|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea000008387b*\id |
|
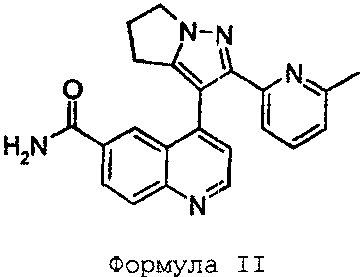
Термины запроса в документе Реферат 1. Соединение формулы II и его фармацевтически приемлемые соли. 2. Фармацевтическая композиция, содержащая соединение по п.1 или его фармацевтически приемлемую соль, вместе с фармацевтически приемлемым разбавителем, наполнителем или носителем. 3. Применение соединения по п.1 или его фармацевтической соли для лечения рака.
Полный текст патента (57) Реферат / Формула: и его фармацевтически приемлемые соли. 2. Фармацевтическая композиция, содержащая соединение по п.1 или его фармацевтически приемлемую соль, вместе с фармацевтически приемлемым разбавителем, наполнителем или носителем. 3. Применение соединения по п.1 или его фармацевтической соли для лечения рака.
008387 Область техники, к которой относится изобретение Настоящее изобретение относится к новым соединениям хинолинилпиразола и к их использованию в качестве фармацевтических средств, в частности к их использованию в качестве ингибиторов ТФР-бета сигнальной трансдукции. Предшествующий уровень техники Полипептиды трансформирующего фактора роста-бета (ТФР-бета) ("ТФР-Р") регулируют рост, дифференцировку и генную экспрессию во многих типах клеток. Первый полипептид из данного семейства, который был охарактеризован, ТФР-Р 1, имеет две идентичные субъединицы из 112 аминокислот, которые ковалентно связаны. ТФР-Р1 представляет собой высоко консервативный белок с единственным аминокислотным различием, отличающим человеческую последовательность от мышиной. Существует два других члена генного семейства ТФР-Р, которые экспрессируются в млекопитающих. ТФР-Р2 представляет собой 71% гомолог ТФР-pi (de Martin, et al. (1987) EMBO J. 6:3673-3677), тогда как ТФР-Р3 является 80% гомологом ТФР-Р1 (Derynck, et al. (1988) EMBO J. 7:3737-3743). Структурные характеристики ТФР-Р1, определенные ядерным магнитным резонансом (Archer, et al. (1993) Biochemistry 32:11641171), соответствуют кристаллической структуре ТФР-Р2 (Daopin, et al. (1992) Science 257:369-374; Schlunegger and Grutter (1992) Nature 358:430-434). Существуют по меньшей мере три различных внеклеточных ТФР-Р рецептора, тип I, II и III, которые вовлечены в биологические функции ТФР-Р1, -Р2 и -Р3 (для ознакомления см. Derynck (1994) TIBS 19:548-553 и Massague (1990) Ann. Rev. Cell Biol. 6:597-641). Рецепторы типа I и типа II представляют собой трансмембранные серин/треонинкиназы, которые в присутствии ТФР-Р формируют гетеромерный сигнальный комплекс (Wrana, et al. (1992) Cell 71:1003-1014). Механизм активации гетеромерного сигнального комплекса на поверхности клетки был объяснен (Wrana, et al. (1994) Nature 370:341-347). Сначала ТФР-Р связывает рецептор типа II, который представляет собой конститутивно активную трансмембранную серин/треонинкиназу. Рецептор типа I затем рекрутируется в комплекс, фосфорилируется на GS домене и активируется для фосфорилирования находящихся далее по последовательности сигнальных компонентов (например, Smad белков), чтобы инициировать внутриклеточный сигнальный каскад. Конститутивно активный рецептор типа I (T204D мутант), как было показано, эффективно преобразовывает реакцию ТФР-Р, таким образом, обходя потребность в ТФР-Р и рецепторе типа II (Wieser, et al. (1995) EMBO J. 14:2199-2208). Хотя никакой сигнальной функции для рецептора типа III не было открыто, он увеличивает сродство ТФР-Р2 к рецептору типа II, делая его, по существу, равносильным ТФР-Р 1 и ТФР-Р3 (Lopez-Casillas, et al. (1993) Cell 73:1435-1444). В сосудистых эндотелиальных клетках рецептор типа III отсутствует. Вместо него эндотелиальные клетки экспрессируют структурно родственный белок, названный эндоглином (Cheifetz, et al. (1992) J. Biol. Chem. 267:19027-19030), который связывает только ТФР-Р1 и ТФР-Р3 с высоким сродством. Таким образом, относительная эффективность ТФР-Р отражает тип рецепторов, экспрессируемых в клеточной системе и системе органа. Кроме регулирования компонентов в многофакторном сигнальном пути, распределение синтеза полипептидов ТФР-Р также воздействует на физиологическую функцию. Распределение ТФР-Р2 и ТФР-Р3 является более ограниченным (Derynck, et al. (1988) EMBO J. 7:3737-3743) по сравнению с ТФР-Р1, например, ТФР-Р3 ограничен тканями мезенхимальной природы, тогда как ТФР-Р1 присутствует как в тканях мезенхимальной, так и эпителиальной природы. ТФР-Р1 представляет собой многофункциональный цитокин, играющий большую роль в заживлении тканей. Высокие концентрации ТФР-Р1 доставляются к поврежденному участку гранулами тромбоцитов (Assoian and Sporn (1986) J. Cell Biol. 102:1217-1223). ТФР-Р1 инициирует серии событий, которые содействуют заживлению, включая хемотаксис клеток, таких как лейкоциты, моноциты и фибробласты, и регуляцию факторов роста