|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea000008409b*\id |
|
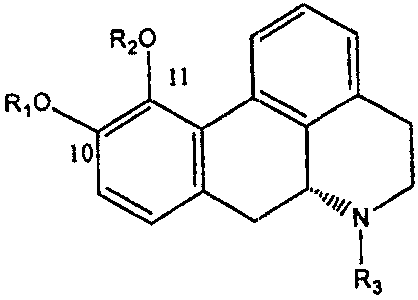
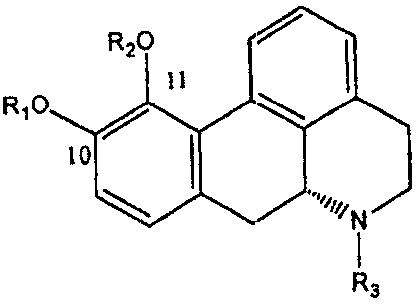
Термины запроса в документе Реферат 1. Фармацевтический состав для лечения заболеваний, выбираемых из группы, состоящей из болезни Паркинсона, синдрома усталых ног и эректильной дисфункции, включающий, как минимум, одно соединение, выбранное из группы, состоящей из A) апоморфина, 6aR-(-)-N-пропил-норапоморфина, их симметричных ди-(С 2 -С 5 )алканоиловых эфиров или дибензоиловых эфиров, в форме основания или фармацевтически приемлемых солей или сольватов, B) пролекарств апорфина, выбранных из группы, состоящей из соединений, имеющих общую формулу где один из R 1 и R 2 является водородом или ацетилом, а другой выбирают из группы, состоящей из (С 3 -С 20 )алканоила; гало(С 3 -С 20 )алканоила; (С 3 -С 20 )алкеноила; (С 4 -С 7 )циклоалканоила; (С 3 -С 6 )циклоалкил(С 2 -С 16 )алканоила; ароила, который является незамещенным или замещенным 1-3 заместителями, выбранными из группы, состоящей из галогена, циано-, трифторметансульфонилокси-, (C 1 -С 3 )алкила и (C 1 -С 3 )алкокси-, где последний может в свою очередь быть замещенным 1-3 атомами галогена; арил(С 2 -С 16 )алканоила, который является незамещенным или замещенным в ариловом компоненте 1-3 заместителями, выбираемыми из группы, состоящей из галогена, (C 1 -С 3 )алкила и (C 1 -С 3 )алкокси-, где последний может в свою очередь быть замещенным 1-3 атомами галогена; и гетероарилалканоила, имеющего 1-3 гетероатома, выбранных из О, S и N в гетероариловом компоненте и 2-10 атомов углерода в алканоиловом компоненте, и который является незамещенным или замещенным в гетероариловом компоненте 1-3 заместителями, выбранными из группы, состоящей из галогена, циано-, трифторметансульфонилокси-, (C 1 -С 3 )алкила и (C 1 -С 3 )алкокси-, где последний может в свою очередь быть замещенным 1-3 атомами галогена; и R 3 является метилом; и их фармацевтически приемлемых солей, в качестве активного ингредиента в фармацевтическом препарате, подходящем для перорального/интрадуоденального введения, причем указанный состав находится в форме прессованной таблетки/гранулы, включающей указанный активный ингредиент вместе с соответствующими наполнителями и адъювантами и имеющей энтеросолюбильное покрытие, растворяемой в тонкой кишке, например, двенадцатиперстной кишке. 2. Фармацевтический состав по п.1, имеющий дополнительный внешний слой, включающий указанный активный ингредиент вместе с соответствующими наполнителями и адъювантами. 3. Фармацевтический состав по п.1, включающий смесь указанных активных ингредиентов и соответствующих наполнителей и адъювантов, помещенных в капсулу, растворяющуюся в тонкой кишке, например, двенадцатиперстной кишке. 4. Фармацевтический состав по п.3, где указанная смесь представлена в форме гранул. 5. Фармацевтический состав по п.1 в форме гранул, имеющих энтеросолюбильное покрытие, помещенных в капсулу, растворяемую в желудке (gastrum) и высвобождающую гранулы, имеющие энтеросолюбильное покрытие, которые имеют оптимальный размер для прохода вместе с содержимым желудка в двенадцатиперстную кишку и дезинтегрируются там или дальше в тонкой кишке с контролируемым высвобождением активного ингредиента. 6. Фармацевтический состав по любому из пп.1-5, где указанный активный ингредиент имеет размер частиц в пределах от 0,1 до 20 мкм, предпочтительно между от 0,1 до 5 мкм. 7. Фармацевтический состав по п.1 в форме, подходящей для введения интрадуоденально при помощи интрадуоденального катетера через брюшную стенку пациента или при помощи назо-дуоденального катетера. 8. Фармацевтический состав по любому из пп.1-7, где активным ингредиентом является фармацевтически приемлемая соль апоморфина или 6aR-(-)-N-пропил-норапоморфина (NPA). 9. Фармацевтический состав по любому из пп.1-8, где активный ингредиент выбран из группы, состоящей из симметричных ди-(С 2 -С 5 )алканоиловых эфиров апоморфина и NPA и их фармацевтически приемлемых солей и дибензоилового эфира апоморфина и NPA и их фармацевтически приемлемых солей. 10. Фармацевтический состав по любому из пп.1-7, где активный ингредиент выбран из группы, состоящей из соединений, имеющих общую формулу где один из R 1 и R 2 является водородом или ацетилом, а другой выбирают из группы, состоящей из (С 3 -С 20 )алканоила; гало(С 3 -С 20 )алканоила; (С 3 -С 20 )алкеноила; (С 4 -С 7 )циклоалканоила; (С 3 -С 6 )циклоалкил(С 2 -С 16 )алканоила; ароила, который является незамещенным или замещенным 1-3 заместителями, выбранными из группы, состоящей из галогена, циано-, трифторметансульфонилокси-, (C 1 -С 3 )алкила и (C 1 -С 3 )алкокси-, где последний может в свою очередь быть замещенным 1-3 атомами галогена; арил(С 2 -С 16 )алканоила, который является незамещенным или замещенным в ариловом компоненте 1-3 заместителями, выбираемыми из группы, состоящей из галогена, (C 1 -С 3 )алкила и (C 1 -С 3 )алкокси-, где последний может в свою очередь быть замещенным 1-3 атомами галогена; и гетероарилалканоила, имеющего 1-3 гетероатома, выбранных из О, S и N в гетероариловом компоненте и 2-10 атомов углерода в алканоиловом компоненте, и который является незамещенным или замещенным в гетероариловом компоненте 1-3 заместителями, выбранными из группы, состоящей из галогена, циано-, трифторметансульфонилокси-, (C 1 -С 3 )алкила и (C 1 -С 3 )алкокси-, где последний может в свою очередь быть замещенным 1-3 атомами галогена; и R 3 является метилом; или их фармацевтически приемлемых солей. 11. Фармацевтический состав по п.10, где активный ингредиент выбран из группы, состоящей из моно-(С 3 -C 5 )алканоиловых эфиров апоморфина и их фармацевтически приемлемых солей. 12. Фармацевтический состав по п.10, где активный ингредиент выбран из группы, состоящей из асимметричных диалканоиловых эфиров апоморфина, где одной из алканоильных групп является ацетильная группа, а другой является (С 3 -С 6 )алканоильная группа, и их фармацевтически приемлемых солей. 13. Способ лечения заболеваний, выбираемых из группы, состоящей из болезни Паркинсона, синдрома усталых ног, мужской эректильной дисфункции и женской сексуальной дисфункции, включающий введение перорально/интрадуоденально пациенту, нуждающемуся в лечении, фармацевтического состава, заявляемого в любом из пп.1-11, в эффективном для улучшения количестве.
