|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea000008137b*\id |
|
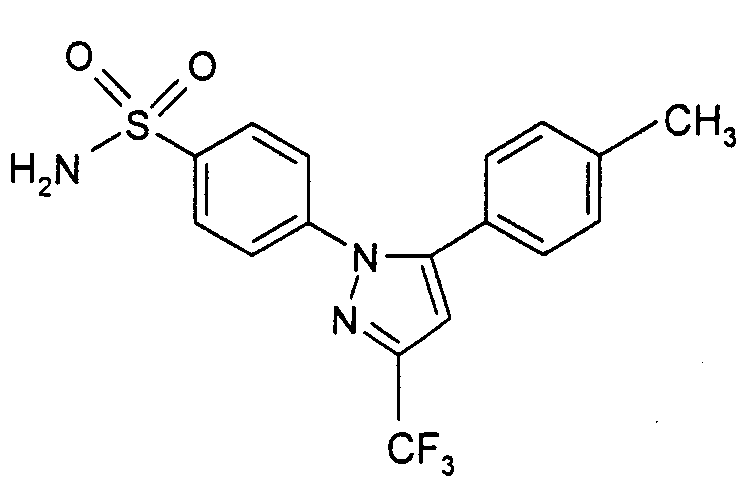
Термины запроса в документе Реферат Способ лечения или предупреждения рака, при котором млекопитающему, нуждающемуся в таком лечении, вводят терапевтически эффективное количество ингибитора протеинкиназы в комбинации с селективным ингибитором циклооксигеназы-2, где указанный ингибитор протеинкиназы представляет собой 5-[фтор-2-оксо-1,2-дигидроиндол-3-илиденметил]-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амид формулы или его фармацевтически приемлемую соль, и указанный селективный ингибитор циклооксигеназы-2 представляет собой 4-[5-(4-метилфенил)-3-(трифторметил)-1Н-пиразол-1-ил]фенилсульфонамид (целекоксиб) формулы или его фармацевтически приемлемую соль.
Полный текст патента (57) Реферат / Формула: или его фармацевтически приемлемую соль, и указанный селективный ингибитор циклооксигеназы-2 представляет собой 4-[5-(4-метилфенил)-3-(трифторметил)-1Н-пиразол-1-ил]фенилсульфонамид (целекоксиб) формулы или его фармацевтически приемлемую соль.
008137 Предпосылки изобретения Область изобретения Настоящее изобретение относится к способам лечения или предупреждения неоплазийных расстройств с использованием ингибиторов протеин-тирозинкиназ в комбинации с ингибиторами циклоок-сигеназ, в частности селективными ингибиторами циклооксигеназы-2. Уровень техники Неоплазма, или опухоль, представляет собой аномальную, нерегулируемую и дезорганизованную пролиферацию роста клеток. Неоплазма является злокачественной, или раковой, если она проявляет свойства деструктивного роста, инвазивности и метастазирования. Под инвазивностью подразумевается локальное распространение неоплазмы путем инфильтрации или разрушения окружающей ткани, обычно с прорывом базальных пластинок, которые определяют границы тканей, и из-за этого часто с прониканием в кровеносную систему организма. Под метастазированием обычно подразумевается диссемина-ция опухолевых клеток по лимфатическим или кровеносным сосудам. Под метастазированием также подразумевается миграция опухолевых клеток путем прямого распространения через серозные полости или субарахноидальные или другие пространства. В процессе метастазирования миграция опухолевых клеток в другие участки организма порождает неоплазмы в областях вдали от места их первоначального возникновения. В настоящее время рак является второй лидирующей причиной смерти в Соединенных Штатах Америки, и у более 8000000 человек в Соединенных Штатах Америки был диагностирован рак. В 1995 г. рак был причиной 23,3% всех смертей в Соединенных Штатах Америки (см. U.S. Dept. of Health and Human Services, National Center for Health Statistics, Health United States 1996-1997 и Injury Chartbook 117(1997)). Рак не вполне осмыслен на молекулярном уровне. Известно, что воздействие канцерогена на клетку, такого как некоторые вирусы, некоторые химические вещества или излучение, приводит к альтерации ДНК (дезоксирибонуклеиновой кислоты), которая инактивирует "супрессивный" ген или активирует "онкоген". Супрессивные гены представляют собой гены-регуляторы роста, которые после мутации больше не могут контролировать рост клеток. Онкогены представляют собой изначально нормальные гены (называемые проонкогенами), которые в результате мутации или изменения окружения экспрессии становятся трансформирующими генами. Продукты трансформирующих генов вызывают неадекватный рост клеток. Более чем двадцать разных нормальных клеточных генов могут стать онкогенами в результате генетической альтерации. Трансформированные клетки отличаются от нормальных клеток во многих отношениях, включая клеточную морфологию, взаимодействие клетка-клетка, состав мембран, цито-скелетную структуру, секрецию белков, экспрессию генов и смертность (трансформированные клетки могут расти бесконечно). В настоящее время рак лечат главным образом одним из или комбинацией трех типов терапии: хирургическое вмешательство, облучение и химиотерапия. Хирургическое вмешательство включает удаление всей массы больной ткани. Хотя хирургическое вмешательство иногда эффективно в удалении опухолей, расположенных в определенных местах, например в молочной железе, толстой кишке и коже, его нельзя применять ни в лечении опухолей, расположенных в других областях, таких как позвоночник, ни в лечении диссеминированных неопластических состояний, таких как лейкемия. Химиотерапия вызывает нарушение репликации клеток или клеточного метаболизма. Ее чаще всего применяют в лечении рака молочной железы, легких и яичников. Неблагоприятные воздействия системной химиотерапии, применяемой при лечении неопластического заболевания, больше всего пугают пациентов, подвергаемых лечению от рака. Из этих неблагоприятных воздействий наиболее распространенными и тяжелыми побочными воздействиями являются тошнота и рвота. Другие неблагоприятные побочные воздействия включают цитопению, инфекцию, кахексию, мукозит у пациентов, получающих высокие дозы химиотерапии с трансплантацией костного мозга или радиотерапию; алопецию (потерю волос); кожные осложнения (см. М. D. Abeloff, et al: Alopecia and Cutaneous Complications. P. 755-756. В Abeloff, M. D. Armitage, J. O. Lichter, A. S. and Niederhuber J. E. (издатели) Clinical Oncology. Churchill Livingston, New York, 1992, относительно кожных реакций на хи-миотерапевтические агенты), такие как зуд, крапивница и ангионевротический отек; неврологические осложнения; легочные и сердечные осложнения у пациентов, получающих облучение или химиотерапию; и репродуктивные и эндокринные осложнения. Побочные воздействия, вызванные химиотерапией, сильно сказываются на качестве жизни пациента и могут драматически влиять на соблюдение пациентом режима лечения. Кроме того, неблагоприятным побочным воздействием, связанным с химиотерапевтическими агентами, обычно является большая, ограничивающая дозу токсичность (dose-limiting toxicity, DLT) при введении этих лекарств. Например, мукозит является одним из проявлений большой ограничивающей дозу токсичности для нескольких противораковых агентов, включая антиметаболические цитотоксические агенты 5-FU (5-фторурацил), метотрексат и противоопухолевые антибиотики, такие как доксорубицин. Многие из этих вызванных химиотерапией побочных воздействий, если они являются тяжелыми, могут привести к госпитализации или требуют лечения анальгетиками для снятия боли. - 1 - 008137 Побочные воздействия, вызываемые химиотерапевтическими агентами и радиотерапией, стали проблемой особой важности в организации лечения раковых пациентов. В настоящее время ученые рассматривают лечение рака путем применения антиангиогенных агентов. Полагают, что ангиогенез является тем механизмом, посредством которого опухоли получают питательные вещества, необходимые для роста и метастазирования в другие части организма. Антиангиоген-ные агенты вмешиваются в эти процессы и разрушают или сдерживают рост опухоли. Например, в патенте США 5854205 описан выделенный белок эндостатин, который является ингибитором пролиферации эндотелиальных клеток и ангиогенеза; в патенте США 5843925 описан способ ингибирования ангиогенеза и пролиферации эндотелиальных клеток с использованием 7-[замещенный амин]-9-[(замещенный глициламидо]-6-деметил-6-дезокситетрациклина; в патенте США 5861372 описано применение сборного эндотелиального ингибитора ангиостатина в ингибировании ангиогенеза; в PCT/GB97/00650 описано применение производных циннолина для продуцирования антиангиогенного воздействия и/или снижения проницаемости сосудов; Tai-Ping D. описывает возможные антиангиоген-ные терапии (смотри Trends Pharmacol. Sci. 16, No. 2, 57-66, 1995); Lode, H. et al. сообщают, что синергия между антиангиогенным антагонистом интегрина альфа v и антителоцитокиновым слитым белком уничтожает спонтанные метастазы опухоли (см. РГОС. Nat. Acad. Sci. USA, 96 (4), 1591-1596, 1999); Giannis A. et al. описывают антагонисты интегринов и другие низкомолекулярные соединения в качестве ингибиторов ангиогенеза (см. New drugs in cancer therapy. Angew. Chem. Int. ed. Engl. 36(6), 588-590, 1997); в WO 97/41844 описан способ применения комбинаций ангиостатических соединений для предупреждения и/или лечения неоваскуляризации у пациентов-людей; в WO 98/22101 описан способ применения [пира-зол-1-ил]бензолсульфонамидов в качестве антиангиогенных агентов; и в патенте США 5792783 описано применение 3-гетероарил-2-индолинонов, которые являются ингибиторами протеинкиназ и антиангио-генными агентами для лечения различных видов рака. Недавно появилось сообщение о том, что лечение колоректального рака селективным ингибитором циклооксигеназы-2, конкретно Celecoxib(r), в комбинации с ингибитором ART-2, конкретно Herceptin(r), является более эффективным, чем если использовать любой из этих агентов по отдельности. Соответственно, существует потребность найти новые комбинации химиотерапевтических агентов, которые, если их использовать вместе, являются более эффективными, чем если их использовать по отдельности. Настоящее изобретение удовлетворяет эту потребность. Сущность изобретения Данное изобретение относится к способу лечения или предупреждения рака, при котором млекопитающему, нуждающемуся в таком лечении, вводят терапевтически эффективное количество ингибитора протеинкиназы в комбинации с селективным ингибитором циклооксигеназы-2, где указанный ингибитор протеинкиназы представляет собой 5-[5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил]-2,4-диметил-1Н-пиррол-3 -карбоновой кислоты (2-диэтиламиноэтил)амид формулы или его фармацевтически приемлемую соль, и указанный селективный ингибитор циклооксигеназы-2 представляет собой 4-[5-(4-метилфенил)-3-(трифторметил)-1Н-пиразол-1-ил]фенилсульфонамид (целе-коксиб) формулы О. о СН3 или его фармацевтически приемлемую соль. Предпочтительно, указанные выше соединения вводят в виде фармацевтической композиции, содержащей одно или более чем одно указанное выше соединение и фармацевтически приемлемый эксци-пиент. В частности, терапевтические агенты по данному изобретению могут быть приготовлены в виде отдельных композиций, которые вводят одновременно или в разное время, или терапевтические агенты можно вводить в виде одной композиции. - 2 - 008137 Способ по настоящему изобретению пригоден для лечения или предупреждения неоплазийных расстройств, включая акральную лентигинозную меланому, актинический кератоз, аденокарциному, адено-кистозную карциному, аденому, аденосаркому, аденосквамозную карциному, астроцитарные опухоли, карциному бартолиновой железы, базально-клеточную карциному, карциномы бронхиальных желез, капиллярные, карциноиды, карциному, карциносаркому, кавернозную, холангиокарциному, хондросарко-му, папиллому/карциному хориоидного сплетения, светлоклеточную карциному, цистаденому, эндодер-мальную опухоль, эндометриальную гиперплазию, эндометриальную стромальную саркому, эндомет-риоидную аденокарциному, эпендимальную, эпителиоидную, саркому Юинга, фиброламеллярную, очаговую узловую гиперплазию, гастриному, опухоли зародышевых клеток, глиобластому, глюкагоному, гемангиобластому, гемангиоэндотелиому, гемангиому, аденому печени, аденоматоз печени, гепатоцел-люлярную карциному, инсулиному, внутриэпителиальную неоплазию, внутриэпителиальную сквамозно-клеточную неоплазию, инвазивную сквамозно-клеточную карциному, крупноклеточную карциному, лейомиосаркому, лентиго злокачественное меланомное, злокачественную меланому, злокачественные мезотелиальные опухоли, медуллобластому, медуллоэпителиому, меланому, менингеальную, мезотели-альную, метастатическую карциному, мукоэпидермоидную карциному, нейробластому, нейроэпители-альную аденокарциному, узловатую меланому, овсяно-клеточную карциному, олигодендроглиальную, остеосаркому, панкреатическую полипептидную, папиллярную серозную аденокарциному, пинеально-клеточные, гипофизарные опухоли, плазмацитому, псевдосаркому, легочную бластому, почечно-клеточную карциному, ретинобластому, рабдомиосаркому, саркому, серозную саркому, мелкоклеточный рак, саркомы мягких тканей, соматостатин-секретирующую опухоль, сквамозную карциному, плоскоклеточный рак, субмезотелиальную, поверхностную меланому, недифференцированную карциному, увеаль-ную меланому, бородавчатый рак, випому, хорошо дифференцированную карциному и опухоль Вильмса. Способы по настоящему изобретению обладают одним или более преимуществами. Одним из этих преимуществ является то, что композиции, агенты и терапевтические средства по настоящему изобретению вводят в комбинации в низкой дозе, т.е. в дозе, более низкой, чем традиционно используемая в клинических случаях доза каждого индивидуального компонента, который вводят один. Преимущество от снижения дозы соединений, композиций, агентов и терапевтических средств по настоящему изобретению, вводимых млекопитающему, включает снижение частоты побочных воздействий, связанных с более высокими дозировками. Например, снижение дозировки химиотерапевтического агента, такого как метотрексат, должно приводить к уменьшению частоты и тяжести тошноты и рвоты по сравнению с наблюдаемыми при более высоких дозировках. Аналогичных преимуществ можно ожидать, если ингибитор протеинкиназы по настоящему изобретению использовать в комбинации с ингибитором циклооксигеназы, в частности селективным ингибитором циклооксигеназы-2. Благодаря снижению частоты побочных воздействий ожидается улучшение качества жизни пациента, подвергающегося лечению от рака. Дополнительные преимущества снижения частоты побочных воздействий включают улучшение соблюдения пациентом режима лечения, снижение количества госпитализаций, необходимых для лечения побочных воздействий, и уменьшение введения анальгетических агентов, необходимых для снятия боли, связанной с побочными воздействиями. Другим ожидаемым преимуществом является то, что ингибирование опухолей сильнее, когда ингибитор протеинкиназы вводят в комбинации с ингибитором циклооксигеназы, предпочтительно селективным ингибитором циклооксигеназы-2, чем в случае, когда любой из этих агентов вводят один. Подробное описание изобретения Определения "Фармацевтическая композиция" относится к смеси одного или более чем одного соединения, описанного в данной заявке, или их физиологически/фармакологически приемлемых солей или пролекарств с другими химическим компонентами, такими как физиологически/фармацевтически приемлемые носители и эксципиенты. Назначение фармацевтической композиции - способствовать введению соединения в организм. Соединения по настоящему изобретению могут также действовать как пролекарства. "Пролекарст-во" относится к агенту, который in vivo превращается в родительское лекарственное средство. Пролекар-ства часто используют, поскольку в некоторых ситуациях их вводить легче, чем родительское лекарственное средство. Например, они могут быть биодоступными при пероральном введении, а родительское лекарственное средство не может. Пролекарство также может иметь улучшенную растворимость в фармацевтических композициях по сравнению с родительским лекарственным средством. Например, без ограничения, пролекарство может представлять собой соединение по настоящему изобретению, которое вводят в виде сложного эфира ("пролекарства"), чтобы способствовать прохождению через клеточную мембрану, где водорастворимость вредна для подвижности, зато потом это пролекарство метаболически гидролизуется до карбоновой кислоты, активного вещества, внутри клетки, где водорастворимость полезна. Еще одним примером пролекарства может быть короткий полипептид, например, без ограничения, полипептид из 2-10 аминокислот, связанный через концевую аминогруппу с карбоксильной группой соединения по данному изобретению, причем этот полипептид гидролизуется или метаболизируется in vivo - 3 - 008137 с высвобождением активной молекулы. Пролекарства соединения 5-[5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил]-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амид формулы входят в объем данного изобретения. Кроме того, предполагается, что соединение 5-[5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил]-2,4-диметил-Ш-пиррол-З-карбоновой кислоты (2-диэтиламиноэтил)амид формулы может быть метаболизирован ферментами в теле организма, такого как человек, с образованием метаболита, который может модулировать активность протеинкиназ. Такие метаболиты входят в объем данного изобретения. Используемый здесь термин "физиологически/фармацевтически приемлемый носитель" относится к носителю или разбавителю, который не вызывает значимого раздражения в организме и не аннулирует биологическую активность и свойства вводимого соединения. " Фармацевтически приемлемый эксципиент" относится к инертному веществу, добавляемому в фармацевтическую композицию для дополнительного облегчения введения соединения. Примеры экс-ципиентов включают, без ограничения, карбонат кальция, фосфат кальция, различные сахара и типы крахмала, производные целлюлозы, желатин, растительные масла и полиэтиленгликоли. Используемый здесь термин "фармацевтически приемлемая соль" относится к тем солям, которые сохраняют биологическую эффективность и свойства родительского соединения. Такие соли включают в себя: 1) соли присоединения кислот, которые получают путем взаимодействия родительского соединения в форме свободного основания с неорганическими кислотами, такими как соляная кислота, бромоводо-родная кислота, азотная кислота, фосфорная кислота, серная кислота и перхлорная кислота и тому подобные, или с органическими кислотами, такими как уксусная кислота, щавелевая кислота, (D) или (L) яблочная кислота, малеиновая кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота, винная кислота, лимонная кислота, янтарная кислота или малоновая кислота и тому подобные, предпочтительно соляная кислота или (Ь)-яблочная кислота, такие как L-малат 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амида; или 2) соли, которые образуются, когда кислотный протон, присутствующий в родительском соединении, либо замещается ионом металла, например ионом щелочного металла, ионом щелочно-земельного металла, либо координируется с органическим основанием. Примерами ионов являются ионы алюминия, кальция, лития, магния, калия, натрия и цинка в их обычных валентностях. Предпочтительные органические основания включают протонированные третичные амины и четвертичные аммониевые катионы, включая частично триэтиламин, диэтиламин, ^№-дибензилэтилендиамин, хлорпрокаин, холин, диэта-ноламин, этилендиамин, меглюмин (N-метилглюкамин) и прокаин. " Способ" относится к методам, средствам, приемам и процедурам для осуществления заданной задачи, включая, но не ограничиваясь ими, те методы, средства, приемы и процедуры, которые либо известны, либо легко могут быть разработаны на основе известных методов, средств, приемов и процедур специалистами в химической, фармацевтической, биологической, биохимической и медицинской областях. "Лечить" и "лечение" относятся к любому процессу, действию, применению, терапии или т.