|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea000008256b*\id |
|
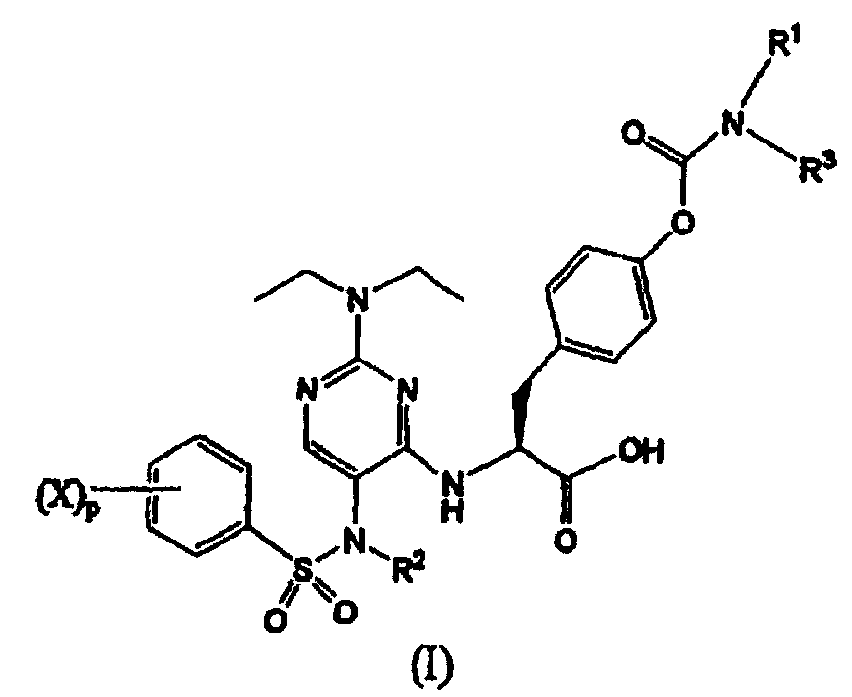
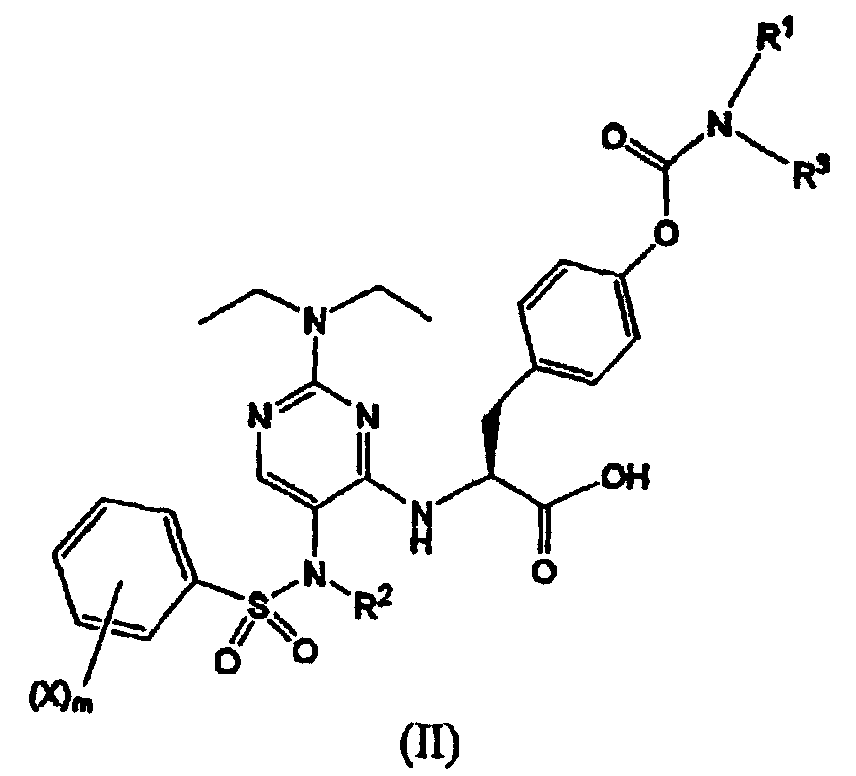
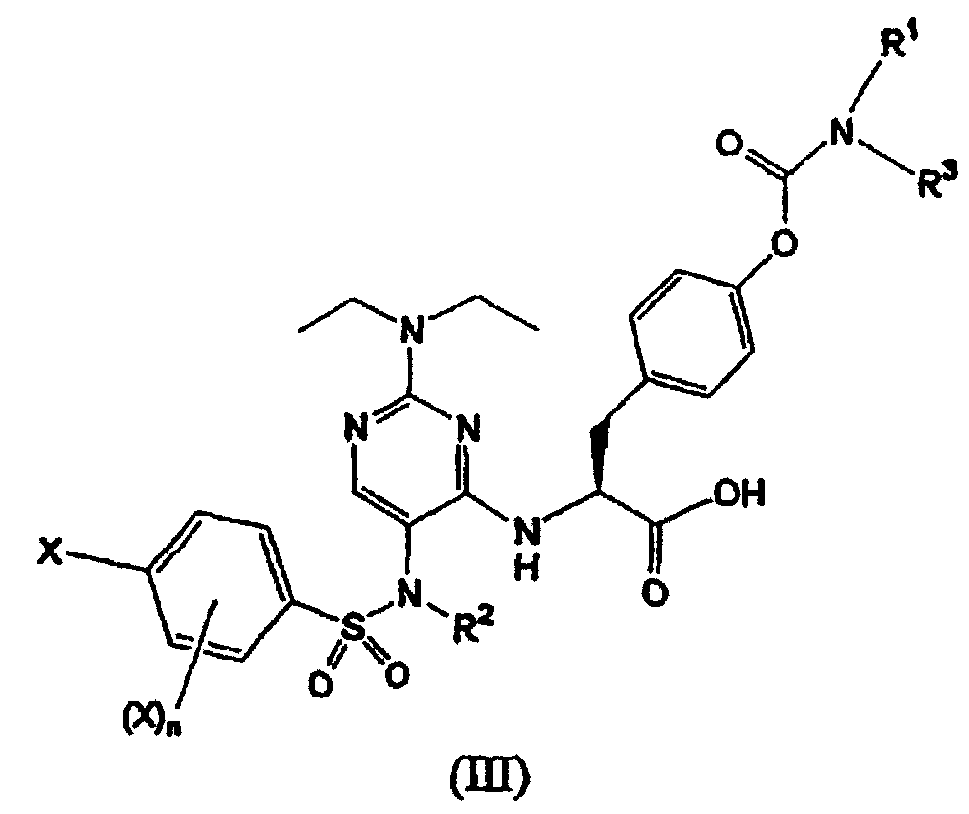
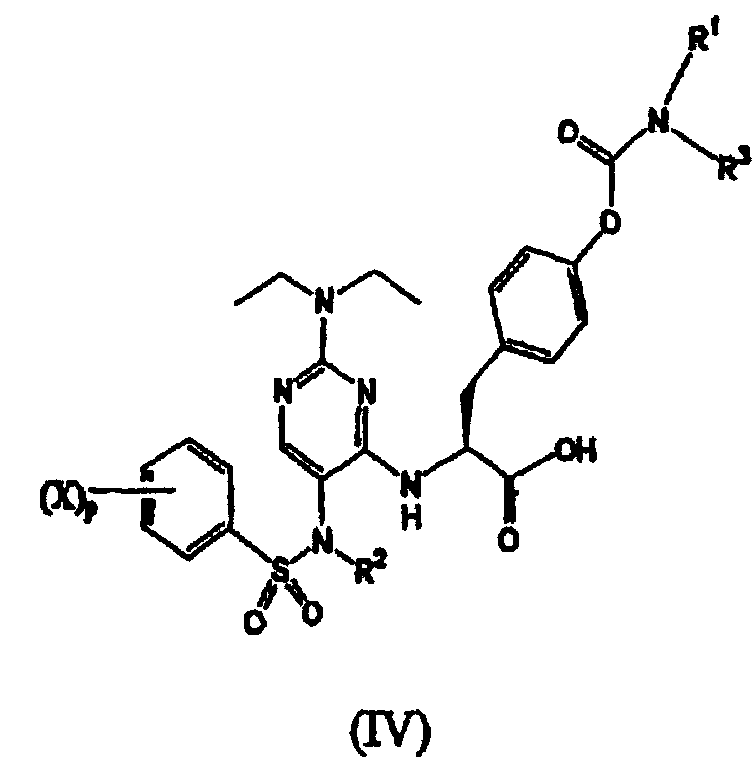
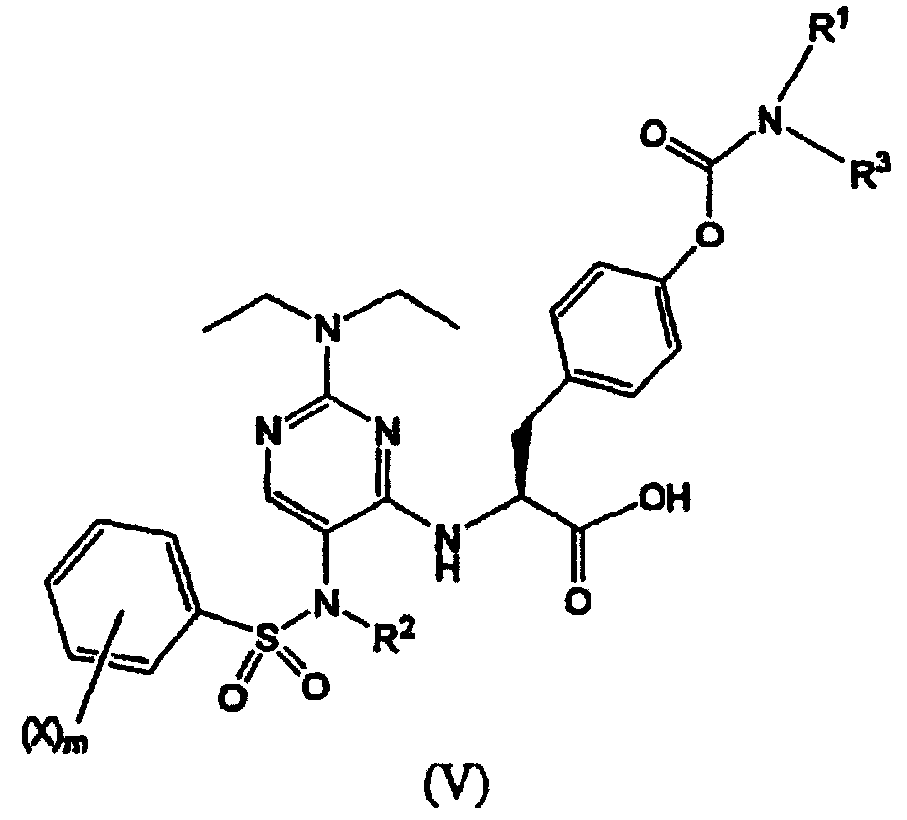
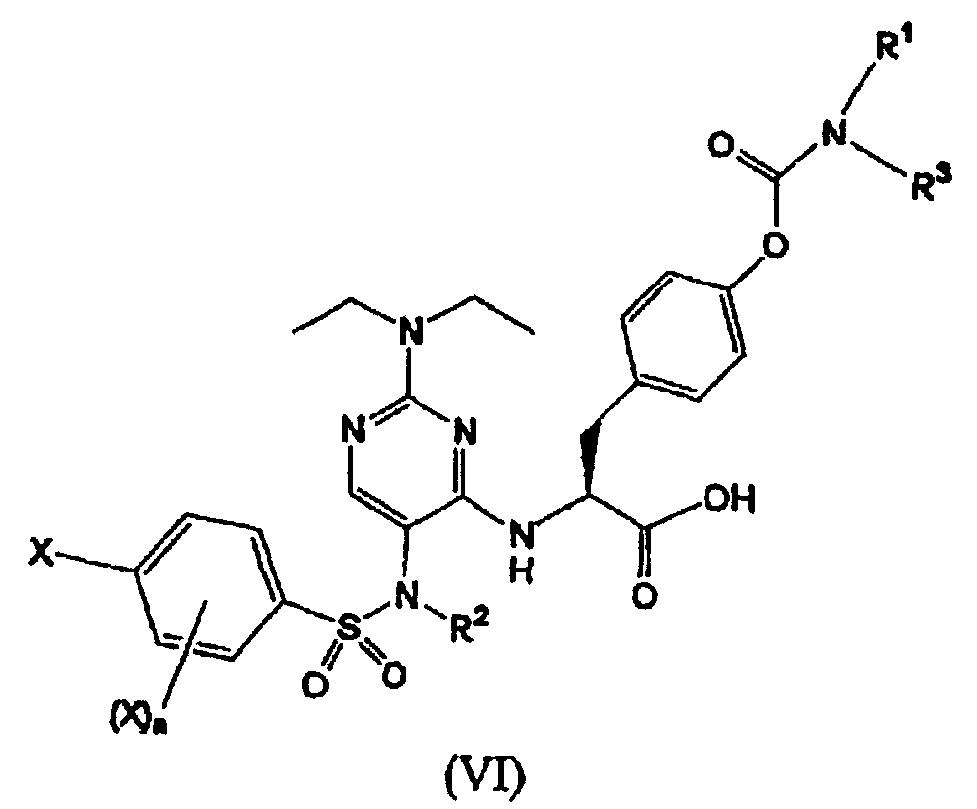
Термины запроса в документе Реферат 1. Соединение формулы (I) где каждый Х независимо представляет собой фтор, хлор или бром; р представляет собой целое число от 0 до 3; R 1 и R 3 вместе с атомом азота, с которым они связаны, образуют азетидинил, пирролидинил, пирролил, 2,5-дигидропиррол-1-ил, пиперидинил или 1,2,3,6-тетрагидропиридин-1-ил; R 2 выбран из группы, состоящей из (С1-С5)алкила, (С2-С6)алкенила и (С3-С6)циклоалкил(С1-С4)алкила; и его фармацевтически приемлемые соли. 2. Соединение формулы (II) где каждый Х независимо выбран из группы, состоящей из фтора и хлора; m представляет собой целое число, равное 1 или 2; R 2 выбран из группы, состоящей из (С1-С5)алкила, (С2-С6)алкенила и (С3-С6)циклоалкил(С1-С4)алкила; R 1 и R 3 вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидинильную или пиперидинильную группу; и его фармацевтически приемлемые соли. 3. Соединение формулы (III) где каждый Х независимо представляет собой фтор или хлор; n равно нулю или единице; R 2 является -CH 2 -R', где R' выбран из группы, состоящей из водорода, метила или -СН=СН 2 ; R 1 и R 3 вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидинильную или пиперидинильную группу; и его фармацевтически приемлемые соли. 4. Соединение по п.1, в котором R 1 и R 3 вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидинильную или пиперидинильную группу. 5. Соединение по одному любому из пп.1, 2 или 3, в котором R 2 является СН 3 . 6. Соединение по п.3, в котором Х является F или Сl и n равно 0. 7. Соединение по п.1, выбранное из группы, содержащей N-(2-[N',N'-диэтиламино]-5-[N"-(4-хлорфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-хлорфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(пиперидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(пиперидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-хлорфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-хлорфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; и его фармацевтически приемлемые соли. 8. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по одному любому из пп.1-4, 6 или 7. 9. Способ лечения заболевания млекопитающего, опосредованного a 4 интегринами, включающий введение фармацевтической композиции, содержащей фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по одному любому из пп.1-4, 6 или 7. 10. Способ по п.9, где заболевание опосредовано VLA-4. 11. Способ по п.9, где заболевание представляет собой воспалительное заболевание. 12. Соединение формулы (IV) где каждый Х независимо представляет собой фтор, хлор или бром; р представляет собой целое число от 0 до 3; R 1 и R 3 вместе с атомом азота, с которым они связаны, образуют азетидинил, пирролидинил, пирролил, 2,5-дигидропиррол-1-ил, пиперидинил или 1,2,3,6-тетрагидропиридин-1-ил; R 2 представляет собой (С2-С6)алкинил; и его фармацевтически приемлемые соли. 13. Соединение формулы (V) где каждый Х независимо выбран из группы, состоящей из фтора и хлора; m представляет собой целое число, равное 1 или 2; R 2 представляет собой (С2-С6)алкинил; R 1 и R 3 вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидинильную или пиперидинильную группу; и его фармацевтически приемлемые соли. 14. Соединение формулы (VI) где каждый X независимо представляет собой фтор или хлор; n равно нулю или единице; R 2 представляет собой (С2-С6)алкинил; R 1 и R 3 вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидинильную или пиперидинильную группу; и его фармацевтически приемлемые соли. 15. Соединение по п.12, в котором R 1 и R 3 вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидинильную или пиперидинильную группу. 16. Соединение по одному любому из пп.12, 13 или 14, в котором R 2 является пропаргилом. 17. Соединение по п.15, в котором Х является F или Сl и n равно нулю. 18. Соединение по п.12, выбранное из группы, содержащей N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-пропаргиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-пропаргиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-пропаргиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-пропаргиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-хлорфенилсульфонил)-N"-пропаргиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; и его фармацевтически приемлемые соли. 19. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по одному любому из пп.12-15, 17 или 18. 20. Способ лечения заболевания млекопитающего, опосредованного a 4 интегринами, включающий введение фармацевтической композиции, содержащей фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по одному любому из пп.12-15, 17 или 18. 21. Способ по п.20, где заболевание опосредовано VLA-4. 22. Способ по п.20, где заболевание представляет собой воспалительное заболевание. 23. Способ по п.20, где заболевание представляет собой ревматоидный артрит.