и цитокинов, вовлеченных в ангиогенез, деление клеток, связанное с заживлением тканей и воспалительные реакции. ТФР-Р1 также стимулирует синтез компонентов внеклеточного матрикса (Roberts, et al. (1986) Proc. Natl. Acad. Sci. USA 83:4167-4171; Sporn, et al. (1983) Science 219:1329-1330; Massague (1987) Cell 49:437-438), и наиболее существенным для понимания патофизиологии ТФР-Р1 является то, что ТФР-Р1 саморегулирует свой собственный синтез (Kim, et al. (1989) J. Biol. Chem. 264:7041-7045). Описанные здесь соединения могут также показывать другую киназную активность, такую как ин-гибирование р38 киназы и/или ингибирование KDR (VEGFR2) киназы. Методы определения такой ки-назной активности известны из уровня техники, и специалист в данной области будет способен протестировать описанные соединения на такую активность. Сущность изобретения Раскрываемое изобретение относится к селективному соединению формулы II - 1 - 008387 Формула II 2-(6-метилпиридин-2-ил)-3-[6-амидохинолин-4-ил]-5,6-дигидро-4Н-пирроло [1,2-Ь]пиразол и его фармацевтически приемлемым солям. Вышеуказанное соединение, в общем, описано и заявлено в заявке на изобретение PCT/US 02/11884, поданной 13 мая 2002 г., для которой испрошен приоритет по заявке на патент США U.S.S.N. 60/293464, поданной 24 мая 2001 г., и включается здесь посредством ссылки. Вышеуказанное соединение было выбрано ввиду того, что обладает удивительно превосходным токсикологическим профилем по сравнению с соединениями, конкретно описанными в цитируемой выше заявке. Детальное описание изобретения Термин "эффективное количество", используемый в "эффективном количестве соединения формулы I", например, относится к количеству соединения по настоящему изобретению, которое способно ин-гибировать ТФР-бета. Термин мкМ относится к микромолярной размерности. Используемые здесь общие химические термины имеют свои обычные значения. В схемах синтеза и примерах использованы следующие сокращения: ДМФА относится к диметилформамиду, ТГФ относится к тетрагидрофурану, Ms относится к мезилу, который представляет собой метилсульфонил, ТГП относится к тетрагидропирану. Описанные здесь соединения можно получить в соответствии со следующими схемами и примерами. Примеры никоим образом не следует понимать как ограничивающие, поскольку они представляют собой иллюстрацию получения указанных соединений. Следующая ниже схема иллюстрирует получение соединения формулы I. Схема I Формула I Следующая ниже схема иллюстрирует получение соединения формулы II. - 2 - 008387 Схема II Формула II Следующие ниже примеры дополнительно иллюстрируют получение соединений по данному изобретению, как схематично показано на схемах I и II. Пример 1. Получение 7-(2-морфолин-4-илэтокси)-4-(2-пиридин-2-ил-5,6-дигидро-4Н-пирроло[1,2-Ъ] пиразол-3-ил)хинолина. A. Получение 4-(2-пиридин-2-ил-5,6-дигидро-4Н-пирроло[1,2-Ъ]пиразол-3-ил)-7-[2-(тетрагидро-пиран-2-илокси)этокси]хинолина. В течение 4 ч нагревают 4-(2-пиридин-2-ил-5,6-дигидро-4Н-пирроло[1,2-Ъ]пиразол-3-ил)хинолин-7-ол (376 мг, 1,146 ммоль), карбонат цезия (826 мг, 2,54 ммоль) и 2-(2-бромэтокси)тетрагидро-2Н-пиран (380 мкл, 2,52 ммоль) в ДМФА (5 мл) при 120°С. Гасят реакцию насыщенным раствором хлорида натрия и затем экстрагируют хлороформом. Органический слой сушат над сульфатом натрия и концентрируют под вакуумом. Реакционную смесь очищают на колонке с силикагелем, элюируя градиентом от дихлор-метана до смеси 10% метанола в дихлометане, получая желаемое промежуточное соединение, указанное в подзаголовке, в виде желтого маслянистого вещества (424 мг, 81%). МС (ES+) m/e 457,0 (M+1). B. Получение 2-[4-(2-пиридин-2-ил-5,6-дигидро-4Н-пирроло[1,2-Ъ]пиразол-3-ил)хинолин-7-илокси]этанола. Нагревают раствор 4-(2-пиридин-2-ил-5,6-дигидро-4Н-пирроло[1,2-Ъ]пиразол-3-ил)-7-[2-(тетра-гидропиран-2-илокси)этокси]хинолина (421 мг, 0,92 ммоль) в смеси уксусная кислота:тетрагидрофуран: вода (4:2:1) (20 мл). Растворитель удаляют под вакуумом и остаток снова растворяют в смеси хлоро-форм:изопропил (3:1). Органический слой промывают насыщенным раствором бикарбоната натрия и сушат над сульфатом натрия. Концентрируют под вакуумом. Остаток является достаточно чистым для следующей стадии в схеме (425 мг, 100%). МС (ES+) m/e 373,1 (M+l). C. Получение 2-[4-(2-пиридин-2-ил-5,6-дигидро-4Н-пирроло[1,2-Ъ]пиразол-3-ил)хинолин-7-илокси] этилового эфира метансульфоновой кислоты. В течение 2 ч перемешивают раствор 2-[4-(2-пиридин-2-ил-5,6-дигидро-4Н-пирроло[1,2-Ъ]пиразол-3-ил)хинолин-7-илокси]этанола (293 мг, 0,78 ммоль) и метансульфонилхлорида (68 мкл, 0,81 мл) в сухом пиридине (5 мл). Пиридин удаляют под вакуумом и повторно растворяют остаток в хлороформе. Органический слой промывают насыщенным раствором бикарбоната натрия и сушат над сульфатом натрия, получая желаемое промежуточное соединение, указанное