Полный текст патента (57) Реферат / Формула: A) апоморфина, 6aR-(-)-N-пропил-норапоморфина, их симметричных ди-(С 2 -С 5 )алканоиловых эфиров или дибензоиловых эфиров, в форме основания или фармацевтически приемлемых солей или сольватов, B) пролекарств апорфина, выбранных из группы, состоящей из соединений, имеющих общую формулу где один из R 1 и R 2 является водородом или ацетилом, а другой выбирают из группы, состоящей из (С 3 -С 20 )алканоила; гало(С 3 -С 20 )алканоила; (С 3 -С 20 )алкеноила; (С 4 -С 7 )циклоалканоила; (С 3 -С 6 )циклоалкил(С 2 -С 16 )алканоила; ароила, который является незамещенным или замещенным 1-3 заместителями, выбранными из группы, состоящей из галогена, циано-, трифторметансульфонилокси-, (C 1 -С 3 )алкила и (C 1 -С 3 )алкокси-, где последний может в свою очередь быть замещенным 1-3 атомами галогена; арил(С 2 -С 16 )алканоила, который является незамещенным или замещенным в ариловом компоненте 1-3 заместителями, выбираемыми из группы, состоящей из галогена, (C 1 -С 3 )алкила и (C 1 -С 3 )алкокси-, где последний может в свою очередь быть замещенным 1-3 атомами галогена; и гетероарилалканоила, имеющего 1-3 гетероатома, выбранных из О, S и N в гетероариловом компоненте и 2-10 атомов углерода в алканоиловом компоненте, и который является незамещенным или замещенным в гетероариловом компоненте 1-3 заместителями, выбранными из группы, состоящей из галогена, циано-, трифторметансульфонилокси-, (C 1 -С 3 )алкила и (C 1 -С 3 )алкокси-, где последний может в свою очередь быть замещенным 1-3 атомами галогена; и R 3 является метилом; и их фармацевтически приемлемых солей, в качестве активного ингредиента в фармацевтическом препарате, подходящем для перорального/интрадуоденального введения, причем указанный состав находится в форме прессованной таблетки/гранулы, включающей указанный активный ингредиент вместе с соответствующими наполнителями и адъювантами и имеющей энтеросолюбильное покрытие, растворяемой в тонкой кишке, например, двенадцатиперстной кишке. 2. Фармацевтический состав по п.1, имеющий дополнительный внешний слой, включающий указанный активный ингредиент вместе с соответствующими наполнителями и адъювантами. 3. Фармацевтический состав по п.1, включающий смесь указанных активных ингредиентов и соответствующих наполнителей и адъювантов, помещенных в капсулу, растворяющуюся в тонкой кишке, например, двенадцатиперстной кишке. 4. Фармацевтический состав по п.3, где указанная смесь представлена в форме гранул. 5. Фармацевтический состав по п.1 в форме гранул, имеющих энтеросолюбильное покрытие, помещенных в капсулу, растворяемую в желудке (gastrum) и высвобождающую гранулы, имеющие энтеросолюбильное покрытие, которые имеют оптимальный размер для прохода вместе с содержимым желудка в двенадцатиперстную кишку и дезинтегрируются там или дальше в тонкой кишке с контролируемым высвобождением активного ингредиента. 6. Фармацевтический состав по любому из пп.1-5, где указанный активный ингредиент имеет размер частиц в пределах от 0,1 до 20 мкм, предпочтительно между от 0,1 до 5 мкм. 7. Фармацевтический состав по п.1 в форме, подходящей для введения интрадуоденально при помощи интрадуоденального катетера через брюшную стенку пациента или при помощи назо-дуоденального катетера. 8. Фармацевтический состав по любому из пп.1-7, где активным ингредиентом является фармацевтически приемлемая соль апоморфина или 6aR-(-)-N-пропил-норапоморфина (NPA). 9. Фармацевтический состав по любому из пп.1-8, где активный ингредиент выбран из группы, состоящей из симметричных ди-(С 2 -С 5 )алканоиловых эфиров апоморфина и NPA и их фармацевтически приемлемых солей и дибензоилового эфира апоморфина и NPA и их фармацевтически приемлемых солей. 10. Фармацевтический состав по любому из пп.1-7, где активный ингредиент выбран из группы, состоящей из соединений, имеющих общую формулу где один из R 1 и R 2 является водородом или ацетилом, а другой выбирают из группы, состоящей из (С 3 -С 20 )алканоила; гало(С 3 -С 20 )алканоила; (С 3 -С 20 )алкеноила; (С 4 -С 7 )циклоалканоила; (С 3 -С 6 )циклоалкил(С 2 -С 16 )алканоила; ароила, который является незамещенным или замещенным 1-3 заместителями, выбранными из группы, состоящей из галогена, циано-, трифторметансульфонилокси-, (C 1 -С 3 )алкила и (C 1 -С 3 )алкокси-, где последний может в свою очередь быть замещенным 1-3 атомами галогена; арил(С 2 -С 16 )алканоила, который является незамещенным или замещенным в ариловом компоненте 1-3 заместителями, выбираемыми из группы, состоящей из галогена, (C 1 -С 3 )алкила и (C 1 -С 3 )алкокси-, где последний может в свою очередь быть замещенным 1-3 атомами галогена; и гетероарилалканоила, имеющего 1-3 гетероатома, выбранных из О, S и N в гетероариловом компоненте и 2-10 атомов углерода в алканоиловом компоненте, и который является незамещенным или замещенным в гетероариловом компоненте 1-3 заместителями, выбранными из группы, состоящей из галогена, циано-, трифторметансульфонилокси-, (C 1 -С 3 )алкила и (C 1 -С 3 )алкокси-, где последний может в свою очередь быть замещенным 1-3 атомами галогена; и R 3 является метилом; или их фармацевтически приемлемых солей. 11. Фармацевтический состав по п.10, где активный ингредиент выбран из группы, состоящей из моно-(С 3 -C 5 )алканоиловых эфиров апоморфина и их фармацевтически приемлемых солей. 12. Фармацевтический состав по п.10, где активный ингредиент выбран из группы, состоящей из асимметричных диалканоиловых эфиров апоморфина, где одной из алканоильных групп является ацетильная группа, а другой является (С 3 -С 6 )алканоильная группа, и их фармацевтически приемлемых солей. 13. Способ лечения заболеваний, выбираемых из группы, состоящей из болезни Паркинсона, синдрома усталых ног, мужской эректильной дисфункции и женской сексуальной дисфункции, включающий введение перорально/интрадуоденально пациенту, нуждающемуся в лечении, фармацевтического состава, заявляемого в любом из пп.1-11, в эффективном для улучшения количестве.
008409 Область техники Настоящее изобретение относится к эффективному введению композиции апоморфина, 6aR-(-)-N-пропил-норапоморфина и их производных и пролекарств для лечения, например, болезни Паркинсона (PD), синдрома усталых ног (RLS), психогенной мужской эректильной дисфункции (MED) и женской сексуальной дисфункции или подобных заболеваний. Предпосылки изобретения Апоморфин применяют для лечения больных паркинсонизмом. Смотри, например, Hagell P. and Odin P., J. Neurosci Nurs Feb, 33(l):21-34, 37-8 (2001); Deffond et al., J.Neurology, Neurosurgery, and Psychiatry 56: 101-103 (1993) and Durif et al., Clinical Neuropharmacology 16(2): 157-166 (1993). Кроме того, апоморфин рассматривают как средство для лечения алкоголизма, шизофрении, деформирующей мышечной дистонии, галлюцинаций, мигрени, икоты, хореи Гентингтона, поздней дискинезии и, в последнее время, мужской эректильной дисфункции. Болезнь Паркинсона является прогрессирующим, нейродегенеративным заболеванием, вызываемым потерей клеточных тел допаминергических (DA-эргических) нейронов в черной субстанции и дегенерацией нервных окончаний в полосатом теле (стриатуме), что приводит к низкому уровню DA в черной субстанции и полосатом теле. Болезнь Паркинсона характеризуется хронической, прогрессирующей моторной дисфункцией, и главными ее симптомами являются тремор в состоянии покоя, мышечная ригидность и снижение частоты произвольных движений (гипокинезия) с затруднениями остановки, начала движения и поворотов при ходьбе. Постоянный тремор накладывается на гипертонус мышечных групп противоположной стороны, и начало движений становится все более затрудненным и медленным. На поздних стадиях, движения пациента становятся практически "замороженными" и пациент становится неспособен обслуживать сам себя. В исследованиях было показано, что симптомы болезни Паркинсона проявляются тогда, когда содержание DA в полосатом теле снижено до 20-40% от нормы. Поскольку болезнь Паркинсона ассоциирована с потерей DA в полосатом теле, обычно данное заболевание лечат с помощью лекарственных препаратов, которые замещают DA, из них наиболее часто используемым является леводопа. Леводопа преобразуется допа-декарбоксилазой в DA в головном мозге и полученный DA проявляет терапевтический эффект. Леводопа необходимо вводить в больших и частых дозах. Кроме того, продукция