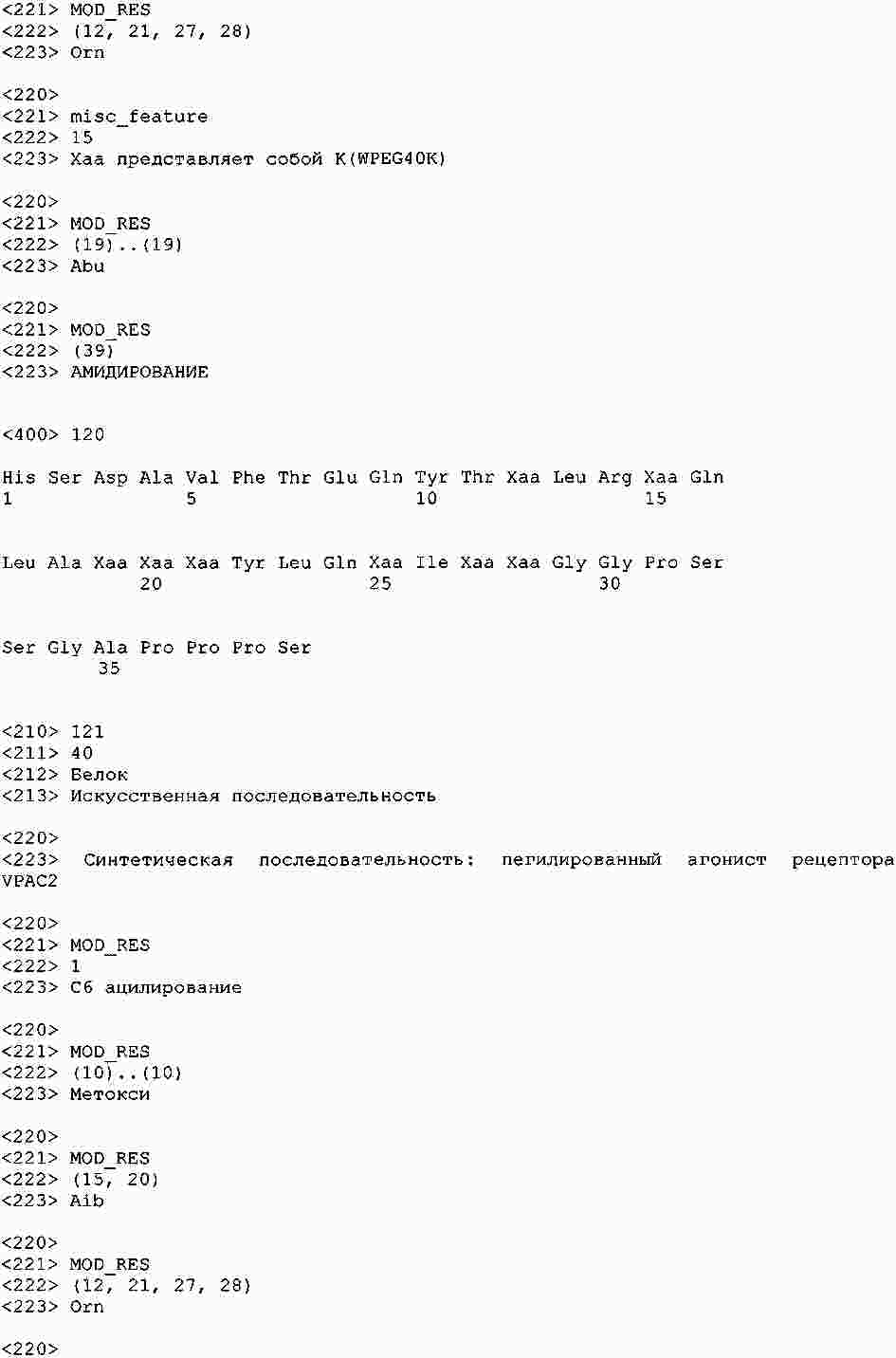
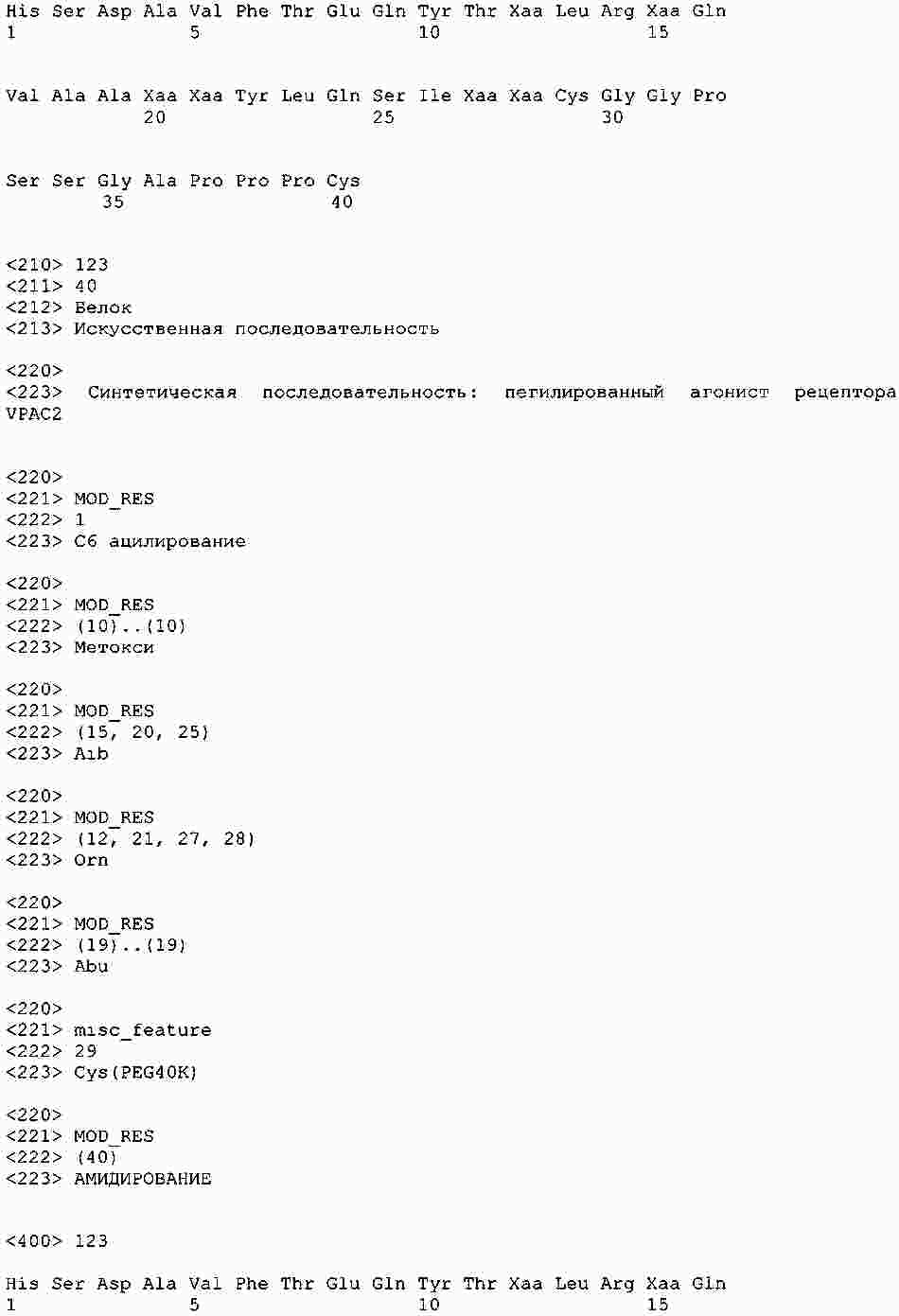
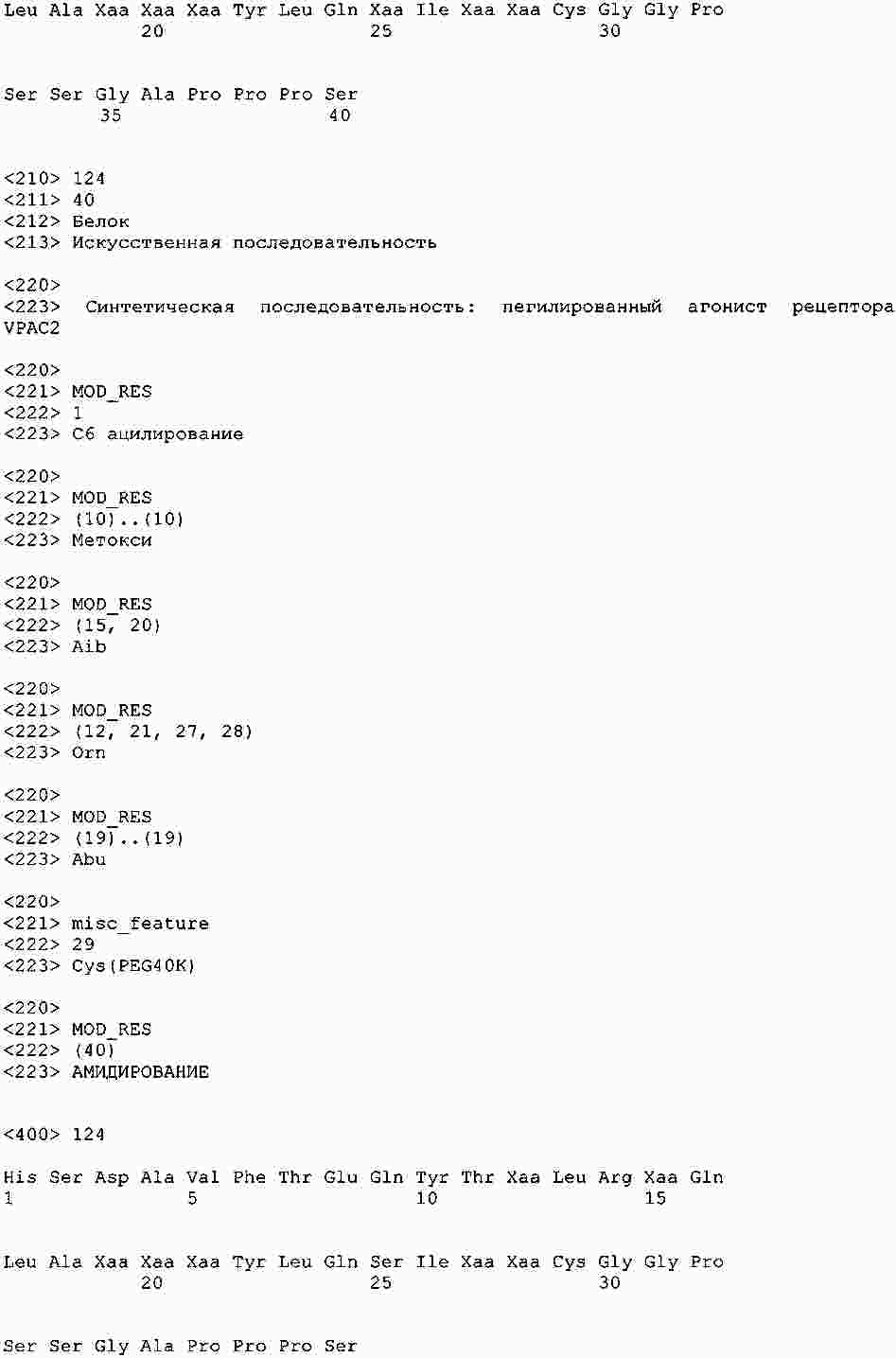
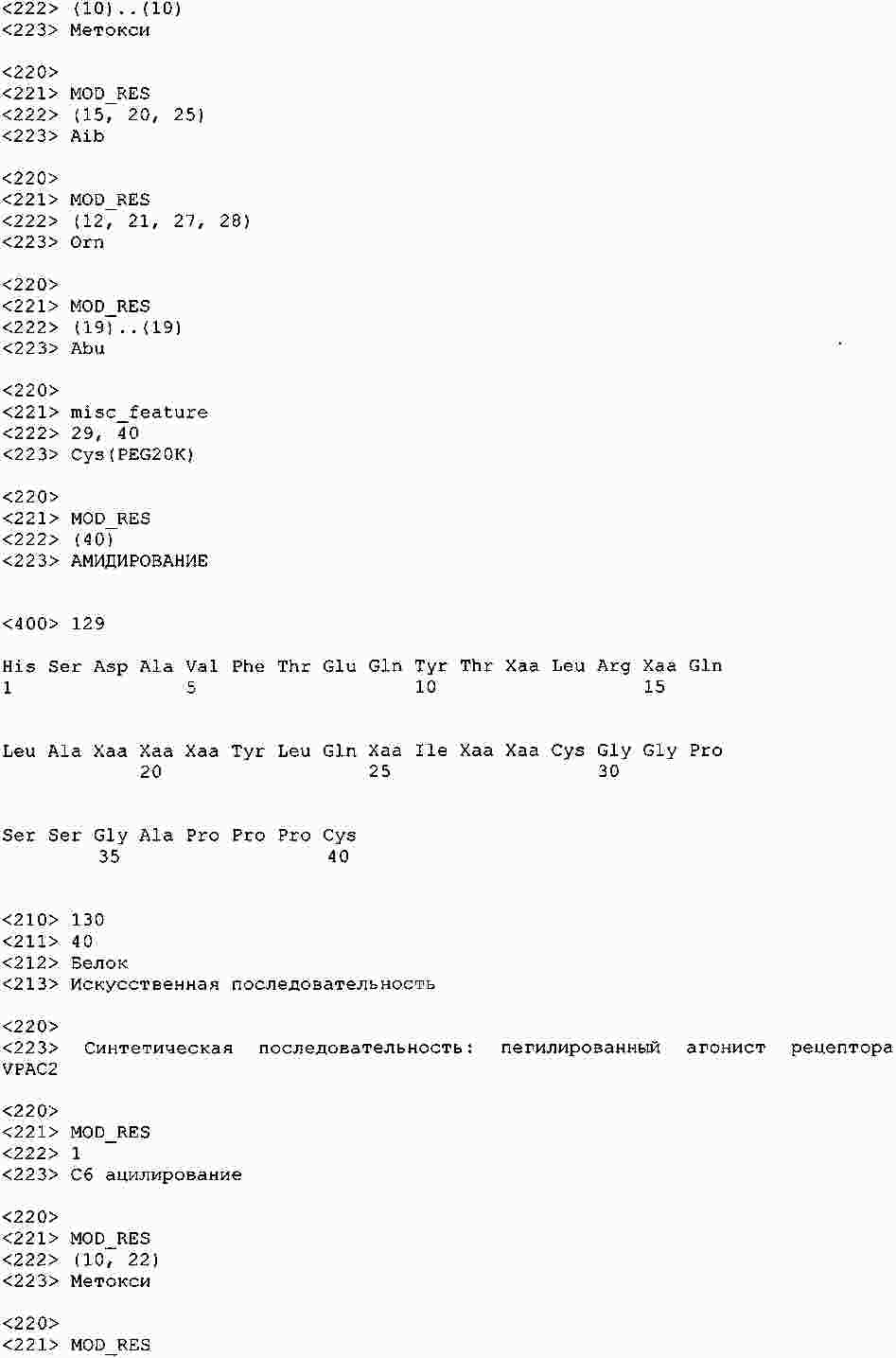
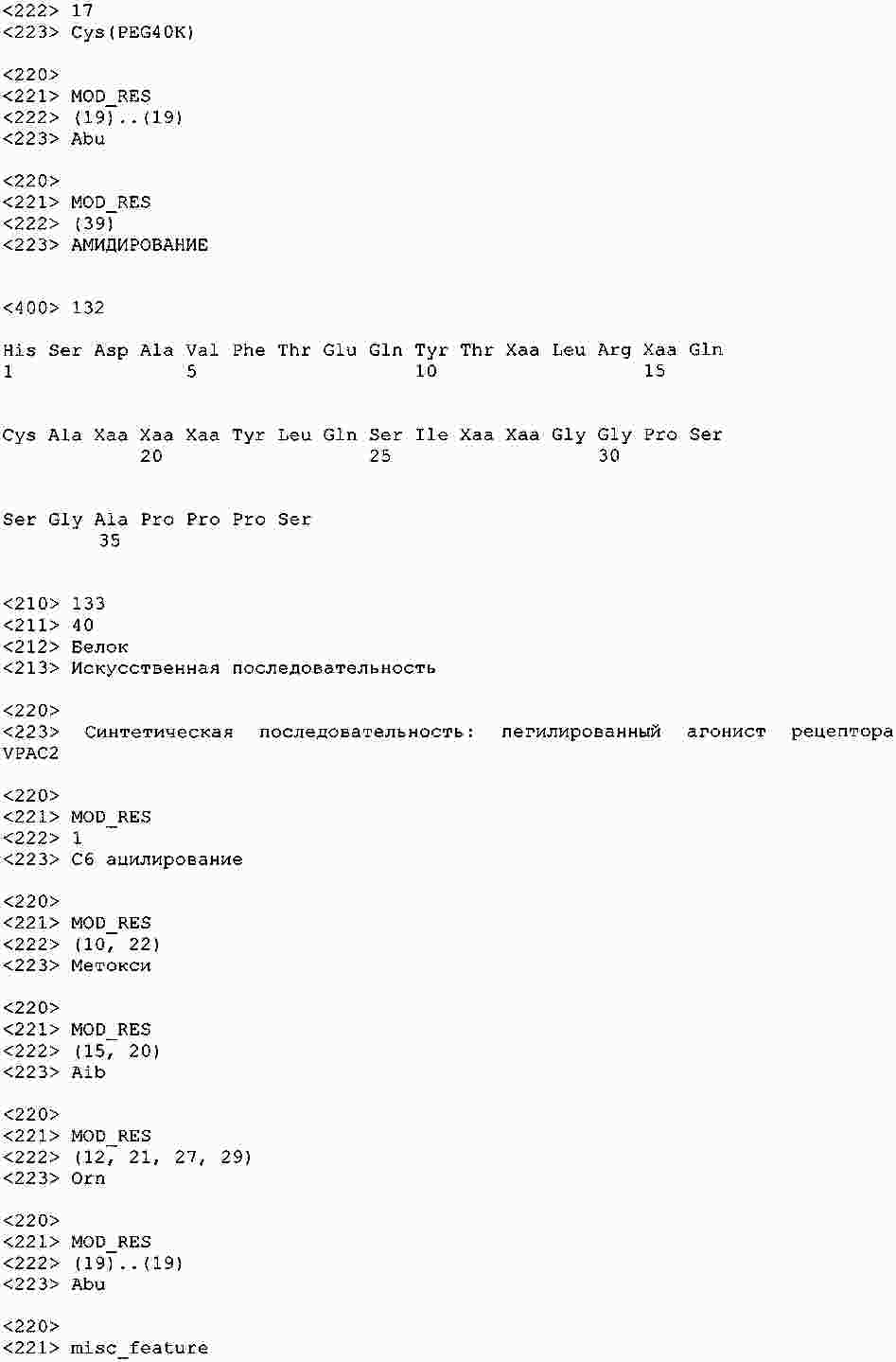
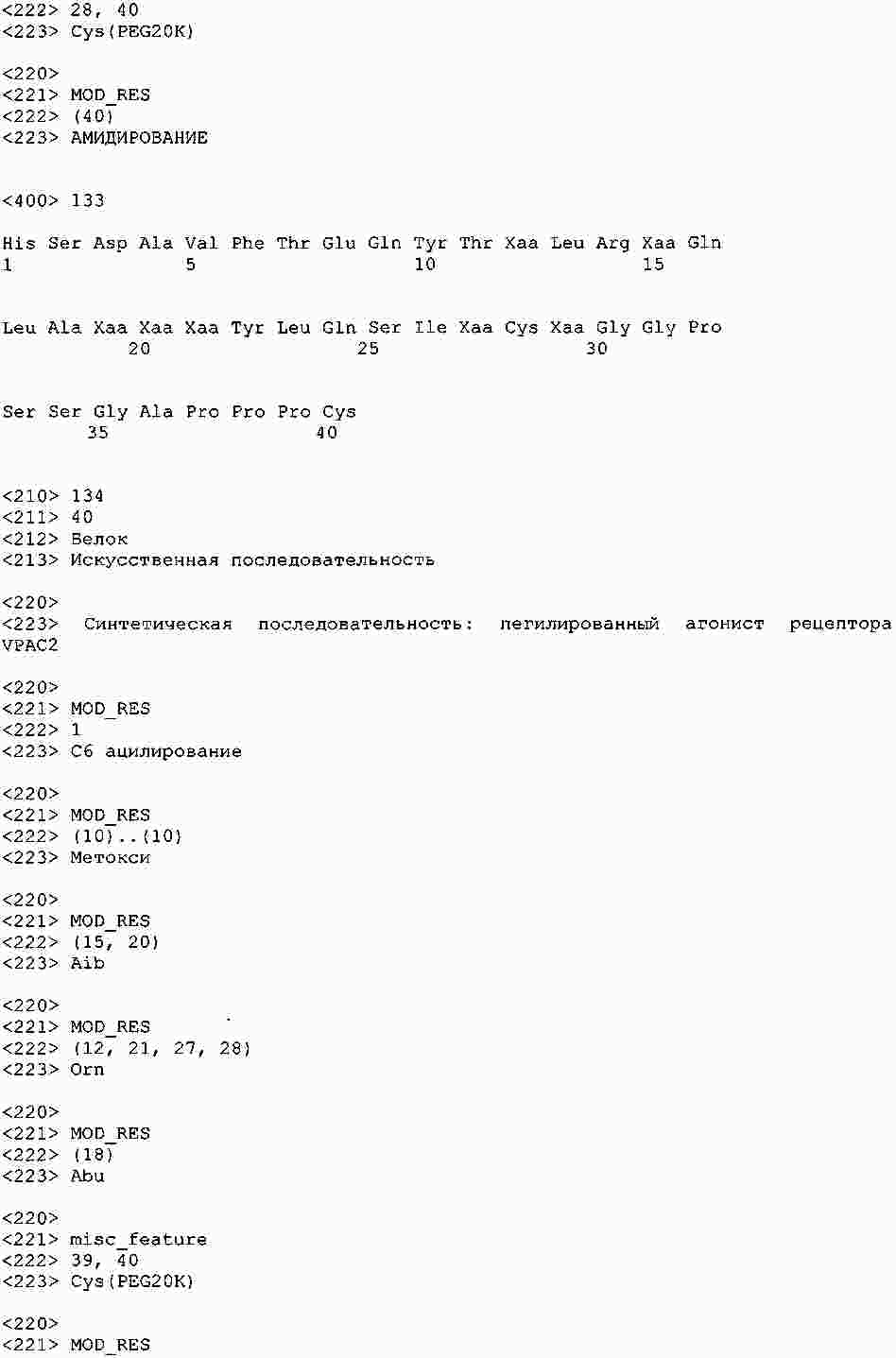
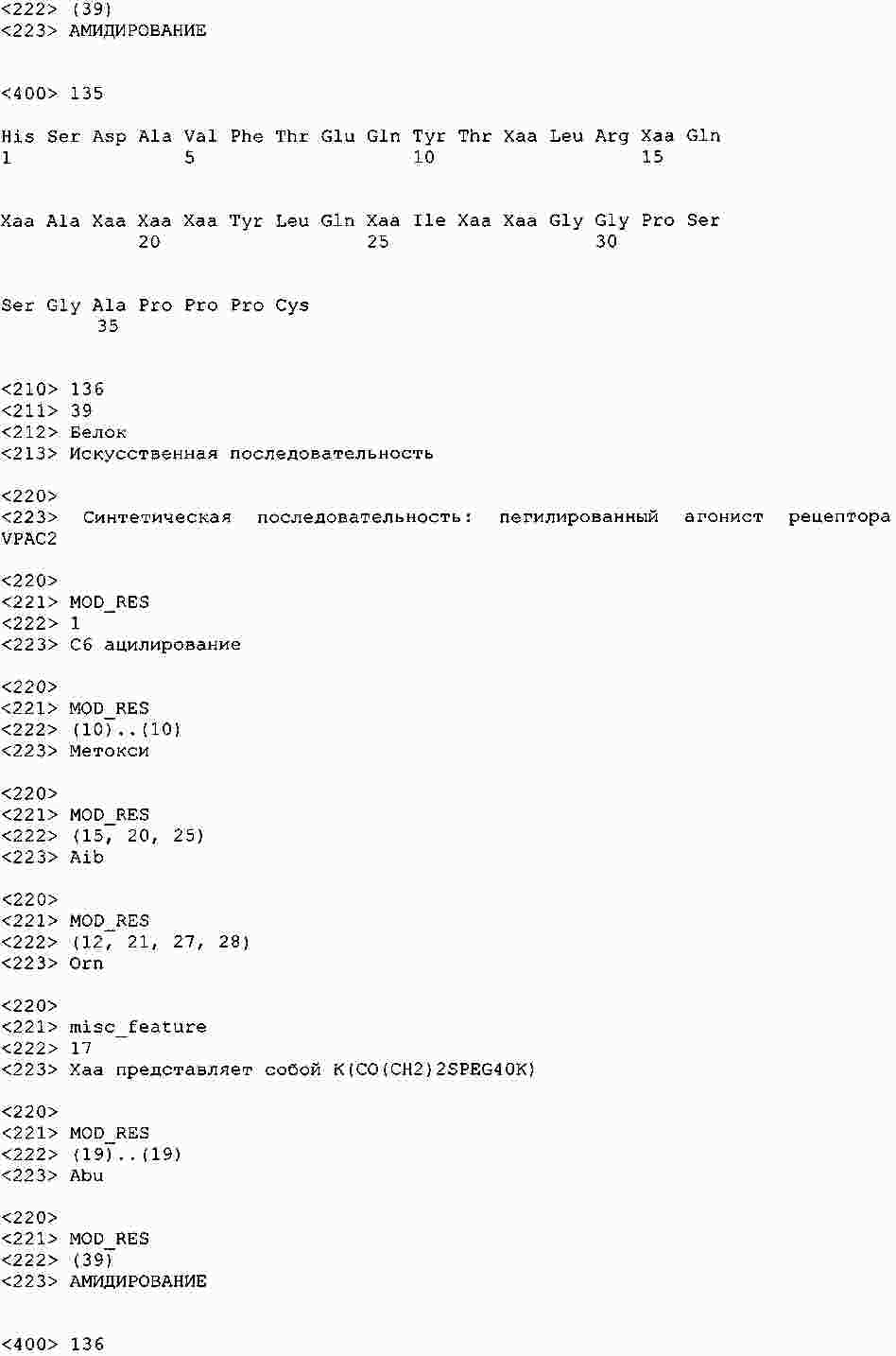
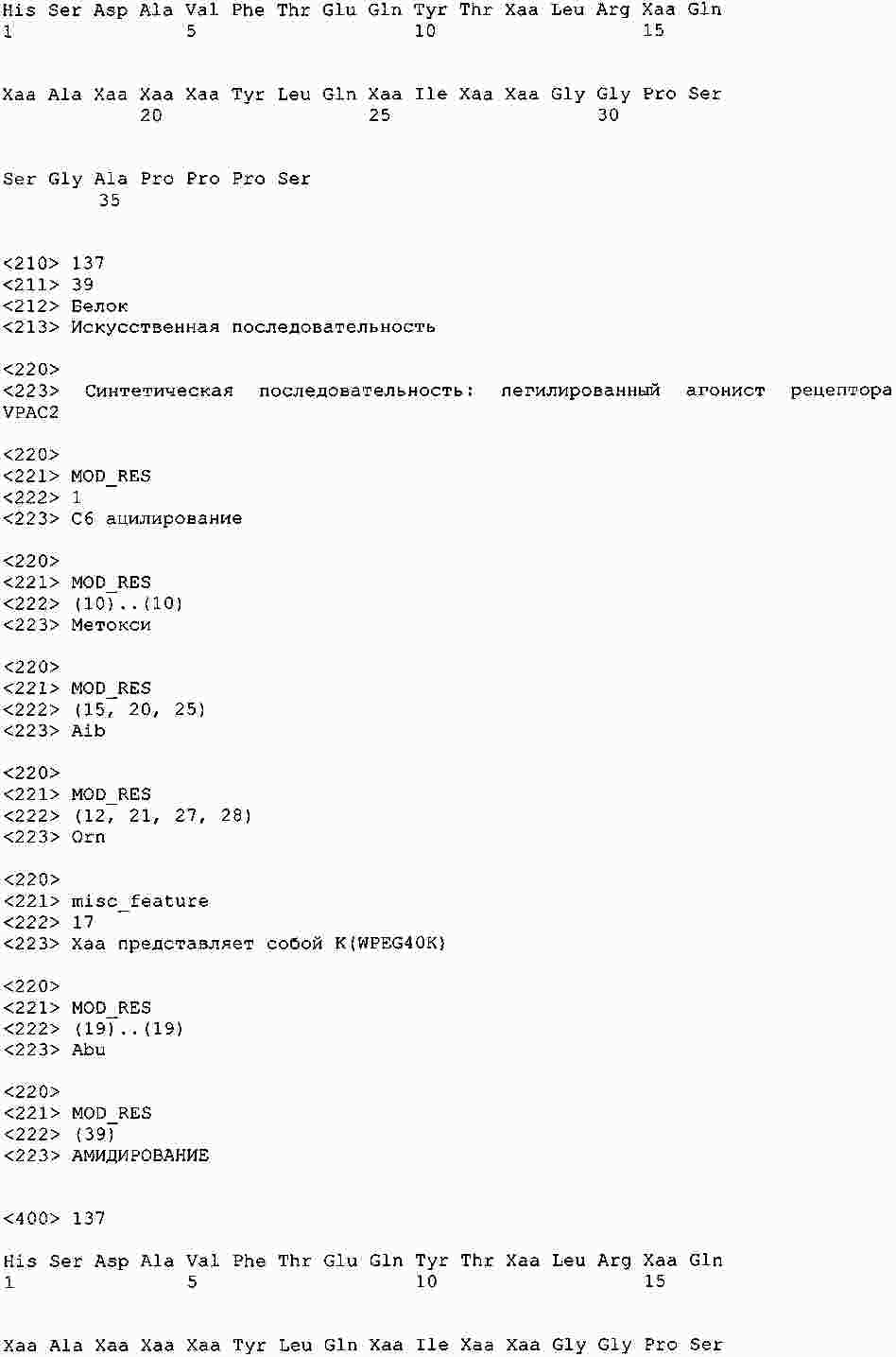
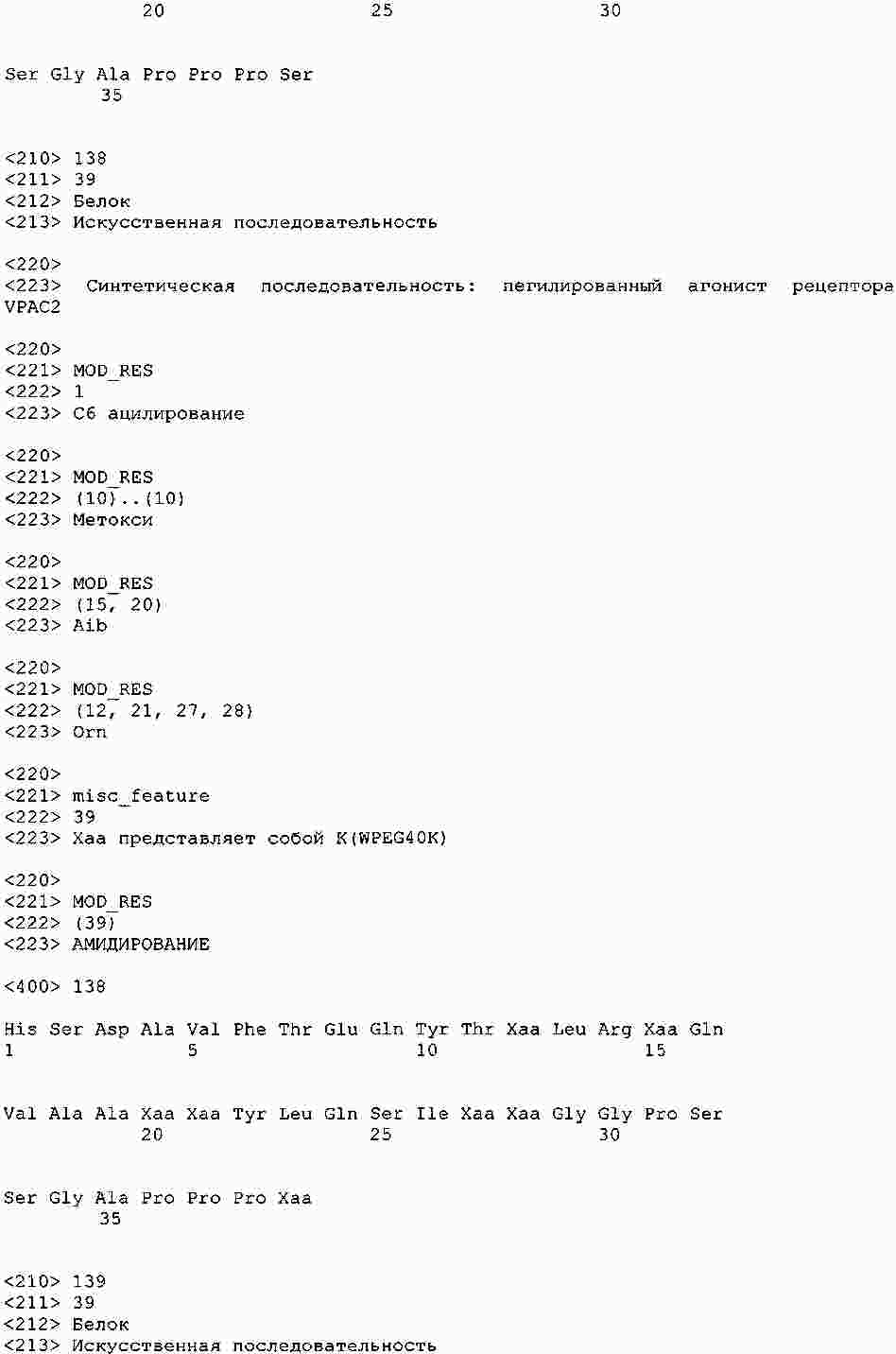
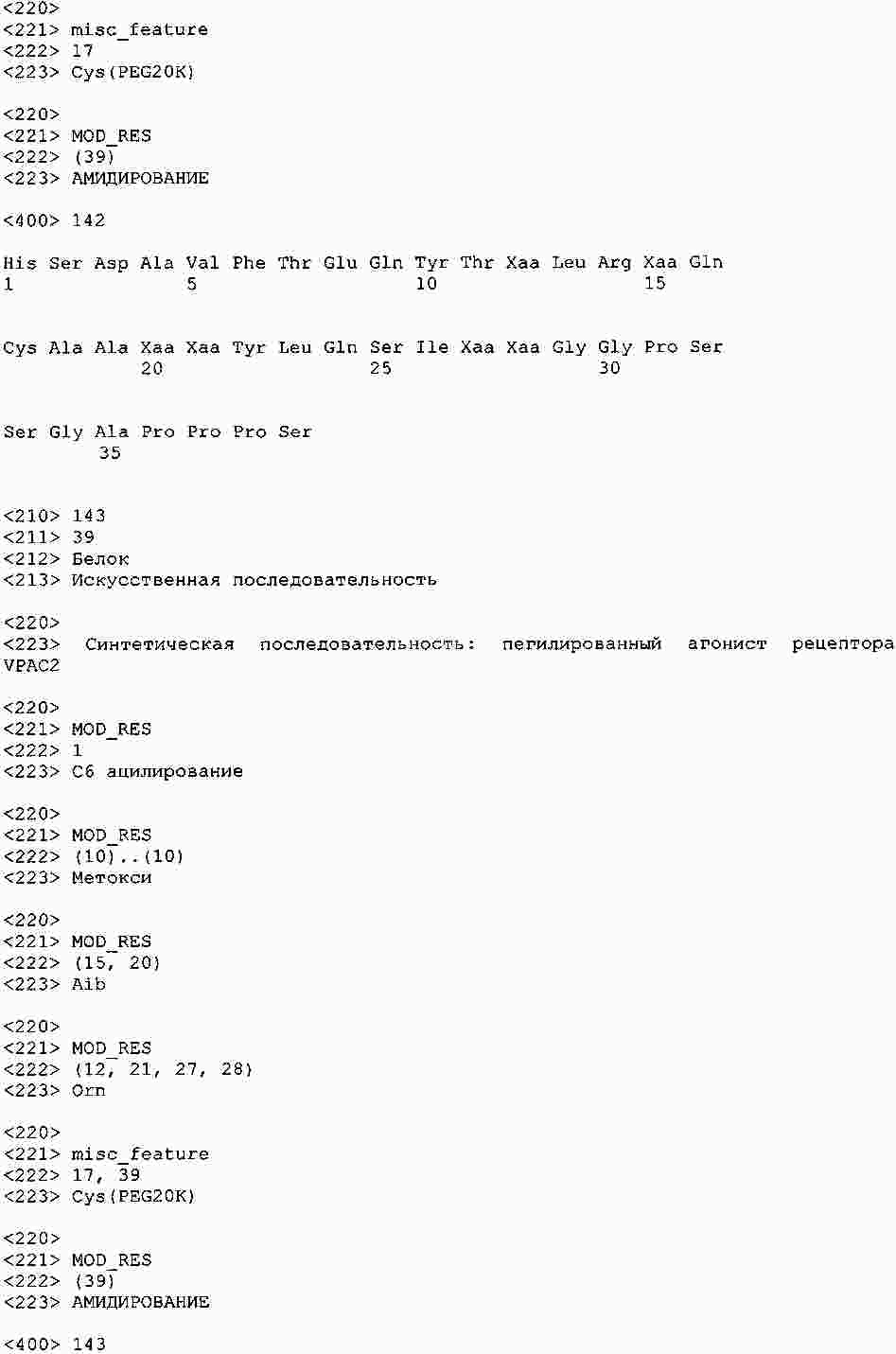
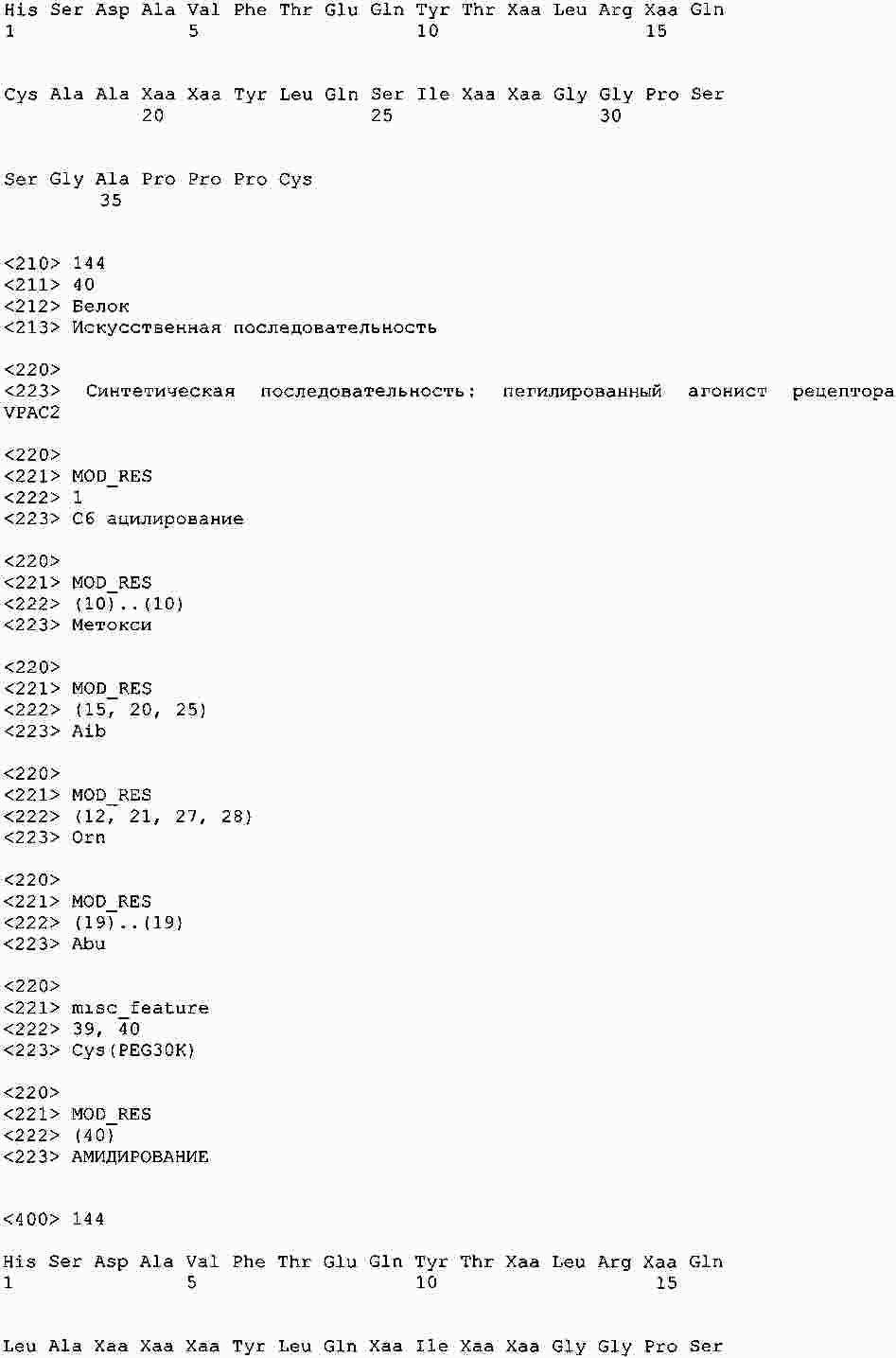
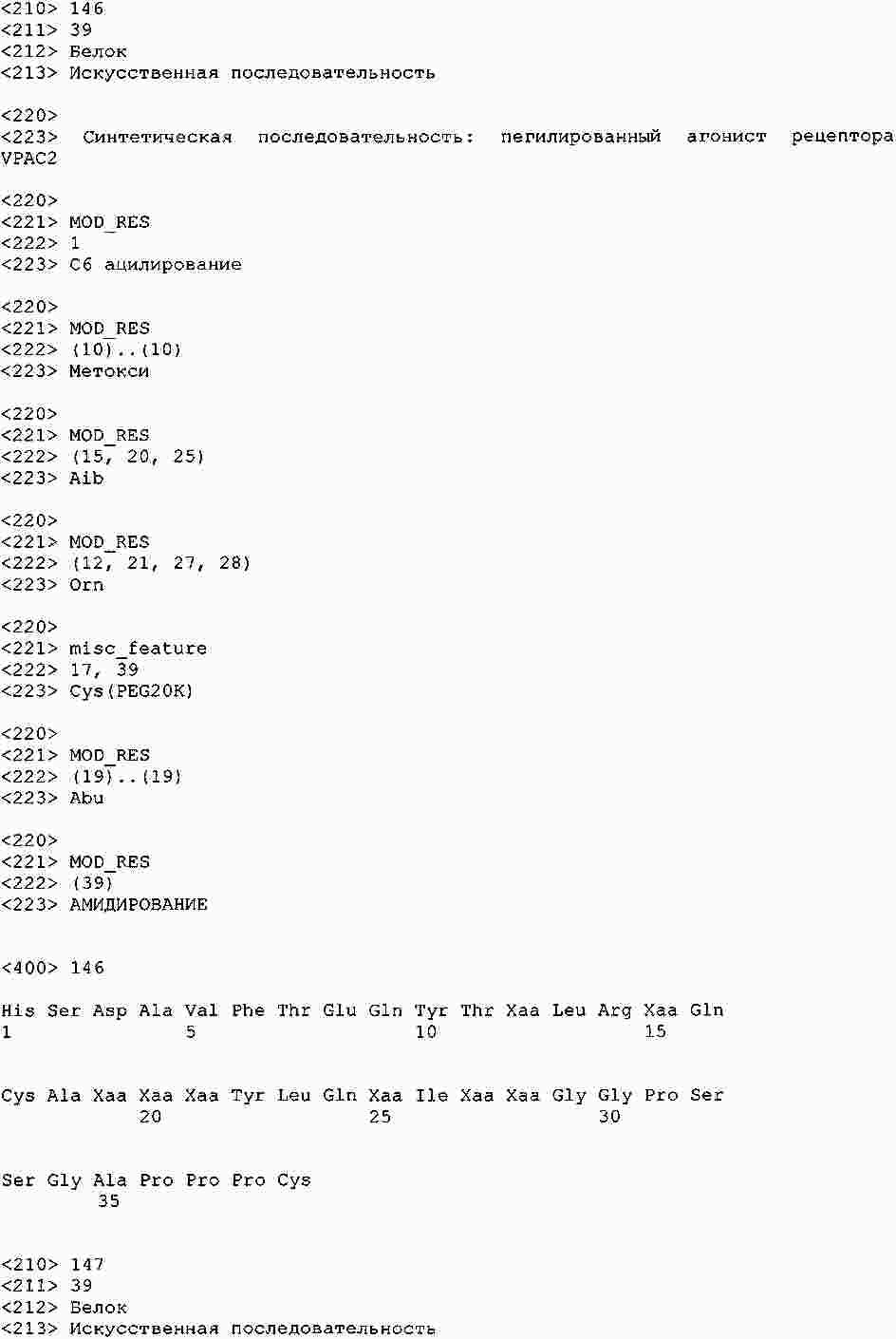
|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea000012930b*\id |
|
Термины запроса в документе Реферат Настоящее изобретение включает пептиды, которые избирательно активируют рецептор VPAC2 и пригодны для лечения диабета. Формула [0001] Пегилированный пептидный агонист рецептора VPAC2, содержащий последовательность, выбранную из [0002] Пегилированный пептидный агонист рецептора VPAC2 по п.1, отличающийся тем, что C-концевой удлиняющий сегмент выбирают из [0003] Пегилированный пептидный агонист рецептора VPAC2 по п.2, отличающийся тем, что C-концевой удлиняющий сегмент представляет собой SEQ ID NO:11 или SEQ ID NO: 12. [0004] Пегилированный пептидный агонист рецептора VPAC2 по п.1, отличающийся тем, что по меньшей мере одна молекула ПЭГ ковалентно присоединена к остатку в C-концевом удлиняющем сегменте. [0005] Пегилированный пептидный агонист рецептора VPAC2 по любому из предыдущих пунктов, отличающийся тем, что молекула ПЭГ является разветвленной. [0006] Пегилированный пептидный агонист рецептора VPAC2 по любому из пп.1-4, отличающийся тем, что молекула ПЭГ является линейной. [0007] Пегилированный пептидный агонист рецептора VPAC2 по любому из предыдущих пунктов, отличающийся тем, что молекулярная масса каждой молекулы ПЭГ составляет 20000, 30000, 40000 или 60000 Да. [0008] Пегилированный пептидный агонист рецептора VPAC2 по любому из предыдущих пунктов, который дополнительно содержит N-концевую модификацию на N-конце пептидного агониста, отличающийся тем, что N-концевую модификацию выбирают из: [0009] Пегилированный пептидный агонист рецептора VPAC2 по п.8, отличающийся тем, что N-концевая модификация представляет собой добавление группы, выбранной из ацетила, пропионила, бутирила, пентаноила, гексаноила, метионина, метионинсульфоксида, 3-фенилпропионила, фенилацетила, бензоила, норлейцина, D-гистидина, изолейцина, 3-меркаптопропионила, биотинил-6-аминогексановой кислоты (6-аминокапроновой кислоты) и -C(=NH)-NH2. [0010] Пегилированный пептидный агонист рецептора VPAC2 по п.9, отличающийся тем, что N-концевая модификация представляет собой добавление ацетила или гексаноила. [0011] Пегилированный пептидный агонист рецептора VPAC2 по п.1, содержащий последовательность аминокислот, выбранную из [0012] Пегилированный пептидный агонист рецептора VPAC2, содержащий последовательность аминокислот, выбранную из [0013] Пегилированный пептидный агонист рецептора VPAC2, содержащий последовательность формулы [0014] Фармацевтическая композиция, содержащая пегилированный пептидный агонист рецептора VPAC2 по любому из пп.1-13 и один или несколько фармацевтически приемлемых наполнителей, носителей и вспомогательных веществ. [0015] Пегилированный пептидный агонист рецептора VPAC2 по любому из пп.1-13 для применения в качестве лекарственного средства. [0016] Применение пегилированного пептидного агониста рецептора VPAC2 по любому из пп.1-13 для производства лекарственного средства для лечения инсулиннезависимого диабета или инсулинзависимого диабета. [0017] Способ лечения инсулиннезависимого диабета или инсулинзависимого диабета у больного, включающий введение указанному больному эффективного количества пегилированного пептидного агониста рецептора VPAC2 по любому из пп.1-13. Полный текст патента Настоящее изобретение относится к селективным пептидным агонистам рецептора VPAC2. В частности, настоящее изобретение относится к селективным пептидным агонистам рецептора VPAC2, к которым ковалентно присоединены одна или несколько молекул полиэтиленгликоля или его производного. Диабет 2-го типа, или инсулиннезависимый сахарный диабет (NIDDM), представляет собой наиболее распространенную форму диабета, которой страдают 90% больных диабетом. При NIDDM у больных нарушена функция β-клеток, что приводит к недостаточной выработке инсулина, и/или снижена чувствительность к инсулину. Если NIDDM не регулировать, избыток глюкозы накапливается в крови, приводя к гипергликемии. Через некоторое время могут развиваться более серьезные осложнения, в том числе почечная дисфункция, проблемы со стороны сердечно-сосудистой системы, ухудшение зрения, язвы нижних конечностей, невропатия и ишемия. Лечение NIDDM включает коррекцию питания, физические нагрузки и контроль массы тела, а также применение разнообразных пероральных лекарственных средств. Больные NIDDM на ранней стадии могут регулировать свой уровень глюкозы в крови, принимая такие пероральные лекарственные средства. Однако такие средства не замедляют прогрессирующего снижения функции β-клеток, которая развивается у больных NIDDM и, таким образом, на более поздних стадиях заболевания их применения недостаточно для регуляции уровней глюкозы в крови. Также лечение существующими на сегодняшний день лекарственными средствами подвергает больных NIDDM риску потенциальных побочных эффектов, таких как гипогликемия, проблемы со стороны желудочно-кишечного тракта, задержка жидкости, отек и/или увеличение массы тела. Гипофизарный пептид, активирующий аденилатциклазу (PACAP) и вазоактивный интестинальный пептид (VIP) принадлежат к тому же семейству пептидов, что секретин и глюкагон. PACAP и VIP действуют посредством трех связанных с G-белком рецепторов, которые оказывают свое действие через цАМФ-опосредованные и другие Са 2+ -опосредованные пути передачи сигнала. Эти рецепторы известны как PACAP-предпочитающий рецептор типа 1 (РАС1) (Isobe, et al., Regul. Pept., 110: 213-217 (2003); Ogi, et al., Biochem. Biophys. Res. Commun., 196: 1511-1521 (1993)) и два VIP-разделяемых рецептора типа 2 (VPAC1 и VPAC2) (Sherwood et al., Endocr. Rev., 21: 619-670 (2000); Hammar et al., Pharmacol Rev, 50: 265-270 (1998); Couvineau, et al., J. Biol. Chem., 278: 24759-24766 (2003); Sreedharan, et al., Biochem. Biophys. Res. Commun., 193: 546-553 (1993); Lutz, et al., FEBS Lett., 458: 197-203 (1999); Adamou, et al., Biochem. Biophys. Res. Commun., 209: 385-392 (1995)). Ряд аналогов PACAP раскрыт в патенте США 6242563 и WO 2000/05260. PACAP обладает сравнимой активностью в отношении всех трех рецепторов, тогда как VIP избирательно активизирует два рецептора VPAC (Tsutsumi et al., Diabetes, 51: 1453-1460 (2002)). Показано, что и VIP (Eriksson et al., Peptides, 10: 481-484 (1989)), и PACAP (Filipsson et al., JCEM, 82: 3093-3098 (1997)) не только стимулируют секрецию инсулина у человека при внутривенном введении, но также повышают секрецию глюкагона и выход глюкозы из печени. Как следствие, PACAP или VIP стимуляция в общем не приводит к фактическому улучшению гликемии. Активация множественных рецепторов PACAP или VIP также вызывает широкий спектр физиологических эффектов на нервную, эндокринную, сердечно-сосудистую, репродуктивную, мышечную и иммунную системы (Gozes et al., Curr. Med. Chem., 6: 1019-1034 (1999)). По-видимому, индуцированная VIP водянистая диарея у крыс опосредуется только одним из рецепторов VPAC - VPAC1 (Ito et al., Peptides, 22: 1139-1151 (2001); Tsutsumi et al., Diabetes, 51: 1453-1460 (2002)). Рецепторы VPAC1 и РАС1 экспрессируются на α-клетках и гепатоцитах и, таким образом, наиболее вероятно принимают участие в воздействии на выход глюкозы из печени. Экзендин-4 найден в слюнных выделениях ящерицы-ядозуба, Heloderma Suspectum (Eng et al., J. Biol.Chem., 267(11): 7402-7405 (1992)). Он представляет собой пептид длиной 39 аминокислот, который обладает глюкозозависимой активностью стимулятора секреции инсулина. Конкретные пептидные агонисты пегилированного экзендина и экзендина описаны в WO 2000/66629. Недавние исследования показали, что пептиды, селективные в отношении рецептора VPAC2, могут стимулировать секрецию инсулина поджелудочной железой без побочного действия на желудочно-кишечный тракт (ЖКТ) и без повышения высвобождения глюкагона и выхода глюкозы из печени (Tsutsumi et al., Diabetes, 51: 1453-1460 (2002)). Пептиды, селективные в отношении рецептора VPAC2, были изначально идентифицированы путем модификации VIP и/или PACAP (см., например, Xia et al., J. Pharmacol Exp Ther., 281: 629-633 (1997); Tsutsumi et al., Diabetes, 51: 1453-1460 (2002); WO 01/23420 и WO 2004/06839.) Однако многие из пептидных агонистов рецептора VPAC2, о которых сообщалось до настоящего времени, обладают менее чем желательными профилями эффективности, селективности и стабильности, что может снижать их целесообразность с клинической точки зрения. Кроме того, многие из этих пептидов не подходят в качестве кандидатов промышленного значения из-за проблем со стабильностью, связанных с полипептидами в препарате, а также из-за проблем с коротким периодом полувыведения указанных полипептидов in vivo. Кроме того, было обнаружено, что некоторые пептидные агонисты рецептора VPAC2 инактивируются дипептидилпептидазой (DPP-IV). Короткий период полувыведения из сыворотки может препятствовать применению данных агонистов в качестве терапевтических агентов. Таким образом, существует потребность в новых терапевтических средствах, которые позволят преодолеть проблемы, связанные с существующими средствами для лечения NIDDM. Настоящее изобретение обеспечивает улучшенные соединения, которые являются селективными в отношении рецептора VPAC2 и которые индуцируют секрецию инсулина из поджелудочной железы только при наличии высоких уровней глюкозы в крови. Соединения по настоящему изобретению представляют собой пептиды, которые, как считается, также улучшают функцию β-клеток. Эти пептиды могут оказывать физиологический эффект индуцирования секреции инсулина без побочного действия на ЖКТ или соответствующего повышения выхода глюкозы из печени, а также в целом обладают повышенной селективностью, эффективностью и/или стабильностью пептида in vivo в сравнении с известными пептидными агонистами рецептора VPAC2. Настоящее изобретение также обеспечивает селективные пептидные агонисты рецептора VPAC2, которые имеют сниженный клиренс и улучшенную стабильность in vivo. Желательно, чтобы агонисты по настоящему изобретению вводились минимальное число раз на протяжении пролонгированного периода времени. В соответствии с первым аспектом изобретения обеспечивается пегилированный пептидный агонист рецептора VPAC2, содержащий последовательность, выбранную из и C-концевой удлиняющий сегмент, где N-конец C-концевого удлиняющего сегмента связан с C-концом пептидной последовательности и где C-концевой удлиняющий сегмент содержит последовательность аминокислот формулы где Xaa 1 представляет собой Gly, Cys или отсутствует; Xaa 2 представляет собой Gly, Arg или отсутствует; Xaa 3 представляет собой Pro, Thr или отсутствует; Xaa 4 представляет собой Ser или отсутствует; Xaa 5 представляет собой Ser или отсутствует; Xaa 6 представляет собой Gly или отсутствует; Xaa 7 представляет собой Ala или отсутствует; Xaa 8 представляет собой Pro или отсутствует; Xaa 9 представляет собой Pro или отсутствует; Xaa 10 представляет собой Pro или отсутствует; Xaa 11 представляет собой Ser, Cys или отсутствует и Xaa 12 представляет собой Cys или отсутствует; где присутствуют по меньшей мере пять из фрагментов Xaa 1 -Xaa 12 C-концевого удлиняющего сегмента, и где, если отсутствует Xaa 1 , Xaa 2 , Xaa 3 , Xaa 4 , Xaa 5 , Xaa 6 , Xaa 7 , Xaa 8 , Xaa 9 , Xaa 10 или Xaa 11 , следующая аминокислота в направлении 5'-3' представляет собой следующую аминокислоту в C-концевом удлиняющем сегменте, и где C-концевая аминокислота может быть амидирована, и где пептидный агонист содержит по меньшей мере один остаток Cys, который ковалентно присоединен к молекуле ПЭГ, или пептидный агонист содержит по меньшей мере один остаток Lys, который ковалентно присоединен к молекуле ПЭГ, или пептидный агонист содержит по меньшей мере один K(W), который ковалентно присоединен к молекуле ПЭГ, или пептидный агонист содержит по меньшей мере один K(CO(CH 2 ) 2 SH), который ковалентно присоединен к молекуле ПЭГ, или карбоксиконцевая аминокислота пептидного агониста ковалентно присоединена к молекуле ПЭГ, или комбинацию указанных характеристик. Предпочтительно присутствуют по меньшей мере шесть из Xaa 1 -Xaa 12 C-концевого удлиняющего сегмента формулы 3. Более предпочтительно присутствуют по меньшей мере семь, восемь, девять, десять, одиннадцать или все Xaa 1 -Xaa 12 C-концевого удлиняющего сегмента. Более предпочтительно C-концевой удлиняющий сегмент пегилированного пептидного агониста рецептора VPAC2 выбран из Еще более предпочтительно C-концевой удлиняющий сегмент пегилированного пептидного агониста рецептора VPAC2 представляет собой SEQ ID NO: 11 или SEQ ID NO: 12. Молекула(ы) ПЭГ может быть ковалентно присоединена к любому из остатков Lys, Cys, K(W) или K(CO(CH 2 ) 2 SH) в любом положении пептидного агониста рецептора VPAC2 в соответствии с первым аспектом настоящего изобретения. Если пегилированный пептидный агонист рецептора VPAC2 содержит последовательность, выбранную из SEQ ID NO: 22, 23, 39, 42, 43, 44, 45, 95, 96, 100, 101, 102, 104, 105, 106, 108 и 109, предпочтительно остаток цистеина является пегилированным. Если пегилированный пептидный агонист рецептора VPAC2 содержит последовательность, выбранную из SEQ ID NO: 19, 34, 35, 36, 38, 41, 99 и 112, предпочтительно остаток K(W) является пегилированным. Если пегилированный пептидный агонист рецептора VPAC2 содержит последовательность, выбранную из SEQ ID NO: 18, 20, 21, 37, 94, 97, 98 и 111, предпочтительно остаток K(CO(CH 2 ) 2 SH) является пегилированным. Если пегилированный пептидный агонист рецептора VPAC2 содержит C-концевой удлиняющий сегмент, молекула(ы) ПЭГ может быть ковалентно присоединена к одному или нескольким остаткам Cys в указанном C-концевом удлиняющем сегменте. Если последовательность, выбранная из SEQ ID NO: 17-45 и 94-112, содержит один или несколько остатков Lys, Cys, K(W) или K(CO(CH 2 ) 2 SH) и C-концевой удлиняющий сегмент содержит один или несколько остатков Cys, может присутствовать один или несколько пегилированных остатков в одной или обеих последовательностях. Предпочтительно по меньшей мере одна молекула ПЭГ ковалентно присоединена к остатку в C-концевом удлиняющем сегменте пептидного агониста рецептора VPAC2. Если присутствует несколько молекул ПЭГ, может присутствовать комбинация пегилирования Lys, Cys, K(CO(CH 2 ) 2 SH), K(W) и карбоксиконцевой аминокислоты. Например, если присутствуют две молекулы ПЭГ, одна может быть присоединена к остатку Lys и другая может быть присоединена к остатку Cys. Предпочтительно молекула ПЭГ является разветвленной. Альтернативно, молекула ПЭГ может быть линейной. Предпочтительно молекулярная масса молекулы ПЭГ составляет от 1000 до 100000 Да. Более предпочтительно молекулярная масса молекулы ПЭГ выбрана из 10000, 20000, 30000, 40000, 50000 и 60000 Да. Еще более предпочтительно молекулярная масса выбрана из 20000, 30000, 40000 или 60000 Да. Если присутствуют две молекулы ПЭГ, ковалентно присоединенные к пептидному агонисту по настоящему изобретению, молекулярная масса каждой составляет 1000-40000 Да, предпочтительно их молекулярная масса составляет 20000 и 20000 Да, 10000 и 30000 Да, 30000 и 30000 Да или 20000 и 40000 Да. Последовательность пегилированного пептидного агониста рецептора VPAC2 может дополнительно содержать остаток гистидина на N-конце пептида перед Xaa 1 . Предпочтительно пегилированный пептидный агонист рецептора VPAC2 в соответствии с первым аспектом настоящего изобретения дополнительно содержит N-концевую модификацию на N-конце пептидного агониста, где N-концевая модификация выбрана из: (a) добавления D-гистидина, изолейцина, метионина или норлейцина; (b) добавления пептида, содержащего последовательность Ser-Trp-Cys-Glu-Pro-Gly-Trp-Cys-Arg (SEQ ID NO: 93), где Arg связан с N-концом пептидного агониста; (c) добавления C 1 -C 16 -алкила, необязательно замещенного одним или несколькими заместителями, независимо выбранными из арила, C 1 -C 6 -алкокси, -NH 2 , -ОН, галогена и -CF 3 ; (d) добавления -C(O)R 1 , где R 1 представляет собой C 1 -C 16 -алкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из арила, C 1 -C 6 -алкокси, -NH 2 , -ОН, галогена, -SH и -CF 3 ; арил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C 1 -C 6 -алкила, C 2 -C 6 -алкенила, C 2 -C 6 -алкинила, C 1 -C 6 -алкокси, -NH 2 , ОН, галогена и -CF 3 ; арил-C 1 -C 4 -алкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C 