п., где млекопитающее, включая человека, является объектом медицинской помощи с целью улучшения его состояния прямо или косвенно. В частности, в отношении рака эти термины означают только то, что ве 008137 роятная продолжительность жизни индивидуума, пораженного раком, будет увеличена или что один или более чем один симптом этого заболевания будет устранен. Контролировать это можно по отсроченному возникновению первичных или вторичных опухолей, замедлению развития первичных или вторичных опухолей, уменьшению распространения первичных и вторичных опухолей, ослаблению или уменьшению тяжести вторичных воздействий заболевания, замедлению роста опухолей и регрессии опухолей. Термин "предупреждение" включает в себя либо предупреждение начала клинически явной неопла-зии в целом, либо предупреждение начала предклинически явной стадии неоплазии у субъектов группы риска. Предполагается также, что это определение охватывает предупреждение зарождения злокачественных клеток или приостановление или реверсирование развития презлокачественных клеток в злокачественные клетки. Это включает в себя профилактическое лечение субъектов с риском развития неопла-зии. Предполагается, что выражение "терапевтически эффективное" определяет количество противоракового агента, которое достигает цели снижения тяжести неопластического заболевания и частоты неопластического заболевания по сравнению с лечением каждым агентом в отдельности с одновременным избежанием нежелательных побочных воздействий, обычно связанных с альтернативными терапиями. Предполагается, что термины "терапевтическое воздействие" или "терапевтически эффективное количество" определяют количество противоракового агента, которое требуется для ослабления до некоторой степени одного или более чем одного симптома неопластического расстройства, включая, но не ограничиваясь ими: 1) уменьшение количества раковых клеток; 2) уменьшение размера опухоли; 3) инги-бирование (т.е. замедление до некоторой степени, предпочтительно остановку) инфильтрации раковых клеток в периферические органы; 3) ингибирование (т.е. замедление до некоторой степени, предпочтительно остановку) метастазирования опухоли; 4) ингибирование до некоторой степени роста опухоли; 5) ослабление или уменьшение до некоторой степени одного или более чем одного симптома, связанного с данным расстройством; и/или 6) ослабление или уменьшение побочных воздействий, связанных с введением противораковых агентов. Выражение "комбинированная терапия" (или "ко-терапия") включает в себя введение ингибитора протеинкиназы и ингибитора циклооксигеназы-2 в рамках конкретного режима лечения, предназначенного для обеспечения полезного эффекта от совместного действия этих терапевтических агентов. Полезный эффект данной комбинации включает, но не ограничивается ими, фармакокинетическое или фарма-кодинамическое совместное действие комбинации терапевтических агентов. Введение этих терапевтических агентов в комбинации в типичном случае осуществляют за определенный период времени (обычно минуты, часы, сутки или недели в зависимости от выбранной комбинации). В общем случае, предполагается, что "комбинированная терапия" не охватывает введение двух или более чем двух этих терапевтических агентов в рамках режимов отдельных монотерапий, которое случайно и произвольно приводит к комбинации по настоящему изобретению. Предполагается, что "комбинированная терапия" охватывает введение этих терапевтических агентов последовательным образом, т.е. каждый терапевтический агент вводят в разное время, а также введение этих терапевтических агентов или по меньшей мере двух терапевтических агентов практически одновременно. В сущности, одновременное введение можно осуществлять, например, путем введения субъекту одной капсулы, имеющей фиксированную пропорцию каждого терапевтического агента, или групп отдельных капсул каждого из этих терапевтических агентов. Последовательное или практически одновременное введение каждого терапевтического агента можно осуществлять любым подходящим путем, включая, но не ограничиваясь ими, пероральные пути, внутривенные пути, внутримышечные пути и прямое всасывание через ткани мембран слизистой. Терапевтические агенты можно вводить одним и тем же путем или разными путями. Например, выбранный первый терапевтический агент данной комбинации можно вводить внутривенной инъекцией, а другие терапевтические агенты данной комбинации можно вводить перорально. Альтернативно, например, оба терапевтических агента можно вводить перорально или оба терапевтических агента можно водить внутривенной инъекцией. Последовательность, в которой терапевтические агенты вводят, не является строго критичной. "Комбинированная терапия" также может включать в себя введение терапевтических агентов, как описано выше, в комбинации с дополнительными другими биологически активными ингредиентами (например, но без ограничения, вторым и другим противоопухолевым агентом) и нелекарственными видами терапии (например, но без ограничения, хирургическим или радиационным лечением). Когда комбинированная терапия дополнительно включает в себя радиационное лечение, это радиационное лечение можно проводить в любое подходящее время при условии, что достигается полезный эффект от совместного действия комбинации терапевтических агентов и радиационного лечения. Например, в соответствующих случаях полезный эффект по-прежнему достигается, когда радиационное лечение отдалено по времени от введения терапевтических агентов, может быть на сутки или даже недели. "Дополнительная терапия" охватывает лечение субъекта агентами, которые уменьшают или позволяют избежать побочных воздействий, связанных с комбинированной терапией по настоящему изобретению, включая, но не ограничиваясь ими, те агенты, которые, например, уменьшают токсическое воздействие противораковых лекарств, например ингибиторы резорбции кости, кардиозащитные агенты; предотвращают или снижают количество случаев тошноты и рвоты, связанных с химиотерапией, радио - 5 - 008137 терапией или операцией; или снижают количество случаев инфекций, связанных с введением миелосу-прессивных противораковых лекарственных средств. Предпочтительные воплощения Целекоксиб, используемый в терапевтических комбинациях по настоящему изобретению, можно получить способом, изложенным в патенте США 5466823. В комбинации по изобретению также включают изомерные формы, пролекарства и таутомеры описанных соединений и их фармацевтически приемлемые соли. В качестве примера, фармацевтически приемлемые соли получают из муравьиной, уксусной, пропионовой, янтарной, гликолевой, глюконовой, молочной, яблочной, винной, лимонной, аскорбиновой, глюкуроновой, малеиновой, фумаровой, пировино-градной, аспарагиновой, глутаминовой, бензойной, антраниловой, мезиловой, стеариновой, салициловой, п-гидроксибензойной, фенилуксусной, миндальной, эмбоновой (памовой), метансульфоновой, этансуль-фоновой, бензолсульфоновой, пантотеновой, толуолсульфоновой, 2-гидроксиэтансульфоновой, сульфа-ниловой, циклогексиламиносульфоновой, альгиновой, бета-гидроксимасляной, галактаровой и галакту-роновой кислот. Полезность Соединения по настоящему изобретению являются ингибиторами протеинкиназ (РК) и фермента циклооксигеназы, в частности фермента циклооксигеназы-2, и, следовательно, полезны в лечении рака. РК, каталитическое действие которых модулируется соединением 5-[5-фтор-2-оксо-1,2-дигидроиндол-3 -илиденметил]-2,4-диметил-1 Н-пиррол-3 -карбоновой кислоты (2-диэтиламиноэтил)амид формулы по данному изобретению, включают протеинтирозинкиназы, такие как рецепторные тирозинкиназы (RTK), клеточные тирозинкиназы (СТК), и серин-треониновые киназы (STK). RTK-опосредованная сигнальная трансдукция инициируется внеклеточным взаимодействием со специфическим фактором роста (лигандом) с последующей димеризацией рецепторов, временной стимуляцией активности внутренних протеинтирозинкиназ и фосфорилированием. В результате, для молекул, передающих внутриклеточный сигнал, создаются сайты связывания, и это ведет к образованию комплексов со спектром цитоплазмати-ческих сигнальных молекул, что облегчает соответствующие клеточные ответы (например деление клеток, метаболические воздействия на внеклеточное микроокружение и так далее) (Schlessinger and Ullrich, 1992, Neuron 9:303-391). Было показано, что сайты фосфорилирования тирозина на рецепторах факторов роста функционируют как сайты связывания с высокой аффинностью к SH2 (scr гомология) доменам сигнальных молекул (Fantl et al., 1992, Cell 69: 413-423; Songyang et al., Mol. Cell. Biol. 14:2777-2785; Songyang et al., 1993, СеИ 72:767-778 и Koch et al., 1991, Science 252:668-678). Было идентифицировано несколько внутриклеточных белков-субстратов, которые связываются с RTK. Их можно разделить на две основные группы: 1) субстраты, которые имеют каталитический домен, и 2) субстраты, у которых отсутствует такой домен, но которые служат адаптерами и связываются с каталитически активными молекулами (Songyang et al., 1993, Cell 72:767-778). Специфичность взаимодействий между рецепторами и SH2 доменами их субстратов определяется аминокислотными остатками, непосредственно окружающими фосфорилированный тирозиновый остаток. Различия в аффинности связывания между SH2 доменами и аминокислотными последовательностями, окружающими фосфотирозиновые остатки на конкретных рецепторах, согласуются с наблюдаемыми различиями в профилях фосфорилирования их субстратов (Songyang et al., 1993, Cell 72:767-778). Эти наблюдения позволяют предполагать, что функция каждой RTK определяется не только характером ее экспрессии и доступностью лиганда, но и порядком путей нисходящей трансдук-ции сигналов, которая активируется конкретным рецептором. Таким образом, фосфорилирование обеспечивает важную регуляторную стадию, которая определяет селективность сигнальных путей, рекрутированных рецепторами специфических факторов роста, а также рецепторами фактора дифференциации. STK, будучи главным образом цитозольными, влияют на внутреннюю биохимию клетки, часто по линии ответа на РТК событие. STK участвуют в сигнальных процессах, которые инициируют синтез ДНК и последующий митоз, приводящий к клеточной пролиферации. Таким образом, РК сигнальная трансдукция приводит, наряду с другими ответами, к клеточной пролиферации, дифференциации, росту и метаболизму. Аномальная клеточная пролиферация может привести к целому ряду расстройств и заболеваний, в том числе к развитию неоплазии, такой как карци - 6 - 008137 нома, саркома, глиобластома и гемангиома, расстройствам, таким как лейкемия, псориаз, атеросклероз, артрит и диабетическая ретинопатия, и другим расстройствам, связанным с неконтролируемым ангиоге-незом и/или васкулогенезом. Чтобы применять на практике настоящее изобретение, не требуется точного понимания механизма, посредством которого соединения по данному изобретению ингибируют РК. Однако без привязки к какому-либо конкретному механизму или теории представляется, что соединение 5-[5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил]-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амид формулы взаимодействуют с аминокислотами в каталитической области РК. РК обычно имеют двудольную структуру, где АТР (аденозинтрифосфат), вероятно, связывается в щели между двумя долями в области, где между РК сохраняются аминокислоты. Представляется, что ингибиторы РК связываются посредством не ковалентных взаимодействий, таких как водородное связывание или Ван-дер-Ваальсовы силы, и ионных взаимодействий в одной и той же общей области, где вышеуказанный АТР связывается с РК. Более конкретно предполагается, что 2-индолиноновый компонент соединения 5-[5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил]-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амид формулы связывается в общем пространстве, обычно занятом адениновым кольцом АТР. Специфичность конкретной молекулы к конкретной РК затем может возрасти в результате дополнительных взаимодействий между различными заместителями на 2-индолиноновым ядре и аминокислотными доменами, специфичными к конкретным РК. Таким образом, разные индолиноновые заместители могут внести вклад в предпочтительное связывание с конкретными РК. Возможность выбора соединений, активных в разных сайтах связывания АТР (или другого нуклеотида), позволяют использовать 5-[5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил]-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амид для направленного воздействия на любой белок с таким сайтом. Таким образом, соединение 5-[5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил]-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амид формулы раскрытое в данной заявке, полезно в in vitro анализах на такие белки, а также как соединение, обладающее in vivo терапевтическими воздействиями через взаимодействие с такими белками. Кроме того, соединение 5-[5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил]-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амид формулы - 7 - 008137 обеспечивают терапевтический подход к лечению многих видов солидных опухолей, включая, но не ограничиваясь ими, карциномы, саркомы, в том числе саркому Капоши, эритробластому, глиобластому, менингиому, астроцитому, меланому и миобластому. Лечение или предупреждение не солидного опухолевого рака, такого как лейкемия, также входит в объем данного изобретения. Показания могут включать, но не ограничиваться ими, рак мозга, рак мочевого пузыря, рака яичника, рак желудка, рак поджелудочной железы, рак прямой кишки, рак крови, рак легкого и рак кости. Дополнительными примерами, без ограничения, типов расстройств, связанных с неадекватной активностью РК, в предупреждении, лечении и исследовании которых могут быть полезны соединения по настоящему изобретению, являются клеточные пролиферативные расстройства, фиброзные расстройства и метаболические расстройства. Клеточные пролиферативные расстройства, которые можно предупреждать, лечить или дополнительно исследовать согласно настоящему изобретению, включают рак, пролиферативные расстройства кровеносных сосудов и пролиферативные расстройства мезангиальных клеток. К пролиферативным расстройствам кровеносных сосудов относятся расстройства, связанные с аномальным васкулогенезом (образование кровеносных сосудов) и ангиогенезом (распространение кровеносных сосудов). Хотя васкулогенез и ангиогенез играют важную роль в ряде нормальных физиологических процессов, таких как развитие эмбриона, образование желтого тела, заживление ран и регенерация органов, они также играют центральную роль в развитии рака, где они приводят к образованию новых капилляров, необходимых для сохранения опухоли живой. Другие примеры расстройств пролиферации кровеносных сосудов включают артрит, где новые капиллярные кровеносные сосуды оккупируют сустав и разрушают хрящ, и глазные заболевания, такие как диабетическая ретинопатия, где новые капилляры в сетчатке вторгаются в стекловидное тело, кровоточат и вызывают слепоту. Было установлено, что две структурно родственные RTK связывают VEGF (васкулярный эндотели-альный фактор роста) с высокой аффинностью: fms-подобный тирозиновый 1 (fit-1) рецептор (Shibuya et al., 1990, Oncogene, 5:519-524; De Vries et al., 1992, Science. 255:989-991) и KDR/FLK-1 рецептор, также известный как VEGF-R2. Известно (Ferrara и Henzel, 1989, Biochen. Biophys. Res. Comm., 161:851-858; Vaisman et al., 1990, J. Biol. Chem., 265:19461-19566), что васкулярный эндотелиальный фактор роста (VEGF) является специфическим митогеном эндотелиальных клеток с in vitro стимулирующей рост эн-дотелиальных клеток активностью. Из информации, изложенной в заявках США 08/193829, 08/038596 и 07/975750, строго следует, что VEGF не только ответственен за пролиферацию клеток эндотелия, но и является первичным регулятором нормального и патологического ангиогенеза (в целом см. Klagsburn и Soker, 1993, Current Biology, 3(10) 699-702; Houck et al., 1992, J. Biol. Chem., 267:26031-26037). Нормальный васкулогенез и нормальный ангиогенез играют важную роль в различных физиологических процессах, таких как развитие эмбриона, заживление ран, регенерация органов и женские репродуктивные процессы, такие как развитие фолликул в желтом теле в процессе овуляции и рост плаценты при беременности (Folkman и Shing, 1992, J. Biological Chem., 267(16): 10931-34). Неконтролируемый васкулогенез и/или ангиогенез связан с такими заболеваниями, как диабет, а также со злокачественными солидными опухолями, рост которых зависит от васкуляризации (Klagsburn и Soker, 1993, Current Biology, 3(10):699-702; Folkham, 1991, J. Natl. Cancer Inst., 82:4-6; Weidner et al., 1991. New Engl. J. Med. 324:1-5). Предполагаемая роль VEGF в пролиферации эндотелиальных клеток и миграции в ходе ангиогенеза и васкулогенеза указывает на важную роль KDR/FLK-1 рецептора в этих процессах. Результатом неконтролируемого васкулогенеза могут быть такие заболевания, как сахарный диабет (Folkman, 198, в XIth Congress of Thrombosis and Haemostasis (Verstraete et al., eds.), pp. 583-596, Leuven University Press, Leuven) и артрит, а также рост злокачественных опухолей (см., например, Folkman 1971, N. Engl. J. Med., 285:1182-186). Рецепторы, с которыми специфически связывается VEGF, являются важной и мощной терапевтической мишенью для регуляции и модуляции васкулогенеза и/или ангиогенеза и ряда тяжелых заболеваний, в которые вовлечен аномальный клеточный рост, вызванный такими процессами (Plowman et al., 1994, DN &P, 7(6):334-339). Более конкретно, весьма специфическая роль KDR/FLK-1 рецептора в неоваскуляризации делает его целью выбора терапевтических подходов к лечению рака и других заболеваний, в которые вовлечено неконтролируемое образование кровеносных сосудов. Таким образом, настоящее изобретение предусматривает использование соединений, способных регулировать и/или модулировать сигнальную трансдукцию тирозинкиназами, включая KGR/FLK-1 рецеп - 8 - 008137 торную сигнальную трансдукцию, чтобы ингибировать или промотировать ангиогенез и/или васкулогенез, т.