Полный текст патента (57) Реферат / Формула: где каждый Х независимо представляет собой фтор, хлор или бром; р представляет собой целое число от 0 до 3; R 1 и R 3 вместе с атомом азота, с которым они связаны, образуют азетидинил, пирролидинил, пирролил, 2,5-дигидропиррол-1-ил, пиперидинил или 1,2,3,6-тетрагидропиридин-1-ил; R 2 выбран из группы, состоящей из (С1-С5)алкила, (С2-С6)алкенила и (С3-С6)циклоалкил(С1-С4)алкила; и его фармацевтически приемлемые соли. 2. Соединение формулы (II) где каждый Х независимо выбран из группы, состоящей из фтора и хлора; m представляет собой целое число, равное 1 или 2; R 2 выбран из группы, состоящей из (С1-С5)алкила, (С2-С6)алкенила и (С3-С6)циклоалкил(С1-С4)алкила; R 1 и R 3 вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидинильную или пиперидинильную группу; и его фармацевтически приемлемые соли. 3. Соединение формулы (III) где каждый Х независимо представляет собой фтор или хлор; n равно нулю или единице; R 2 является -CH 2 -R', где R' выбран из группы, состоящей из водорода, метила или -СН=СН 2 ; R 1 и R 3 вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидинильную или пиперидинильную группу; и его фармацевтически приемлемые соли. 4. Соединение по п.1, в котором R 1 и R 3 вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидинильную или пиперидинильную группу. 5. Соединение по одному любому из пп.1, 2 или 3, в котором R 2 является СН 3 . 6. Соединение по п.3, в котором Х является F или Сl и n равно 0. 7. Соединение по п.1, выбранное из группы, содержащей N-(2-[N',N'-диэтиламино]-5-[N"-(4-хлорфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-хлорфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(пиперидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(пиперидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-хлорфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-хлорфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; и его фармацевтически приемлемые соли. 8. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по одному любому из пп.1-4, 6 или 7. 9. Способ лечения заболевания млекопитающего, опосредованного a 4 интегринами, включающий введение фармацевтической композиции, содержащей фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по одному любому из пп.1-4, 6 или 7. 10. Способ по п.9, где заболевание опосредовано VLA-4. 11. Способ по п.9, где заболевание представляет собой воспалительное заболевание. 12. Соединение формулы (IV) где каждый Х независимо представляет собой фтор, хлор или бром; р представляет собой целое число от 0 до 3; R 1 и R 3 вместе с атомом азота, с которым они связаны, образуют азетидинил, пирролидинил, пирролил, 2,5-дигидропиррол-1-ил, пиперидинил или 1,2,3,6-тетрагидропиридин-1-ил; R 2 представляет собой (С2-С6)алкинил; и его фармацевтически приемлемые соли. 13. Соединение формулы (V) где каждый Х независимо выбран из группы, состоящей из фтора и хлора; m представляет собой целое число, равное 1 или 2; R 2 представляет собой (С2-С6)алкинил; R 1 и R 3 вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидинильную или пиперидинильную группу; и его фармацевтически приемлемые соли. 14. Соединение формулы (VI) где каждый X независимо представляет собой фтор или хлор; n равно нулю или единице; R 2 представляет собой (С2-С6)алкинил; R 1 и R 3 вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидинильную или пиперидинильную группу; и его фармацевтически приемлемые соли. 15. Соединение по п.12, в котором R 1 и R 3 вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидинильную или пиперидинильную группу. 16. Соединение по одному любому из пп.12, 13 или 14, в котором R 2 является пропаргилом. 17. Соединение по п.15, в котором Х является F или Сl и n равно нулю. 18. Соединение по п.12, выбранное из группы, содержащей N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-пропаргиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-пропаргиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-пропаргиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-пропаргиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-хлорфенилсульфонил)-N"-пропаргиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; и его фармацевтически приемлемые соли. 19. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по одному любому из пп.12-15, 17 или 18. 20. Способ лечения заболевания млекопитающего, опосредованного a 4 интегринами, включающий введение фармацевтической композиции, содержащей фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по одному любому из пп.12-15, 17 или 18. 21. Способ по п.20, где заболевание опосредовано VLA-4. 22. Способ по п.20, где заболевание представляет собой воспалительное заболевание. 23. Способ по п.20, где заболевание представляет собой ревматоидный артрит.
008256 Предшествующий уровень техники Область изобретения Данное изобретение относится к соединениям, которые ингибируют адгезию лейкоцитов, и в особенности адгезию лейкоцитов, опосредованную а4интегринами, где а4интегрин предпочтительно представляет собой VLA-4. Ссылки Следующие публикации, патенты и заявки на патент цитированы в данной заявке в соответствии со следующими индексами: 1 Hemler and Takada, заявка на Европейский патент №330506, опубликованная 30 августа 1989 г. 2 Elices, et al., Cell, 60:577-584 (1990) 3 Springer, Nature, 346:425-434 (1990) 4 Osborne, Cell, 62:3-6 (1990) 5 Vedder, et al., Surgery, 106:509 (1989) 6 Pretolani, et al., J. Exp. Med., 180:795 (1994) 7 Abraham, et al., J. Clin. Invest., 93:776 (1994) 8 Mulligan, et al., J. Immunology, 150:2407 (1993) 9 Cybulsky, et al., Science, 251:788 (1991) 10 Li, et al., Arterioscler. Thromb., 13:197 (1993) 11 Sasseville, et al., Am. J. Path., 144:27 (1994) 12 Yang, et al., Proc. Nat. Acad. Science (USA), 90:10494 (1993) 13 Burkly, et al., Diabetes, 43:529 (1994) 14 Baron, et al., J. Clin. Invest., 93:1700 (1994) 15 Hamann, et al., J. Immunology, 152:3283 (1994) 16 Yednock, et al., Nature, 356:63 (1992) 18 Baron, et al., J. Exp. Med., 177:57 (1993) 18 van Dinther-Janssen, et al., J. Immunology, 147:4207 (1991) 19 van Dinther-Janssen, et al., Annals. Rheumatic Dis., 52:672 (1993) 20 Elices, et al., J. Clin. Invest., 93:405 (1994) 21 Postigo, et al., J. Clin. Invest., 89:1445 (1991) 22 Paul, et al., Transpl. Proceed., 25:813 (1993) 23 Okarhara, et al., Can. Res., 54:3233 (1994) 24 Paavonen, et al., Int. J. Can., 58:298 (1994) 25 Schadendorf, et al., J. Path., 170:429 (1993) 2267 Bao, et al., Diff., 52:239 (1993) 27 Lauri, et al., British J. Cancer, 68:862 (1993) 29 Kawaguchi, et al., Japanese J. Cancer Res., 83:1304 (1992) 29 Konradi, et al., PCT/US00/01686, дата подачи 21 января 2000 г. Все из вышеуказанных публикаций включены в данное описание во всей своей полноте в качестве ссылок до такой степени, как если бы каждая отдельная публикация была специально и отдельно включена в качестве ссылки во всей полноте. Уровень техники VLA-4 (также именуемый а4р1интегрином и CD49d/CD29), идентифицированный впервые Hemler и Takada1, является членом р1интегринового семейства рецепторов клеточной поверхности, каждый из которых включает две субъединицы: а-цепь и в-цепь. VLA-4 содержит а4-цепь и р1-цепь. Существует по меньшей мере девять р1интегринов, имеющих одну и ту же р1-цепь и каждый из которых имеет разную а-цепь. Все указанные девять рецепторов связывают различный комплемент разных молекул клеточной матрицы, таких как фибронектин, ламинин и коллаген. VLA-4, например, связывается с фибронектином. VLA-4 связывает также нематричные молекулы, которые экспрессируются эндотелиальными и другими клетками. Указанные нематричные молекулы включают VCAM-1, который экспрессируется в культуре активированных цитокином эндотелиальных клеток человеческой пупочной вены. Различные эпитопы VLA-4 ответственны за фибронектин и связующие активности к VCAM-1, и было показано, что каждая активность угнетается независимо2. Межклеточная адгезия, опосредованная VLA-4 и другими рецепторами на поверхности клеток, связана с рядом воспалительных реакций. На участке поражения или другого раздражителя, вызывающего воспаление, активированные васкулярные эндотелиальные клетки экспрессируют молекулы, которые являются связующими для лейкоцитов. Механизмы адгезии лейкоцитов к эндотелиальным клеткам включают частью распознавание и связывание рецепторов на клеточной поверхности лейкоцитов с соответствующими молекулами на клеточной поверхности эндотелиальных клеток. После связывания лейкоциты мигрируют через стенки кровеносных сосудов, поступая в поврежденный участок и высвобождая химические медиаторы, противостоящие инфекции. Для обзора рецепторов адгезии в иммунной системе смотри, например, Springer3 и Osborn4. - 1 - 008256 Воспалительные заболевания головного мозга, такие как аутоиммунный энцефаломиелит (ЕАЕ), рассеянный склероз (MS) и менингит, являются примерами расстройств центральной нервной системы, при которых механизм адгезии эндотелия и лейкоцитов приводит к разрушению здоровой ткани головного мозга. Большое число лейкоцитов мигрирует через гематоэнцефалический барьер (ВВВ) субъектов, страдающих указанными воспалительными заболеваниями. Лейкоциты высвобождают токсичные медиаторы, которые вызывают интенсивное повреждение ткани, что приводит к нарушению нервной проводимости и параличу. В системах других органов повреждение ткани происходит также через механизм адгезии, приводящий к миграции или активации лейкоцитов. Так например, было показано, что начальная стадия инсульта с последующей миокардиальной ишемией сердечной ткани может быть дополнительно осложнена проникновением лейкоцитов в поврежденную ткань, вызывающим дополнительное поражение (Ved-der et al.)5. Другие воспалительные или болезненные состояния, опосредованные механизмом адгезии, включают, например, астму6-8, болезнь Альцгеймера, атеросклероз9-10, вызванное СПИДом слабоумие11, диабет12-14 (включающий острый юношеский диабет), воспалительные заболевания пищеварительного тракта15 (включающие язвенный колит и болезнь Крона), рассеянный склероз16-17, ревматоидный арт- 18-21 22 23-28 рит18-21, трансплантацию тканей22, опухолевый метастаз23-28, менингит, энцефалит, инсульт и другие церебральные повреждения, нефрит, ретинит, атопический дерматит, псориаз, миокардиальную ишемию и опосредованное лейкоцитами острое поражение легких, которое, например, встречается при респираторном дистресс-синдроме у взрослых людей. Раскрыты как класс замещенные аминопиримидины, которые ингибируют связывание VLA-4 с VCAM-1 и соответственно показывают противовоспалительные свойства29. В то время как указанные соединения обладают антагонистическими свойствами к данному связыванию, повышенная биодоступность представленных соединений будет усиливать их действенность. Сущность изобретения В данном изобретении обнаружено, что некоторые производные №[2-№,№-диэтиламино-5-фенилсульфониламинопиримидин-4-ил]-п-карбомилоксифенилаланина неожиданно обладают превосходной биодоступностью, которая измеряется их AUC, по сравнению с другими, прежде раскрытыми замещенными производными аминопиримидина. В соответствии с одним из составляющих его аспектов, данное изобретение относится к соединению формулы (I) где каждый Х независимо представляет собой фтор, хлор или бром; р представляет собой целое число от 0 до 3; R1 и R3 вместе с атомом азота, с которым они связаны, образуют азетидинил, пирролидинил, пирро-лил, 2,5-дигидропиррол-1-ил, пиперидинил или 1,2,3,6-тетрагидропиридин-1-ил; R2 выбран из группы, состоящей из низшего алкила, низшего алкенила и низшего алкиленциклоал- кила; и к его фармацевтически приемлемым солям. В предпочтительном варианте R1 и R3 вместе с атомом азота, с которым они связаны, образуют азе-тидинильную, пирролидинильную или пиперидинильную группу. В предпочтительном варианте данного изобретения предлагаются соединения формулы (II) - 2 - 008256 где каждый Х независимо выбран из группы, состоящей из фтора и хлора; m представляет собой целое число, равное 1 или 2; R2 выбран из группы, состоящей из низшего алкила, низшего алкенила и низшего алкиленциклоал- кила; R1 и R3 вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидиниль-ную или пиперидинильную группу; и их фармацевтически приемлемые соли. В особенно предпочтительном варианте данного изобретения предлагаются соединения формулы (III) где каждый Х независимо представляет собой фтор или хлор; n равно нулю или единице; R2 является -СН2-R', где R' выбран из группы, состоящей из водорода, метила или -СН=СН2; R1 и R3 вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидиниль-ную или пиперидинильную группу; и их фармацевтически приемлемые соли. В соответствии с другим составляющим его аспектом, данное изобретение относится к соединению формулы (IV) - 3 - 008256 OV) где каждый Х независимо представляет собой фтор, хлор или бром; р представляет собой целое число от 0 до 3; R1 и R3 вместе с атомом азота, с которым они связаны, образуют азетидинил, пирролидинил, пирро-лил, 2,5-дигидропиррол-1-ил, пиперидинил или 1,1,3,6-тетрагидропиридин-1-ил; R2 представляет собой низший алкинил; и к его фармацевтически приемлемым солям. В предпочтительном варианте R1 и R3 вместе с атомом азота, с которым они связаны, образуют азе-тидинильную, пирролидинильную или пиперидинильную группу и R2 представляет собой пропаргил. В предпочтительном варианте данного изобретения предлагаются соединения формулы (V) (V) где каждый Х независимо выбран из группы, состоящей из фтора и хлора; m представляет собой целое число, равное 1 или 2; R2 представляет собой низший алкинил; R1 и R3 вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидиниль-ную или пиперидинильную группу; и их фармацевтически приемлемые соли. В особо предпочтительном варианте данного изобретения предлагаются соединения