1 -C 6 -алкила, C 2 -C 6 -алкенила, C 2 -C 6 -алкинила, C 1 -C 6 -алкокси, -NH 2 , ОН, галогена и -CF 3 ; -NR 2 R 3 , где R 2 и R 3 независимо представляют собой водород, C 1 -C 6 -алкил, арил или арил-C 1 -C- 4 алкил; -OR 4 , где R 4 представляет собой C 1 -C 16 -алкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из арила, C 1 -C 6 -алкокси, -NH 2 , -ОН, галогена и -CF 3 ; арил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C 1 -C 6 -алкила, C 2 -C 6 -алкенила, C 2 -C 6 -алкинила, C 1 -C 6 -алкокси, -NH 2 , -ОН, галогена и -CF 3 ; или арил-C 1 -C 4 -алкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C 1 -C 6 -алкила, C 2 -C 6 -алкенила, C 2 -C 6 -алкинила, C 1 -C 6 -алкокси, -NH 2 , -ОН, галогена и -CF 3 ; или 5-пирролидин-2-он; (e) добавления -SO 2 R 5 , где R 5 представляет собой арил, арил-C 1 -C 4 -алкил или C 1 -C 16 -алкил; (f) образования сукцинимидной группы, необязательно замещенной C 1 -C 6 -алкилом или -SR 6 , где R 6 представляет собой водород или C 1 -C 6 -алкил; (g) добавления метионинсульфоксида; (h) добавления биотинил-6-аминогексановой кислоты (6-аминокапроновая кислота) и (i) добавления -C(=NH)-NH 2 . Предпочтительно N-концевая модификация представляет собой добавление группы, выбранной из ацетила, пропионила, бутирила, пентаноила, гексаноила, метионина, метионинсульфоксида, 3-фенилпропионила, фенилацетила, бензоила, норлейцина, D-гистидина, изолейцина, 3-меркаптопропионила, биотинил-6-аминогексановой кислоты (6-аминокапроновой кислоты) и -C(=NH)-NH 2 . Особенно предпочтительно N-концевая модификация представляет собой добавление ацетила или гексаноила. Специалисту в данной области будет понятно, что пегилированные пептидные агонисты рецептора VPAC2, содержащие различные комбинации пептидной последовательности, выбранной из SEQ ID NO: 17-45 и 94-112, C-концевые удлиняющие сегменты и N-концевые модификации, раскрытые в данном описании, могут быть осуществлены на основании приведенного выше раскрытия. Предпочтительно пегилированный пептидный агонист рецептора VPAC2 в соответствии с первым аспектом настоящего изобретения содержит последовательность аминокислот, выбранную из Более предпочтительно пегилированный пептидный агонист рецептора VPAC2 в соответствии с первым аспектом настоящего изобретения содержит последовательность аминокислот, выбранную из SEQ ID NO: 47, 64, 66, 115, 119, 122, 126, 130 и 144. В соответствии со вторым аспектом настоящего изобретения предлагается пегилированный пептидный агонист рецептора VPAC2, содержащий последовательность аминокислот, выбранную из Более предпочтительно пегилированный пептидный агонист рецептора VPAC2 в соответствии со вторым аспектом настоящего изобретения содержит последовательность аминокислот SEQ ID NO: 80 или SEQ ID NO: 83. В соответствии с третьим аспектом настоящего изобретения предлагается пегилированный пептидный агонист рецептора VPAC2, содержащий последовательность аминокислот формулы где Xaa 1 представляет собой His, dH или отсутствует; Xaa 2 представляет собой dA, Ser, Val, Gly, Thr, Leu, dS, Pro или Aib; Xaa 3 представляет собой Asp или Glu; Xaa 4 представляет собой Ala, Ile, Tyr, Phe, Val, Thr, Leu, Trp, Gly, dA, Aib или NMeA; Xaa 5 представляет собой Val, Leu, Phe, Ile, Thr, Trp, Tyr, dV, Aib или NMeV; Xaa 6 представляет собой Phe, Ile, Leu, Thr, Val, Trp или Tyr; Xaa 8 представляет собой Asp, Glu, Ala, Lys, Leu, Arg или Tyr; Xaa 9 представляет собой Asn, Gln, Asp, Glu, Ser, Cys, Lys или K(CO(CH 2 ) 2 SH); Xaa 10 представляет собой Tyr, Trp, Tyr(OMe), Ser, Cys или Lys; Xaa 12 представляет собой Arg, Lys, Glu, hR, Orn, Lys (изопропил), Aib, Cit, Ala, Leu, Gln, Phe, Ser или Cys; Xaa 13 представляет собой Leu, Phe, Glu, Ala, Aib, Ser, Cys, Lys или K(CO(CH 2 ) 2 SH); Xaa 14 представляет собой Arg, Leu, Lys, Ala, hR, Orn, Lys (изопропил), Phe, Gln, Aib, Cit, Ser или Cys; Xaa 15 представляет собой Lys, Ala, Arg, Glu, Leu, hR, Orn, Lys (изопропил), Phe, Gln, Aib, K(Ac), Cit, Ser, Cys, K(W) или K(CO(CH 2 ) 2 SH); Xaa 16 представляет собой Gln, Lys, Glu, Ala, hR, Orn, Lys (изопропил), Cit, Ser, Cys, K(CO(CH 2 ) 2 SH) или K(W); Xaa 17 представляет собой Val, Ala, Leu, Ile, Met, Nle, Lys, Aib, Ser, Cys, K(CO(CH 2 ) 2 SH) или K(W); Xaa 18 представляет собой Ala, Ser, Cys, Lys, K(CO(CH 2 ) 2 SH), K(W), Abu или Nle; Xaa 20 представляет собой Lys, Gln, hR, Arg, Ser, His, Orn, Lys (изопропил), Ala, Aib, Trp, Thr, Leu, Ile, Phe, Tyr, Val, K(Ac), Cit, Cys, K(CO(CH 2 ) 2 SH) или K(W); Xaa 21 представляет собой Lys, His, Arg, Ala, Phe, Aib, Leu, Gln, Orn, hR, K(Ac), Cit, Ser, Cys, Val, Tyr, Ile, Thr, Trp, K(W) или K(CO(CH 2 ) 2 SH); Xaa 22 представляет собой Tyr, Trp, Phe, Thr, Leu, He, Val, Tyr(OMe), Ala, Aib, Ser, Cys, Lys, K(W) или K(CO(CH 2 ) 2 SH); Xaa 23 представляет собой Leu, Phe, Ile, Ala, Trp, Thr, Val, Aib, Ser, Cys, Lys, K(W) или K(CO(CH 2 ) 2 SH); Xaa 24 представляет собой Gln, Glu, Asn, Ser, Cys, Lys, K(CO(CH 2 ) 2 SH) или K(W); Xaa 25 представляет собой Ser, Asp, Phe, Ile, Leu, Thr, Val, Trp, Gln, Asn, Tyr, Aib, Glu, Cys, Lys, K(CO(CH 2 ) 2 SH) или K(W); Xaa 26 представляет собой Ile, Leu, Thr, Val, Trp, Tyr, Phe, Aib, Ser, Cys, Lys, K(CO(CH 2 ) 2 SH) или K(W); Xaa 27 представляет собой Lys, hR, Arg, Gln, Ala, Asp, Glu, Phe, Gly, His, Ile, Met, Asn, Pro, Ser, Thr, Val, Trp, Tyr, Lys (изопропил), Cys, Leu, Orn, dK, K(W) или K(CO(CH 2 ) 2 SH); Xaa 28 представляет собой Asn, Asp, Gln, Lys, Arg, Aib, Orn, hR, Cit, Pro, dK, Ser, Cys, K(CO(CH 2 ) 2 SH) или K(W); Xaa 29 представляет собой Lys, Ser, Arg, Asn, hR, Ala, Asp, Glu, Phe, Gly, His, Ile, Leu, Met, Pro, Gln, Thr, Val, Trp, Tyr, Cys, Orn, Cit, Aib, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; Xaa 30 представляет собой Arg, Lys, Ile, Ala, Asp, Glu, Phe, Gly, His, Leu, Met, Asn, Pro, Gln, Ser, Thr, Val, Trp, Tyr, Cys, hR, Cit, Aib, Orn, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; Xaa 31 представляет собой Tyr, His, Phe, Thr, Cys, Ser, Lys, Gln, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; Xaa 32 представляет собой Ser, Cys, Lys или отсутствует; Xaa 33 представляет собой Trp или отсутствует; Xaa 34 представляет собой Cys или отсутствует; Xaa 35 представляет собой Glu или отсутствует; Xaa 36 представляет собой Pro или отсутствует; Xaa 37 представляет собой Gly или отсутствует; Xaa 38 представляет собой Trp или отсутствует; Xaa 39 представляет собой Cys или отсутствует; и Xaa 40 представляет собой Arg или отсутствует, где, если Xaa 29 , Xaa 30 , Xaa 31 , Xaa 32 , Xaa 33 , Xaa 34 , Xaa 35 , Xaa 36 , Xaa 37 , Xaa 38 или Xaa 39 отсутствует, следующая аминокислота в направлении 5'-3' представляет собой следующую аминокислоту в последовательности пептидного агониста, и C-концевой удлиняющий сегмент, где N-конец C-концевого удлиняющего сегмента связан с C-концом пептида формулы 4, и где C-концевой удлиняющий сегмент содержит последовательность аминокислот формулы где Xaa 1 представляет собой Gly, Cys или отсутствует; Xaa 2 представляет собой Gly, Arg или отсутствует; Xaa 3 представляет собой Pro, Thr или отсутствует; Xaa 4 представляет собой Ser или отсутствует; Xaa 5 представляет собой Ser или отсутствует; Xaa 6 представляет собой Gly или отсутствует; Xaa 7 представляет собой Ala или отсутствует; Xaa 8 представляет собой Pro или отсутствует; Xaa 9 представляет собой Pro или отсутствует; Xaa 10 представляет собой Pro или отсутствует; Xaa 11 представляет собой Ser, Cys или отсутствует; и Xaa 12 представляет собой Cys или отсутствует; где присутствуют по меньшей мере пять из Xaa 1 -Xaa 12 C-концевого удлиняющего сегмента, и где, если Xaa 1 , Xaa 2 , Xaa 3 , Xaa 4 , Xaa 5 , Xaa 6 , Xaa 7 , Xaa 8 , Xaa 9 , Xaa 10 или Xaa 11 отсутствует, следующая аминокислота в направлении 5'-3' представляет собой следующую аминокислоту в C-концевом удлиняющем сегменте, и где C-концевая аминокислота может быть амидирована, и где пептидный агонист содержит по меньшей мере один остаток Cys, который ковалентно присоединен к молекуле ПЭГ, или пептидный агонист содержит по меньшей мере один остаток Lys, который ковалентно присоединен к молекуле ПЭГ, или пептидный агонист содержит по меньшей мере один K(W), который ковалентно присоединен к молекуле ПЭГ, или пептидный агонист содержит по меньшей мере один K(CO(CH 2 ) 2 SH), который ковалентно присоединен к молекуле ПЭГ, или карбоксиконцевая аминокислота пептидного агониста ковалентно присоединена к молекуле ПЭГ, или комбинацию указанных характеристик. Предпочтительно пегилированный пептидный агонист рецептора VPAC2 в соответствии с третьим аспектом настоящего изобретения содержит последовательность формулы 4 (SEQ ID NO: 4), где Xaa 3 представляет собой Asp или Glu; Xaa 8 представляет собой Asp или Glu; Xaa 9 представляет собой Asn или Gln; Xaa 10 представляет собой Tyr или Tyr(OMe); Xaa 12 представляет собой Arg, hR, Lys или Orn; Xaa 14 представляет собой Arg, Gln, Aib, hR, Orn, Cit, Lys, Ala или Leu; Xaa 15 представляет собой Lys, Aib, Orn или Arg; Xaa 16 представляет собой Gln или Lys; Xaa 17 представляет собой Val, Leu, Ala, Ile, Lys или Nle; Xaa 20 представляет собой Lys, Val, Leu, Aib, Ala, Gln или Arg; Xaa 21 представляет собой Lys, Aib, Orn, Ala, Gln или Arg; Xaa 23 представляет собой Leu или Aib; Xaa 25 представляет собой Ser или Aib; Xaa 27 представляет собой Lys, Orn, hR или Arg; Xaa 28 представляет собой Asn, Gln, Lys, hR, Aib, Orn или Pro и Xaa 29 представляет собой Lys, Orn, hR или отсутствует. Предпочтительно пегилированный пептидный агонист рецептора VPAC2 в соответствии с третьим аспектом настоящего изобретения содержит последовательность формулы 4 (SEQ ID NO: 4), где либо Xaa 23 , либо Xaa 25 представляет собой Aib. Еще более предпочтительно Xaa 23 и Xaa 25 , оба, представляют собой Aib. Предпочтительно пегилированный пептидный агонист рецептора VPAC2 в соответствии с третьим аспектом настоящего изобретения содержит последовательность формулы 4, где либо Xaa 14 , либо Xaa 15 представляет собой Aib. Альтернативно, пегилированный пептидный агонист рецептора VPAC2 в соответствии с третьим аспектом настоящего изобретения содержит последовательность формулы 4, где либо Xaa 20 , либо Xaa 21 представляет собой Aib. Более предпочтительно либо Xaa 14 , либо Xaa 15 представляет собой Aib и либо Xaa 20 , либо Xaa 21 представляет собой Aib. Особенно предпочтительно Xaa 15 представляет собой Aib и Xaa 20 представляет собой Aib. Предпочтительно пегилированный пептидный агонист рецептора VPAC2 в соответствии с третьим аспектом настоящего изобретения содержит последовательность формулы 4, где Xaa 15 представляет собой Aib; Xaa 20 представляет собой Aib и Xaa 12 , Xaa 21 , Xaa 27 и Xaa 28 , все, представляют собой Orn. Более предпочтительно Xaa 15 представляет собой Aib, Xaa 20 представляет собой Aib, Xaa 12 , Xaa 21 , Xaa 27 и Xaa 28 , все, представляют собой Orn, Xaa 8 представляет собой Glu, Xaa 9 представляет собой Gln и Xaa 10 представляет собой Tyr(OMe). Еще более предпочтительно Xaa 15 представляет собой Aib, Xaa 20 представляет собой Aib, Xaa 12 , Xaa 21 , Xaa 27 и Xaa 28 , все, представляют собой Orn, Xaa 8 представляет собой Glu, Xaa 9 представляет собой Gln, Xaa 10 представляет собой Tyr(OMe) и Xaa 23 и/или Xaa 25 представляет собой Aib. Любой один или несколько из Xaa 8 , Xaa 9 , Xaa 10 , Xaa 12 , Xaa 15 , Xaa 20 , Xaa 21 , Xaa 23 , Xaa 25 , Xaa 27 и Xaa 28 может представлять собой пегилированный Lys, Cys, K(CO(CH 2 ) 2 SH) или K(W), тогда как все остальные положения содержат предпочтительные замены аминокислот, как было описано. Предпочтительно присутствуют по меньшей мере шесть из Xaa 1 -Xaa 12 C-концевого удлиняющего сегмента формулы 3. Более предпочтительно присутствуют семь, восемь, девять, десять, одиннадцать или все Xaa 1 -Xaa 12 C-концевого удлиняющего сегмента. Предпочтительно C-концевой удлиняющий сегмент пегилированного пептидного агониста рецептора VPAC2 в соответствии с третьим аспектом настоящего изобретения выбран из SEQ ID NO: 5, 6, 7, 8, 9, 10, 11 и 12. Более предпочтительно C-концевой удлиняющий сегмент пегилированного пептидного агониста рецептора VPAC2 в соответствии с третьим аспектом настоящего изобретения представляет собой SEQ ID NO: 11 или SEQ ID NO: 12. Молекула(ы) ПЭГ может быть ковалентно присоединена к любому остатку Lys, Cys, K(W) или K(CO(CH 2 ) 2 SH) в любом положении пептидного агониста рецептора VPAC2 в соответствии с третьим аспектом настоящего изобретения. C-концевой удлиняющий сегмент может содержать один или несколько остатков Cys, которые могут быть пегилированными. Если последовательность в соответствии с формулой 4 содержит один или несколько остатков Lys, Cys, K(W) или K(CO(CH 2 ) 2 SH) и C-концевой удлиняющий сегмент содержит один или несколько остатков Cys, одна или обе последовательности могут содержать один или несколько пегилированных остатков. Предпочтительно присутствует по меньшей мере одна молекула ПЭГ, ковалентно присоединенная к остатку в формуле 4. Более предпочтительно присутствует молекула ПЭГ, ковалентно присоединенная к остатку в одном или нескольких из следующих положений формулы 4: 9, 13, 15, 16, 17, 18, 20, 21, 24, 25, 26 и 28. Предпочтительно присутствует по меньшей мере одна молекула ПЭГ, ковалентно присоединенная к остатку в C-концевом удлиняющем сегменте пептидного агониста рецептора VPAC2. Если присутствует несколько молекул ПЭГ, может присутствовать комбинация пегилирования Lys, Cys, K(CO(CH 2 ) 2 SH), K(W) и карбоксиконцевой аминокислоты. Например, если присутствуют две молекулы ПЭГ, одна из них может быть присоединена к остатку Lys, а другая может быть присоединена к остатку Cys. Предпочтительно молекула ПЭГ является разветвленной. Альтернативно, молекула ПЭГ может быть линейной. Предпочтительно молекулярная масса молекулы ПЭГ составляет от 1000 до 100000 Да. Более предпочтительно молекулярная масса молекулы ПЭГ выбрана из 10000, 20000, 30000, 40000, 50000 и 60000 Да. Еще более предпочтительно молекулярная масса выбрана из 20000, 30000, 40000 или 60000 Да. Если присутствуют две молекулы ПЭГ, ковалентно присоединенные к пептидному агонисту по настоящему изобретению, молекулярная масса каждой составляет 1000-40000 Да, предпочтительно их молекулярная масса составляет 20000 и 20000 Да, 10000 и 30000 Да, 30000 и 30000 Да или 20000 и 40000 Да. Предпочтительно пегилированный пептидный агонист рецептора VPAC2 в соответствии с третьим аспектом настоящего изобретения дополнительно содержит N-концевую модификацию на N-конце пептидного агониста, где N-концевая модификация выбрана из: (a) добавления D-гистидина, изолейцина, метионина или норлейцина; (b) добавления пептида, содержащего последовательность Ser-Trp-Cys-Glu-Pro-Gly-Trp-Cys-Arg (SEQ ID NO: 93), где Arg связан с N-концом пептидного агониста; (c) добавления C 1 -C 16 -алкила, необязательно замещенного одним или несколькими заместителями, независимо выбранными из арила, C 1 -C 6 -алкокси, -NH 2 , -ОН, галогена и -CF 3 ; (d) добавления -C(O)R 1 , где R 1 представляет собой C 1 -C 16 -алкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из арила, C 1 -C 6 -алкокси, NH 2 , -ОН, галогена, -SH и -CF 3 ; арил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C 1 -C 6 -алкила, C 2 -C 6 -алкенила, C 2 -C 6 -алкинила, C 1 -C 6 -алкокси, -NH 2 , ОН, галогена и -CF 3 ; и арил-C 1 -C 4 -алкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C 1 -C 6 -алкила, C 2 -C 6 -алкенила, C 2 -C 6 -алкинила, C 1 -C 6 -алкокси, -NH 2 , ОН, галогена и -CF 3 ; -NR 2 R 3 , где R 2 и R 3 независимо представляют собой водород, C 1 -C 6 -алкил, арил или арил-C 1 -C 4 -алкил; -OR 4 , где R 4 представляет собой C 1 -C 16 -алкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из арила, C 1 -C 6 -алкокси, -NH 2 , -ОН, галогена и -CF 3 ; арил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C 1 -C 6 -алкила, C 2 -C 6 -алкенила, C 2 -C 6 -алкинила, C 1 -C 6 -алкокси, -NH 2 , -ОН, галогена и -CF 3 ; или арил-C 1 -C 4 -алкил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из C 1 -C 6 -алкила, C 2 -C 6 -алкенила, C 2 -C 6 -алкинил, C 1 -C 6 -алкокси, -NH 2 , -ОН, галогена и -CF 3 ; или 5-пирролидин-2-он; (e) добавления -SO 2 R 5 , где R 5 представляет собой арил, арил-C 1 -C 4 -алкил или C 1 -C 16 -алкил; (f) образования сукцинимидной группы, необязательно замещенной C 1 -C 6 -алкилом или -SR 6 , где R 6 представляет собой водород или C 1 -C 6 -алкил; (g) добавления метионинсульфоксида; (h) добавления биотинил-6-аминогексановой кислоты (6-аминокапроновая кислота) и (i) добавления -C(=NH)-NH 2 . Предпочтительно N-концевая модификация представляет собой добавление группы, выбранной из ацетила, пропионила, бутирила, пентаноила, гексаноила, метионинсульфоксида, метионина, 