е. соединения, которые ингибируют, предупреждают или препятствуют сигналу, передаваемому KDR/FLK-1, при активации лигандами, такими как VEGF. Хотя считается, что соединения по настоящему изобретению действуют на рецептор или другой компонент по пути тирозинкиназной передачи сигнала, они также могут действовать непосредственно на клетки опухоли, которые являются результатом неконтролируемого ангиогенеза. Хотя номенклатура человеческой и мышиной копий общего "flk-1" рецептора различается, они во многих аспектах являются взаимозаменяемыми. Мышиный рецептор, FLK-1, и его человеческая копия, KDR, имеют общую гомологию последовательности 93,4% в пределах внутриклеточного домена. Аналогично, мышиный FLK-1 связывает человеческий VEGF с такой же аффинностью, как и мышиный VEGF, и, соответственно, активируется лигандом, производным от обоих видов (Millauet et al., 1993, Cell, 72:835-846; Quinn et al., 1993, Proc. Natl. Acad. Sci. USA, 90:7533-7537). FLK-1 также связывается с, а затем фосфорилирует по тирозину человеческие RTK субстраты (например, PLC-y (фосфолипаза С гамма) или р85), при совместной экспрессии в 293 клетках (фибробласты почки человеческого эмбриона). Таким образом, модели, рассчитанные на FLK-1 рецептор, непосредственно применимы для распознавания KDR рецептора. Например, использование мышиного FLK-1 рецептора в способах идентификации соединений, регулирующих пути сигнальной трансдукции у мышей, непосредственно применимо к идентификации соединений, которые можно использовать для регуляции пути сигнальной трансдукции у человека, т.е. соединений, которые регулируют активность, связанную с KDR рецептором. Так, химические соединения, идентифицированные как ингибиторы KDR/FLK-1 in vitro, могут быть утверждены в подходящих in vivo моделях. Было показано, что in vivo как мышиная, так и крысиная животные модели имеют исключительное значение для исследования клинического потенциала агентов, действующих на KDR/FLK-1-индуцируемый путь сигнальной трансдукции. Таким образом, настоящее изобретение предусматривает использование соединений, которые регулируют, модулируют и/или ингибируют васкулогенез и/или ангиогенез, воздействуя на ферментативную активность KDR/FLK-1 рецептора и препятствуя сигналу, передаваемому KDR/FLK-1. Таким образом, согласно настоящему изобретению предложен терапевтический подход к лечению многих видов солидных опухолей, включая, но не ограничиваясь ими, глиобластому, меланому и саркому Капоши, и рак яичника, легкого, молочной железы, простаты, поджелудочной железы, прямой кишки и эпидермиса. Кроме того, данные дают основание ожидать, что введение соединений, которые ингибируют KDR/FLK-1-опосредованный путь сигнальной трансдукции, можно применять также в лечении гемангиомы, рестеноза и диабетической ретинопатии. Кроме того, данное изобретение предусматривает ингибирование васкулогенеза и ангиогенеза другими рецептор-опосредованными путями, в том числе путем, включающим в себя flt-1 рецептор. Сигнальная трансдукция, опосредованная рецепторной тирозинкиназой, инициируется внеклеточным взаимодействием со специфическим фактором роста (лигандом) с последующей димеризацией, временной стимуляцией активности внутренней протеинтирозинкиназы и аутофосфорилированием. В результате этого, для молекул внутриклеточной сигнальной трансдукции создаются сайты связывания, что приводит к образованию комплексов с рядом цитоплазматических сигнальных молекул, которые облегчают соответствующий клеточный ответ, например деление клеток и метаболические воздействия на внеклеточное микроокружение (см. Schlessinger and Ullrich, 1992, Neuron 9:1-20). Близкая гомология внутриклеточных областей KDR/FLK-1 с областью рецептора PDGF-в (тромбо-цитарный фактор роста в) (гомология 50,3%) и/или родственного рецептора flt-1 указывает на индуцирование перекрывающихся путей сигнальной трансдукции. Например, для рецептора PDGF-в было показано, что члены src семейства (Twamley et al., 1993, Proc. Natl. Acad. Sci. USA, 90:7696-7700), фосфатиди-линозитол-3'-киназа (Hu et al., 1992, Mol. Cell. Biol., 12:981-990), фосфолипаза cy (Kashishian и Cooper, 1993, Mol. Cell. Biol., 4:49-51), ras-GTPаза-активирующий белок (GTP - гуанитрифосфатаза) (Kashishian ef al., 1992, EMBO J., 11:1373-1382), PTP-ID/syp (Kazlauskas et al., 1993, Proc. Natl. Acad. Sci. USA. 10 90:6939-6943), Grb2 (белок, связывающийся с фактором роста) (Arvidsson et al., 1994, Mol. Cell. Biol., 14:6715-6726) и адаптерные молекулы Shc и Nck (Nishimura et al., 1993, Mol. Cell. Biol., 13:6889-6896) связываются с областями, включающими в себя разные сайты аутофосфорилирования (в общем см. Claesson-Welsh, 1994, Prog. Growth Factor Res., 5:37-54). Так, пути сигнальной трансдукции, активируемые KDR/FLK-1, включают ras путь (Rozakis et al., 1992, Nature, 360:689-692), Pl (фосфатидилинозитол)-3'-киназный, src-опосредованный и plcy-опосредованный пути. Каждый из этих путей может играть критическую роль в ангиогенном и/или васкулогенном эффекте KDR/FLK-1 в эндотелиальных клетках. Следовательно, органические соединения, описанные здесь, могут применяться для модулирования ангиоге-неза и васкулогенеза, так как эти процессы регулируются этими путями. Расстройства, относящиеся к сморщиванию, сужению или смыканию кровеносных сосудов, такие как рестеноз, также подразумеваются и могут быть подвергнуты лечению или могут быть предотвращены способом по данному изобретению. Фиброзные расстройства относятся к аномальному образованию внеклеточных матриксов. Примерами фиброзных расстройств являются цирроз печени и пролиферативные расстройства мезангиальных - 9 - 008137 клеток. Цирроз печени характеризуется увеличением составляющих внеклеточного матрикса, приводящих к образованию рубцов на печени. Увеличение составляющих внеклеточного матрикса, приводящее к образованию рубцов на печени, также может быть вызвано вирусной инфекцией, такой как гепатит. Представляется, что в циррозе печени важную роль играют липоциты. Другие подразумеваемые фиброзные расстройства включают атеросклероз. Пролиферативные расстройства мезангиальных клеток относятся к расстройствам, вызываемым аномальной пролиферацией мезангиальных клеток. Мезангиальные пролиферативные расстройства включают различные почечные заболевания человека, такие как гломерулонефрит, диабетическая неф-ропатия и злокачественный нефросклероз, а также такие расстройства, как тромботические микроангио-патические синдромы, отторжение трансплантата и гломерулопатии. RTK PDGF-R вовлечен в поддержание пролиферации мезангиальных клеток (Floege et al., 1993, Kidney International 43:47S-54S). Многие виды рака представляют собой клеточные пролиферативные расстройства и, как указывалось ранее, РК связаны с клеточными пролиферативными расстройствами. Таким образом, не удивительно, что РК, такие как, например, члены RTK семейства, связаны с развитием рака. Некоторые из этих рецепторов, такие как EGFR (Tuzi ef al., 1991, Br. J. Cancer 63:227-233, Torp et al., 1992, APMIS 100:713719), HER2 (рецептор человеческого эпидермального фактора роста типа 2 )/neu (Slamon et al., 1989, Science 244:707-712) и PDGF-R (рецептор PDGF) (Kumabe et al., 1992, Oncogene, 7:627-633) сверхэкспресси-руются во многих опухолях и/или постоянно активируются аутокринными петлями. Действительно, при большинстве распространенных и тяжелых видов рака были продемонстрированы эти сверхэкспрессии рецепторов (Akbasak and Suner-Akbasak et al., 1992, J. Neurol. Sci 111:119-133, Dickson et al.,1992, Cancer Treatment Res. 61:249-273, Korc et al., 1992, J. Clin. Invest. 90:1352-1360) и аутокринные петли (Lee and Donoghue, 1992, J. Cell. Biol., 118:1057-1070, Korc et al., выше, Akbasak and Suner-Akbasak et al., выше). Например, EGFR связан со сквамозно-клеточной карциномой, астроцитомой, глиобластомой, раком в области головы и шеи, раком легкого и раком мочевого пузыря. HER2 связан с раком молочной железы, яичника, желудка, легкого, поджелудочной железы и мочевого пузыря. PDGF-R связан с глиобластомой и меланомой, а также раком легкого, яичника и простаты. RTK c-met также связан с образованием злокачественных опухолей. Например, c-met связан, наряду с другими видами рака, с колоректальной, тирео-идной, панкреатической, желудочной и гепатоцеллюляной карциномами и лимфомами. Кроме того, c-met также связан с лейкемией. Сверхэкспрессия c-met гена также обнаружена у пациентов с болезнью Ходжкина и болезнью Беркита. IGF-IR (рецептор инсулиноподобного фактора роста-I), в дополнение к тому, что он вовлечен в обеспечение питания и диабет типа II, также связан с некоторыми видами рака. Например, IGF-I задействован как аутокринный стимулятор роста для некоторых видов опухолей, например карциномных клеток рака молочной железы человека (Arteaga et al., 1989, J. Clin. Invest. 84:1418-1423) и мелких клеток рака легкого (Macauley et al., 1990, Cancer Res., 50:2511-2517). Кроме того, оказывается, что IGF-I, полностью вовлеченный нормальный рост и дифференциацию нервной системы, одновременно является также ау-токринным стимулятором глиом человека (Sandberg-Nordqvist et al., 1993, Cancer Res. 53:2475-2478). Важность IGF-IR и его лигандов в клеточной пролиферации подтверждается также тем фактом, что рост многих типов клеток в культуре (фибробласты, эпителиальные клетки, клетки гладких мышц, Т-лимфоциты, миелоидные клетки, хондроциты и остеобласты (стволовые клетки костного мозга) стимулируется IGF-I (Goldring ana Goldring, 1991, Eukaryotic Gene Expression, 1:301-326). Basera and Coppola высказывают предположение, что IGF-IR играет центральную роль в механизме трансформации и, как таковые, могут быть предпочтительной мишенью терапевтических вмешательств в широкий спектр злокачественных образований человека (Baserga, 1995, Cancer Res., 55:249-252; Baserga, 1994, Cell 79:927930; Coppola et al., 1994, Mol. Cell. Biol.. 14:4588-4595). STK вовлечены во многие виды рака, в том числе в частности рак молочной железы (Cance et al., Int. J. Cancer, 54:571-77 (1993)). Связь между аномальной активностью РК и заболеванием не ограничена раком. Например, RTK связаны с такими заболеваниями, как псориаз, сахарный диабет, эндометриоз, ангиогенез, развитие ате-роматозных бляшек, болезнь Альцгеймера, рестеноз, болезнь Гиппеля-Линдау, эпидермальная гиперпролиферация, нейродегенеративные заболевания, возрастная дегенерация желтого пятна и гемангиома. Например, EGFR участвует в заживлении ран роговицы и кожи. Дефекты в инсулин-R и IGF-1R служат признаком сахарного диабета II типа. Более полная корреляция между специфическими RTK и их терапевтическими признаками изложена в Plowman et al., 1994, DN &P 7:334-339. Как отмечалось ранее, не только RTK, но и СТК, включая, но не ограничиваясь ими, src, abl, fps, yes, fyn, lyn, lck, blk, hck, fgr и yrk (обзор Bolen et al., 1992, FASEB J., 6:3403-3409) вовлечены в пролифе-ративный и метаболический путь сигнальной трансдукции и, таким образом, можно ожидать, и это было показано, что они вовлечены во многие РТК-опосредованные расстройства, на которые направлено настоящее изобретение. Например, было показано, что мутировавший src (v-src) является онкопротеином (pp60v-src) у цыплят. Более того, его клеточный гомолог, протоонкоген pp60c-src передает онкогенные сигналы многих рецепторов. Сверхэкспрессия EGFR или HER-2/neu в опухолях приводит к конститутивной активации pp60c-src, что характерно для злокачественных клеток, но отсутствует в нормальных клетках. С - 10 - 008137 другой стороны, мыши с дефицитом экспрессии c-src обладают остеопетрозным фенотипом, что указывает на ключевое участие с-src в функционировании остеокластов и возможное его вовлечение в связанные с этим расстройства. Аналогично, Zap70 вовлечен в Т-клеточную сигнализацию, которая может быть связана с аутоиммунными заболеваниями. STK связаны с воспалением, аутоиммунным заболеванием, иммунореакциями и гиперпролифера-тивными расстройствами, такими как рестеноз, фиброз, псориаз, остеоартрит и ревматоидный артрит. РК также вовлечены в имплантацию эмбриона. Поэтому соединения по настоящему изобретению могут обеспечить эффективный способ предупреждения такой имплантации эмбриона и, тем самым, быть полезными в качестве агентов контроля рождаемости. Дополнительными расстройствами, которые можно лечить или предупреждать, используя соединения по данному изобретению, являются иммунологические расстройства, такие как аутоиммунное заболевание, СПИД (синдром приобретенного иммунодефицита) и сердечно-сосудистые расстройства, такие как атеросклероз. Наконец, в настоящее время существует мнение, что как RTK, так и СТК вовлечены в гипериммунные расстройства. Пример воздействия соединения 5-[5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил]-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амид по изобретению на некоторые РТК приведен в табл. 2 ниже. Ингибитор циклооксигеназы-2, или ингибитор СОХ-2, включает агенты, которые специфически ин-гибируют один класс ферментов, циклооксигеназу-2, при менее значительном ингибировании циклоок-сигеназы-1. Предпочтительно, он включает соединения, которые имеют IC50 (концентрацию, вызывающую 50% ингибирование) циклооксигеназы-2 менее 0,2 мкМ, а также имеют отношение селективности ингибирования циклооксигеназы-2 к ингибированию циклооксигеназы-1 по меньшей мере 50, а более предпочтительно по меньшей мере 100. Еще более предпочтительно эти соединения имеют IC50 циклоок-сигеназы-1 более примерно 1 мкМ, а более предпочтительно более 10 мкМ. Исследования показывают, что простагландины, синтезируемые циклооксигеназами, играют критическую роль в инициировании и активизации рака. Кроме того, СОХ-2 сверхэкспрессируется в неопластических поражениях толстой кишки, молочной железы, легкого, простаты, пищевода, поджелудочной железы, кишечника, шейки, яичников, мочевого пузыря и головы и шеи. В некоторых in vitro и животных моделях ингибиторы СОХ-2 ингибировали рост опухолей и метастазирование. Кроме рака как такового, СОХ-2 также экспрессируется в ангиогенной сосудистой сети в пределах и по соседству с гиперпластическими и неопластическими поражениями, что указывает на то, что СОХ-2 участвует в ангиогенезе. И у мыши, и у крысы ингибиторы СОХ-2 значительно ингибировали bFGF-индуцированную неоваскулярязацию (bFGF - основной фактор роста фибробластов). Полезность ингибиторов СОХ-2 в качестве химиопревентивных, антиангиогенных и химиотерапевтических агентов также описана в литературе (Koki et al., Potential utility of COX-2 inhibitors in chemoprevention and chemotherapy. Exp. Opin. Invest. Grugs (1999) 8(10) pp. 1623-1638, включен в данное описание изобретения ссылкой). Амплификация и/или сверхэкспресиия HER-2/neu (ErbB2) происходит в 20-30% случаев рака молочной железы и яичника человека, а также в 5-15% случаев рака желудка и пищевода и ассоциируется с плохим прогнозом. Кроме того, недавно было обнаружено in vitro, что экспрессия СОХ-2 позитивно регулируется в клетках, сверхэкспрессирующих HER-2/neu онкоген (Subbaramaiah et al., Increased expression of cyclooxygenase-2 in HER-2/neu-overexpressing breast cancer. Cancer Research (представлено в 1999), включено в данное описание изобретения ссылкой). В этом исследовании существенно повышенные уровни продукции PGE2 (простагландина Е2), СОХ-2 белка и тРНК определяли в HER-2/neu трансформированных эпителиальных клетках млекопитающих по сравнению с нетрансформированной партнерской клеточной линией. Продукты активности СОХ-2, т.е. простагландины, стимулируют пролиферацию, увеличивают инвазивность злокачественных клеток и усиливают продукцию васкулярного эндоте-лиального ростового фактора, который стимулирует ангиогенез. Кроме того, HER-2/neu индуцирует продукцию ангиогенных факторов, таких как васкулярный эндотелиальный ростовой фактор. Следовательно, введение ингибитора СОХ-2 в комбинации с антиHER-2/neu антителами, такими как трастузумаб (Herceptin(r)) и другими терапевтическими средствами, направленными на ингибирова-ние HER-2/neu, предполагается для лечения рака, в котором сверхэкспресируется HER-2/neu. Предполагается также, что уровни СОХ-2 повышаются в опухолях с амплификацией и/или сверхэкспрессией других онкогенов, включая, но не ограничиваясь ими, c-myc, N-myc, L-myc, K-ras, H-ras, N-ras. Продукты активности СОХ-2 стимулируют клеточную пролиферацию, ингибируют иммунный контроль, увеличивают инвазивность злокачественных клеток и стимулируют ангиогенез. Следовательно, введение ингибитора протеинкиназы в комбинации с ингибитором СОХ-2 по настоящему изобретению полезно для предупреждения или лечения рака, в котором сверхэкспрессируются онкогены. Специфические ингибиторы СОХ-2 полезны для лечения рака (WO 98/16227) и в некоторых животных моделях уменьшают ангиогенез, управляемый различными факторами роста (WO 98/22101). Анти-ангиогенез был достигнут с использованием ингибитора СОХ-2 у крыс с имплантированным bFGF, вас-кулярного эндотелиального фактора роста (VEGF) или каррагенана, белков с общеизвестными ангиоген - 11 - 008137 ными свойствами (Masferrer et al., 89th Annual Meeting of the American Association for the Cancer Research, March 1998). Фармацевтическая композиция и введение Соединение по настоящему изобретению или его фармацевтически приемлемую соль можно вводить пациенту-человеку само по себе или в фармацевтической композиции, в которой вышеупомянутые вещества смешаны с подходящими носителями или эксципиентом(ами). Методики приготовления препаратов и введения лекарственных средств можно найти в "Remington's Pharmacological Sciences", Mack Publishing Co., Easton, PA., последнее издание. Используемые здесь термины "вводят" или "введение" относятся к доставке соединения по изобретению или его фармацевтически приемлемой соли, или фармацевтически композиции, содержащей соединение по изобретению или его фармацевтически приемлемую соль, в организм с целью предупреждения или лечения РК-связанного расстройства. Подходящие пути введения могут включать, без ограничения, пероральное, ректальное, местное, трансмукозальное или кишечное введение или внутримышечные, подкожные, внутримозговые, внутри-трахеальные, прямые внутрижелудочковые, внутривенные, в стекловидное тело, внутрибрюшинные, ин-траназальные или внутриглазные инъекции. Предпочтительными путями введения являются перораль-ный и парентеральный. Альтернативно, соединение можно вводить местным, а не системным путем, например инъекцией соединения непосредственно в солидную опухоль, часто в виде депо или препарата с замедленным высвобождением. Кроме того, лекарственное средство можно вводить в нацеленной на мишень системе доставки лекарственного средства, например в липосомах, покрытых специфичным к опухоли антителом. Липосомы будут нацелены на опухоль и селективно захвачены ею. Фармацевтические композиции по настоящему изобретению