DA в периферических тканях способствует увеличению частоты нежелательных побочных эффектов. Следовательно, леводопа обычно вводят в комбинации с другими лекарственными препаратами для увеличения эффектов леводопа в головном мозге и минимизации его периферических эффектов. В частности, леводопа обычно применяют в комбинации с периферическими ингибиторами допа-декарбоксилазы, которые не могут проходить через гематоэнцефалический барьер, такими как карбидо-па, который ингибирует расщепление леводопа до DA вне головного мозга, таким образом, уменьшая периферические нежелательные эффекты. Ингибитор также обеспечивает тот факт, что относительно большее количество пероральной дозы леводопа достигает головного мозга и вследствие этого обеспечивает снижение дозы леводопа, что также уменьшает периферические побочные эффекты. Кроме того, периферический антагонист DA, который не проникает через гематоэнцефалический барьер, такой как домперидон, также можно вводить для уменьшения таких побочных эффектов леводопа, как тошнота и рвота. Помимо побочных эффектов, упомянутых выше, дополнительные нежелательные эффекты ассоциированы с длительным применением леводопа. В частности, у многих пациентов развиваются непроизвольные хорееформные движения, которые являются результатом чрезмерной активации DA-рецепторов. Данные движения обычно затрагивают лицо и конечности и могут становиться очень тяжелыми. Подобные движения исчезают, если дозу леводопа уменьшают, что, однако, вызывает возвращение ригидности. Более того, грань между полезными и нежелательными эффектами постепенно становится более узкой по мере увеличения периода лечения с применением леводопа. Традиционным способом борьбы с таким эффектом является увеличение частоты введения леводопа, в то время как суммарная доза остается стабильной. Данный подход уменьшает разрушающее действие конечной дозы и сокращает вероятность развития у пациента дискинезий, которые появляются при высших пиковых дозах. Следующим осложнением длительного лечения с применением леводопа является развитие частых колебаний в клиническом состоянии, когда состояние пациента внезапно меняется от мобильности к неподвижности на периоды, колеблющиеся от нескольких минут до нескольких часов. Данный феномен известен как "эффект включения-выключения", состояние "включения" является предпочтительным, во время которого может быть достигнута практически нормальная моторная функция, а состояние "выключения" характеризуется дистоническими позами во время периода сниженной мобильности. Действительно, данный эффект может способствовать такой неожиданной потере мобильности, что пациент может внезапно остановиться при ходьбе или не сможет подняться со стула, на котором он нормально сидел некоторое время назад. Данный эффект обычно не подвергается влиянию манипуляций с дозой леводопа и может потребовать лечения с помощью других лекарственных препаратов. В добавление к вышеуказанным длительным побочным эффектам лечения с применением леводопа, было обнаружено, что эффективность леводопа постепенно снижается со временем, до тех пор, пока он не становится не - 1 - 008409 эффективным. Также наблюдали повышенную частоту развития злокачественной меланомы у пациентов, подвергавшихся лечению с применением леводопа и предполагают, что лечение с применением леводопа может быть связано с развитием злокачественной меланомы. Соответственно, применение леводопа в лечении болезни Паркинсона является далеким от идеала. Альтернативным подходом к лечению болезни Паркинсона является применение лекарственных препаратов, которые имитируют действие DA. Такие лекарственные препараты вместе известны как DA-агонисты, так как они непосредственно стимулируют DA-рецепторы в рамках DA-дефицитного пути связи черной субстанции с полосатым телом. В отличие от леводопа, DA-агонисты не нуждаются в преобразовании в головном мозге в активные соединения. Также, DA-агонисты являются эффективными у пациентов с поздними стадиями болезни Паркинсона, когда леводопа становится неэффективным, так как они действуют непосредственно на DA-рецепторы и вследствие этого не подвержены влиянию отсутствия DA-продуцирующих нервных клеток у таких пациентов. Однако действие таких DA-агонистов на DA-рецепторы также вызывает нежелательные DA-эргические эффекты, такие как тошнота, рвота и экстрапирамидные эффекты, которые могут ослаблять организм, и некоторые DA-агонисты, такие как апоморфин, ассоциированы с другими нежелательными побочными эффектами, особенно при применении высоких доз, такими как седация, угнетение дыхания, гипотония, брадикардия, потливость и зевота. На тяжесть и характер таких побочных эффектов можно воздействовать путем способа введения лекарственного препарата. Например, в исследованиях апоморфина изучали множество путей введения данного лекарственного препарата. Однако пероральное введение таблеток апоморфина требует введения более высоких доз для достижения необходимого терапевтического эффекта. Также, длительные исследования таких пероральных форм прекращали через 7-10 дней из-за невыясненного повышения в крови азота мочевины. Введение таблеток апоморфина сублингвально вызывало развитие тяжелого стоматита, при пролонгированном применении, у половины пациентов, с развитием изъязвления слизистой оболочки щек. Интраназальное введение вызывало преходящую заложенность носа, ощущение жжения и припухлость носа и губ, и, у некоторых пациентов, участвовавших в исследовании, было по необходимости прекращено из-за предположительного химического воспаления слизистой оболочки носа (Zaleska, В. et al., Neurol. Neurochir. Pol. 33:1297-1303, 1999). Следовательно, до настоящего момента, единственным удовлетворительным путем введения апо-морфина для лечения болезни Паркинсона, который позволяет избежать высокого метаболизма первого пассажа, как было обнаружено, является подкожное введение и, следовательно, единственной коммерчески доступной препаративной формой апоморфина является жидкая форма для подкожных инъекций и инфузий. Даже при этих условиях подкожное введение не позволяет избежать побочных эффектов обычных DA-агонистов, таких как тошнота и рвота, а подкожное введение путем инъекции или инфузии не легко осуществлять, особенно у пациентов, у которых моторная функция уже нарушена и, следовательно, требует обучения пациентов и тех, кто за ними ухаживает. Также, место инъекции необходимо менять каждые 12 часов для минимизации риска изменения цвета кожи и образования узлов. В свете данных проблем, не является неожиданным, что применение DA-агонистов, таких как апоморфин, в лечении болезни Паркинсона значительно ограничено лечением периодов "выключения", вызываемых терапией с использованием леводопа, несмотря на очевидные клинические выгоды таких лекарственных препаратов по сравнению с леводопа. Из вышеизложенного становится очевидным, что особенно желаемым с клинической точки зрения является поиск пути введения DA-агонистов, таких как апоморфин, 6aR-(-)-N-пропил-норапоморфин и их производные и пролекарства, который будет эффективным и легким для применения пациентами. Синдром усталых ног (RLS; см. также Glasauer FE, Spinal Cord 2001 Mar; 39(3):125-33) является четким симптомокомплексом и часто ассоциирован с нарушениями сна и признанным семейным анамнезом. Он существует как идиопатический RLS или в связи с множеством