3-фенилпропионила, фенилацетила, бензоила, норлейцина, D-гистидина, изолейцина, 3-меркаптопропионила, биотинил-6-аминогексановой кислоты (6-аминокапроновая кислота) и -C(=NH)-NH 2 . Особенно предпочтительно N-концевая модификация представляет собой добавление ацетила или гексаноила. Специалисту в данной области будет понятно, что пегилированные пептидные агонисты рецептора VPAC2, содержащие различные комбинации пептидной последовательности в соответствии с формулой 4, C-концевые удлиняющие фрагменты и N-концевые модификации, как раскрыто в данном описании, могут быть осуществлены на основании приведенного выше раскрытия. Предпочтительно пегилированный пептидный агонист рецептора VPAC2 в соответствии с третьим аспектом настоящего изобретения содержит последовательность аминокислот, выбранную из SEQ ID NO: 59, 62, 64, 65, 66, 71, 72, 73, 74, 115, 116, 117, 118, 119, 120, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 135, 136, 137, 141, 144, 146 и 148. В соответствии с четвертым аспектом настоящего изобретения обеспечивается фармацевтическая композиция, содержащая пегилированный пептидный агонист рецептора VPAC2 по настоящему изобретению и один или несколько фармацевтически приемлемых наполнителей носителей и/или вспомогательных веществ. В соответствии с пятым аспектом настоящего изобретения предлагается пегилированный пептидный агонист рецептора VPAC2 по настоящему изобретению для применения в качестве лекарственного средства. В соответствии с шестым аспектом настоящего изобретения предлагается применение пегилированного пептидного агониста рецептора VPAC2 по настоящему изобретению для производства лекарственного средства для лечения инсулиннезависимого диабета. В соответствии со следующим аспектом настоящего изобретения предлагается применение пегилированного пептидного агониста рецептора VPAC2 по настоящему изобретению для производства лекарственного средства для лечения инсулинзависимого диабета. Настоящее изобретение обеспечивает способ лечения диабета у пациента, который в этом нуждается, который включает введение пегилированного пептидного агониста рецептора VPAC2 по настоящему изобретению, где диабет может представлять собой инсулиннезависимый диабет или инсулинзависимый диабет. Настоящее изобретение дополнительно предлагает фармацевтическую композицию, содержащую пегилированный пептидный агонист рецептора VPAC2 по настоящему изобретению, для лечения инсулиннезависимого диабета или инсулинзависимого диабета. В соответствии с альтернативным вариантом настоящего изобретения предлагается пегилированный пептидный агонист рецептора VPAC2, содержащий последовательность, выбранную из SEQ ID NO: 17-45 и 94-112; и C-концевой удлиняющий сегмент, где N-конец C-концевого удлиняющего сегмента связан с C-концом пептидной последовательности и где C-концевой удлиняющий сегмент содержит последовательность аминокислот формулы где Xaa 1 представляет собой Gly, Cys, Lys, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; Xaa 2 представляет собой Gly, Arg, Cys, Lys, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; Xaa 3 представляет собой Pro, Thr, Ser, Ala, Cys, Lys, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; Xaa 4 представляет собой Ser, Pro, His, Cys, Lys, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; Xaa 5 представляет собой Ser, Arg, Thr, Trp, Lys, Cys, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; Xaa 6 представляет собой Gly, Ser, Cys, Lys, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; Xaa 7 представляет собой Ala, Asp, Arg, Glu, Lys, Gly, Cys, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; Xaa 8 представляет собой Pro, Ser, Ala, Cys, Lys, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; Xaa 9 представляет собой Pro, Ser, Ala, Cys, Lys, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; Xaa 10 представляет собой Pro, Ser, Ala, Arg, Lys, His, Cys, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; Xaa 11 представляет собой Ser, Cys, His, Pro, Lys, Arg, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; Xaa 12 представляет собой His, Ser, Arg, Lys, Cys, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; и Xaa 13 представляет собой His, Ser, Arg, Lys, Cys, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; при условии, что, если Xaa 1 , Xaa 2 , Xaa 3 , Xaa 4 , Xaa 5 , Xaa 6 , Xaa 7 , Xaa 8 , Xaa 9 , Xaa 10 , Xaa 11 или Xaa 12 отсутствует, следующая аминокислота в направлении 5'-3' представляет собой следующую аминокислоту в C-концевом удлиняющем сегменте, где C-концевая аминокислота может быть амидирована и где пептидный агонист содержит по меньшей мере один остаток Cys, который ковалентно присоединен к молекуле ПЭГ, или пептидный агонист содержит по меньшей мере один остаток Lys, который ковалентно присоединен к молекуле ПЭГ, или пептидный агонист содержит по меньшей мере один K(W), который ковалентно присоединен к молекуле ПЭГ, или пептидный агонист содержит по меньшей мере один K(CO(CH 2 ) 2 SH), который ковалентно присоединен к молекуле ПЭГ, или карбоксиконцевая аминокислота пептидного агониста ковалентно присоединена к молекуле ПЭГ, или комбинацию указанных характеристик. Предпочтительно C-концевой удлиняющий сегмент формулы 1 содержит не более трех любых из следующих остатков: Cys, Lys, K(W) или K(CO(CH 2 ) 2 SH). Более предпочтительно C-концевой удлиняющий сегмент содержит не более двух любых из указанных остатков. Если присутствуют два остатка Cys в C-концевом удлиняющем сегменте, предпочтительно остатки Cys находятся на C-конце. Еще более предпочтительно C-концевой удлиняющий сегмент содержит не более одного любого из указанных остатков. Если присутствует только один остаток Cys в C-концевом удлиняющем сегменте, предпочтительно остаток Cys находится на C-конце. Предпочтительно C-концевой удлиняющий сегмент пегилированного пептидного агониста рецептора VPAC2 в соответствии с указанным выше альтернативным вариантом содержит последовательность аминокислот формулы где Xaa 1 представляет собой Gly, Cys, Lys или отсутствует; Xaa 2 представляет собой Gly, Arg, Cys, Lys или отсутствует; Xaa 3 представляет собой Pro, Thr, Ser, Ala, Cys, Lys или отсутствует; Xaa 4 представляет собой Ser, Pro, His, Cys, Lys или отсутствует; Xaa 5 представляет собой Ser, Arg, Thr, Trp, Lys, Cys или отсутствует; Xaa 6 представляет собой Gly, Ser, Cys, Lys или отсутствует; Xaa 7 представляет собой Ala, Asp, Arg, Glu, Lys, Gly, Cys или отсутствует; Xaa 8 представляет собой Pro, Ser, Ala, Cys, Lys или отсутствует; Xaa 9 представляет собой Pro, Ser, Ala, Cys, Lys или отсутствует; Xaa 10 представляет собой Pro, Ser, Ala, Arg, Lys, His, Cys или отсутствует; Xaa 11 представляет собой Ser, Cys, His, Pro, Lys, Arg или отсутствует; Xaa 12 представляет собой His, Ser, Arg, Lys, Cys или отсутствует; и Xaa 13 представляет собой His, Ser, Arg, Lys, Cys или отсутствует; при условии, что, если Xaa 1 , Xaa 2 , Xaa 3 , Xaa 4 , Xaa 5 , Xaa 6 , Xaa 7 , Xaa 8 , Xaa 9 , Xaa 10 , Xaa 11 или Xaa 12 отсутствует, следующая аминокислота в направлении 5'-3' представляет собой следующую аминокислоту в C-концевом удлиняющем сегменте, и где C-концевая аминокислота может быть амидирована. Предпочтительно присутствует по меньшей мере одна из Xaa 1 -Xaa 13 C-концевого удлиняющего сегмента формулы 1 или 2. Более предпочтительно присутствуют по меньшей мере две, три, четыре, пять, шесть, семь, восемь, девять, десять, одиннадцать, двенадцать или все Xaa 1 -Xaa 13 C-концевого удлиняющего сегмента. Более предпочтительно C-концевой удлиняющий сегмент пегилированного пептидного агониста рецептора VPAC2 в соответствии с описанным выше альтернативным вариантом содержит последовательность аминокислот формулы где Xaa 1 представляет собой Gly, Cys или отсутствует; Xaa 2 представляет собой Gly, Arg или отсутствует; Xaa 3 представляет собой Pro, Thr или отсутствует; Xaa 4 представляет собой Ser или отсутствует; Xaa 5 представляет собой Ser или отсутствует; Xaa 6 представляет собой Gly или отсутствует; Xaa 7 представляет собой Ala или отсутствует; Xaa 8 представляет собой Pro или отсутствует; Xaa 9 представляет собой Pro или отсутствует; Xaa 10 представляет собой Pro или отсутствует; Xaa 11 представляет собой Ser, Cys или отсутствует; и Xaa 12 представляет собой Cys или отсутствует; при условии, что, если Xaa 1 , Xaa 2 , Xaa 3 , Xaa 4 , Xaa 5 , Xaa 6 , Xaa 7 , Xaa 8 , Xaa 9 , Xaa 10 или Xaa 11 отсутствует, следующая аминокислота в направлении 5'-3' представляет собой следующую аминокислоту в C-концевом удлиняющем сегменте, и где C-концевая аминокислота может быть амидирована. Предпочтительно присутствует по меньшей мере одна из Xaa 1 -Xaa 12 C-концевого удлиняющего сегмента формулы 3. Более предпочтительно присутствуют по меньшей мере две, три, четыре, пять, шесть, семь, восемь, девять, десять, одиннадцать или все Xaa 1 -Xaa 12 C-концевого удлиняющего сегмента. Альтернативный C-концевой удлиняющий сегмент, который может использоваться в любом из аспектов и вариантов настоящего изобретения, содержит последовательность аминокислот формулы где Xaa 1 представляет собой Ser, Cys, Lys, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; Xaa 2 представляет собой Arg, Ser, hR, Orn, His, Cys, Lys, K(W), K(CO(CH 2 ) 2 SH) или отсутствует, Xaa 3 представляет собой Thr, Cys, Lys, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; Xaa 4 представляет собой Ser, Cys, Lys, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; Xaa 5 представляет собой Pro, Ser, Ala, Cys, Lys, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; Xaa 6 представляет собой Pro, Ser, Ala, Arg, Cys, Lys, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; Xaa 7 представляет собой Pro, Ser, Ala, Cys, Lys, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; Xaa 8 представляет собой Lys, K(W), Pro, Cys, K(CO(CH 2 ) 2 SH) или отсутствует; Xaa 9 представляет собой K(E-C 16 ), Ser, Cys, Lys, K(W), K(CO(CH 2 ) 2 SH) или отсутствует; и Xaa 10 представляет собой Ser, Cys, Lys, K(W), K(CO(CH 2 ) 2 SH) или отсутствует. Предпочтительно, если Xaa 1 , Xaa 2 , Xaa 3 , Xaa 4 , Xaa 5 , Xaa 6 , Xaa 7 , Xaa 8 или Xaa 9 формулы 13 отсутствует, следующая аминокислота в направлении 5'-3' представляет собой следующую аминокислоту в C-концевом удлиняющем сегменте. C-концевая аминокислота может быть амидирована. Предпочтительно присутствует по меньшей мере одна из Xaa 1 -Xaa 10 C-концевого удлиняющего сегмента формулы 13. Более предпочтительно присутствуют по меньшей мере две, три, четыре, пять, шесть, семь, восемь, девять или все Xaa 1 -Xaa 10 C-концевого удлиняющего сегмента. Более предпочтительно альтернативный C-концевой удлиняющий сегмент, который может использоваться в любом из аспектов и вариантов настоящего изобретения, выбран из Пептидные агонисты рецептора VPAC2 по настоящему изобретению обладают преимуществом повышенной селективности, эффективности и/или стабильности по сравнению с известными петидными агонистами рецептора VPAC2. В частности, добавление C-концевой последовательности экзендина-4 или варианта этой C-концевой последовательности в качестве C-кэппинга последовательности неожиданно увеличило селективность в отношении рецептора VPAC2, а также увеличило стабильность в протеолитических условиях. Ковалентное присоединение одной или нескольких молекул ПЭГ к конкретным остаткам пептидного агониста рецептора VPAC2 дает биологически активный, пегилированный пептидный агонист рецептора VPAC2 с увеличенным периодом полувыведения и сниженным клиренсом по сравнению с не-пегилированными пептидными агонистами рецептора VPAC2. Термин "VPAC2" используется для обозначения и в связи с конкретным рецептором (Lutz, et al., FEBS Lett., 458: 197-203 (1999); Adamou, et al., Biochem. Biophys. Res. Commun., 209: 385-392 (1995)), который активируют агонисты по настоящему изобретению. Данный термин также используется для обозначения и в связи с агонистами по настоящему изобретению. "Селективный пептидный агонист рецептора VPAC2" или "пептидный агонист рецептора VPAC2" по настоящему изобретению представляет собой пептид, который избирательно активизирует рецептор VPAC2 для индукции секреции инсулина. Предпочтительно последовательность для селективного пептидного агониста рецептора VPAC2 по настоящему изобретению содержит от 28 до 40 природных и/или неприродных аминокислот и может дополнительно содержать или не содержать C-концевой удлиняющий сегмент. "Селективный пегилированный пептидный агонист рецептора VPAC2" или "пегилированный пептидный агонист рецептора VPAC2" представляет собой селективный пептидный агонист рецептора VPAC2, ковалентно связанный с одной или несколькими молекулами полиэтиленгликоля (ПЭГ, PEG) или его производного, где каждая молекула ПЭГ присоединена к аминокислоте цистеину или лизину к K(W) или K(CO(CH 2 ) 2 SH) или карбоксильному концу пептида. Селективные пегилированные пептидные агонисты рецептора VPAC2 могут содержать C-концевой удлиняющий сегмент. "C-концевой удлиняющий сегмент" по настоящему изобретению содержит последовательность, включающую от 1 до 13 природных или неприродных аминокислот, связанных с C-концом последовательности на N-конце C-концевого удлиняющего сегмента посредством пептидной связи. Любые остатки Cys, Lys, K(W) или K(CO(CH 2 ) 2 SH) в C-концевом удлиняющем сегменте могут быть ковалентно связаны с молекулой ПЭГ и/или карбоксиконцевая аминокислота C-концевого удлиняющего сегмента может быть ковалентно присоединена к молекуле ПЭГ. В данном описании термин "связанный с" в связи с термином "C-концевой удлиняющий сегмент" включает добавление или присоединение аминокислот или химических групп непосредственно к C-концу пептидной последовательности. Селективный пегилированный пептидный агонист рецептора VPAC2 также может необязательно содержать N-концевую модификацию. Термин "N-концевая модификация" в данном описании включает добавление или присоединение аминокислот или химических групп непосредственно к N-концу пептида и образование химических групп, которые включают азот на N-конце пептида. N-концевая модификация может включать добавление одной или нескольких природных или неприродных аминокислот к последовательности пептидного агониста рецептора VPAC2, предпочтительно присутствует не более десяти аминокислот, причем присутствие одной аминокислоты является более предпочтительным. Природные аминокислоты, которые могут быть добавлены к N-концу, включают метионин и изолейцин. Модифицированная кислота для добавления к N-концу может быть D-гистидином. Альтернативно, следующие аминокислоты могут быть добавлены к N-концу: SEQ ID NO: 93 Ser-Trp-Cys-Glu-Pro-Gly-Trp-Cys-Arg, где Arg связан с N-концом пептидного агониста. Предпочтительно любые аминокислоты, добавленные к N-концу, связаны с N-концом посредством пептидной связи. Термин "связанный с" в данном описании в связи с термином "N-концевая модификация", включает добавление или присоединение аминокислот или химических групп непосредственно к N-концу агониста рецептора VPAC2. Добавление упомянутых выше N-концевых модификаций может быть достигнуто в нормальных условиях соединения для образования пептидной связи. N-конец пептидного агониста также может быть модифицирован путем добавления алкильной группы (R), предпочтительно группы C 1 -C 16 -алкил для образования (R)NH-. Альтернативно, N-конец пептидного агониста может быть модифицирован добавлениям группы формулы -C(O)R 1 с образованием амида формулы R 1 C(O)NH-. Добавление группы формулы -C(O)R 1 может быть осуществлено с помощью реакции с органической кислотой формулы R 1 COOH. Модификация N-конца последовательности аминокислот с помощью ацилирования продемонстрирована в уровне техники (например, Gozes et al., J. Pharmacol. Exp. Ther., 273: 161-167 (1995)). Добавление группы формулы -C(O)R 1 может приводить к образованию группы мочевины (см. WO 01/23240, WO 2004/06839) или группы карбамата на N-конце. Также N-конец может быть модифицирован добавлениям пироглутаминовой кислоты или 6-аминогексановой кислоты. N-конец пептидного агониста может быть модифицирован добавлениям группы формулы -SO 2 R 5 для образования сульфонамидной группы на N-конце. N-конец пептидного агониста также может быть модифицирован реакцией с ангидридом янтарной кислоты для образования сукцинимидной группы на N-конце. Сукцинимидная группа включает азот на N-конце пептида. Альтернативно, N-конец может быть модифицирован добавлениям метионинсульфоксида, биотинил-6-аминогексановой кислоты или -C(=NH)-NH 2 . Добавление -С(=NH)-NH 2 представляет собой модификация гуанидирования, когда концевая группа NH 2 N-концевой аминокислоты превращается в -NH-C(=NH)-NH 2 . Большинство последовательностей по настоящему изобретению, в том числе N-концевые модификации и C-концевые удлиняющие фрагменты, содержат стандартные однобуквенные или трехбуквенные коды для 20 природных аминокислот. Другие используемые коды определены следующим образом: С6 = гексаноил; d = D изоформа (неприродная) соответствующей аминокислоты, например dA = D-аланин, dS = D-серин, dK = D-лизин; hR = гомоаргинин; Aib = аминоизомасляная кислота; ОМе = метокси; Nle = норлейцин; NMe = N-метил, присоединенный к α-аминогруппе аминокислоты, например NMeA = N-метилаланин, NMeV = N-метилвалин; Orn = орнитин; K(CO(CH 2 ) 2 SH) = ε-(3'-меркаптопропионил)лизин; K(W) = ε-(L-триптофил)лизин; Abu = α-амино-н-масляная кислота или 2-аминобутановая кислота; Cit = цитруллин; K(Ас) = ε-ацетиллизин; ПЭГ = полиэтиленгликоль; PEG 40 К = ПЭГ с молекулярной массой 40000 Да; PEG 30 К = ПЭГ с молекулярной массой 30000 Да; PEG 20 К = ПЭГ с молекулярной массой 20000 Да. VIP встречается в природе в виде единой последовательности, имеющей 28 аминокислот. Однако PACAP существует или в виде пептида длиной 38 аминокислот (PACAP-38), или в виде пептида длиной 27 аминокислот (PACAP-27) с амидированным карбоксилом (Miyata, et al., Biochem Biophys Res Commun, 170: 643-648 (1990)). Последовательности VIP, PACAP-27 и PACAP-38 приведены ниже. Термин "природная аминокислота" в данном описании означает 20 аминокислот, кодируемых генетическим кодом человека (т.