в подзаголовке, в виде белого пенообразного вещества (425 мг, 100%). МС (ES+) m/e 451,1 (M+1). D. Получение 7-(2-морфолин-4-илэтокси)-4-(2-пиридин-2-ил-5,6-дигидро-4Н-пирроло[1,2-Ъ]пира-зол-3-ил)хинолина. - 3 - 008387 Нагревают 2-[4-(2-пиридин-2-ил-5,6-дигидро-4Н-пирроло [1,2-Ъ]пиразол-3 -ил)хинолин-7-илокси] этиловый эфир метансульфоновой кислоты (87 мг, 0,19 ммоль) с морфолином (1 мл) при 50°С в течение 4 ч. Морфолин удаляют лод вакуумом и затем продукт экстрагируют смесью изопропиловый спирт: хлороформ (1:3). Органический слой промывают хлоридом натрия и сушат над сульфатом натрия. Концентрируют под вакуумом, получая желаемый указанный в заголовке продукт в виде светло-желтого твердого вещества (83 мг, 100%). МС (ES+) m/e 442,0 (M+1). Пример 2. Получение 2-(6-метилпиридин-2-ил)-3-[6-амидохинолин-4-ил]-5,6-дигидро-4Н-пирроло [1,2-Ъ]пиразола. A. Получение 6-бром-4-метилхинолина. Перемешивают раствор 4-бромфениламина (1 экв.) в 1,4-диоксане и охлаждают примерно до 12°С. Медленно добавляют серную кислоту (2 экв.) и кипятят с обратным холодильником. По каплям добавляют метилвинилкетон (1,5 экв.) в раствор, кипящий при температуре флегмообразования. Нагревают раствор в течение 1 ч после завершения добавления. Реакционный раствор выпаривают до сухого состояния и остаток растворяют в метиленхлориде. Доводят рН раствора, до 8 1 М раствором карбоната натрия и три раза экстрагируют водой. Остаток подвергают хроматографии на SiO2 (70/30 гексан/ этилацетат), получая желаемое промежуточное соединение, указанное в подзаголовке. МС (ES+) m/e = 158,2 (M+1). B. Получение метилового эфира 6-метилпиридин-2-карбоновой кислоты. Суспендируют 6-метилпиридин-2-карбоновую кислоту (10 г, 72,9 ммоль) в метиленхлориде (200 мл). Охлаждают до 0°С. Добавляют метанол (10 мл), 4-диметиламинопиридин (11,6 г, 94,8 ммоль) и 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид (EDC) (18,2 г, 94,8 ммоль). Смесь перемешивают при комнатной температуре в течение 6 ч, промывают водой и насыщенным раствором соли и сушат над сульфатом натрия. Смесь фильтруют и концентрируют под вакуумом. Остаток подвергают хроматографии на SiO2 (50% этилацетат/гексан), получая желаемое промежуточное соединение, указанное в подзаголовке, 9,66 г (92%) в виде бесцветной жидкости. 1Н ЯМР (CDCl3) 8 7,93-7,88 (м, 1Н), 7,75-7,7 (м, 1Н), 7,35-7,3 (м, 1Н), 4,00 (с, 3Н), 2,60 (с, 3Н). C. Получение 2-(6-бромхинолин-4-ил)-1-(6-метилпиридин-2-ил)этанона. 6-Бром-4-метилхинолин (38,5 г, 153 ммоль) растворяют в 600 мл сухого ТГФ. Охлаждают до -70°С и обрабатывают, добавляя по каплям 0,5М гексаметилдисилазан калия (KN(SiMe3)2 (400 мл, 200 ммоль) в течение 2 ч, в то же время поддерживая температуру ниже -65°С. Полученный в результате раствор перемешивают при -70°С в течение 1 ч и по каплям добавляют раствор метилового эфира 6-метилпиридин-2-карбоновой кислоты (27,2, 180 ммоль) в 100 мл сухого ТГФ в течение 15 мин. В ходе добавления смесь меняет цвет от темно-красного до горохово-зеленого, и образуется осадок. Смесь перемешивают при -70°С в течение 2 ч, затем дают нагреться до комнатной температуры при перемешивании в течение 5 ч. Смесь охлаждают, затем гасят 12н. НС1 до рН=1. Повышают рН до 9 твердым карбонатом калия. Сливают раствор с твердого вещества и дважды экстрагируют 200 мл этилацетата. Органические экстракты объединяют, промывают водой и сушат над карбонатом калия. Твердое вещество перемешивают в смеси 200 мл воды и 200 мл этилацетата и обрабатывают дополнительным количеством карбоната калия. Органическую часть отделяют и сушат с предшествующими экстрактами этилацетата. Раствор концентрируют под вакуумом до темного маслянистого вещества. Маслянистое вещество пропускают через 300 мл слой силикагеля с метиленхлоридом и затем с этилацетатом. Соответствующие фракции объединяют и концентрируют под вакуумом, получая маслянистое вещество цвета янтаря. Смывают маслянистое вещество со стенок колбы метиленхлоридом, затем разбавляют гексаном, вращая колбу, в результате получая 38,5 г (73,8%) желаемого указанного в подзаголовке промежуточного соединения в виде твердого желтого вещества. МС (ES+) m/e = 341 (M+1). D. Получение 1-[2-(6-бромхинолин-4-ил)-1-(6-метилпиридин-2-ил)этилиденамино]пирролидин-2- она. Перемешивают смесь 2-(6-бромхинолин-4-ил)-1-(6-метилпиридин-2-ил)этанона (38,5 г, 113 ммоль) и 1-аминопирролидинон гидрохлорида (20 г, 147 ммоль) в 115 мл пиридина при комнатной температуре в течение 10 ч. Добавляют примерно 50 г 4 А неактивированных молекулярных сит. Продолжают перемешивание в течение дополнительных 13 ч, добавляют 10-15 г силикагеля и фильтруют смесь через 50 г слой силикагеля. Элюируют слой силикагеля 3 л этилацетата. Объединяют фильтраты и концентрируют под вакуумом. Фильтрованием собирают гидразоновый осадок и сушат отсасыванием, получая 33,3 г (69,7%) желаемого указанного в подзаголовке промежуточного соединения в виде твердого грязно-белого вещества. - 4 - 008387 МС (ES+) m/e = 423 (M+1). E. Получение 6-бром-4-[2-(6-метилпиридин-2-ил)-5,6-дигидро-4Н-пирроло[1,2-b]пиразол-3-ил] хинолина. К смеси (1,2 экв.) карбоната цезия и 1-[2-(6-бромхинолин-4-ил)-1-(6-метилпиридин-2-ил) этилиденамино]пирролидин-2-она (33,3 г, 78,7 ммоль) добавляют 300 мл сухого ^^диметилформамида. Смесь перемешивают в течение 20 ч при 100°С. Смесь может стать темной В ходе реакции. Удаляют ^^диметилформамид под вакуумом. Остаток распределяют между водой и метиленхлоридом. Водную часть экстрагируют дополнительным количеством метиленхлорида. Органические растворы фильтруют через 300 мл слой силикагеля, элюируя смесью 1,5 л метиленхлорида, 1,5 л этилацетата и 1,5 л ацетона. Объединяют соответствующие фракции и концентрируют под вакуумом. Фильтрованием собирают полученный в результате осадок, получая 22,7 г (71,2%) желаемого указанного в подзаголовке промежуточного соединения в виде твердого грязно-белого вещества. МС (ES+) = 405 (М+1). F. Получение метилового эфира 4-[2-(6-метилпиридин-2-ил)-5,6-дигидро-4Н-пирроло[1,2-b] пиразол-3 -ил]хинолин-6-карбоновой кислоты. К смеси ацетата натрия (19 г, 230 ммоль) и палладиевого катализатора [1,1'-бис (дифенилфосфино)ферроцен]дихлорпалладия(П), комплекс с дихлорметаном, (1:1) (850 мг, 1,04 ммоль) в 130 мл метанола добавляют 6-бром-4-[2-(6-метилпиридин-2-ил)-5,6-дигидро-4Н-пирроло[1,2-b]пиразол-3-ил]хинолин (22,7 г, 45 ммоль). Смесь помещают в атмосферу монооксида углерода под давлением 50 фунт/кв. дюйм, перемешивают при нагревании до 90°С в течение 1 ч и с постоянной загрузкой дополнительного количества монооксида углерода. Смеси дают охладиться в течение 8 ч, снова загружают монооксид углерода и нагревают до 90°С. Давление может возрасти примерно до 75 фунт/кв.дюйм. Реакция завершается примерно за час, когда давление становится постоянным, и ТСХ (1:1 толуол/ацетон) показывает отсутствие остаточного бромида. Смесь распределяют между метиленхлоридом (600 мл) и водой (1 л). Водную часть экстрагируют дополнительной порцией метиленхлорида (400 мл). Органический раствор фильтруют через 300 мл слой силикагеля и промывают смесью 500 мл метиленхлорида, 1200 мл этилацетата и 1500 мл ацетона. Сливают ацетоновую часть. Объединяют соответствующие фракции и концентрируют, получая 18,8 г (87,4%) желаемого указанного в подзаголовке промежуточного соединения в виде розового порошка. МС (ES+) = 385 (М+1). G. Получение 2-(6-метилпиридин-2-ил)-3-[6-амидохинолин-4-ил]-5,6-дигидро-4Н-пирроло[1,2-b] пиразола. Нагревают смесь метилового эфира 4-[2-(6-метилпиридин-2-ил)-5,6-дигидро-4Н-пирроло[1,2-b]пиразол-3-ил]хинолин-6-карбоновой кислоты в 60 мл 7н. аммиака в метаноле до 90°С в сосуде из нержавеющей стали для работы под давлением в течение 66 ч. Давление будет возрастать примерно до 80 фунт/кв. дюйм. Поддерживают давление в течение реакции. Сосуд охлаждают и концентрируют коричневую смесь под вакуумом. Очищают оставшееся твердое вещество на двух 12 г картриджах Redi-Pak, соединенных последовательно, элюируя ацетоном. Объединяют соответствующие фракции и концентрируют под вакуумом. Суспендируют полученное в результате желтовато-белое вещество в метиленхлори-де, разбавляют гексаном и фильтруют. Собирают твердое грязно-белое вещество, получая 1,104 г (63,8%) желаемого указанного в заголовке продукта. МС (ES+) = 370 (М+1). Описанные здесь соединения тестировали с помощью следующих протоколов ингибирования ТФР-Р, как описано ниже в описании протокола. Очистка ТФР-Р рецептора I и киназные реакции in vitro Для ТФР-Р рецепторов типа I (RIT204D): 6X-HIS меченный цитоплазматический киназный домен каждого рецептора экспрессируют и очищают от лизатов клеток насекомых Sf9, как кратко описано ниже. Клеточный дебрис после 48-72 ч инфицирования лизируют в буфере для лизиса (LB: 50 мМ Трис рН 7,5, 150 мМ NaCl, 50 мМ NaF, 0,5% NP40 с только что добавленным 20 мМ Р-меркаптоэтанолом, 10 мМ имидазола, 1 мМ PMSF, 1X не содержащий ЭДТУ полный ингибитор протеазы (Boehringer Mannheim). Клеточные лизаты очищают центрифугированием и 0,45 мкМ фильтруют перед очисткой Ni/NTA афинной хроматографией (Qiagen). - 5 - 008387 Хроматографический протокол Уравновешивают 10 КО LB, загружают образец, промывают 10 КО буфера RIPA (50 мМ Трис рН 7,5, 150 мМ NaCl, 1% NP40, 1 мМ ЭДТУ, 0,25% деоксихолата натрия, добавляют свежий 20 мМ Р-меркаптоэтанол, 1 мМ PMSF), промывают 10 КО LB, промывают 10 КО 1X KB (50 мМ Трис рН 7,5, 150 мМ NaCl, 4 мМ МgQ2, 1 мМ NaF, 2 мМ Р-меркаптоэтанол), элюируют линейным градиентом 1X KB, содержащим 200 мМ имидазола. Оба фермента имели чистоту примерно 90% и обладали активностью аутофосфорилирования. Реакционные смеси: 170-200 нМ фермента в 1X KB, серии разбавления соединения