медицинских, неврологических или сосудистых расстройств. Неврологический осмотр и обычное обследование при идиопатическом RLS соответствуют норме. Полисомнография подтверждает диагноз RLS путем документирования связанных нарушений сна и периодических движений конечностей во время сна (PLMS). Существуют убедительные доказательства того, что RLS представляет собой дисфункцию центральной нервной системы (ЦНС), что предполагает широкое вовлечение нисходящих допаминергических проводящих путей, возможно, происходящих из промежуточного мозга или верхней части ствола мозга. Данный факт подтверждает успешный результат лечения RLS с помощью DA-агентов, седативных препаратов и нейротранс-миттеров. Однако RLS также может сочетаться со спинальными нарушениями и повреждениями спинного мозга, подразумевая существование спинального генератора. Частота случаев RLS при беременности хорошо известна, и его ассоциация с сосудистыми расстройствами подтверждает другой механизм развития у некоторых пациентов. Первичное лечение RLS является в большей степени симптоматическим и вполне эффективным с помощью DA-агентов, DA-агонистов, опиоидов и других лекарственных препаратов, действующих на различные нейротрансмиттеры. Лечение RLS, ассоциированного с различными заболеваниями, направлено на коррекцию лежащих в его основе патологических или дефицитных состояний. Антидепрессантные лекарственные препараты часто усугубляют или ухудшают состояние RLS. Сообщают, что ночная подкожная инфузия апоморфина оказывает положительный эффект на качество - 2 - 008409 сна как при болезни Паркинсона, так и при синдроме усталых ног (RLS) (см. Reuter I., Ellis СМ., Ray Chaudhuri K., Acta Neurol Scand 1999 Sep; 100 (3): 163-7). В своем исследовании Reuter et al. предполагают, что инфузия апоморфина в течение ночи может быть эффективной для преодоления стойкой неспособности к ночному сну у определенных пациентов с болезнью Паркинсона и синдромом усталых ног. Импотенцию или мужскую эректильную дисфункцию (ED) определяют, как неспособность достичь и поддерживать эрекцию, достаточную для полового сношения. Импотенция в любом представленном случае может развиваться в результате психологических расстройств (психогенная), общих физиологических нарушений (органическая), неврологических нарушений (нейрогенная), дефицита гормонов (эндокринная) или комбинации перечисленного. Однако данные описания не являются точными. В настоящее время не существует стандартизованного метода диагностики или лечения. Как применяется в данном описании, психогенную импотенцию определяют как функциональную импотенцию без очевидной преобладающей органической основы. Ее можно характеризовать способностью иметь эрекцию в ответ на некоторые стимулы (например, мастурбация, спонтанная ночная, спонтанная в ранние утренние часы, эротическое видео и др.), но не другие (например, внимание партнера или супруга). Однако, специфические механизмы, путем которых действует апоморфин, вызывая эректильный ответ у пациентов-людей, к настоящему моменту полностью неясны. Сублингвальная форма апоморфина (Uprima(r)) в настоящее время имеется в продаже в некоторых европейских странах для лечения мужской эректильной дисфункции. Было показано, что апоморфин обладает плохой биодоступностью при пероральном применении (см., например, Baldessarini et al., in Gessa et al., eds., Apomorphine and other Dopaminomimetics, Basic Pharmacology, Vol.1, Raven Press, N.Y. (1981), pp. 219-228). Следовательно, продолжается поиск эффективного лечения апоморфином перорально PD, RLS и психогенной импотенции у пациентов-мужчин, также как и методов диагностики для выявления таких пациентов. Краткое описание изобретения В данном изобретении обеспечивается фармацевтическая композиция для введения апоморфина, 6aR-(-)-N-пропил-норапоморфина и их производных и пролекарств, при помощи которой удастся преодолеть низкую биодоступность при пероральном введении апоморфина, 6aR-(-)-N-пропил-норапоморфина и их производных и пролекарств. Данное изобретение основано на неожиданном обнаружении в экспериментах на животных того, что апоморфин, вводимый в двенадцатиперстную кишку, является фармацевтически более сильным по сравнению с апоморфином, вводимым традиционным пероральным путем, оканчивающимся в желудке. То же самое показано для NPA. На основании этого данное изобретение обеспечивает фармацевтическую композицию, содержащую апоморфин, 6aR-(-)-N-пропил-норапоморфин и их производные и про-лекарства в форме их основания или фармацевтически приемлемой соли или сольвата в качестве активного ингредиента в фармацевтической композиции для перорального/интрадуоденального введения как непосредственно, так и проведением через гастральный отдел (желудок = gastrum) неизмененной, что обеспечивается покрытием оболочкой для растворения в кишечнике и быстрым растворением и абсорбцией в двенадцатиперстной/тонкой кишке, или в композиции с контролируемым высвобождением активного ингредиента (например, путем инкапсулирования в пластиковой оболочке, которая может быть биоразлагаемой). В некоторых сообщениях было показано, что интрадуоденальное введение водных растворов лекарственных препаратов имеет несколько преимуществ по сравнению с пероральным введением (в желудок) как таблеток, так суспензий и растворов (например, Watari et al., J. Pharmacokinet. Biopharm, Oct. 1983 11 (5), p. 529-545). В особенности, колебания концентрации лекарственного препарата в плазме крови были существенно ниже при применении интрадуоденального пути введения, в основном за счет избежания эффекта колебаний во времени опустошения желудка. Более того, соединение апоморфина является чрезмерно чувствительным к окислению и разлагается в растворах при контакте с атмосферным воздухом. Посредством настоящего изобретения недостатки, упомянутые выше, в значительной мере устранены. Подробное описание изобретения Как указано выше, настоящее изобретение обеспечивает фармацевтическую композицию для лечения болезни Паркинсона, синдрома усталых ног, мужской эректильной дисфункции, которая включает по меньшей мере одно соединение, выбранное из группы, состоящей из апоморфина, 6aR-(-)-N-пропил-норапоморфина, их производных и пролекарств в форме их основания, фармацевтически приемлемой соли или сольвата в качестве активного ингредиента в фармацевтическом составе, подходящем для пе-рорального/интрадуоденального введения. Согласно предпочтительному варианту воплощения изобретения, фармацевтический состав по данному изобретению представлен в форме прессованных таблеток или гранул для перорального введения, включающих указанный активный ингредиент вместе с соответствующими наполнителями и адъюван-тами, и обеспеченных энтеросолюбильным покрытием, растворяющимся в тонкой кишке (двенадцатиперстная, тощая и/или подвздошная кишка), например, в двенадцатиперстной кишке. Апоморфин является агонистом рецепторов допамина D1 и D2, который имеет установленное при - 3 - 008409 менение, как антипаркинсонический лекарственный препарат, применяемый при введении подкожно в дозе около 5 мг. Для целей настоящего изобретения апоморфин вводили перорально в количестве, достаточном для лечения PD, RLS и/или ED у людей. Доза, необходимая для лечения таких различных состояний, могла различаться в зависимости от состояния и с учетом индивидуальных особенностей каждого пациента. Для апоморфина, 