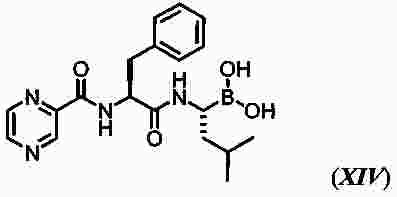
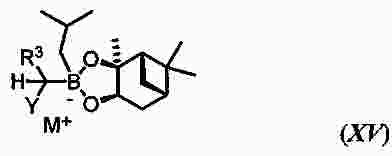
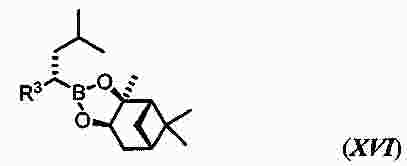
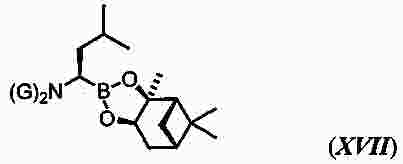
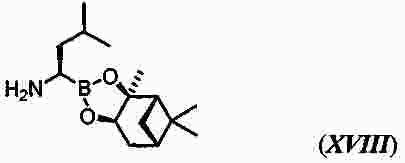
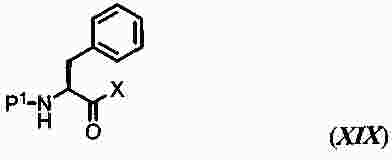
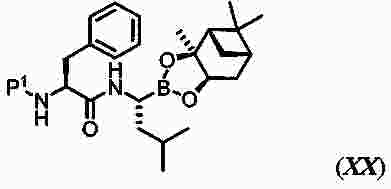
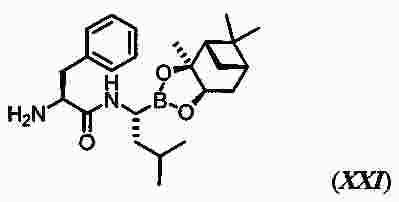
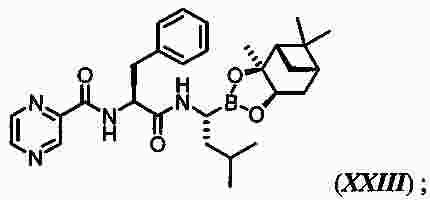
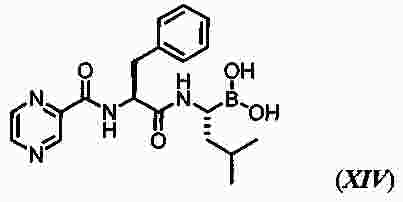
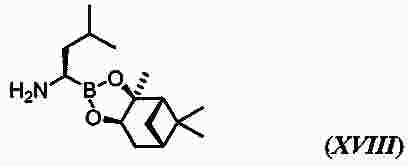
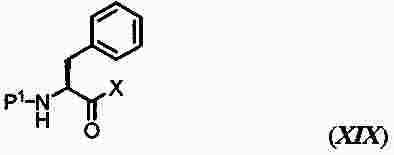
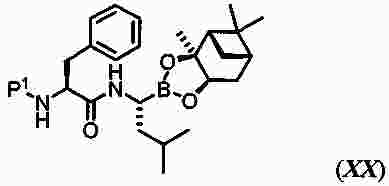
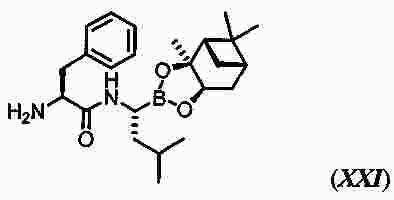
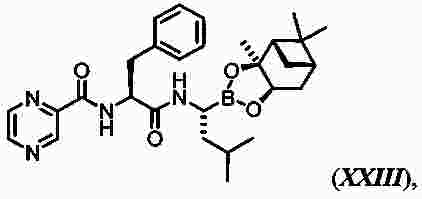
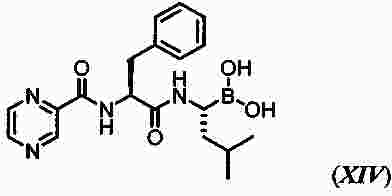
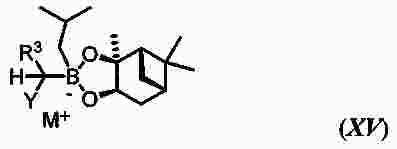
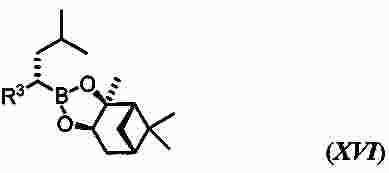
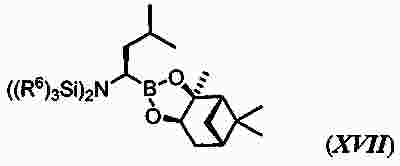
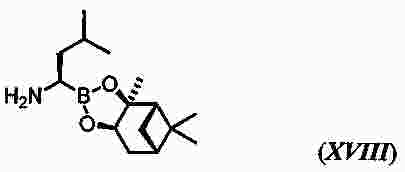
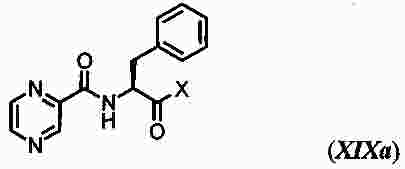
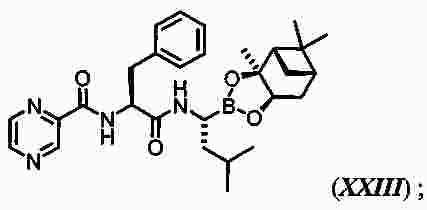
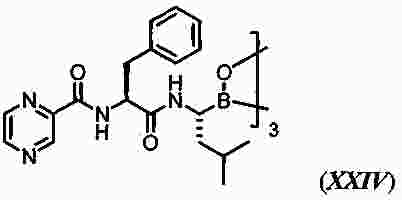
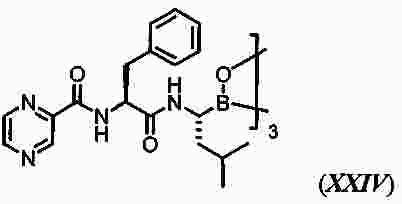
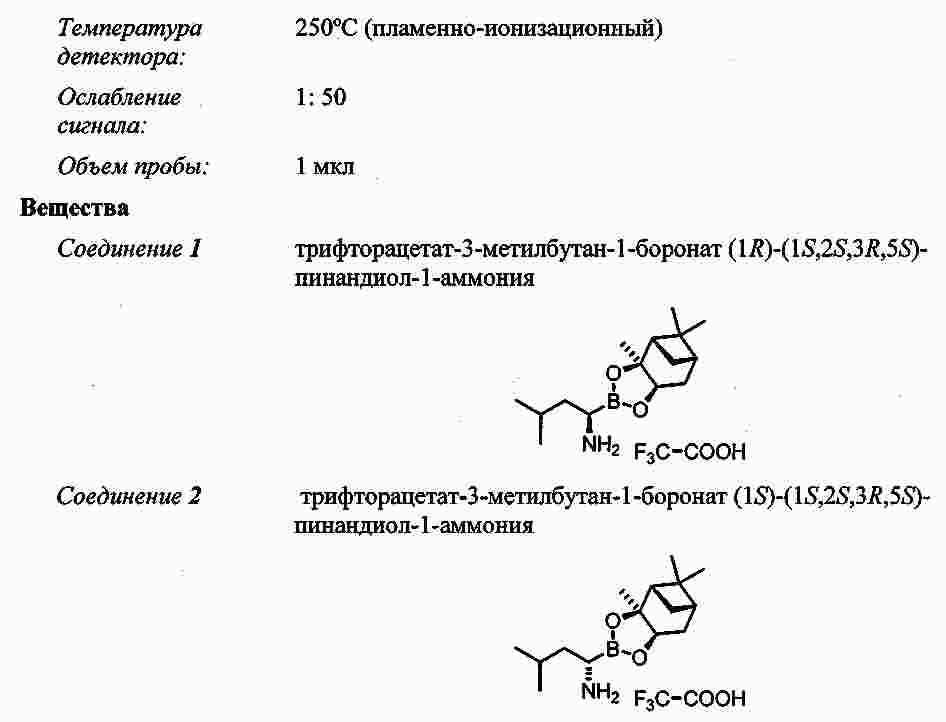
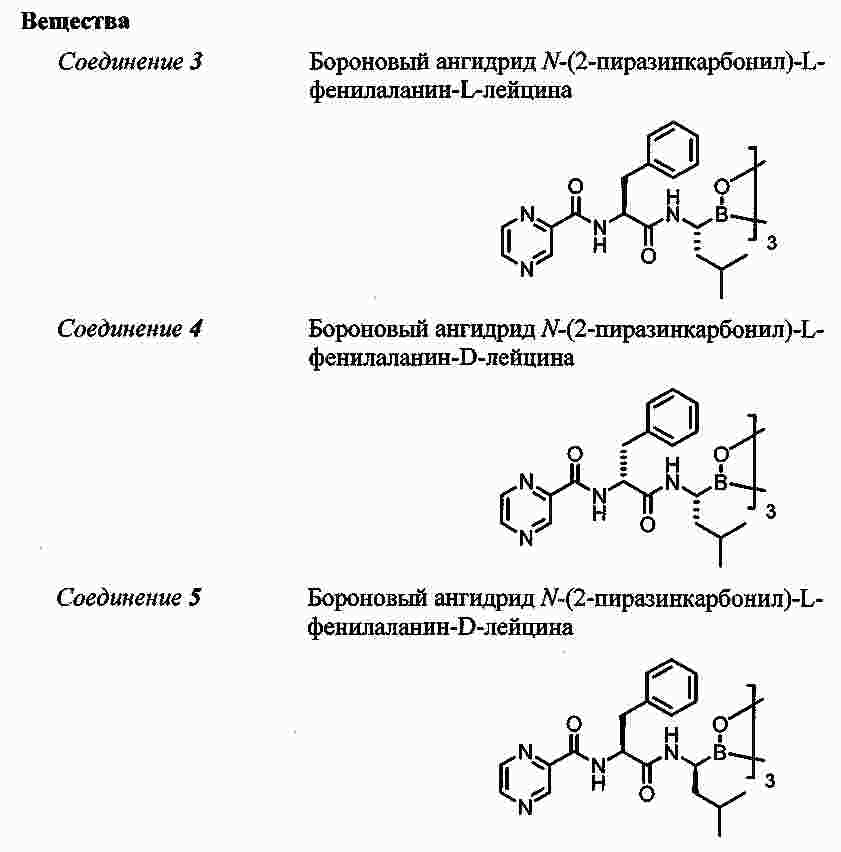
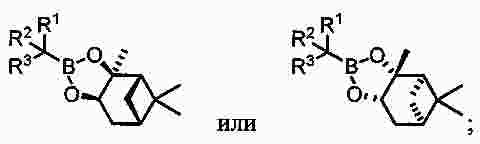|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea000012927b*\id |
|
Термины запроса в документе Реферат Изобретение относится к синтезу эфиров бороновой кислоты и кислотных соединений. Более конкретно, изобретение касается усовершенствованных способов синтеза для крупномасштабного производства эфиров бороновой кислоты и кислотных соединений, включая ингибитор протеазомы на основе пептидной бороновой кислоты - бортезомиб. Формула [0001] Крупномасштабный способ получения эфирного соединения бороновой кислоты формулы (I) [0002] Способ по п.1, в котором реакционная смесь содержит координационный сорастворитель. [0003] Способ по п.2, в котором координационный сорастворитель выбирают из группы, состоящей из тетрагидрофурана, диоксана, воды и их смесей. [0004] Способ по п.3, в котором координационный сорастворитель составляет не больше чем приблизительно 15% от объема реакционной смеси. [0005] Способ по п.1, в котором растворимость воды в растворителе - простом эфире, который плохо смешивается с водой, составляет меньше чем 2 мас.%. [0006] Способ по п.5, в котором растворитель - простой эфир, который плохо смешивается с водой, выбирают из группы, состоящей из трет-бутилметилового эфира, трет-бутилэтилового эфира, трет-амилметилового эфира, изопропилового эфира и их смесей. [0007] Способ по п.6, в котором растворитель - простой эфир, который плохо смешивается с водой, составляет по меньшей мере около 70% от объема реакционной смеси. [0008] Способ по п.1, в котором на стадии (а) получается по меньшей мере около 5 моль боронатного комплекса формулы (II). [0009] Способ по п.1, в котором на стадии (а) получается по меньшей мере около 20 моль боронатного комплекса формулы (II). [0010] Способ по п.1, в котором на стадии (а) получается по меньшей мере около 50 моль боронатного комплекса формулы (II). [0011] Способ по п.1, в котором на стадии (а) получается по меньшей мере около 100 моль боронатного комплекса формулы (II). [0012] Способ по п.1, в котором кислота Льюиса является влажной. [0013] Способ по п.1, в котором на стадии (а) боронатный комплекс формулы (II) получается в растворе, содержащем растворитель - простой эфир, который плохо смешивается с водой, и стадия контактирования (b) включает в себя этапы: [0014] Способ по п.1, в котором на стадии (а) боронатный комплекс формулы (II) получается в растворе, содержащем растворитель - простой эфир, который плохо смешивается с водой, и стадия контактирования (b) включает в себя этапы: [0015] Способ по п.1, в котором R1 представляет собой C1-8-алифатическую группу. [0016] Способ по п.1, в котором R4 и R5 вместе представляют собой хиральную группу. [0017] Способ по п.16, в котором боронатный комплекс формулы (II) представляет собой [0018] Способ по п.16, в котором на стадии (b) получают эфирное соединение бороновой кислоты формулы (I), где атом углерода, связанный с радикалами R1, R2 и R3, является хиральным центром, имеющим соотношение диастереоизомеров по меньшей мере около 96:4 относительно хирального центра в хиральной группе R4-R5. [0019] Способ по п.16, в котором на стадии (b) получают эфирное соединение бороновой кислоты формулы (I), где атом углерода, связанный с радикалами R1, R2 и R3, является хиральным центром, имеющим соотношение диастереоизомеров по меньшей мере около 97:3 относительно хирального центра в R4-R5 хиральной группе. [0020] Способ по п.16, отличающийся по меньшей мере одним из следующих признаков: [0021] Способ по п.20, отличающийся по меньшей мере двумя признаками из (a)-(g). [0022] Способ по п.20, отличающийся по меньшей мере тремя признаками из (a)-(g). [0023] Способ по п.20, отличающийся всеми семью признаками (a)-(g). [0024] Способ по п.18, который дополнительно включает: [0025] Способ по п.24, в котором остаток содержит по меньшей мере около 5 моль эфирного соединения бороновой кислоты формулы (I). [0026] Способ по п.25, в котором эфирное соединение бороновой кислоты формулы (I), присутствующее в остатке, имеет соотношение диастереоизомеров при атоме углерода, связанном с радикалами R1, R2 и R3, по меньшей мере около 96:4 относительно хирального центра в R4-R5 хиральной группы. [0027] Способ по п.25, в котором эфирное соединение бороновой кислоты формулы (I), присутствующее в остатке, имеет соотношение диастереоизомеров при атоме углерода, связанном с радикалами R1, R2 и R3, по меньшей мере около 97:3 относительно хирального центра в R4-R5 хиральной группы. [0028] Крупномасштабный способ получения эфирного соединения бороновой кислоты формулы (I) [0029] Крупномасштабный способ получения эфирного соединения бороновой кислоты формулы (I) [0030] Крупномасштабный способ получения эфирного соединения бороновой кислоты формулы (I) [0031] Способ по п.29 или 30, в котором пространственно затрудненным основанием является диалкиламид щелочного металла формулы M2N(R#)2, в которой М2 выбирают из группы, состоящей из Li, Na и K, и каждый R#, независимо, является разветвленной или циклической С3-6-алифатической группой. [0032] Способ по п.30, в котором органический растворитель на стадии (а) выбирают из группы, состоящей из ацетонитрила, толуола, гексана, гептана и их смесей. [0033] Способ по п.30, в котором органический растворитель на стадии (а) представляет собой простой эфир, который плохо смешивается с водой. [0034] Способ по п.33, в котором каждый раствор на стадиях (а) и (с) содержит один и тот же растворитель - простой эфир. [0035] Способ по п.34, в котором на стадии (b) получают раствор продукта, содержащий эфир бороновой кислоты формулы (III), и раствор продукта со стадии (b) используется на стадии (с) без выделения эфира бороновой кислоты формулы (III). [0036] Крупномасштабный способ получения аминоэфирного соединения бороновой кислоты формулы (VII) [0037] Способ по п.36, в котором реакционная смесь на стадии (с) содержит органический растворитель, в котором побочный продукт M1-R3 имеет низкую растворимость. [0038] Способ по п.37, в котором М1 означает Li и R3 - это Cl. [0039] Способ по п.38, в котором реакционная смесь на стадии (с) содержит органический растворитель, выбранный из группы, состоящей из метилциклогексана, циклогексана, гептана, гексана, толуола и их смесей. [0040] Способ по п.36, в котором взаимодействие на стадии (с) проводят при температуре реакции в диапазоне приблизительно от -100 до 50 °С. [0041] Способ по п.40, в котором температура реакции находится в диапазоне приблизительно от -50 до 25 °С. [0042] Способ по п.40, в котором температура реакции находится в диапазоне приблизительно от -30 до 0 °С. [0043] Способ по п.36, в котором стадия (d) включает обработку соединения формулы (VIII) кислотой и выделение соединения формулы (VII) в виде аддитивной соли с кислотой. [0044] Способ по п.43, в котором кислота означает трифторуксусную кислоту. [0045] Способ по п.36, в котором стадия (с) дополнительно включает фильтрацию реакционной смеси, чтобы получить фильтрат, содержащий соединение формулы (VIII). [0046] Способ по п.45, в котором на стадии (с) реагент формулы M1-N(Si(R6)3)2 добавляют в реакционную смесь в виде раствора, содержащего тетрагидрофуран, и стадия (с) дополнительно включает удаление тетрагидрофурана до фильтрации реакционной смеси. [0047] Способ по п.45, в котором фильтрат используют непосредственно на стадии (d). [0048] Способ по п.36, который дополнительно включает в себя стадию: [0049] Способ по п.48, в котором Р1 является отщепляемой защитной группой. [0050] Способ по п.49, который дополнительно включает следующие стадии: [0051] Крупномасштабный способ получения аминоэфирного соединения бороновой кислоты формулы (VIIa) или (VIIb) [0052] Крупномасштабный способ получения соединения формулы (XIV) [0053] Способ по п.52, отличающийся по меньшей мере одним из следующих признаков (1)-(4): [0054] Способ по п.53, отличающийся всеми признаками (1)-(4). [0055] Способ по п.53, в котором стадии (h)(iii) включают следующие стадии: [0056] Способ по п.55, в котором на стадии (h)(iii)(3) водный слой промывают дихлорметаном. [0057] Способ по п.56, в котором на стадии (h)(iv) соединение формулы (XIV) или бороновый ангидрид этого соединения, экстрагируют дихлорметаном, растворитель заменяют на этилацетат, и соединение формулы (XIV) или бороновый ангидрид этого соединения кристаллизуется за счет добавления гексана или гептана. [0058] Способ по п.57, в котором добавление гексана или гептана приводит к кристаллизации циклического ангидрида тримерной бороновой кислоты формулы (XXIV) [0059] Крупномасштабный способ получения соединения формулы (XIV) [0060] Способ по п.59, который на стадии (dd)(iii) включает следующие стадии: [0061] Способ по п.60, в котором на стадии (dd)(iv) соединение формулы (XIV) или бороновый ангидрид этого соединения экстрагируют дихлорметаном, растворитель заменяют на этилацетат, и соединение формулы (XIV) или бороновый ангидрид этого соединения кристаллизуется путем добавления гексана или гептана. [0062] Способ по п.61, в котором добавление гексана или гептана приводит к кристаллизации циклического ангидрида тримерной бороновой кислоты формулы (XXIV) [0063] Крупномасштабный способ получения соединения формулы (XIV) [0064] Способ по п.63, отличающийся по меньшей мере одним из следующих признаков (1), (2): [0065] Способ по п.64, в котором стадия (f')(iii) включает следующие стадии: [0066] Способ по п.65, в котором на стадии (f')(iii)(3) водный слой промывают дихлорметаном. [0067] Способ по п.65, в котором на стадии (f')(iv) соединение формулы (XIV) или бороновый ангидрид этого соединения экстрагируют дихлорметаном, растворитель заменяют на этилацетат, и соединение формулы (XIV) или бороновый ангидрид этого соединения кристаллизуется путем добавления гексана или гептана. [0068] Способ по п.67, в котором добавление гексана или гептана приводит к кристаллизации циклического ангидрида тримерной бороновой кислоты формулы (XXIV) Полный текст патента Область техники Настоящее изобретение относится к синтезу эфиров бороновой кислоты и кислотных соединений. Более конкретно, изобретение относится к способам синтеза для крупномасштабного производства эфиров бороновой кислоты и кислотных соединений путем перегруппировки боронатных комплексов, промотируемой кислотой Льюиса. Бороновая кислота и ее сложные эфиры демонстрируют разнообразную биологическую активность, находящую фармацевтическое применение. В патенте US № 4499082 (1985) Shenvi и др. описано, что пептидные бороновые кислоты являются ингибиторами определенных протеолитических ферментов. В патентах US № № 5187157 (1993), 5242904 (1993) и 5250720 (1993) Kettner и Shenvi описан класс пептидных бороновых кислот, которые ингибируют трипсиноподобные протеазы. Патент US № 5169841 (1992) Kleeman и др. описывает N-терминально модифицированные пептидные бороновые кислоты, которые ингибируют действие ренина. В патенте US № 5106948 (1992) Kinder и др. раскрыто, что определенные трипептидные бороновые кислотные соединения ингибируют рост раковых клеток. Сравнительно недавно было продемонстрировано, что бороновая кислота и ее эфирные соединения являются весьма перспективными ингибиторами протеазомы - мультикаталитической протеазы, ответственной за большинство внутриклеточного круговорота протеинов. В работе Ciechanover (Cell, 79: 13-21 (1994)) описано, что протеазома представляет собой протеолитический компонент убиквитин-протеазомного маршрута, в котором протеины являются целью для разложения путем конъюгации с множеством молекул убиквитина. Кроме того, Ciechanover показал, что убиквитин-протеазомный маршрут играет существенную роль в разнообразных важных физиологических процессах. В патентах US № № 5780454 (1998), 6066730 (2000), 6083903 (2000), 6297217 (2001), 6548668 и 6617317 (2003) Adams и др., которые приведены в настоящем изобретении в качестве ссылок, описаны пептидные эфиры бороновой кислоты и кислотных соединений, используемых в качестве ингибиторов протеазомы. Кроме того, в этих ссылках описано применение эфиров бороновой кислоты и кислотных соединений для уменьшения интенсивности деградации мышечного протеина, уменьшения активности NF- κВ в клетке, уменьшения интенсивности деградации протеина р53 в клетке, для подавления деградации циклина в клетке, для подавления роста раковых клеток, для подавления воспроизведения антигена в клетке, для ингибирования клеточной адгезии, зависящей от NF- κВ, и для ингибирования репликации ВИЧ. В работе Drugs of the Future 27: 1079-1092 (2002) Albanell и Adams описано, что один такой ингибитор протеазомы на основе пептидной бороновой кислоты, бортезомиб (N-2-пиразинкарбонил-L-фенилаланин-L-лейцинбороновая кислота), проявляет значительную противоопухолевую активность на ксенотрансплантатных моделях опухоли человека и подвергается клинической оценке. New Engl. J. Med, 348:2609 (2003) Richardson и др., сообщает результаты исследования второй фазы бортезомиба, продемонстрировав его эффективность при лечении рецидивной и резистентной множественной миеломы. Исследования ингибиторов протеазы на основе бороновой кислоты были значительно усовершенствованы за счет развития химического способа получения функционализированных соединений бороновой кислоты, особенно альфа-галоид- и альфа-аминобороновых кислот. В работе J. Am. Chem. Soc, 102:7590 (1980) Matteson и Majumdar описан способ получения альфа-хлорэфиров бороновой кислоты путем гомологизации эфиров бороновой кислоты, а в Am. Chem. Soc, 102:7591 (1980) Matteson и Ray, J. указано, что хиральный контроль реакции гомологизации может быть достигнут за счет использования пинандиоловых эфиров бороновой кислоты. Кроме того, известно получение альфа-аминобороновой кислоты и сложноэфирных соединений из соответствующих альфа-хлорэфиров бороновой кислоты (см. J. Am. Chem. Soc, 103:5241 (1981) Matteson и др.; патент US № 4537773 (1985) Shenvi). В патенте US № 4525309 (1985) Matteson и Sadhu предложен усовершенствованный способ гомологизации эфиров бороновой кислоты путем перегруппировки промежуточного боронатного комплекса в присутствии катализатора - кислоты Льюиса. Известно, что кислота Льюиса промотирует процесс перегруппировки и сводит к минимуму эпимеризацию при альфа-атоме углерода. Однако для получения оптимальных результатов требуется строгое исключение воды и тщательный контроль стехиометрии кислоты Льюиса. Эти требования приводят к тому, что эффективное осуществление этой реакции в производственном масштабе