формулы (VI) - 4 - 008256 где каждый Х независимо представляет собой фтор или хлор; n равно нулю или единице; R2 представляет собой низший алкинил; R1 и R3 вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидиниль-ную или пиперидинильную группу; и их фармацевтически приемлемые соли. Входящие в область данного изобретения производные №[2-1ЧР,№-диэтиламино-5-аминосульфонилфенилпиримидин-4-ил]-п-карбомилоксифенилаланина включают такие, которые перечислены в табл. 1. Таблица 1 R1 и R3 Соединение № Пирролидинил Этил 4-фторфенил - 5 - 008256 Пирролидинил Метил -фторфенил Пирр олидинил Метил -хлорфенил Пирр олидинил Этил -хлорфенил Пиперидинил Метил -фторфенил Азетидинил Этил -фторфенил Азетидинил Метил -фторфенил Азетидинил Метил -хлорфенил Азетидинил Этил -хлорфенил Пиперидинил Этил -фторфенил Азетидинил Этил -дифторфенил Пирролидинил Метил -дифторфенил Пирролидинил Этил -дифторфенил Азетидинил Метил -дифторфенил Пирр олидинил Пропаргил -фторфенил Пирролидинил Пропаргил -дифторфенил Азетидинил Пропаргил -дифторфенил Азетидинил Пропаргил -фторфенил Пирролидинил Пропаргил -хлорфенил Отдельные соединения, входящие в область данного изобретения, включают следующие соединения. Представленные ниже соединения названы как производные фенилаланина, но альтернативно данные соединения могут быть названы как производные №[2-1ЧГДЧ'-диэтиламино-5-аминосуль-фонилфенилпиримидин-4-ил]-п-карбомилоксифенилаланина или производные 2-({2-диэтиламино-5-[(бензолсульфонил)метиламино]пиримидин-4-иламино}-п-карбамоилоксифенил)пропионовой кислоты. N-(2-[N',N'-диэтиламино]-5-[N"-(4-хлорфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(пирролидин-1 -илкарбонилокси)-Ь-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(пирролидин-1 -илкарбонилокси)-Ь-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(пирролидин-1 -илкарбонилокси)-Ь-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-хлорфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(пирролидин-1 -илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(пиперидин-1 -илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(пиперидин-1 -илкарбонилокси)^-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(азетидин-1 -илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(азетидин-1 -илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-хлорфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(азетидин-1 -илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(4-хлорфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(азетидин-1 -илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(пирролидин-1 -илкарбонилокси)-Ь-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(пирролидин-1 -илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(азетидин-1 -илкарбонилокси)-L-фенилаланин; N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(азетидин-1 -илкарбонилокси)-L-фенилаланин; - 6 - 008256 М-(2-[М',М'-диэтиламино]-5-[М"-(4-фторфенилсульфонил)-М"-пропаргиламино]пиримидин-4-ил)-4'-(пирролидин-1 -илкарбонилокси)-L-фенилаланин; М-(2-[М',М'-диэтиламино]-5-[М"-(2,4-дифторфенилсульфонил)-М"-пропаргиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; М-(2-[М',М'-диэтиламино]-5-[М"-(2,4-дифторфенилсульфонил)-М"-пропаргиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; М-(2-[М',М'-диэтиламино]-5-[М"-(4-фторфенилсульфонил)-М"-пропаргиламино]пиримидин-4-ил)-4'-(азетидин-1 -илкарбонилокси)-L-фенилаланин; М-(2-[М',М'-диэтиламино]-5-[М"-(4-хлорфенилсульфонил)-М"-пропаргиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; и их фармацевтически приемлемые соли. В соответствии с другим аспектом в данном изобретении предлагаются фармацевтические композиции, содержащие фармацевтически приемлемый носитель и терапевтически эффективное количество представленных в данном описании соединений. В соответствии с одним из аспектов, касающихся способа, данное изобретение относится к способу лечения заболевания, опосредованного, по меньшей мере, частично а4интегринами, предпочтительно VLA-4, включающему введение фармацевтической композиции, содержащей фармацевтически приемлемый носитель и терапевтически эффективное количество соединения данного изобретения. Соединения и фармацевтические композиции данного изобретения полезны для лечения болезненных состояний, опосредованных, по меньшей мере, частично а4интегринами, предпочтительно VLA-4, или адгезией лейкоцитов. Такие болезненные состояния включают в качестве примера астму, болезнь Альцгеймера, атеросклероз, вызванное СПИДом слабоумие, диабет (включающий острый юношеский диабет), воспалительные заболевания пищеварительного тракта (включающие язвенный колит и болезнь Крона), рассеянный склероз, ревматоидный артрит, трансплантацию тканей, опухолевый метастаз, менингит, энцефалит, инсульт и другие церебральные повреждения, нефрит, ретинит, атонический дерматит, псориаз, миокардиальную ишемию и опосредованное лейкоцитами острое поражение легких, которое, например, встречается при респираторном дистресс-синдроме у взрослых людей. Другие болезненные состояния включают, но без ограничения, воспалительные состояния, такие как нодозная эритема, аллергический конъюнктивит, ретробульбарный неврит, увеит, аллергический ринит, анкилозирующий спондилоартрит, псориатический артрит, васкулит, синдром Рейтера, системная красная волчанка, прогрессирующий системный склероз, полимиозит, дерматомиозит, некротический неинфекционный гранулематоз, аортит, саркоидоз, лимфопения, височный артериит, перикардит, миокардит, застойная сердечная недостаточность, нодозный полиартериит, синдром повышенной чувствительности, аллергия, гиперэозинофильные синдромы, синдром Херг-Страусса, хроническое обструктив-ное легочное заболевание, пневмонию, с повышенной чувствительностью, хронический активный гепатит, интерстициальный цистит, аутоиммунная эндокринная недостаточность, первичный биллиарный цирроз печени, аутоиммунная гипопластическая анемия, хронический прогрессирующий гепатит и ти-реоидит. В предпочтительном варианте болезненное состояние представляет собой воспаление. Подробное описание изобретения Как указывалось выше, данное изобретение относится к соединениям, которые ингибируют адгезию лейкоцитов, и в особенности адгезию лейкоцитов, опосредованную, по меньшей мере, частично а4интегринами, предпочтительно VLA-4. Однако перед подробным описанием изобретения сначала будут определены следующие термины. Определения Если не указано иначе, использованные в описании и формуле изобретения следующие термины имеют указанные ниже значения. Использованный в данном описании термин "низший алкил" относится к одновалентным алкиль-ным группам, имеющим от 1 до 5 атомов углерода, включающим алкильные группы с прямой и разветвленной цепью. В качестве примера данный термин включает группы, такие как метильная, этильная, изопропильная, н-пропильная, н-бутильная, изобутильная, втор-бутильная, трет-бутильная, н-пентильная и подобные. Термин "низший алкилен" относится к двухвалентным алкиленовым группам, имеющим от 1 до 4 атомов углерода, включающим алкиленовые группы с прямой и разветвленной цепью. В качестве примера данный термин включает группы, такие как метиленовая, этиленовая, н-пропиленовая, изопропиле-новая (-СН2СН(СН3)- и -СН(СН3)СН2-) и подобные. В соответствии с рекомендациями IUPAC термину "низший алкенил" отвечает термин "алкандиил". Термин "низший алкенил" относится к алкенильной группе, предпочтительно имеющей от 2 до 6 атомов углерода и имеющей по меньшей мере 1 положение и предпочтительно только 1 положение с алкенильной ненасыщенностью (то есть > С=С <). В качестве примера данный термин включает группы, такие как аллильная, этенильная, пропенильная, бутенильная и подобные. - 7 - 008256 Термин "низший алкинил" относится к алкинильной группе, предпочтительно имеющей от 2 до 6 атомов углерода и имеющей по меньшей мере 1 положение и предпочтительно только 1 положение с алкинильной ненасыщенностью (то есть -С=С-). В качестве примера данный термин включает группы, такие как ацетиленильная (-С=СН), пропаргильная (-СН2-С=СН), 3-бутинильная (-СН2СН2С=СН3) и подобные. Термин "низший циклоалкил" относится к циклическим алкильным группам, содержащим от 3 до 6 атомов углерода, имеющим одно циклическое кольцо, включающее, например, циклопропильное, цик-лобутильное, циклопентильное и циклогексильное. Термин "низший алкиленциклоалкил" относится к группе, состоящей из низшего алкилена и низшего циклоалкила, определенных в данном описании. В качестве примера данный термин включает группы, такие как метиленциклопропильная (-СН2-циклопропил), этиленциклопропильная и подобные. В соответствии с рекомендациями IUPAC термину "низший алкиленциклоалкил" отвечает термин "цикло-алкилалкил". Термин "фармацевтически приемлемый носитель" означает носитель, применимый при приготовлении фармацевтической композиции, который обычно является безопасным, нетоксичным и биологически или иным образом желательным, и включает носитель, являющийся приемлемым для применения в ветеринарии, а также для использования при получении фармацевтических препаратов для человека. Использованный в данном описании и формуле изобретения термин "фармацевтически приемлемый носитель" включает как один, так и несколько таких носителей. Термины "терапия" или "лечение" заболевания включают: (1) профилактику заболевания, то есть создание условий, при которых не происходит развитие клинических симптомов заболеваний у млекопитающего, которое может быть подвержено или предрасположено к заболеванию, но симптомы заболевания не проявляются и не обнаруживаются, (2) ингибирование заболевания, то есть остановку или уменьшение развития заболевания или его клинических симптомов или (3) ослабление заболевания, то есть регрессию заболевания или его клинических симптомов. Термин "терапевтически эффективное количество" означает количество соединения, которое при введении млекопитающему для лечения заболевания является достаточным для осуществления такого лечения заболевания. "Терапевтически эффективное количество" будет зависеть от соединения, заболевания и его тяжести, возраста и массы тела подвергаемого лечению млекопитающего и т.д. Термин "фармацевтически приемлемая соль" относится к фармацевтически приемлемым солям соединения формулы I, соли которого образованы множеством органических и неорганических противоио-нов, хорошо известных в данной области, и включают, только в качестве примера, ионы натрия, калия, кальция, магния, аммония, тетраалкиламмония и подобные; и когда молекула содержит основную функциональную группу - соли органических или неорганических кислот, такие как гидрохлорид, гидробромид, тартрат, мезилат, ацетат, малеат, оксалат, и подобные соли. Интегрины представляют собой большое семейство гомологичных трансмембранных линкерных белков, которые являются основными рецепторами в клетках животных, связывающими большинство белков внеклеточной матрицы, таких как коллаген, фибронектин и ламинин. Интегрины представляют собой гетеродимеры, состоящие из а-цепи и в-цепи. На сегодняшний день идентифицировано двадцать различных интегриновых гетеродимеров, состоящих из 9 различных а-субъединиц и 14 различных 0-субъединиц. Термин "а4интегрины" относится к классу гетеродимерных, связанных ферментом рецепторов на поверхности клеток, которые содержат а4-субъединицу, спаренную с любой из 0-субъединиц. VLA-4 является примером а4интегрина и представляет собой гетеродимер, состоящий из а4- и 01-субъединиц, который также именуется а401интегрином. Получение соединений Соединения данного изобретения могут быть получены из легкодоступных исходных продуктов с использованием способов и методик, представленных ниже в примерах. Представленные способы и методики в общих чертах раскрывают протоколы специальных реакций, предназначенных для получения производных М-[2-М',М'-диэтиламино-5-аминосульфонилфенилпиримидин-4-ил]-п-карбомилоксифенил-аланина. Соединения, входящие в область данного изобретения, не приведенные в данных примерах и способах, можно легко получить соответствующей заменой исходных продуктов, которые являются коммерчески доступными или хорошо известны в данной области. Другие методики и условия реакций получения соединения данного изобретения описаны в ниже-представленных примерах. Кроме того, другие методики получения соединений, применимых в некоторых аспектах данного изобретения, раскрыты в патенте США №6492372, опубликованном 10 декабря 2002 г., описание которого во всей его полноте включено в данное изобретение в качестве ссылки. Фармацевтические препараты При использовании в виде фармацевтических препаратов соединения данного изобретения вводят обычно в форме фармацевтических композиций. Указанные композиции могут быть введены множеством путей, включающих пероральный, ректальный, чрескожный, подкожный, внутривенный, внутри - 8 - 008256 мышечный и интраназальный. Указанные композиции являются эффективными при их доставке путем инъекции или пероральным путем. Такие композиции приготавливают способом, хорошо известным в области фармации, и они содержат по меньшей мере одно активное соединение. Данное изобретение включает также фармацевтические композиции, содержащие в качестве активного ингредиента одно или более указанных выше соединений формулы I, ассоциированных с фармацевтически приемлемыми носителями. При приготовлении композиций данного изобретения активный ингредиент обычно смешивают с эксципиентом, разбавляют эксципиентом или заключают в такой носитель, который может быть в форме капсулы, саше, бумаги и другого контейнера. Используемый эксци-пиент обычно представляет собой эксипиент, подходящий для введения людям или другим млекопитающим. Когда эксипиент служит в качестве разбавителя, он может быть твердым, полутвердым или жидким материалом, который действует в качестве наполнителя, носителя или среды для активного ингредиента. Таким образом, композиции могут быть в форме таблеток, пилюль, порошков, лепешек, саше, крахмальных капсул, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозолей (в виде твердого вещества или в жидкой среде), мазей, содержащих, например, до 10 мас.