6aR-(-)-N-пропил-норапоморфина, их производных и пролекарств характерна предпочтительная абсорбция в ограниченном сегменте желудочно-кишечного тракта человека, а именно, в тонком кишечнике (например, в двенадцатиперстной кишке). Настоящее изобретение обеспечивает дозированную форму апоморфина, 6aR-(-)-N-пропил-норапоморфина, их производных и пролекарств, в которой используется энтеросолюбильное покрытие, и которая представляет собой быстро разлагающуюся/растворяющуюся таблетку, состоящую из апомор-фина, 6aR-(-)-N-пропил-норапоморфина, их производных и пролекарств. Такая дозированная форма обеспечивает удобный способ дозирования пациенту один или два раза в день в сочетании с обычными дозированными формами апоморфина, 6aR-(-)-N-пропил-норапоморфина, их производных и проле-карств. Состав по настоящему изобретению может содержать другой дополнительный агент, хорошо известный специалисту в данной области техники, в связи с фармацевтической композицией, содержащей апоморфин. В качестве примера таких агентов могут быть упомянуты противорвотные средства (например, домперидон), прокинетические агенты (например, домперидон), стабилизаторы, антиоксиданты, консервирующие агенты и агенты регулирования рН. Наполнители и адъюванты, применяемые в фармацевтических составах согласно настоящему изобретению в форме прессованных таблеток или гранул, могут включать (1) наполнители для добавления объема и улучшения прессуемости, например лактозу, крахмал, сахарные спирты, производные целлюлозы, сульфат или фосфат кальция, (2) дезинтегрирующие агенты, для дезинтеграции дозированной формы, например крахмал, гликолат натриевого крахмала, производные целлюлозы, альгинаты, камеди, шипучие смеси, (3) связующие вещества для образования гранул или улучшения прессуемости, например камеди, сахара, крахмал, производные целлюлозы, альгинаты, поливинилпирролидон, (4) смазывающие агенты для уменьшения трения, например стеариновая кислота, стеараты металлов, воски, имеющие высокую температуру плавления, тальк, (5) агенты для улучшения растворимости, например поверхностно-активные вещества, щелочные буферы и (6) глиданты для улучшения текучести, например крахмал, тальк, силикат. При получении таблеток/гранул основу таблеток/гранул вначале получают путем прессования смеси активного(ых) ингредиента(ов), добавок, адъювантов или возможных других дополнительных веществ. Затем наносят слой энтеросолюбильного покрытия на указанную основу таблетки/гранулы путем обычных технологий нанесения покрытия, таких как, например, покрытие погружением или покрытие в псевдоожиженном слое с применением растворов пленкообразующих полимеров в воде и/или подходящих органических растворителях или с использованием суспензий таких полимеров. Примерами таких пленкообразующих полимеров являются шеллак, фталат ацетата целлюлозы, гидроксипропилметилцел-люлоза, фталат поливинилацетата, карбоксиметилэтилцеллюлоза и сополимеры, синтезированные из метакриловой кислоты и метилового эфира метакриловой кислоты, такие как продукты, продаваемые под торговым наименованием Eudragit(r)S, Rohm Pharma, Darmstadt, Germany. Растворители, применяемые в этой связи, включают, например, метанол, этанол, изопропанол и ме-тиленхлорид. Растворы или суспензии пленкообразующих агентов могут необязательно содержать фармацевтически приемлемые пластификаторы, такие как, например, полиэтиленгликоль, касторовое масло, глицерин, пропиленгликоль и эфиры фталевой кислоты. Диспергаторы, такие как тальк, также могут быть включены в слой энтеросолюбильного покрытия. Согласно разновидности данного варианта воплощения изобретения, прессованные таблетки/гранулы, снабженные энтеросолюбильным покрытием, растворяющимся в двенадцатиперстной/тонкой кишке, имеют дополнительный внешний слой, включающий указанный активный ингредиент вместе с соответствующими наполнителями и адъювантами для обеспечения немедленного высвобождения дозы в комбинации с замедленным высвобождением дозы. В соответствии с другим вариантом воплощения настоящего изобретения, фармацевтический состав включает смесь указанного активного ингредиента и соответствующих наполнителей и адъювантов, помещенную в капсулу, растворимую в двенадцатиперстной/тонкой кишке. Предпочтительно, указанная смесь представлена в форме раствора активного ингредиента в растворителе, таком как вода или фармацевтически приемлемый органический растворитель или масло, вместе с, например, противорвотным агентом, стабилизатором, антиоксидантом, консервирующим агентом и/или агентом регулирования рН. Капсула сама по себе должна состоять из материала, который является устойчивым к действию желудочного сока, но быстро растворим при приближении и попадании в двенадцатиперстную кишку. В соответствии со следующим вариантом воплощения настоящего изобретения фармацевтический состав представлен в форме гранул с энтеросолюбильным покрытием, заключенных в капсулу, раство - 4 - 008409 римую в желудке (gastrum), с высвобождением гранул с энтеросолюбильным покрытием, которые имеют оптимальный размер для прохождения вместе с желудочным содержимым в двенадцатиперстную кишку и дезинтеграции там или ниже в тонкой кишке, при контролируемом высвобождении активного ингредиента. Активный ингредиент при применении в фармацевтическом составе, в котором он представлен не в растворе, должен быть представлен в тонкоизмельченном виде, например, с размером частиц в пределах от 0,1 до 20 мкм, предпочтительно от 0,1 до 5 мкм. Такие частицы с энтеросолюбильным покрытием могут предпочтительно быть заключены в капсулу, которая быстро разлагается в желудочном соке. Высвободившиеся частицы, которые являются устойчивыми к желудочному соку из-за их энтеросолюбильного покрытия, имеют оптимальный размер для протекания в двенадцатиперстную кишку вместе с желудочным содержимым при освобождении желудка. В двенадцатиперстной кишке данные частицы дезинтегрируются с контролируемой скоростью, которая зависит от состава, выбранного для покрытия оболочкой таких частиц. Согласно другому варианту воплощения настоящего изобретения фармацевтический состав представлен в форме подходящей для интрадуоденального введения с помощью интрадуоденального катетера через брюшную стенку пациента или с помощью назо-дуоденального катетера. В данном варианте воплощения изобретения активный ингредиент или ингредиенты предпочтительно растворены в носителе, таком как вода или фармацевтически приемлемый органический растворитель или масло. Однако также предполагаются возможные суспензии активных ингредиента(ов) в носителе. В свете того факта, что апоморфин и его производные являются чувствительными к окислению, составы по настоящему изобретению необходимо получать и хранить при исключении доступа кислорода, включая предотвращение контакта с атмосферным воздухом. Фармацевтические составы по данному изобретению содержат в качестве активного ингредиента или ингредиентов, как минимум, один представитель следующих групп веществ: A) Апоморфин, 6aR-(-)-N-пропил-норапоморфин (NPA), симметричные ди-(С2-С5)алканоиловые эфиры апорфинов и NPA и их фармацевтически приемлемые соли, и дибензоиловые эфиры апоморфина и NPA и их фармацевтически приемлемые соли. B) Пролекарства апорфина, описанные в международной патентной заявке № PCT/SE01/01658(WO02/014279 от 21.02.2002) (приоритет заявлен на основании патентной заявки Швеции № 