становится затруднительным, что ограничивает возможность получения фармацевтически важных эфиров бороновой кислоты и кислотных соединений, таких как бортезомиб. Таким образом, в этой области существует потребность в усовершенствованных способах крупномасштабного производства эфиров бороновой кислоты и кислотных соединений. Настоящее изобретение предлагает усовершенствованные способы синтеза для крупномасштабного производства эфиров бороновой кислоты и кислотных соединений. Эти способы обеспечивают повышенный выход и степень чистоты, повышенную производительность и упрощение процесса по сравнению с известными из уровня техники способами. Описанные в настоящем изобретении способы особенно применимы для серийного производства в большом масштабе (нескольких килограмм), который ограничивается только размером доступных производственных мощностей. Способы, согласно изобретению, являются особенно эффективными для синтеза хиральных эфиров бороновой кислоты и кислотных соединений, в том числе альфа-аминоэфиров бороновой кислоты и кислотных соединений. Независимо от масштаба производства получаются целевые продукты, имеющие очень высокую химическую и стереохимическую чистоту. Ссылки на патентную и научную литературу, сделанные в данном изобретении, определяют объем знаний, который доступен для специалистов в этой области. Если не оговорено другое, все технические и научные термины, использованные в описании, имеют те же самые значения, которые общеизвестны обычному специалисту в области, к которой относится настоящее изобретение. Хотя при практическом осуществлении или тестировании настоящего изобретения могут быть использованы методы и материалы, аналогичные или эквивалентные описанным в настоящем изобретении, здесь описаны предпочтительные методы и материалы. Таким образом, выданные патенты, заявки и ссылки, которые процитированы в этом описании, приведены в данном изобретении специально индивидуально или совместно и предназначены для их использования в качестве ссылок. В случае противоречивости терминов могут быть использованы определения настоящего изобретения. Кроме того, материалы, методы и примеры являются только иллюстративными и не предназначены для ограничения изобретения. Используемый здесь термин "около" означает приблизительно, в диапазоне, ориентировочно или вблизи. Когда термин "около" используется в сочетании с числовым значением, это значение видоизменяется за счет расширения диапазона выше и ниже приведенного числового значения. Обычно термин "около" используется в описании, чтобы видоизменить числовое значение выше и ниже приведенной величины приблизительно на 10%. Используемый здесь термин "включает в себя" используется для обозначения "включает, но не ограничивается этим". Используемый здесь термин "алифатический" означает углеводород C 1 -С 12 с неразветвленной или разветвленной цепью, или циклический углеводород, который является полностью насыщенным или который имеет одно или несколько ненасыщенных звеньев, но который не является ароматическим. Например, подходящие алифатические группы включают замещенные или незамещенные, линейные, разветвленные или циклические алкильные, алкенильные, алкинильные группы и их сочетания, такие как (циклоалкил)алкил, (циклоалкенил)алкил или (циклоалкил)алкенил. В различных вариантах воплощения алифатическая группа имеет 1-12, 1-8, 1-6 или 1-4 атомов углерода. Термины "алкил", "алкенил" и "алкинил", используемые отдельно или как часть более крупной функциональной группы, относятся к алифатической группе с неразветвленной и разветвленной цепью, имеющей от 1 до 12 атомов углерода, которые необязательно замещены одним, двумя или тремя заместителями. В рамках настоящего изобретения термин "алкил" будет использоваться, когда атом углерода, присоединяющий алифатическую группу к остальной части молекулы, является насыщенным атомом углерода. Однако алкильная группа может включать ненасыщенность при других атомах углерода. Таким образом, алкильные группы включают, без ограничения, метил, этил, пропил, аллил, пропаргил, бутил, пентил и гексил. В рамках настоящего изобретения термин "алкенил" будет использоваться, когда атом углерода, присоединяющий алифатическую группу к остальной части молекулы, образует часть двойной углерод-углеродной связи. Алкенильные группы включают, без ограничения, винил, 1-пропенил, 1-бутенил, 1-пентенил и 1-гексенил. В рамках настоящего изобретения термин "алкинил" будет использоваться, когда атом углерода, присоединяющий алифатическую группу к остальной части молекулы, образует часть тройной углерод-углеродной связи. Алкинильные группы включают, без ограничения, этинил, 1-пропинил, 1-бутинил, 1-пентинил и 1-гексинил. Термины "циклоалкил", "карбоцикл", "карбоциклический радикал", "карбоцикло" или "карбоциклический", используемые отдельно или как часть более крупной функциональной группы, означают насыщенную или частично ненасыщенную циклическую алифатическую кольцевую систему, имеющую от 3 до приблизительно 14 членов, где алифатическая кольцевая система необязательно является замещенной. Циклоалкильные группы включают, без ограничения, циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогептил, циклогептенил, циклооктил, циклооктенил и циклооктадиенил. В некоторых вариантах циклоалкил имеет 3-6 атомов углерода. Термины "циклоалкил", "карбоцикл", "карбоциклический радикал", "карбоцикло" или "карбоциклический" также включают алифатические кольца, которые конденсированы с одним или несколькими ароматическими или неароматическими кольцами, такими как декагидронафтил или тетрагидронафтил, где радикал или место присоединения находятся в алифатическом кольце. Термины "галоидалкил", "галоидалкенил" и "галоидалкокси" относятся к алкильной, алкенильной или алкоксигруппе, которые, в зависимости от обстоятельств, могут быть замещены одним или несколькими атомами галогена. Используемый здесь термин "галоген" или "галоид" означает F, С, Br или I. Если не оговорено другое, термины "алкил", "алкенил" и "алкокси" включают галоидалкил, галоидалкенил и галоидалкоксигруппы, которые, в частности, включают группы с 1-5 атомами фтора. Термины "арил" и "ар-", используемые отдельно или как часть более крупной функциональной группы, например "аралкил", "аралкокси" или "арилоксиалкил", относятся к ароматическим фрагментам С 6-14 , содержащим от одного до трех ароматических колец, которые необязательно замещены. Предпочтительной арильной группой является арильная группа С 6 -С 10 . Арильные группы включают, без ограничения, фенил, нафтил и антраценил. Используемый здесь термин "арил" также включает группы, в которых ароматическое кольцо конденсировано с одним или несколькими неароматическими кольцами, такими как инданил, фенантридинил или тетрагидронафтил, где радикал или место присоединения находятся в ароматическом кольце. Термин "арил" может использоваться попеременно с термином "арильное кольцо". "Аралкильная" или "арилалкильная" группы включают в себя арильную группу, которая ковалентно связана с алкильной группой, каждая из которых независимо является необязательно замещенной. Предпочтительно аралкильная группа представляет собой С 6-10 -арил(С 1-6 )алкил, включая, без ограничения, бензил, фенетил и нафтилметил. Термины "гетероарил" и "гетероар-", используемые отдельно или как часть более крупной функциональной группы, например гетероаралкил или "гетероаралкокси", относятся к группам, имеющим от 5 до 14 атомов в кольце. Предпочтительно 5, 6, 9 или 10 атомов в кольце, имеющем 6, 10 или 14 π-электронов, обобщенных в составе цикла; и имеющем, кроме атомов углерода, от одного до четырех гетероатомов, которые выбирают из группы, состоящей из атомов N, О и S. Гетероарильные группы включают, без ограничения, тиенил, фуранил, пирролил, имидазолил, пиразолил, триазолил, тетразолил, оксазолил, изоксазолил, оксадиазолил, тиазолил, изотиазолил, тиадиазолил, пиридил, пиридазинил, пиримидинил, пиразинил, индолил, изоиндолил, бензотиенил, бензофуранил, дибензофуранил, индазолил, бензимидазолил, бензотиазолил, пуринил, хинолил, изохинолил, циннолинил, фталазинил, хиноксалинил, нафтиридинил, птеридинил, карбазолил, акридинил и феназинил. Используемые здесь термины "гетероарил" и "гетероар-" также включают группы, в которых гетероароматическое кольцо конденсировано с одним или несколькими неароматическими кольцами, в которых радикал или место его присоединения находится в гетероароматическом кольце. Не ограничивающие примеры включают тетрагидрохинолинил, тетрагидроизохинолинил и пиридо[3,4-d]пиримидинил. Термин "гетероарил" может быть использован попеременно с термином "гетероарильное кольцо" или с термином "гетероароматический", причем любые из этих терминов включают кольца, которые необязательно замещены. Термин "гетероаралкил" относится к алкильной группе, замещенной гетероарилом, в котором алкильные и гетероарильные части независимо являются необязательно замещенными. Используемые здесь термины "гетероцикл", "гетероциклический" или "гетероциклический радикал" относятся к стабильной моноциклической 5-7-членной или 7-10-членной бициклической гетероциклической группе, которая является или насыщенной, или частично ненасыщенной и имеет, кроме атомов углерода, один или несколько, предпочтительно от одного до четырех, гетероатомов, которые выбирают из группы, состоящей из N, О и S; причем гетероатомы азота и серы необязательно являются окисленными, и атомы азота необязательно являются кватернизованными. Гетероциклическое кольцо может быть присоединено к собственной висячей группе при любом гетероатоме или атоме углерода, что приводит к стабильной структуре, и любой атом в кольце может быть необязательно замещенным. Примеры таких насыщенных или частично ненасыщенных гетероциклических радикалов включают, без ограничения, тетрагидрофуранил, тетрагидротиенил, пирролидинил, пирролидонил, пиперидинил, пирролинил, тетрагидрохинолинил, тетрагидроизохинолинил, декагидрохинолинил, оксазолидинил, пиперазинил, диоксанил, диоксоланил, диазепинил, оксазепинил, тиазепинил и морфолинил. Используемые здесь термины "гетероцикл", "гетероциклический" и "гетероциклический радикал", кроме того, включают группы, в которых неароматическое кольцо, содержащее гетероатом, является конденсированным с одним или несколькими ароматическими или неароматическими кольцами, такими как индолинил, хроманил, фенантридинил или тетрагидрохинолинил, где радикал или место его присоединения находится в неароматическом кольце, содержащем гетероатом. Термин "гетероциклический алкил" относится к алкильной группе, замещенной гетероциклическим радикалом, в котором алкильная и гетероциклическая части независимо являются необязательно замещенными. Используемый здесь термин "частично ненасыщенный" относится к фрагменту кольца, который включает по меньшей мере одну двойную или тройную связь между атомами кольца. Термин "частично ненасыщенный" обобщенно предназначается для колец, имеющих одно или несколько мест ненасыщенности, но не предполагается, что термин включает арильные или гетероарильные функциональные группы, которые определены в описании. Используемый здесь термин "замещенный" означает, что один или несколько атомов водорода обозначенной функциональной группы замещены, при условии, что это замещение приводит к стабильному или химически возможному соединению. Стабильным или химически возможным соединением является одно из соединений, химическая структура которого существенно не изменяется при выдерживании при температуре 40 °С или ниже, в отсутствие влаги или в других химически реакционноспособных условиях, по меньшей мере, в течение недели, или соединение, которое достаточно долго сохраняет свою целостность, чтобы его можно было использовать для способов синтеза этого изобретения. Используемое здесь выражение "один или несколько заместителей" относится к ряду заместителей, число которых равно от одного до максимально возможного числа заместителей, в расчете на число имеющихся мест для связывания, при условии, что выполняются указанные выше условия стабильности и химической выполнимости. Арильная (в том числе арильный фрагмент в аралкильной, аралкокси, арилоксиалкильной и тому подобным группам) или гетероарильная (в том числе гетероарильный фрагмент в гетероаралкильной и гетероарилалкокси и тому подобным) группа может содержать один или несколько заместителей. Примеры подходящих заместителей при ненасыщенном атоме углерода арильной или гетероарильной группы включают: галоид, -NO 2 , -CN, -R*, -OR*, -SR o , -N(R + ) 2 , -NR + C(O)R*, -NR + C(O)N(R + ) 2 , -NR + CO 2 R o , -O-CO 2 R*, -O-C(O)R*, -CO 2 R*, -C(O)R*, -C(O)N(R + ) 2 , -OC(O)N(R + ) 2 , -S(O) 2 R o , -SO 2 N(R + ) 2 , -S(O)R ° и -NR + SO 2 N(R + ) 2 . Каждый радикал R + независимо выбирают из группы, состоящей из R*, -C(O)R*, -CO 2 R* и -SO 2 R*, или два радикала R + при одном и том же атоме азота, взятые вместе с атомом азота, образуют 5-8-членное ароматическое или неароматическое кольцо, имеющее, кроме атома азота, 0-2 кольцевых гетероатома, которые выбирают из N, О и S. Каждый R* независимо представляет собой водород или необязательно замещенную алифатическую, арильную, гетероарильную или гетероциклическую группу. Каждый R o независимо является необязательно замещенной алифатической или арильной группой. Кроме того, алифатическая группа может быть замещена одним или несколькими заместителями. Примеры подходящих заместителей при насыщенном атоме углерода алифатической группы или неароматическом гетероциклическом кольце включают, без ограничения, перечисленные выше заместители при ненасыщенном атоме углерода арильной или гетероарильной группы. В настоящем изобретении обнаружено, что требования тщательной осушки оборудования, растворителей и реагентов, которые характерны для описанных ранее способов перегруппировки боронатных комплексов, промотируемой кислотой Льюиса, могут быть исключены за счет применения растворителя - простого эфира, который плохо смешивается с водой. Примечательно, что применение такого растворителя обеспечивает проведение процесса в масштабе нескольких килограмм без ухудшения показателей выхода или чистоты. Фактически, масштаб процесса ограничивается только размером имеющихся производственных мощностей. Следовательно, одним аспектом этого изобретения является крупномасштабный способ получения эфирных соединений бороновой кислоты формулы (I) в которой R 1 представляет собой необязательно замещенную алифатическую, ароматическую или гетероароматическую группу; R 2 означает водород, нуклеофугическую группу или необязательно замещенную алифатическую, ароматическую или гетероароматическую группу; R 3 является нуклеофугической группой или необязательно замещенной алифатической, ароматической или гетероароматической группой; и каждый R 4 и R 5 , независимо, представляет собой необязательно замещенную алифатическую, ароматическую или гетероароматическую группу, или R 4 и R 5 , взятые вместе с промежуточными атомами кислорода и бора, образуют необязательно замещенное 5-10-членное кольцо, имеющее 0-2 дополнительных кольцевых гетероатомов, выбранных из N, О или S. Способ включает следующие стадии: (а) приготовление боронатного комплекса формулы (II) где Y означает нуклеофугическую группу; М + представляет собой катион; и каждый радикал R 1 -R 5 является таким, как определено выше; и (b) контактирование боронатного комплекса формулы (II) с кислотой Льюиса в условиях, в которых образуется эфирное соединение бороновой кислоты формулы (I), причем указанную стадию контактирования