% активного соединения, мягких и твердых желатиновых капсул, суппозиториев, стерильных инъецируемых растворов и стерильных упакованных порошков. При приготовлении препарата может стать необходимым измельчение активного соединения перед объединением его с другими ингредиентами для обеспечения подходящего размера частиц. Если активное соединение является по существу нерастворимым, его обычно измельчают до размера частиц менее 200 меш. Если активное соединение является по существу водорастворимым, размер частиц обычно регулируют измельчением, например до около 40 меш, для обеспечения по существу однородного распределения в препарате. Некоторые примеры подходящих эксципиентов включают лактозу, декстрозу, сахарозу, сорбит, маннит, крахмалы, аравийскую камедь, фосфат кальция, альгинаты, трагакант, желатин, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, воду, сироп и метилцеллю-лозу. Препараты могут дополнительно включать смазывающие вещества, такие как тальк, стеарат магния и минеральное масло; увлажняющие средства; эмульгирующие и суспендирующие средства; консерванты, такие как метил- и пропилгидроксибензоаты; подсластители и корригенты. Композиции изобретения могут быть приготовлены таким образом, чтобы после введения больному с использованием методик, известных в данной области, было обеспечено быстрое, пролонгированное или замедленное высвобождение активного ингредиента. Композиции предпочтительно приготавливают в виде стандартной лекарственной формы, при этом каждая доза содержит от около 5 до около 100 мг, более предпочтительно от около 10 до около 30 мг активного ингредиента. Термин "единичные дозированные формы" относится к физически дискретным единицам, подходящим в качестве единичных доз для людей и других млекопитающих, причем каждая единица содержит заданное количество активного вещества, рассчитанное на получение требуемого терапевтического действия, в ассоциации с подходящим фармацевтическим эксципиентом. Активное соединение является эффективным в широком диапазоне доз и его обычно вводят в фармацевтически эффективном количестве. Однако, понятно, что количество вводимого соединения в действительности будет определяться лечащим врачом с точки зрения конкретных обстоятельств, включающих состояние больного, выбранный путь введения, данное вводимое соединение, возраст, массу тела и реакцию отдельного больного, тяжесть симптомов больного и подобные факторы. Для получения твердых композиций, таких как таблетки, основной активный ингредиент смешивают с фармацевтическим эксципиентом с образованием твердой предварительной композиции, содержащей гомогенную смесь соединения настоящего изобретения. Ссылка на "гомогенные предварительные композиции" означает, что активный ингредиент однородно диспергирован в композиции с тем, чтобы композиция могла быть легко разделена на равные эффективные единичные дозированные формы, такие как таблетки, пилюли и капсулы. Затем полученную предварительную композицию разделяют на единичные дозированные формы указанного выше типа, содержащие, например, от 0,1 до около 500 мг активного ингредиента настоящего изобретения. Таблетки или пилюли настоящего изобретения могут быть покрыты оболочкой или иным образом приготовлены с получением лекарственной формы, дающей преимущества пролонгированного действия. Так например, таблетка или пилюля может содержать находящийся внутри компонент и находящийся снаружи компонент, при этом последний из указанных присутствует в форме оболочки, покрывающей первый из указанных компонентов. Два компонента могут быть разделены энтеросолюбильным слоем, который служит для противостояния распадаемости в желудке и дает возможность внутреннему компоненту поступать в неизменной форме в двенадцатиперстную кишку или высвобождаться пролонгиро-ванно. Для указанных энтеросолюбильных слоев может быть использовано множество материалов, которые включают ряд полимерных кислот и смесей полимерных кислот с такими материалами, как шеллак, цетиловый спирт и ацетат целлюлозы. Жидкие формы для перорального введения или инъекции, в которые могут быть включены новые композиции настоящего изобретения, включают водные растворы, предпочтительно имеющие приятный - 9 - 008256 вкус сиропы, водные или масляные суспензии и имеющие приятный вкус эмульсии со съедобными маслами, такими как хлопковое масло, кунжутное масло, кокосовое масло или арахисовое масло, а также эликсиры и подобные фармацевтические наполнители. Композиции для ингаляции или инсуффляции включают растворы и суспензии в фармацевтически приемлемых водных или органических растворителях или их смесях и порошки. Жидкие или твердые композиции могут содержать указанные выше подходящие фармацевтически приемлемые эксципиенты. Композиции предпочтительно вводят оральным путем или путем вдыхания для оказания местного или системного действия. Композиции в предпочтительных фармацевтически приемлемых растворителях могут быть распылены с использованием инертных газов. Распыленные растворы можно вдыхать непосредственно из ингалятора, или ингалятор может быть прикреплен к тампону лицевой маски, или из дыхательного периодически действующего аппарата для вентиляции легких положительного давления. Композиции в виде раствора, суспензии или порошка могут быть предпочтительно введены перорально или назально из приспособлений, которые доставляют препарат соответствующим путем. Фармацевтические композиции настоящего изобретения иллюстрируют следующие примеры препаратов. Пример 1 препарата. Получают твердые желатиновые капсулы, содержащие следующие ингредиенты: Ингредиент Количество (мг/капсулу) Активный ингредиент 30,0 Крахмал 305,0 Стеарат магния 5,0 Вышеуказанные ингредиенты смешивают и помещают в количестве 340 мг в твердые желатиновые капсулы. Пример 2 препарата. Таблетированный препарат получают с использованием нижеуказанных ингредиентов: Ингредиент Количество (мг/таблетку) Активный ингредиент 25,0 Целлюлоза микрокристаллическая 200,0 Коллоидный диоксид кремния 10,0 Стеариновая кислота 5,0 Компоненты смешивают и прессуют с образованием таблеток, масса каждой из которых составляет 240 мг. Пример 3 препарата. Получают лекарственный препарат для ингаляции сухого порошка, содержащий следующие компоненты: Ингредиент Мас.% Активный ингредиент 5 Лактоза 95 Активную смесь перемешивают с лактозой и смесь вводят в приспособление для вдыхания сухого порошка. Пример 4 препарата. Таблетки, каждая из которых содержит 30 мг активного ингредиента, получают следующим образом: Ингредиент Количество (мг/таблетку) Активный ингредиент 30,0 Крахмал 45,0 Микрокристаллическая целлюлоза 35,0 Поливинилпирролидон (в виде 10% раствора в воде) 4,0 Натрийкарбоксиметиловый крахмал 4,5 Стеарат магния 0,5 Тальк 1,0 Всего 120 Активный ингредиент, крахмал и целлюлозу пропускают через сито с размером ячеек 20 меш США и тщательно перемешивают. С образовавшимися порошками смешивают раствор поливинилпирролидо-на и затем пропускают через сито с размером ячеек 16 меш США. Полученные таким образом гранулы сушат при температуре 50-60°С и пропускают через сито с размером ячеек 16 меш США. Натрийкарбок-симетиловый крахмал, стеарат магния и тальк предварительно пропускают через сито с размером ячеек 30 меш США и затем добавляют к гранулам, которые после перемешивания прессуют на таблетирующей машине с получением таблеток, каждая из которых имеет массу 120 мг. Пример 5 препарата. Капсулы, каждая из которых содержит 40 мг лекарственного средства, изготавливают следующим образом: - 10 - 008256 Ингредиент Количество (мг/капсулу) Активный ингредиент 40,0 Крахмал 109,0 Стеарат магния 1,0 Всего 150,0 Активный ингредиент, крахмал и стеарат магния смешивают и пропускают через сито с размером ячеек 20 меш США и помещают в количестве 150 мг в твердые желатиновые капсулы. Пример 6 препарата. Суппозитории, каждый из которых содержит 25 мг активного ингредиента, изготавливают следующим образом: Ингредиент Количество Активный ингредиент 25 мг Глицериды насыщенной жирной кислоты 2,000 мг Активный ингредиент пропускают через сито с размером ячеек 60 меш США и суспендируют в глицеридах насыщенной жирной кислоты, предварительно расплавленных с использованием минимально необходимого количества тепла. Затем смесь вливают в форму для отливки суппозиториев емкостью 2,0 г и дают возможность охладиться. Пример 7 препарата. Суспензии, каждая из которых содержит 50 мг лекарственного средства в 5,0 мл дозе, получают следующим образом: Ингредиент Количество Активный ингредиент 50,0 мг Ксантановая смола 4,0 мг Натрийкарбоксиметилцеллюлоза (11%) Микрокристаллическая целлюлоза (89%) 50,0 мг Сахароза 1,75 г Бензоат натрия 10,0 мг Корригент и красящее вещество q.v. Дистиллированная вода До 5,0 мл Лекарственное средство, сахарозу и ксантановую смолу смешивают, пропускают через сито с размером ячеек 10 меш США и затем смешивают с предварительно приготовленным раствором микрокристаллической целлюлозы и натрийкарбоксиметилцеллюлозой в воде. Бензоат натрия, корригент и красящее вещество разбавляют некоторым количеством воды и добавляют при перемешивании. Затем для получения требуемого объема добавляют достаточное количество воды. Пример 8 препарата. Ингредиент Количество (мг/капсулу) Активный ингредиент 15,0 Крахмал 407,0 Стеарат магния 3,0 Всего 425,0 Активный ингредиент, крахмал и стеарат магния смешивают, пропускают через сито с размером ячеек 20 меш США и помещают в количестве 425 мг в твердые желатиновые капсулы. Пример 9 препарата. Препарат для внутривенного введения может быть получен следующим образом: Ингредиент Количество Активный ингредиент 250,0 мг Изотонический солевой раствор 1000 мл Пример 10 препарата. Препарат для местного введения может быть получен следующим образом: Ингредиент Количество Активный ингредиент 1-10 г Эмульгирующий воск 30 г Жидкий вазелин 20 г Белый мягкий парафин До 100 г Белый мягкий парафин нагревают до расплавления. Вводят жидкий вазелин и эмульгирующий воск и перемешивают до растворения. Добавляют активный ингредиент и продолжают перемешивание до образования суспензии. Затем смесь охлаждают до получения твердого вещества. Другой предпочтительный препарат, используемый в способах настоящего изобретения, применяется в приспособлениях для чрескожной доставки ("пластыря"). Такие чрескожные пластыри могут быть использованы для непрерывной или прерывистой инфузии соединений настоящего изобретения в регулируемых количествах. Строение и применение чрескожных пластырей для доставки лекарственных средств хорошо известно в данной области. Смотри, например, патент США №5023252, опубликованный - 11 - 008256 11 июня 1991 г., включенный в данное описание в качестве ссылки. Такие пластыри могут быть созданы для непрерывной, пульсирующей или требуемой доставки лекарственных средств. Когда фармацевтические композиции желательно или необходимо ввести в головной мозг, могут быть использованы прямые или непрямые методики введения. Прямые методики обычно включают помещение катетера для доставки лекарственного средства в вентрикулярную систему хозяина для обхода гематоэнцефалического барьера. Одна такая система имплантируемой доставки, используемая для переноса биологических факторов в специфические анатомические области тела, описана в патенте США №5011472, включенном в данное описание в качестве ссылки. Непрямые методики, которые обычно являются предпочтительными, включают обычно приготовление композиций с обеспечением латентного состояния лекарственного средства за счет превращения гидрофильных лекарственных средств в растворимые в липидах лекарственные средства. Латентное состояние достигается обычно через блокирование присутствующих в лекарственном средстве гидро-ксильной, карбонильной, сульфатной и первичной аминогруппы, делающее лекарственное средство более растворимым в липидах и поддающимся переносу через гематоэнцефалический барьер. Альтернативно, доставка гидрофильных лекарственных средств может быть ускорена за счет внутриартериально-го вливания гипертонических растворов, которые могут временно открывать гематоэнцефалический барьер. Применение Соединения данного изобретения ингибируют in vivo адгезию лейкоцитов к эндотелиальным клеткам, опосредованную, по меньшей мере, частично а4интегринами, предпочтительно VLA-4, за счет конкурентного связывания с а4интегринами, предпочтительно VLA-4. Соответственно, соединения данного изобретения могут быть использованы при лечении заболеваний млекопитающих, опосредованных, по меньшей мере, частично а4интегринами, предпочтительно VLA-4, или адгезией лейкоцитов. Такие заболевания включают воспалительные заболевания млекопитающих больных, такие как астма, болезнь Альцгеймера, атеросклероз, вызванное СПИДом слабоумие, диабет (включающий острый юношеский диабет), воспалительные заболевания пищеварительного тракта (включающие язвенный колит и болезнь Крона), рассеянный склероз, ревматоидный артрит, трансплантация тканей, опухолевый метастаз, менингит, энцефалит, инсульт и другие церебральные повреждения, нефрит, ретинит, атопический дерматит, псориаз, миокардиальная ишемия и опосредованное лейкоцитами острое поражение легких, такое как то, которое встречается при респираторном дистресс-синдроме у взрослых людей. Количество лекарственного средства, вводимое млекопитающему больному, будет изменяться в зависимости от самого лекарственного средства, цели введения, такой как профилактика или терапия, состояния больного, способа введения и подобных факторов. При терапевтических