в 1X КВ/16% ДМСО (от 20 мкМ до 1 нМ конечной концентрации с 4% конечной концентрации ДМСО), реакцию начинают, добавляя АТФ смесь (конечные концентрации 4 мкМ АТФ/1 мкКи 33Р-у-АТФ) в 1X КВ. Реакционные смеси инкубировали при 30°С в течение 1 ч. Реакции останавливали и проводили количественную оценку, используя стандартное TCA/BSA осаждение на фильтровальных пластинах Milli-pore FB из стекловолокна и жидкостный сцинтилляционный счетчик MicroBeta JET. Описанные здесь соединения ингибируют киназный домен рецептора ТФР-Р типа I (RIT204D) со значениями ИК50 <20 мкМ, в то же время показывая меньшую токсичность in vivo по сравнению со структурно родственными соединениями, которые описаны в РСТ заявке на патент PCT/US 02/11884, идентифицированной выше. Состояния "характеризующиеся увеличенной активностью ТФР-Р" включают состояния, в которых синтез ТФР-Р стимулируется таким образом, что ТФР-Р присутствует при повышенных уровнях, или в которых латентный белок ТФР-Р нежелательно активирован или конвертирован в активный белок ТФР-Р, или в которых ТФР-Р рецептор активирован, или в которых ТФР-Р белок показывает увеличенное связывание с клетками или внеклеточным матриксом на патологических участках. Таким образом, в любом случае "увеличенная активность" относится к любому состоянию, в котором биологическая активность ТФР-Р является нежелательно высокой, независимо от причины. Ряд болезней связан с сверхпродукцией ТФР-Р1. Ингибиторы внутриклеточного сигнального пути ТФР-Р являются применимыми для терапии фибрознопролиферативных заболеваний. Конкретно, фиб-рознопролиферативные заболевания включают заболевания почек, связанные с неконтролируемой активностью ТФР-Р и избыточным фиброзом, включая гломерулонефрит (GN), такой как мезангиально-пролиферативный GN, иммунный GN и GN с полулуниями. Другие почечные состояния включают диабетическую нефропатию, почечный интерстициальный фиброз, почечный фиброз у пациентов с трансплантатом, получающих циклоспорин, и вызванную ВИЧ нефропатию. Коллагеновые сосудистые заболевания включают прогрессирующий системный склероз, полимиозит, склеродерму, дерматомиозит, эозинофильный фасцит, кольцевидную склеродерму или состояния, связанные с возникновением синдрома Рейно. Фиброзы легкого в результате избыточной активности ТФР-Р включают респираторный дистресс-синдром взрослых, идиопатический легочный фиброз и интерстициальный легочный фиброз, часто связанный с аутоиммунными расстройствами, такими как системная эритематозная волчанка и склеродерма, химическим контактом или аллергией. Другим аутоиммунным заболеванием, связанным с фибропролиферативными характеристиками, является ревматоидный артрит. Связанные с фибропролиферативным состоянием заболевания глаз, включающие хирургическое лечение отслоения сетчатки, сопровождающейся пролиферативной витреоретинопатией, удаление катаракты с имплантацией внутриглазной линзы и последствия дренирования при хирургии глаукомы, связаны с сверхпродукцией ТФР-Р1. Фиброзные заболевания, связанные со сверхпродукцией ТФР-Р1 можно разделить на хронические состояния, такие как фиброз почки, легкого и печени, и более острые состояния, такие как рубцевание кожи и рестеноз (Chamberlain, J. Cardiovascular Drug Reviews, 19 (4):329-344). Синтез и секреция ТФР-Р1 клетками опухоли также может вести к подавлению иммунного ответа, как отмечается у пациентов с агрессивной опухолью головного мозга или молочной железы (Arteaga, et al. (1993) J. Clin. Invest. 92:2569-2576). Течение лейшманиоза у мышей резко изменяется с помощью ТФР-Р1 (Barral-Netto, et al. (1992) Science 257:545-547). ТФР-Р1 обостряет течение болезни, тогда как антитела ТФР-Р1 останавливают развитие болезни у генетически восприимчивых мышей. Генетически устойчивые мыши становятся восприимчивыми к лейшманиозу при введении ТФР-Р1. Был сделан обзор сильного воздействия ТФР-Р1 на осаждение внеклеточного матрикса (Rocco and Ziyadeh (1991) in Contemporary Issues in Nephrology, v. 23, Hormones, autocoids and kidney, ed. Jay Stein, Churchill Livingston, New York pp.391-410; Roberts, et al. (1988) Rec. Prog. Hormone Res. 44:157-197), и оно включает в себя стимуляцию синтеза и ингибирование распада компонентов внеклеточного матрик-са. Поскольку структура и фильтрационные свойства гломерул в значительной степени определяются составом внеклеточного матрикса мезангия и гломерулярной мембраны, неудивительно, что ТФР-Р1 оказывает сильное воздействие на почку. Накопление мезангиального матрикса при пролиферативном гло-мерулонефрите (Border, et al. (1990) Kidney Int. 37:689-695) и диабетической нефропатии (Mauer, et al. (1984) J. Clin. Invest. 