могут быть изготовлены способами, общеизвестными в данной области техники, например традиционными способами смешивания, растворения, гранулирования, изготовления драже, растирания в порошок, эмульгирования, инкапсулирования, включения или лиофилизации. Фармацевтические композиции для применения в соответствии с настоящим изобретением можно приготовить традиционным способом, используя один или более чем один физиологически приемлемый носитель, включая эксципиенты и вспомогательные вещества, которые облегчают переработку активных соединений в препараты, которые можно применять фармацевтически. Подходящий препарат зависит от выбранного пути введения. Для инъецирования соединения по изобретению могут быть приготовлены в водных растворах, предпочтительно в физиологически совместимых буферах, таких как раствор Хэнкса, раствор Рингера или физиологический солевой буфер. Для трансмукозального введения в препарате используют подходящие пенетранты - вещества, улучшающие проникание через барьер. Такие пенетранты известны в данной области техники. Для перорального введения препарат соединения может быть приготовлен путем объединения активных соединений с фармацевтически приемлемыми носителями, общеизвестными в данной области техники. Такие носители дают возможность изготовлять препарат соединения по данному изобретению в форме таблеток, пилюль, леденцов, драже, капсул, жидкостей, сиропов, взвесей, суспензий и тому подобного для перорального приема пациентами. Фармацевтические препараты для перорального применения можно приготовить, используя твердый эксципиент, возможно измельчая полученную смесь и обрабатывая смесь гранул после добавления других подходящих вспомогательных веществ, если это требуется, с получением таблеток или ядер драже. Пригодными эксципиентами являются, в частности, наполнители, такие как сахара, включая лактозу, сахарозу, маннит или сорбит, препараты целлюлозы, такие как, например, маисовый крахмал, пшеничный крахмал, рисовый крахмал и картофельный крахмал, и другие вещества, такие как желатин, трагакантовая камедь, метилцеллюлоза, гидроксипропилме-тилцеллюлоза, карбоксиметилцеллюлоза натрия и/или поливинилпирролидон (PVP). Если требуется, можно добавлять разрыхляющие агенты, такие как поперечно-сшитый поливинилпирролидон, агар или альгиновая кислота. Также можно добавлять соль, такую как альгинат натрия. Ядра драже снабжают подходящими оболочками. Для этой цели можно использовать концентрированные растворы сахара, которые возможно могут содержать гуммиарабик, тальк, поливинилпирроли-дон, гель карбопол, полиэтиленгликоль и/или диоксид титана, растворы лака и подходящие органические растворители или смеси растворителей. В таблетки или оболочки драже можно добавлять красители или пигменты для идентификации или чтобы отличать разные комбинации доз активных компонентов. Фармацевтические композиции, которые можно применять перорально, включают вставные капсулы, изготовленные из желатина, а также мягкие, герметичные капсулы, изготовленные из желатина и пластификатора, такого как глицерин или сорбит. Вставные капсулы могут содержать активные ингредиенты в смеси с наполнителем, таким как лактоза, связывающим веществом, таким как крахмал, и/или смазывающим веществом, таким как тальк или стеарат магния, и, возможно, стабилизаторами. В мягких капсулах активные соединения могут быть растворены или суспендированы в подходящих жидкостях, - 12 - 008137 таких как жирные масла, вазелиновое масло или жидкие полиэтиленгликоли. В эти препараты также можно добавлять стабилизаторы. Капсулы могут быть упакованы в сосуды из коричневого стекла или пластика для защиты активного соединения от света. Контейнеры, содержащие капсульный препарат активного соединения, необходимо хранить при контролируемой комнатной температуре (15-30°С). Для введения ингаляцией соединения для использования по настоящему изобретению удобно доставлять в форме аэрозольного спрея, используя упаковки под давлением или распылитель и подходящий пропеллент, например, без ограничения, дихлордифторметан, трихлорфторметан, дихлортетрафторэтан или диоксид углерода. В случае аэрозоля под давлением дозировочную единицу можно регулировать при помощи клапана для доставки отмеренного количества. Капсулы и картриджи, например желатиновые, для использования в ингаляторе или инсуффляторе могут содержать порошковую смесь соединения и подходящей порошковой основы, такой как лактоза или крахмал. Данные соединения могут быть приготовлены также в виде препарата для парентерального введения, например болюсной инъекцией или длительной инфузией. Препараты для инъекции могут быть представлены в стандартной лекарственной форме, например в ампулах или многодозовых контейнерах с добавленным консервантом. Композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных разбавителях, и могут содержать препаратообразующие вещества, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты. Фармацевтические композиции для парентерального введения включают водные растворы водорастворимой формы, такой как, без ограничения, соль активного соединения. Кроме того, могут быть приготовлены суспензии активных соединений в липофильном носителе. Подходящие липофильные носители включают жирные масла, такие как кунжутное масло, сложные эфиры синтетических жирных кислот, такие как этилолеат и триглицериды, или такие вещества, как липосомы. Водные инъекционные суспензии могут содержать вещества, которые увеличивают вязкость суспензии, такие как карбоксиме-тилцеллюлозу натрия, сорбит или декстран. Суспензия возможно может содержать также подходящие стабилизаторы и/или агенты, которые увеличивают растворимость соединений, чтобы обеспечить получение высококонцентрированных растворов. Альтернативно, активный ингредиент может быть в порошковой форме для разведения перед использованием подходящим разбавителем, например стерильной, апирогенной водой. Соединения также могут быть приготовлены в виде ректальных композиций, таких как суппозитории или удерживающие клизмы, с использованием, например, стандартных суппозиторных основ, таких как масло какао или другие глицериды. Кроме препаратов, описанных ранее, соединения могут быть приготовлены также в виде депо-препаратов. Такие длительно действующие препараты можно вводить имплантацией (например подкожной или внутримышечной) или внутримышечной инъекцией. Препарат соединения по данному изобретению можно приготовить для этого пути введения с использованием подходящих полимерных или гидрофобных веществ (например в эмульсии с фармакологически приемлемым маслом), с ионообменными смолами или в виде трудно растворимого производного, такого как, без ограничения, трудно растворимая соль. Не ограничивающим примером фармацевтического носителя для гидрофобных соединений по изобретению является система со-растворителей, содержащая бензиловый спирт, неполярное поверхностно-активное вещество, смешиваемый с водой органический полимер и водную фазу, такая как система со-растворителей VPD. VPD представляет собой раствор 3% мас./об. бензилового спирта, 8% мас./об. неполярного поверхностно-активного вещества Polysorbate 80 и 65% мас./об. полиэтиленгликоля 300, доведенный до нужного объема абсолютным этанолом. Система сорастворителей VPD (VPD:D5W) состоит из VPD, разбавленного 1:1 5% раствором декстрозы в воде. Эта система сорастворителей хорошо растворяет гидрофобные соединения, и сама имеет низкую токсичность при систематическом введении. Естественно, соотношения сорастворителей в такой системе могут существенно меняться с сохранением ее свойств растворимости и токсичности. К тому же, индивидуальные компоненты системы со-растворителей можно варьировать: например, другие низкотоксичные неполярные поверхностно-активные вещества можно использовать вместо Polysorbate 80, можно варьировать фракционный размер полиэтиленгликоля, полиэтиленгликоль можно заменить другими биосовместимыми полимерами, например поливинилпирролидоном, а декстрозу можно заменить другими сахарами или полисахаридами. Альтернативно, можно использовать другие системы доставки гидрофобных фармацевтических соединений. Липосомы и эмульсии являются общеизвестными примерами средств доставки или носителей для гидрофобных лекарственных средств. Кроме того, можно использовать некоторые органические растворители, такие как диметилсульфоксид, хотя часто ценой большей токсичности. Кроме того, соединения можно доставлять, используя систему пролонгированного высвобождения, такую как полупроницаемые матрицы из твердых гидрофобных полимеров, содержащие терапевтический агент. Были разработаны различные материалы, обеспечивающие пролонгированное высвобождение, и они общеизвестны специалистам в данной области техники. Капсулы пролонгированного высвобождения в зависимости от их химической природы могут вы - 13 - 008137 свобождать соединения в течение времени от нескольких недель до более 100 суток. В зависимости от химической природы и биологической стабильности терапевтического реагента можно использовать дополнительные методики для стабилизации белка. Фармацевтические композиции могут содержать также подходящие твердофазные и гель-фазные носители и эксципиенты. Примеры таких носителей и эксципиентов включают, но не ограничиваются ими, карбонат кальция, фосфат кальция, различные сахара, крахмалы, производные целлюлозы, желатин и полимеры, такие как полиэтиленгликоли. Многие из РК модулирующих соединений по изобретению могут быть предоставлены в виде физиологически приемлемых солей, где заявленное соединение может образовывать отрицательно или положительно заряженные частицы. Примерами солей, в которых соединение может образовывать положительно заряженную группировку, включают, без ограничения, четвертичный аммоний (определенный в другом месте в данном описании изобретения), соли, такие как гидрохлорид, сульфат, карбонат, лактат, тартрат, малат, малеат, сукцинат, где атом азота четвертичной аммониевой группы представляет собой азот выбранного соединения по данному изобретению, который прореагировал с соответствующей кислотой. Соли, в которых соединение по данному изобретению образует отрицательно заряженные частицы, включают, без ограничения, натриевые, калиевые, кальциевые и магниевые соли, образованные путем взаимодействия карбоксильной кислотной группы соединения с соответствующим основанием (например гидроксидом натрия (NaOH), гидроксидом калия (KOH), гидроксидом кальция (Са(ОН)2 и т.д.). Фармацевтические композиции, пригодные для использования в настоящем изобретении, включают композиции, в которых активные ингредиенты содержатся в количестве, достаточном для достижения намеченной цели, например модулирования активности РК или лечения или предупреждения связанного с РК расстройства. Более конкретно, терапевтически эффективное количество означает количество соединения, эффективное для предупреждения, ослабления или улучшения симптомов заболевания или продления жизни субъекта, которого лечат. Определение терапевтически эффективного количества находится в рамках компетентности специалистов в данной области техники, в частности в свете подробного описания, представленного в данной заявке. Для любого соединения, используемого в способах по данному изобретению, терапевтически эффективное количество или дозу можно оценить первоначально из анализов клеточных культур. Затем эту дозу можно приготовить в виде препарата для использования в животной модели так, чтобы достичь интервала циркулирующей в системе кровообращения концентрации, которое включает ГС50, как она определена в клеточной культуре (т.е. концентрацию тестируемого соединения, при которой достигается половина от максимального ингибирования активности РК). Эту информацию затем можно использовать для более точного определения доз, пригодных для человека. Токсичность и терапевтическую эффективность соединений, описанных в заявке, можно определить по стандартным фармацевтическим методикам в клеточных культурах или на экспериментальных животных, например путем определения Ю5о и LD50 (LD50 - доза, при которой летальность составляет 50%) (обе из которых обсуждаются в другом месте данного описания изобретения) подвергнутого испытанию соединения. Данные, полученные из этих анализов клеточных культур и исследований на животных, можно использовать для определения интервала дозировки для применения у людей. Дозировка может меняться в зависимости от используемой лекарственной формы и применяемого пути введения. Точный состав, путь введения и дозировку может выбрать лечащий врач с учетом состояния пациента (см., например, Fingl et а!, 1975 в "The Pharmacological Basis of Therapeutics", Ch. 1, p. 1). Дозировочное количество и интервал можно регулировать индивидуально для обеспечения уровня активных компонентов в плазме, который достаточен для поддержания модулирующих эффектов в отношении киназ. Этот уровень в плазме называется минимальной эффективной концентрацией (МЕС). МЕС будет различаться для каждого соединения, но ее можно оценить, основываясь на in vitro данных, например концентрация, необходимая для достижения 50-90% ингибирования киназы, может быть установлена с использованием анализов, описанных в данной заявке. Дозировки, необходимые для достижения МЕС будут зависеть от индивидуальных особенностей и пути введения. Для определения концентрации в плазме можно использовать ВЭЖХ (высокоэффективная жидкостная хроматография) анализы или биоанализы. Интервалы дозировок также можно определить, используя значения МЕС. Соединения следует вводить, используя схему, которая обеспечивает поддержание уровней в плазме выше МЕС в течение 1090% времени, предпочтительно 30-90% и наиболее предпочтительно 50-90%. На данный момент терапевтически эффективное количество 5-[5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил]-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амида может находиться в пределах от приблизительно 0,25 до 1500 мг/мл2 в сутки; предпочтительно около 3 мг/м2 в сутки. Еще более предпочтительно от 50 мг/каждое утро ежедневно до 400 мг/ежедневно. Терапевтически эффективное количество ингибитора циклооксигеназы составляет от приблизительно 0,1 до приблизительно 10000 мг для лечения указанных выше состояний, с предпочтительными - 14 - 008137 уровнями от приблизительно 1,0 до приблизительно 1000 мг. Количество активного ингредиента, которое можно объединять с другими противораковыми агентами для получения единичной лекарственной формы, будет подбираться в зависимости от подвергаемого лечению хозяина и конкретного способа введения. В случаях местного введения или селективного захвата эффективную местную концентрацию лекарственного средства можно не связывать с концентрацией в плазме, и для определения точного количества дозировки и интервала можно использовать другие способы, известные в данной области техники. Разумеется, количество вводимого соединения будет зависеть от субъекта, которого лечат, тяжести болезни, способа введения, мнения лечащего врача и т.д. Композиция может, если требуется, быть представлена в упаковке или дозирующем устройстве, например в наборе, одобренном FDA (Управлением по контролю за качеством пищевых продуктов, медикаментов и косметических средств (США)), который может содержать одну или более чем одну стандартную лекарственную форму, содержащую активный ингредиент. Например, упаковка может содержать металлическую или пластиковую фольгу, такую как блистерная упаковка. Упаковка или дозирующее устройство может сопровождаться инструкциями по введению. Упаковка или дозирующее устройство может также сопровождаться извещением, объединенным с контейнером, в форме, предписанной правительственным органом, регулирующим изготовление, применение или продажу фармацевтических средств, которое отражает одобрение этим органом формы данной композиции или ее введения человеку или животному. Такое извещение может представлять собой, например, этикетку, одобренную FDA для предписываемого лекарственного средства, или одобренный вкладыш. Также можно изготавливать композиции, содержащие соединение по изобретению, в совместимом фармацевтическом носителе, поместить их в подходящий контейнер и маркировать для лечения указанного состояния. Подходящие состояния, указанные на маркировке, могут включать лечение опухоли, ингибирование ангиогенеза, лечение фиброза, диабета и т.п. Соединение, описанное в данной заявке, или его соль или пролекарство можно объединять с другими химиотерапевтическими агентами для лечения заболеваний или расстройств, описанных выше. Например, соединение, соль или пролекарство по данному изобретению можно объединять с алкилирую-щими агентами, такими как фторурацил (5-FU), один или дополнительно в комбинации с лейковорином; или другими алкилирующими агентами, такими как, без ограничения, другие пиримидиновые аналоги, например UFT, капецитабин, гемцитабин и цитарабин, алкилсульфонаты, например бусульфан (используемый в лечении хронической гранулоцитарной лейкемии), импросульфан и пипосульфан; азиридины, например бензодепа, карбоквион, метуредепа и уредепа; этиленимины и метилмеламины, например ал-третамин, триэтиленмеламин, триэтиленфосфорамид, триэтилентиофосфорамид и триметилолмеламин; и азотистые аналоги иприта, например хлорамбуцил (применяемый в лечении хронической лимфоцитар-ной лейкемии, первичной макроглобулинемии и неходжкинской лимфомы), циклофосфамид (применяемый в лечении болезни Ходжкина, множественной миеломы, нейробластомы, рака молочной железы, рака яичника, рака легкого, опухоли Вильмса и рабдомиосаркомы), эстрамустин, ифосфамид, новембри-цин, преднимустин и урациловый аналог иприта (используемый при лечении первичного тромбоцитоза, неходжкинской лимфомы, болезни Ходжкина и рака яичников); и триазины, например дакарбазин (применяемый при лечении саркомы мягких тканей). Соединение, соль или пролекарство по данному изобретению можно применять также в комбинации с другими антиметаболическими химиотерапевтическими агентами, такими как, без ограничения, аналоги фолиевой кислоты, например метотрексат (применяемый в лечении острой лимфоцитарной лейкемии, хориокарциномы, фунгоидной гранулемы, рака молочной железы, рака в области головы и шеи и остеогенной саркомы) и птероптерин; и пуриновые аналоги, такие как меркаптопурин и тиогуанин, которые находят применение в лечении острой гранулоцитарной, острой лимфацитарной и хронической гра-нулоцитарной лейкемий. Предполагается, что соединение, соль или пролекарство по данному изобретению также можно применять в комбинации с химиотерапевтическими агентами на основе природных продуктов, такими как, без ограничения, винка-алкалоиды, например винбластин (применяемый в лечении рака молочной железы или яичка), винкристин и виндезин; эпиподофилотоксины, например этопозид и тенипозид, которые оба используют в лечении рака яичка и саркомы Капоши; антибиотические химиотерапевтические агенты, например даунорубицин, доксорубицин, эпирубицин, митомицин (применяемые в лечении рака желудка, шейки, прямой кишки, молочной железы, мочевого пузыря и поджелудочной железы), дакти-номицин, темизоломид, пликамицин, блеомицин (применяемые в лечении рака кожи, пищевода и мочеполового тракта); и энзиматические химиотерапевтические агенты, такие как L-аспарагиназа. В дополнение к перечисленному выше соединение, соль или пролекарство по данному изобретению можно применять также в комбинации с координационными комплексами платины (цисплатин и т.