е. 20 стандартных аминокислот). Эти 20 аминокислот представляют собой: аланин, аргинин, аспарагин, аспарагиновую кислоту, цистеин, глутамин, глутаминовую кислоту, глицин, гистидин, изолейцин, лейцин, лизин, метионин, фенилаланин, пролин, серин, треонин, триптофан, тирозин и валин. Примеры "неприродных аминокислот" включают как синтетические аминокислоты, так и аминокислоты, модифицированные организмом. Они включают D-аминокислоты, аргининподобные аминокислоты (например, гомоаргинин), и другие аминокислоты, содержащие дополнительный остаток метилена в боковой цепи ("гомо" аминокислоты), а также модифицированные аминокислоты (например, норлейцин, лизин [изопропил], где амин боковой цепи лизина модифицирован изопропильной группой). Также включены такие аминокислоты, как орнитин, аминоизомасляная кислота и 2-аминобутановая кислота. Термин "селективный" в данном описании означает пептидный агонист рецептора VPAC2 с повышенной селективностью в отношении рецептора VPAC2 по сравнению с другими известными рецепторами. Степень селективности определяется соотношением сродства связывания с рецептора VPAC2 к сродству связывания с рецептором VPAC1 или соотношением сродства связывания с рецептором VPAC2 к сродству связывания с рецептором РАС1. Сродство связывания определяется, как описано ниже в примере 4. Термин "инсулинотропная активность" означает способность стимулировать секрецию инсулина в ответ на повышение уровней глюкозы, таким образом вызывая захват глюкозы клетками и снижение уровней глюкозы в плазме крови. Инсулинотропная активность может быть оценена по методам, известным из уровня техники, в том числе с использованием экспериментов, в ходе которых измеряется активность связывания с рецептором VPAC2 или активация рецептора (например, секреция инсулина линиями клеток инсулиномы или островками, тест переносимости глюкозы при внутривенном введении (IVGTT), тест переносимости глюкозы при внутрибрюшинном введении (IPGTT) и тест переносимости глюкозы при пероральном введении (OGTT)). Инсулинотропную активность у человека обычно измеряют путем измерения уровней инсулина или уровней C-пептида. Селективные пегилированные пептидные агонисты рецептора VPAC2 по настоящему изобретению обладают инсулинотропной активностью. Термин "эффективность in vitro " в данном описании представляет собой меру способности пептида активировать рецептор VPAC2 в клеточном анализе. Эффективность in vitro выражается как "EC 50 ", которая представляет собой эффективную концентрацию соединения, которая приводит к 50% увеличению активности в эксперименте "одна доза-реакция" по сравнению с максимальным. Для целей настоящего изобретения эффективность in vitro определяют с использованием двух различных анализов: DiscoveRx и Alpha Screen. Подробное описание этих анализов см. в примерах 3 и 5. Хотя указанные анализы выполняются различными способами, результаты демонстрируют общую корреляцию между двумя анализами. Термин "период полувыведения из плазмы" означает время, за которое выводится половина соответствующих молекул, циркулирующих в плазме. Альтернативный термин - "элиминационный период полувыведения". Термин "увеличенный" или "удлиненный" в контексте периода полувыведения из плазмы или элиминационного периода полувыведения указывает на присутствие статистически значимого увеличения периода полувыведения пегилированного пептидного агониста рецептора VPAC2 по отношению к молекуле сравнения (например, непегилированная форма пептида или природный пептид), определенного в сравнимых условиях. Период полувыведения, приведенный в данном описании, представляет собой элиминационный период полувыведения; он является таким, который соответствует терминальной логарифмически линейной скорости элиминации. Специалисту в данной области будет понятно, что период полувыведения является производным параметром, который изменяется как функция клиренса и объема распределения. Клиренс представляет собой меру способности организма к элиминации лекарственного средства. Поскольку клиренс снижается в результате, например, модификации лекарственного средства, ожидается, что период полувыведения будет увеличиваться. Однако данная взаимосвязь точно соблюдается только тогда, когда объем распределения остается неизменным. Полезное приближенное соотношение между терминальным логарифмическим периодом полувыведения (t 1/2 ), клиренсом (С) и объемом распределения (V) дает уравнение: t 1/2 ≈ 0,693 (V/C). Клиренс не показывает, сколько лекарственного средства выведено, скорее, он показывает объем биологической жидкости, такой как кровь или плазма, которая должна быть полностью свободной от лекарственного средства, чтобы объяснить элиминацию. Клиренс выражают как объем за единицу времени. Термин "процент (%) идентичности последовательностей" в данном описании используется для обозначения последовательностей, которые при выравнивании содержат сходные (идентичные остатки или консервативные замены) аминокислоты в подобных положениях или участках, где идентичные аминокислоты или консервативные замены аминокислот являются такими, которые не изменяют активности или функции белка в сравнении с исходным белком. Например, две последовательности аминокислот по меньшей мере с 85% идентичностью по отношению друг к другу содержат по меньшей мере 85% сходных (идентичных остатков или консервативных замен) в сходном положении при выравнивании, оптимально разрешая до 3 промежутков, при условии, что в отношении промежутков затрагивается не более 15 остатков аминокислот. Пептид сравнения, используемый для вычисления процента идентичности последовательностей в данном описании: Процент идентичности последовательностей может быть вычислен путем определения количества остатков, которые отличаются в пептиде, включенном в настоящее изобретение, и пептиде сравнения, таком как Р487 (SEQ ID NO: 84), деления полученного числа на количество аминокислот в пептиде сравнения (например, 39 аминокислот для Р487), умножения результата на 100 и вычитания полученного числа из 100. Например, последовательность, имеющая 39 аминокислот, с четырьмя аминокислотами, которые отличаются от Р487, имела бы процент (%) идентичности последовательностей 90% (например, 100 ((4/39) ×100)). Для последовательности, которая длиннее 39 аминокислот, количество остатков, которые отличаются от последовательности Р487, будет включать дополнительные аминокислоты (свыше 39) для целей вышеупомянутого вычисления. Например, последовательность, имеющая 40 аминокислот, с четырьмя аминокислотами, отличающимися от 39 аминокислот в последовательности Р487, и с одной дополнительной аминокислотой на карбоксильном конце, которая отсутствует в последовательности Р487, имела бы в целом 5 аминокислот, которые отличаются от Р487. Таким образом, данная последовательность демонстрировала бы процент (%) идентичности последовательностей 87% (например, 100 - (5/39) ×100)). Степень идентичности последовательностей может быть определена с использованием способов, хорошо известных из уровня техники (см., например, Wilbur, W.J. and Lipman, D.J., Proc. Natl. Acad. Sci. USA 80: 726-730 (1983) и Myers E. and Miller W., Comput. Appl. Biosci. 4: 11-17 (1988)). Одна из программ, которая может использоваться для определения степени сходства, - метод одной пары MegAlign Липмана-Пирсона (Lipman-Pearson) (применение параметров, используемых по умолчанию), которая может быть получена от DNAstar Inc, 1128, Селфпарк стрит, Мэдисон, Висконсин, 53715, США, как часть системы Lasergene. Другая программа, которая может использоваться, - Clustal W. Это пакет выравнивания нескольких последовательностей, разработанный Thompson et al (Nucleic Acids Research, 22 (22): 4673-4680 (1994)) для последовательностей ДНК или белка. Данный инструмент пригоден для перекрестного сравнения родственных последовательностей и просмотра сохранности последовательностей. Clustal W представляет собой универсальную программу выравнивания нескольких последовательностей для ДНК или белков. Она производит биологически значимое выравнивание нескольких последовательностей для дивергентных последовательностей. Она рассчитывает наилучшую совместимость для отобранных последовательностей и располагает их таким образом, чтобы можно было увидеть идентичность, сходство и различия. Эволюционные соотношения можно увидеть с помощью просмотра Cladograms или Phylograms. Последовательность селективного пегилированного пептидного агониста рецептора VPAC2 по настоящему изобретению является селективной в отношении рецептора VPAC2 и предпочтительно демострирует идентичность последовательностей с Р487 (SEQ ID NO: 84) в интервале 60-70%, 60-65%, 65-70%, 70-80%, 70-75%, 75-80%, 80-90%, 80-85%, 85-90%, 90-97%, 90-95% или 95-97%. Предпочтительно последовательность демострирует идентичность последовательностей с Р487 (SEQ ID NO: 84) более 82%. Более предпочтительно последовательность демострирует идентичность последовательностей с Р487 (SEQ ID NO: 84) более 90%. Еще более предпочтительно последовательность демострирует идентичность последовательностей с Р487 (SEQ ID NO: 84) более 92%. Еще более предпочтительно последовательность демострирует идентичность последовательностей с Р487 (SEQ ID NO: 84) более 95 % или 97%. Термин "C 1 -C 16 -алкил" в данном описании означает одновалентный насыщенный углеводородный радикал с неразветвленной, разветвленной или циклической цепью, содержащий от 1 до 16 атомов углерода или в случае циклического радикала, содержащий от 3 до 16 атомов углерода. Таким образом, термин "C 1 -C 16 -алкил" включает, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-гептил, н-октил, циклопропил, циклобутил, циклопентил и циклогексил. Группа C 1 -C 16 -алкил необязательно может быть замещена одним или несколькими заместителями, включая, например, арил, C 1 -C 6 -алкокси, -ОН, галоген, -CF 3 и -SH. Термин "C 1 -C 6 -алкил" в данном описании обозначает одновалентный насыщенный углеводородный радикал с неразветвленной, разветвленной или циклической цепью, содержащий от 1 до 6 атомов углерода или в случае циклического радикала, содержащий от 3 до 6 атомов углерода. Таким образом, термин "C 1 -C 6 -алкил" включает, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, циклопропил, циклобутил, циклопентил и циклогексил. Группа C 1 -C 6 -алкил необязательно может быть замещена одним или несколькими заместителями. Термин "C 2 -C 6 -алкенил" в данном описании обозначает одновалентный углеводородный радикал с неразветвленной, разветвленной или циклической цепью, содержащий по меньшей мере одну двойную связь и от 2 до 6 атомов углерода или в случае циклического радикала, содержащий от 3 до 6 атомов углерода. Таким образом, термин "C 2 -C 6 -алкенил" включает винил, проп-2-енил, бут-3-енил, пент-4-енил и изопропенил. Группа C 2 -C 6 -алкенил необязательно может быть замещена одним или несколькими заместителями. Термин "C 2 -C 6 -алкинил" в данном описании обозначает одновалентный углеводородный радикал с неразветвленной или разветвленной цепью, содержащий по меньшей мере одну тройную связь и от 2 до 6 атомов углерода. Таким образом, термин "C 2 -C 6 -алкинил" включает проп-2-инил, бут-3-инил и пент-4-инил. С 2 -С 6 -алкинил может быть необязательно замещен одним или несколькими заместителями. Термин "C 1 -C 6 -алкокси" в данном описании обозначает одновалентный незамещенный насыщенный углеводородный радикал с неразветвленной или разветвленной цепью, содержащий от 1 до 6 атомов углерода, присоединенных в точке замещения через радикал -O-. Таким образом, термин "C 1 -C 6 -алкокси" включает, например, метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси и трет-бутокси. Группа C 1 -C 6 -алкокси необязательно может быть замещена одним или несколькими заместителями. Термин "гало" или "галоген" обозначает фтор, хлор, бром или йод. Термин "арил", который используется отдельно или как часть группы, обозначает 5-10-членную ароматическую или гетероароматическую группу, в том числе фенильную группу, 5- или 6-членные моноциклические гетероароматические группы, которые в любом положении могут быть необязательно замещены 1, 2, 3, 4 или 5 заместителями (в зависимости от количества доступных для замещения положений), нафтильную группу или 8-, 9- или 10-членные бициклические гетероароматические группы, которые необязательно могут быть замещены в любом положении 1, 2, 3, 4, 5 или 6 заместителями (в зависимости от количества доступных для замещения положений). В пределах данного определения арила подходящие заместители включают С 1 -С б -алкил, C 2 -C 6 -алкенил, C 2 -C 6 -алкинил, -NH 2 , -ОН, галоген, -SH и CF 3 Термин "арил-C 1 -C 4 -алкил" в данном описании обозначает группу C 1 -C 4 -алкил, замещенную арилом. Таким образом, термин "арил-C 1 -C 4 -алкил" включает бензил, 1-фенилэтил ( α-метилбензил), 2-фенилэтил, 1-нафталинметил или 2-нафталинметил. Термин "нафтил" включает 1-нафтил и 2-нафтил. 