применениях композиции вводят больному, уже страдающему заболеванием, в количестве, достаточном для лечения, или, по меньшей мере, для частичной остановки развития симптомов заболевания и его осложнений. Количество, адекватное для осуществления этого, определяется как "терапевтически эффективная доза". Количество, эффективное для указанного применения, будет зависеть от подвергаемого лечению болезненного состояния, а также от оценки лечащего врача, зависящей от таких факторов, как тяжесть воспаления, возраст, масса тела и общее состояние больного, и подобных факторов. Вводимые больному композиции находятся в форме вышеуказанных фармацевтических композиций. Данные композиции могут быть стерилизованы традиционными методиками стерилизации или могут быть стерильно отфильтрованы. Полученные водные растворы могут быть упакованы для применения как таковых или лиофилизованы, при этом лиофилизованный препарат перед введением объединяют со стерильным водным носителем. рН препаратов лекарственного соединения обычно составляет между 3 и 11, более предпочтительно от 5 до 9 и наиболее предпочтительно от 7 до 8. Понятно, что использование некоторых из вышеуказанных эксципиентов, носителей или стабилизаторов будет приводить к образованию фармацевтических солей. Терапевтическая доза соединений настоящего изобретения будет изменяться в соответствии, например, с конкретным использованием, для которого проводится лечение, путем введения соединения, здоровьем и состоянием больного и оценкой лечащего врача. Так, например, для внутривенного введения доза будет обычно находится в диапазоне от около 20 мкг до около 500 мкг на килограмм массы тела, предпочтительно от около 100 мкг до около 300 мкг на килограмм массы тела. Подходящие дозы для интраназального введения обычно находятся в диапазоне от около 0,1 пг до 1 мг на килограмм массы тела. Эффективные дозы могут быть экстраполированы из кривых зависимости "доза-эффект", полученных в испытании in vitro, или из систем испытаний моделей заболеваний на животных. Для иллюстрации данного изобретения предлагаются следующие примеры синтеза и биологические примеры, которые не следует истолковывать как примеры, ограничивающие каким-либо образом область изобретения. Все температуры приведены в градусах Цельсия, если не указано иначе. Примеры В представленных ниже примерах последующие сокращения имеют следующие значения. Если сокращение не определено, оно имеет его общепринятое значение. - 12 - 008256 AUC = площадь под кривой bd = широкий дублет bs = широкий синглет BSA = бычий сывороточный альбумин d = дублет DMAP = гидрохлорид 4-М,М-диметиламинопиридинэтилкарбодиимида EDTA = этилендиаминтетрауксусная кислота EtOAc = этилацетат EtOH = этанол eq = эквивалент FACS = сортировщик клеток, активированных флуоресценцией FITC = флуоресцеинизотиоцианат г = граммы i. р. = внутрибрюшинный ч = час HBSS = сбалансированный солевой раствор Хэнка Hct = гематокрит или измерение эритроцитарной массы, полученной центрифугированием в объеме пробы крови НВ или НЬ = гемоглобин HEPES = 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота IgGFc = связующий домен иммуноглобулина кг = килограмм л = литр m = мультиплет (когда используется с данными ЯМР) М = молярный МСН = среднее содержание корпускулярного гемоглобина; Hb/RBC МСНС = подсчет среднего содержания корпускулярного гемоглобина, выраженного в процентах; Hb/Hct MCV = средний корпускулярный объем; средний объем эритроцитов, обычно выраженный в кубических микрометрах на эритроцит МеОН = метанол мг = миллиграмм мл = миллилитр мм = миллиметр мМ = миллимолярный моль = моли ммоль = миллимоль mpk = миллиграммы на килограмм N = нормальный нг = нанограммы РВ8++ = фосфатный буферный солевой раствор psi = фунты на квадратный дюйм q.s. или Q.S. = доведение до объема Rfs или Rf = фактор удерживания rpm = число оборотов в минуту к. т. или КТ = комнатная температура s = синглет t = триплет TFA = трифторуксусная кислота THF = тетрагидрофуран TLC или tlc = тонкослойная хроматография мкл = микролитр мкг = микрограмм мкм = микрометр Vt = общий объем WBC = лейкоциты w/v = мас./об. Соединения настоящего изобретения могут быть получены как показано на схеме 1 и как описано в представленных ниже способах. - 13 - 008256 Пример 1. Получение N-(2-[N',N'-диэтиламино]-5-[N"-(4-хлорфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)^-фенилаланина. Стадия 1. Получение 2,4-дихлор-5-нитропиримидина (2). 5-Нитроурацил (1) обрабатывали оксихлоридом фосфора (РОС13) и N,N-диметиланилином (PhNMe2) в соответствии с методикой Whittaker (J. Chem. Soc. 1951, 1565) с получением соединения 2. Соединение 2 также доступно от City Chemical (West Haven, CT). Стадия 2. Получение сложного трет-бутилового эфира №(2-[№,№-диэтиламино]-5-нитропиримидин-4-ил)^-тирозина (3). К раствору сложного трет-бутилового эфира L-тирозина (Н-Туг(ОН)-ОШи) (30,6 г, 0,129 моль) в ТГФ (250 мл) при -10°С добавляли 2,4-дихлор-5-нитропиримидин (25 г, 0,129 моль), поддерживая во время добавления температуру ниже 5°С. Сразу после завершения добавления добавляли по каплям N,N-диизопропилэтиламин (EtiPr2N) (33,7 мл, 0,194 моль). После перемешивания в течение 1 ч при -10°С медленно добавляли диэтиламин (Et2NH) (66,73 мл, 0,645 моль) и затем реакционная смесь нагревалась всю ночь до комнатной температуры. Реакционную смесь разбавляли диэтиловым эфиром (500 мл) и органический слой промывали 0,2N лимонной кислотой (3x150 мл), водой (1x150 мл) и 10% К2СО3 (3x150 мл). Органическую фазу сушили (Na2SO4), фильтровали и концентрировали в вакууме с получе - 14 - 008256 нием желтого остатка. Остаток очищали флэш-хроматографией (смесь 20% EtOAc/гексан на силикагеле) с получением 37,39 г (67%) соединения 3 в виде желтой пены. Rf = 0,21 (смесь 25% EtOAc/гексан на си-ликагеле). Стадия 3. Получение сложного трет-бутилового эфира №(2-[№,№-диэтиламино]-5-нитропиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланина (4). К раствору сложного трет-бутилового эфира №(2-[№,№-диэтилшино]-5-нитропиримидин-4-ил)^-тирозина (37,39 г, 0,087 моль) в СН2С12 (150 мл) добавляли DMAP (10,59 г, 0,087 моль). Через 5 мин добавляли по каплям триэтиламин (TEA) (18,19 мл, 0,131 моль). Добавляли по каплям 1-пирролидинкарбамоилхлорид (14,42 мл, 0,131 моль) и реакционную смесь кипятили с обратным холодильником (40°С) всю ночь. Реакционную смесь концентрировали в вакууме и переносили в EtOAc (300 мл). Органическую фазу промывали 0,2N лимонной кислотой (3x150 мл), водой (1x150 мл), насыщенным раствором NaHCO3 (3x150 мл), насыщенным солевым раствором (1х150 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением 43,07 г (94%) соединения 4 в виде желтого твердого вещества. Rf = 0,5 (смесь 50% EtOAc/гексан на силикагеле). Стадия 4. Получение сложного трет-бутилового эфира №(2-[№,№-диэтиламино]-5-аминопиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланина (5). Смесь сложного трет-бутилового эфира №(2-[№,№-диэтиламино]-5-нитропиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)^-фенилаланина (43,07 г, 0,081 моль) и 10% Pd/C (4,3 г, 10 мас.% Pd) в EtOH (200 мл) встряхивали под давлением водорода 45 фунтов/дюйм (310,3 кПа) до тех пор, пока TLC (смесь 50% EtOAc/гексан на силикагеле) стала показывать 100% превращение в продукт (48 ч). Затем реакционную смесь фильтровали через рыхлый слой целита и концентрировали в вакууме с получением 40,29 г (100%) соединения 5 в виде пурпурной пены. Rf = 0,11 (смесь EtOAc/гексан 6:1 на силикагеле). Стадия 5. Получение сложного трет-бутилового эфира №(2-[№,№-диэтиламино]-5-[№'-(4-хлорфенилсульфонил)амино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланина (6). Пиридиновый раствор (160 мл) №(2-[№,№-диэтилшино]-5-аминопиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)^-фенилаланина (40,29 г, 0,081 моль) охлаждали до -20°С на бане со смесью сухой лед/СН3СК Смесь перемешивали в течение 30 мин и затем медленно добавляли 4-хлорбензолсульфонилхлорид (17,06 г, 0,081 моль). Реакционную смесь перемешивали при температуре от -20 до -15°С в течение 4 ч и затем ей давали возможность нагреваться до комнатной температуры всю ночь. Реакционную смесь разбавляли EtOAc (400 мл) и органическую фазу промывали 0,2N лимонной кислотой (3x150 мл), водой (1x150 мл), насыщенным раствором NaHCO3 (3x150 мл), насыщенным солевым раствором (1 х150 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением коричневого остатка. Остаток очищали флэш-хроматографией (смесь 50% EtOAc/гексан на силикагеле) с получением 43,49 г (80%) соединения 6 в виде желтой пены. Rf = 0,35 (смесь 50% EtOAc/гексан на сили-кагеле). Стадия 6. Получение сложного трет-бутилового эфира №(2-[№,№-диэтиламино]-5-[№'-(4-хлорфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланина (7). К раствору сложного трет-бутилового эфира №(2-[№,№-диэтиламино]-5-[№'-(4-хлорфенил-сульфонил)амино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланина (42,92 г, 0,064 моль) в ацетоне (Мe2СО) (600 мл) добавляли К2СО3 (12,75 г, 0,096 моль) и смесь перемешивали в течение 1 ч при комнатной температуре. Затем медленно добавляли иодэтан (EtI) (7,73 мл, 0,096 моль) и реакционную смесь перемешивали всю ночь при комнатной температуре. Реакционную смесь концентрировали в вакууме, и остаток переносили в EtOAc (300 мл). Органическую фазу промывали водой (2x300 мл), насыщенным солевым раствором (1 x100 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток очищали флэш-хроматографией (смесь гексан/EtOAc 2:1 на силикагеле) с получением 37,36 г (85%) соединения 7 в виде белого твердого вещества. Rf = 0,53 (смесь 50% EtOAc/гексан на сили-кагеле). Стадия 7. Получение гидрохлорида №(2-[№,№-диэтилшино]-5-[№-(4-хлорфенилсульфонил)-№'-этиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланина (8). Раствор (500 мл) сложного трет-бутилового эфира №(2-[№,№-диэтиламино]-5-[№'-(4-хлорфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилала-нина (36,21 г, 0,052 моль) в муравьиной кислоте нагревали при 70°С в течение 2 ч и затем концентрировали в вакууме. Остаток опять растворяли в муравьиной кислоте (500 мл) и опять нагревали при 70°С в течение 2 ч. Объем раствора уменьшали на 80% и затем обрабатывали по меньшей мере 1 экв. 1,0N HCl (52 мл, 0,052 моль) и затем дистиллированной водой (100 мл). Образовавшуюся гетерогенную смесь концентрировали в вакууме. Добавляли дистиллированную воду (100 мл) и гетерогенную смесь концентрировали в вакууме. Последние стадии повторяли дважды с получением влажного белого порошка. Порошок сушили, помещая в высокий вакуум при 40°С (7 дней), с получением 32,8 г (93%) соединения 8 в виде свободнотекущего белого твердого вещества. Rf = 0,25 (смесь MeOH/H2O 7/3+0,1% TFA, хроматография с обращенной фазой). - 15 - 008256 1H ЯМР (CD3OD) 8 8,22 (шир.с, 1Н), 7,82-7,79 (м, 1Н), 7,64-7,60 (м, 2Н), 7,36-7,33 (м, 1Н), 7,22-7,13 (м, 2Н), 7,07-6,98 (м, 2Н), 4,91-4,90 (м, 1Н), 4,80-4,79 (м, 1Н), 4,12-4,10 (м, 1Н), 3,87-3,75 (м, 1Н), 3,553,53 (м, 4Н), 3,41-3,40 (м, 3Н), 3,26-3,19 (м, 2Н), 2,03 (шир.с, 1Н), 1,97-1,89 (м, 3H), 1,27-1,15 (м, 6Н), 1,10-1,05 (т, 1,5Н), 0,97-0,92 (т, 1,5Н). 13С ЯМР ^D^D) 8 175,8, 175,7, 166,5, 162,7, 162,2, 155,8, 155,7, 155,7, 152,6, 148,1, 147,7, 142,0, 138,5, 136,2, 132,6, 132,3, 131,9, 131,7, 123,7, 111,8, 111,5, 62,3, 57,8, 44,9, 38,7, 38,0, 27,4, 26,6, 15,3, 14,9, 14.7, 14,0, 13,9. Пример 2. Получение №(2-[№,№-диэтиламино]-5-[№' -(4-фторфенилсульфонил)-№' -этиламино] пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)^-фенилаланина. Стадии 1, 2, 3, 4, 6 и 7 осуществляли, следуя методикам примера 1. Стадию 5 осуществляли с использованием 4-фторбензолсульфонилхлорида вместо 4-хлорбензолсульфонилхлорида. 1H ЯМР ^D^D) 8 8,17 (шир.с, 1Н), 7,90-7,87 (м, 2Н), 7,40-7,34 (м, 2Н), 7,20-7,16 (м, 1Н), 7,08-7,00 (м, 3Н), 5,52-5,51 (м, 1Н), 4,96-4,93 (м, 2Н), 5,78-5,70 (м, 1Н), 3,85-3,75 (м, 1Н), 3,59-3,53 (м, 4Н), 4,474,43 (м, 2Н), 3,44-3,24 (м, 2Н), 2,02-1,94 (м, 3H), 1,24-1,16 (м, 6Н), 1,10-1,05 (т, 1,5Н), 0,99-0,94 (т, 1,5Н). 13С ЯМР (CD3OD) 8 133,0, 132,9, 132,5, 132,2, 123,7, 123,6, 118,6, 57,1, 44,3, 38,3, 27,3, 26,6, 14,7, 14,1. МС м/z 629,5 (МН+). Пример 3. Получение N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)^-фенилаланина. Стадии 1, 2, 3, 4, 5 и 7 осуществляли, следуя методикам примера 2. Стадию 6 осуществляли с использованием диметилсульфата вместо этилиодида. 1H ЯМР ^D^D) 8 8,16 (шир.с, 1Н), 7,89-7,88 (м, 1Н), 7,39-7,35 (м, 3H), 7,20-7,13 (м, 1Н), 7,05-7,00 (м, 2Н), 4,85-4,84 (м, 1Н), 4,14-4,12 (м, 1Н), 3,59-3,54 (м, 5Н), 3,45-3,44 (м, 2Н), 3,45-3,33 (м, 3H), 3,133,12 (м, 1Н), 3,02-3,01 (м, 1Н), 2,04-1,95 (м, 4Н), 1,29-1,18 (м, 6Н). 13С ЯМР (CD3OD) 8 176,5, 169,8, 166,9, 166,4, 156,2, 152,7, 151,8, 150,4, 136,8, 133,3, 133,2, 132,5, 123,7, 118,8, 118,5, 57,8, 57,1, 48,3, 44,5, 41,0, 38,8, 27,5, 26,7, 14,1. МС м/z 615,2 (МН+). Пример 4. Получение N-(2-[N',N'-диэтиламино]-5-[N"-(4-хлорфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)^-фенилаланина. Стадии 1, 2, 3, 4, 5 и 7 осуществляли, следуя методикам примера 1. Стадию 6 осуществляли с использованием диметилсульфата вместо этилиодида. 1H ЯМР (CD3OD) 8 8,20 (шир.с, 1Н), 7,83-7,80 (м, 2Н), 7,67-7,64 (м, 2Н), 7,37-7,34 (м, 1Н), 7,21-7,18 (м, 1Н), 7,10-7,03 (м, 2Н), 4,88-4,87 (м, 1Н), 4,13-4,10 (м, 1Н), 3,55-3,45 (м, 6Н), 3,42-3,40 (м, 2Н), 3,243,23 (м, 2Н), 3,11-3,10 (м, 1Н), 3,02-3,01 (м, 1Н), 2,04-2,03 (м, 1Н), 1,98-1,90 (м, 3H), 1,28-1,18 (м, 6Н). 13С ЯМР (CD3OD) 8 176,0, 166,4, 161,8, 155,9, 155,4, 152,6, 146,5, 142,2, 137,6, 137,4, 136,4, 132,5, 131,9, 123,7, 114,6, 62,4, 58,1, 57,7, 45,0, 40,8, 38,6, 38,3, 27,4, 26,6, 15,3, 13,9. Пример 5. Получение N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(пиперидин-1-илкарбонилокси)^-фенилаланина. Стадии 1, 2, 4, 5, 6 и 7 осуществляли, следуя методикам примера 3. Стадию 3 осуществляли с использованием 1-пиперидинкарбонилхлорида вместо 1-пирролидинкарбонилхлорида. 