74:1143-1155) является четкой и доминирующей патологической особенностью - 6 - 008387 данных заболеваний. Уровни ТФР-Р1 являются повышенными при диабетическом гломерулосклерозе у человека (прогрессирующая нейропатия) (Yamamoto, et al. (1993) Proc. Natl. Acad. Sci. 90:1814-1818). ТФР-Р1 является важным посредником в генезисе почечного фиброза в ряде экспериментальных моделей на животных (Phan, et al. (1990) Kidney Int. 37:426; Okuda, et al. (1990) J. Clin. Invest. 86:453). Подавление экспериментально вызванного гломерулонефрита у крыс было продемонстрировано с помощью антисыворотки против ТФР-Р1 (Border, et al. (1990) Nature 346:371) и белка внеклеточного матрикса, де-корина, который может связать ТФР-Р1 (Border, et al. (1992) Nature 360:361-363). Слишком большое количество ТФР-Р 1 приводит к образованию кожной рубцовой ткани. Было показано, что нейтрализующие ТФР-Р1 антитела, введенные в границы заживающих ран у крыс, подавляют рубцевание, не мешая заживлению раны или растяжимости раны (Shah, et al. (1992) Lancet 339:213-214). В то же время присутствовали сниженный ангиогенез, пониженное количество макрофагов и моноцитов в ране и сниженное осаждение неупорядоченного коллагенового волокна в рубцовой ткани. ТФР-Р1 может являться движущей силой при прогрессирующем утолщении стенки артерии, которое является результатом пролиферации гладкомышечных клеток и осаждения внеклеточного матрикса в артерии после баллонной ангиопластики. Диаметр артерии, подверженной рестенозу, может быть уменьшен на 90% из-за данного утолщения, и поскольку наибольшая часть снижения диаметра обусловлена скорее внеклеточным матриксом, чем тельцами гладкомышечных клеток, может являться возможным открыть данные сосуды до 50%, просто снижая обширное осаждение внеклеточного матрикса. В неповрежденных артериях свиньи, трансфицированных in vivo геном ТФР-Р1, экспрессия гена ТФР-Р1 была связана как с синтезом внеклеточного матрикса, так и гиперплазией (Nabel, et al. (1993) Proc. Natl. Acad. Sci. USA 90:10759-10763). Гиперплазия, вызванная ТФР-Р1, была не такой обширной, как вызванная PDGF-BB, но внеклеточный матрикс был более распространен у трансфектантов ТФР-Р 1. Никакое осаждение внеклеточного матрикса не было связано с гиперплазией, вызванной FGF-1 (секретированная форма FGF), в данной модели переноса гена на свиньях (Nabel (1993) Nature 362:844-846). Существует несколько типов рака, где продуцируемый опухолью ТФР-Р может являться вредным. Клетки рака предстательной железы крысы MATLyLu (Steiner and Barrack (1992) Mol. Endocrinol. 6:1525) и клетки рака молочной железы человека MCF-7 (Arteaga, et al. (1993) Cell Growth and Differ. 4:193201) становились более онкогенными и метастатическими после трансфекции вектором, экспрессирую-щим мышиный ТФР-Р1. ТФР-Р1 был связан с ангиогенезом, метастазом и плохим прогнозом при раке предстательной железы и запущенном раке желудка у человека (Wikstrom, P., et al. (1998) Prostate 37:1929; Saito, H. et al. (1999) Canser 86:1455-1462). При раке молочной железы плохой прогноз связан с повышенным уровнем ТФР-Р (Dickson, et al., (1987) Proc. Natl. Acad. Sci. USA 84:837-841; Kasid, et al. (1987) Canser Res. 47:5733-5738; Daly, et al. (1990) J. Cell. Biochem. 43:199-211; Barrett-Lee, et al. (1990) Br. J. Cancer 61:612-617; King, et al. (1989) J. Steroid Biochem. 34:133-138; Welch, et al. (1990) Proc. Natl. Acad. Sci. USA 87:7678-7682; Walker, et al. (1992) Eur. J. Cancer 238: 641-644) и индукция ТФР-Р 1 терапией тамоксифеном (Butta, et al. (1992) Cancer Res. 52:4261-4264) была связана с недостаточностью терапии темоксифеном для лечения рака молочной железы (Thompson, et al. (1991) Br. J. Cancer 63:609-614). Анти ТФР-Р1 антитела подавляют рост клеток опухоли молочной железы человека MDA-231 в "атимических" мышах (Arteaga, et al. (1993) J. Clin. Invest. 92:2569-2576), терапия которыми коррелирует с увеличением в селезенке активности естественных клеток-киллеров. СНО клетки, трансфицированные латентным ТФР-Р1, также показали пониженную NK активность и увеличенный рост опухоли у "голых" мышей (Wallick, et al. (1990) J. Exp. Med. 172:1777-1784). Таким образом, ТФР-Р, выделяемый опухолями молочной железы, может вызвать эндокринное подавление иммунного ответа. Высокие концентрации ТФР-Р1 в плазме, как было показано, свидетельствуют о плохом прогнозе для пациентов, страдающих запущенным раком молочной железы (Anscher, et al. (1993) N. Engl. J. Med. 328:1592-1598). Пациенты с высокой циркуляцией ТФР-Р до химиотерапии высокими дозами и аутологической трансплантации костного мозга имеют высокий риск возникновения облитерирующего эндофлебита печеночных вен (15-50% от общего количества пациентов с процентом смертности до 50%) и интерстициальной идиопатической пневмонии (40-60% от общего количества пациентов). Следствием данных открытий является то, что 1) повышенные уровни ТФР-Р1 в плазме можно использовать для идентификации пациентов, находящихся в группе риска, и 2) что уменьшение ТФР-Р1 могло бы