1-Нафтил является предпочтительным. Термин "бензил" в данном описании обозначает одновалентный незамещенный фенильный радикал, присоединенный к положению замещения через группу -CH 2 -. Термин "5- или 6-членная моноциклическая гетероароматическая группа" в данном описании обозначает моноциклическую ароматическую группу, содержащую в целом 5 или 6 атомов в кольце, где от 1 до 4 указанных атомов, каждый независимо, выбраны из N, О и S. Предпочтительные группы содержат в кольце 1 или 2 атома, каждый из которых независимо выбран из N, О и S. Примеры 5-членных моноциклических гетероароматических групп включают пирролил (также носит название азолил), фуранил, тиенил, пиразолил (также носит название 1Н-пиразолил и 1,2-диазолил), имидазолил, оксазолил (также носит название 1,3-оксазолил), изоксазолил (также носит название 1,2-оксазолил), тиазолил (также носит название 1,3-тиазолил), изотиазолил (также носит название 1,2-тиазолил), триазолил, оксадиазолил, тиадиазолил, тетразолил, оксатриазолил и тиатриазолил. Примеры 6-членных моноциклических гетероароматических групп включают пиридинил, пиримидил, пиразинил, пиридазинил и триазинил. Термин "8-, 9- или 10-членная бициклическая гетероароматическая группа" в данном описании обозначает конденсированную бициклическую ароматическую группу, содержащую в целом 8, 9 или 10 атомов в системе колец, где от 1 до 4 указанных атомов, каждый независимо, выбраны из N, О и S. Предпочтительные группы содержат в системе колец от 1 до 3 атомов, каждый из которых независимо выбран из N, О и S. Подходящие 8-членные бициклические гетероароматические группы включают имидазо[2,1-b][1,3]тиазолил, тиено[3,2-b]тиенил, тиено[2,3-d][1,3]тиазолил и тиено[2,3-d]имидазолил. Подходящие 9-членные бициклические гетероароматические группы включают индолил, изоиндолил, бензофуранил (также носит название бензо[b]фуранил), изобензофуранил (также носит название бензо[с]фуранил), бензотиенил (также носит название бензо[b]тиенил), изобензотиенил (также носит название бензо[с]тиенил), индазолил, бензимидазолил, 1,3-бензоксазолил, 1,2-бензизоксазолил, 2,1-бензизоксазолил, 1,3-бензотиазолил, 1,2-бензоизотиазолил, 2,1-бензоизотиазолил, бензотриазолил, 1,2,3-бензоксадиазолил, 2,1,3-бензоксадиазолил, 1,2,3-бензотиадиазолил, 2,1,3-бензотиадиазолил, тиенопиридинил, пуринил и имидазо[1,2-а]пиридин. Подходящие 10-членные бициклические гетероароматические группы включают хинолинил, изохинолинил, циннолинил, хиназолинил, хиноксалинил, 1,5-нафтиридил, 1,6-нафтиридил, 1,7-нафтиридил и 1,8-нафтиридил. Термин "ПЭГ" в данном описании обозначает молекулу полиэтиленгликоля. В типичной форме ПЭГ представляет собой линейный полимер с концевыми гидроксильными группами, который имеет формулу HO-CH 2 CH 2 -(CH 2 CH 2 O) n -CH 2 CH 2 -OH, где n равно от приблизительно 8 до приблизительно 4000. Концевой водород может быть замещен защитной группой, такой как алкильная или алканольная группа. Предпочтительно ПЭГ содержит по меньшей мере одну гидроксигруппу, более предпочтительно она является концевой гидроксигруппой. Именно эта гидроксигруппа предпочтительно активизируется для реакции с пептидом. Существует множество форм ПЭГ, пригодных в соответствии с данным изобретением. Многочисленные производные ПЭГ известны из уровня техники и пригодны для применения в изобретении (см., например, патенты США № 5445090; 5900461; 5932462; 6436386; 6448369; 6437025; 6448369; 6495659; 6515100 и 6514491 и Zalipsky, S. Bioconjugate Chem. 6: 150-165, 1995). Молекула ПЭГ, ковалентно присоединенная к пептидным агонистам рецептора VPAC2 по настоящему изобретению, не предназначена ограничиваться конкретным видом. Молекулярная масса молекулы ПЭГ предпочтительно составляет 500-100000 Да. ПЭГ может быть линейным или разветвленным. Пегилированные пептидные агонисты рецептора VPAC2 по изобретению могут содержать одну, две или три молекулы ПЭГ, присоединенные к пептиду. Более предпочтительно на пегилированный пептидный агонист рецептора VPAC2 приходится одна или две молекулы ПЭГ, однако, если присутствует более одной молекулы ПЭГ на молекулу пептида, предпочтительно их количество составляет не более трех. Кроме того, предусмотрено, что оба конца молекулы ПЭГ могут быть гомо- или гетерофункциональными для поперечного сшивания двух или более пептидных агонистов рецептора VPAC2 вместе. Если присутствуют две молекулы ПЭГ, молекулы ПЭГ будут предпочтительно представлять собой молекулы ПЭГ по 20000 Да каждая или молекулы ПЭГ по 30000 Да каждая. Однако могут использоваться молекулы ПЭГ с другой молекулярной массой, например одна молекула ПЭГ с молекулярной массой 10000 Да и одна молекула ПЭГ с молекулярной массой 30000 Да или одиа молекула ПЭГ с молекулярной массой 20000 Да и одна молекула ПЭГ с молекулярной массой 40000 Да. В настоящем изобретении молекула ПЭГ может быть ковалентно присоединена к остатку Cys или Lys или к C-концевому остатку. Молекула ПЭГ также может быть ковалентно присоединена к остатку Trp, который связан с боковой цепью остатка Lys (K(W)). Альтернативно, группа K(CO(CH 2 ) 2 SH) может быть пегилированной для образования K(CO(CH 2 ) 2 S-PEG). Любой остаток Lys в пептидном агонисте может быть замещен K(W) или K(CO(CH 2 ) 2 SH), который затем может быть пегилирован. Кроме того, любой остаток Cys в пептидном агонисте может быть замещен модифицированным остатком цистеина, например hC. Модифицированный остаток Cys может быть ковалентно присоединен к молекуле ПЭГ. Термин "пегилирование" в данном описании обозначает присоединение ковалентно одной или нескольких молекул ПЭГ, как описано выше, к пептидным агонистам рецептора VPAC2 по настоящему изобретению. В соответствии с предпочтительным вариантом настоящего изобретения предлагается пегилированный пептидный агонист рецептора VPAC2, содержащий пептидную последовательность, выбранную из SEQ ID NO: 17-45 и 94-112, и C-концевой удлиняющий сегмент, выбранный из SEQ ID NO: 5, 6, 7, 8, 9, 10, 11 и 12. Особенно предпочтительно C-концевой удлиняющий сегмент представляет собой SEQ ID NO: 11 или SEQ ID NO: 12. В соответствии с более предпочтительным вариантом настоящего изобретения предлагается пегилированный пептидный агонист рецептора VPAC2, содержащий пептидную последовательность, выбранную из SEQ ID NO: 17-45 и 94-112, и C-концевой удлиняющий сегмент, выбранный из SEQ ID NO: 5, 6, 7, 8, 9, 10, 11 и 12, где пептидный агонист рецептора VPAC2 дополнительно содержит N-концевую модификацию, которая представляет собой добавление ацетила, пропионила, бутирила, пентаноила, гексаноила, метионинсульфоксида, метионина, 3-фенилпропионила, фенилацетила, бензоила, норлейцина, D-гистидина, изолейцина, 3-меркаптопропионила, биотинил-6-аминогексановой кислоты (6-аминокапроновая кислота) и -C(=NH)-NH 2 . В данном варианте более предпочтительно N-концевая модификация представляет собой добавление ацетила или гексаноила. В соответствии с другим предпочтительным вариантом настоящего изобретения предлагается пегилированный пептидный агонист рецептора VPAC2, содержащий последовательность аминокислот формулы 4 (SEQ ID NO: 4) и C-концевой удлиняющий сегмент, выбранный из SEQ ID NO: 5, 6, 7, 8, 9, 10, 11 и 12, где пегилированный пептидный агонист рецептора VPAC2 дополнительно содержит N-концевую модификацию, которая представляет собой добавление ацетила, пропионила, бутирила, пентаноила, гексаноила, метионинсульфоксида, метионина, 3-фенилпропионила, фенилацетила, бензоила, норлейцина, D-гистидина, изолейцина, 3-меркаптопропионила, биотинил-6-аминогексановой кислоты (6-аминокапроновая кислота) и -C(=NH)-NH 2 . В данном варианте более предпочтительно N-концевая модификация представляет собой добавление ацетила или гексаноила. В соответствии с более предпочтительным вариантом настоящего изобретения предлагается пегилированный пептидный агонист рецептора VPAC2, содержащий последовательность аминокислот формулы 4 (SEQ ID NO: 4), где Xaa 15 представляет собой Aib, Xaa 20 представляет собой Aib, и Xaa 12 , Xaa 21 , Xaa 27 и Xaa 28 , все, представляют собой Orn, и C-концевой удлиняющий сегмент, выбранный из SEQ ID NO: 5, 6, 7, 8, 9, 10, 11 и 12, где пегилированный пептидный агонист рецептора VPAC2 дополнительно содержит N-концевую модификацию, которая представляет собой добавление ацетила, пропионила, бутирила, пентаноила, гексаноила, метионинсульфоксида, метионина, 3-фенилпропионила, фенилацетила, бензоила, норлейцина, D-гистидина, изолейцина, 3-меркаптопропионила, биотинил-6-аминогексановой кислоты (6-аминокапроновая кислота) и -C(=NH)-NH 2 . В данном варианте более предпочтительно Xaa 15 представляет собой Aib, Xaa 20 представляет собой Aib, Xaa 12 , Xaa 21 , Xaa 27 и Xaa 28 все представляют собой Orn, Xaa 8 представляет собой Glu, Xaa 9 представляет собой Gln, и Xaa 10 представляет собой Tyr(OMe). Особенно предпочтительно Xaa 15 представляет собой Aib, Xaa 20 представляет собой Aib, Xaa 12 , Xaa 21 , Xaa 27 и Xaa 28 все представляют собой Orn, Xaa 8 представляет собой Glu, Xaa 9 представляет собой Gln, Xaa 10 представляет собой Tyr(OMe), и Xaa 23 и/или Xaa 25 представляет собой Aib. Пегилирование белков может преодолевать многие фармакологические и токсикологические/иммунологические проблемы, связанные с применением пептидов или белков в качестве терапевтических средств. Однако для любого конкретного пептида неизвестно, будет ли пегилированная форма пептида демонстрировать значительное снижение биологической активности по сравнению с непегилированной формой пептида. Биологическая активность пегилированных белков может ухудшаться под воздействием таких факторов, как: i) размер молекулы ПЭГ; ii) конкретные положения присоединения; iii) степень модификации; iv) условия побочного связывания; v) использование линкера для присоединения или непосредственного присоединения полимера; vi) образование вредных побочных продуктов; vii) повреждение, вызванное активированным полимером; или viii) удержание заряда. Работа, проведенная по пегилированию цитокинов, например, показывает влияние, которое может оказывать пегилирование. В зависимости от используемой реакции соединения модификация цитокинов с помощью полимера приводила к резкому уменьшению биологической активности (Francis, G.E., et al., (1998), PEGylation of cytokines and other therapeutic proteins and peptides: the importance of biological optimization of coupling techniques, Intl. J. Hem. 68: 1-18). Сохранение биологической активности в случае пегилированных пептидов еще более проблематично, чем в случае белков. Поскольку пептиды имеют меньший размер, чем белки, модификация пегилированием потенциально может оказывать большее влияние на биологическую активность. Пептидные агонисты рецептора VPAC2 по настоящему изобретению модифицированы ковалентным присоединением одной или нескольких молекул ПЭГ и в целом обладают улучшенным фармакокинетическим профилем за счет замедленного протеолитического разложения и сниженного почечного клиренса. Пегилирование увеличивает размер пептидных агонистов рецептора VPAC2, таким образом уменьшая почечную фильтрацию и изменяя распределение в биологической системе. Пегилирование может экранировать антигенные эпитопы пептидных агонистов рецептора VPAC2, таким образом уменьшая ретикулоэндотелиальный клиренс и распознавание иммунной системой и также уменьшая разложение протеолитическими ферментами, такими как DPP-IV. Ковалентное присоединение одной или нескольких молекул ПЭГ к низкомолекулярному, биологически активному пептидному агонисту рецептора VPAC2 содержит в себе риск побочного влияния на агонист, например, путем дестабилизации свойственной ему вторичной структуры и биологически активной конформации со снижением биологической активности, что может сделать агонист непригодным для применения в качестве терапевтического средства. Настоящее изобретение, однако, основано на том открытии, что ковалентное присоединение одной или нескольких молекул ПЭГ к конкретным остаткам пептидного агониста рецептора VPAC2 неожиданно дает биологически активный, пегилированный пептидный агонист рецептора VPAC2 с удлиненным периодом полувыведения и сниженным клиренсом по сравнению с непегилированными пептидными агонистами рецептора VPAC2. Соединения по настоящему изобретению включают селективные пегилированные пептидные агонисты рецептора VPAC2. Для того чтобы определить потенциальные сайты пегилирования в молекуле пептидного агониста рецептора VPAC2, может быть проведено сканирование серина. Остаток Ser замещается в конкретном положении пептида, и Ser-модифицированный пептид исследуют на предмет активности и селективности. Если замещение Ser оказывает минимальное влияние на активность и Ser-модифицированный пептид является селективным в отношении рецептора VPAC2, далее остаток Ser замещают на остаток Cys или Lys, который служит сайтом прямого или непрямого пегилирования. Непрямое пегилирование остатка представляет собой пегилирование химической группы или остатка, который связан с остатком сайта пегилирования. Косвенное пегилирование Lys включает пегилирование K(W) и K(CO(CH 2 ) 2 SH). Изобретение, раскрытое в данном описании, предлагает пегилированные пептидные агонисты рецептора VPAC2. Пегилирование может увеличить период полувыведения селективных пептидных агонистов рецептора VPAC2, что дает пегилированные пептидные агонисты рецептора VPAC2 с элиминационным периодом полувыведения по меньшей мере 1 ч, предпочтительно по меньшей мере 3, 5, 7, 10, 15, 20 или 24 ч и наиболее предпочтительно по меньшей мере 48 ч. Пегилированные пептидные агонисты рецептора VPAC2 по настоящему изобретению предпочтительно демонстрируют значение клиренса 200 мл/ч/кг или менее, более предпочтительно 180, 150, 120, 100, 80, 60 мл/ч/кг или менее, наиболее предпочтительно менее 50, 40 или 20 мл/ч/кг. Настоящее изобретение включает открытие того, что конкретные аминокислоты, добавленные к C-концу пептидной последовательности пептидного агониста рецептора VPAC2, могут защищать пептид, а также могут увеличивать активность, селективность и/или выраженность эффекта действия. Например, такие C-концевые удлиняющие фрагменты могут стабилизировать спиральную структуру пептида и стабилизировать сайты, размещенные поблизости C-конца, которые склонны к ферментативному расщеплению. Кроме того, многие пептиды с C-концевым удлиняющим фрагментом, раскрытые в данном описании, могут обладать большей селективностью в отношении рецептора VPAC2 и могут быть более активными, чем VIP, PACAP и другие известные пептидные агонисты рецептора VPAC2. Примером предпочтительного C-концевого удлиняющего сегмента является удлиняющий сегмент пептида экзендин-4 как последовательность C-кэппинга. Экзендин-4 найден в слюнных выделениях ящерицы-ядозуба Heloderma Suspectum, (Eng et al., J. Biol. Chem., 267 (11): 7402-7405 (1992)). Другими примерами C-концевых удлиняющих сегментов являются C-концевые последовательности гелодермина и гелоспектина. Гелодермин и гелоспектин также найдены в слюнных выделениях ящерицы-ядозуба. Кроме того, было обнаружено, что модификация N-конца пептидного агониста рецептора VPAC2 может увеличивать активность и/или обеспечивать устойчивость к расщеплению DPP-IV. VIP и некоторые известные пептидные агонисты рецептора VPAC2 восприимчивы к расщеплению различными ферментами и, таким образом, демонстрируют короткий период полувыведения in vivo. Ниже обсуждаются различные сайты ферментативного расщепления в пептидных агонистах рецептора VPAC2. Сайты расщепления обсуждаются относительно положений аминокислот в VIP (SEQ ID NO: 14) и применимы к последовательностям, приведенным в данном описании. Расщепление пептидного агониста ферментом дипептидилпептидазой-IV (DPP-IV) происходит между положением 2 (серии в VIP) и положением 3 (аспарагиновая кислота в VIP). Агонисты по настоящему изобретению могут быть сделаны более устойчивыми к расщеплению DPP-IV в данном участке путем добавления N-концевой модификации. Примеры N-концевых модификаций, которые могут повышать устойчивость к расщеплению DPP-IV, включают добавление ацетила, пропионила, бутирила, пентаноила, гексаноила, метионинсульфоксида, метионина, 3-фенилпропионила, фенилацетила, бензоила, норлейцина, D-гистидина, изолейцина, 3-меркаптопропионила, биотинил-6-аминогексановой кислоты или -C(=NH 2 )-NH 2 . Предпочтительно N-концевая модификация представляет собой добавление ацетила или гексаноила. В VIP дикого типа существуют сайты расщепления химотрипсином между аминокислотами 10 и 11 (тирозин и треонин), а также 22 и 23 (тирозин и лейцин). Замены в положениях 10 и/или 11 и положениях 22 и/или 23 могут повышать устойчивость пептида в этих сайтах. Например, замена тирозина в положении 10 и/или положении 22 на Tyr(OMe) может повышать устойчивость. Существует сайт расщепления трипсином между аминокислотами в положениях 12 и 13 VIP дикого типа. Некоторые аминокислоты делают пептид менее восприимчивым к расщеплению в указанном сайте, например орнитин в положении 12 и аминоизомасляная кислота в положении 13. В VIP дикого типа, а также в многочисленных пептидных агонистах рецептора VPAC2, известных из уровня техники, присутствуют сайты расщепления между основными аминокислотами в положениях 14 и 15, а также в положениях 20 и 21. Устойчивость к протеолитическому расщеплению in vivo селективных пегилированных пептидных агонистов рецептора VPAC2 по настоящему изобретению может быть улучшена за счет замен на этих сайтах. Предпочтительными заменами на этих сайтах являются такие, которые делают пептид менее восприимчивым к расщеплению трипсин-одобными ферментами, в том числе трипсином. Например, изомасляная аминокислота в положении 15, изомасляная аминокислота в положении 20 и орнитин в положении 21, все, являются предпочтительными заменами, которые могут давать повышенную устойчивость. Повышенная устойчивость репрезентативного количества селективных пегилированных пептидных агонистов рецептора VPAC2 с сопротивлением к расщеплению пептидазой и включенных в настоящее изобретение продемонстрирована в примере 7. Существует также сайт расщепления между аминокислотами в положениях 25 и 26 VIP дикого типа. Участок пептидного агониста рецептора VPAC2, включающий аминокислоты в положениях 27, 28, 29, 30 и 31, также восприимчив к ферментативному расщеплению. Добавление C-концевого удлиняющего сегмента может делать пептидный агонист более устойчивым к нейроэндопептидазе (NEP), а также это может увеличивать селективность в отношении рецептора VPAC2. Данный участок также могут атаковать трипсинподобные ферменты. Если это происходит, пептидный агонист может утрачивать C-концевой удлиняющий сегмент при дальнейшей активности карбоксипептидазы, что дает неактивную форму пептида. Предпочтительные замены, которые могут увеличивать устойчивость к расщеплению на данном сайте, включают орнитин в положении 27, орнитин или аминоизомасляную кислоту в положении 28 и орнитин в положении 29. В дополнение к селективным пегилированным пептидным агонистам рецептора VPAC2 с устойчивостью к расщеплению различными пептидазами селективные пегилированные пептидные агонисты рецептора VPAC2 по настоящему изобретению могут также включать пептиды с повышенной селективностью в отношении рецептора