1H ЯМР (CD3OD) 8 8,16 (шир.с, 1Н), 7,90-7,88 (м, 2Н), 7,40-7,35 (м, 2Н), 7,21-7,20 (м, 1Н), 7,14-7,13 (м, 1Н), 7,02-7,01 (м, 2Н), 5,51 (шир.с, 1Н), 4,83-4,77 (м, 1Н), 3,64-3,53 (м, 6Н), 3,34-3,33 (м, 2Н), 3,20-3,17 (м, 1Н), 3,12-3,11 (м, 2Н), 3,02-3,01 (м, 1H), 1,68-1,65 (м, 6Н), 1,19-1,17 (м, 6Н). 13С ЯМР (CD3OD) 8 185,0, 169,7, 166,3, 152,7, 136,6, 135,0, 133,2, 133,0, 132,5, 131,8, 126,3, 123,6, 121,7, 118,6, 118,3, 57,6, 54,5, 46,9, 44,3, 39,6, 38,7, 27,6, 25,9, 14,0. Пример 6. Получение N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(пиперидин-1-илкарбонилокси)^-фенилаланина. Стадии 1, 2, 4, 5, 6 и 7 осуществляли, следуя методикам примера 2. Стадию 3 осуществляли с использованием 1-пиперидинкарбонилхлорида вместо 1-пирролидинкарбонилхлорида. 1H ЯМР ^D^D) 8 8,17 (шир.с, 1H), 7,91-7,85 (м, 2Н), 7,39-7,31 (м, 3H), 7,20-7,16 (м, 1H), 7,05-6,97 (м, 2Н), 4,88-4,69 (м, 2Н), 4,71-4,69 (м, 1H), 3,80-3,75 (м, 1H), 3,62-3,39 (м, 6Н), 3,34-3,32 (м, 2Н), 3,303,16 (м, 3H), 1,68-1,65 (м, 4Н), 1,23-1,17 (м, 6Н), 1,10-1,05 (т, 1,5Н), 0,99-0,94 (т, 1,5Н). 13С ЯМР (CD3OD) 8 199,9, 187,6, 183,1, 176,2, 169,7, 166,3, 163,0, 162,7, 153,9, 152,9, 136,5, 133,1, 133,0, 132,7, 132,4, 123,8, 118,8, 118,4, 111,1, 110,6, 102,8, 79,4, 57,3, 55,4, 44,4, 38,9, 38,4, 27,7, 26,1, 15,1, 14.8, 14,3, 14,2. Пример 7. - 16 - 008256 Получение N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)^-фенилаланина. Стадии 1, 2, 4, 5, 6 и 7 осуществляли, следуя методикам примера 2. Стадию 3 осуществляли в соответствии со следующей методикой. 1H ЯМР ^D^D) 8 7,92-7,86 (м, 2Н), 7,41-7,32 (м, 3H), 7,22 (д, 1H), 7,04-6,91 (м, 3H), 4,29-3,98 (м, 4Н), 3,88-3,72 (м, 1H), 3,69-3,37 (м, 4Н), 2,40-2,24 (м, 2Н), 1,28-1,11 (м, 6Н), 1,10-1,00 (т, 1,5Н), 1,01-0,89 (т, 1,5Н). 13С ЯМР (CD3OD) 8 174,2, 169,7, 166,4, 163,2, 162,8, 157,0, 153,3, 153,2, 152,4, 144,3, 143,8, 136,1, 135,6, 135,5, 133,2, 133,1, 132,5, 132,2, 123,7, 118,9, 118,6, 112,9, 112,6, 57,5, 38,1, 37,7, 17,4, 14,7, 14,5, 13,8, 13,7. МС м/z 615 (МН+). Альтернативное получение сложного трет-бутилового эфира №(2-[№,№-диэтиламино]-5-нитропиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланина. К находящемуся при -15°С перемешиваемому раствору соединения 3 (24,9 г, 0,0578 моль) и 4-нитрофенилхлорформиата (11,7 г, 0,0578 ммоль) в CH2Cl2 (300 мл) добавляли триэтиламин (24,2 мл, 0,173 моль) с такой скоростью, чтобы температура реакционной смеси не превышала -10°С. После перемешивания в течение 20 мин добавляли по каплям азетидин (3,30 г, 0,0578 ммоль) и реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали всю ночь. Реакционную смесь разбавляли EtOAc (100 мл) и гексаном (100 мл) и затем повторно экстрагировали 10% водным раствором К2СО3 до тех пор, пока в водной фазе не исчезало желтое окрашивание (4-нитрофенол). Органический слой промывали насыщенным солевым раствором (75 мл), сушили MgSO4, фильтровали и выпаривали с получением 28,5 г (96%) сложного трет-бутилового эфира №(2-[№,№-диэтилшино]-5-нитропиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)^-фенилаланина в виде желтого твердого вещества, которое использовали без очистки. Rf = 0,17 (смесь EtOAc/гексан, 2:5 на силикагеле). Пример 8. Получение N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)^-фенилаланина. Стадии 1, 2, 3, 4, 5 и 7 осуществляли, следуя методикам примера 7. Стадию 6 осуществляли с использованием диметилсульфата вместо этилиодида. 1H ЯМР ^D^D) 8 7,95-7,76 (м, 2Н), 7,44-7,11 (м, 4Н), 7,01-6,83 (м, 3H), 4,30-3,93 (м, 4Н), 3,66-3,41 (м, 4Н), 3,14-2,92 (м, 3H), 2,42-2,21 (м, 2Н), 1,32-1,01 (м, 6Н). 13С ЯМР (CD3OD) 8 152,3, 136,3, 133,4, 133,2, 132,4, 123,6, 118,8, 118,5, 38,2, 17,4, 13,8. МС м/z 601 (МН+). Пример 9. Получение N-(2-[N',N'-диэтиламино]-5-[N"-(4-хлорфенилсульфонил)-N"-метиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)^-фенилаланина. Стадии 1, 2, 3, 4, 6 и 7 осуществляли, следуя методикам примера 8. Стадию 5 осуществляли с использованием 4-хлорбензолсульфонилхлорида вместо 4-фторбензолсульфонилхлорида. 1H ЯМР (CD3OD) 8 7,83 (д, 2Н), 7,67 (д, 2Н), 7,36-7,18 (м, 2Н), 7,06-6,86 (м, 3H), 4,29-3,97 (м, 4Н), 3,66-3,34 (м, 5Н), 3,15-2,95 (м, 4Н), 2,41-2,22 (м,2Н), 1,26-1,06 (м,6Н). 13С ЯМР (CD3OD) 8 157,2, 153,0, 152,5, 142,9, 142,5, 136,4, 132,5, 132,1, 132,0, 123,8, 57,9, 52,2, 40,7, 38,0, 17,4, 13,6. МС м/z 617 (МН+). Пример 10. Получение N-(2-[N',N'-диэтиламино]-5-[N"-(4-хлорфенилсульфонил)-N"-этиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)^-фенилаланина. Стадии 1, 2, 3, 4, 6 и 7 осуществляли, следуя методикам примера 7. Стадию 5 осуществляли с использованием 4-хлорбензолсульфонилхлорида вместо 4-фторбензолсульфонилхлорида. 1H ЯМР (CD3OD) 8 7,86-7,76 (м, 2Н), 7,70-7,60 (м, 2Н), 7,32 (шир.д, 1Н), 7,21 (шир.д, 1Н), 7,03-6,97 (м, 2Н), 6,90 (шир.с, 1Н), 4,29-4,00 (м, 4Н), 3,89-3,72 (м, 1Н), 3,70-3,36 (м, 5Н), 3,28-3,10 (м, 2Н), 2,42-2,24 (м, 2Н), 1,28-1,13 (м, 6Н), 1,11-1,02 (т, 1,5Н), 1,01-0,90 (т, 1,5Н). МС м/z 631 (МН+). Пример 11. Получение N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-метиламино]пирими-дин-4-ил)-4'-(пирролидин-1 -илкарбонилокси)^-фенилаланина. Стадии 1, 2, 3, 4, 6 и 7 осуществляли, следуя методикам примера 3. Стадию 5 осуществляли с использованием 2,4-дифторбензолсульфонилхлорида вместо 4-фторбензолсульфонилхлорида. 1H ЯМР (CDC13) 8 1,16 (шир.с, 6Н), 1,93 (шир.с, 4Н), 2,50-3,75 (м, 13Н), 4,83 (шир.с, 1Н), 6,60-7,40 (м, 7Н), 7,60 (шир.с, 1Н), 7,77 (м, 1Н), 9,41 (шир.с, 1Н). Пример 12. - 17 - 008256 Получение N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-этиламино]пирими-дин-4-ил)-4'-(пирролидин-1 -илкарбонилокси)-L-фенилаланина. Стадии 1, 2, 3, 4, 6 и 7 осуществляли, следуя методикам примера 2. Стадию 5 осуществляли с использованием 2,4-дифторбензолсульфонилхлорида вместо 4-фторбензолсульфонилхлорида. 1H ЯМР (CDC13) 8 0,91 (т, J=6,9, 1,8Н), 1,12 (м, 7,2Н), 1,92 (шир.с, 4Н), 2,50-4,00 (м, 13Н), 4,78 (м, 0,6Н), 4,88 (м, 0,4Н), 6,55 (д, J=6,9, 0.4H), 6,77 (д, J=6,3, 0,6H), 6,80-7,38 (м, 6Н), 7,51 (с, 0,4Н), 7,58 (с, 0,6Н), 7,74 (м, 1Н), 9,33 (м, 1Н). Пример 13. Получение N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-метиламино]пирими-дин-4-ил)-4'-(азетидин-1-ижарбонилокси)^-фенилаланина. Стадии 1, 2, 4, 5, 6 и 7 осуществляли, следуя методикам примера 11. Стадию 3 осуществляли, следуя методике примера 7. 1H ЯМР (CDC13) 8 1,14 (т, J=6,6, 6Н), 2,32 (м, 2Н), 2,50-3,80 (м, 9Н), 4,13 (м, 4Н), 4,62 (м, 0,6Н), 4,81 (м, 0,4Н), 5,81 (шир.д, 0,6Н), 5,90 (шир.д, 0,4Н), 6,90-7,40 (м, 7Н), 7,77 (м, 1Н). МС м/z 619,2 (МН+). Пример 14. Получение N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-этиламино]пирими-дин-4-ил)-4'-(азетидин-1-ижарбонилокси)^-фенилаланина. Стадии 1, 2, 4, 5, 6 и 7 осуществляли, следуя методикам примера 12. Стадию 3 осуществляли, следуя методике примера 7. 1H ЯМР (CDC13) 8 0,89 (т, J=6,7, 1,8H), 1,16 (м, 7,2Н), 2,28 (м, 2Н), 3,00-4,00 (м, 8Н), 4,09 (шир.с, 4Н), 4,79 (м, 0,6Н), 4,88 (м, 0,4Н), 6,80-7,30 (м, 7Н), 7,57 (с, 0,4Н), 7,62 (с, 0,6Н), 7,75 (м, 1Н), 11,9 (шир.с, 1Н). МС м/z 633,2 (МН+). Пример 15. Получение N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-пропаргиламино]пирими-дин-4-ил)-4'-(пирролидин-1 -илкарбонилокси)^-фенилаланина. Стадии 1, 2, 3, 4, 5 и 7 осуществляли, следуя методикам примера 2. Стадию 6 осуществляли с использованием пропаргилбромида вместо этилиодида. 1H ЯМР (CDC13) 8 1,18 (м, 6Н), 1,93 (шир.с, 4Н), 2,37 (с, 1Н), 3,00-3,70 (м, 10H), 3,80 (д, J=21,3, 0,6H), 3,98 (д, J=18,3, 0,4Н), 4,51 (м, 1Н), 4,88 (м, 1Н), 6,75-7,35 (м, 7Н), 7,58 (с, 0,6Н), 7,63 (с, 0,4Н), 7,86 (м, 2Н), 9,71 (шир.с, 1Н). Пример 16. Получение N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-пропаргиламино]пи-римидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланина. Стадии 1, 2, 3, 4, 5 и 7 осуществляли, следуя методикам примера 11. Стадию 6 осуществляли с использованием пропаргилбромида вместо диметилсульфата. 1H ЯМР ^DCb) 8 1,17 (м, 6Н), 1,94 (м, 4Н), 2,40 (м, 1Н), 3,00-3,75 (м, 10Н), 3,99 (д, J=18,0, 0,6Н), 4,18 (д, J=18,0, 0,4Н), 4,50 (м, 1Н), 4,90 (м, 1Н), 6,75-7,35 (м, 7Н), 7,81 (м, 2Н), 10,0 (шир.с, 1Н). Пример 17. Получение N-(2-[N',N'-диэтиламино]-5-[N"-(2,4-дифторфенилсульфонил)-N"-пропаргиламино]пи-римидин-4-ил)-4'-(азетидин-1 -илкарбонилокси)^-фенилаланина. Стадии 1, 2, 4, 5, 6 и 7 осуществляли, следуя методикам примера 16. Стадию 3 осуществляли, следуя методике примера 7. 1H ЯМР (CDC13) 8 1,18 (м, 6Н), 2,34 (м, 3H), 3,00-3,75 (м, 6Н), 3,80-4,25 (м, 5Н), 4,47 (м, 1Н), 4,89 (м, 1Н), 6,75-7,35 (м, 7Н), 7,79 (м, 2Н), 10,3 (шир.с, 1Н). МС м/z 643,2 (МН+). Пример 18. Получение N-(2-[N',N'-диэтиламино]-5-[N"-(4-фторфенилсульфонил)-N"-пропаргиламино]пири-мидин-4-ил)-4'-(азетидин-1 -илкарбонилокси)^-фенилаланина. Стадии 1, 2, 3, 4, 5 и 7 осуществляли, следуя методикам примера 7. Стадию 6 осуществляли с использованием пропаргилбромида вместо этилиодида. 1H ЯМР (CDC13) 8 1,25 (м, 6Н), 2,28 (м, 3H), 3,00-3,75 (м, 6Н), 3,80-4,25 (м, 5Н), 4,47 (м, 1Н), 4,89 (м, 1Н), 6,75-7,35 (м, 7Н), 7,57 (с, 0,6Н), 7,62 (с, 0,4Н), 7,79 (м, 2Н), 10,6 (шир.с, 1Н). МС м/z 625,2 (МН+). Пример 19. Получение N-(2-[N',N'-диэтиламино]-5-[N"-(4-хлорфенилсульфонил)-N"-пропаргиламино] пири-мидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланина. Стадии 1, 2, 3, 4, 5 и 7 осуществляли, следуя методикам примера 1. Стадию 6 осуществляли с использованием пропаргилбромида вместо этилиодида. - 18 - 008256 1H ЯМР (CD3OD) 8 8,13 (с, 1Н), 7,86-7,82 (м, 2Н), 7,62-7,58 (м, 2Н), 7,32-7,28 (м, 2Н), 7,19-7,17 (м, 1Н), 7,04-6,98 (м, 2Н), 4,83-4,5 (м, 2Н), 4,12-3,82 (м, 1Н), 3,63-3,37 (м, 8Н), 3,27-3,08 (м, 2Н), 2,72 (шир.с, 1Н), 2,04-1,86 (м, 4Н), 1,24-1,07 (м, 6Н). 13С ЯМР (CD3OD) 8 177,2, 176,5, 162,7, 156,7, 155,7, 154,5, 153,2, 142,6, 140,3, 137,4, 137,3, 133,1, 132,9, 132,8, 132,7, 132,2, 132,1, 124,3, 111,3, 80,5, 80,3, 77,7, 58,2, 57,7, 44,9, 43,4, 28,1, 27,3, 14,8, 14,7. МС м/z 655 (МН+). Для испытания соединений данного изобретения могут быть использованы следующие методы. Пример А. Анализ адгезии а4р1интегрина: адгезия клеток Jurkat(tm) к фибронектину человеческой плазмы. Методика 96-луночные планшеты (планшеты Costar 3590 EIA) заполняли при 4°С человеческим фибронекти-ном (Gibco/BRL, № кат. 33016-023) с концентрацией 10 мкг/мл на всю ночь. Затем планшеты блокировали раствором бычьего сывороточного амбумина (BSA; 0,3%) в физиологическом растворе. Клетки Jurkat(tm) (сохраняемые в фазе логарифмического роста) метили Ca1cein AM в соответствии с инструкциями производителя и суспендировали при концентрации 2x106 клеток/мл в смеси Hepes/солевой рас-твор/BSA. Затем клетки подвергали действию тестируемого и контрольного соединений в течение 30 мин при комнатной температуре перед их переносом в отдельные лунки планшетов, покрытых фибро-нектином. Давали возможность происходить адгезии в течение 35 мин при 37°С. После этого лунки промывали медленной аспирацией и промыванием из пипетки свежего солевого раствора. Флуоресценцию, связанную с остальными слипшимися клетками, подсчитывали количественно с использованием пластинчатого устройства для считывания флуоресценции EX 485/EM 530. Культуры клеток приготавливали сначала разделением клеток Jurkat(tm) в стационарной фазе при соотношении 1:10 в первый день и при соотношении 1:2 на второй день для проведения анализа на третий день. Клетки, разделенные при соотношении 1:10 в первый день, разделяли при соотношении 1:4 на третий день для анализа на 4-ый день. Подготавливали планшеты для анализа, при этом сначала приготавливали рабочий раствор человеческого фибронектина Gibco/BRL (№ кат. 33016-023) в PBS^ с концентрацией 10 мкг/мл. Планшеты Co-star 3590 EIA заполняли затем 50 мкл/лунку в течение 2 ч при комнатной температуре (однако они могли быть также оставлены на ночь при 4°С). И наконец, планшеты аспирировали и блокировали смесью Hepes/солевой буферный раствор, 100 мкл/лунку, в течение 1 ч при к.т. с последующей промывкой 3 раза 150 мл PBS++. Разбавления соединения осуществляли приготовлением последовательно разбавленных 1:3 соединений следующим образом. На каждый планшет (4 соединения/планшет) в 4 пробирки для титрования Bio-Rad, находящиеся на штативе для пробирок для титрования, добавляли 600 мкл. B каждую соответствующую пробирку добавляли количество соединения, достаточное для получения 2x концентрации, с использованием методик, хорошо известных в данной области. С использованием гибких пластин Fa1con в ряды B-G добавляли 100 мкл смеси Hepes/солевой буферный раствор или человеческой сыворотки. Для распределения использовали рассчитанный на 180 мкл многоканальный пипеточный прибор с четырьмя равномерно установленными наконечниками. Каждый ряд из четырех пробирок перемешивали 5 раз и в первую колонку, состоящую из каждого разбавленного соединения в ряду B, переносили 180 мкл 2x соединения, оставляя при этом ряд А пустым. B другие лунки в ряду А добавляли 180 мкл. Осуществляли последовательные разбавления переносом вниз по планшету 50 мкл в следующее разбавление и перемешиванием 5 раз, меняя при этом наконечники каждый раз после перемешивания. B ряду F разбавления прекращали. B ряду G соединение отсутствовало. Раствор антитела 21/6 с концентрацией 20 мкг/мл в смеси Hepes/солевой буферный раствор или человеческой сыворотке представлял собой положительный контроль и был помещен в реагентный лоток для добавления на планшет с суспензией клеток. Окрашивание клеток осуществляли сбором клеток Jurkat(tm) в логарифмической фазе роста центрифугированием в 50 мл пробирках (1100 об./мин в течение 5 мин). Клетки ресуспендировали в 50 мл PBS++, вращали и ресуспендировали в 20 мл PBS++. Клетки окрашивали добавлением 20 мкл Ca1cein AM в течение 30 мин при к.