д.); замещенными мочевинами, такими как гидроксимочевина; производными метилгидразина, например прокарбазином; аденокортикоидными супрессивными средствами, например митотаном, аминоглутети-мидом; гормоном и антагонистами гормона, такими как адренокортикостероиды (например, преднизон), прогестины (например, гидроксипрогестерона капроат); эстрогенами (например, диэтилстилбестеролом); - 15 - 008137 антиэстрогенами, такими как тамоксифен; андрогенами, например тестостерона пропионатом; и ингибиторами ароматазы, такими как анастрозол. Другие противораковые агенты, известные в данной области техники, можно применять параллельно с комбинированной терапией по настоящему изобретению. Примеры этих и других антинеопластиче-ских агентов приведены в заявке на патент США 60/113786, поданной 23 декабря 1998 г., и в международной публикации РСТ WO 00/38716, описание которых включено в данное описание изобретения ссылкой. Наконец, предполагается также, что комбинация соединения по данному изобретению будет эффективной в комбинации с митоксантроном или паклитакселем для лечения солидноопухолевого рака или лейкемий, таких как, без ограничения, острая миелогенная (нелимфоцитарная) лейкемия. Примеры Следующие ниже подготовительные примеры и примеры приведены, чтобы дать возможность специалистам в данной области техники более ясно понять настоящее изобретение и применять его на практике. Они должны рассматриваться не как ограничивающие объем изобретения, а только как иллюстративные и типичные. Синтез ингибитора протеинкиназ по настоящему изобретению Общая методика синтеза Следующую общую методологию можно использовать для получения соединений по данному изобретению. Соответственно замещенный 2-оксиндол (1 эквивалент), соответственно замещенный альдегид (1,2 эквивалента) и основание (0,1 эквивалент) смешивают в растворителе (1-2 мл/ммоль 2-оксиндола), и эту смесь затем нагревают в течение от 2 до приблизительно 12 ч. После охлаждения отфильтровывают образовавшийся осадок, промывают его холодным этанолом или эфиром и сушат вакуумом с получением твердого продукта. Если осадок не образуется, реакционную смесь концентрируют, и остаток растирают со смесью дихлорметан/эфир, полученное в результате твердое вещество собирают фильтрацией и затем сушат. Продукт возможно очищают хроматографией. Основание может представлять собой органическое или неорганическое основание. Если используют органическое основание, оно предпочтительно представляет собой азотистое основание. Примеры органических азотистых оснований включают, но не ограничиваются ими, диизопропиламин, тримети-ламин, триэтиламин, анилин, пиридин, 1,8-диазабицикло[5.4.1]ундец-7-ен, пирролидин и пиперидин. Примерами неорганических оснований являются, без ограничения, гидроксиды аммония, щелочных металлов или щелочно-земельных металлов, фосфаты, карбонаты, бикарбонаты, бисульфаты и амиды. Щелочные металлы включают литий, натрий и калий, а щелочно-земельные земли включают кальций, магний и барий. В предпочтительном на данный момент воплощении данного изобретения, если растворитель является протонным растворителем, таким как вода или спирт, то основание представляет собой неорганическое основание щелочного металла или щелочно-земельного металла, предпочтительно гидроксид щелочного или щелочно-земельного металла. Специалистам в данной области техники должно быть ясно, на основе известных общих принципов органического синтеза и на основе описаний данного изобретения, какое основание будет наиболее подходящим для предполагаемого взаимодействия. Растворитель, в котором проводят взаимодействие, может быть протонным или апротонным, предпочтительно он является протонным растворителем. "Протонный растворитель" - это растворитель, который имеет атом(ы) водорода, ковалентно связанный(е) с атомами кислорода или азота, что делает атомы водорода существенно кислотными и, соответственно, способными быть "общими" с растворенным веществом благодаря образованию водородных связей. Примеры протонных растворителей включают, без ограничения, воду и спирты. " Апротонный растворитель" может быть полярным или неполярным, но в любом случае он не содержит кислотных атомов водорода и, следовательно, не способен образовывать водородные связи с растворенными веществами. Примерами неполярных апротонных растворителей являются, без ограничения, пентан, гексан, бензол, толуол, метиленхлорид и четыреххлористый углерод. Примерами полярных ап-ротонных растворителей являются хлороформ, тетрагидрофуран, диметилсульфоксид и диметилформа-мид. В предпочтительном на данный момент воплощении данного изобретения растворитель представляет собой протонный растворитель, предпочтительно воду или спирт, такой как этанол. Взаимодействие осуществляют при температуре выше комнатной температуры. Температура в общем случае составляет от примерно 30 до примерно 150°С, предпочтительно от примерно 80 до примерно 100°С, наиболее предпочтительно от примерно 75 до примерно 85°С, что является примерно точкой кипения этанола. Под "примерно" подразумевается, что интервал температур предпочтительно находится в пределах 10°C от указанной температуры, более предпочтительно в интервале 5°C от указанной температуры и наиболее предпочтительно в пределах 2°C от указанной температуры. Так, например, под "примерно 75°С" подразумевается 75±10°С, предпочтительно 75±5°С и наиболее предпочтительно - 16 - 008137 75±2°С. 2-Оксиндолы и альдегиды легко можно синтезировать, используя методики, общеизвестные в химической области. Специалистам в данной области техники должно быть ясно, что для образования соединений по данному изобретению доступны другие пути синтеза, и что нижеследующее предложено в качестве примера, а не ограничения. Способ А: Форматирование пирролов POCl3 (1,1 эквивалент) добавляют по каплям к диметилформамиду (3 эквивалента) при -10°С, затем добавляют соответствующий пиррол, растворенный в диметилформамиде. После перемешивания в течение двух часов реакционную смесь разбавляют H2O и подщелачивают до рН 11 10 н KOH. Образовавшийся осадок собирают фильтрацией, промывают H2O и сушат в вакуум-сушильном шкафу с получением целевого альдегида. Способ Б: Омыление эфиров пирролкарбоновой кислоты Смесь эфира пирролкарбоновой кислоты и KOH (2-4 эквивалента) в EtOH кипятят с обратным холодильником до тех пор, пока тонкослойная хроматография (ТХС) не укажет на окончание реакции. Охлажденную реакционную смесь подкисляют 1 н. HCl до рН 3. Образовавшийся осадок собирают фильтрацией, промывают H2O и сушат в вакуум-сушильном шкафу с получением целевой пирролкарбоновой кислоты. Способ В: Амидирование К перемешиваемому раствору пирролкарбоновой кислоты, растворенной в диметилформамиде (0,3 М), добавляют 1-этил-3-(3-диметиламинопропил)карбодиимид (1,2 эквивалента), 1-гидроксибензотриазол (1,2 эквивалента) и триэтиламин (2 эквивалента). Добавляют соответствующий амин (1 эквивалент), и эту реакционную смесь перемешивают до тех пор, пока ТХС не укажет на окончание реакции. Затем к реакционной смеси добавляют этилацетат и раствор промывают насыщенным NaHCO3 и рассолом (с солью высшего качества), сушат над безводным MgSO4 и концентрируют с получением целевого амида. Способ Г: Конденсация альдегидов и оксиндолов, содержащих карбоновокислотные заместители Смесь оксиндола (1 эквивалент), 1 эквивалента альдегида и 1-3 эквивалентов пиперидина (или пир-ролидина) в этаноле (0,4 М) перемешивают при 90-100°С до тех пор, пока ТХС не укажет на окончание реакции. Смесь затем концентрируют, и остаток подкисляют 2 н. HCl. Образовавшийся осадок промывают H2O и EtOH и затем сушат в вакуум-сушильном шкафу с получением продукта. Способ Д: Конденсация альдегидов и оксиндолов, не содержащих карбоновокислотных заместителей Смесь оксиндола (1 эквивалент), 1 эквивалента альдегида и 1-3 эквивалента пиперидина (или пир-ролидина) в этаноле (0,4 М) перемешивают при 90-100°С до тех пор, пока ТХС не укажет на окончание реакции. Смесь охлаждают до комнатной температуры, и образовавшееся твердое вещество собирают вакуумной фильтрацией, промывают этанолом и сушат с получением продукта. Если при охлаждении реакционной смеси осадок не образуется, эту смесь концентрируют и очищают колоночной хроматографией. Синтез 2-оксииндолов 5-фтор-2-оксиндол 5-Фторизатин (8,2 г) растворяли в 50 мл гидразина гидрата и кипятили с обратным холодильником в течение 1 ч. Реакционную смесь затем вливали в воду. Осадок затем отфильтровывали, промывали водой и сушили в вакуумной сушилке с получением указанного в заголовке соединения. Синтез пиррол-замещенных 2-индолинонов Пример 80. 5-(5-Фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1H-пиррол-3-карбоновой кислоты (2-диэтиламино-этил)амид 5-Фтор-1,3-дигидроиндол-2-он (0,54 г, 3,8 ммоль) конденсировали с 5-формил-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амидом с получением 0,83 мг (55%) указанного в заголовке соединения в виде желто-зеленого твердого вещества. 1Н ЯМР (300 МГц, ДМСО-d; ) 8 13,66 (s, 1H, NH), 10,83 (s, br, 1H, NH), 7,73 (dd, J=2,5 и 9,4 Гц, 1H), 7,69 (s, 1H, Н-винил), 7,37 (t, 1H, СОNHСH2СH2), 6,91 (m, 1H), 6,81-6,85 (m, 1H), 3,27 (m, 2H, СН2), 2,51 (m, 6H, 3хСН2), 2,43 (s, 3H, СН3), 2,41 (s, 3H, СН3), 0,96 (t, J=6,9 Гц, 6H, N(CH2 CH3)2). МС-ЭУ m/z 398 [М+]. Пример 80 (Альтернативный синтез) 5-[5-Фтор-2-оксо-1,2-дигидро-индол-(3Z)-илиденметил]-2,4-метил-1H-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амид Гидразина гидрат (55%, 3000 мл) и 5-фторизатин (300 г) нагревали до 100°С. Порциями (100 г) добавляли дополнительное количество 5-фторизатина (500 г) за 120 мин при перемешивании. Смесь нагревали до 110°С и перемешивали в течение 4 ч. Смесь охлаждали до комнатной температуры, и твердые вещества собирали вакуумной фильтрацией с получением сырого (2-амино-5-фторфенил)уксусной ки - 17 - 008137 слоты гидразида (748 г). Гидразид суспендировали в воде (700 мл), и рН смеси доводили 12 н соляной кислотой до значения рН менее 3. Смесь перемешивали в течение 12 ч при комнатной температуре. Твердые вещества собирали вакуумной фильтрацией и дважды промывали водой. Продукт сушили под вакуумом с получением 5-фтор-1,3-дигидроиндол-2-она (600 г, выход 73%) в виде коричневого порошка. 1Н ЯМР (диметилсульфоксид-d6) 8 3,46 (s, 2Н, СН2), 6,75, 6,95, 7,05 (3 х m, 3Н, ароматический), 10,35 (s, 1H, NH). MC m/z 152 [М+1]. 3,5-Диметил-1 Н-пиррол-2,4-дикарбоновой кислоты 2-трет-бутиловый эфир 4-этиловый эфир (2600 г) и этанол (7800 мл) интенсивно перемешивали, одновременно медленно добавляя 10 н соляную кислоту (3650 мл). Температура повышалась с 25 до 35°С, и начиналось газовыделение. Смесь нагревали до 54°С и перемешивали при дальнейшем нагревании в течение одного часа, при этом температура была 67°С. Смесь охлаждали до 5°С и при перемешивании медленно добавляли 32 л льда и воды. Твердое вещество собирали вакуумной фильтрацией и промывали три раза водой. Твердое вещество сушили воздухом до постоянной массы с получением 2,4-диметил-Ш-пиррол-3-карбоновой кислоты этилового эфира (1418 г, выход 87%) в виде розоватого твердого вещества. 1Н ЯМР (диметилсульфоксид^6) 8 2,10, 2,35 (2xs, 2х3Н, 2хСН3), 4,13 (q, 2H, СН2), 6,37 (s, 1H, СН), 10,85 (s, 1H, NH). MC m/z 167 [М+1]. Диметилформамид (322 г) и дихлорметан (3700 мл) охлаждали в ледяной бане до 4°С и при перемешивании добавляли оксихлорид фосфора (684 г). Медленно добавляли твердый 2,4-диметил-Ш-пиррол-3-карбоновой кислоты этиловый эфир (670 г) аликвотами за 15 мин. Максимальная достигнутая температура была 18°С. Смесь нагревали до образования флегмы в течение 1 ч, охлаждали до 10°С в ледяной бане, и быстро добавляли 1,6 л ледяной воды при интенсивном перемешивании. Температура возрастала до 15°С. При интенсивном перемешивании добавляли 10 н. соляную кислоту (1,6 л). Температура поднималась до 22°С. Смесь оставляли стоять в течение 30 мин и давали возможность слоям разделиться. Температура достигала максимума 40°С. Водный слой доводили 10 н. гидроксидом калия (3,8 л) до рН 12-13 со скоростью, которая позволяла температуре во время добавления достичь и оставаться при 55°С. После окончания добавления смесь охлаждали до 10°С и перемешивали в течение 1 ч. Твердое вещество собирали вакуумной фильтрацией и промывали четыре раза с получением 5-формил-2,4-диметил-Ш-пиррол-3-карбоновой кислоты этилового эфира (778 г, выход 100%) в виде желтого твердого вещества. 1Н ЯМР (ДМСОч16) 8 1,25 (t, 3H, СН3), 2,44, 2,48 (2xs, 2х3^ 2хСН3), 4,16 (q, 2Н, СН2), 9,59 (s, 1H, СНО), 12,5 (br s, 1H, NH). MC m/z 195 [M+1]. 5-Формил-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты этиловый эфир (806 г), гидроксид калия (548 г), воду (2400 мл) и метанол (300 мл) кипятили с обратным холодильником в течение 2 ч при перемешивании, а затем охлаждали до 8°С. Смесь экстрагировали дважды дихлорметаном. Водный слой доводили 1000 мл 10 н. соляной кислоты до рН 4, поддерживая температуру ниже 15°С. Для облегчения перемешивания добавляли воду. Твердое вещество собирали вакуумной фильтрацией, промывали три раза водой и сушили под вакуумом при 50°С с получением 5-формил-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (645 г, выход 93,5%) в виде желтого твердого вещества. ЯМР (ДМСО-d6) 8 2,40, 2,43 (2xs, 2х3H, 2хСН3), 9,57 (s, 1H, СНО), 12,07 (br s, 2H, NH+COOH). MC m/z 168 [М+1]. 5-Формил-2,4-диметил-Ш-пиррол-3-карбоновую кислоту (1204 г) и 6020 мл диметилформамида перемешивали при комнатной температуре, одновременно добавляя 1-(3-диметиламинопропил-3-этилкарбодиимида гидрохлорид (2071 г), гидроксибензотриазол (1460 г), триэтиламин (2016 мл) и диэти-лэтилендиамин (1215 мл). Смесь перемешивали в течение 20 ч при комнатной температуре. Смесь разбавляли 3000 мл воды, 2000 мл рассола и 3000 мл насыщенного раствора бикарбоната натрия и рН доводили до значения выше 10 с использованием 10 н. гидроксида натрия. Смесь экстрагировали дважды, каждый раз 5000 мл 10% метанола в дихлорметане, и экстракты объединяли, сушили над безводным сульфатом магния и досуха упаривали на роторном испарителе. Смесь затем разбавляли 1950 мл толуола и снова досуха упаривали на роторном испарителе. Остаток растирали со смесью 3:1 гексан:диэтиловый эфир (4000 мл). Твердые вещества собирали вакуумной фильтрацией, дважды промывали 400 мл этил-ацетата и сушили под вакуумом при 34°С в течение 21 ч с получением 5-формил-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амида (819 г, выход 43%) в виде светло-коричневого твердого вещества. 1Н ЯМР (диметилсульфоксид^6) 8 0,96 (t, 6Н, 2хСН3), 2,31, 2,38 (2xs, 2 х СН3), 2,51 (т, 6Н, 3хСН2), 3,28 (т, 2Н, СН2), 7,34 (m, 1H, амидный NH), 9,56 (s, 1H, СНО), 11,86 (s, 1H, пиррольный NH). MC m/z 266 [M+1]. 5-Формил-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амид (809 г), 5-фтор-1,3-дигидроиндол-2-он (438 г), этанол (8000 мл) и пирролидин (13 мл) нагревали при 78°С в течение 3 ч. Смесь охлаждали до комнатной температуры и твердые вещества собирали вакуумной фильтрацией и промывали этанолом. Твердые вещества перемешивали с этанолом (5900 мл) при 72°С в течение 30 мин. Смесь охлаждали до комнатной температуры. Твердые вещества собирали вакуумной фильтрацией, промывали этанолом и сушили под вакуумом при 54°С в течение 130 ч с получением 5-[5-фтор-2-оксо-1,2-дигидроиндол-(31)-илиденметил]-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил) - 18 - 008137 амида (1013 г, выход 88%) в виде оранжевого твердого вещества. 1Н ЯМР (диметилсульфоксид^6) 8 0,98 (t, 6H, 2хСН3), 2,43, 2,44 (2xs, 6H, 2хСН3), 2,50 (m, 6Н, 3хСН2), 3,28 (q, 2H, СН2), 6,84, 6,92, 7,42, 7,71, 7,50 (5xm, 5H, ароматический, винил, CONH), 10,88 (s, 1H, CONH), 13,68 (s, 1H, пиррольный NH). MC m/z 397 [M-1]. Биологические примеры Ингибиторы протеинкиназы: Следующие анализы можно использовать для определения уровня активности и воздействия ингибиторов протеинкиназ на одну или более чем одну протеинкиназу (ниже РК). Аналогичные анализы могут быть разработаны по тем же направлениям для любой РК с использованием методик, общеизвестных в данной области техники. А. Методики анализа Некоторые описанные здесь анализы осуществляют в формате ELISA (твердофазный иммунофер-ментный анализ) (Voller et al., 1980, "Enzyme-Linked Immunosorbent Assay," Manual of Clinical Immunology, 2d ed., Rose and Friedman, Am. Soc. of Microbiology, Washington D. C., pp. 359-371). Общая методика представляет собой следующее: соединение вводят в клетки, экспрессирующие тестируемую киназу, либо естественным образом, либо рекомбинантно, в течение выбранного периода времени, после которого, если тестируемая киназа является рецептором, добавляют лиганд, который, как известно, активирует этот рецептор. Клетки лизируют и лизат переносят в лунки ELISA планшета, предварительно покрытого специфическим антителом, распознающим субстрат реакции ферментного фосфорилирования. Несубстратные компоненты клеточного лизата вымывают, и количество фосфорилирования на субстрате определяют с использованием антитела, специфически распознающего фосфотирозин, по сравнению с контрольными клетками, которые не контактировали с тестируемым соединением. Предпочтительные на данный момент протоколы проведения ELISA экспериментов в отношении конкретных РК предложены ниже. Однако адаптация этих протоколов для определения активности соединений в отношении других рецепторных тирозинкиназ (RTK), а также цитоплазматических тирозин-киназ (СТК) и серин-треониновых тирозинкиназ (STK) находится полностью в пределах компетенции специалистов в данной области техники. Другие описанные здесь анализы позволяют измерять количество ДНК, продуцированной в ответ на активацию тестируемой киназы, которое является главным критерием пролиферативного ответа. Общая методика этого анализа представляет собой следующее: соединение вводят в клетки, экспрессирующие тестируемую киназу, либо естественным образом, либо реком-бинантно, в течение выбранного периода времени, после которого, если исследуемая киназа является рецептором, добавляют лиганд, который, как известно, активирует этот рецептор. После инкубации в течение, по меньшей мере, ночи добавляют реагент, вводящий метку в ДНК, такой как 5-бромдезоксиуридин (BrdU) или Н3-тимидин. Количество меченой ДНК определяют либо при помощи анти-BrdU антитела, либо путем измерения радиоактивности, и его сравнивают с контрольными клетками, не контактировавшими с тестируемым соединением. Биоанализ GST-FLK-1 В этом анализе анализируют тирозинкиназное действие GST-FIM (GST -глутатион^-трансфераза) на поли^ш^^пептиды. Материалы и реагенты: 1. 