осуществляют в реакционной смеси, содержащей: (i) координационный растворитель - простой эфир, который плохо смешивается с водой; или (ii) растворитель - простой эфир, который плохо смешивается с водой и координационным со-растворителем. В описанных ранее способах перегруппировки боронатных комплексов, промотируемой кислотой Льюиса, применяется тетрагидрофуран - растворитель типа простого эфира, который полностью смешивается с водой. Невыполнение условий строгой осушки оборудования, растворителей и реагентов в этих процессах приводит к поразительному уменьшению соотношения диастереоизомеров. В частности, кислоты Льюиса обычно должны быть осушены в пламени непосредственно до использования в этом процессе. Хотя специалистам в этой области техники известны методики проведения реакций, чувствительных к влаге, и они хорошо освоены при работе в лабораторном масштабе, такие процессы являются дорогостоящими, и их трудно осуществлять в укрупненном масштабе. Более того, попытки осуществления способов уровня техники в укрупненном масштабе часто приводят к дальнейшему уменьшению соотношения диастереоизомеров в ходе обработки и выделения продукта - эфирного соединения бороновой кислоты. Organometallics, 2, с.1083 (1983) Matteson и Erdiik указывают, что при воздействии на альфа-галоидэфирные продукты бороновой кислоты свободного галогенидного иона происходит эпимеризация при альфа-углеродном центре. Не желая быть связанными теорией, авторы настоящего изобретения полагают, что проблема эпимеризации возникает особенно в процессе обработки реакционной смеси и/или последующих стадий. Например, полагают, что эпимеризация происходит в ходе концентрирования реакционной смеси с целью удаления тетрагидрофуранового растворителя и замены его на растворитель, не смешивающийся с водой. Неспособность полностью удалить тетрагидрофуран также оказывает отрицательное воздействие на соотношение диастереоизомеров в ходе последующих стадий промывки водой. В ходе этих стадий трудно избежать воздействия галогенидного иона на продукт, особенно когда процесс осуществляется в укрупненном масштабе. Авторы настоящего изобретения обнаружили, что перегруппировку боронатных комплексов выгодно осуществлять в растворителе - простом эфире, который плохо смешивается с водой. При использовании таких растворителей отпадает необходимость замены растворителя до стадии промывки водой, и растворимый органический продукт эффективно защищен от воздействия иона галогенида в воде в ходе промывки, даже если процесс осуществляется в укрупненном масштабе. Предпочтительно растворимость воды в растворителе - простом эфире - составляет меньше чем приблизительно 5 мас.%, более предпочтительно меньше чем приблизительно 2 мас.%. В различных вариантах растворитель - простой эфир, который плохо смешивается с водой, составляет по меньшей мере около 70, по меньшей мере около 80, по меньшей мере около 85, по меньшей мере около 90 или по меньшей мере около 95% от объема реакционной смеси. Предпочтительно простым эфирным растворителем является растворитель, который подходит для стандартного применения в крупномасштабном производстве. Используемый здесь термин "крупномасштабный" относится к процессу, в котором используются по меньшей мере около 5 моль по меньшей мере одного исходного материала. Предпочтительно в крупномасштабном процессе используются по меньшей мере около 10, 20, 50 или 100 моль по меньшей мере одного исходного материала. В рамках этого изобретения термин "простой эфир" относится к любому классу химических соединений, который характеризуется наличием атома кислорода, связанного с двумя атомами углерода. "Растворитель - простой эфир" представляет собой простое эфирное соединение, которое находится в жидком состоянии при желаемой температуре реакции и способно растворять исходный материал (материалы) и/или продукт (продукты) реакции. Не ограничивающие примеры растворителей - простых эфиров, подходящих для применения в способе изобретения, включают трет-бутилметиловый эфир, трет-бутилэтиловый эфир, трет-амилметиловый эфир и изопропиловый эфир. В одном варианте изобретения реакционная смесь дополнительно содержит координационный сорастворитель. В другом варианте растворитель - простой эфир, который плохо смешивается с водой, обладает достаточной координационной способностью, так что отпадает необходимость в координационном сорастворителе. В рамках этого изобретения термины "координационный сорастворитель" и "координационный растворитель" относятся к растворителю, который обеспечивает координацию кислоты Льюиса и сольватацию ионных компонентов процесса. Пространственно затрудненные растворители - простые эфиры, такие как трет-бутилметиловый эфир, обладают плохой координационной способностью и предпочтительно используются с координационным сорастворителем. Не ограничивающие примеры координационных сорастворителей, подходящих для практического осуществления этого изобретения, включают тетрагидрофуран, диоксан, воду и их смеси. В некоторых вариантах изобретения реакционная смесь содержит по меньшей мере около 5 или по меньшей мере около 10 об.% координационного сорастворителя. Предпочтительно количество смешивающегося с водой координационного сорастворителя, находящегося в реакционной смеси, не настолько велико, чтобы мешать разделению фаз в ходе процесса или обработки реакционной смеси. В различных вариантах изобретения координационный сорастворитель составляет не больше чем приблизительно 20, приблизительно 15 или около 10% от объема реакционной смеси. Используемый здесь термин "нуклеофугический" относится к любой группе, которая может подвергаться нуклеофильному замещению в условиях процесса перегруппировки настоящего изобретения. Такие нуклеофугические группы известны из уровня техники. Предпочтительно нуклеофугической группой является галоид, более предпочтительно хлор или бром. В ходе процесса перегруппировки с превращением боронатного комплекса формулы (II) в эфирное соединение бороновой кислоты формулы (I) нуклеофугическая группа Y выделяется в виде Y - . Например, когда Y означает хлор, на стадии (b) выделяется хлоридный ион. Переменный катион М + означает любой катионный противоион для отрицательно заряженного четырехвалентного атома бора в боронатном комплексе формулы (II). В некоторых предпочтительных вариантах изобретения М + выбирают из группы, состоящей из Li + , Na + и K + . Специалист в этой области техники понимает, что на стадии (b) процесса перегруппировки в качестве побочного продукта образуется соль M + Y - . Переменный радикал R 1 предпочтительно означает группу с хорошей мигрирующей способностью. В некоторых вариантах изобретения R 1 представляет собой С 1-8 -алифатическую, С 6-10 -арильную или (С 6-10 -арил)(С 1-6 -алифатическую) группу, каждая из которых необязательно является замещенной. В определенных вариантах изобретения R 1 является С 1-4 -алифатической группой, особенно изобутилом. Переменный радикал R 2 предпочтительно означает водород, нуклеофугическую группу или необязательно замещенную С 1-8 -алифатическую, С 6-10 -арильную или (С 6-10 -арил)(С 1-6 -алифатическую) группу. Переменный радикал R 3 предпочтительно является нуклеофугической группой или необязательно замещенной С 1-8 -алифатической, С 6-10 -арильной, или (С 6-10 -арил)(С 1-6 -алифатической) группой. Специалист в этой области техники может признать, что в каждом из R 1 , R 2 или R 3 могут присутствовать функциональные заместители при условии, что функциональный заместитель не мешает образованию боронатного комплекса формулы (II). Один вариант изобретения относится к способу получения эфирного соединения бороновой кислоты формулы (I), в которой R 3 представляет собой нуклеофугическую группу. Такие соединения применяются в качестве промежуточных соединений при синтезе альфа-замещенных эфиров бороновой кислоты и кислотных соединений, в том числе альфа-аминоэфиров бороновой кислоты и кислотных соединений, которые описаны ниже. В определенных предпочтительных вариантах изобретения R 3 означает нуклеофугическую группу и R 2 является водородом. Переменные радикалы R 4 и R 5 могут быть одинаковыми или различными. В некоторых вариантах изобретения R 4 и R 5 связаны непосредственно, так что R 4 и R 5 , взятые вместе с промежуточными атомами кислорода и бора, образуют необязательно замещенное 5-10-членное кольцо, которое может содержать 0-2 дополнительных кольцевых гетероатома, выбранных из N, О или S. В некоторых вариантах изобретения это кольцо является 5- или 6-членным кольцом, предпочтительно пятичленным кольцом. Настоящее изобретение является особенно выгодным для промотируемой кислотами Льюиса перегруппировки боронатных комплексов формулы (II), в которой R 4 и R 5 связаны непосредственно и вместе представляют собой хиральную функциональную группу. Один вариант изобретения относится к перегруппировке таких хиральных боронатных комплексов с образованием эфирного соединения бороновой кислоты формулы (I), в которой атом углерода, связанный с радикалами R 1 , R 2 и R 3 , является хиральным центром. Предпочтительно процесс перегруппировки протекает с высокой степенью стереоспецифичности относительно хиральной функциональной группы R 4 -R 5 , чтобы получить эфирное соединение бороновой кислоты формулы (I), где атом углерода, связанный с радикалами R 1 , R 2 и R 3 , является хиральным центром, имеющим соотношение диастереоизомеров по меньшей мере около 96:4, относительно хирального центра в хиральной функциональной группе R 4 -R 5 . Предпочтительно соотношение диастереоизомеров составляет по меньшей мере около 97:3. Термины "стереоизомер", "энантиомер", "диастереомер", "эпимер" и "хиральный центр" используются здесь в соответствии с каждым значением, которое обычно придается при использовании обычным специалистом в этой области техники. Таким образом, стереоизомеры являются соединениями, которые имеют такую же связность атомов, но отличаются пространственным расположением атомов. Энантиомеры представляют собой стереоизомеры, которые находятся в соотношении зеркального отображения, то есть противоположную стереохимическую конфигурацию при всех соответствующих хиральных центрах. Диастереомеры представляют собой стереоизомеры, имеющие больше одного хирального центра, которые отличаются друг от друга тем, что стереохимическая конфигурация по меньшей мере одного, но не всех соответствующих хиральных центров, является противоположной. Эпимеры представляют собой диастереомеры, которые отличаются по стереохимической конфигурации только в одном хиральном центре. Используемый здесь термин "соотношение диастереоизомеров" относится к отношению между диастереомерами, которые отличаются по стереохимической конфигурации при одном хиральном центре, относительно второго хирального центра в той же самой молекуле. В качестве примера химическая структура с двумя хиральными центрами обеспечивает четыре возможных стереоизомера: R*R, R*S, S*R и S*S, в которых звездочкой отмечен соответствующий хиральный центр в каждом стереоизомере. Соотношение диастереоизомеров в такой смеси стереоизомеров означает отношение диастереомера и его энантиомера к другому диастереомеру и его энантиомеру, т.е. (R*R + S*S): (R*S + S*R). Обычный специалист в этой области техники может признать, что возможны дополнительные стереоизомеры, когда молекула имеет больше двух хиральных центров. В рамках настоящего изобретения термин "соотношение диастереоизомеров" имеет одинаковое значение при ссылке на соединения с множеством хиральных центров, а также при ссылке на соединения, имеющие два хиральных центра. Таким образом, термин "соотношение диастереоизомеров" относится к соотношению всех соединений, имеющих R*R или S*S конфигурацию, при указанных хиральных центрах ко всем соединениям, имеющим R*S или S*R конфигурации, при указанных хиральных центрах. Для удобства это соотношение называется в этом изобретении как отношение диастереоизомеров при атоме углерода, отмеченным звездочкой, относительно второго указанного хирального центра. Соотношение диастереоизомеров можно определить любым аналитическим методом, подходящим для различения между диастереоизомерными соединениями, имеющими различную относительную стереохимическую конфигурацию при указанных хиральных центрах. Такие методы включают, без ограничения, ядерный магнитный резонанс (ЯМР), газовую хроматографию (ГХ) и жидкостную хроматографию высокого разрешения (ЖХВР). Как уже обсуждалось ранее, один вариант изобретения посвящен способу получения эфирного соединения бороновой кислоты формулы (I), имеющего соотношение диастереоизомеров при атоме углерода, связанном с радикалами R 1 , R 2 и R 3 , по меньшей мере около 96:4 относительно хирального центра в хиральной функциональной группе R 4 -R 5 . Специалист в этой области техники может признать, что сама хиральная группа R 4 -R 5 может содержать больше одного хирального центра. Когда группа R 4 -R 5 действительно содержит больше одного хирального центра, предпочтительно она имеет высокую диастереоизомерную чистоту, и соотношение диастереоизомеров при атоме углерода, связанном с радикалами R 1 , R 2 и R 3 , можно измерить относительно любого одного хирального центра в группе R 4 -R 5 . В способах данного изобретения хиральная группа R 4 -R 5 предпочтительно имеет высокий уровень энантиомерной чистоты. В рамках изобретения используемый термин "энантиомерная чистота" означает "энантиомерный избыток", который представляет собой избыточное количество основного энантиомера относительно второстепенного энантиомера, выраженное в процентах от суммарного количества. Предпочтительно хиральная группа R 4 -R 5 имеет энантиомерную чистоту по меньшей мере около 98%, более предпочтительно по меньшей мере около 99%, еще более предпочтительно по меньшей мере около 99,5% и наиболее предпочтительно по меньшей мере около 99,9%. Когда хиральная группа R 4 -R 5 имеет весьма высокую энантиомерную чистоту, соотношение диастереоизомеров при атоме углерода, связанном с радикалами R 1 , R 2 , R 3 , приблизительно равняется эпимерному отношению при этом центре, т.