0002934-8, поданной 17 августа, 2000) и имеющие общую формулу где один из R1 и R2 является водородом или ацетилом и другой выбирают из группы, состоящей из (С3-С20)алканоил-; гало(С3-С20)алканоил-; (С3-С20)алкеноил-; (С4-С7)циклоалканоил-; (С3-С6)циклоалкил(С2-С16)алканоил-; ароила, который является незамещенными или замещенными 1-3 заместителями, выбираемыми из группы, состоящей из галогена, цианотрифторметансульфонилокси-, (Q-С^алкила и (C1-С3)алкокси-, из которых последний может, в свою очередь, быть замещенным 1-3 атомами галогена; арил(С2-С16)алканоила, который является незамещенным или замещенным в ариловом компоненте 1-3 заместителями, выбираемыми из группы, состоящей из галогена, (Q-С^алкила и (Q-С^алкокси-, из которых последний может, в свою очередь, быть замещенным 1-3 атомами галогена; и гетероарилалканои-ла, имеющего один-три гетероатома, выбираемые из О, S и N, в гетероариловом компоненте и 2-10 атомов углерода в алканоиловом компоненте, и который является незамещенным или замещенным в гете-роариловом компоненте 1-3 заместителями, выбираемыми из группы, состоящей из галогена, циано-, трифторметансульфонилокси-, (Q-С^алкила и (С1-С3)алкокси-, из которых последний, в свою очередь, является замещенным 1-3 атомами галогена; и R3 является метилом; и их физиологически приемлемые соли. Описаны симметричные ди-(С2-С5)алканоиловые эфиры и дибензоиловый эфир апорфинов и представлены данные о биодоступности таких эфиров, однако, итоговый результат оказался разочаровывающим. Так, например, дипивалоиловый эфир пролекарства оказался менее активным, чем исходное соединение апоморфин сам по себе. Алканоильные группы симметричных ди-(С2-С5)алканоиловых эфиров апоморфина могут быть представлены линейными или разветвленными цепями. Такие симметричные диалканоиловые эфиры включают, например, диацетиловые, дипропиониловые, дибутириловые и дипивалоиловые эфиры апо-морфина. - 5 - 008409 Одна предпочтительная группа пролекарств апорфина для применения в настоящем изобретении, описанная в WO 02/014279, включает моно-(С2-С5)алканоиловые эфиры апоморфина, в которых алкано-ильная группа может быть представлена линейной или разветвленной цепью. Примеры таких эфиров включают моноацетил-, монобутирил- и монопивалоил-апоморфин. Другая предпочтительная группа пролекарств апорфина для применения в настоящем изобретении, описанная в WO 02/014279, включает асимметричные диалканоиловые эфиры апоморфина, где одна из алканоильных групп является ацетильной, а другие являются (С3-С5)алканоилом, цепь которого может быть линейной или разветвленной. Примеры таких эфиров включают пропионил-ацетилапоморфин, бу-тирил-ацетилапоморфин, изобутирил-ацетилапоморфин, изопропаноил-ацетилапоморфин и пивалоил-ацетилапоморфин. Согласно следующему аспекту настоящего изобретения, обеспечивается способ лечения заболеваний, выбираемых из группы, состоящей из болезни Паркинсона, синдрома усталых ног, мужской эрек-тильной дисфункции и женской сексуальной дисфункции, данный способ включает введение перораль-но/интрадуоденально пациенту, нуждающемуся в таком лечении, фармацевтического состава по настоящему изобретению, как определено выше, в эффективном для улучшения количестве. Данное изобретение далее будет описано посредством некоторого количества примеров, которые не должны истолковываться как ограничивающие объем настоящего изобретения. Пример 1. Получение таблеток, содержащих гидрохлорид апоморфина. Основу таблеток получают путем смешивания гидрохлорида апоморфина с микрокристаллической целлюлозой, гликолатом натриевого крахмала, кукурузным крахмалом, тальком и стеаратом магния в подходящих пропорциях согласно приемлемым фармацевтическим способам получения. Полученную смесь просеивают и прессуют выпуклую основу таблеток/гранул путем прямого прессования с применением подходящего таблетировочного пресса, формируя таблетки/гранулы. Прессованную основу таблеток/гранул, полученную таким образом, покрывают энтеросолюбиль-ным покрытием посредством суспензии, полученной из Eudragit(r)S, 12,5% суспензии в изопропаноле; полиэтиленгликоля 6000, 33% водный раствор; талька и изопропанола/ацетона 1:1. Основу таблеток/гранул покрывают энтеросолюбильным покрытием путем распыления вышеуказанной суспензии Eudragit-S на их поверхность при вращении таблеток/гранул в обычном резервуаре для покрытия оболочкой для получения ровной, непрерывной поверхности распределения оболочки. Пример 2. Получение таблеток, содержащих производные апоморфина. Микрокристаллическую целлюлозу (МКЦ) (РН 112, Eur. Ph; from OPG Groothandel B.V., Utrecht, The Netherlands) смешивали с гидрохлоридом апоморфина (АРО), монопивалоил-апоморфином (МРА) (полученным в соответствии с WO 02/14279A1) (UVPA) (из Sigma) соответственно. В смесях соотношение МКЦ/апоморфин составило 5/1 мас./мас. (т.е. 83% МКЦ/17% производных апоморфина). Смеси гомогенизировали с помощью встряхивания и перемешивания. Прессование смесей в округлые двояковыпуклые таблетки (12 таблеток) с диаметром 4 мм и весом 25-30 мг проводили с применением ESH гидравлического пресса (Hydro Mooi, Appingedam, The Netherlands). Для всех таблеток применяли давление прессования приблизительно 100 МПа. После прессования таблетки покрывали слоями энтеросолюбильного покрытия. Данное покрытие состояло из Eudragit(r)L30 (от Rohm, Darmstadt, Germany), который представлен 30% мас./об. суспензии сополимера метакриловой кислоты/метилметакрилата. Данное вещество является нерастворимым при кислом рН, но легко растворимым при нейтральном или основном рН. 5 г данной суспензии смешивали с водой (4 г), тальком (0,75 г), Citroflex(r) (триэтилцитрат от Fluka, Buchs, Switzerland) (0,15 г) и противо-пенным раствором кремния (от Boom, Meppel, The Netherlands) (0,05 г). Данную смесь взбалтывали приблизительно в течение 1 ч перед применением. Процесс нанесения покрытия затем проводили, как описано ниже. Таблетки помещали в плоское круглое сито с диаметром 45 мм. Таблетки предварительно нагревали до температуры приблизительно 40-45°С с применением сушильного аппарата для волос. Затем каплю (30-50 мкл) жидкости для покрытия добавляли в сито и таблетки перемешивали с помощью стеклянного стержня в потоке горячего воздуха, пока испарялась вода. Данный процесс повторяли 8 раз, что приводило к покрытию таблеток однородным слоем энтеросолюбильного покрытия. Затем таблетки оставляли на ночь для высыхания. Таблица 1 Смеси, применяемые для таблеток и средняя масса таблеток до и после нанесения покрытия. Производные апоморфина (иг) МКЦ (кг) Масса таблетки До нанесения покрытия (иг) После нанесения покрытия (кг) АРО (67,9) 335 29, 6 37,4 NPA (66,5) 336 29, 9 38,1 МРА (65,2) 336 29,5 39,3 Пример 3. Получение таблеток, содержащих гидрохлорид апоморфина (АРО) (12%) в биоразлагае- - 6 - 008409 мом полимере PLG и монопивалоил-М-пропил-норапорфин (MPNPA). 