96-луночные планшеты Corning (Corning Catalog № 5805-96). 2. Поли^ш, tyr) 4:1, лиофилизат (Sigma Catalog № Р0275). 3. Подготовка покрытых поли(glu,tyr)(pEY) аналитических планшетов: покрывают 2 мкг/лунку по-ли(glu,tyr)(pEY) в 100 мкл PBS (фосфатный буферный раствор), выдерживают при комнатной температуре в течение 2 ч или при 4°С в течение ночи. Хорошо закрывают планшеты, чтобы предотвратить испарение. 4. PBS буфер: на 1 л смешивают 0,2 г 1,15 г Na2HPO4, 0,2 г KCl и 8 г NaCl в приблизительно 900 мл dH2O. Когда все реагенты растворятся, доводят рН до 7,2 с использованием HCl. Доводят общий объем до 1 л dH2O. 5. PBST буфер: к 1 л PBS буфера добавляют 1,0 мл TWEEN-20. 6. ТВВ-блокирующий буфер: на 1 л смешивают 1,21 г TRIS (трис(гидроксиметил)аминометана), 8,77 г NaCl, 1 мл TWEEN-20 в приблизительно 900 мл dH2 O. Доводят рН до 7,2 с использованием HCl. Добавляют 10 г BSA (бычьего сывороточного альбумина), перемешивают до растворения. Доводят общий объем до 1 л dH2O. Фильтруют для удаления частиц вещества. 7. 1% BSA в PBS: Для приготовления 1х рабочего раствора добавляют 10 г BSA к приблизительно 990 мл PBS буфера, перемешивают до растворения. Доводят общий объем до 1 л PBS буфером, фильтруют для удаления частиц вещества. 8. 50 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота) рН 7,5. 9. GST-FLK1 cd, очищенный из sf9 рекомбинантной бакуловирусной трансформации (SUGEN, Inc.). 10. 4% ДМСО в dH2O. 11. 10 мМ АТР (аденозин 5'-трифосфат) в dH2O. 12. 40 мМ MnCl2 - 19 - 008137 13. Буфер для разведения киназы (KDB): смешивают 10 мл HEPES (рН 7,5), 1 мл 5М NaCl, 40 мкл 100 мМ ортованадата натрия и 0,4 мл 5% BSA в dH2O с 88,56 мл dH2O. 14. 96-луночные полипропиленовые планшеты NUNC с V-образным дном, Applied Scientific Catalog № AS-72092 15. EDTA: смешивают 14,12 г этилендиаминтетрауксусной кислоты (EDTA) с приблизительно 70 мл dH2O. Добавляют 10 н. NaOH, пока EDTA не растворится. Доводят рН до 8,0. Доводят общий объем до 100 мл dH2O. 16. 1° буфер для разведения антител: смешивают 10 мл 5% BSA в PBS буфере с 89,5 мл TBST. 17. Антифосфотирозиновое моноклональное антитело, конъюгированное с пероксидазой хрена (PY99 HRP, Santa Cruz Biotech). 18. 2,2'-Азинобис(3-этилбензтиазолин-6-сульфоновая кислота (ABTS, Moss, Cat. No. ABST). 19. 10% SDS (додецилсульфат натрия). Методика: 1. 96-луночные ELISA планшеты Corning покрывают 2 мкг полй^ пептидом в стерильном PBS, как описано в п.3 в разделе "Материалы и реагенты". 2. Удаляют несвязанную жидкость из лунок переворачиванием планшета. Промывают один раз TBST. Похлопывают планшет по бумажной салфетке для удаления избытка жидкости. 3. Добавляют по 100 мкл 1% BSA в PBS в каждую лунку. Инкубируют, при встряхивании, в течение 1 ч при комнатной температуре. 4. Повторяют стадию 2. 5. Пропитывают лунки 50 мМ HEPES (рН 7,5) (150 мкл/лунку). 6. Разводят тестируемое соединение смесью dH2O/4% ДМСО до 4-кратной желаемой конечной аналитической концентрации в 96-луночных полипропиленовых планшетах. 7. Добавляют 25 мкл разведенного тестируемого соединения в ELISA планшет. В контрольные лунки помещают по 25 мкл dH2O/4% ДМСО. 8. Добавляют по 25 мкл 40 мМ MnCl2 с 4х АТР (2мкМ) в каждую лунку. 9. Добавляют по 25 мкл 0,5М EDTA в лунки с отрицательным контролем. 10. Разбавляют GST-FLK1 до 0,005 мкг (5 нг)/лунку KDB. 11. Добавляют по 50 мкл разбавленного фермента в каждую лунку. 12. Инкубируют при встряхивании в течение 15 мин при комнатной температуре. 13. Останавливают реакцию добавлением 50 мкл 250 мМ EDTA (рН 8,0). 14. Промывают трижды TBST и похлопывают планшет по бумажной салфетке для удаления избытка жидкости. 15. Добавляют по 100 мкл на лунку антифосфотирозинового HRP конъюгата (конъюгата с перокси-дазой хрена), разведение 1:5000 в буфере для разведения антител. Инкубируют при встряхивании в течение 90 мин при комнатной температуре. 16. Промывают, как на стадии 14. 17. Добавляют по 100 мкл ABTS раствора комнатной температуры в каждую лунку. 18. Инкубируют при встряхивании в течение 10-15 мин. Удаляют все пузырьки. 19. Останавливают реакцию добавлением 20 мкл 10% SDS в каждую лунку. 20. Считывают результаты на Dynatech MR7000 ELISA считывающем устройстве с тест-фильтром при 410 нм и фильтром сравнения при 630 нм. Биоанализ PYK2 Этот анализ используют для определения in vitro киназной активности НА эпитоп-меченного полноразмерного pyk2 (FL.pyk2-HA) в ELISA анализе. Материалы и реагенты: 1. 96-луночные ELISA планшеты от Corning. 2. 12СА5 моноклональное анти-НА антитело (SUGEN, Inc.) 3. PBS (фосфатный буферный раствор по Дульбекко (Gibco Catalog № 450-1300ЕВ) 4. TBST буфер: на 1 л смешивают 8,766 г NaCl, 6,057 г TRIS и 1 мл 0,1% Triton Х-100 в приблизительно 900 мл dH2O. Доводят рН до 7,2, доводят объем до 1 л. 5. Блокирующий буфер: на 1 л смешивают 100 г 10% BSA, 12,1 г 100 мМ TRIS, 58,44 г 1М NaCl и 10 мл 1% TWEEN-20. 6. FL.pyk2-HA из sf9 клеточных лизатов (SUGEN, Inc.). 7. 4% ДМСО в MilliQue H2O. 8. 10 мM ATP в dH2O. 9. 1M MnCl2 10. 1M MgCl2. 11. 1М дитиотрейтол (DTT). 12. 10х киназный буфер для фосфорилирования: смешивают 5,0 мл HEPES (рН 7,5), 0,2 мл 1М MnCl2, 1,0 мл 1 М MgCl2, 1,0 мл 10% Triton Х-100 в 2,8 мл dH2O. Непосредственно перед использованием добавляют 0,1 мл 1 М DTT. - 20 - 008137 13. 96-луночные полипропиленовые планшеты NUNC с V-образным дном. 14. 500 мМ EDTA в dH2O. 15. Буфер для разведения антител: на 100 мл 1 мл 5% смеси BSA/PBS и 1 мл 10% TWEEN-20 в 88 мл TBS. 16. HRP-конъюгированный анти-Ptyr PY99, Santa Cruz Biotech Cat. № SC-7020. 17. ABTS, Moss, каталожный № ABST-2000. 18. 10% SDS. Методика 1. 96-луночные ELISA планшеты Corning покрывают 0,5 мкг на лунку 12СА5 анти-НА антителом в 100 мкл PBS. Хранят в течение ночи при 4°С. 2. Удаляют несвязанное НА антитело из ячеек путем переворачивания планшета. Промывают планшет один раз dH2O. Похлопывают планшет по бумажной салфетке для удаления избытка жидкости. 3. Добавляют по 150 мкл блокирующего буфера в каждую лунку. Инкубируют при встряхивании в течение 30 мин при комнатной температуре. 4. Промывают планшет 4 раза TBS-T. 5. Разводят лизат в PBS (1,5 мкг лизата/100 мкл PBS). 6. Добавляют по 100 мкл разведенного лизата в каждую лунку. Встряхивают при комнатной температуре в течение 1 ч. 7. Промывают как на стадии 4. 8. Добавляют 50 мкл 2х киназного буфера в ELISA планшет, содержащий захваченный pyk2-HА. 9. Добавляют по 25 мкл 400 мкМ тестируемого соединения в 4% ДМСО в каждую лунку. Для контрольных лунок используют только 4% ДМСО. 10. Добавляют 25 мкл 0,5М EDTA в лунки с отрицательным контролем. 11. Добавляют 25 мкл 20 мкМ АТР во все лунки. Инкубируют при встряхивании в течение 10 мин. 12. Останавливают реакцию добавлением 25 мкл 500 мМ EDTA (рН 8,0) во все лунки. 13. Промывают как на стадии 4. 14. Добавляют в каждую лунку по 100 мкл HRP конъюгированного анти-Ptyr, разведенного 1:6000 в буфере для разведения антител. Инкубируют при встряхивании в течение 1 ч при комнатной температуре. 15. Промывают планшет 3 раза TBST и 1 раз PBS. 16. Добавляют по 100 мкл ABTS раствора в каждую лунку. 17. Если требуется, останавливают развитие реакции добавлением 20 мкл 10% SDS в каждую лунку. 18. Считывают планшет на ELISA считывающем устройстве с тест-фильтром при 410 нм и фильтром сравнения при 630 нм. Биоанализ FGFR1 Этот анализ используют для измерения in vitro киназной активности FGF1-R в ELISA анализе. Материалы и реагенты: 1. 96-луночные ELISA планшеты Costar (Corning Catalog № 3369). 2. ПОЛИ^Ш^Г) (Sigma Catalog № P0275). 3. PBS (Каталог Gibco, № 450-1300EB). 4. 50 мМ HEPES буферный раствор. 5. Блокирующий буфер (5% BSA/PBS). 6. Очищенный GST-FGFR1 (SUGEN, Inc.). 7. Буфер для разведения киназ. Смешивают 500 мкл 1М HEPES (GIBCO), 20 мкл 5% BSA/PBS, 10 мкл 100 мМ ортованадата натрия и 50 мкл 5М NaCl. 8. 10 мМ АТР. 9. Смесь для фосфорилирования ATP/MnCl2: смешивают 20 мкл АТР, 400 мкл 1 М MnCl2 и 9,56 мл dH2O. 10. 96-луночные полипропиленовые планшеты NUNC с V-образным дном (Applied Scientific Catalog № AS-72092). 11. 0,5M EDTA. 12. 0,05% TBST. Добавляют 500 мкл TWEEN к 1 л TBS. 13. Кроличья поликлональная антифосфотирозиновая сыворотка (SUGEN, Inc.). 14. Конъюгат козьих антител к IgG кролика с пероксидазой (Biosource, Catalog № ALI0404). 15. ABTS раствор. 16. ABST/H2O2 раствор. Методика 1. 96-луночные ELISA планшеты Costar покрывают 1 мкг на лунку ПОЛИ^Ш^Г) в 100 мкл PBS. Хранят в течение ночи при 4°С. 2. Промывают покрытые планшеты один раз PBS. - 21 - 008137 3. Добавляют по 150 мкл 5% BSA/PBS блокирующего буфера в каждую лунку. Инкубируют при встряхивании в течение 1 ч при комнатной температуре. 4. Промывают планшет 2 раза PBS, затем один раз 50 мМ HEPES. Похлопывают планшеты по бумажной салфетке для удаления избытка жидкости и пузырьков. 5. Добавляют 25 мкл 0,4 мМ тестируемого соединения в 4% ДМСО или 4% ДМСО один (контроль) в планшет. 6. Разбавляют очищенный GST-FGFR1 в буфере для разведения киназ (5 нг киназы/50 мкл KDB/лунку). 7. Добавляют по 50 мкл разведенной киназы в каждую лунку. 8. Начинают киназную реакцию добавлением 25 мкл/лунку свежеприготовленного АТР/Mn++ (0,4 мл 1 М MnCl2, 40 мкл 10 мМ АТР, 9,56 мл dH2O), свежеприготовленный). 9. Это быстрая киназная реакция, и ее следует остановить 25 мкл 0,5М EDTA способом, аналогичным добавлению АТР. 10. Промывают планшет 4 раза свежим TBST. 11. Готовят буфер для разведения антител: на 50 мл: Смешивают 5 мл 5% BSA, 250 мкл 5% молока и 50 мкл 100 мМ ванадата натрия, доводят до конечного объема 0,05% TBST. 12. Добавляют 100 мкл на лунку антифосфотирозина (1:10000 разведение в ADB). Инкубируют при встряхивании в течение 1 ч при комнатной температуре. 13. Промывают как на стадии 10. 14. Добавляют по 100 мкл на лунку Biosource конъюгата козьих антител к IgG кролика с пероксида-зой (1:6000 разведение в ADB). Инкубируют при встряхивании в течение 1 ч при комнатной температуре. 15. Промывают как на стадии 10, а затем PBS для удаления пузырей и избытка TWEEN. 16. Добавляют по 100 мкл ABTS/H2O2 раствора в каждую лунку. 17. Инкубируют при встряхивании в течение 10-20 мин. Удаляют все пузырьки. 18. Считывают анализ на Dynatech MR7000 ELISA считывающем устройстве: тест-фильтр при 410 нм и фильтр сравнения при 630 нм. Биоанализ EGFR Этот анализ используют для определения in vitro киназной активности FGF1-R в ELISA анализе. Материалы и реагенты: 1. 96-луночные ELISA планшеты Corning. 2. SUMO1 моноклональное анти-EGFR антитело (SUGEN, Inc.). 3. PBS. 4. TBST буфер. 5. Блокирующий буфер: на 100 мл смешивают 5,0 г Carnation Instant Nonfat Milk(r) с 100 мл PBS. 6. А431 клеточный лизат (SUGEN, Inc.). 7. TBS буфер. 8. TBS+10% ДМСО: на 1 л смешивают 1,514 г TRIS, 2,192 г NaCl и 25 мл ДМСО; доводят до общего объема 1 л dH2O. 9. ATP (аденозин-5'-трифосфат, из конской мышцы, Sigma Catalog № A-5394), 1,0 мМ раствор в dH2O. Этот реагент следует готовить непосредственно перед использованием и хранить на льду. 10. 1,0 мл MnCl2. 11. ATP/MnCl2 смесь для фосфорилирования: для приготовления 10 мл смешивают 300 мкл 1 мМ АТР, 500 мкл MnCl2 и 9,2 мл dH2O. Готовят непосредственно перед использованием, хранят на льду. 12. 96-луночные полипропиленовые планшеты NUNC с V-образным дном. 13. EDTA. 14. Кроличья поликлональная антифосфотирозиновая сыворотка (SUGEN, Inc.). 15. Конъюгат козьих антител к IgG кролика с пероксидазой (Biosource Cat. № ALI0404). 16. ABTS. 17. 30% перекись водорода. 18. ABST/H2O2. 19. 0,2M HCl. Методика 1. 96-луночные ELISA планшеты Corning покрывают 0,5 мкг SUMO1 в 100 мкл PBS на лунку, хранят в течение ночи при 4°С. 2. Удаляют несвязанный SUMO1 из лунок путем переворачивания планшета для удаления жидкости. Промывают 1 раз dH2O. Похлопывают планшетом по бумажной салфетке для удаления избытка жидкости. 3. Добавляют по 150 мкл блокирующего буфера в каждую лунку. Инкубируют при встряхивании в течение 30 мин при комнатной температуре. 4. Промывают планшет 3 раза деионизированной водой, затем один раз TBST. Похлопывают план - 22 - 008137 шет по бумажной салфетке для удаления избытка жидкости и пузырьков. 5. Разводят лизат в PBS (7 мкг лизата/100 мкл PBS). 6. Добавляют по 100 мкл разведенного лизата в каждую лунку. Встряхивают при комнатной температуре в течение 1 ч. 7. Промывают планшеты как на стадии 4. 8. Добавляют 120 мкл TBS в ELISA планшет, содержащий захваченный EGFR. 9. Разводят тестируемое соединение 1:10 в TBS, помещают в лунку. 10. Добавляют 13,5 мкл разведенного тестируемого соединения в ELISA планшет. В контрольные лунки добавляют 13,5 мкл TBS в 10% ДМСО. 11. Инкубируют при встряхивании в течение 30 мин при комнатной температуре. 12. Добавляют 15 мкл смеси для фосфорилирования во все лунки за исключением лунки с отрицательным контролем. Конечный объем в лунке должен составлять приблизительно 150 мкл с 3 мкМ АТР/5 мМ MnCl2 конечной концентрации в каждой лунке. Инкубируют при встряхивании в течение 5 мин. 13. Останавливают реакцию путем добавления 16,5 мкл EDTA раствора при встряхивании. Встряхивают в течение еще 1 мин. 14. Промывают 4 раза деионизированной водой, 2 раза TBST. 15. Добавляют по 100 мкл антифосфотирозина (1:3000 разведение в TBST) на лунку. Инкубируют при встряхивании в течение 30-45 мин при комнатной температуре. 16. Промывают как на стадии 4. 17. Добавляют по 100 мкл Biosource конъюгата козьих антител к IgG кролика с пероксидазой (1:2000 разведение в TBST) в каждую лунку. Инкубируют при встряхивании в течение 30 мин при комнатной температуре 18. Промывают как на стадии 4. 19. Добавляют по 100 мкл ABTS/H2O2 раствора в каждую лунку. 20. Инкубируют от 5 до 10 мин при встряхивании. Удаляют все пузырьки. 21. Если необходимо, останавливают реакцию добавлением 100 мкл 0,2М HCl на лунку. 22. Считывают анализ на Dynatech MR7000 ELISA считывающем устройстве: тест-фильтром при 410 нм и фильтр сравнения при 630 нм. Биоанализ PDGFR Этот анализ используют для определения in vitro киназной активности FGF1-R в ELISA анализе. Материалы и реагенты: 1. 96-луночные ELISA планшеты Corning. 2. 28D4C10 моноклональное анти-PDGFR антитело (SUGEN, Inc.). 3. PBS. 4. TBST буфер. 5. Блокирующий буфер (такой же, как для EGFR биоанализа). 6. PDGFR-в экспрессирующий NIH 3Т3 клеточный лизат (SUGEN, Inc.). 7. TBS буфер. 8. TBS+10% ДМСО. 9. ATP. 10. MnCl2. 11. Киназная буферная смесь для фосфорилирования: на 10 мл смешивают 250 мкл 1М TRIS, 200 мкл 5М NaCl, 100 мкл 1М MnCl2 и 50 мкл 100 мМ Triton Х-100 в количестве dH2O, достаточном для приготовления 10 мл. 12. 96-луночные полипропиленовые планшеты NUNC с V-образным дном. 13. EDTA. 14. Кроличья поликлональная антифосфотирозиновая сыворотка (SUGEN, Inc.). 15. Конъюгат козьих антител к IgG кролика с пероксидазой (Biosource Cat. № ALI0404). 16. ABTS. 17. Перекись водорода, 30% раствор. 18. ABST/H2O2. 19. 0,2M HCl. Методика 1. 96-луночные ELISA планшеты Corning покрывают 0,5 мкг 28D4C10 в 100 мкл PBS на лунку, хранят в течение ночи при 4°С. 2. Удаляют несвязанный 28D4C10 из лунок путем переворачивания планшета для удаления жидкости. Промывают 1 раз dH2O. Похлопывают планшетом по бумажной салфетке для удаления избытка жидкости. 3. Добавляют по 150 мкл блокирующего буфера в каждую лунку. Инкубируют в течение 30 мин при комнатной температуре при встряхивании. 4. Промывают планшет 3 раза деионизированной водой, затем один раз TBST. Похлопывают планшетом по бумажной салфетке для удаления избытка жидкости и пузырьков. - 23 - 008137 5. Разводят лизат в HNTG (10 мкг лизата/100 мкл HNTG). 6. Добавляют по 100 мкл разведенного лизата в каждую лунку. Встряхивают при комнатной температуре в течение 60 мин. 7. Промывают планшеты как описано на стадии 4. 8. Добавляют 80 мкл рабочей киназной буферной смеси к ELISA планшету, содержащему захваченный PDGFR. 9. Разводят тестируемое соединение 1:10 в TBS в 96-луночных полипропиленовых планшетах. 10. Добавляют 10 мкл разведенного тестируемого соединения в ELISA планшет. В контрольные лунки добавляют 10 мкл TBS+10% ДМСО. Инкубируют при встряхивании в течение 30 мин при комнатной температуре. 11. Добавляют 10 мкл ATР прямо во все лунки за исключением лунки с отрицательным контролем (конечный объем в лунке должен составлять приблизительно 100 мкл с 20 мкМ АТР в каждой лунке). Инкубируют 30 мин при встряхивании. 12. Останавливают реакцию путем добавления 10 мкл EDTA раствора в каждую лунку. 13. Промывают 4 раза деионизированной водой, 2 раза TBST. 14. Добавляют 100 мкл антифосфотирозина (1:3000 разбавление в TBST) на лунку. Инкубируют при встряхивании в течение 30-45 мин при комнатной температуре. 15. Промывают как на стадии 4. 16. Добавляют 100 мкл Biosource конъюгата козьих антител к IgG кролика с пероксидазой (1:2000 разведение в TBST) в каждую лунку. Инкубируют при встряхивании в течение 30 мин при комнатной температуре. 17. Промывают как на стадии 4. 18. Добавляют по 100 мкл ABTS/H2O2 раствора в каждую лунку. 19. Инкубируют 10-30 мин при встряхивании. Удаляют все пузырьки. 20. Если необходимо, останавливают реакцию добавлением 100 мкл 0,2М HCl на лунку. 21. Считывают анализ на Dynatech MR7000 ELISA считывающем устройстве с тест-фильтром при 410 нм и фильтром сравнения при 630 нм. Анализ клеточной HER-2 КИНАЗЫ Этот анализ используют для измерения HER-2 киназной активности в цельных клетках в ELISA формате. Материалы и реагенты: 1. DMEM (среда Игла, модифицированная по Дульбекко) (GIBCO Catalog №11965-092). 2. Сыворотка плода коровы (FBS, GIBCO Catalog № 16000-044), инактивированная нагреванием в водяной бане в течение 30 мин при 56°С. 3. Трипсин (GIBCO Catalog № 25200-056). 4. L-глутамин (GIBCO Catalog № 25030-081). 5. HEPES (GIBCO Catalog № 15630-080). 6. Среда для выращивания. Смешивают 500 мл DMEM, 55 мл инактивированной нагреванием FBS, 10 мл HEPES и 5,5 мл L-глутамина. 7. Голодная среда. Смешивают 500 мл DMEM, 2,5 мл инактивированной нагреванием FBS, 10 мл HEPES и 5,5 мл L-глутамина. 8. PBS. 9. 