т. Объем доводили до 50 мл с использованием смеси Нереs/солевой буферный раствор и клетки подсчитывали, вращали и ресуспендировали до получения 2x106 клеток/мл в смеси Hepes/солевой буферный раствор или человеческой сыворотке. Соединения инкубировали с использованием следующей методики. На новый гибкий планшет в ряды В-Н добавляли 65 мкл окрашенных клеток. Затем в соответствующие ряды добавляли 65 мкл 2x соединений, после чего планшет устанавливали и перемешивали три раза. B ряд Н добавляли 65 мкл антитела 2Х-21/6 и перемешивали 3 раза. И наконец, планшет инкубировали при комнатной температуре в течение 30 мин. Адгезию фибронектина измеряли с использованием устройства EX 485/EM 530 для считывания флуоресценции с пластин после обработки с использованием следующей методики. После инкубирования клетки три раза перемешивали, переносили 100 мкл на планшеты, покрытые фибронектином, и ин - 19 - 008256 кубировали при 37°С в течение около 35 мин. Каждый планшет ряд за рядом промывали медленным прибавлением из пипетки 100 мкл при комнатной температуре. Для аспирации добавляли РВ8^ по боковым сторонам лунок, планшет поворачивали на 90°. Данную методику повторяли в течение в целом 3-х промывок. После промывки прибавлением из пипетки по боковым сторонам лунок каждую лунку заполняли 100 мкл. Рассчитывали значение IC50 для каждого соединения, как в присутствии человеческой сыворотки, так и в отсутствии человеческой сыворотки. IC50 представляет собой концентрацию, при которой рост или активность угнетается на 50%. Данные представлены в следующих таблицах. Адгезия клеток к фибронектину человеческой плазмы (без человеческой сыворотки) Пример Ш 1С5о (уг/мл) 0, 011 0, 001 0,004 0,012 0,00377 0, 003 0, 001 0,001 0,002 0,00256 0,005293 0,005632 0,001515 Пример № 1С5о (уг/мл) 0, 002146 0, 063 0, 009 0,00337 0,003663 0,004538 Адгезия клеток к фибронектину человеческой плазмы (в присутствии человеческой сыворотки) Пример Ш 1С5о (уг/мл) 0,264 0,38 1,062 1,437 0, 987 0, 451 0,053 0,135 0,128 0,052332 0,305436 0,147085 0,055391 0,03259 1,102 0,371 0,080426 0,3 4,114982 - 20 - 008256 Пример В. Анализ насыщения in vitro для определения связывания соединений-кандидатов с а4р\. Далее в описании представлен анализ in vitro для определения уровней в плазме, необходимых для того, чтобы соединение было активно на модели экспериментального аутоиммунного энцефаломиелита ("ЕАЕ"), описанной в следующем примере, или на других моделях in vivo. Клетки Jurkat в логарифмической фазе роста промывали и ресуспендировали в обычной животной плазме, содержащей 20 мкг/мл антитела 15/7 (Yednock et al., J. Biol. Chem., (1995) 270 (48) : 28740). Клетки Jurkat разбавляли два раза или в пробах обычной плазмы, содержащей известные количества соединения-кандидата при различных концентрациях в диапазоне от 66 до 0,01 мкМ, полученных с использованием стандартного 12-разового последовательного разбавления для построения стандартной кривой, или в пробах плазмы, полученных из периферической крови животных, обработанной соединением-кандидатом. Затем клетки инкубировали в течение 30 мин при комнатной температуре, дважды промывали фосфатным буферным солевым раствором ("PBS"), содержащим 2% фетальную бычью сыворотку и 1 мМ каждого из хлорида кальция и хлорида магния (среда для анализа), для удаления антитела 15/7. Клетки затем подвергали действию F(ab')2 козла против мышиного IgGFc (Immunotech, Westbrook, ME), конъюгированного с фикоэритрином, который поглощался за счет неспецифической перекрестной реакционноспособности при совместном инкубировании с 5% сывороткой, полученной от подвергаемых исследованию видов животных, при 1:200 и инкубировали в темноте при 4°С в течение 30 мин. Клетки дважды промывали средой для анализа и ресуспендировали в ней. Затем их анализировали стандартным анализом с использованием сортировщика клеток, активированных флуоресценцией ("FACS"), как описано в Yednock et al., J. Biol. Chem., 1995, 270 : 28740. Затем данные выражали графически в виде кривой зависимости флуоресценции от дозы, например в виде кривой зависимости "доза-ответ". Уровни доз, которые приводили к подъему кривой, представляли собой уровни, необходимые для получения действенности соединений на модели in vivo. Пример С. Кассетное дозирование и анализ сыворотки для определения биодоступности. Пероральную биодоступность проверяли дозированием крысам кассеты, то есть смеси 6 соединений в дозируемом растворе. Кассета включала 5 тестируемых соединений и стандартное соединение, общая доза которых составляла 10 мг/кг. Каждое соединение/тестируемое соединение превращали в натриевую соль с использованием эквимолярного 1N NaOH и растворяли в воде с получением концентрации 2 мг/мл. Кассету приготавливали смешиванием равных объемов каждого из шести растворов. Кассетный дозируемый раствор хорошо перемешивали и затем доводили рН до 7,5-9. Дозируемый раствор приготавливали за день до исследования и перемешивали всю ночь при комнатной температуре. В данном испытании использовали крыс-самцов Sprague Dawley (SD) возрастом 6-8 недель из лаборатории Charles River. Крыс подвергали карантину в течение по меньшей мере одного дня и они имели постоянный доступ к пище и воде. За ночь до введения кассеты крысы голодали примерно 16 ч. Каждую кассету вводили четырем крысам SD. Каждой крысе вводили перорально разовую дозу дозируемого раствора. Регистрировали дозируемый объем (5 мл/кг) и время и через 2 ч после дозирования крыс кормили. Собирали пробы крови сердечной пункцией при следующих временных точках: 4 часа, 8 часов и 12 часов. Непосредственно перед сбором крови крыс анестезировали ТО2 газом в течение 10-20 с. Через 12 ч собирали пробы, крыс умерщвляли асфиксией в атмосфере OO2 с последующей цервикальной дислокацией. Пробы крови перед их обработкой сохраняли в гепаринизованных микроцентрифужных пробирках для крови при температуре ниже комнатной (4°С). Пробы крови центрифугировали (10000 об./мин, в течение 5 мин) и пробы плазмы удаляли и хранили в холодильнике при -20°С до проведения анализа на уровни лекарственного средства. Уровни лекарственного средства в плазме анализировали с использованием следующего протокола прямого осаждения плазмы. Пробы плазмы in vivo приготавливали в 1,5 мл 96-луночном планшете добавлением по порядку 100 мкл тестируемой плазмы, 150 мкл метанола и затем осуществляли вращение в течение 10-20 с. Прибавляли 150 мкл внутреннего стандартного эталонного вещества с концентрацией 0,05 нг/мкл в ацетонитри-ле и вращали в течение 30 с. Пробы плазмы для построения стандартной кривой получали в 1,5 мл 96-луночном планшете добавлением по порядку 100 мкл контрольной плазмы мыши, 150 мкл метанола и вращением в течение 1020 с. Прибавляли 150 мкл внутреннего стандартного эталонного вещества с концентрацией 0,05 нг/мкл в ацетонитриле и вращали в течение 30 с. В пробы шприцем вводили представляющее интерес соединение в количестве 0-200 нг (10 концентраций) в 50% метаноле с получением диапазона для стандартной кривой 0,5-2000 нг/мл. Пробу опять вращали в течение 30 с. Затем пробы опять вращали в течение 20-30 мин со скоростью 3000 об./мин в микрофуге Эппен-дорфа (Eppendorf) перед переносом 80-90% супернатанта в чистый 96-луночный планшет. Затем выпари - 21 - 008256 вали органический растворитель до тех пор, пока пробы стали сухими (в атмосфере N2 при 40°С/30-60 мин (Zymark Turbovap)). Остаток затем растворяли в 200-600 л подвижной фазы (смесь 50% СН3ОН/0,1% TFA). После этого проводили анализы ЖХ/МС/МС с использованием масс-спектрометра с тройным квадруполем PE-Sciex API 3000 (SNO 749707), автопробоотборника Перкина-Элмера серии 200 и насоса Shimadzu 10A. Оценку проводили с помощью анализатора PE-Sciex (v 1.1) и осуществляли анализ данных и количественное определение с использованием анализатора PE-Sciex (v 1.1). Пробу объемом 5-50 мкл впрыскивали в колонку с обращенной фазой ThermoHypersil DASH-18 (Keystone с размерами 2,0x20 мм, 5 мкм, PN: 8823025-701) с использованием в качестве подвижной фазы 25% СН3ОН, 0,1% TFA - 100% СН3ОН, 0,1% TFA. Время опыта составляло около 8 мин при объемном расходе около 300 мкл/мин. Площадь под кривой (AUC) вычисляли с использованием правила трапеций для t=0 до последнего времени отбора пробы tx (см. Handbook of Basic Pharmacokinetics, Wolfgang A. Ritschel and Gregory L. Kearns, 5th ed., 1999). AUC0^tx=S ((Cn+Cn+1)/2)-(tn+1-tn) [(мкг/мл)ч] В случае примера дозирования кассеты, когда пробы взяты после внесосудистого дозирования в 4, 8 и 12 часов, определяли AUC в диапазоне времени от t=0 до t=12 ч. Значения AUC °^12 час вычисляли для каждого отдельного животного и ниже в таблице представлены средние значения AUC °^12 час. Пример № AUC 14,798 15,971 22,271 13,829 2,1654 0,5125 0,8979 2,4082 2,0774 2,1113 14,818 4,7816 1,283 0,3566 90,317 23,808 0,8628 4,7528 Пример D. Модели астмы. Воспалительные состояния, опосредованные а4р1интегрином, включают, например, эозинофиль-ный лейкоцитоз, повышенную чувствительность дыхательных путей и окклюзию, которые встречаются при хронической астме. Далее в описании раскрыты модели астмы на животных, которые использовались для изучения действий соединений данного изобретения in vivo, предназначенных для использования при лечении астмы. Модель астмы у крыс. Данная модель следует методикам, описанным Chapman et al., Am. J. Resp. Crit. Care Med., 153:4, A219 (1996) и Chapman et al. Am. J. Resp. Crit. Care Med. 155:4, A881 (1997), обе из которых включены в данное описание в качестве ссылки во всей своей полноте. Яичный альбумин (ОА; 10 мг/мл) смешивали с гидроксидом алюминия (10 мг/мл) и впрыскивали (i.p.) крысам Brown Norway в 0 день. Инъекции ОА вместе с адъювантом повторяли на 7 и 14 дни. На 21 день сенсибилированных животных удерживали в пластиковых трубках и подвергали (60 мин) действию аэрозоля ОА (10 мг/кг) в системе, воздействующей только на нос. Через 72 ч животных умерщвляли пентобарбиталом (250 мг/кг, i.p.). Проводили ла-важ легких через трахеотомическую трубку с использованием 3-х аликвот (4 мл) раствора Хэнка (Hank) (HBSS x 10, 100 мл; EDTA 100 мМ, 100 мл; HEPES 1M, 25 мл; доведенный до 1 л Н20); извлеченные - 22 - 008256 клетки объединяли и общий объем извлеченной жидкости доводили до 12 мл добавлением раствора Хэнка. Подсчитывали общее количество клеток (счетчик микроклеток Sysmex F-500, TOA Medical Electronics Ltd., Япония) и делали мазки разбавлением извлеченной жидкости (примерно до 106 клеток/мл) и прибавлением из пипетки аликвоты (100 мкл) в центрифугу (Cytospin, Shandon, U.K.). Мазки сушили на воздухе, связывали с использованием раствора прочного зеленого в метаноле (2 мг/мл) в течение 5 с и окрашивали эозином G (5 с) и тиазином (5 с) (Diff-Quick, Browne Ltd. U.K.) для дифференциации эози-нофилов, нейтрофилов, макрофагов и лимфоцитов. Подсчитывали суммарное количество клеток в мазке световой микроскопией (x100) при погружении в масло, которое было равно 500. Соединения данного изобретения смешивали с 0,5% карбоксиметилцеллюлозой и 2% суспензией Tween 80 и вводили перо-рально крысам, которые были сенсибилизированы к воздействию аллергена, которым являлся яичный альбумин. Соединения, которые угнетали индуцированное аллергеном накопление лейкоцитов в дыхательных путях активно сенсибилизированных крыс Brown Norway, считались активными для данной модели. Модель астмы у мышей. Соединения также оценивали на модели острого воспаления легких у мышей, следуя методикам, описанным Kung et al., Am. J. Respir. Cell Mol. Biol. 13:360-365, (1995) и Schneider et al., (1999) и Am. J. Respir. Cell Mol. Biol. 20:448-457 (1999), обе из которых включены в данное описание в качестве ссылки во всей своей полноте. 6 черных мышей-самок (возрастом 8-12 недель) сенсибилизировали в 1 день внутрибрюшинной инъекцией (i.p.) 0,2 мл смеси яичный альбумин/гидроксид алюминия, содержащей 20 мкг яичного альбумина (4 сорт, Sigma) и 2 мг Alum (гидроксида алюминия) (Pierce). На 14-ый день вводили инъекцию бустера. На 28-ой и 29-ый дни мышам в течение 20 мин вводили провокационную пробу в виде превращенного в аэрозоль 1% яичного альбумина (в 0,9% солевом растворе). Мышей умерщвляли и на 30-ый день через 48 ч после введения первой провокационной пробы собирали пробы бронхоальве-олярного лаважа. Подсчитывали число эозинофилов методом окрашивания FACs/FITC. Соединения данного изобретения смешивали с 0,5% карбоксиметилцеллюлозой и 2% суспензией Tween 80 и вводили перорально мышам, которые были сенсибилизированы к воздействию аллергена, которым являлся яичный альбумин. Соединения, которые угнетали индуцированное аллергеном накопление лейкоцитов в дыхательных путях активно сенсибилизированных мышей C57BL/6, считались активными для данной модели. Модель астмы у овец. Данная модель следует методикам, описанным Abraham et al., J. Clin, Invest., 93:776-787 (1994) и Abraham et al., J. Respir. Crit Care Med. 156:696-703 (1997), обе из которых включены в данное описание в качестве ссылки во всей своей полноте. Соединения данного изобретения оценивали внутривенным (солевой водный раствор), пероральным (2% Tween 80, 0,5% карбоксиметилцеллюлоза) и аэрозольным введением овцам, которые являются сверхчувствительными к антигену Ascaris suum. Соединения, которые ослабляют индуцированную антигеном раннюю бронхиальную реакцию и/или блокируют реакцию дыхательных путей в поздней фазе, например, обладают защитным действием против индуцированных антигеном поздних реакций и сверхчувствительности дыхательных путей ("AHR"), считаются активными для данной модели. Больную аллергией овцу, у которой, как было показано, развивается как ранняя, так и поздняя бронхиальная реакция на ингаляционный антиген Ascaris suum, использовали для изучения действия соединений-кандидатов на дыхательные пути. После местной анестезии носовых ходов 2% лидокаином катетер-баллон продвигали через одну ноздрю в нижний отдел пищевода. Затем животных интубировали через другую ноздрю с помощью интубационной трубки с надувной манжетой с использованием в качестве зонда гибкого фибробронхоскопа. Плевральное давление оценивали по Абрахаму (1994). Генерировали аэрозоли (смотри представленный ниже препарат) с использованием передвижного медицинского ингалятора, который обеспечивая аэрозоль со срединным аэродинамическим диаметром подачи массы 3,2 мкм, определенным с помощью каскадного импактора Андерсена. Ингалятор соединяли с дозиметрической системой, состоящей из соленоидного клапана и источника сжатого воздуха (20 фунтов/дюйм2, 137,9 кПа). Выходной конец ингалятора направляли в пластмассовую Т-образную трубку, один конец которой соединяли с дыхательным отверстием поршневого респиратора. В начале дыхательного цикла из респиратора соленоидный клапан активировали в течение 1 с. Аэрозоли доставлялись при VT 500 мл и скорости 20 вдохов/мин. В качестве контроля использовался только 0,5% раствор бикарбоната натрия. Для оценки бронхиальной восприимчивости к карбаколу строили кривые зависимости "кумулятивная концентрация-ответ" по Абрахаму (1994). Проводили бронхиальные биопсии до и после начала терапии и через 24 ч после введения антигена. Бронхиальные биопсии осуществляли по Абрахаму (1994). Исследование адгезии альвеолярных макрофагов in vitro осуществляли также по Абрахаму (1994) и вычисляли процентное содержание слипшихся клеток. Аэрозольный препарат. Раствор соединения-кандидата в смеси 0,5% бикарбонат натрия/солевой раствор (мае/об.) с концентрацией 30,0 мг/мл получали с использованием следующей методики. - 23 - 008256 А. Приготовление смеси 0,5% бикарбонат натрия/исходный солевой раствор: 100 мл ИнтредиенФ Грамм/100,0 мл Конечная концентрация Бикарбонат натрия 0,5 г 0,5% Солевой раствор q.s. до 100 мл q.s. до 100% Методика 1. В 100 мл мерную колбу добавляют 0,5 г бикарбоната натрия. 