снизить болезненность и летальность данных общих терапий для пациентов, страдающих раком молочной железы. Многие злокачественные клетки выделяют трансформирующий фактор роста-Р (ТФР-Р), мощный иммуносупрессант, наводя на мысль, что продукция ТФР-Р может представлять собой существенный механизм, посредством которого опухоль избегает воздействия иммунной системы хозяина. Создание субпопуляции лейкоцитов с нарушенной передачей сигнала ТФР-Р в организме хозяина, пораженного опухолью, предоставляет потенциальное средство иммунотерапии рака. Трансгенная модель на животных с нарушенной передачей сигнала ТФР-Р в Т-клетках способна уничтожить обычно летальную сверх-экспрессирующую ТФР-Р опухоль лимфатической ткани, EL4 (Gorelik and Flavell, (2001) Nature Medicine 7(10): 1118-1122). Супрессирующая регуляция секреции ТФР-Р в клетках опухоли приводит к восстанов - 7 - 008387 лению иммуногенности у хозяина, в то время как нечувствительность Т-клетки к ТФР-Р приводит к ускоренной дифференцировке и аутоиммунности, элементы которых могут потребоваться для борьбы с аутоантиген-экспрессирующими опухолями в толерантном хозяине. Иммунодепрессивное действие ТФР-Р также подразумевалось в субпопуляции ВИЧ-пациентов с более низкой по сравнению с прогнозируемой иммунной реакцией на основании их CD4/CD8 счета Т-клеток (Garba, et al. J. Immunology (2002) 168: 2247-2254). ТФР-Р нейтрализующее антитело было способно изменить направление эффекта на обратное в культуре, показывая, что ингибиторы передачи сигнала ТФР-Р могут быть полезными для реверсирования подавления иммунного ответа в данной подгруппе ВИЧ-пациентов. На ранних стадиях канцерогенеза ТФР-Р может действовать как эффективный суппрессор опухоли и может опосредовать действие некоторых химиотерапевтических агентов. Однако в некоторый момент в течение развития и продвижения злокачественной опухоли, клетки опухоли, по-видимому, избегают подавления ТФР-Р-зависимого роста параллельно с появлением биоактивного ТФР-Р в микросреде. Двойная роль ТФР-Р подавления опухоли/активации опухоли наиболее четко была объяснена в трансгенной системе, сверхэкспрессирующей ТФР-Р в кератиноцитах. В то время как трансгенные системы были более устойчивы к образованию доброкачественных поражений кожи, скорость метастатической конверсии в трансгенных системах резко увеличивалась (Cui, et al. (1996) Cell 86 (4) :531-42). Продукция ТФР-Р1 злокачественными клетками в первичных опухолях, по-видимому, увеличивается на прогрессирующих стадиях развития опухоли. Исследования многих основных типов эпителиальных опухолей говорят о том, что увеличенная продукция ТФР-Р при раке у человека происходит в виде относительно позднего события в течение развития опухоли. Более того, данный связанный с опухолью ТФР-Р предоставляет клеткам опухоли селективное преимущество и содействует развитию опухоли. Воздействие ТФР-Р на взаимодействие клетка/клетка и клетка/строма приводит к большей предрасположенности к инвазии и метастазированию. Связанный с опухолью ТФР-Р может дать возможность клеткам опухоли избежать иммунного контроля, поскольку он является мощным ингибитором клональной экспансии активированных лимфоцитов. Также было показано, что ТФР-Р подавляет продукцию ангиостатина. Терапевтические методы лечения рака, такие как радиационная терапия и химиотерапия, вызывают продукцию активированного ТФР-Р в опухоли, тем самым обеспечивая отбор тех злокачественных клеток, которые устойчивы к эффектам подавления роста ТФР-Р. Таким образом, данные противораковые терапии увеличивают риск и ускоряют развитие опухолей с увеличенным ростом и инвазивностью. В данной ситуации вещества, нацеленные на сигнальную трансдукцию, посредником в которой является ТФР-Р, могут являться очень эффективной терапевтической стратегией. Устойчивость клеток опухоли к ТФР-Р, как было показано, существенно противоречит цитотоксическим эффектам радиотерапии и химиотерапии, и зависимая от терапии активация ТФР-Р в строме может быть даже вредной, поскольку это может создать микросреду более способствующую для развития опухоли и внести вклад в повреждение тканей, ведущее к фиброзу. Разработка ингибиторов сигнальной трансдукции ТФР-Р, вероятно, поможет лечению прогрессирующего рака сама по себе или в комбинации с другими терапиями. Соединения применимы для лечения рака и других патологических состояний, на которые оказывает влияние ТФР-Р, ингибируя ТФР-Р у пациента, при необходимости этого, посредством введения указанного соединения(й) указанному пациенту. Ингибиторы рецептора ТФР-Р также применимы против атеросклероза (ТА. McCaffrey: TGF^s and TGF-Р Receptors in Atherosclerosis: Cytokine and Growth Factor Reviews 2000, 11, 103-114) и болезней Альцгеймера (Masliah, E.; Ho, G.; Wyss-Coray, Т.: Functional Role of TGF-Р in Alzheimer's Disease Microvascular