96-луночные микротитровальные планшеты для культуры ткани с плоским дном (Corning Catalog № 25860). 10. 15 см чашки для культуры ткани (Corning Catalog № 08757148). 11. 96-луночные ELISA планшеты Corning. 12. 96-луночные полипропиленовые планшеты NUNC с V-образным дном. 13. Переносные картриджи Costar для Transtar 96 (Costar Catalog № 7610). 14. SUMO 1: моноклональное анти-EGFR антитело (SUGEN, Inc.). 15. TBST буфер. 16. Блокирующий буфер: 5% Carnation Instant Milk(r) в PBS. 17. EGF лиганд: EGF-201, Shinko American, Japan. Суспендируют порошок в 100 мкл 10 мМ HCl. Добавляют 100 мкл 10 мМ NaOH. Добавляют 800 мкл PBS и переносят в пробирку Эппендорфа, хранят при -20°С до готовности к использованию. 18. HNTG буфер для лизиса. Для исходного 5-кратного раствора HNTG смешивают 23,83 г HEPES, 43,83 г NaCl, 500 мл глицерина и 100 мл Triton Х-100 и достаточное количество dH2O для приготовления 1 л общего раствора. Для 1-кратного HNTG* смешивают 2 мл HNTG, 100 мкл 0,1М Na3VO4, 250 мкл 0,2М Na4P2O7 и 100 мкл EDTA. 19. EDTA. - 24 - 008137 20. Na3VO4. Чтобы приготовить исходный раствор, смешивают 1,84 г Na3VO4 с 90 мл dH2O. Доводят рН до 10. Кипятят в микроволновой печи в течение 1 мин (раствор становится прозрачным). Охлаждают до комнатной температуры. Доводят рН до 10. Повторяют цикл нагревание/охлаждение, пока рН остается 10. 21. 200 мМ Na4P2O7. 22. Кроличья поликлональная антисыворотка, специфичная к фосфотирозину (анти-Ptyr антитело, SUGEN, Inc.). 23. Аффинно очищенная антисыворотка, конъюгат антител козы к IgG кролика с пероксидазой (Biosource Catalog № ALI0404). 24. ABTS раствор. 25. 30% раствор перекиси водорода. 26. ABTS/H2O2. 27. 0,2М HCl. Методика 1. Покрывают Corning 96-луночные ELISA планшеты SUMO1 в количестве 1,0 мкг на лунку в PBS, 100 мкл конечного объема/лунку. Хранят в течение ночи при 4°С. 2. В день использования удаляют покрывающий буфер и промывают планшет 3 раза dH2O и один раз TBST буфером. Все промывания в данном анализе следует выполнять этим способом, если не указано иное. 3. Добавляют по 100 мкл блокирующего буфера в каждую лунку. Инкубируют планшет при встряхивании в течение 30 мин при комнатной температуре. Непосредственно перед использованием планшет промывают. 4. Для этого анализа используют EGFr/HER-2 химерную/3Т3-С7 клеточную линию 5. Выбирают чашки, имеющие 80-90% конфлюэнтность. Клетки собирают трипсинизацией и центрифугируют при 1000 об./мин при комнатной температуре в течение 5 мин. 6. Ресуспендируют клетки в голодной среде и подсчитывают с использованием трипанового синего. Требуется жизнеспособность выше 90%. Клетки засевают в голодную среду с плотностью 2500 клеток на лунку, 90 мкл на лунку, в 96-луночный микротитровальный планшет. Инкубируют засеянные клетки в течение ночи при 37°С под 5% С32. 7. Начинают анализ через 2 суток после посева. 8. Тестируемые соединения растворяют в 4% ДМСО. Образцы затем дополнительно разбавляют на планшетах с голодной-DMEM. Обычно это разбавление составляет 1:10 или более. Все лунки затем переносят на клеточный планшет при дополнительном разведении 1:10 (10 мкл образца и среды в 90 мкл голодной среды. Конечная концентрация ДМСО должна составлять 1% или ниже. Также можно использовать стандартное серийное разведение. 9. Инкубируют под 5% ТО2 при 37°С в течение 2 ч. 10. Готовят EGF лиганд путем разведения исходного EGF (16,5 мкМ) в теплой DMEM до 150 нМ. 11. Готовят свежий HNTG* в количестве, достаточном для 100 мкл на лунку; помещают на лед. 12. После 2 ч инкубации с тестируемым соединением добавляют приготовленный EGF лиганд к клеткам, 50 мкл на лунку, до конечной концентрации 50 нМ. В лунки с положительным контролем добавляют такое же количество EGF. В отрицательные контроли не добавляют EGF. Инкубируют при 37°С в течение 10 мин. 13. Удаляют тестируемое соединение, EGF и DMEM. Промывают клетки один раз PBS. 14. Переносят HNTG* к клеткам, 100 мкл на лунку. Помещают на лед на 5 мин. Тем временем, удаляют блокирующий буфер из ELISA планшета и промывают. 15. Выскабливают клетки с планшета микропипеткой и гомогенизируют клеточный материал, многократно отсасывая и выпуская HNTG* буфер для лизиса. Переносят лизат в покрытый, закрытый, промытый ELISA планшет. Или используют картридж для переноса Costar для переноса лизата в планшет. 16. Инкубируют при встряхивании при комнатной температуре в течение 1 ч. 17. Удаляют лизат, промывают. Переносят свежеразведенное анти-Ptyr антитело (1:3000 в TBST) в ELISA планшет, 100 мкл на лунку. 18. Инкубируют при встряхивании при комнатной температуре в течение 30 мин. 19. Удаляют анти-Ptyr антитело, промывают. Переносят свежеразведенное BIOSOURCE антитело в ELISA планшет (1:8000 в TBST, 100 мкл на лунку). 20. Инкубируют при встряхивании при комнатной температуре в течение 30 мин. 21. Удаляют BIOSOURCE антитело, промывают. Переносят свежеприготовленный ABTS/H2O2 раствор в ELISA планшет, 100 мкл на лунку. 22. Инкубируют при встряхивании в течение 5-10 мин. Удаляют всякие пузырьки. 23. Останавливают реакцию добавлением 100 мкл 0,2 М HCl на лунку. 24. Считывают анализ на Dynatech MR7000 ELISA считывающем устройстве с тест-фильтром при 410 нм и фильтром сравнения при 630 нм. - 25 - 008137 Анализ циклинзависимой киназы (CDK2)/циклина А Этот анализ используют для измерения in vitro серин/треонин киназной активности cdk2/циклина А человека в анализе сцинтилляционной близости (SPA). Материалы и реагенты: 1. 96-луночные полиэтилен-терфталатные (гибкие) планшеты Wallac (Wallac Catalog № 1450-401). 2. Amersham Redivue [y33P] ATP (Amersham Catalog № AH 9968). 3. Покрытые стрептавидином поливинилтолуоловые SPA гранулы Amersham (Amersham Catalog № RPNQ0007). Гранулы следует растворить в PBS без магния или кальция в концентрации 20 мг/мл. 4. Активированный cc^/innunni А ферментный комплекс, очищенный, из Sf9 клеток (SUGEN, Inc.). 5. Биотинилированный пептидный субстрат (Debtide). Пептидный биотин-X-PKTPKKAKKL растворяют в dH2O в концентрации 5 мг/мл. 6. Смесь пептид/ATP: на 10 мл смешивают 9,979 мл dH2O, 0,00125 мл "холодного" АТР, 0,010 мл Debtide и 0,010 мл у33Р АТР. Окончательная концентрация на лунку будет 0,5 мкМ "холодного" АТР, 0,1 мкг Debtide и 0,2 мкКи у33Р АТР. 7. Киназный буфер: на 10 мл смешивают 8,85 мл dH2O, 0,625 мл TRIS (рН 7,4), 0,25 мл 1М MgCl2, 0,25 мл 10% NP40 и 0,025 мл 1М DTT, добавляют свежим непосредственно перед использованием. 8. 10 мМ АТР в dH2O. 9. 1 М TRIS, рН доводят до 7,4, используя HCl. 10. 1М MgCl2. 11.1M DTT. 12. PBS (Gibco Catalog № 14190-144). 13. 0,5M EDTA. 14. Стоп-раствор: на 10 мл смешивают 9,25 мл PBS, 0,005 мл 100 мМ АТР, 0,1 мл 0,5М EDTA, 0,1 мл 10% Triton X-100 и 1,25 мл 20 мг/мл SPA гранул. Методика: 1. Готовят растворы тестируемых соединений в 5-кратной требуемой конечной концентрации в 5% ДМСО. Добавляют 10 мкл в каждую лунку. Для отрицательных контролей используют 10 мкл 5% ДМСО одного в лунках. 2. Разводят 5 мкл раствора cdk2AQnunm А в 2,1 мл 2х киназного буфера. 3. Добавляют по 20 мкл фермента в каждую лунку. 4. Добавляют 10 мкл 0,5М EDTA в лунки с отрицательным контролем. 5. Чтобы запустить киназную реакцию, добавляют по 20 мкл смеси пептид/ATP в каждую лунку. Инкубируют в течение 1 ч без встряхивания. 6. Добавляют по 200 мкл стоп-раствора в каждую лунку. 7. Выдерживают по меньшей мере 10 мин. 8. Вращают планшет при приблизительно 2300 об./мин в течение 3-5 мин. 9. Считывают планшет, используя Trilux или аналогичное считывающее устройство. Анализ меттрансфосфорилирования Этот анализ используют для измерения уровней фосфотирозина на субстрате поли(глутаминовая кислота: тирозин (4:1)) как средства идентификации агонистов/антагонистов меттрансфосфорилирования субстрата. Материалы и реагенты: 1. 96-луночные ELISA планшеты Corning (Corning Catalog № 25805-96). 2. ПОЛИ^ШДУГ) 4:1, Sigma Catalog № P0275. 3. PBS, Gibco Catalog № 450-1300EB. 4. 50 мM HEPES. 5. Блокирующий буфер: растворяют 25 г бычьего сывороточного альбумина, Sigma Catalog № А-7888, в 500 мл PBS, фильтруют через 4 мкм фильтр. 6. Очищенный GST слитый белок, содержащий Met киназный домен, Sugen, Inc. 7. TBST буфер. 8. 10% водный (MilliQue H2O) ДМСО. 9. 10 мМ водный (dH2O) аденозин-5'-трифосфат, Sigma Catalog № A-5394. 10. 2х буфер для разбавления киназы: на 100 мл смешивают 10 мл 1 М HEPES с рН 7,5 с 0,4 мл 5% BSA/PBS, 0,2 мл 0,1М ортованадата натрия и 1 мл 5М хлорида натрия в 88,4 мл dH2O. 11. 4х АТР реакционная смесь: на 10 мл смешивают 0,4 мл 1М хлорида марганца и 0,02 мл 0,1 М АТР в 9,56 мл dH2O. 12. 4х смесь для отрицательного контроля: на 10 мл смешивают 0,4 мл 1 М хлорида марганца в 9,56 мл dH2O. 13. 96-луночные полипропиленовые планшеты NUNC с V-образным дном, Applied Scientific Catalog № S-72092 - 26 - 008137 14. 500 мM EDTA. 15. Буфер для разведения антител: на 100 мл смешивают 10 мл 5% BSA/PBS, 0,5 мл 5% Carnation Instant Milk(r) в PBS и 0,1М ортованадата натрия в 88,4 мл TBST. 16. Кроличье поликлональное антифосфотирозиновое антитело, Sugen, Inc. 17. Конъюгат антикроличьих антител с пероксидазой хрена, Biosource, Inc. 18. ABST раствор: на 1 л смешивают 19,21 г лимонной кислоты, 35,49 г Na2HPO4 и 500 мг ABTS с количеством dH2O, достаточным для приготовления 1 л. 19. ABTS/H2O: смешивают 15 мл ABST раствора с 2 мкл H2O2 за 5 мин до использования. 20. 0,2M HCl. Методика: 1. Покрывают ELISA планшеты 2 мкг ПОЛИ^Ш^Г) в 100 мкл PBS, хранят в течение ночи при 4°С. 2. Блокируют планшет 150 мкл 5% BSA/PBS в течение 60 мин. 3. Промывают планшет дважды PBS, один раз 50 мМ HEPES буфера с рН 7,4. 4. Добавляют 50 мкл разведенной киназы во все ячейки (Очищенную киназу разводят в буфере для разведения киназы. Конечная концентрация должна составлять 10 нг/лунку). 5. Добавляют 25 мкл тестируемого соединения (в 4% ДМСО) или ДМСО один (4% в dH2O) для контроля на планшете. 6. Инкубируют смесь киназа/соединение в течение 15 мин. 7. Добавляют 25 мкл 40 мМ MnCl2 в лунки с отрицательным контролем. 8. Добавляют 25 мкл смеси ATP/MnCl2 во все остальные лунки (за исключением отрицательных контролей). Инкубируют в течение 5 мин. 9. Добавляют 25 мкл 500 мМ EDTA, чтобы остановить реакцию. 10. Промывают планшет 3х TBST. 11. Добавляют по 100 мкл кроличьего поликлонального анти-Ptyr, разведенного 1:10000 в буфере для разведения антител, в каждую лунку. Инкубируют при встряхивании при комнатной температуре в течение 1 ч. 12. Промывают планшет 3х TBST. 13. Разводят конъюгат антикроличьих антител с пероксидазой хрена (Biosource) 1:6000 в буфере для разведения антител. Добавляют по 100 мкл на лунку и инкубируют при комнатной температуре при встряхивании в течение 1 ч. 14. Промывают планшет 1 х PBS. 15. Добавляют по 100 мкл ABTS/H2O2 раствора в каждую лунку. 16. Если необходимо, останавливают развитие реакции добавлением 100 мкл 0,2М HCl на лунку. 17. Считывают результаты на Dynatech MR7000 ELISA считывающем устройстве с тест-фильтром при 410 нм и фильтром сравнения при 630 нм. Анализ IGF-1 трансфорилирования Этот анализ использован для измерения уровня фосфотирозина в поли(глутаминовая кисло-та:тирозин (4:1) для определения агонистов/антагонистов gst-IGF-1 трансфосфорилирования субстрата. Материалы и реагенты: 1. 96-луночные ELISA планшеты Corning. 2. ПОЛИ^ШДУГ) 4:1, Sigma Catalog № P0275. 3. PBS, Gibco Catalog № 450-1300ЕВ. 4. 50 мМ HEPES. 5. TBB блокирующий буфер: на 1 л смешивают 100 г BSA, 12,1 г TRIS (рН 7,5), 58,44 г хлорида натрия и 10 мл 1% TWEEN-20. 6. Очищенный GST слитый белок, содержащий IGF-1 киназный домен (Sugen, Inc). 7. TBST буфер: на 1 л смешивают 6,075 г TRIS, 8,766 г хлорида натрия и 0,5 мл TWEEN-20 с количеством dH2O, достаточным для приготовления 1 л. 8. 4% ДМСО в Milli-Q H2O. 9. 10 мМ АТР в dH2O. 10. 2х буфер для разведения киназы: на 100 мл смешивают 10 мл 1М HEPES (рН 7,5), 0,4 мл 5% BSA в dH2O, 0,2 мл 0,1М ортованадата натрия и 1 мл 5М хлорида натрия с количеством dH2O, достаточным для приготовления 100 мл. 11. 4х АТР реакционная смесь: на 10 мл смешивают 0,4 мл 1 М MnCl2 и 0,008 мл 0,01 М АТР и 9,56 мл dH2O. 12. 4х смесь для отрицательного контроля: смешивают 0,4 мл 1 М хлорида марганца в 9,60 мл dH2O. 13. 96-луночные полипропиленовые планшеты NUNC с V-образным дном. 14. 500 мМ EDTA в dH2O. 15. Буфер для разведения антител: на 100 мл смешивают 10 мл 5% BSA в PBS, 0,5 мл 5% Carnation Instant Non-fat Milk(r) в PBS и 0,1 мл 0,1М ортованадата натрия в 88,4 мл TBST. 16. Кроличье поликлональное антифосфотирозиновое антитело, Sugen, Inc. 17. Конъюгат козьих антикроличьих антител с пероксидазой хрена, Biosource. - 27 - 008137 18. ABST раствор. 19. ABTS/H2O2: смешивают 15 мл ABST с 2 мкл Н2О2 за 5 мин до использования. 20. 0,2М HCl в dH2O. Методика: 1. Покрывают ELISA планшет 2 мкг/лунку ПОЛИ^Ш^Г) 4:1 (Sigma Р0275) в 100 мкл PBS. Планшет хранят в течение ночи при 4°С. 2. Промывают планшет один раз PBS. 3. Добавляют по 100 мкл ТВВ блокирующего буфера в каждую лунку. Инкубируют планшет в течение 1 ч при встряхивании при комнатной температуре. 4. Промывают планшет один раз PBS, затем дважды 50 мМ HEPES буфера с рН 7,5. 5. Добавляют 25 мкл тестируемого соединения в 4% ДМСО (получают путем разведения исходного раствора 10 мМ тестируемого соединения в 100% ДМСО dH2O) в планшет. 6. Добавляют 10,0 нг gst-IGF-1 киназы в 50 мкл буфера для разведения киназы) во все ячейки. 7. Запускают киназную реакцию добавлением 25 мкл 4х АТР реакционной смеси во все тестируемые лунки и лунки с положительным контролем. Добавляют 25 мкл 4х смеси для отрицательных контролей во все лунки с отрицательным контролем. Инкубируют в течение 10 мин при встряхивании при комнатной температуре. 8. Добавляют 25 мкл 0,5М EDTA (рН 8,0) во все лунки. 9. Промывают планшет 4 раза TBST буфером. 10. Добавляют кроличью поликлональную антифосфотирозиновую антисыворотку в разведении 1:10000 в 100 мкл буфера для разведения антител во все лунки. Инкубируют при встряхивании при комнатной температуре в течение 1 ч. 11. Промывают планшет как на стадии 9. 12. Добавляют 100 мкл конъюгат антикроличьих антител с пероксидазой хрена Biosource в разведении 1:10000 в буфере для разведения антител во все лунки. Инкубируют при встряхивании при комнатной температуре в течение 1 ч. 13. Промывают планшет как на стадии 9, затем один раз промывают PBS для сокращения пузырьков и избытка TWEEN-20. 14. Проявляют добавлением 100 мкл/лунку ABTS/H2O2 раствора в каждую лунку. 15. Через примерно 5 мин считывают на ELISA считывающем устройстве с тест-фильтром при 410 нм и фильтром сравнения при 630 нм. Анализы включения BRDU В следующих анализах использованы клетки, сконструированные для экспрессии выбранного рецептора и последующей оценки воздействия интересующего соединения на активность лиганд-индуцированного синтеза ДНК путем определения включения BrdU в ДНК. Следующие материалы, реагенты и методика являются общими для каждого из следующих анализов включения BrdU. Отмечены различия в конкретных анализах. Материалы и реагенты: 1. Подходящий лиганд. 2. Подходящие сконструированные клетки. 3. BrdU меченый реагент: 10 мМ в PBS (рН 7,4) (Boehringer Mannheim, Germany). 4. FixDenat: фиксирующий раствор (готовый) (Boehringer Mannheim, Germany). 5. Anti-BrdU-POD: конъюгат мышиных моноклональных антител с пероксидазой (Boehringer Mannheim, Germany). 6. Раствор ТМВ субстрата:тетраметилбензидин (ТМВ, Boehringer Mannheim, Germany). 7. PBS промывочный раствор: 1х PBS, pH 7,4. 8. Альбумин, бычий (BSA), порошок фракции V (Sigma Chemical Co., USA). Общая методика 1. Клетки засеивают в количестве 8000 клеток/лунку в 10% CS, 2 мМ Gln в DMEM в 96-луночном планшете. Клетки инкубируют в течение ночи при 37°С в 5% СO2. 2. Через 24 ч клетки промывают PBS, а затем выдерживают в не содержащей сыворотку среде (0% CS DMEM с 0,1% BSA) в течение 24 ч. 3. На день 3 к клеткам одновременно добавляют подходящий лиганд и тестируемое соединение. В лунки с отрицательным контролем добавляют только не содержащую сыворотку DMEM с 0,1% BSA; в лунки с положительным контролем добавляют лиганд, но не добавляют тестируемое соединение. Тестируемые соединения готовят в не содержащей сыворотку DMEM с лигандом в 96-луночном планшете и серийно разводят с получением 7 тестируемых концентраций. 4. После 18 ч активации лигандом, добавляют разведенный BrdU меченный реагент (1:100 в DMEM, 0,1% BSA), и клетки инкубируют с BrdU (конечная концентрация = 10 мкМ) в течение 1,5 ч. 5. После инкубирования с меченым реагентом среду удаляют декантацией и постукиванием перевернутого планшета по бумажной салфетке. Добавляют FixDenat раствор (50 мкл/лунку) и планшеты инкубируют при комнатной температуре в течение 45 мин на шейкере для планшетов. - 28 - 008137 6. FixDenat раствор аккуратно удаляют декантацией и постукиванием перевернутого планшета по бумажной салфетке. Добавляют молоко (5% обезвоженное молоко в PBS, 200 мкл/лунку) в качестве блокирующего раствора и планшеты инкубируют в течение 30 мин при комнатной температуре на шейкере для планшетов. 7. Блокирующий раствор удаляют декантацией, и клетки промывают один раз PBS. Добавляют ан-ти-Brd-U-POD раствор (разведение 1:200 в PBS, 1% BSA) (50 мкг/лунку) и планшет инкубируют в течение 90 мин при комнатной температуре на шейкере для планшетов. 8. Конъюгат антител аккуратно удаляют декантацией и ополаскиванием лунок PBS 5 раз и планшет сушат путем переворачивания и постукивания по бумажной салфетке. 9. Добавляют раствор ТМВ субстрата (100 мкг/лунку) и инкубируют в течение 20 мин при комнатной температуре на шейкере для планшетов, пока проявление цвета не станет достаточным для фотометрического определения. 10. Поглощение образцов измеряют при 410 нм (в режиме "двойная длина волны" со считыванием через фильтр при 490 нм в качестве длины волны сравнения) на Dynatech ELISA считывающем устройстве для планшетов. Анализ EGF-индуцированного BrdU включения Материалы и реагенты: 1. Мышиный EGF, 201 (Toyobo Co., Ltd., Япония). 2. 3E3/EGFRc7. Анализ EGF-индуцированного Неr-2-управляемого BrdU включения Материалы и реагенты: 1. Мышиный EGF, 201 (Toyobo Co., Ltd., Япония). 2. 3E3/EGFr/Her2/EGFr (EGFr c Her-2 киназным доменом). Анализ EGF-индуцированного Не^4-управляемого BrdU включения Материалы и реагенты: 1. Мышиный EGF, 201 (Toyobo Co., Ltd., Япония). 2. 3E3/EGFr/Her4/EGFr (EGFr c Her-4 киназным доменом). Анализ PDGF-индуцированного BrdU включения Материалы и реагенты: 3. Человеческий PDGF В/В (Boehringer Mannheim, Германия). 4. 3T3/EGFRc7. Анализ FDF-индуцированного BrdU включения Материалы и реагенты: 5. Человеческий FGF2/bFGF (Gibco BRL, США). 6. 3T3c7/EGFr. Анализ IGFI-индуцированного BrdU включения Материалы и реагенты: 7. Человеческий, рекомбинантный (G511, Promega Corp., США). 