2. Добавляют примерно 90,0 мл солевого раствора и воздействуют ультразвуком до растворения. 3. Объем раствора доводят до 100 мл солевым раствором и тщательно перемешивают. В. Приготовление соединения-кандидата с концентрацией 30,0 мг/мл: 10,0 мл Ингредиент Грамм/10,0 мл Конечная концентрация Соединение-кандидат 0,300 г 30,0 мг/мл Смесь 0,5% бикарбонат натрия/исходный солевой раствор q.s. до 10 мл q.s. до 100% Методика 1. В 10,0 мл мерную колбу добавляют 0,300 г соединения-кандидата. 2. Добавляют примерно 9,7 мл смеси 0,5% бикарбонат натрия/исходный солевой раствор. 3. Воздействуют ультразвуком до полного растворения соединения-кандидата. 4. Объем раствора доводят до 10,0 мл смесью 0,5% бикарбонат натрия/исходный солевой раствор и тщательно перемешивают. Пример Е. 10-дневное исследование токсичности на мышах С57В6. Для оценки токсичности соединений настоящего изобретения проводили 10-дневное исследование мышей-самок С57В56. Соединение вводили желудочным зондом при пяти уровнях дозы: 0 (наполнитель в качестве контроля), 10, 30, 100, 300 и 1000 мг/кг, при этом каждый уровень дозы вводили пяти мышам. Объем дозы во всех уровнях составлял 10 мл/кг. Растворы или суспензии доз приготавливали в 2% Tween 80, в 0,5% карбоксиметилцеллюлозе (CMC) и каждые два-три дня приготавливали свежие растворы или суспензии доз. Наблюдения за жизненными функциями включали измерения массы тела (1, 2, 3, 5, 7, 8 и 11 дни исследований), ежедневные клинические наблюдения через боковую сторону клетки (1-2 дни) и набор тестов для периодического функционального исследования (1, 2 и 9 дни исследований). В завершение собирали пробы крови сердечной пункцией для оценки клинической патологии (гематология и клиническая химия) и уровней лекарственного средства. Анализировали пробы крови в EDTA на общее содержание лейкоцитов, эритроцитов, гемоглобина, гематокритного числа, эритроцит-ных индексов (MCV, МСН, МСНС), тромбоцитов и на состоящий из пяти частей дифференциал WBC (нейтрофилы, лимфоциты, моноциты, эозинофилы и базофилы). Пробы гепаринизованной плазмы анализировали на аланин-трансаминазу, аспартат-трансаминазу, щелочную фосфатазу, общее содержание билирубина, альбумина, белка, кальция, глюкозы, мочевинного азота, креатинина, холестерина и триглице-ридов. После сбора крови вскрывали трупы и взвешивали органы (печень, селезенку, почки, сердце и ви-лочковую железу). Собирали образцы ткани; мозг, слюнные железы, вилочковую железу, сердце, легкие, печень, почки, надпочечники, селезенку, желудок, двенадцатиперстную кишку, подвздошную кишку, толстую кишку и матку/яичники и фиксировали формалином. Ткани групп животных, которым вводили наполнитель в качестве контроля и уровни доз, равные 300 и 1000 mpk, обрабатывали до получения окрашенных препаратов на предметном стекле Н &Е и оценивали на гистопатологические поражения. Изменения массы тела, абсолютные и относительные массы органов и результаты клинической патологии анализировали для получения статистических значительных отличий, по сравнению с используемыми в качестве контроля наполнителями, посредством многократных сравнительных испытаний с использованием компьютерного обеспечения Prism. Анализировали результаты набора тестов для функционального наблюдения с целью получения отличий с использованием точных испытаний по Дуннету (Dunnet), Фишеру (Fisher) и эффектов направления изменения доз, определяемых коррелятивным испытанием по Cochran-Mantel-Haenszel с использованием компьютерного обеспечения SAS. При использовании в традиционном препарате для перорального введения соединения данного изобретения будут активными в данной модели. Пример F. Индуцированный адъювантом артрит у крыс. Индуцированный адъювантом артрит ("AIA") представляет собой модель на животном, применимую при исследовании ревматоидного артрита (РА), вызванного инъекцией М. tuberculosis в основание - 24 - 008256 хвоста крыс Льюиса (Lewis). После инъекции между 10 и 15 днями у животных развивался тяжелый прогрессирующий артрит. Обычно соединения испытывают на их способность изменять припухлость задней лапы и повреждение костей в результате индуцированной адъювантом водянки у крыс. Для количественного определения ингибирования припухлости задней лапы в результате AIA были установлены две стадии воспаления: (1) задняя лапа после первичной и вторичной инъекции; и (2) задняя лапа без вторичной инъекции, которая обычно начинается развитием воспаления в инъецированной лапе примерно через одиннадцать дней после его индуцирования. Уменьшение последнего типа воспаления является показателем иммуно-супрессорной активности. Cf. Chang, Arth. Rheum., 20, 1135-1141 (1977). Использование модели РА на животных, такого как AIA, дает возможность исследовать клеточные события, включенные в ранние стадии заболевания. Экспрессия CD 44 на макрофагах и лимфоцитах дополнительно регулируется во время ранней стадии развития артрита, индуцированного адъювантом, тогда как экспрессия LFA-1 дополнительно регулируется позже при развитии заболевания. Понимание взаимодействий между слипшимися молекулами и эндотелием на самых ранних стадиях индуцированного адъювантом артрита может привести к значительным успехам в способах, используемых при лечении РА. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I) 6 о (I) где каждый Х независимо представляет собой фтор, хлор или бром; р представляет собой целое число от 0 до 3; R1 и R3 вместе с атомом азота, с которым они связаны, образуют азетидинил, пирролидинил, пирро-лил, 2,5-дигидропиррол-1-ил, пиперидинил или 1,2,3,6-тетрагидропиридин-1-ил; R2 выбран из группы, состоящей из (С1-С5)алкила, (С2-С6)алкенила и (С3-С6)циклоалкил(С1-С4)алкила; и его фармацевтически приемлемые соли. 2. Соединение формулы (II) (Х)п (П) где каждый Х независимо выбран из группы, состоящей из фтора и хлора; m представляет собой целое число, равное 1 или 2; R2 выбран из группы, состоящей из (С1-С5)алкила, (С2-С6)алкенила и (С3-С6)циклоалкил(С1-С4)алкила; - 25 - 008256 R1 и R3 вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидиниль-ную или пиперидинильную группу; и его фармацевтически приемлемые соли. 3. Соединение формулы (III) (Ш) где каждый Х независимо представляет собой фтор или хлор; n равно нулю или единице; R2 является -CH2-R', где R' выбран из группы, состоящей из водорода, метила или -СН=СН2; R1 и R3 вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидиниль-ную или пиперидинильную группу; и его фармацевтически приемлемые соли. 4. Соединение по п.1, в котором R1 и R3 вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидинильную или пиперидинильную группу. 5. Соединение по одному любому из пп.1, 2 или 3, в котором R2 является СН3. 6. Соединение по п.3, в котором Х является F или С1 и n равно 0. 7. Соединение по п.1, выбранное из группы, содержащей К-(2-[К',К'-диэтиламино]-5-[К"-(4-хлорфенилсульфонил)-К"-этиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; К-(2-[К',К'-диэтиламино]-5-[К"-(4-фторфенилсульфонил)-К"-этиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; К-(2-[К',К'-диэтиламино]-5-[К"-(4-фторфенилсульфонил)-К"-метиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)^-фенилаланин; К-(2-[К',К'-диэтиламино]-5-[К"-(4-хлорфенилсульфонил)-К"-метиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; К-(2-[К',К'-диэтиламино]-5-[К"-(4-фторфенилсульфонил)-К"-метиламино]пиримидин-4-ил)-4'-(пиперидин-1-илкарбонилокси)-L-фенилаланин; К-(2-[К',К'-диэтиламино]-5-[К"-(4-фторфенилсульфонил)-К"-этиламино]пиримидин-4-ил)-4'-(пиперидин-1-илкарбонилокси)-L-фенилаланин; К-(2-[К',К'-диэтиламино]-5-[К"-(4-фторфенилсульфонил)-К"-этиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; К-(2-[К',К'-диэтиламино]-5-[К"-(4-фторфенилсульфонил)-К"-метиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)^-фенилаланин; К-(2-[К',К'-диэтиламино]-5-[К"-(4-хлорфенилсульфонил)-К"-метиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; К-(2-[К',К'-диэтиламино]-5-[К"-(4-хлорфенилсульфонил)-К"-этиламино]пиримидин-4-ил)-4'-(азетидин-1 -илкарбонилокси)^-фенилаланин; К-(2-[К',К'-диэтиламино]-5-[К"-(2,4-дифторфенилсульфонил)-К"-метиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; К-(2-[К',К'-диэтиламино]-5-[К"-(2,4-дифторфенилсульфонил)-К"-этиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; К-(2-[К',К'-диэтиламино]-5-[К"-(2,4-дифторфенилсульфонил)-К"-метиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; К-(2-[К',К'-диэтиламино]-5-[К"-(2,4-дифторфенилсульфонил)-К"-этиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; и его фармацевтически приемлемые соли. 8. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по одному любому из пп.1-4, 6 или 7. - 26 - 008256 9. Способ лечения заболевания млекопитающего, опосредованного а4интегринами, включающий введение фармацевтической композиции, содержащей фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по одному любому из пп.1-4, 6 или 7. 10. Способ по п.9, где заболевание опосредовано VLA-4. 11. Способ по п.9, где заболевание представляет собой воспалительное заболевание. 12. Соединение формулы (IV) где каждый Х независимо представляет собой фтор, хлор или бром; р представляет собой целое число от 0 до 3; R1 и R3 вместе с атомом азота, с которым они связаны, образуют азетидинил, пирролидинил, пирро-лил, 2,5-дигидропиррол-1-ил, пиперидинил или 1,2,3,6-тетрагидропиридин-1-ил; представляет собой (С2-С6)алкинил; и его фармацевтически приемлемые соли. 13. Соединение формулы (V) (V) где каждый Х независимо выбран из группы, состоящей из фтора и хлора; m представляет собой целое число, равное 1 или 2; представляет собой (С2-С6)алкинил; R1 и R3 вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидиниль-ную или пиперидинильную группу; и его фармацевтически приемлемые соли. 14. Соединение формулы (VI) (VI) - 27 - 008256 где каждый X независимо представляет собой фтор или хлор; n равно нулю или единице; R2 представляет собой (С2-С6)алкинил; R1 и R3 вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидиниль-ную или пиперидинильную группу; и его фармацевтически приемлемые соли. 15. Соединение по п.12, в котором вместе с атомом азота, с которым они связаны, образуют азетидинильную, пирролидинильную или пиперидинильную группу. 16. Соединение по одному любому из пп.12, 13 или 14, в котором R2 является пропаргилом. 17. Соединение по п.15, в котором Х является F или С1 и n равно нулю. 18. Соединение по п.12, выбранное из группы, содержащей М-(2-[М',М'-диэтиламино]-5-[М"-(4-фторфенилсульфонил)-М"-пропаргиламино]пиримидин-4-ил)-4'-(пирролидин-1 -илкарбонилокси)^-фенилаланин; М-(2-[М',М'-диэтиламино]-5-[М"-(2,4-дифторфенилсульфонил)-М"-пропаргиламино]пиримидин-4-ил)-4'-(пирролидин-1-илкарбонилокси)-L-фенилаланин; М-(2-[М',М'-диэтиламино]-5-[М"-(2,4-дифторфенилсульфонил)-М"-пропаргиламино]пиримидин-4-ил)-4'-(азетидин-1-илкарбонилокси)-L-фенилаланин; М-(2-[М',М'-диэтиламино]-5-[М"-(4-фторфенилсульфонил)-М"-пропаргиламино]пиримидин-4-ил)-4'-(азетидин-1 -илкарбонилокси)-L-фенилаланин; М-(2-[М',М'-диэтиламино]-5-[М"-(4-хлорфенилсульфонил)-М"-пропаргиламино]пиримидин-4-ил)-4'-(пирролидин-1 -илкарбонилокси)-L-фенилаланин; и его фармацевтически приемлемые соли. 19. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по одному любому из пп.12-15, 17 или 18. 20. Способ лечения заболевания млекопитающего, опосредованного а4интегринами, включающий введение фармацевтической композиции, содержащей фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по одному любому из пп.12-15, 17 или 18. 21. Способ по п.20, где заболевание опосредовано VLA-4. 22. Способ по п.20, где заболевание представляет собой воспалительное заболевание. 23. Способ по п.20, где заболевание представляет собой ревматоидный артрит. Евразийская патентная организация, ЕАПВ Россия, 109012, Москва, Малый Черкасский пер., 2/6 - 28 - |