Injury: Lessons from Transgenic Mice: Neurochemistry International 2001, 39, 393-400). Фармацевтические композиции Композиции по настоящему изобретению представляют собой терапевтически эффективные количества антагонистов ТФР-Р, отмеченных ранее. Композиции могут быть составлены с обычными наполнителями, разбавителями или носителями, и спрессованы в таблетки, или составлены в виде эликсиров или растворов для удобного орального введения или введения внутримышечным, внутривенным путями. Соединения можно вводить трансдермально, или они могут быть составлены в виде лекарственных форм пролонгированного действия и аналогичных. Способ лечения человека по настоящему изобретению включает введение антагонистов ТФР-Р. Из антагонистов ТФР-Р составляют рецептуры, которые можно вводить оральным или ректальным путями, топически, парентерально, например, инъекцией или непрерывной или прерывистой внутриартериальной инфузией, в форме, например, таблеток, лепешек, подъязычных таблеток, саше, облаток, эликсиров, гелей, суспензий, аэрозолей, мазей, например, содержащих от 1 до 10 мас.% активного соединения в подходящей основе, мягких или твердых желатиновых капсул, суппозиториев, растворов для инъекций и суспензий в физиологически приемлемой среде, и стерильных упакованных порошков, адсорбированных на материале носителя для изготовления растворов для инъекций. Преимущественно, для данной задачи композиции можно изготовить в виде стандартной лекарственной формы, причем, предпочтительно, ка - 8 - 008387 ждая стандартная лекарственная форма содержит примерно от 5 до 500 мг (примерно от 5 до 50 мг в каждом случае парентерального введения или введения ингаляцией, и примерно от 25 до 500 мг в случае орального или ректального введения) соединений. Можно вводить дозировки примерно от 0,5 до 300 мг/кг в сутки, предпочтительно от 0,5 до 20 мг/кг активного ингредиента, хотя конечно будет понятно, что количество соединения, которое действительно необходимо вводить, будет определяться врачом в свете всех относящихся к делу обстоятельств, включая состояние, которое необходимо лечить, выбор соединения, которое следует вводить, и выбор пути введения, и, следовательно, вышеуказанный предпочтительный диапазон дозировки никоим образом не имеет намерения ограничивать объем патентной охраны настоящего изобретения. Композиции, пригодные для раздельного введения антагонистов ТФР-Р, обычно будут состоять по меньшей мере из одного соединения, выбранного из описанных здесь соединений, смешанного с носителем, или разбавленного носителем, или заключенного или включенного в капсулу с помощью носителя, который можно глотать, в форме капсулы, саше, облатки, бумаги или другого контейнера или одноразового контейнера, такого как ампула. Носитель или разбавитель может быть твердым, полутвердым или жидким веществом, которое служит в качестве растворителя, наполнителя или среды для активного терапевтического вещества. Некоторыми примерами разбавителей или носителей, которые можно использовать в фармацевтических композициях по настоящему изобретению, являются лактоза, декстроза, сахароза, сорбит, маннит, полипропиленгликоль, жидкий парафин, белый мягкий парафин, каолин, коллоидальный диоксид кремния, микрокристаллическая целлюлоза, силикат кальция, диоксид кремния, поливинилпирролидон, цетостеариловый спирт, крахмал, модифицированные крахмалы, аравийская камедь, фосфат кальция, масло какао, этоксилированные сложные эфиры, масло теобромы, арахисовое масло, альгинаты, трагакантовая камедь, желатин, сироп, метилцеллюлоза, полиоксиэтиленсорбит моно-лаурат, этиллактат, метил- и пропилгидроксибензоат, триолеат сорбита, сесквиолеат сорбита, олеиловый спирт и газы-вытеснители, такие как трихлормонофторметан, дихлордифторметан и дихлортетрафторэ-тан. В случае таблеток можно ввести скользящее вещество для предотвращения слипания и связывания порошкообразных ингредиентов в пресс-форме и штампе машины для изготовления таблеток. Для данной цели можно использовать, например, стеараты алюминия, магния или кальция, тальк или минеральное масло. Предпочтительными фармацевтическими формами по настоящему изобретению являются капсулы, таблетки, суппозитории, растворы для инъекций, кремы и мази. Особенно предпочтительными являются рецептуры для ингаляций, такие как аэрозоли, для инъекций и для орального приема внутрь. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы II H2N Формула II и его фармацевтически приемлемые соли. 2. Фармацевтическая композиция, содержащая соединение по п.1 или его фармацевтически приемлемую соль, вместе с фармацевтически приемлемым разбавителем, наполнителем или носителем. 3. Применение соединения по п.1 или его фармацевтической соли для лечения рака. Евразийская патентная организация, ЕАПВ Россия, 109012, Москва, Малый Черкасский пер., 2/6 - 9 - |