е. соотношение диастереоизомеров ≅ (R*R):(S*R) или (R*S):(S*S) ≅ (R*):(S*). Используемый здесь термин "эпимерное отношение" относится к отношению количества продукта, имеющего одну абсолютную стереохимическую конфигурацию при данном хиральном центре, к количеству продукта, имеющего противоположную абсолютную стереохимическую конфигурацию при соответствующем хиральном центре. Предпочтительно эти продукты имеют одинаковую стереохимическую конфигурацию при всех других соответствующих хиральных центрах. Поэтому в одном варианте изобретение относится к перегруппировке хирального боронатного комплекса формулы (II), чтобы получить эфирное соединение бороновой кислоты формулы (I), в которой эпимерное отношение при атоме углерода, связанном с радикалами R 1 , R 2 и R 3 , составляет по меньшей мере около 96:4, более предпочтительно по меньшей мере около 97:3. Кислоты Льюиса, подходящие для практического осуществления изобретения, представляют собой те, которые способны образовать комплекс с нуклеофугической группой, что способствует замещению при миграции радикала R 1 . Предпочтительно кислота Льюиса способна к дополнительной координации с атомом кислорода, связанным с бором. Не ограничивающие примеры подходящих кислот Льюиса включают бромид цинка, хлорид цинка, бромид железа (III) и хлорид железа (III). В определенных предпочтительных вариантах изобретения кислота Льюиса представляет собой хлорид цинка. Предпочтительно стадию контактирования проводят при низкой температуре, однако взаимодействие может быть осуществлено при температуре окружающей среды или повышенной температуре. Выбор подходящей температуры процесса будет зависеть, главным образом, от применяемой кислоты Льюиса, а также от мигрирующей способности группы R 1 . Специалист в этой области техники будет в состоянии подобрать подходящую температуру с учетом используемых условий процесса. В некоторых вариантах изобретения стадию контактирования проводят при температуре по меньшей мере около -100, -78 или -60 °С. В некоторых вариантах изобретения стадию контактирования проводят при температуре, которая не превышает приблизительно 80, 40 или 30 °С. В объем изобретения включен любой диапазон, охватывающий эти высокие и низкие температуры. Предпочтительно стадию контактирования проводят при температуре в диапазоне приблизительно от -100 до 80 °С, приблизительно от -70 до 40 °С, приблизительно от -60 до 30 °С или приблизительно от -50 до 30 °С. В определенных предпочтительных вариантах изобретения стадию контактирования начинают при низкой температуре предпочтительно в диапазоне приблизительно от -70 до -30 °С и затем дают реакционной смеси нагреться предпочтительно до температуры окружающей среды. Неожиданно оказалось, что в способе настоящего изобретения не требуются специальные меры предосторожности, чтобы исключить наличие воды в ходе самой стадии перегруппировки. В некоторых вариантах изобретения применяется влажная кислота Льюиса с минимальным ухудшением в соотношении диастереоизомеров. Когда термин "влажная" используется в связи с кислотой Льюиса, это означает, что содержание воды в кислоте Льюиса больше чем приблизительно 100, 200, 500 или 1000 ч./млн. Примечательно, что кислоту Льюиса можно добавлять в реакционную смесь даже в виде водного раствора без вредного влияния на соотношение диастереоизомеров. Следовательно, в некоторых вариантах способ согласно изобретению включает следующие стадии: (a) приготовление раствора, содержащего боронатный комплекс формулы (II), и (i) координационный растворитель - простой эфир, который плохо смешивается с водой; или (ii) растворитель - простой эфир, который плохо смешивается с водой, и координационный сорастворитель и (b) добавление к раствору со стадии (а) раствора кислоты Льюиса, содержащего воду и кислоту Льюиса. В некоторых других вариантах изобретения раствор кислоты Льюиса содержит тетрагидрофуран и кислоту Льюиса. Таким образом, в отличие от способа, известного из уровня техники, способ, согласно изобретению, может быть легко приспособлен к крупномасштабному производству. В различных вариантах изобретения по меньшей мере около 5, 10, 20, 50, 100, 500 или 1000 моль боронатного комплекса формулы (II) контактируют с кислотой Льюиса в условиях, в которых образуется эфирное соединение бороновой кислоты формулы (I). Кроме того, изобретение предоставляет композицию, содержащую эфирное соединение бороновой кислоты формулы (I), которое описано здесь, и растворителя - простого эфира, который плохо смешивается с водой. Предпочтительно эта композиция содержит по меньшей мере около 5, 10, 20, 50, 100, 500 или 1000 моль эфирного соединения бороновой кислоты формулы (I). В определенных вариантах изобретения R 4 и R 5 вместе представляют собой хиральную группу, причем соединение формулы (I), присутствующее в композиции, имеет соотношение диастереоизомеров по меньшей мере около 96:4 при атоме углерода, связанном с радикалами R 1 , R 2 и R 3 , относительно хирального центра в хиральной группе R 4 -R 5 . Предпочтительно обработка реакционной смеси включает в себя промывку этой смеси водным раствором и концентрирование промытой реакционной смеси путем удаления растворителей, чтобы получить остаток, содержащий эфирное соединение бороновой кислоты формулы (I). Предпочтительно этот остаток содержит по меньшей мере около 5, 10, 20, 50, 100, 500 или 1000 моль эфирного соединения бороновой кислоты формулы (I). В тех вариантах изобретения, в которых R 4 -R 5 представляет собой хиральную группу, присутствующее в остатке эфирное соединение бороновой кислоты формулы (I) предпочтительно имеет соотношение диастереоизомеров по меньшей мере около 96:4 при атоме углерода, связанном с радикалами R 1 , R 2 и R 3 , относительно хирального центра в хиральной группе R 4 -R 5 . Более предпочтительно соотношение диастереоизомеров составляет по меньшей мере около 97:3. Боронатный комплекс формулы (II) может быть получен любым известным способом, но предпочтительно его получают путем взаимодействия эфира бороновой кислоты формулы (III) с реагентом формулы (IV) в которой каждый из М + , Y и R 1 -R 5 определен выше для боронатного комплекса формулы (II). В некоторых вариантах изобретения это взаимодействие проводят при реакционной температуре по меньшей мере около -100, -78 или -60 °С. В некоторых вариантах изобретения это взаимодействие проводят при температуре не выше чем приблизительно 0, -20 или -40 °С. В объем изобретения включен любой диапазон, охватывающий эти высокие и низкие температуры. Предпочтительно взаимодействие проводят при реакционной температуре в диапазоне приблизительно от -100 до 0 °С, приблизительно от -78 до -20 °С или приблизительно от -60 до -40 °С. В некоторых вариантах изобретения боронатный комплекс формулы (II) получают в растворе, содержащем растворитель - простой эфир, обладающий плохой смешиваемостью с водой, и реакционную смесь непосредственно обрабатывают кислотой Льюиса для того, чтобы осуществить перегруппировку в сложноэфирное соединение бора формулы (I). В некоторых вариантах изобретения реагент формулы (IV) образуется in situ. Такие варианты изобретения включают стадии: (i) предоставление раствора, содержащего эфир бороновой кислоты формулы (III), который определен выше, и соединение формулы (V) в которой радикалы R 2 и R 3 определены выше для реагента формулы (IV); и (ii) обработки раствора сильным, пространственно затрудненным основанием для того, чтобы образовался боронатный комплекс формулы (II). В некоторых вариантах изобретения пространственно затрудненное основание представляет собой диалкиламид щелочного металла формулы M 2 N(R # ) 2 , где М 2 означает Li, Na или K, и каждый R # независимо является разветвленной или цикло(С 3 -С 6 )алифатической группой. Образование реагента формулы (IV) in situ является особенно привлекательным в тех вариантах изобретения, в которых Y представляет собой нуклеофугическую группу в связи с неустойчивостью реагента формулы (IV). Эфир бороновой кислоты формулы (III) может быть получен любым известным способом, но обычно этот эфир получают путем этерификации соответствующего соединения бороновой кислоты, например, по методу, описанному в статье Brown и др., Organometallics, 2, с.1311-1316 (1983). Предпочтительно циклические эфиры бороновой кислоты формулы (III) получают следующим образом: (a) приготавливают раствор, содержащий: (i) соединение бороновой кислоты формулы R 1 -B(OH) 2 ; (ii) соединение формулы HO-R 4 -R 5 -OH, в которой радикалы R 4 и R 5 , взятые вместе, представляют собой необязательно замещенную связывающую цепочку, содержащую 2-5 атомов углерода и 0-2 гетероатома, которые выбирают из группы, состоящей из О, N и S; и (iii) органический растворитель, который образует азеотроп с водой; и (b) нагревают раствор при температуре кипения с азеотропным удалением воды. Термин "связывающая цепочка", используемый в связи с радикалами R 4 и R 5 , относится к самой короткой линейной цепи атомов, соединяющей атомы кислорода, с которыми связаны радикалы R 4 и R 5 . Связывающая цепочка необязательно является замещенной при любом атоме в цепи, причем один или несколько атомов в цепи также могут образовать часть кольцевой системы, которая является спирогруппой, конденсированной или мостиковой группой для линейной связывающей цепочки. С целью иллюстрации, но не ограничения, в некоторых вариантах изобретения соединением формулы HO-R 4 -R 5 -OH является пинандиол, имеющий структуру В таких вариантах изобретения связывающая цепочка R 4 -R 5 содержит два атома углерода, которые вместе образуют одну сторону кольцевой системы бицикло[3.1.1]гептана, причем один из них дополнительно замещен метильной группой. В некоторых вариантах изобретения соединение формулы HO-R 4 -R 5 -OH является хиральным диолом, предпочтительно диолом, имеющим высокую диастереоизомерную и энантиомерную чистоту. Специалист в этой области поймет, что в таких вариантах изобретения соединение формулы HO-R 4 -R 5 -OH используется в качестве хирального вспомогательного агента, который ориентирует стереохимическую конфигурацию при атоме углерода, связанном с радикалами R 1 , R 2 и R 3 . Хиральные диолы, используемые в качестве хиральных вспомогательных агентов в органическом синтезе, хорошо известны из уровня техники. Не ограничивающие примеры включают 2,3-бутандиол, предпочтительно (2R,3R)-(-)-2,3-бутандиол или (2S,3S)-(+)-2,3-бутандиол; пинандиол, предпочтительно (1R,2R,3R,5S)-(-)-пинандиол или (1S,2S,3S,5R)-(+)-пинандиол; 1,2-циклопентандиол, предпочтительно (1S,2S)-(+)-транс-1,2-циклопентандиол или (1R,2R)-(-)-транс-1,2-циклопентандиол; 2,5-гександиол, предпочтительно (2S,5S)-2,5-гександиол или (2R,5R)-2,5-гександиол; 1,2-дициклогексил-1,2-этандиол, предпочтительно (1R,2R)-1,2-дициклогексил-1,2-этандиол или (1S,2S)-1,2-дициклогексил-1,2-этандиол; гидробензоин, предпочтительно (S,S)-(-)-гидробензоин или (R,R)-(+)-гидробензоин; 2,4-пентандиол, предпочтительно (R,R)-(-)-2,4-пентандиол или (S,S,)-(+)-2,4-пентандиол; эритронный γ-лактон, предпочтительно D-эритронный γ-лактон. Кроме того, в качестве хиральных диолов могут быть использованы углеводы, например 1,2,5,6-симметрично защищенный маннит. Не ограничивающие примеры органических растворителей, подходящие для использования в процессе этерификации, включают ацетонитрил, толуол, гексан, гептан и их смеси. В некоторых вариантах изобретения органический растворитель является простым эфирным растворителем, предпочтительно простым эфиром, который плохо смешивается с водой. В определенных предпочтительных вариантах изобретения процесс этерификации проводят в растворителе - простом эфире, который плохо смешивается с водой, и полученный раствор, который содержит эфир бороновой кислоты формулы (III), используется непосредственно на следующей стадии без выделения эфира бороновой кислоты. Как указано выше, способ согласно данному изобретению впервые обеспечивает крупномасштабную обработку реакционных смесей без существенного ухудшения соотношения диастереоизомеров. Следовательно, другим аспектом этого изобретения является композиция, содержащая по меньшей мере около 5, 10, 20, 50, 100, 500 или 1000 моль эфирного соединения бороновой кислоты формулы (7) в которой R 1 представляет собой необязательно замещенную алифатическую, ароматическую или гетероароматическую группу; R 2 означает водород, нуклеофугическую группу или необязательно замещенную алифатическую, ароматическую или гетероароматическую группу; R 3 означает нуклеофугическую группу или необязательно замещенную алифатическую, ароматическую или гетероароматическую группу и R 4 и R 5 , взятые вместе с промежуточными атомами кислорода и бора, образуют необязательно замещенное 5-10-членное хиральное кольцо, имеющее 0-2 дополнительных кольцевых гетероатомов, которые выбирают из N, О или S, где атом углерода, с которым связаны радикалы R 1 , R 2 и R 3 , представляет собой хиральный центр, имеющий соотношение диастереоизомеров по меньшей мере около 96:4, предпочтительно по меньшей мере около 97:3, относительно хирального центра в хиральной группе R 4 -R 5 . Предпочтительные значения для радикалов R 1 -R 3 указаны выше. Предпочтительно растворители составляют меньше чем приблизительно 30, 20, 10 или 5 мас.% композиции в соответствии с этим замыслом изобретения. В некоторых вариантах изобретения эфирное соединение бороновой кислоты формулы (I) составляет по меньшей мере около 70, 80, 90 или 95 мас.% композиции. Один вариант изобретения относится к описанной выше композиции, в которой имеется по меньшей мере один из следующих признаков: (a) R 3 означает хлор; (b) эфирное соединение бороновой кислоты (I) представляет собой (c) R 2 означает водород и (d) R 1 является С 1-4 -алифатическим радикалом. Все эфирные соединения бороновой кислоты формулы (I), присутствующие в композиции, могут быть получены в одном периодическом процессе. В рамках этого изобретения термин " периодический процесс" относится к осуществлению процесса синтеза, в котором каждую стадию процесса проводят только один раз. Предпочтительно эфирное соединение бороновой кислоты формулы (I), присутствующее в композиции, получают в одном периодическом процессе в соответствии с первым замыслом изобретения. Обычный специалист в этой области техники может признать, что получение заданного количества продукта в одном периодическом цикле крупномасштабного процесса является более эффективным и обеспечивает более однородный продукт, чем получение такого же количества продукта за счет повторного осуществления процесса в малом масштабе. Эфирные соединения бороновой кислоты формулы (I), в которой R 3 означает нуклеофугическую группу, являются полезными промежуточными соединениями для синтеза альфа-аминоэфирных соединений бороновой кислоты. Поэтому в других аспектах настоящего изобретения обеспечивают крупномасштабный способ получения альфа-аминоэфира бороновой кислоты, причем предпочтительно этот способ включает следующие стадии: (а) приготовление боронатного комплекса формулы (II) где Y означает нуклеофугическую группу; М + представляет собой катион; R 1 является необязательно замещенной алифатической, ароматической или гетероароматической группой; R 2 - это водород; R 3 означает нуклеофугическую группу и каждый радикал R 4 и R 5 , независимо, является необязательно замещенной алифатической, ароматической или гетероароматической группой или R 4 и R 5 , взятые вместе с промежуточными атомами кислорода и бора, образуют необязательно замещенное 5-10-членное кольцо, имеющее 0-2 дополнительных кольцевых гетероатома, выбранных из N, О или S; (b) контактирование боронатного комплекса формулы (II) с кислотой Льюиса в условиях, в которых образуется эфирное соединение бороновой кислоты формулы (I) где каждый из радикалов R 1 -R 5 определен выше, причем указанную стадию контактирования проводят в реакционной смеси, содержащей: (i) координационный растворитель - простой эфир, который плохо смешивается с водой; или (ii) растворитель - простой эфир, который плохо смешивается с водой, и координационный сорастворитель; и (с) обработка эфирного соединения бороновой кислоты формулы (I) реагентом формулы M 1 -N(G) 2 , в которой М 1 означает щелочной металл и каждая группа G индивидуально, или обе вместе, представляет собой защитную группу для аминогруппы, чтобы образовался побочный продукт формулы М 1 -R 3 и соединение формулы (VIII) в которой каждая группа G и от R 1 до R 5 определены выше; и (d) удаление групп G с образованием соединения формулы (VII) или его аддитивной соли с кислотой. В некоторых вариантах изобретения на стадии (с) эфирное соединение бороновой кислоты формулы (I) обрабатывают реагентом формулы M 1 -N(Si(R 6 ) 3 ) 2 , где М 1 означает щелочной металл и каждый R 6 независимо выбирают из группы, состоящей из алкила, аралкила и арила, где арил или арильный фрагмент аралкила необязательно являются замещенными. Предпочтительно взаимодействие эфирного соединения бороновой кислоты формулы (I) с реагентом