VPAC2, с повышенной эффективностью и/или повышенной стабильностью по сравнению с некоторыми пептидами, известными из уровня техники. Повышенная эффективность и селективность различных пегилированных пептидных агонистов рецептора VPAC2 по настоящему изобретению продемонстрированы в примерах 3-5. В табл. 1 (пример 3) приведен перечень селективных пегилированных пептидных агонистов рецептора VPAC2 и соответствующие результаты их активности in vitro. Предпочтительно селективные пегилированные пептидные агонисты рецептора VPAC2 по настоящему изобретению демонстрируют значение ЕС 50 менее 200 нМ. Более предпочтительно значение ЕС 50 составляет менее 100 нМ. Еще более предпочтительно значение ЕС 50 составляет менее 50 нМ. Еще более предпочтительно значение ЕС 50 составляет менее 30 нМ. В табл. 2 (пример 4) приведен перечень пегилированных пептидных агонистов рецептора VPAC2 и соответствующие результаты их связывания с рецепторами VPAC2, VPAC1 и РАС1 человека. Подробное описание этих анализов см. в примере 4. Степень селективности определяют по соотношению сродства связывания с рецептором VPAC2 к сродству связывания с рецептором VPAC1 и соотношению сродства связывания с рецептором VPAC2 к сродству связывания с рецептором РАС1. Предпочтительно агонисты по настоящему изобретению демонстрируют соотношение селективности, где сродство к рецептору VPAC2 по меньшей мере в 50 раз превышает сродство к рецепторам VPAC1 и/или РАС1. Более предпочтительно данное сродство в отношении VPAC2 по меньшей мере в 100 раз больше, чем в отношении VPAC1 и/или РАС1. Еще более предпочтительно сродство в отношении VPAC2 по меньшей мере в 200 раз больше, чем в отношении VPAC1 и/или РАС1. Еще более предпочтительно сродство в отношении VPAC2 по меньшей мере в 500 раз больше, чем в отношении VPAC1 и/или РАС1. Еще более предпочтительно соотношение для VPAC2 по меньшей мере в 1000 раз больше, чем для VPAC1 и/или РАС1. В данном описании "селективные пептидные агонисты рецептора VPAC2" также включают фармацевтически приемлемые соли агонистов, раскрытых в данном описании. Селективный пептидный агонист рецептора VPAC2 по данному изобретению может содержать в достаточной степени кислые, в достаточной степени основные или оба вида функциональных групп и соответственно реагировать с любым из целого ряда неорганических оснований, а также неорганических и органических кислот с образованием солей. Кислоты, которые обычно применяются для образования кислотно-аддитивных солей, представляют собой неорганические кислоты, такие как соляная, бромисто-водородная, йодисто-водородная, серная, фосфорная кислоты и т.п., и органические кислоты, такие как п-толуолсульфоновая, метансульфоновая, щавелевая, п-бромфенилсульфоновая, угольная, янтарная, лимонная, бензойная, уксусная, трифторуксусная кислоты и т.п. Примеры таких солей включают сульфат, пиросульфат, бисульфат, сульфит, бисульфит, фосфат, моногидрофосфат, дигидрофосфат, мета-фосфат, пирофосфат, хлорид, бромид, йодид, ацетат, пропионат, деканоат, каприлат, акрилат, формиат, изобутират, капроат, гептаноат, пропиолят, оксалат, малонат, сукцинат, суберат, себацат, фумарат, малеат, бутин-1,4-диоат, гексин-1,6-диоат, бензоат, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат, метоксибензоат, фталат, сульфонат, ксиленсульфонат, фенилацетат, фенилпропионат, фенилбутират, цитрат, лактат, гамма-гидроксибутират, гликолят, тартрат, метансульфонат, пропансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат, манделат и т.п. Основно-аддитивные соли включают полученные от неорганических оснований, таких как аммиак, гидроксиды щелочных или щелочно-земельных металлов, карбонат, бикарбонат и т.п. Такие основания, пригодные для получения солей по данному изобретению, таким образом, включают гидроксид натрия, гидроксид калия, гидроксид аммония, карбонат калия и т.п. Селективные пегилированные пептидные агонисты рецептора VPAC2 по настоящему изобретению предпочтительно вводят в фармацевтические композиции. Могут применяться стандартные методы приготовления фармацевтических препаратов, таких как описаны в Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA. Селективные пегилированные пептидные агонисты рецептора VPAC2 по настоящему изобретению могут быть введены в препараты для буккального, местного, перорального, трансдермального, назального или легочного введения, а также для парентерального введения. Парентеральное введение может включать, например, системное введение, такое как внутримышечную, внутривенную, подкожную, внутрикожную или внутрибрюшинную инъекцию. Селективные пегилированные пептидные агонисты рецептора VPAC2 могут вводиться субъекту в сочетании с приемлемым фармацевтическим носителем, наполнителем или вспомогательным веществом как часть фармацевтической композиции для лечения NIDDM или нарушений, которые обсуждаются ниже. Фармацевтическая композиция может представлять собой раствор или, в случае парентерального введения, суспензию пегилированного пептидного агониста рецептора VPAC2 или суспензию комплекса пегилированного пептидного агониста рецептора VPAC2 с двухвалентным металлическим катионом, таким как цинк. Подходящие фармацевтические носители могут содержать инертные ингредиенты, которые не взаимодействуют с пептидом или производным пептида. Подходящие фармацевтические носители для парентерального введения включают, например, стерильную воду, физиологический солевой раствор, бактериостатический солевой раствор (солевой раствор, содержащий около 0,9% мг/мл бензилового спирта), буферизованный фосфатом солевой раствор, раствор Хэнкса, раствор Рингера с лактатом и т.п. Некоторые примеры подходящих вспомогательных веществ включают лактозу, глюкозу, сахарозу, трегалозу, сорбитол и маннитол. Пегилированные пептидные агонисты рецептора VPAC2 по изобретению могут быть введены в препараты для введения таким образом, чтобы поддерживать уровни в плазме крови в эффективном интервале на протяжении длительных периодов времени. Главным барьером на пути к эффективному пероральному введению лекарственных средств пептидной природы является низкая биодоступность из-за разложения пептидов кислотами и ферментами, низкая абсорбция через эпителиальные мембраны, а также переход пептидов в нерастворимую форму после контакта с кислой средой в пищеварительном тракте. Системы перорального введения пептидов, таких как включены в настоящее изобретение, известны из уровня техники. Например, пегилированные пептидные агонисты рецептора VPAC2 могут быть инкапсулированы с использованием микросфер, а затем введены перорально. Например, пегилированные пептидные агонисты рецептора VPAC2 могут быть инкапсулированы в микросферы, состоящие из коммерчески доступного, биосовместимого, полимера, который разлагается в биологических системах, такого как поли (лактид-согликолид)-COOH, с оливковым маслом в качестве наполнителя (см. Joseph, et al. Diabetologia, 43: 1319-1328 (2000)). Другие виды технологии микросфер также являются коммерчески доступными, например полимеры Medisorb ® и Prolease ® от Alkermes, которые разлагаются в биологических системах. Полимеры Medisorb ® могут быть произведены с использованием любого изомера лактида. Соотношение лактид/гликолид может варьировать в пределах от 0:100 до 100:0, позволяя изменять свойства полимера в широком интервале. Это позволяет спроектировать системы доставки и устройств для имплантации со временем резорбции от недель до месяцев. Emisphere также опубликовано множество статей, где обсуждается технология перорального введения пептидов и белков. Например, см. WO 95/28838 Leone-bay et al., где раскрыты конкретные носители для облегчения абсорбции, состоящие из модифицированных аминокислот. Селективные пегилированные пептидные агонисты рецептора VPAC2, раскрытые в данном описании, могут применяться для лечения субъектов с широким спектром заболеваний и состояний. Агонисты, включенные в настоящее изобретение, оказывают свое биологическое действие на уровне рецептора под названием рецептор VPAC2. Субъекты с состояниями и/или заболеваниями, которые реагируют на стимуляцию рецептора VPAC2 или на введение пептидных агонистов рецептора VPAC2, могут, таким образом, получать лечение пегилированными агонистами VPAC2 по настоящему изобретению. Говорят, что такие субъекты «нуждаются в лечении агонистами VPAC2 » или «нуждаются в стимуляции рецептора VPAC2 ». Селективные пегилированные пептидные агонисты рецептора VPAC2 по настоящему изобретению могут применяться для лечения диабета, в том числе диабета типа 1 и типа 2 (инсулиннезависимый сахарный диабет или NIDDM). Агонисты также могут применяться для лечения субъектов, нуждающихся в профилактическом лечении агонистом рецептора VPAC2, например субъектов, подвергающихся риску развития NIDDM. Такое лечение может также задерживать развитие диабета и его осложнений. Другие субъекты, которых можно лечить агонистами по настоящему изобретению, включают субъекты с нарушенной переносимостью глюкозы (IGT) (Expert Committee on Classification of Diabetes Mellitus, Diabetes Care 22 (Supp. 1): S5, 1999) или отклонениями уровня глюкозы в крови натощак (IFG) (Charles, et al., Diabetes 40: 796, 1991); субъекты, масса тела которых приблизительно на 25% превышает нормальную массу тела для их роста и конституции субъекта; субъекты, у которых один или оба родителя больны NIDDM; субъекты с гестационным диабетом и субъекты с метаболическими расстройствами, такими как расстройства в результате сниженной эндогенной секреции инсулина. Селективные пегилированные пептидные агонисты рецептора VPAC2 могут применяться для профилактики развития NIDDM у субъектов с нарушенной переносимостью глюкозы, предупреждения ухудшения функции панкреатических β-клеток, индукции пролиферации β-клеток, улучшения функции β-клеток, активации бездействующих β-клеток, дифференциации клеток в β-клетки, стимуляции репликации β-клеток и торможения апоптоза β-клеток. Другие заболевания и состояния, которые можно лечить или предупреждать с применением агонистов по изобретению в способах по изобретению, включают: диабет зрелого возраста у молодых людей (MODY) (Herman, et al., Diabetes 43: 40, 1994); латентный аутоиммунный диабет взрослых (LADA) (Zimmet, et al., Diabetes Med. 11: 299, 1994); гестационный диабет (Metzger, Diabetes, 40: 197, 1991); метаболический синдром X, дислипидемию, гипергликемию, гиперинсулинемию, гипертриглицеридемию и инсулинорезистентность. Селективные пегилированные пептидные агонисты рецептора VPAC2 по изобретению также могут применяться для лечения причин вторичного диабета (Expert Committee on Classification of Diabetes Mellitus, Diabetes Care 22 (Supp. 1): S5, 1999). Такие вторичные причины включают избыток глюкокортикоидов, избыток гормона роста, феохромоцитому и медикаментозный диабет. Лекарственные средства, которые могут индуцировать диабет включают, не ограничиваясь ими, пириминил, никотиновую кислоту, глюкокортикоиды, фенитоин, гормон щитовидной железы, β-адренергические агенты, α-интерферон и лекарственные средства, применяемые для лечения ВИЧ-инфекции. Селективные пегилированные пептидные агонисты рецептора VPAC2 по настоящему изобретению могут быть эффективными для подавления потребления пищи и лечения ожирения. Селективные пегилированные пептидные агонисты рецептора VPAC2 по настоящему изобретению также могут быть эффективными для профилактики или лечения таких нарушений, как атеросклеротическое заболевание, гиперлипидемия, гиперхолестеринемия, низкий уровень ЛПВП, гипертензия, первоначальная легочная гипертензия, сердечно-сосудистое заболевание (в том числе атеросклероз, заболевание коронарных сосудов сердца и заболевание коронарных артерии), заболевание сосудов мозга и заболевание периферических сосудов; а также для лечения волчанки, синдрома поликистоза яичников, канцерогенеза и гиперплазии, нарушений со стороны мужской и женской репродуктивной системы, сексуальных расстройств, язв, расстройств сна, расстройств липидного и углеводного обмена, циркадной дисфункции, расстройств роста, расстройств гомеостаза энергии, иммунных заболеваний, в том числе аутоиммунных заболеваний (например, системная красная волчанка), а также острых и хронических воспалительных заболеваний, ревматоидного артрита и септического шока. Селективные пегилированные пептидные агонисты рецептора VPAC2 по настоящему изобретению также могут быть пригодными для лечения физиологических нарушений, связанных, например, с дифференциацией клеток для продуцирования клеток, аккумулирующих липиды, регуляцией чувствительности к инсулину и уровней глюкозы в крови, которые вовлечены, например, в аномальную функцию β-клеток поджелудочной железы, секретирующих инсулин опухолей и/или аутоиммунной гипогликемии в результате аутоантител к инсулину, аутоантител к рецептору инсулина или ацтоантител, которые стимулируют панкреатические β-клетки, дифференциацию макрофага, которая приводит к образованию атеросклеротических бляшек, воспалительную реакцию, канцерогенез, гиперплазию, экспрессию гена адипоцитов, дифференциацию адипоцитов, уменьшение массы панкреатических β-клеток, секрецию инсулина, чувствительность тканей к инсулину, рост клеток липосаркомы, поликистозное заболевание яичников, хроническую ановуляцию, гиперандрогенизма, выработку прогестерона, стероидогенез, окислительно-восстановительный потенциал и окислительный стресс в клетках, выработку синтетазы оксида азота (NOS), повышение уровней гамма-глутамилтранспептидазы, каталазы, триглицеридов плазмы, холестерина ЛПВП и ЛПНП и т.п. Кроме того, селективные пегилированные пептидные агонисты рецептора VPAC2 по изобретению могут применяться для лечения астмы (Bolin, et al., Biopolymer 37: 57-66 (1995); патент США 5677419 показывает, что полипептид R3PO активно расслабляет гладкие мышцы трахеи морских свинок); индукции гипотензии (VIP индуцирует гипотензию, тахикардию и приливы к лицу у больных астмой (Morice, et al., Peptides 7: 279-280 (1986); Morice, et al., Lancet 2: 1225-1227 (1983)); для лечения нарушений со стороны мужской репродуктивной системы (Siow, et al., Arch. Androl. 43 (1): 67-71 (1999)); в качестве антиапоптотического/нейропротекторного средства (Brenneman, et al., Ann. N.Y. Acad. Sci. 865: 207-12 (1998)); для кардиопротекции при ишемических событиях (Kalfin, et al., J. Pharmacol. Exp. Ther. 1268 (2): 952-8 (1994); Das, et al., Ann. N.Y. Acad. Sci. 865: 297-308 (1998)); для манипуляции циркадными часами и родственными расстройствами (Hamar, et al., Cell 109: 497-508 (2002); Shen, et al., Proc. Natl. Acad. Sci. 97: 11575-80, (2000)); а также в качестве противоязвенного средства (Tuncel, et al., Ann. N.Y. Acad. Sci. 865: 309-22, (1998)). "Эффективное количество" селективного пегилированного пептидного агониста рецептора VPAC2 представляет собой количество, которое приводит к желаемому терапевтическому и/или профилактическому эффекту, не вызывая неприемлемых побочных эффектов, при введении субъекту, который нуждается в стимуляции рецептора VPAC2. "Желаемый терапевтический эффект" включает одно или несколько из следующего: 1) облегчение симптома(ов), связанного с заболеванием или состоянием; 2) задержку развития симптомов, связанных с заболеванием или состоянием; 3) увеличение продолжительности жизни по сравнению с отсутствием лечения и 4) увеличение качества жизни по сравнению с отсутствием лечения. Например, "эффективное количество" пегилированного агониста VPAC2 для лечения NIDDM представляет собой количество, которое давало бы большую степень контроля концентрации глюкозы в крови по сравнению с отсутствием лечения, таким образом приводя к задержке развития осложнений диабета, таких как ретинопатия, невропатия или заболевание почек. "Эффективное количество" селективного пегилированного пептидного агониста рецептора VPAC2 для предотвращения NIDDM представляет собой количество, которое задерживало бы, по сравнению с отсутствием лечения, развитие повышенных уровней глюкозы в крови, которые требуют лечения гипогликемическими лекарственными средствами, такими как производные сульфонилмочевины, тиазолидиндионы, инсулин и/или бисгуанидинов. "Эффективное количество" селективного пегилированного пептидного агониста рецептора VPAC2, вводимое субъекту, также будет зависеть от вида и тяжести заболевания, а также от характеристик субъекта, таких как общее состояние здоровья, возраст, пол, масса тела и переносимость лекарственных средств. Эффективная доза селективного пегилированного пептидного агониста рецептора VPAC2 для нормализации глюкозы крови пациента будет зависеть от целого ряда факторов, которые включают, не ограниваясь ими, пол, массу тела и возраст субъекта, выраженность неспособности регулировать уровень глюкозы в крови, способа введения и биодоступности, фармакокинетического профиля пептида, эффективности и препарата. Типичный интервал доз селективных пегилированных пептидных агонистов рецептора VPAC2 по настоящему изобретению варьирует от приблизительно 1 до приблизительно 5000 мкг/сутки. Предпочтительным является интервал доз от приблизительно 1 до приблизительно 2500 мкг/сутки, более предпочтительно от приблизительно 1 до приблизительно 1000 мкг/сутки. Еще более предпочтительным является интервал дозы от приблизительно 5 до приблизительно 100 мкг/сутки. Кроме того, предпочтительный интервал доз составляет от приблизительно 10 до приблизительно 50 мкг/сутки. Наиболее предпочтительно доза составляет приблизительно 20 мкг/сутки. Термин "субъект" представляет собой млекопитающее, предпочтительно человека, но может также быть животным, например домашним животным (например, собаки, кошки и т.