96 мг микрокристаллической целлюлозы (МКЦ) (РН 112, Eur.Ph.) (от OPG Groothandel B.V., Utrecht, The Netherlands) смешивали с 4,2 мг MPNPA. Смесь гомогенизировали путем встряхивания и перемешивания. Прессование смеси в три округлые двояковыпуклые таблетки с диаметром 4 мм и массой 25-30 мг проводили с помощью применения гидравлического пресса ESH (Hydro Mooi, Appingedam, The Netherlands). Подобным образом получали из АРО и полимера PLG таблетки с примерной массой 40 мг. Для всех таблеток применяли давление прессования приблизительно 100 МПа. Массу таблеток определяли с помощью аналитических весов (Mettler-Toledo). После прессования таблетки покрывали слоями энтеросолюбильного покрытия. Данное покрытие состояло из Eudragit(r) L30 (от Rohm, Darmstadt, Germany), который является 30% мас./об. суспензией сополимера метакриловой кислоты/метилметакрилата. Данное вещество является нерастворимым при кислом рН, но легко растворимым при нейтральном и основном рН. 5 г данной суспензии смешивали с водой (4 г), тальком (0,75 г), Citroflex(r) (триэтилцитрат от Fluka, Buchs, Switzerland) (0,15 г) и противо-пенным раствором кремния (от Boom, Meppel, The Netherlands) (0,05 г). Эту смесь перемешивали приблизительно в течение одного часа перед применением. Процесс нанесения покрытия затем проводили, как описано ниже. Таблетки помещали в плоское круглое сито с диаметром 45 мм. Таблетки предварительно нагревали до температуры приблизительно 40-45°С с применением сушильного аппарата для волос. Затем каплю (30-50 мкл) жидкости для покрытия добавляли в сито и таблетки перемешивали с помощью стеклянного стержня в потоке горячего воздуха, пока испарилась вода. Данный процесс повторяли 8 раз, что приводило к покрытию таблеток однородным слоем энтеросолюбильного покрытия. Затем данные таблетки оставляли на ночь для высыхания. Массу таблеток определяли с помощью аналитических весов (Mettler-Toledo). Таблица 2 Масса таблеток до и после нанесения покрытия, соответственно. Тип таблетки Масса до нанесения покрытия иг) Масса после нанесения покрытия (иг) MPNPA 28,32+1,16 34,39; 35,92 APO+PLG 36, 97±0,88 44,06±1,23 Фармакологические эксперименты 1. Поведенческий эксперимент - инъекция в двенадцатиперстную кишку. Гидрохлорид апоморфина (4 мг/кг или 5 мг/кг) и его монопивалоиловый эфир (4,6 или 4,9 мг/кг) и N-пропилнорапорфин (NPA; 5 мг/кг) вводили путем болюсной инъекции в двенадцатиперстную кишку крыс. Данных крыс оперировали за 1-14 дней перед экспериментом. Им помещали пластиковую трубку, входящую в стенку двенадцатиперстной кишки примерно в средней части и изогнутую таким образом, что ее канал направлен книзу (т.е. направлена к тощей кишке и имеет длину около 2 см). Опытный исследователь наблюдал животных в течение всего времени фармакологической активности, оценивая поведение и акцентируя внимание на следующих деталях: зевание, фырканье, жевание, лизание, способность становиться на задние лапы, способность ухода за собой, уход за мужскими половыми органами, локомоторная активность и стереотипия. Оценивали общую продолжительность действия, когда присутствовали одна или несколько из данных поведенческих реакций. Соединение Доаа (мг/кг) Длительность допаминергического поведения (мин) Гидрохлорид апоморфина 4,0 Интенсивная стереотипия (5-75), менее интенсивная стереотипия (7585); длит. (90) Гидрохлорид апоморфина 5,0 Интенсивная стереотипия (5-40), менее интенсивная стереотипия (4090); длит. (95) Гидрохлорид апоморфина 5,0 Интенсивная стереотипия (5-45), менее интенсивная стереотипия (4555); длит. (60) Моно-Пив-Аро 4,6 Интенсивная стереотипия (5-105), менее интенсивная стереотипия (105115) ,- длит. (120) - 7 - 008409 Моно-Пив-Аро 5,9 Интенсивная стереотипия (5-105), менее интенсивная стереотипия (105130); длит. (135) Моно-Пив-Аро 5,9 Интенсивная стереотипия (5-80), менее интенсивная стереотипия (8090); дгоге. (120) NPA 5,0 Интенсивная стереотипия (5 мин - 9 ч) ; длит. (> 9 ч) В качестве контрольного эксперимента гидрохлорид апоморфина (4 мг/кг) вводили перорально таким же крысам. Наблюдали очень слабую допаминергическую стимуляцию и временной период, в течение которого наблюдали данные эффекты, составлял 10-20 мин. 2. Поведенческий эксперимент - таблетки, покрытые энтеросолюбильным покрытием. Одну таблетку, покрытую энтеросолюбильным покрытием, полученную как описано в примере 1 и содержащую около 5 мг гидрохлорида NPA, помещали под анестезией (изофлуран) в горло крысы и проталкивали в глотку с помощью тупого инструмента. В течение пяти минут крысу пробуждали и помещали в клетку. Через приблизительно 3-4 ч у крысы начинали проявляться знаки допаминергической стимуляции, такие как фырканье, жевание, уход за мужскими половыми органами, уход за собой и стереотипия в отношении вставания на задние лапы, локомоторной активности, интенсивного фырканья, а также лизания. Данные стереотипии продолжались в течение более чем 24 ч. 3. Эксперимент по микродиализу (полосатое тело) с одной таблеткой с энтеросолюбильным покрытием, содержащей гидрохлорид NPA. Одну таблетку с энтеросолюбильным покрытием, полученную как описано в примере 1 и содержащую около 5 мг гидрохлорида NPA, вводили крысе таким же образом, как описано в фармакологическом эксперименте 2 и проводили стандартный микродиализ. После начального снижения высвобождения допамина через приблизительно два часа высвобождение допамина уменьшается. Однако через приблизительно четыре часа, что составляет приблизительное время, необходимое для прохождения через желудок и растворения оболочки в тонкой кишке, высвобождение допамина максимально снижается приблизительно до 20% по сравнению с контрольными значениями. Данный эффект продолжается в течение нескольких часов до тех пор, пока эксперимент не останавливают. В это время крыса проявляла стереотипическое поведение, которое по опыту являлось соответствующим максимальному снижению высвобождения допамина. 4. Эксперимент по микродиализу (полосатое тело) с одной таблеткой с энтеросолюбильным покрытием, содержащим основание монопивалоил-апоморфина. Фармакологический эксперимент 2 повторяли с применением таблетки с энтеросолюбильным покрытием, содержащей около 5 мг основания монопивалоил-апоморфина вместо гидрохлорида NPA. Через приблизительно 60 мин высвобождение допамина снижалось до максимального снижения 20% (т.е. 80% от контрольных значений). Высвобождение допамина возвращалось к контрольным значениям через приблизительно восемь часов. 5. Эксперимент по микродиализу (полосатое тело) с одной таблеткой с энтеросолюбильным покрытием, содержащей около 1 мг основания монопивалоил-N-пропил-норапорфина (MPNPA). Фармакологический эксперимент 2 повторяли с применением таблетки с энтеросолюбильным покрытием, полученной, как описано в примере 3, содержащей около 1 мг основания монопивалоил-N-пропил-норапорфина (MPNPA) вместо гидрохлорида NPA. Высвобождение допамина непрерывно снижалось между одним часом и четырьмя часами (максимальное снижение до 30% от контроля) и затем медленно повышалось до уровня 80% от контрольных значений через 18 ч после введения таблетки. Была отмечена интенсивная стереотипия между одним часом и восемью часами после введения. 