формулы M 1 -N(G) 2 проводят при реакционной температуре в диапазоне приблизительно от -100 до 50 °С, предпочтительно приблизительно от -50 до 25 °С и более предпочтительно приблизительно от -30 до 0 °С. В некоторых вариантах изобретения R 3 является галоидом, предпочтительно хлором, и М 1 означает Li. Для облегчения выделения продукта формулы (VIII) реакционная смесь предпочтительно содержит органический растворитель, в котором побочный продукт M 1 -R 3 плохо растворяется. Не ограничивающие примеры подходящих органических растворителей включают метилциклогексан, циклогексан, гептан, гексан и толуол. В некоторых вариантах изобретения стадия (с) дополнительно включает в себя фильтрацию реакционной смеси для того, чтобы удалить M 1 -R 3 и обеспечить получение фильтрата, содержащего соединение формулы (VIII). Предпочтительно фильтрат используют непосредственно на стадии (d). В тех вариантах изобретения, когда реакционная смесь содержит органический растворитель, в котором побочный продукт M 1 -R 3 плохо растворяется, реакционная смесь может дополнительно содержать растворитель, в котором побочный продукт М 1 -R 3 имеет высокую растворимость. В таких случаях растворитель, в котором побочный продукт M 1 -R 3 имеет высокую растворимость, предпочтительно удаляется до фильтрации реакционной смеси. С целью иллюстрации в некоторых вариантах изобретения в реакционную смесь добавляют реагент формулы М 1 -N(Si(R 6 ) 3 ) 2 в виде раствора, содержащего тетрагидрофуран. В таких вариантах изобретения предпочтительно стадия (с) дополнительно включает в себя удаление тетрагидрофурана до фильтрации реакционной смеси. Специалисты в этой области техники могут признать, что могут быть использованы различные методы для того, чтобы удалить защитные группы G в соединении формулы (VIII), которые включают, например, водный гидролиз или обработку кислотой. Образующийся альфа-аминоэфир бороновой кислоты формулы (VII) обладает низкой стабильностью и предпочтительно подвергается незамедлительной дериватизации (см. Matteson и др., J. Am. Chem. Soc, 103:5241 (1981)) или его выделяют в виде аддитивной соли с кислотой. В некоторых вариантах изобретения стадия (d) включает в себя обработку соединения формулы (VIII) кислотой и выделение соединения формулы (VII) в виде аддитивной соли с кислотой. В определенных предпочтительных вариантах изобретения эта кислота представляет собой трифторуксусную кислоту, и соединение формулы (VII) выделяют в виде аддитивной соли с трифторуксусной кислотой. Как отмечено выше, способы изобретения особенно хорошо подходят для получения альфа-аминоэфирных соединений бороновой кислоты формулы (VII), в которой атом альфа-углерода представляет собой хиральный центр. Таким образом, один вариант изобретения относится к крупномасштабному способу получения альфа-аминоэфирного соединения бороновой кислоты формулы (VIIa) или (VIIb) или его аддитивной соли с кислотой, где R 1 является необязательно замещенной алифатической, ароматической или гетероароматической группой и R 4 и R 5 , взятые вместе с промежуточными атомами кислорода и бора, образуют необязательно замещенный хиральный циклический эфир бороновой кислоты и указанный способ включает следующие стадии: (а) предоставление боронатного комплекса формулы (IIa) или (IIb) где Y представляет собой нуклеофугическую группу; М + - это катион; R 2 - водород; R 3 является нуклеофугической группой и R 4 и R 5 имеют указанные выше значения; (b) контактирование боронатного комплекса формулы (IIa) или (IIb) с кислотой Льюиса в условиях, в которых образуется эфирное соединение бороновой кислоты формулы (Ia) или (Ib) где каждый из радикалов R 1 -R 5 имеет указанные выше значения, причем указанную стадию контактирования проводят в реакционной смеси, содержащей (i) координационный растворитель - простой эфир, который плохо смешивается с водой; или (ii) растворитель - простой эфир, который плохо смешивается с водой, и координационный сорастворитель и (с) обработка эфирного соединения бороновой кислоты формулы (Ia) или (Ib) реагентом формулы M 1 -N(G) 2 , где М 1 представляет собой щелочной металл и G является защитной группой для аминогруппы, с образованием соединения формулы (VIIIa) или (VIIIb) где каждый из G и R 1 -R 5 имеют указанные выше значения; и (d) удаление групп G с образованием соединения формулы (VIIa) или (VIIb) или его аддитивной соли с кислотой. Предпочтительные значения для групп Y, М + , R 1 -R 5 и G описаны выше. Предпочтительно соединение формулы (VIIa) или (VIIb) имеет соотношение диастереоизомеров при альфа-углеродном атоме по меньшей мере около 96:4, более предпочтительно по меньшей мере около 97:3, относительно хирального центра в R 4 -R 5 хиральной группе. Альфа-аминоэфирные соединения бороновой кислоты формулы (VII) являются полезными синтетическими промежуточными соединениями для получения пептидиловых эфирных соединений бороновой кислоты. Поэтому в некоторых вариантах изобретения способ согласно этому замыслу изобретения дополнительно включает в себя сочетание соединения формулы (VII) с соединением формулы (IX) в которой Р 1 означает группу, блокирующую аминогруппу; R 7 выбирают из группы, состоящей из водорода, С 1-10 -алифатической, необязательно замещенной С 6-10 -арильной или С 1-6 -алифатической-R 8 групп; и R 8 выбирают из группы, состоящей из алкокси, алкилтио, необязательно замещенного арила, гетероарила и гетероциклической группы и необязательно защищенных амино, гидрокси и гуанидиновых групп; и X означает ОН или отщепляемую группу; с образованием соединения формулы (X) в которой каждый из заместителей Р 1 , R 1 , R 4 , R 5 и R 7 имеет указанные выше значения. Отщепляемая группа X представляет собой любую группу, способную к нуклеофильному замещению альфа-аминогруппой соединения формулы (VII). В некоторых вариантах изобретения функциональная группа -С(О)-Х означает активированный сложный эфир, такой как сложный эфир O-(N-гидроксисукцинимида). В некоторых вариантах изобретения активированный сложный эфир образуется in situ путем контактирования соединения формулы (IX), в которой X представляет собой ОН, с пептидным сочетающим реагентом. Примеры подходящих пептидных сочетающих реагентов включают, без ограничения, карбодиимидные реагенты, например дициклогексилкарбодиимид (DCC) или 1-(3-диметиламинопропил)-3-этилкарбодиимид (EDC); фосфониевые реагенты, например гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония (ВОР реагент); и урониевые реагенты, например тетрафторборат О-(1Н-бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (TBTU). Кроме того, специалистам в этой области известны методики, которые обеспечивают непосредственное сочетание силилзащищенных аминов, без предварительной стадии удаления защиты. В таких методиках силильные группы удаляются in situ в условиях реакции сочетания. Следовательно, в некоторых вариантах настоящего изобретения соединение формулы (VIII) контактирует с соединением формулы (IX) в условиях, в которых группы (R 6 ) 3 Si удаляются in situ, и образуется соединение формулы (X). В рамках этого изобретения термин "группа, блокирующая аминогруппу" относится к любой группе, используемой для дериватизации аминогруппы, особенно N-терминальной аминогруппы пептида или аминокислоты. Термин "группа, блокирующая аминогруппу" включает (но не ограничивается) защитные группы, которые обычно применяются в органическом синтезе, особенно в пептидном синтезе (см., например, книги The Peptides Vol. 3, Academic Press, New York, 1981, с.3-88 Gross и Mienhoffer, ред.; Protective Groups in Organic Synthesis, 3 е издание, John Wiley & Sons, Inc., New York, 1999 Green и Wuts). Однако если не указано другое, необязательно, чтобы группу, блокирующую аминогруппу, можно было легко отщепить. Группы, блокирующие аминогруппу, включают, например, алкильную, ацильную, алкоксикарбонильную, аминокарбонильную и сульфонильную группы. В некоторых вариантах изобретения группа, блокирующая аминогруппу, является ацильной группой, произведенной из аминокислоты, или пептида, или их производных, или их аналогов. Используемый здесь термин "аминокислота" включает аминокислоты природного, а также искусственного происхождения. В рамках этого изобретения термин "производная" аминокислоты или пептида означает соединение, в котором функциональная группа, например гидрокси, амино, карбокси или гуанидиновая группы на N-конце или в боковой цепи, модифицирована блокирующей группой. Используемый здесь термин "аналог" аминокислоты или пептида означает соединение, которое включает модифицированную основную или боковую цепь молекулы. Предполагается, что термин "пептидный аналог" включает пептиды, в которых инвертированы один или несколько стереоцентров, причем одна или несколько пептидных связей замещены пептидной изостерой. В некоторых вариантах изобретения Р 1 представляет собой отщепляемую защитную группу. Примеры отщепляемых защитных групп включают, без ограничения, ацильные защитные группы, например формил, ацетил (Ас), сукцинил (Suc) или метоксисукцинил (MeOSuc), и уретановые защитные группы, например трет-бутоксикарбонил (BOK), бензилоксикарбонил (Cbz) или флуоренилметоксикарбонил (Fmoc). В некоторых таких вариантах изобретения способ в соответствии с этим аспектом изобретения дополнительно включает следующие стадии: (f) удаление защитной группы Р 1 с образованием соединения формулы (XI) или его аддитивной соли с кислотой, где каждый из радикалов R 1 , R 4 , R 5 и R 7 имеет указанные выше значения; и (g) сочетание соединения формулы (XI) с реагентом формулы Р 2 -Х, в которой Р 2 представляет собой любую группу, блокирующую аминогруппу, как описано выше, и X означает отщепляемую группу с образованием соединения формулы (XII) в которой каждый из заместителей Р 2 , R 1 , R 4 , R 5 и R 7 имеет указанные выше значения. Специалисту в этой области понятно, что в тех вариантах изобретения, в которых Р 2 означает ацильную группу, в том числе, например, ацильную группу, произведенную из аминокислоты, или пептида, или их аналогов, или производных, отщепляемая группа X может образоваться in situ, как рассмотрено выше для соединения формулы (IX). В каждом из соединений (X) и (XII) функциональная группа бороновой кислоты защищена как эфир бороновой кислоты. Если это желательно, защита функциональной группы бороновой кислоты может быть удалена любым методом, который известен из уровня техники. Предпочтительно защиту функциональной группы бороновой кислоты удаляют с помощью реакции трансэтерификации в двухфазной смеси. Более предпочтительно стадия удаления защиты бороновой кислоты включает следующие стадии: (i) приготовление двухфазной смеси, содержащей эфирное соединение бороновой кислоты формулы (X) или (XII), акцептора органической бороновой кислоты, низшего алканола, углеводородного растворителя C 5 -C 8 и водной неорганической кислоты; (ii) перемешивание двухфазной смеси, чтобы получить соответствующее незащищенное соединение бороновой кислоты формулы (Ха) или (XIII) (iii) разделение слоев растворителей и (iv) экстракция органическим растворителем соединения формулы (Ха), (XIII) или боронового ангидрида этого соединения. Предпочтительным акцептором органической бороновой кислоты на стадии (i) является алифатическая, арильная или ар(алифатическая)бороновая кислота. В некоторых вариантах изобретения акцептор бороновой кислоты выбирают из группы, состоящей из фенилбороновой кислоты, бензилбороновой кислоты, бутилбороновой кислоты, пентилбороновой кислоты, гексилбороновой кислоты и циклогексилбороновой кислоты. В определенных вариантах изобретения акцептором бороновой кислоты является изобутилбороновая кислота. В некоторых вариантах изобретения акцептор бороновой кислоты выбирают таким образом, чтобы эфирное соединение бороновой кислоты формулы (III) получалось в виде побочного продукта реакции удаления защиты. Затем эфирное соединение бороновой кислоты формулы (III) может быть использовано в другой загрузке описанного выше периодического процесса. В таких вариантах изобретения функциональную группу R 4 -R 5 эффективно возвращают в цикл, что может быть особенно выгодно, если группа R 4 -R 5 является дорогой хиральной группой. Предпочтительно для увеличения степени чистоты продукта водный слой, содержащий соединение формулы (Xa) или (XIII), промывают, чтобы удалить нейтральные органические примеси до стадии экстракции (iv). В таких вариантах изобретения стадия (iii) предпочтительно включает следующие стадии: (1) разделение слоев растворителей; (2) добавление основания в водный слой до щелочного рН; (3) промывки водного слоя органическим растворителем и (4) подкисление водного слоя до рН меньше чем приблизительно 6. В некоторых вариантах воплощения изобретение относится к усовершенствованному способу получения ингибитора протеазомы - бортезомиба. Таким образом, в одном варианте изобретение обеспечивает крупномасштабный способ получения соединения формулы (XIV) боронового ангидрида этого соединения. Этот способ включает следующие стадии: (а) приготовление боронатного комплекса формулы (XV) в которой R 3 представляет собой нуклеофугическую группу; Y - это нуклеофугическая группа и М + является щелочным металлом; (b) контактирование боронатного комплекса формулы (XV) с кислотой Льюиса в условиях, в которых образуется эфирное соединение бороновой кислоты формулы (XVI) причем указанную стадию контактирования проводят в реакционной смеси, содержащей (i) координационный растворитель - простой эфир, который плохо смешивается с водой; или (ii) растворитель - простой эфир, который плохо смешивается с водой, и координационный сорастворитель; (с) обработка эфирного соединения бороновой кислоты формулы (XVI) реагентом формулы M 1 -N(G) 2 , где М 1 представляет собой щелочной металл и каждый заместитель G, индивидуально или оба вместе, является защитной группой для аминогруппы с образованием соединения формулы (XVII) (d) удаление G групп с образованием соединения формулы (XVIII) или его аддитивной соли с кислотой; (е) сочетание соединения формулы (XVIII) с соединением формулы (XIX) в которой Р 1 является отщепляемой защитной группой для аминогруппы; и X означает ОН или отщепляемую группу; с образованием соединения формулы (XX) в которой Р 1 имеет указанные выше значения; (f) удаление защитной группы Р 1 с образованием соединения формулы (XXI) или его аддитивной соли с кислотой; (g) сочетание соединения формулы (XXI) реагентом формулы (XXII) в которой X представляет собой ОН или отщепляемую группу, с образованием соединения формулы (XXIII) и (h) удаление защиты из функциональной группы бороновой кислоты с образованием соединения формулы (XIV) или боронового ангидрида этого соединения. В некоторых вариантах изобретения способ отличается по крайней мере одним из следующих признаков (1)-(5). В определенных предпочтительных вариантах изобретения этот способ отличается всеми пятью признаками (1)-(5), указанными ниже. (1) В боронатном комплексе формулы (XV) оба заместителя R 3 и Y представляют собой хлор. (2) Стадия сочетания (е) включает следующие этапы: (i) сочетание соединения формулы (XVIII) с соединением формулы (XIX), в которой X означает -ОН, в присутствии тетрафторбората 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (TBTU) и третичного амина в дихлорметане; (ii) осуществление обмена растворителем с заменой дихлорметана на этилацетат и (iii) осуществление водной промывки этилацетатного раствора. (3) Стадия (f) удаления защитной группы включает следующие этапы: (i) обработка соединения формулы (XX) раствором HCl в этилацетате; (ii) добавление гептана в реакционную смесь и (iii) выделение соединения формулы (XXI) в виде аддитивной соли с HCl путем кристаллизации. (4) Стадия сочетания (g) включает следующие этапы: (i) сочетание соединения формулы (XXI) с 2-пиразинкарбоновой кислотой в присутствии TBTU и третичного амина в дихлорметане; (ii) осуществление обмена растворителем, чтобы заменить дихлорметан на этилацетат; и (iii) осуществление водной промывки этилацетатного раствора. (5) Стадия удаления защиты (h) из бороновой кислоты включает следующие этапы: (i) приготовление двухфазной смеси, содержащей соединение формулы (XXIII), акцептор органической бороновой кислоты, низший алканол, углеводородный растворитель C 5 -C 8 и водную неорганическую кислоту; (ii) перемешивание двухфазной смеси, чтобы получить соединение формулы (XIV); (iii) разделение слоев растворителей и (iv) экстракцию органическим растворителем соединения формулы (XIV) или боронового ангидрида этого соединения. Предпочтительно стадия (h) (iii) включает следующие этапы: (1) разделение слоев растворителей; (2) добавление основания в водный слой до щелочного рН; (3) промывка водного слоя органическим растворителем и (4) подкисление водного слоя до рН меньше чем приблизительно 6. В другом варианте воплощения изобретение относится к крупномасштабному способу получения соединения формулы (XIV) или боронового ангидрида этого соединения, который включает следующие стадии: (аа) сочетание соединения формулы (XVIII) или его аддитивной соли с кислотой, с соединением формулы (XIX) в которой Р 1 является отщепляемой защитной группой для аминогруппы; и X означает ОН или отщепляемую группу; с образованием соединения формулы (XX) в которой Р 1 имеет указанные выше значения, причем указанная стадия сочетания (аа) включает следующие этапы: (i) сочетание соединения формулы (XVIII) с соединением формулы (XIX), в которой X означает -ОН, в присутствии тетрафторбората 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (TBTU) и третичного амина в дихлорметане; (ii) осуществление обмена растворителем, чтобы заменить дихлорметан на этилацетат; и (iii) осуществление водной промывки этилацетатного раствора; (bb) удаление защитной группы Р 1 с образованием соединения формулы (XXI) или его аддитивной соли с кислотой, причем указанная стадия (bb) удаления защитной группы включает следующие этапы: (i) обработка соединения формулы (XX) раствором HCl в этилацетате; (ii) добавление гептана к реакционной смеси и (iii) выделение путем кристаллизации соединения формулы (XXI) в виде аддитивной соли с HCl; (cc) сочетание соединения формулы (XXI) с реагентом формулы (XXII) в которой X означает ОН или отщепляемую группу, с образованием соединения формулы (XXIII) указанная стадия сочетания (cc) включает следующие этапы: (i) сочетание соединения формулы (XXI) с 2-пиразинкарбоновой кислотой в присутствии TBTU и третичного амина в дихлорметане; (ii) осуществление обмена растворителем, чтобы заменить дихлорметан на этилацетат; и (iii) осуществление водной промывки этилацетатного раствора; и (dd) удаление защиты из функциональной группы бороновой кислоты с образованием соединения формулы (XIV) или боронового ангидрида этого соединения, причем указанная стадия (dd) удаления защиты включает следующие этапы: (i) приготовление двухфазной смеси, содержащей соединение формулы (XXIII), акцептор органической бороновой кислоты, низший алканол, углеводородный растворитель C 5 -C 8 и водную неорганическую кислоту; (ii) перемешивание двухфазной смеси, чтобы получить соединение формулы (XIV); (iii) разделение слоев растворителей и (iv) экстракция органическим растворителем соединения формулы (XIV) или боронового ангидрида этого соединения. Предпочтительно стадия (dd) (iii) включает следующие этапы: (1) разделение слоев растворителей; (2) добавление основания в водный слой до щелочного рН; (3) промывка водного слоя органическим растворителем и (4) подкисление водного слоя до рН меньше чем приблизительно 6 Эффективность описанных выше способов дополнительно увеличивается за счет сокращения продолжительности стадий, например, путем переноса реакционной смеси или обработанного раствора продукта из одной реакции непосредственно в следующую реакцию без выделения промежуточного продукта. Например, в некоторых вариантах изобретения на стадии (е)(iii) или (аа)(iii) получают этилацетатный раствор, содержащий соединение формулы (XX), и этот этилацетатный раствор непосредственно обрабатывают на стадии (f) или (bb) в условиях, в которых эффективно удаляется защитная группа Р 1 . В некоторых таких вариантах изобретения защитная группа Р 1 является лабильной в кислоте защитной группой, например трет-бутоксикарбонилом (BOK), и этилацетатный раствор со стадии (е)(iii) или (aa)(iii) обрабатывают кислотой. В определенных предпочтительных вариантах изобретения этилацетатный раствор со стадии (е)(iii) или (аа)(iii) сушат путем азеотропной перегонки и затем обрабатывают газообразным HCl. Когда удаление защиты на стадии (f) или (bb) проводят в безводных условиях, как описано выше, продукт формулы (XXI) может быть выделен путем кристаллизации из реакционной смеси в виде аддитивной соли с HCl. Кристаллизация соли продукта стимулируется путем добавления углеводородного растворителя, такого как н-гептан. В некоторых вариантах изобретения реакционную смесь частично концентрируют до добавления углеводородного растворителя. Авторы настоящего изобретения обнаружили, что при кристаллизации соединения формулы (XXI) таким образом эффективно удаляются любые трипептидные примеси, которые могут образоваться в ходе стадии сочетания (е) или (аа). Такие примеси трудно удаляются на более поздних стадиях синтеза. Кроме того, сокращение продолжительности процесса возможно за счет переноса смеси продуктов со стадии сочетания (g) или (cc) непосредственно на стадию (h) или (dd) удаления защиты из группы бороновой кислоты. Предпочтительно сначала органический растворитель из реакции сочетания заменяют этилацетатом с целью облегчения промывки водой. Затем вторая замена растворителя на углеводородный растворитель позволяет использовать раствор продукта со стадии (g) или (cc) непосредственно на стадии (h) или (dd) удаления защиты в двухфазной бороновой кислоте без выделения соединения формулы (XXIII). В качестве альтернативы для синтеза соединения формулы (XIV) может быть выбран более рациональный подход. Таким образом, в еще одном варианте изобретение обеспечивает крупномасштабный способ получения соединения формулы (XIV) или боронового ангидрида этого соединения. Этот способ включает следующие стадии: (а) приготовление боронатного комплекса формулы (XV) в которой R 3 означает нуклеофугическую группу; Y - это нуклеофугическая группа и М + является щелочным металлом; (b) контактирование боронатного комплекса формулы (XV) с кислотой Льюиса в условиях, в которых образуется эфирное соединение бороновой кислоты формулы (XVI) причем указанная стадия контактирования осуществляется в реакционной смеси, содержащей: (i) координационный растворитель - простой эфир, который плохо смешивается с водой; или (ii) растворитель - простой эфир, который плохо смешивается с водой, и координационный сорастворитель; (с) обработка эфирного соединения бороновой кислоты формулы (XVI) реагентом формулы M 1 -N(Si(R 6 ) 3 ) 2 , в которой М 1 представляет собой щелочной металл и каждый R 6 независимо выбирают из группы, состоящей из алкила, аралкила и арила, где арил или арильный фрагмент аралкила необязательно является замещенным, с образованием соединения формулы (XVII) (d) удаление (R 6 ) 3 Si групп с образованием соединения формулы (XVIII) или его аддитивной соли с кислотой; (е') сочетание соединения формулы (XVIII) с соединением формулы (XIXa) в которой X представляет собой ОН или отщепляемую группу, с образованием соединения формулы (XXIII) и (f) удаление защиты из группы бороновой кислоты с образованием соединения формулы (XIV) или боронового ангидрида этого соединения. В некоторых вариантах изобретения способ характеризуется по меньшей мере одним из следующих признаков (1)-(3). В определенных предпочтительных вариантах изобретения этот способ характеризуется всеми тремя признаками (1)-(3), указанными ниже. (1) В боронатном комплексе формулы (XV) оба заместителя R 3 и Y означают хлор. (2) Стадия сочетания (е') включает следующие этапы: (i) сочетание соединения формулы (XVIII) с соединением формулы (XIXa), в которой X представляет собой ОН, в присутствии тетрафторбората 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония (TBTU) и третичного амина в дихлорметане; (ii) осуществление обмена растворителем, чтобы заменить дихлорметан на этилацетат и (iii) осуществление водной промывки этилацетатного раствора. (3) Стадия (f) удаления защиты бороновой кислоты включает следующие этапы: (i) предоставление двухфазной смеси, содержащей соединение формулы (XXIII), акцептор органической бороновой кислоты, низший алканол, углеводородный растворитель C 5 -C 8 и водную неорганическую кислоту; (ii) перемешивание двухфазной смеси, чтобы получить соединение формулы (XIV); (iii) разделение слоев растворителей и (iv) экстракция органическим растворителем соединения формулы (XIV) или боронового ангидрида этого соединения. Предпочтительно стадия (f)(iii) включает следующие этапы: (1) разделения слоев растворителей; (2) добавления основания в водный слой до щелочного рН; (3) промывки водного слоя органическим растворителем и (4) подкисления водного слоя до рН меньше чем приблизительно 6. На стадии (h)(iv), (dd)(iv) или (f)(iv) описанных выше способов предпочтительно соединение формулы (XIV) или бороновый ангидрид этого соединения экстрагируется этилацетатом и кристаллизуется путем добавления гексана или гептана. В некоторых вариантах изобретения способ дополнительно включает в себя выделение боронового ангидрида соединения формулы (XIV), предпочтительно тримерного ангидрида бороновой кислоты формулы (XXIV) Способы изобретения обеспечивают крупномасштабное производство бортезомиба, имеющего очень высокую химическую и стереохимическую чистоту. Способы уровня техники имели ограниченный масштаб производства и давали продукт с пониженной общей чистотой. Поэтому в другом аспекте настоящее изобретение обеспечивает композицию, содержащую по меньшей мере один килограмм соединения формулы (XXIV) Предпочтительно соединение формулы (XXIV) получают в соответствии с описанным способом, и предпочтительно составляет по меньшей мере 99% от массы композиции в соответствии с этим аспектом изобретения. Сокращения Пример 1. Способ получения трифторацетата 1-аммоний 3-метилбутан-1-бороната(1R)-(S)пинандиола. 1-Хлор-3-метилбутан-1-боронат(1S)-(S)пинандиола. 1. (S)-Пинандиол-2-метилпропан-1-боронат (12,0 кг, 50,8 моль) загружают в реакционный сосуд, в котором находится газообразный азот. 2. Загружают трет-бутил метиловый эфир (53 кг) и дихлорметан (22,5 кг) и полученную смесь охлаждают до -57 °С при перемешивании. 3. В другой реакционный сосуд, в котором находится газообразный азот, загружают диизопропиламин (6,7 кг). 4. В диизопропиламин добавляют трет-бутилметиловый эфир (27 кг) и полученную смесь охлаждают до -10 °С при перемешивании. 5. Раствор н-гексиллития в гексане (33,2 мас.%, 17,6 кг) добавляют к смеси диизопропиламина в течение 57 мин, поддерживая температуру реакции равной от -10 до -7 °С. 6. Эту смесь (смесь ДАЛ) перемешивают в течение 33 мин при температуре от -9 до -7 °С до ее использования. 7. В третий реакционный сосуд, в котором находится газообразный азот, загружают хлорид цинка (12,1 кг). 8. К хлориду цинка добавляют трет-бутилметиловый эфир (16 кг) и полученную смесь нагревают до 30 °С при перемешивании. 9. К суспензии хлорида цинка добавляют тетрагидрофуран (53 кг) в течение 18 мин, поддерживая температуру реакции равной от 35 до 40 °С. 10. Эту смесь (смесь ZnCl 2 ) перемешивают в течение 4 ч и 28 мин при температуре от 38 до 39 °С до ее использования. 11. Смесь ДАЛ (из пп.3-6) добавляют в течение 60 мин в реакционный сосуд, содержащий (S)-пинандиол-2-метилпропан-1-боронат, поддерживая температуру реакции равной от -60 до -55 °С. 12. Для завершения добавления используют промывку трет-бутилметиловым эфиром (10 кг). 13. Реакционную смесь перемешивают дополнительно в течение 20 мин при температуре от -59 до -55 °С. 14. Реакционную смесь нагревают до -50 °С в течение 11 мин. 15. Смесь ZnCl 2 (из пп.7-10) добавляют в течение 48 мин в реакционный сосуд, содержащий (S)-пинандиол-2-метилпропан-1-боронат и смесь ДАЛ, поддерживая температуру реакции равной от -50 до -45 °С. 16. Для завершения добавления используют промывку трет-бутилметиловым эфиром (10 кг). 17. Реакционную смесь перемешивают дополнительно в течение 30 мин при температуре от -45 до -40 °С и затем нагревают до 10 °С в течение 81 мин. 18. Добавляют 10%-ный раствор серной кислоты (72 кг) в течение 40 мин в реакционный сосуд, поддерживая температуру реакции равной от 10 до 21 °С. 19. Реакционную смесь перемешивают в течение 16 мин при температуре окружающей среды до выделения водной фазы. 20. Органическую фазу последовательно промывают деионизированной водой (ДИ, 32 кг) и 10%-ным раствором хлорида натрия (26,7 кг), причем каждая промывка включает интенсивное перемешивание в течение 15-17 мин при температуре окружающей среды. 21. Реакционную смесь концентрируют при пониженном давлении (Р мин = 81 мбар), поддерживая внешнюю температуру равной 50-55 °С (с помощью рубашки и бани), получая остаток, который растворяют в метилциклогексане (56 кг). 22. Реакционную смесь подвергают дефлегмации (с ловушкой типа Дина-Старка для удаления воды) при пониженном давлении (Р мин = 67 мбар), поддерживая внешнюю температуру равной 50-55 °С (рубашка и баня) в течение 2 ч и 7 мин до прекращения выделения воды. 23. При пониженном давлении (Р мин = 81 мбар) отгоняют приблизительно 35 л растворителей, поддерживая внешнюю температуру от 50 до 55 °С (рубашка и баня). 24. Полученную осушенную смесь в метилциклогексане, содержащую 1-хлор-3-метилбутан-1-боронат(1S)-(S)пинандиола, охлаждают до 14 °С. 1-бис(Триметилсилил)амино-3-метилбутан-1-боронат(1R)-(S)пинандиола. 1. Раствор бис(триметилсилил)амид лития в тетрагидрофуране (19,4 мас.%, 41,8 кг) загружают в реакционный сосуд, в котором находится газообразный азот, и охлаждают до -19 °С при перемешивании. 2. Добавляют метилциклогексановую смесь, содержащую 1-хлор-3-метилбутан-1-боронат(1S)-(S)-пинандиола, в течение 55 мин, поддерживая температуру реакции равной от -19 до -13 °С. 3. Для завершения добавления используют промывку метилциклогексаном (5 кг). 4. Реакционную смесь перемешивают дополнительно в течение 65 мин при температуре от -13 до -12 °С и затем нагревают до 25 °С в течение 25 мин. 5. В реакционную смесь добавляют суспензию Целита (2,5 кг) в метилциклогексане (22 кг). 6. Реакционную смесь концентрируют при пониженном давлении (Р мин = 25 мбар), поддерживая внешнюю температуру от 45 до 50 °С (рубашка и баня), получая остаток, который растворяют в метилциклогексане (36 кг). 7. Затем отбирают пробу для определения содержания тетрагидрофурана в ходе процесса методом ГХ. 8. Содержание тетрагидрофурана по данным анализа равно 0,58%. 9. Твердое вещество удаляют с помощью фильтрации, причем смесь фильтруют через слой силикагеля (2,0 кг). 10. Оба фильтрующих блока промывают изопропиловым эфиром (30 кг). 