п.), сельскохозяйственным животным (например, коровы, овца, свиньи, лошади и т.п.) и лабораторным животным (например, крысы, мыши, морские свинки и т.п.). Селективные пегилированные пептидные агонисты рецептора VPAC2 по настоящему изобретению могут быть получены с применением стандартных способов твердофазного синтеза пептидов. Синтезаторы пептидов коммерчески доступны, например, от Rainin-PTI Symphony Peptide Synthesizer (Tucson, AZ). Реактивы для твердофазного синтеза коммерчески доступны, например, от Glycopep (Chicago, IL). Твердофазные синтезаторы пептидов могут применяться в соответствии с инструкциями производителей для блокирования перекрывания групп, защиты аминокислоты, которая должна вступать в реакцию, соединения, разъединения и кэппингя не вступивших в реакцию аминокислот. Обычно, α-N-защищенная аминокислота и N-концевая аминокислота на растущей пептидной цепи на смоле сочетаются при комнатной температуре в инертном растворителе, таком как диметилформамид, N-метилпирролидон или метиленхлорид в присутствии агентов соединения, таких как дициклогексилкарбодиимид и 1-гидроксибензотриазол, и основания, такого как диизопропилэтиламин. α-N-защитную группу удаляют со смолы полученного пептида с использованием таких реактивов, как трифторуксусная кислота или пиперидин, и реакцию соединения повторяют со следующей целевой N-защищенной аминокислотой, которая должна быть добавлена к пептидной цепи. Подходящие защитные группы для амина хорошо известны из уровня техники и описаны, например, в Green and Wuts, "Protecting Groups in Organic Synthesis", John Wiley &Sons, 1991. Примеры включают трет-бутоксикарбонил (tBoc) и флуоренилметоксикарбонил (Fmoc). Селективные пептидные агонисты рецептора VPAC2 также могут быть синтезированным с применением стандартных автоматизированных протоколов твердофазного синтеза с использованием трет-бутоксикарбонил- или флуоренилметоксикарбонил- α-аминокислот с подходящей защитой боковой цепи. После завершения синтеза может быть осуществлена модификация N-конца путем реакции α-аминогруппы, например, с: (i) активными эфирами (с применением протоколов, сходных с описанными выше, для введения α-N-защищенной аминокислоты); (ii) альдегидами в присутствии восстанавливающего агента (процедура восстановительного аминирования) и (iii) реактивами для гуанидирования. Затем пептиды отщепляют от твердофазной подложки с одновременным снятием защиты с боковой цепи с применением стандартных способов фторида водорода или трифторуксусной кислоты (ТФУ). Сырые пептиды далее дополнительно очищают, используя обращенно-фазовую хроматографию на колонках VYDAC C18 с градиентами ацетонитрила в 0,1% ТФУ. Для удаления ацетонитрила пептиды лиофилизируют из раствора, содержащего 0,1% ТФУ, ацетонитрил и воду. Чистота может быть подтверждена аналитической обращенно-фазовой хроматографией. Идентичность пептидов может быть подтверждена масс-спектрометрией. Пептиды могут быть солюбилизированы в водных буферных растворах при нейтральных значениях рН. Пептидные агонисты по настоящему изобретению также могут быть получены рекомбинантными способами, известными из уровня техники, с использованием как эукариотных, так и прокариотных клеток-хозяев. Как только пептид получен и очищен, его модифицируют путем ковалентного связывания по меньшей мере одной молекулы ПЭГ с остатками Cys или Lys, с K(W) или K(CO(CH 2 ) 2 SH) или с карбоксиконцевой аминокислотой. В уровне техники описано широкое разнообразие способов получения пептидов, ковалентно конъюгированных с ПЭГ и конкретный способ, используемый в настоящем изобретении, не должен рассматриваться как ограничивающий (см. обзорную статью в Roberts, M. et al. Advanced Drug Delivery Reviews, 54: 459-476, 2002). Примером молекулы ПЭГ, которая может использоваться, является метокси-PEG2-MAL-40 К, раздвоенный ПЭГ малеинимид (Nektar, Huntsville, Алабама). Другие примеры включают, не ограничиваясь ими, насыпной mPEG-SBA-20 К (Nektar), mPEG2-ALD-40 К (Nektar) и метокси-PEG-MAL-30 К (Dow). Карбоксиконцевое присоединение ПЭГ может быть осуществлено посредством ферментативного соединения с использованием рекомбинантного пептидного агониста рецептора VPAC2 в качестве предшественника или альтернативных способов, известных из уровня техники и описанных, например, в патенте США 4343898 или Intl. J. Pept. & Prot. Res. 43: 127-38 (1994). Один из способов получения пегилированных пептидных агонистов рецептора VPAC2 по настоящему изобретению включает использование ПЭГ-малеинимида для непосредственного присоединения ПЭГ к тиольной группе пептида. Введение тиольной функциональной группы может быть осуществлено путем добавления или вставки остатка Cys или hC на или в пептид в положениях, описанных выше. Тиольная функциональная группа также может быть введена на боковую цепь пептида (например, ацилирование ε-аминогруппы лизина тиолсодержащей кислотой, такой как меркаптопропионовая кислота). В процессе пегилирования по настоящему изобретению используется реакция Михаэля для образования стойкого линкера тиоэфира. Реакция является высокоспецифичной и протекает в мягких условиях в присутствии других функциональных групп. ПЭГ малеинимид применяют в качестве реакционно-способного полимера для получения четких, биологически активных конъюгатов ПЭГ-белок. Предпочтительно в методике используют молярный избыток, предпочтительно молярный избыток от 1 до 10 тиолсодержащего пептидного агониста рецептора VPAC2 относительно ПЭГ малеинимида для завершения реакции. Реакции предпочтительно осуществляют при рН от 4,0 до 9,0 при комнатной температуре в течение периода от 10 мин до 40 ч. Избыток тиолсодержащего пегилированного пептида легко отделяется от пегилированного продукта обычными способами разделения. Пегилированный пептидный агонист рецептора VPAC2 предпочтительно выделяют с использованием обращенно-фазовой ВЭЖХ или эксклюзионной хроматографии. Конкретные условия, необходимые для пегилирования пептидных агонистов рецептора VPAC2, сформулированы в примере 8. Может осуществляться пегилирование цистеина с использованием ПЭГ малеинимида или раздвоенного ПЭГ малеинимида. Альтернативный способ получения пегилированных пептидных агонистов рецептора VPAC2 по изобретению включает пегилирование остатка лизина с использованием ПЭГсукцинимидильного производного. Для того чтобы достичь сайт-специфического пегилирования, остатки Lys, которые не используются для пегилирования, заменяют остатками Arg. Другой подход к пегилированию осуществляется посредством реакции Пиктета-Спенглера (Pictet-Spengler). Остаток Trp со свободной аминогруппой необходим для введения молекулы ПЭГ на селективный в отношении рецептора VPAC2 пептид. Один из подходов для достижения этого состоит в сайт-специфическом введении остатка Trp на амин боковой цепи Lys с помощью амидной связи в ходе твердофазного синтеза (см. пример 10). Различные предпочтительные особенности и варианты настоящего изобретения будут описаны ниже посредством примеров со ссылкой на приведенную фигуру, на которой показано увеличение инсулинового ответа на в/в нагрузку глюкозой у животных, получавших Р505 или Р525 за 24 ч до инъекции глюкозы. Инсулиновый ответ у получавших растворители животных приведен для сравнения. Пример 1. Получение селективных пептидных агонистов рецептора VPAC2 способом твердофазной химии t-Boc. Приблизительно 0,5-0,6 г (0,38-0,45 ммоль) смолы Boc Ser(Bzl)-PAM помещают в стандартную реакционную емкость объемом 60 мл. Двойное связывание осуществляют на синтезаторе пептидов Applied Biosystems ABI430A. Следующие аминокислоты с защищенной боковой цепью (картридж по 2 ммоль Boc аминокислот) получены от Midwest Biotech (Fishers, IN) и использованы в синтезе Arg-тозил (TOS), Asp- δ-циклогексиловый эфир (OcHx), Glu- δ-циклогексиловый эфир (OcHx), His-бензилоксиметил (BOM), Lys-2-хлорбензилоксикарбонил (2Cl-Z), Ser-O-бензиловый эфир (OBzl), Thr-O-бензиловый эфир (OBzl), Trp-формил (CHO) и Tyr-2-бромбензилоксикарбонил (2Br-Z). Трифторуксусная кислота (ТФУ), диизопропилэтиламин (ДИЭА), 0,5 М гидроксибензотриазол (HOBt) в ДМФА и 0,5 М дициклогексилкарбодиимид (ДЦК) в дихлорметане были приобретены у PE-Applied Biosystems (Foster City, CA). Диметилформамид (DMF-Burdick и Jackson) и дихлорметан (DCM-Mallinkrodt) приобретены у Mays Chemicals Co. (Индианаполис, IN). Стандартные двойные связывания осуществляют с использованием симметричного ангидрида или эфиров HOBt, оба образуются с использованием ДЦК. После завершения синтеза N-концевую группу Boc удаляют и пептидильные смолы обрабатывают 20% пиперидином в ДМФА для деформилирования боковой цепи Trp, если Trp присутствует в последовательности. Для N-концевого ацилирования добавляют четырехкратный избыток симметричного ангидрида соответствующей кислоты на пептидную смолу. Симметричный ангидрид получают активацией диизопропилкарбодиимидом (ДИК) в ДХМ. Реакции дают протекать в течение 4 ч и контролируют ее протекание с помощью нингидринового теста. После промывания ДХМ смолы переносят в реакционную емкость TEFLON и сушат под вакуумом. Расщепление осуществляют, присоединяя реакционные емкости к аппарату HF (фтористо-водородная кислота) (Penninsula Laboratories). Добавляют 1 мл м-крезола на 1 г смолы и 10 мл HF (приобретен у AGA, Индианаполис, IN), конденсируют в предварительно охлажденную емкость. Добавляют 1 мл ДМС (DMS) на 1 г смолы, если присутствует метионин. Реакционную смесь перемешивают в течение 1 ч на ледяной бане. HF удаляют под вакуумом. Остаток суспендируют в этиловом эфире. Твердые вещества отфильтровывают и промывают эфиром. Каждый пептид экстрагируют в водную уксусную кислоту и сушат вымораживанием или загружают непосредственно на колонку обращенно-фазовой хроматографии. Очистку проводят на колонках 2,2 ×25 см VYDAC C18 в буфере (0,1% ТФУ в воде). Осуществляют ВЭЖХ (Waters) с градиентом 20-90% В (0,1% ТФУ в ацетонитриле) на протяжении 120 мин со скоростью потока 10 мл/мин с УФ-мониторингом на длине волны 280 нм (4,0 А) и собирают фракции каждую минуту. Соответствующие фракции объединяют, замораживают и лиофилизируют. Высушенные продукты анализируют с помощью ВЭЖХ (0,46 ×15 см METASIL AQ C18) и масс-спектрометрии MALDI. Пример 2. Получение селективных пептидных агонистов рецептора VPAC2 методом твердофазной химии Fmoc. Приблизительно 114 мг смолы (50 ммоль) Fmos Ser(tBu) WANG (приобретена у GlycoPep, Чикаго, IL) помещают в каждую реакционную емкость. Синтез проводят на синтезаторе пептидов Rainin Symphony. Аналоги с C-концевым амидом получают с использованием 75 мг (50 мкмоль) смолы Rink Amide AM (Rapp Polymere. Tuebingen, Германия). Следующие Fmos аминокислоты были приобретены у GlycoPep (Чикаго, IL) и NovaBiochem (La Jolla, CA): Arg-2,2,4,6,7-пентаметилдигидробензофуран-5-сульфонил (Pbf), Asn-тритил (Trt), Asp- β-трет-бутиловый эфир (tBu), Glu- δ-трет-бутиловый эфир (tBu), Gln-тритил (Trt), His-тритил (Trt), Lys-трет-бутоксикарбонил (Boc), Ser-трет-бутиловый эфир (OtBu), Thr-трет-бутиловый эфир (OtBu), Trp-трет-бутоксикарбонил (Вос), Tyr-трет-бутиловый эфир (OtBu). Растворители диметилформамид (DMF-Burdick и Jackson), N-метилпирролидон (NMP-Burdick и Jackson) и дихлорметан (DCM-Mallinkrodt) были приобретены у Mays Chemicals Co. (Индианаполис, IN). Гидроксибензотризол (HOBt), диизопропилкарбодиимид (ДИК), диизопропилэтиламин (ДИЭА) и пиперидин (Pip) были приобретены у Aldrich Chemical Co. Milwaukee, WI). Все аминокислоты растворяют в ДМФА в концентрации 0,3 М. 3-часовое соединение активированных соединений ДИК/НОВТ выполняют после 20 мин снятия защиты с использованием 20% Pip/ДМФА. Каждую смолу промывают ДМФА после снятия защиты и соединения. После последнего соединения и снятия защиты пептидильные смолы промывают ДХМ и сушат под вакуумом в реакционной емкости. Для N-концевого ацилирования добавляют четырехкратный избыток симметричного ангидрида соответствующей кислоты на пептидную смолу. Симметричный ангидрид получают активацией ДИК в ДХМ. Реакции дают возможность протекать в течение 4 ч и протекание реакции контролируют с помощью нингидринового теста. Затем пептидную смолу промывают ДХМ и сушат под вакуумом. Реакционную смесь реакции расщепления перемешивают в течение 2 ч с коктейлем расщепления, состоящим из 0,2 мл тиоанизола, 0,2 мл метанола, 0,4 мл триизопропилсилана на 10 мл ТФУ, все приобретены у Aldrich Chemical Co., Milwaukee, WI. Если в последовательности присутствует Cys, добавляют 2% этандитиола. Фильтраты ТФУ добавляют к 40 мл этилового эфира. Осадок центрифугируют в течение 2 мин при 2000 об/мин. Супернатанты декантируют. Гранулы ресуспендируют в 40 мл эфира, снова центрифугируют, повторно декантируют, сушат в атмосфере азота, а затем под вакуумом. 0,3-0,6 мг каждого продукта растворяют в 1 мл 0,1% ТФУ/ацетонитрил (ACN) и 20 мкл анализируют с помощью ВЭЖХ [0,46 ×15 см Metasil AQ C18, скорость потока 1 мл/мин, 45 °С, 214 нМ (0,2А), А=0,1% ТФУ, В=0,1% ТФУ/50% ацетонитрила. Градиент = 50-90% В в течение 30 мин]. Очистку проводят на колонке 2,2 ×25 см Vydac C18 в буферном растворе (0,1% ТФУ в воде). Используют градиент 20-90% В (0,1% ТФУ в ацетонитриле), хроматограмму ВЭЖХ (Waters) регистрируют на протяжении 120 мин со скоростью потока 10 мл/мин с УФ-мониторингом на длине волны 280 нм (4,0 А) и собирают фракции каждую минуту. Соответствующие фракции объединяют, замораживают и лиофилизируют. Высушенные продукты анализируют с помощью ВЭЖХ (0,46 ×15 см Metasil AQ C18) и масс-спектрометрии MALDI. Предшественник Р521 (C6-HSDAVFTEQY(ОМе) TOrnLRAibQLAAbuAibOrnYAibQSIOrnOrnGGPSSGAPPPCC-NH 2 ). Синтез осуществляют с применением протоколов Fmoc, описанныех выше. Пептид характеризуют с помощью аналитической ВЭЖХ: t R =10,9 мин, условия ВЭЖХ описаны выше, и MALDI-TOF: вычисленное соотношение массы к заряду = 4290,0, измеренное соотношение массы к заряду = 4290,8 [М+Н + ]. После очистки с использованием обращенно-фазовой препаративной ВЭЖХ чистые фракции объединяют и лиофилизируют. Выход: 14,8 мг конечного лиофилизированного порошка. Предшественник Р525 (C6-HSDAVFTEQY(ОМе) TOrnLRAibQLAAbuAibOrnYAibQAibIOrnOrnGGPSSGAPPPCC-NH 2 ). Получают, как описано для предшественника Р521. Аналитическая ВЭЖХ: t R =11,0 мин. MALDI-TOF: вычисленное соотношение массы к заряду = 4288,0, измеренное соотношение массы к заряду = 4288,8 [М+Н + ]. Выход: 18,7 мг конечного лиофилизированного порошка. Предшественник Р574 (C6-HSDAVFTEQY(ОМе) TOrnLRAibQLAAbuAibOrnY(OMe)LQAibIOrnOrnGGPSSGAPPPCC-NH 2 ). Получают, как описано для предшественника Р521. Аналитическая ВЭЖХ: t R =11,7 мин. MALDI-TOF: вычисленное соотношение массы к заряду = 4330,1, измеренное соотношение массы к заряду = 4330,7 [М+Н + ]. Выход: 46,2 мг конечного лиофилизированного порошка. Пример 3. Эффективность в отношении рецепторов VPAC2 человека in vitro. Alpha screen. Клетки один раз промывают в колбе для культивирования фосфатным буферным раствором. Далее клетки промывают не содержащим фермента буфером диссоциации. Диссоциированные клетки удаляют. Затем клетки собирают и промывают в стимулирующем буфере. Для каждой точки данных используют 50000 клеток, суспендированных в буфере стимуляции. К данному буферу добавляют гранулы акцептора Alpha screen наряду со стимулами. Полученную смесь инкубируют в течение 60 мин. Добавляют буфер лизиса и гранулы донора Alpha screen, затем инкубируют в течение 60-120 мин. Сигнал Alpha screen (показывает уровни внутриклеточного цАМФ) считывают на подходящем приборе (например, AlphaQuest от Perkin-Elmer). Стадии, в том числе донор и акцептор Alpha screen, выполняют при сниженном освещении. ЕС 50 для генерации цАМФ вычисляют на основе сырого сигнала или на основе абсолютных уровней цАМФ, определенных по стандартной кривой, построенной для каждого планшета. Результаты для каждого агониста представляют собой результаты по меньшей мере двух анализов, выполненных в