6. Поведенческий эксперимент. Поведенческий эксперимент проводили с применением трех таблеток (каждая содержала около 5 мг гидрохлорида апоморфина, помещенного в биоразлагаемую пластиковую матрицу PLG, и была получена, как описано в примере 3), которые помещали под анестезией в горло крысы и проталкивали в глотку и помощью тупого инструмента. Слабые проявления стимуляции поведения, такие как жевание, фырканье, уход за собой, лизание мужских половых органов и некоторая моторная активность, были отмечены в течение времени эксперимента (10 ч). Поэтому было очевидно, что небольшие количества апоморфина высвобождались из таблетки и абсорбировались в тонкой кишке. Эксперимент заканчивали путем анестезии крысы с помощью изофлурана и забора образцов крови непосредственно из сердца крысы. Также извлекали головной мозг и гомогенизировали в 60% СНзСN/вода и сухой остаток удаляли путем центрифугирования. С целью - 8 - 008409 изучения, могут ли все еще быть обнаружены таблетки, тщательно исследовали кишечную систему от желудка до нисходящей ободочной кишки. Две таблетки были обнаружены в ободочной кишке, и одна таблетка была обнаружена в нисходящей ободочной кишке, заключенными в сформированные части испражнений. Эти три таблетки высушивали в течение ночи в вакуумном сушильном шкафу и взвешивали (34,6, 35,5 и 35,6 мг). Перед введением средняя масса данных таблеток составляла около 37 мг, что означает, что масса после прохождения через кишечную систему является приблизительно такой же, как и масса перед покрытием оболочкой. Следовательно, наиболее эффективной препаративной формой является применение покрытой эн-теросолюбильным покрытием капсулы, наполненной апоморфином, производными апоморфина или биораспадающейся композицией, такой как применялась для таблеток в поведенческих экспериментах, описанных выше. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Фармацевтический состав для лечения заболеваний, выбираемых из группы, состоящей из болезни Паркинсона, синдрома усталых ног и эректильной дисфункции, включающий, как минимум, одно соединение, выбранное из группы, состоящей из A) апоморфина, 6aR-(-)-N-пропил-норапоморфина, их симметричных ди-(С2-С5)алканоиловых эфи-ров или дибензоиловых эфиров, в форме основания или фармацевтически приемлемых солей или сольва-тов, B) пролекарств апорфина, выбранных из группы, состоящей из соединений, имеющих общую формулу где один из R1 и R2 является водородом или ацетилом, а другой выбирают из группы, состоящей из (С3-С20)алканоила; гало(С3-С20)алканоила; (С3-С20)алкеноила; (С4-С7)циклоалканоила; (С3-С6)циклоалкил(С2-С16)алканоила; ароила, который является незамещенным или замещенным 1-3 заместителями, выбранными из группы, состоящей из галогена, циано-, трифторметансульфонилокси-, (C1-С3)алкила и (C1-С3)алкокси-, где последний может в свою очередь быть замещенным 1-3 атомами галогена; арил(С2-С16)алканоила, который является незамещенным или замещенным в ариловом компоненте 1-3 заместителями, выбираемыми из группы, состоящей из галогена, (Q-С^алкила и (Q-С^алкокси-, где последний может в свою очередь быть замещенным 1-3 атомами галогена; и гетероарилалканоила, имеющего 1-3 гетероатома, выбранных из О, S и N в гетероариловом компоненте и 2-10 атомов углерода в алканоило-вом компоненте, и который является незамещенным или замещенным в гетероариловом компоненте 1-3 заместителями, выбранными из группы, состоящей из галогена, циано-, трифторметансульфонилокси-, (C1-С3)алкила и (Q-С^алкокси-, где последний может в свою очередь быть замещенным 1-3 атомами галогена; и R3 является метилом; и их фармацевтически приемлемых солей, в качестве активного ингредиента в фармацевтическом препарате, подходящем для перорально-го/интрадуоденального введения, причем указанный состав находится в форме прессованной таблетки/гранулы, включающей указанный активный ингредиент вместе с соответствующими наполнителями и адъювантами и имеющей энтеросолюбильное покрытие, растворяемой в тонкой кишке, например, двенадцатиперстной кишке. 2. Фармацевтический состав по п.1, имеющий дополнительный внешний слой, включающий указанный активный ингредиент вместе с соответствующими наполнителями и адъювантами. 3. Фармацевтический состав по п.1, включающий смесь указанных активных ингредиентов и соответствующих наполнителей и адъювантов, помещенных в капсулу, растворяющуюся в тонкой кишке, например, двенадцатиперстной кишке. 4. Фармацевтический состав по п.3, где указанная смесь представлена в форме гранул. 5. Фармацевтический состав по п.1 в форме гранул, имеющих энтеросолюбильное покрытие, помещенных в капсулу, растворяемую в желудке (gastrum) и высвобождающую гранулы, имеющие энтеросо-любильное покрытие, которые имеют оптимальный размер для прохода вместе с содержимым желудка в двенадцатиперстную кишку и дезинтегрируются там или дальше в тонкой кишке с контролируемым высвобождением активного ингредиента. 6. Фармацевтический состав по любому из пп.1-5, где указанный активный ингредиент имеет размер частиц в пределах от 0,1 до 20 мкм, предпочтительно между от 0,1 до 5 мкм. 7. Фармацевтический состав по п.1 в форме, подходящей для введения интрадуоденально при по - 9 - 008409 мощи интрадуоденального катетера через брюшную стенку пациента или при помощи назо-дуоденального катетера. 8. Фармацевтический состав по любому из пп.1-7, где активным ингредиентом является фармацевтически приемлемая соль апоморфина или 6aR-(-)-N-пропил-норапоморфина (NPA). 9. Фармацевтический состав по любому из пп.1-8, где активный ингредиент выбран из группы, состоящей из симметричных ди-(С2-С5)алканоиловых эфиров апоморфина и NPA и их фармацевтически приемлемых солей и дибензоилового эфира апоморфина и NPA и их фармацевтически приемлемых солей. 10. Фармацевтический состав по любому из пп.1-7, где активный ингредиент выбран из группы, состоящей из соединений, имеющих общую формулу где один из R1 и R2 является водородом или ацетилом, а другой выбирают из группы, состоящей из (С3-С20)алканоила; гало(С3-С20)алканоила; (С3-С20)алкеноила; (С4-С7)циклоалканоила; (С3-С6)циклоалкил(С2-С16)алканоила; ароила, который является незамещенным или замещенным 1-3 заместителями, выбранными из группы, состоящей из галогена, циано-, трифторметансульфонилокси-, (Q-С^алкила и (C1-С3)алкокси-, где последний может в свою очередь быть замещенным 1-3 атомами галогена; арил(С2-С16)алканоила, который является незамещенным или замещенным в ариловом компоненте 1-3 заместителями, выбираемыми из группы, состоящей из галогена, (Q-С^алкила и (Q-С^алкокси-, где последний может в свою очередь быть замещенным 1-3 атомами галогена; и гетероарилалканоила, имеющего 1-3 гетероатома, выбранных из О, S и N в гетероариловом компоненте и 2-10 атомов углерода в алканоило-вом компоненте, и который является незамещенным или замещенным в гетероариловом компоненте 1-3 заместителями, выбранными из группы, состоящей из галогена, циано-, трифторметансульфонилокси-, (Q-С^алкила и (Q-С^алкокси-, где последний может в свою очередь быть замещенным 1-3 атомами галогена; и R3 является метилом; или их фармацевтически приемлемых солей. 11. Фармацевтический состав по п.10, где активный ингредиент выбран из группы, состоящей из моно-(С3-C5)алканоиловьгх эфиров апоморфина и их фармацевтически приемлемых солей. 12. Фармацевтический состав по п.10, где активный ингредиент выбран из группы, состоящей из асимметричных диалканоиловых эфиров апоморфина, где одной из алканоильных групп является ацетильная группа, а другой является (С3-С6)алканоильная группа, и их фармацевтически приемлемых солей. 13. Способ лечения заболеваний, выбираемых из группы, состоящей из болезни Паркинсона, синдрома усталых ног, мужской эректильной дисфункции и женской сексуальной дисфункции, включающий введение перорально/интрадуоденально пациенту, нуждающемуся в лечении, фармацевтического состава, заявляемого в любом из пп.1-11, в эффективном для улучшения количестве. Евразийская патентная организация, ЕАПВ Россия, 109012, Москва, Малый Черкасский пер., 2/6 - 10 - |