11. Полученную смесь в метилциклогексане/изопропиловом эфире, содержащую 1-бис(триметилсилил)амино-3-метилбутан-1-боронат(1R)-(S)пинандиола, сохраняют в контейнере при температуре окружающей среды до ее использования на следующей стадии. Трифторацетат-3-метилбутан-1-боронат(1R)-(S)пинандиол-1-аммония. 1. Трифторуксусную кислоту (12 кг) загружают в другой реакционный сосуд, в котором находится газообразный азот. 2. Изопропиловый эфир (78 кг) добавляют к трифторуксусной кислоте и полученную смесь охлаждают до -10 °С при перемешивании. 3. Добавляют смесь в метилциклогексане/изопропиловом эфире, содержащую 1-бис(триметилсилил)амино-3-метилбутан-1-боронат(1R)-(S)пинандиола, в течение 53 мин, что вызывает осаждение продукта при поддержании температуры реакции от -10 до -5 °С. 4. Для завершения добавления используют промывку изопропиловым эфиром (5 кг). 5. Реакционную смесь перемешивают дополнительно в течение 8 ч и 20 мин при температуре от -9 до -7 °С. 6. Твердое вещество собирают с помощью фильтрации, промывают изопропиловым эфиром (70 кг) двумя порциями и сушат при пониженном давлении (Р мин = 56 мбар) при температуре 41-42 °С в течение 2 ч и 15 мин. 7. Твердое вещество перемешивают с деионизированной водой (60 кг) в течение 24 мин при температуре окружающей среды и затем деионизированную воду удаляют с помощью фильтрации. 8. Твердое вещество промывают ДИ водой (12 кг). 9. Затем твердое вещество сушат в вакууме (Р мин = 4 мбар) при 40-44 °С в течение 9 ч и 22 мин; за это время потери при сушке составляют 0,51%, что соответствует требованию ≤ 1%. 10. Затем промежуточное соединение, неочищенный трифторацетат-3-метилбутан-1-боронат(1R)-(S)пинандиол-1-аммония, упаковывают в отдельные полиэтиленовые пакеты в полипропиленовых барабанах и снабжают этикеткой. Выход составляет 72%. Перекристаллизация неочищенного трифторацетат-3-метилбутан-1-бороната(1R)-(S)пинандиол-1-аммония. 1. Неочищенный трифторацетат-3-метилбутан-1-боронат(1R)-(S)пинандиол-1-аммония (13 кг) загружают в реакционный сосуд, в котором находится газообразный азот. 2. Трифторуксусную кислоту (31 кг) загружают в реакционный сосуд и полученную смесь охлаждают до 4 °С при перемешивании. 3. После растворения всего твердого вещества с образованием слегка мутной смеси добавляют изопропиловый эфир (29 кг) в течение 57 мин, поддерживая температуру реакции при 2-3 °С. 4. После завершения добавления смесь фильтруют через фильтр в приемник, в котором находится газообразный азот. 5. Реактор и фильтр промывают смесью трифторуксусной кислоты (3,8 кг) и изопропилового эфира (5 кг). Промывку добавляют к фильтрату. 6. Поддерживая температуру реакции равной 16-18 °С, добавляют в течение 15 мин изопропиловый эфир (126 кг), что вызывает осаждение продукта. 7. Смесь перемешивают 15 мин при температуре 16-18 °С, затем охлаждают до -5 °С в течение 67 мин и перемешивают при температуре от -3 до -5 °С в атмосфере азота в течение 89 мин. 8. Затем твердое вещество, выделенное с помощью фильтрации, промывают изопропиловым эфиром (48 кг) двумя порциями и сушат в вакууме (Р мин = 2 мбар) при 34-40 °С в течение 2 ч и 55 мин, за это время потери при сушке составляют 0,32%, что соответствует требованию ≤ 0,5%. 9. Затем продукт, трифторацетат-3-метилбутан-1-боронат(1R)-(S)пинандиол-1-аммония, упаковывают в двойные полиэтиленовые пакеты, в картонные барабаны и снабжают этикеткой. Выход составляет 86%. Пример 2. Способ получения боронового ангидрида н-(2-пиразинкарбонил)-L-фенилаланин-L-лейцина. N-BOK-L-фенилаланин-L-лейцин-боронат(1S,2S,3R,5S)пинандиола. 1. В вытяжном шкафу продувают азотом трехгорлую реакционную колбу из стекла, снабженную насадкой Кляйзена с термографом и мешалкой с приводом. 2. В эту колбу загружают трифторацетат-3-метилбутан-1-боронат(1R)-(S)пинандиол-1-аммония (2,0 кг). 3. В колбу загружают BOK-L-фенилаланин (1,398 кг). 4. В колбу добавляют тетрафторборат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония, TBTU (1,864 кг). 5. В колбу добавляют дихлорметан (15,8 л). 6. Регулируют привод мешалки, чтобы обеспечить скорость перемешивания 260 об/мин. 7. Реакционную смесь, выдерживаемую в атмосфере азота, охлаждают до 1,0 °С, используя охлаждающую баню с ледяной водой. 8. В стеклянную колбу загружают N,N-диизопропилэтиламин (2,778 л) и перекачивают в реакционную смесь в течение 117 мин, используя перистальтический насос и поддерживая температуру реакции в диапазоне от 0,7 до 2,1 °С. Общая скорость добавления составляет 23,7 мл/мин. 9. Для завершения добавления в реакционную смесь используют промывку колбы дихлорметаном (0,2 л). 10. Реакционную смесь перемешивают дополнительно в течение 35 мин. В начале перемешивания температура смеси составляет 1,8 °С и в конце - 2,5 °С. 11. Затем в ходе процесса отбирают пробу для анализа методом жидкостной хроматографии высокого разрешения с обращенной фазой (ЖХВР-ОФ). Найденная степень превращения составляет 99,3%. 12. Реакционную смесь, разделенную на две приблизительно равные части, переносят в две колбы роторного испарителя. Реакционную смесь концентрируют при пониженном давлении, используя роторный испаритель и поддерживая температуру внешней бани 29-30 °С. 13. Этилацетат (4,0 л), разделенный на две приблизительно равные части, загружают в две колбы роторного испарителя. 14. Смеси в каждой колбе снова концентрируют при пониженном давлении, используя роторный испаритель, поддерживая температуру внешней бани 29-30 °С. 15. Затем остаток из каждой колбы роторного испарителя переносят обратно в реакционную колбу, используя этилацетат (13,34 л). 16. В стеклянной колбе, снабженной мешалкой, готовят 1%-ный водный раствор фосфорной кислоты, смешивая ДИ воду (13,18 л) и фосфорную кислоту (0,160 кг). 17. В стеклянной колбе, снабженной мешалкой, готовят 2%-ный водный раствор карбоната калия (12,0 л), смешивая ДИ воду (11,76 л) и карбонат калия (0,24 кг). 18. В стеклянной колбе, снабженной мешалкой, готовят 10%-ный водный раствор хлорида натрия (13,34 л), смешивая ДИ воду (13,34 л) и хлорид натрия (1,334 кг). 19. В реакционную колбу, содержащую этилацетатный раствор, добавляют ДИ воду (13,34 л) и смесь перемешивают 380 об/мин в течение 7 мин. Дают слоям разделиться и водную фазу (нижний слой) переносят в вакууме в подходящую колбу и выливают. 20. Снова загружают в реакционную колбу, содержащую этилацетатный раствор, ДИ воду (13,34 л) и смесь перемешивают 7 мин со скоростью 385 об/мин. Дают слоям разделиться и водную фазу (нижний слой) переносят в вакууме в подходящую колбу и выливают. 21. Приготовленный на стадии 16 раствор фосфорной кислоты (1%) загружают в реакционную колбу, содержащую этилацетатный раствор, и смесь перемешивают 7 мин со скоростью 365 об/мин. Дают слоям разделиться и кислотную водную фазу (нижний слой) переносят в подходящую колбу и выливают. 22. Приготовленный на стадии 17 раствор карбоната калия (2%) загружают в реакционную колбу, содержащую этилацетатный раствор, и смесь перемешивают 7 мин со скоростью 367 об/мин. Дают слоям разделиться и щелочную водную фазу (нижний слой) переносят в подходящую колбу и выливают. 23. Приготовленный на стадии 18 раствор хлорида натрия (10%) загружают в реакционную колбу, содержащую этилацетатный раствор, и смесь перемешивают 6 мин со скоростью 373 об/мин. Дают слоям разделиться и водную фазу (нижний слой) переносят в подходящую колбу и выливают. 24. Этилацетатный раствор переносят в колбу роторного испарителя и концентрируют при пониженном давлении, используя роторный испаритель и поддерживая температуру бани 29-30 °С, чтобы получить остаток. 25. Затем остаток снова растворяют в этилацетате (4,68 л). 26. Этот раствор концентрируют в вакууме, используя роторный испаритель и поддерживая температуру бани 29-30 °С, чтобы еще получить остаток. 27. Затем остаток снова растворяют в этилацетате (4,68 л) и отбирают две пробы для определения содержания воды методом титрования по Карлу Фишеру. Найдено, что содержание воды в этих двух пробах составляет 0,216 и 0,207%. 28. Используя дополнительное количество этилацетата (12,66 л), смесь переносят из колбы роторного испарителя в сухую реакционную колбу, снабженную термографом, мешалкой с приводом и фриттованной трубкой для диспергирования газа, и продувают азотом. L-Фенилаланин-L-лейцин-боронат(1S,2S,3R,5S)пинандиола, HCl-соль. 1. Охлаждают до температуры -0,9 °С этилацетатный раствор, содержащий N-BOK-L-фенилаланин-L-лейцин-боронат(1S,2S,3R,5S)пинандиола, используя охлаждающую баню с ледяной водой. 2. Газообразный хлористый водород (1,115 кг) барботируют в реакционную смесь в течение 1,48 ч. В начале добавления температура равна -0,9 °С, а в конце добавления равна 6,8 °С. 3. Затем реакционной смеси дают нагреться до 14,4 °С в течение 50 мин, поддерживая атмосферу азота. 4. В ходе процесса отбирают пробу для анализа методом ЖХВР-ОФ. Степень превращения составляет 68,9% (по площади пика). 5. Реакционную смесь перемешивают в течение 35 мин. В начале добавления температура равна 14 °С, а в конце добавления - 14,8 °С. 6. В ходе процесса отбирают пробу для анализа методом ЖХВР-ОФ. Степень превращения составляет 94,7% (по площади пика). 7. Реакционную смесь дополнительно перемешивают приблизительно в течение 50 мин, поддерживая температуру 10 ± 5 °С. 8. В ходе процесса отбирают пробу для анализа методом ЖХВР-ОФ. Степень превращения составляет 97,3%. 9. Реакционную смесь дополнительно перемешивают приблизительно в течение 50 мин, поддерживая температуру 10 ± 5 °С. Конечная температура равна 14,6 °С. 10. В ходе процесса отбирают пробу для анализа методом ЖХВР-ОФ. Суммарное время взаимодействия после добавления газообразного хлористого водорода составляет четыре (4) часа. 11. Степень превращения составляет 99%. 12. Наблюдается образование суспензии. 13. В реакционную смесь загружают н-гептан (8,8 л). 14. Суспензию перемешивают в течение 2 ч. В начале перемешивания температура равна 12,7 °С, а в конце процесса - 15,3 °С. 15. Твердое вещество выделяют с помощью фильтрации на воронке Бюхнера, покрытой фильтровальной подушкой из полипропиленового войлока. 16. Твердое вещество промывают н-гептаном (4,68 л). 17. Это твердое вещество переносят в три сушильных поддона глубиной не более 25,4 мм и сушат на воздухе в течение 1 ч под тягой. 18. Затем твердое вещество сушат при температуре ≤35 °С в вакууме 685,8 мм рт. ст. (27") в течение 16 ч 28 мин в вакуумном шкафу, снабженном вакуумметром и термографом. 19. Из каждого сушильного поддона отбирают пробы твердого вещества, чтобы определить потери при сушке (%). Найдено, что для трех отобранных проб ППС составляет 0, 0,02 и 0,02 %. 20. Затем упаковывают HCl-соль L-фенилаланин-L-лейцин-боронат(1S,2S,3R,5S)пинандиола в двойные полимерные пакеты и в картонные барабаны, снабжают этикеткой и отбирают образцы. 21. Выход выделенного продукта 1,87 кг, 79,1%. Это промежуточное соединение сохраняют при 2-8 °С до использования в дальнейшем получении. N-(2-Пиразинкарбонил)-L-фенилаланин-L-лейцин-боронат(1S,2S,3R,5S)пинандиола. 1. В вытяжном шкафу продувают азотом трехгорлую стеклянную реакционную колбу, снабженную насадкой Кляйзена, термографом и мешалкой с приводом. 2. В эту колбу загружают (1S,2S,3R,5S)пинандиол L-фенилаланин-L-лейцин боронат, HCl-соль (1,85 кг). 3. В колбу загружают 2-пиразинкарбоновую кислоту (0,564 кг). 4. В колбу добавляют тетрафторборат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония, TBTU (1,460 кг). 5. В колбу добавляют дихлорметан (18,13 л). 6. Регулируют привод мешалки, чтобы обеспечить скорость перемешивания 272 об/мин. 7. Реакционную смесь охлаждают до -1,2 °С, используя охлаждающую баню. 8. В стеклянную колбу загружают N,N-диизопропилэтиламин (1,865 кг) и перекачивают в реакционную смесь в течение 50 мин, используя перистальтический насос и поддерживая температуру реакции в диапазоне от -1,2 до 2,8 °С. 9. Для завершения добавления в реакционную смесь используют промывку колбы дихлорметаном (0,37 л). 10. Реакционной смеси дают нагреться и ее дополнительно перемешивают в течение 81 мин. 11. В начале перемешивания температура смеси составляет 15 °С и в конце температура равна 24,9 °С. 12. Затем в ходе процесса отбирают пробу для анализа методом ЖХВР-ОФ. Найденная степень превращения составляет 99,9%. 13. Реакционную смесь (две приблизительно равные части) переносят в две колбы роторного испарителя. Реакционную смесь концентрируют при пониженном давлении, используя роторный испаритель и поддерживая температуру внешней бани 33-34 °С. 14. Этилацетат (12,95 л), разделенный на две приблизительно равные части, загружают в две колбы роторного испарителя. 15. Затем смеси в каждой колбе концентрируют при пониженном давлении, используя роторный испаритель, поддерживая температуру внешней бани равной 33-34 °С. 16. Затем остаток из каждой колбы роторного испарителя переносят обратно в реакционную колбу, используя этилацетат (12,95 л). 17. В стеклянной колбе, снабженной мешалкой, готовят 1%-ный водный раствор фосфорной кислоты (12,34 л), смешивая ДИ воду (12,19 л) и фосфорную кислоту (0,148 кг). 18. В стеклянной колбе, снабженной мешалкой, готовят 2%-ный водный раствор карбоната калия (12,34 л), смешивая ДИ воду (12,09 л) и карбонат калия (0,247 кг). 19. В стеклянной колбе, снабженной мешалкой, готовят 10%-ный водный раствор хлорида натрия (12,34 л), смешивая ДИ воду (12,34 л) и хлорид натрия (1,234 кг). 20. В реакционную колбу, содержащую этилацетатный раствор, добавляют ДИ воду (12,34 л) и смесь перемешивают 382 об/мин в течение 7 мин. Дают слоям разделиться и водную фазу (нижний слой) переносят в вакууме в подходящую колбу и выливают. 21. Снова загружают в реакционную колбу, содержащую этилацетатный раствор, ДИ воду (12,34 л), и смесь перемешивают 7 мин со скоростью 398 об/мин. Дают слоям разделиться и водную фазу (нижний слой) переносят в подходящую колбу и выливают. 22. Приготовленный на стадии 17 раствор фосфорной кислоты (1%) загружают в реакционную колбу, содержащую этилацетатный раствор, и смесь перемешивают 8 мин со скоростью 364 об/мин. Дают слоям разделиться и кислотную водную фазу (нижний слой) переносят в подходящую колбу и выливают. 23. Приготовленный на стадии 18 раствор карбоната калия (2%) загружают в реакционную колбу, содержащую этилацетатный раствор, и смесь перемешивают 8 мин со скоростью 367 об/мин. Дают слоям разделиться и щелочную водную фазу (нижний слой) переносят в подходящую колбу и выливают. 24. Приготовленный на стадии 19 раствор хлорида натрия (10%) загружают в реакционную колбу, содержащую этилацетатный раствор, и смесь перемешивают 8 минут со скоростью 374 об/мин. Дают слоям разделиться и водную фазу (нижний слой) переносят в подходящую колбу и выливают. 25. Этилацетатный раствор (приблизительно две равные части) переносят в вакууме в две колбы роторного испарителя и концентрируют при пониженном давлении, используя роторный испаритель и поддерживая внешнюю температуру бани 34 °С. 26. н-Гептан (14,8 л) разделяют на две приблизительно равные части и загружают в две колбы роторного испарителя. Затем смеси в каждой колбе концентрируют при пониженном давлении, используя роторный испаритель и поддерживая внешнюю температуру бани равной 34 °С. Бороновый ангидрид N-(2-пиразинкарбонил)-L-фенилаланин-L-лейцина, неочищенный. 1. В стеклянной колбе, снабженной мешалкой, готовят 1н. раствор соляной кислоты (22,2 л), смешивая ДИ воду (20,36 л) и концентрированную соляную кислоту (1,84 кг). 2. В стеклянной колбе, снабженной мешалкой, готовят 2н. раствор гидроксида натрия (12,03 л), смешивая ДИ воду (12,03 л) и гидроксид натрия (0,962 кг). 