едином цикле. Для некоторых агонистов результаты представляет собой среднее значение более чем одного определения. Исследуемые концентрации пептида составляли 10000, 1000, 100, 10, 3, 1, 0,1, 0,01, 0,003, 0,001, 0,0001 и 0,00001 нМ. DiscoveRx. Линию клеток CHO-S, стабильно экспрессирующих рецептор VPAC2 человека, засевают в 96-луночный планшет для микротитрования с плотностью 50000 клеток/лунку накануне анализа. Клеткам дают возможность прикрепиться на протяжении 24 ч в 200 мкл среды для культивирования. В день эксперимента среду удаляют. Клетки дважды промывают. Клетки инкубируют в буфере анализа плюс 3-изобутил-1-метилксантин (IBMX) в течение 15 мин при комнатной температуре. Впоследствии добавляют стимулы и растворяют в буфере анализа. Стимулы присутствуют в течение 30 мин. Затем буфер анализа осторожно удаляют. Добавляют реактив для лизиса клеток из набора DiscoveRx цАМФ. С этого момента используют стандартный протокол для образования сигнала цАМФ, описанный производителем (DiscoveRx Inc., США). Значения ЕС 50 для генерации цАМФ рассчитывают на основе сырого сигнала или на основе абсолютных уровней цАМФ, определенных по стандартной кривой для каждого планшета. Результаты для каждого агониста представляют собой среднее значение двух независимых определений. Исследуемые концентрации пептида обычно составляют 1000, 300, 100, 10, 1, 0,3, 0,1, 0,01, 0,001, 0,0001 и 0 нМ. Активность (ЕС 50 , нМ) в отношении человеческого рецептора VPAC2 приведена в табл. 1 для различных форматов анализа. Таблица 1 Пример 4. Селективность. Анализы связывания. Используют мембрану, приготовленную из стабильной линии клеток VPAC2 (см. пример 3) или из клеток, временно трансфицированных человеческим VPAC1 или РАС1. Анализ связывания на фильтре выполняют с использованием меченого 125 I VIP для VPAC1 и VPAC2 и меченого 125 I PACAP-27 для РАС1 в качестве маркеров. Растворы и оборудование для данного анализа включают в себя следующее: раствор для предварительного пропитывания: 0,5% полиэтиленамина в воде дистиллированной: буфер для промывания фильтровальных пластин: 25 мМ HEPES, рН 7,4; блокирующий буфер: 25 мМ HEPES, рН 7,4; 0,2% не содержащего протеазы бычьего сывороточного альбумина; буфер анализа: 25 мМ HEPES, рН 7,4; 0,5 % не содержащего протеазы бычьего сывороточного альбумина; планшет для разведения и анализа: PS-Microplate, U-форма; планшет для фильтрования: непрозрачный планшет FB Opaque; фильтр Glasfiber типа В размером пор 1,0 мкм. Чтобы подготовить планшеты для фильтрования, раствор для предварительного пропитывания отсасывают с помощью вакуумной фильтрации. Планшеты дважды промывают по 200 мкл буфера для промывания; 200 мкл блокирующего буфера добавляют на планшет для фильтрования. Затем планшет для фильтрования инкубируют с 200 мкл раствора для предварительного пропитывания в течение 1 ч при комнатной температуре. На планшет для анализа помещают 25 мкл буфера анализа, 25 мкл мембран (2,5 мкг), суспендированных в буфере анализа, 25 мкл агониста в буфере анализа и 25 мкл маркера (приблизительно 40000 импульсов/мин) в буфере анализа. Заполненный планшет инкубируют в течение 1 ч при встряхивании. Осуществляют перенос с планшета анализа на планшет для фильтрования. Блокирующий буфер удаляют вакуумной фильтрацией и дважды промывают буфером для промывания. 90 мкл переносят с планшета для анализа на планшет для фильтрования. Аликвоту 90 мкл, перенесенную с планшета для анализа, отсасывают и трижды промывают 200 мкл буфера для промывания. Пластиковую основу удаляют. Ее сушат в течение 1 ч при температуре 60 °С. Добавляют 30 мкл Microscint. Выполняют подсчет. Связывание с рецептором (IC 50 ) для VPAC2, VPAC1 и РАС1 человека приведено в табл. 2. Таблица 2 Пример 5. Эффективность в отношении рецепторов VPAC1 и VPAC2 крыс in vitro. DiscoveRx. Клетки CHO-PO временно трансфицируют ДНК рецептора VPAC1 или VPAC2 крыс, используя коммерчески доступные реактивы для трансфекции (Lipofectamine от Invitrogen). Клетки засевают с плотностью 10000/лунку в 96-луночный планшет и дают расти на протяжении 3 дней в 200 мл среды культивирования. В день 3 проводят анализ. В день эксперимента среду удаляют. Клетки дважды промывают. Клетки инкубируют в буфере анализа плюс 3-изобутил-1-метилксантин (IBMX) в течение 15 мин при комнатной температуре. Далее добавляют стимулы и растворяют в буфере анализа. Стимулы присутствуют на протяжении 30 мин. Затем буфер анализа осторожно удаляют. Добавляют реактив для лизиса клеток из набора DiscoveRx цАМФ. Далее используют стандартный протокол генерации сигнала цАМФ, как описано производителем (DiscoveRx Inc., США). Значения ЕС 50 для генерации цАМФ вычисляют на основании сырого сигнала или на основании абсолютных уровней цАМФ, как определяется стандартной кривой для каждого планшета. Результаты для каждого агониста представляют собой среднее значение двух независимых измерений. Результаты для VPAC1 и VPAC2 крыс генерируются только с использованием анализа DiscoveRx. Обычно исследуемые концентрации пептида представляют собой 1000, 300, 100, 10, 1, 0,3, 0,1, 0,01, 0,001, 0,0001 и 0 нМ. Активность (ЕС 50 , нМ) в отношении крысиных рецепторов VPAC2 и VPAC1 приведена в табл. 3. Таблица 3 Пример 6. Анализы in vivo. Тест на переносимость глюкозы при внутривенном введении (IVGTT). Нормальные крысы Вистар голодают на протяжении ночи, после чего животным проводят анестезию перед экспериментом. Крысам устанавливают катетеры для забора крови. Агонист вводят подкожно, обычно за 24 ч до нагрузки глюкозой. Образцы крови отбирают из сонной артерии. Образец крови отбирают непосредственно перед инъекцией глюкозы вместе с агонистом. После отбора образца крови в начальной точке глюкозу смешивают и вводят внутривенно. Нагрузку глюкозой 0,5 г/кг массы тела осуществляют путем инъекционного введения общего количества растворителя с глюкозой и агонистом 1,5 мл/кг массы тела. Концентрации пептида варьируют таким образом, чтобы получить желательную дозу в мкг/кг. Образцы крови отбирают через 2, 4, 6 и 10 мин после введения глюкозы. Животные из контрольной группы получают такой же растворитель вместе с глюкозой, но без добавления агониста. В некоторых случаях отбирали образцы крови через 20 и 30 мин после введения глюкозы. К образцу крови добавляют апротинин (250-500 кМЕ/мл крови). Затем плазму анализируют на содержание глюкозы и инсулина с применением стандартных методик. В анализе используют приготовленный и откалиброванный запасной раствор пептида в фосфатном буферном растворе. Обычно такой запасной раствор представляет собой предварительно разбавленный запасной раствор с концентрацией 100 мкМ. Однако используют более концентрированный запасной раствор с концентрацией приблизительно 1 мг агониста/мл. Всегда известна конкретная концентрация. Вариабельность максимальной реакции по большей части возникает за счет вариабельности дозы растворителя. Подробное описание протокола приведено ниже. Таблица 4а Площадь под кривой = область под кривой (инсулин, 0-10 мин после введения глюкозы) Фармакокинетические профили пегилированных пептидов. Здоровым крысам (по 3 животного на группу) Фишер 344 инъекционно вводят 100 мкг/кг массы тела агонист (количество агониста, основано на содержании пептида и растворенное в фосфатном буферном растворе). Образцы крови отбирают через 3, 12, 24, 48, 72, 96 и 168 ч после введения дозы и содержание пептида в плазме анализируют радиоиммуноанализом (RIA), направленным против N-конца пептида. Далее вычисляют параметры фармакокинетики с использованием независимого от модели способа (WinNonlin Pro, Pharsight Corp., Pharsight Corp., Mountain View, CA, США). Таблица 4b *NC = не вычислено из-за недостаточности данных. Пример 7. Исследование стабильности в сыворотке крыс. Для определения стабильности пептидных агонистов рецептора VPAC2 в сыворотке крыс получали клетки CHO-VPAC2 клон #6 (96-луночные планшеты/50000 клеток/лунку, культивируют в течение 1 дня), фосфатный буферный раствор 1X (Gibco), пептиды для анализа в виде 100 мкМ запасного раствора, сыворотку крыс, полученную от умерщвленных нормальных крыс Вистар, апротинин и набор для анализа DiscoveRx. Сыворотку крыс хранят при температуре 4 °С до использования (не более 2 недель). В день 0 готовят две аликвоты по 100 мкл 10 мкМ раствора пептида в сыворотке крыс, добавляя 10 мкл запасного раствора пептида к 90 мкл сыворотки крыс для каждой аликвоты. Добавляют 250 кМЕ апротинина на 1 мл в одну из полученных аликвот. Аликвоту с апротинином хранят при температуре 4 °С. Аликвоту без апротинина хранят при температуре 37 °С. Аликвоты инкубируют в течение 24 или 72 ч. В день 1 после инкубации аликвот, приготовленных в день 0, в течение 24 или 72 ч готовят буфер для инкубации, содержащий фосфатный буферный раствор + 1,3 мМ CaCl 2 , 1,2 мМ MgCl 2 , 2 мМ глюкозы и 0,25 мМ 3-изобутил-1-метилксантина (IBMX). Готовят планшет с 11 серийными 5Х разведениями пептида для аликвот 4 и 37 °С для каждого исследуемого пептида. 2000 нМ используют в качестве максимальной концентрации, если ЕС 50 пептида составляет более 1 нМ, и 1000 нМ используют в качестве максимальной концентрации, если ЕС 50 пептида составляет менее 1 нМ по данным первичного скрининга (см. пример 3). Планшет(ы) с клетками дважды промывают в буфере для инкубации. В планшетах оставляют 50 мкл среды для инкубации на лунку на протяжении 15 мин. 50 мкл раствора на лунку переносят к клеткам с планшета, подготовленного с 11 серийными 5Х разведениями пептида для аликвот 4 и 37 °С для каждого исследуемого пептида, используя максимальные концентрации, определенные по данным первичного скрининга, в двойном повторении. На данной стадии концентрация пептида разбавляется с фактором 2. Клетки инкубируют при комнатной температуре в течение 30 мин. Супернатант удаляют. Добавляют 40 мкл/лунку буфера антитела/экстракта DiscoveRx. Клетки инкубируют на шейкере (300 об/мин) в течение 1 ч. Следуют обычной методике с набором DiscoveRx. Стандарты цАМФ включают в колонку 12. Значения ЕС 50 определяют на основе данных анализа цАМФ. Оставшееся количество активного пептида оценивают по формуле ЕС 50 , 4 °С /ЕС 50 , 37 °С для каждого состояния. Таблица 5 1 Значения > 100% могут представлять высвобождение интактного пептида из ПЭГ конъюгата. Пример 8. Пегилирование селективных пептидных агонистов рецептора VPAC2 с использованием химии на основе тиола. В целом, реакции пегилирования проводят в условиях, которые позволяют образование тиоэфирной связи. Конкретно, рН раствора находится в интервале приблизительно от 4 до 9, и интервал концентраций тиолсодержащего пептида составляет от 0,7 до 10 молярного избытка концентрации ПЭГ малеинимида. Реакции пегилирования обычно проводят при комнатной температуре. Пегилированный пептидный агонист рецептора VPAC2 далее выделяют с использованием обращенно-фазовой ВЭЖХ или эксклюзионной хроматографии (size exclusion, SEC). Пегилированные пептидные агонисты характеризуют с использованием аналитической обращенно-фазовой ВЭЖХ, эксклюзионной ВЭЖХ, SDS-PAGE и/или масс-спектрометрии MALDI. Обычно тиольную функцию вводят в или на селективный пептидный агонист рецептора VPAC2, добавляя цистеин, или гомоцистеин, или тиолсодержащий фрагмент на один или оба конца или путем вставки цистеина, или гомоцистеина, или тиолсодержащего фрагмента в последовательность. Тиолсодержащие пептидные агонисты рецептора VPAC2 реагируют с ПЭГ малеинимидом молекулярной массой 40, 30 или 20 кДа для получения производных, где ПЭГ присоединен ковалентно посредством тиоэфирной связи. Синтез Р505. 19 мг пептидного предшественника (непегилированный Р505) 162 мг метокси-ПЭГ малеинимида (NOF, Япония) со средней молекулярной массой 20000 Да растворяют в 2 мл буфера 100 мМ NH 4 Ac, содержащего 10 мМ ЭДТА (рН 6,8), и реакции позволяют протекать в течение 4 ч. Получают 97 мг продукта в виде лиофилизированного порошка после очистки препаративной обращенно-фазовой ВЭЖХ. Пегилированный пептидный агонист характеризуют с помощью обращенно-фазовой ВЭЖХ и эксклюзионной ВЭЖХ и исследуют его активность in vitro. Синтез Р525. 18,7 мг пептидного предшественника (непегилированный Р525) и 157 мг метокси-ПЭГ малеинимида со средней молекулярной массой 20000 Да растворяют в 2 мл буфера 100 мМ NH 4 Ac, содержащего 10 мМ ЭДТА (рН 6,8), и реакции позволяют протекать в течение 4 ч. Получают 114 мг продукта в виде лиофилизированного порошка после очистки препаративной обращенно-фазовой ВЭЖХ. Пегилированный пептидный агонист характеризуют с помощью обращенно-фазовой ВЭЖХ и эксклюзионной ВЭЖХ и исследуют его активность in vitro. Синтез Р572. 22,3 мг пептидного предшественника (непегилированный Р572) и 177 мг метокси-ПЭГ малеинимида со средней молекулярной массой 20000 Да растворяют в 2 мл буфера 100 мМ NH 4 Ac, содержащего 10 мМ ЭДТА (рН 6,8), и реакции позволяют протекать в течение 4 ч. Получают 137 мг продукта в виде лиофилизированного порошка после очистки препаративной обращенно-фазовой ВЭЖХ. Пегилированный пептидный агонист характеризуют с помощью обращенно-фазовой ВЭЖХ и эксклюзионной ВЭЖХ и исследуют их активность in vitro. Синтез Р574. 31,9 мг пептидного предшественника (непегилированный Р574) и 283 мг метокси-ПЭГ малеинимида со средней молекулярной массой 20000 Да растворяют в 3 мл буфера 100 мМ NH 4 Ac, содержащего 10 мМ ЭДТА (рН 6,8), и реакции позволяют протекать в течение 4 ч. Получают 171 мг продукта в виде лиофилизированного порошка после очистки препаративной обращенно-фазовой ВЭЖХ. Пегилированный пептидный агонист характеризуют с помощью обращенно-фазовой ВЭЖХ и эксклюзионной ВЭЖХ и исследуют их активность in vitro. Синтез Р602. 20 мг пептидного предшественника (непегилированный Р602) и 223 мг метоксиполи(этиленгликоль)малеинимид-пропиоамида (Chirotech Technology Ltd., Великобритания) со средней молекулярной массой 30000 Да растворяют в 2 мл буфера 100 мМ NH 4 Ac, содержащего 10 мМ ЭДТА (рН 6,8), и реакции позволяют протекать в течение 4 ч. Получают 132 мг продукта в виде лиофилизированного порошка после очистки препаративной обращенно-фазовой ВЭЖХ. Пегилированный пептидный агонист характеризуют с помощью обращенно-фазовой ВЭЖХ и эксклюзионной ВЭЖХ и исследуют их активность in vitro. Пример 9. Пегилирование посредством ацилирования в боковой цепи лизина. Для того чтобы осуществить сайт-специфическое пегилирование селективных пептидных агонистов рецептора VPAC2, все остатки Lys заменяют на остатки Arg, за исключением остатков Lys, где предусмотрено пегилирование. Молекула ПЭГ, которая может использоваться, представляет собой mPEG-SBA-20 К (Nektar, серия РТ-04Е-11). Реакцию пегилирования предпочтительно проводят при комнатной температуре в течение 2-3 ч. Пептид очищают препаративной ВЭЖХ. Пример 10. Пегилирование посредством реакции Пиктета-Спенглера (Pictet-Spengler). Для пегилирования посредством реакции Пиктета-Спенглера (Pictet-Spengler) необходим остаток Trp со свободным амином для введения молекулы ПЭГ на селективный пептидный агонист рецептора VPAC2. Для присоединения остатка Trp на боковую цепь остатка Lys экстенсивные SAR исследования показывают, что данная модификация не изменяет свойств исходного пептида с точки зрения его эффективности и селективности in vitro. Для реакции используют ПЭГ с альдегидной функцией, например mPEG2-BUTYRALD-40 К (Nektar, США). Сайт-специфическое пегилирование включает образование тетракарболинового кольца между ПЭГ и пептидом. Пегилирование осуществляют в ледяной уксусной кислоте при комнатной температуре в течение 1-48 ч. В реакции используют молярный избыток от 1 до 10 ПЭГ альдегида. После удаления уксусной кислоты пегилированный пептидный агонист рецептора VPAC2 выделяют с помощью препаративной обращенно-фазовой ВЭЖХ. Синтез Р535. 27,7 мг пептидного предшественника (непегилированный Р535) и 590 мг mPEG2-BUTYRALD-40 К растворяют в 3 мл уксусной кислоты и реакции позволяют протекать в течение 2 дней. Продукт выделяют препаративной обращенно-фазовой ВЭЖХ с получением 94 мг пегилированного пептидного агониста в виде лиофилизированного порошка. Пегилированный пептидный агонист характеризуют обращенно-фазовой ВЭЖХ и эксклюзионной ВЭЖХ и исследуют in vitro. Синтез Р557. 23 мг пептидного предшественника (непегилированный Р557) и 460 мг mPEG2-BUTYRALD-40 К растворяют в 3 мл уксусной кислоты и реакции позволяют протекать в течение 2 дней. Продукт выделяют препаративной обращенно-фазовой ВЭЖХ с получением 50 мг пегилированного пептидного агониста в виде лиофилизированного порошка. Пегилированный пептидный агонист характеризуют обращенно-фазовой ВЭЖХ и эксклюзионной ВЭЖХ и исследуют in vitro. Другие модификации настоящего изобретения будут очевидны для специалиста в данной области без выхода за границы изобретения. |