3. Затем переносят остаток из каждой колбы роторного испарителя, содержащий N-(2-пиразинкарбонил)-L-фенилаланин-L-лейцин-боронат(1S,2S,3R,5S)пинандиола, в трехгорлую стеклянную реакционную колбу, снабженную термографом и мешалкой с приводом, используя н-гептан (14,8 л) и метанол (14,8 л). 4. Регулируют привод мешалки, чтобы обеспечить скорость перемешивания 284 об/мин. 5. Загружают в колбу 2-метилпропанбороновую кислоту (0,672 кг). 6. Загружают в колбу 1н. соляную кислоту, приготовленную на стадии 1 (11,2 л). 7. Регулируют привод мешалки, чтобы обеспечить скорость перемешивания 326 об/мин. 8. Реакционную смесь перемешивают в течение 16,38 ч. В начале перемешивания температура смеси составляет 28,6 °С и в конце температура смеси равна 21,6 °С. 9. Затем в ходе процесса отбирают пробу для анализа методом ЖХВР-ОФ. 10. Найденная степень превращения составляет 100%. 11. Перемешивание прекращают и двухфазной смеси дают разделиться. 12. н-Гептановый слой (верхний слой) переносят в подходящую колбу и выливают. 13. н-Гептан (5,37 л) загружают в реакционную колбу и смесь перемешивают 6 мин со скоростью 381 об/мин. Дают слоям разделиться и гептановую фазу (верхний слой) переносят в подходящую колбу и выливают. 14. В реакционную колбу снова загружают н-гептан (5,37 л) и смесь перемешивают 6 мин со скоростью 340 об/мин. Дают слоям разделиться и гептановую фазу (верхний слой) переносят в подходящую колбу и выливают. 15. Водный метанольный раствор, разделенный приблизительно на две равные части, переносят в две колбы роторного испарителя и концентрируют при пониженном давлении, используя роторный испаритель и поддерживая внешнюю температуру бани 33-34 °С. Отгоняют 15 л метанола. 16. Для переноса остатка из колб роторного испарителя обратно в реакционную колбу используют дихлорметан (5,37 л). 17. В реакционную колбу загружают 2н. раствор гидроксида натрия (11,2 л), приготовленный на стадии 2. 18. Дихлорметановый слой (нижний слой) переносят в подходящую колбу и выливают. 19. Дихлорметан (5,37 л) загружают в колбу и смесь перемешивают 6 мин со скоростью 374 об/мин. Фазам дают разделиться и дихлорметановый слой (нижний слой) переносят в подходящую колбу и выливают. 20. В колбу снова загружают дихлорметан (5,37 л) и смесь перемешивают 8 мин со скоростью 368 об/мин. Фазам дают разделиться и дихлорметановый слой (нижний слой) переносят в подходящую колбу и выливают. 21. Дихлорметан (5,37 л) загружают в колбу. 22. При перемешивании загружают в колбу 1н. соляную кислоту (10,7 л). Определяют значение рН водной фазы, равное 6. 23. Перемешивание прекращают и фазам дают разделиться. 24. Дихлорметановую фазу (нижний слой) переносят в вакууме в стеклянный приемник. 25. Дихлорметан (5,37 л) загружают в колбу и смесь перемешивают 6 мин со скоростью 330 об/мин. Фазам дают разделиться и дихлорметановый слой (нижний слой) переносят в стеклянный приемник. 26. В колбу снова загружают дихлорметан (5,37 л) и смесь перемешивают 6 мин со скоростью 335 об/мин. Фазам дают разделиться и дихлорметановый слой (нижний слой) переносят в стеклянный приемник. 27. Дихлорметановые экстракты объединяют, переносят две приблизительно равные части в две колбы роторного испарителя и концентрируют при пониженном давлении, используя роторный испаритель, поддерживая внешнюю температуру бани равной 33-34 °С. 28. Этилацетат (12,95 л) разделяют на две приблизительно равные части и загружают в две колбы роторного испарителя. Затем концентрируют смеси в каждой колбе при пониженном давлении, используя роторный испаритель и поддерживая внешнюю температуру бани равной 45-46 °С. 29. Этилацетат (12,95 л) снова разделяют на две приблизительно равные части и загружают в две колбы роторного испарителя. Затем концентрируют смеси в каждой колбе при пониженном давлении, используя роторный испаритель и поддерживая внешнюю температуру бани равной 45-46 °С, пока не останется приблизительно 10% от исходного объема. 30. н-Гептан (10,2 л) разделяют на две приблизительно равные части и загружают в две колбы роторного испарителя и суспензию перемешивают 2,67 ч в атмосфере азота при температуре 22-23 °С. 31. Твердое вещество выделяют с помощью фильтрации на воронке Бюхнера, покрытой фильтровальной подушкой из полипропиленового войлока. 32. Твердое вещество промывают н-гептаном (2,96 л). 33. Это твердое вещество переносят в четыре сушильных поддона и сушат на воздухе в течение 1,25 ч под тягой. 34. Затем твердое вещество сушат при температуре 36-50 °С в вакууме 685,8 мм рт. ст. (27") в течение 18 ч 27 мин в вакуумном шкафу, снабженном вакуумметром и термографом. 35. Из каждого сушильного поддона отбирают пробы твердого вещества, чтобы определить потери при сушке (ППС, %). Найдено, что для четырех отобранных проб ППС составляют 0,38, 0,62, 0,71 и 0,63%. 36. Неочищенный бороновый ангидрид N-(2-пиразинкарбонил)-L-фенилаланин-L-лейцина упаковывают в две широкогорлые бутыли (по 5 л) из ПЭВП, защищенные от удара, и снабжают этикеткой. 37. Выход выделенного продукта составляет 1,314 кг, 83%. Перекристаллизация неочищенного боронового ангидрида N-(2-пиразинкарбонил)-L-фенилаланин-L-лейцина. 1. Под тягой продувают азотом стеклянную реакционную колбу, снабженную мешалкой с приводом, обратным холодильником и термографом. 2. Загружают в колбу этилацетат (21 л). 3. Нагревают этилацетат до 66,8 °С в атмосфере азота, используя горячую водно/паровую баню. 4. В реакционную колбу медленно загружают неочищенный бороновый ангидрид N-(2-пиразинкарбонил)-L-фенилаланин-L-лейцина (1,311 кг). Операция загрузки занимает около 3 мин. 5. Смесь перемешивают в течение 1 мин, пока не растворится все твердое вещество. Температура раствора равна 64 °С. 6. Удаляют источник тепла и смесь медленно охлаждают до 60 °С, используя охлаждающую баню. 7. Горячий этилацетатный раствор переносят в приемник по полимерной трубке и через встроенную полипропиленовую фильтрующую капсулу, используя перистальтический насос. 8. Смеси дают охладиться до 27,2 °С и выдерживают ее в атмосфере азота без перемешивания в течение 17,75 ч. Зарегистрированная конечная температура составляет 20,5 °С. 9. Смесь охлаждают, используя баню с ледяной водой, при перемешивании в течение 2,33 ч. В начале перемешивания температура равна 3,8 °С, а в конце процесса -2,8 °С. 10. Твердое вещество выделяют с помощью фильтрации на воронке Бюхнера, покрытой фильтровальной подушкой из полипропиленового войлока. Фильтрат собирают в колбу-приемник. 11. Твердое вещество промывают этилацетатом (2,62 л), охлажденным до 4,7 °С. 12. Это твердое вещество переносят в два сушильных поддона под тягой. 13. Затем твердое вещество сушат при 51-65 °С в вакууме 685,8 мм рт. ст. (27") в течение 19 ч 10 мин в вакуумном шкафу, снабженном вакуумметром и термографом. 14. Отбирают пробы твердого вещества, чтобы определить потери при сушке (ППС, %). Найдено, что для двух отобранных проб ППС составляют 0,65 и 0,62%. 15. Бороновый ангидрид N-(2-пиразинкарбонил)-L-фенилаланин-L-лейцина упаковывают в четыре широкогорлые бутыли янтарного цвета (по 1 л), тип 3 с крышкой, покрытой тефлоном, и снабжают этикеткой. 16. Выход выделенного продукта составляет 1,132 кг, 86,3%. 17. Бороновый ангидрид N-(2-пиразинкарбонил)-L-фенилаланин-L-лейцина хранят при температуре от -25 до -15 °С. Пример 3. Рациональный синтез боронового ангидрида N-(2-пиразинкарбонил)-L-фенилаланин-L-лейцина. N-(2-Пиразинкарбонил)-L-фенилаланин-L-лейцин-боронат(1S,2S,3R,5S)пинандиола. Раствор трифторацетат-3-метилбутан-1-бороната(1R)-(S)пинандиол-1-аммония (13,97 г) и N-гидроксисукцинимида (6,23 г) в 66 мл ДМФ охлаждают до -5 °С с последующим добавлением дициклогексилкарбодиимида (10,83 г). Образовавшуюся суспензию перемешивают в течение 1 ч при температуре от -5 до 0 °С. К раствору N-(2-пиразинкарбонил)-L-фенилаланина (19,52 г; получен сочетанием предварительно приготовленного сукцинимидного сложного эфира пиразинкарбоновой кислоты с L-фенилаланином в водном диоксане) в 62 мл ДМФ добавляют N-метилморфолин (5,7 мл) при температуре 0 °С и образовавшийся раствор добавляют к суспензии. Доводят суспензию до значения рН 7 за счет добавления другой порции (5,7 мл) N-метилморфолина и перемешивают в течение ночи, медленно повышая температуру до 21 °С. После фильтрации осадок на фильтре промывают 2 раза МТБЭ и объединенные фильтраты разбавляют 950 мл эфира МТБЭ. Органические слои промывают 20%-ным водным раствором лимонной кислоты (3 раза по 150 мл), 20% водным раствором NaHCO 3 (3 раза по 150 мл) и 2 раза рассолом. Органические слои сушат над Na 2 SO 4 , фильтруют и концентрируют, получая 25,5 г (95,5 %) указанного в заголовке соединения в виде пены. Методом тонкослойной хроматографии показано, что этот материал содержит некоторые второстепенные примеси, в том числе около 2% циклогексилмочевины. Бороновый ангидрид N-(2-пиразинкарбонил)-L-фенилаланин-L-лейцина. Раствор N-(2-пиразинкарбонил)-L-фенилаланин-L-лейцин-бороната(1S,2S,3R,5S)пинандиола (25,2 г) в 207 мл метанола и 190 мл гексана охлаждают до 15 °С и по частям добавляют 109,4 мл 1н. HCl-кислоты, поддерживая температуру между 15 и 25 °С. Затем при интенсивном перемешивании добавляют 2-метилпропанбороновую кислоту (8,67 г) и продолжают перемешивание двухфазной смеси в течение ночи. После разделения этих двух фаз нижний слой экстрагируют 1 раз гексаном (75 мл). Затем нижний слой концентрируют в вакууме до появления помутнения с последующим добавлением 109,4 мл 2н. раствора NaOH и 100 мл диэтилового эфира. Две фазы разделяют, нижний слой экстрагируют эфиром (4 раза по 100 мл) и затем подкисляют до рН 6,0, добавляя 109 мл 1н. HCl-кислоты. После экстракции этилацетатом (100 мл) нижний слой подкисляют до рН 6,0 соляной кислотой (1н.) и экстрагируют еще раз этилацетатом (75 мл). Объединенные этилацетатные слои промывают полунасыщенным рассолом (2 раза по 25 мл) и рассолом (2 раза по 25 мл), сушат над Na 2 SO 4 , фильтруют и концентрируют, чтобы получить 15,3 г (81,8%) сырого боронового ангидрида N-(2-пиразинкарбонил)-L-фенилаланин-L-лейцина в виде пены. Этот сырой материал растворяют в 150 мл этилацетата и концентрируют в вакууме до суспензии с последующим добавлением 150 мл МТБЭ. Суспензию хранят при температуре между 2 и 8 °С в течение ночи, фильтруют, промывают два раза эфиром МТБЭ и сушат в высоком вакууме, получая 10,69 г (57,2%) боронового ангидрида N-(2-пиразинкарбонил)-L-фенилаланин-L-лейцина в виде белого твердого вещества. Пример 4. Определение соотношения диастереоизомеров трифторацетат-3-метилбутан-1-бороната(1R)-(1S,2S,3R,5S)пинандиол-1-аммония. Диастереоизомерную чистоту трифторацетат-3-метилбутан-1-бороната(1R)-(1S,2S,3R,5S)пинандиол-1-аммония (соединение 1) определяют методом нехиральной газовой хроматографии (ГХ). Параметры газовой хроматографии Стабильность раствора. Готовят исходный раствор соединения 1 путем взвешивания 150,13 мг соединения 1 в мерной колбе на 10 мл и доведения объема до 10 мл, добавляя растворитель А. Стабильность этого раствора определяют при температуре окружающей среды в течение 48 ч. Исходным раствором заполняют 6 отдельных ГХ ампул. Инжекцию проб в систему ГХ проводят из этих ампул, спустя 0, 12, 24, 48 и 72 ч (из каждой ампулы вводят две пробы). Определяют площади пиков (%) для соединения 1 и соединения 2. Изменения площади пиков не наблюдались, что указывает на стабильность этого раствора в течение 72 частиц при температуре окружающей среды. Специфичность. Растворяют пробу, содержащую приблизительно 150 мг соединения 1 и соединения 2, в растворителе А и вводят в хроматографическую систему ГХ. Пик, соответствующий соединению 1, хорошо разделяется с пиком соединения 2. Чистоту соединений контролируют методом ГХ-масс-спектрометрии: не наблюдаются другие компоненты, элюирующиеся вместе с соединением 1 или соединением 2. Предел обнаружения. Предел обнаружения (ПО) определяется как концентрация соединения 1, при которой отношение сигнал/шум составляет по меньшей мере 3:1. В проведенном ранее холостом измерении было показано, что отсутствуют другие мешающие пики. Отношение сигнал/шум рассчитывают по уравнению S/N = отношение сигнал/шум; Н(с) = высота сигнала для соединения 1 [мм]; Н(b) = высота сигнала фона [мм]. Вводят пробу стандартного опытного образца с концентрацией 0,05% и определяют отношение сигнал/шум, равное 4,3. Следовательно, предел обнаружения составляет 0,0075 мг/мл. Предел количественного анализа. Предел количественного анализа (ПКА) определяется как концентрация соединения 1, при которой отношение сигнал/шум составляет по меньшей мере 10:1. Отношение сигнал/шум рассчитывают, как описано выше. Вводят пробу стандартного опытного образца с концентрацией 0,1% и определяют отношение сигнал/шум, равное 10,1. Следовательно, предел количественного анализа составляет 0,015 мг/мл. Пример 5. Анализ чистоты боронового ангидрида N-(2-пиразинкарбонил)-L-фенилаланин-L-лейцина. Степень чистоты боронового ангидрида N-(2-пиразинкарбонил)-L-фенилаланин-L-лейцина (соединение 3) оценивают методом ЖХВР на обращенной фазе. Реагенты: Вода, чистая для ЖХВР. Ацетонитрил, чистый для ЖХВР. Муравьиная кислота, сорт «ч.д.а. », чистота ≥ 98%. Пероксид водорода (3%), «ч.д.а. » или эквивалент. Обычно при использовании системы ЖХВР время удерживания соединения 3 составляет от 10 до 14 мин, и время задержки, связанное с объемом колонки, - 1,3 мин. Соединения 4 и 5 элюируются совместно при большем времени удерживания с разрешением ≥ 2,0. Относительное удерживание соединения 3 в хроматограмме пробы по сравнению с временем удерживания в стандартной хроматограмме рассчитывают по следующему уравнению: где R r = относительное удерживание; t sam = время удерживания соединения 3 по его пику в хроматограмме пробы, мин; t std = время удерживания лекарственного соединения по пику в наиболее близкой стандартной хроматограмме, мин. Результаты анализа (%) рассчитывают для каждой пробы в соответствии со следующим уравнением: где A sam = площадь пика, соответствующего соединению 3 в препарате пробы; A std = средняя площадь пика, соответствующего соединению 3 в рабочем стандартном препарате; W std = масса стандартного препарата, мг; Р = оценка чистоты стандарта (десятичный формат); W sam = масса пробы, мг; М = содержание влаги в пробе, %; 100 = коэффициент перевода в проценты. Относительное удерживание и содержание примесей в каждой пробе рассчитывают в соответствии со следующими уравнениями: где R r = относительное удерживание; t i = время удерживания индивидуальной примеси; t ds = время удерживания по пику соединения 3. где I i = содержание индивидуальной примеси; A i = площадь пика, соответствующего индивидуальной примеси в препарате пробы; A std,1% = средняя площадь пика, соответствующего соединению 3 в 1%-ном стандартном препарате; W std = масса стандарта, мг; W sam = масса пробы, мг; Р = оценка чистоты стандарта (десятичный формат); DF = коэффициент разбавления, 1/100; RF i = относительный коэффициент отклика для индивидуальной примеси; 100 = коэффициент перевода в проценты. При анализе указанным методом найдено, что бороновый ангидрид N-(2-пиразинкарбонил)-L-фенилаланин-L-лейцина из примера 2 имеет суммарное содержание примесей меньше чем 1%. Хотя вышеизложенное изобретение описано до некоторой степени детально с целью большей ясности и понимания, приведенные конкретные варианты изобретения следует рассматривать как иллюстрирующие изобретение, а не как ограничивающие его. Прочитав описание, специалист в этой области может понять, что могут быть осуществлены различные изменения по форме и в деталях, в объеме данного изобретения и прилагаемой формулы изобретения. |