8. 3T3/IGF1r. Анализ инсулин-индуцированного BrdU включения Материалы и реагенты: 1. Цинк-инсулин, кристаллический, бычий (13007 Gibco BRL, США). 2. 3Т3/Н25. Анализ HGF-индуцированного BrdU включения Материалы и реагенты: 1. Рекомбинантный человеческий HGF (Cat. No. 249-HG, R &D Systems, Inc. США). 2. BxPC-3 клетки (ATCC CRL-1687). Методика: 1. Клетки засеивают в количестве 9000 клеток/лунку в RPMI 10% FBS в 96-луночном планшете. Клетки инкубируют в течение ночи при 37°С в 5% CO2. 2. Через 24 ч клетки промывают PBS, а затем их держат без сыворотки в 100 мкл не содержащей сыворотку среде (RPMI с 0,1% BSA) в течение 24 ч. 3. На день 3 к клеткам добавляют 25 мкл, содержащие лиганд (приготовленный в концентрации 1 мкг/мл в RPMI с 0,1% BSA; конечная концентрация HGF составляет 200 нг/мл) и тестируемые соединения. В лунки с отрицательным контролем добавляют только 25 мкл не содержащей сыворотку RPMI с 0,1% BSA; в лунки с положительным контролем добавляют лиганд (HGF), но не добавляют тестируемое соединение. Тестируемые соединения готовят в их 5-кратной конечной концентрации в не содержащей сыворотку RPMI с лигандом в 96-луночном планшете и серийно разводят с получением 7 тестируемых концентраций. Обычно самая высокая конечная концентрация тестируемого соединения составляет 100 мкМ, и используют разведения 1:3 (т.е. интервал конечных концентраций тестируемых соединений составляет 0,137-100 мкМ). 4. После 18 ч активации лигандом в каждую лунку добавляют 12,5 мкл разведенного BrdU меченно - 29 - 008137 го реагента (1:100 в RPMI, 0,1% BSA) и клетки инкубируют с BrdU (конечная концентрация составляет 10 мкМ) в течение 1 ч. 5. Так же, как в общей методике. 6. Так же, как в общей методике. 7. Блокирующий раствор удаляют декантацией, и лунки промывают один раз PBS. Добавляют анти-Brd-U-POD раствор (разведение 1:100 в PBS, 1% BSA) (100 мкг/лунку) и планшет инкубируют в течение 90 мин при комнатной температуре на шейкере для планшетов. 8. Так же, как в общей методике. 9. Так же, как в общей методике. 10. Так же, как в общей методике. Анализ HUV-EC-C Этот анализ используется для измерения активности соединения по отношению к PDGF-R, FGF-R, VEGF, aFGF или Flk-1/KDR, которые все естественно экспрессируются HUV-EC клетками. День 0 1. Промывают и трипсинизируют HUV-EC-C клетки (эндотелиальные клетки пупочной вены человека, Американская коллекция типовых культур, каталожный № 1730 CRL). Промывают фосфатным буферным солевым раствором Дульбекко (D-PBS, приобретенным у Gibco BRL, каталожный № 14190-029) 2 раза в количестве примерно 1 мл/10 см2 культурального флакона. Трипсинизируют 0,05%-ным трип-син-EDTA в неферментном растворе для диссоциации клеток (Sigma Chemical Company, каталожный № С-1544). 0,05% трипсин готовят путем разведения 0,25% трипсина/1 мМ EDTA (Gibco, каталожный № 25200-049) в растворе для диссоциации клеток. Трипсинизируют примерно 1 мл/25-30 см2 культурально-го флакона в течение примерно 5 мин при 37°С. После отсоединения клеток от флакона добавляют равный объем аналитической среды и переносят в стерильную центрифужную пробирку емкостью 50 мл (Fisher Scientific, каталожный № 05-539-6). 2. Промывают клетки примерно 35 мл аналитической среды в стерильной центрифужной пробирке емкостью 50 мл путем добавления этой аналитической среды, центрифугируют в течение 10 мин при приблизительно 200хg, отсасывают супернатант и ресуспендируют с 35 мл D-PBS. Повторяют промывание D-PBS еще два раза, ресуспендируют клетки в примерно 1 мл аналитической среды/15 см2 культу -рального флакона. Аналитическая среда состоит из среды F12K (Gibco BRL, каталожный № 21127-014) и 0,5% инактивированной фетальной бычьей сыворотки. Подсчитывают клетки на Coulter Counter(r) (Coulter Electronics, Inc.) и к этим клеткам добавляют аналитическую среду до получения концентрации 0,8-1,0х105 клеток/мл. 3. Добавляют клетки в 96-луночные планшеты с плоским дном в количестве 100 мкл/лунку или 0,8-1,0x104 клеток/лунку, инкубируют примерно 24 ч при 37°С, 5% ТО2. День 1 1. Выполняют двукратные титрования тестируемого соединения в отдельных 96-луночных планшетах, обычно с 50 мкМ с понижением до 0 мкМ. Используют такую же аналитическую среду, как в день 0 на стадии 2 выше. Титрования выполняют путем добавления 90 мкл/лунку тестируемого соединения при 200 мкМ (4-кратная конечная концентрация в лунке) в верхнюю лунку индивидуальной колонки планшета. Поскольку исходная концентрация тестируемого соединения обычно составляет 20 мМ в ДМСО, лекарственное средство в концентрации 200 мкМ содержит 2% ДМСО. Разбавитель, приготовленный для 2% ДМСО в аналитической среде (F12K+0,5% фетальной бычьей сыворотки), используют в качестве разбавителя для титрований тестируемых соединений, чтобы разбавлять тестируемое соединение, но поддерживать концентрацию ДМСО постоянной. Добавляют этот разбавитель в остальные лунки в колонке в количестве 60 мкл/лунку. Отбирают 60 мкл из 120 мкл 200 мкМ разведения тестируемого соединения в верхней лунке колонки и смешивают с 60 мкл во второй лунке колонки. Отбирают 60 мкл из этой лунки и смешивают с 60 мкл в третьей лунке данной колонки и так далее, пока двукратные титрования не будут окончены. После смешивания содержимого предпоследней лунки отбирают 60 мкл из 120 мкл этой лунки и их отбрасывают. Оставляют последнюю лунку с 60 мкл смеси ДМСО/среда-разбавитель в качестве контроля, не содержащего тестируемого соединения. Готовят 9 колонок титрованного тестируемого соединения, достаточных для лунок в трех повторах, каждая для: (1) VEGF (приобретенного у Pepro Tech Inch., каталожный № 100-200, (2) фактора роста эндотелиальных клеток (ECGF) (также известного как кислотный фактор роста фибробластов, или aFGF) (приобретенного у Boehringer Mannheim Biochemica, каталожный № 1439 600), или (3) PDGF В/В человека (1276-956, Boehringer Mannheim, Germany) и аналитической среды-контроля. ECGF поступает в виде препарата с натрий-гепарином. 2. Переносят 50 мкл/лунку разведений тестируемого соединения в 96-луночные аналитические планшеты, содержащие 0,8-1,0x104 клеток/100 мкл/лунку HUV-EC-C клеток с дня 0 и инкубируют примерно 2 ч при 37°С, 5% Ш2. 3. В трех повторах добавляют 50 мкл/лунку 80 мкг/мл VEGF, 20 нг/мл ECGF или среду-контроль к тестируемому соединению в каждом состоянии. Как и в случае тестируемых соединений концентрации факторов роста в 4 раза больше требуемой конечной концентрации. Используют аналитическую среду со - 30 - 008137 стадии 2 дня 0 для приготовления концентраций факторов роста. Инкубируют приблизительно 24 ч при 37°С, 5% CO2. В каждой лунке будет 50 мкл разведения тестируемого соединения, 50 мкл фактора роста или среды и 100 мкл клеток, что составляет в сумме 200 мкл/лунку. Таким образом, 4-кратные концентрации тестируемого соединения и факторов роста становятся 1-кратными, как только их добавят в лунки. День 2 1. Добавляют 3Н-тимидин (Amersham, каталожный № TRK-686) в количестве 1 мкКи/лунку (10 мкл/лунку 100 мкКи/мл раствора в среде RPMI + 10% инактивированная нагреванием сыворотка плода коровы) и инкубируют примерно 24 ч при 37°С, 5% ОД2. RPMI приобретают у Gibco BRL, каталожный № 11875-051. День 3 1. Замораживают планшеты в течение ночи при -20°С. День 4 1. Размораживают планшеты и собирают при помощи харвестера для 96-луночных планшетов (Tomtec Harvester 96(r)) на фильтровальные маты (Wallac, каталожный № 1205-401), считывают импульсы на жидкостном сцинтилляционном счетчике Wallac Betaplate(tm). В табл. 2 приведены результаты биологических испытаний 5-[5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил]-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амида (соединение 80). Результаты представлены в виде IC50, причем тестировали микромолярную (мкМ) концентрацию соединения, которая вызывает 50% изменение активности РТК-мишени по сравнению с активностью РТК в контроле, к которому тестируемое соединение не было добавлено. Конкретно, представленные результаты указывают концентрацию тестируемого соединения, необходимую, чтобы вызвать 50% снижение активности РТК-мишени. Биоанализы, которые были использованы или которые можно использовать для оценки этого соединения, подробно описаны ниже. Таблица 2 Пример био био био био клеточный Нег2 cdk2spa био flkGST FGFR1 PDGF EGF EGF киназы С50 рук2 1С50 1С50 1С50 1С50 1С50 1С50 (мкМ) IC50 (мкМ) (мкМ) (мкМ) (мкМ) (мкМ) (мкМ) (мкМ) 0,13 4,29 0,001 > 100 50,19 17,19 0,28 In vivo животные модели Животные модели ксенотрансплантата Способность опухолей человека расти как ксенотрансплантаты у бестимусных мышей (например Balb/c, nu/nu) обеспечивает пригодную in vivo модель для исследования биологического ответа на терапевтические средства для человеческих опухолей. Со времени первой успешной ксенотрансплантации человеческих опухолей в бестимусных мышей (Rygaard and Povlsen, 1969, Acta Pathol. Microbial. Scand. 77:758-760) много разных линий человеческих опухолевых клеток (например молочной железы, легочных, мочеполовых, желудочно-кишечных, головы и шеи, глиобластомы, костных и злокачественных ме-ланом) было трансплантировано и успешно выращено на голых мышах. Следующие ниже анализы можно использовать для определения уровня активности, специфичности и воздействия различных ингибиторов протеинкиназ. Для оценки ингибиторов протеинкиназ пригодны три основных типа анализов: клеточный/каталитический, клеточный/биологический и in vivo. Целью клеточных/каталитических анализов является определение воздействия 5-[5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил]-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амида на способность ТК фосфорилировать тирози-ны на известном субстрате в клетке. Целью клеточных/биологических анализов является определение воздействия 5-[5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил]-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амида на биологический ответ, стимулируемый ТК в клетке. Целью in vivo анализов является определение воздействия 5-[5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил]-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амида в животной модели конкретного расстройства, такого как рак. Подходящие клеточные линии для экспериментов по подкожной ксенотрансплантации включают клетки С6 (глиома, АТСС № CCL 107), клетки А375 (меланома, АТСС № CRL 1619), клетки А431 (эпи-дермоидная карцинома, АТСС № CRL 1555), клетки Calu 6 (легкое, АТСС № НТВ 56), клетки РС3 (простата, АТСС № CRL 1435), клетки SKOV3TP5 и фибробласты NIH 3Т3, сконструированные методом генной инженерии для сверхэкспресии EGFR, PDGFR, IGF-1R или любой другой тестируемой киназы. Для осуществления экспериментов по ксенотрансплантации можно использовать следующий протокол: Самок бестимусных мышей (Balb/c, nu/nu) доставляют из Simonsen Laboratories (Gilroy, CA). Всех животных содержат в условиях чистой комнаты в клетках Microisolator с подстилкой Alpha-dri. Они по - 31 - 008137 лучают стерильную пищу для грызунов и воду по своему желанию. Клеточные линии выращивают в подходящей среде (например MEM, DMEM, Ham's F10 или Ham's F12 плюс 5-10% сыворотка плода коровы (FBS) и 2 мМ глутамина (GLN)). Все среды для культуры клетки, глутамин и сыворотку плода коровы приобретают у Gibco Life Technologies (Grand Island, NY), если не указано иное. Все клетки вьгращивают во влажной атмосфере 90-95% воздуха и 5-10% СO2 при 37°С. Все клеточные линии регулярно инокулируют два раза в неделю, и они отрицательны на микоплазму при определении методом Mycotec (Gibco). Клетки собирают при или почти при конфлюэнтности 0,05% трипсин-EDTA и гранулируют при 450хg в течение 10 мин. Гранулы ресуспендируют в стерильном PBS или среде (без FBS) до конкретной концентрации, и эти клетки имплантируют в заднюю боковую область мышей (8-10 мышей на группу, 2-10х106 клеток/животное). Рост опухоли измеряют от 3 до 6 недель, используя штангенциркуль. Объемы опухолей рассчитывают как произведение длина х ширина х высота, если не указано иное. Значения Р вычисляют, используя t-критерий Стьюдента. Тестируемое соединение 5-[5-фтор-2-оксо-1,2-дигидроиндол-3 -илиденметил]-2,4-диметил-1Н-пиррол-3 -карбоновой кислоты (2-диэтиламиноэтил)амид в 50-100 мкл эксципиента (ДМСО или VPD:D5W) можно доставлять путем в.б. (внутрибрюшинной) инъекции в различных концентрациях, обычно начиная с первого дня после имплантации. Модель инвазии опухолей Следующая модель инвазии опухолей была разработана, и ее можно использовать для оценки терапевтического значения и эффективности 5-[5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил]-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амида, идентифицированного для селективного ингибирования KDR/FLK-1 рецептора. Методика В качестве экспериментальных животных используют голых мышей (самок) (Simonsen Inc.) возрастом восемь недель. Имплантацию опухолевых клеток можно осуществить в верх ламинарного потока. Для анестезии внутрибрюшинно вводят смесь ксилазин/кетамин (100 мг/кг кетамина и 5 мг/кг ксилази-на). Делают срединный разрез для вскрытия брюшной полости (приблизительно 1,5 см длиной) для инъекции 107 опухолевых клеток в объеме 100 мкл среды. Клетки инъецируют либо в дуоденальную долю поджелудочной железы, либо под серозную оболочку толстой кишки. Брюшину и мышцы зашивают непрерывным швом шелком 6-0 и кожу закрывают, используя зажимы для ран. За животными ежедневно наблюдают. Анализ Через 2-6 недель, в зависимости от суммарных наблюдений за животными, мышей умерщвляют и местные опухолевые метастазы в различные органы (легкое, печень, мозг, желудок, селезенка, сердце, мышцы) вырезают и анализируют (измерение размера опухоли, степень инвазии, иммунохимия, in situ определение гибридизации и так далее). Анализ С-К1Т Этот анализ используют для определения уровня c-kit фосфорилирования тирозина. Клетки MO7E (острая миелоидная лейкемия человека) выдерживали в условиях сывороточного голодания в течение ночи в 0,1% сыворотке. Перед стимуляцией лигандами клетки предварительно обрабатывали 5-[5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил]-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амидом (одновременно с сывороточным голодом). Клетки стимулировали 250 нг/мл rh-SCF (rh-фактор стволовых клеток) в течение 15 мин. После стимуляции клетки лизировали и иммунологически осаждали анти-c-kit антителом. Уровни фосфотирозина и белка определяли Вестерн-блоттингом. Анализ МТТ пролиферации Клетки MO7E выдерживали в условиях сывароточного голода и предварительно обрабатывали 5-[5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил]-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амидом, как описано для экспериментов по фосфорилированию. Клетки помещали в 96-луночный планшет в количестве 4x105 клеток/лунку, в 100 мкл RPMI+10% сыворотка. Добавляли rh-SCF (100 нг/мл) и планшет инкубировали в течение 48 ч. Через 48 ч добавляли 10 мкл 5 мг/мл МТТ [3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолий бромид) и оставляли инкубироваться в течение 4 ч. Добавляли кислотный изопропанол (100 мкл 0,04 н HCl в изопропаноле) и измеряли оптическую плотность при длине волны 550 нм. Анализ апоптоза Клетки MO7E ±SCF и ±5-[5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил]-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амид инкубировали в 10% FBS с rh-GM-CSF (10 нг/мл) и rh-IL-3 (10 нг/мл). Образцы анализировали через 24 и 48 ч. Для измерения активированной кас-пазы-3 образцы промывали PBS и проницаемость мембран нарушали ледяным 70% этанолом. Клетки затем окрашивали с РЕ-конъюгированной поликлональной кроличьей антиактивной каспазой-3 и анализировали при помощи FACS (активируемого флюоресценцией анализатора клеток). Для определения расщепленной PARP (поли-АБР(аденозиндифосфат)рибозилполимеразы) образцы лизировали и анализировали вестерн-блоттингом с использованием анти-PARP антитела. - 32 - 008137 Дополнительные анализы Дополнительные анализы, которые можно использовать для оценки 5-[5-фтор-2-оксо-1,2-дигидроиндол-3 -илиденметил]-2,4-диметил-1Н-пиррол-3 -карбоновой кислоты (2-диэтиламиноэтил)ами-да, включают, без ограничения, bio-flk-1 анализ, анализ EGF рецептор-НER2 химерного рецептора в целых клетках, bio-lck анализ и анализ, измеряющий фосфорилирующую функцию raf. Протоколы каждого из этих анализов можно найти в заявке на патент США серийный № 09/099842, который включен в данное описание изобретения ссылкой, включая любые графические материалы. Измерение клеточной токсичности Терапевтические соединения должны быть более сильнодействующими в ингибировании активности рецепторных тирозинкиназ, чем в оказании цитотоксического воздействия. Оценку эффективности и клеточной токсичности соединения можно получить путем определения терапевтического индекса, т.е. IC50/LD50-IC50, дозу, необходимую для достижения 50% ингибирования, можно определить, используя стандартные методики, такие как методики, описанные в данной заявке. LD50, дозу, которая приводит к 50% летальности, также можно определить стандартными методами (Mossman, 1983, J. Immunol. Methods., 65:55-63) путем измерения количества высвобожденной LDH (лактатдегидрогеназы) (Kor-zeniewski and Callewaert, 1983, J. Immunol. Methods, 64:313, Decker and Lohmann-Matthes, 1988, J. Immunol. Methods, 115:61) или путем определения летальной дозы в животных моделях. Предпочтительными являются соединения с большим терапевтическим индексом. Терапевтический индекс должен быть выше 2, предпочтительно по меньшей мере 10, более предпочтительно по меньшей мере 50. Б. Пример результатов клеточного анализа с использованием 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амида (Соединение 80). Для подтверждения силы действия 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламино-этил)амида (соединение 80), определенной биохимическими анализами (см. ниже), способность указанного соединения ингибировать лиганд-зависимое RTK фосфорилирование оценивали в анализах, основанных на клетках, используя клетки мыши NIH 3Т3, сконструированные для сверхэкспрессии Flk-1 или человеческого PDGFRp. 5-(5-Фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламино-этил)амид (соединение 80) ингибировал VEGF-зависимое Flk-1 фосфорилирование тирозина со значением IC50 приблизительно 0,03 мкМ. Это значение соответствует значению Ki 0,009 мкМ, определенному в отношении ингибирования Flk-1 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амидом (соединение 80) в биохимических анализах. Это указывает на то, что 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амид (соединение 80) легко проникает в клетки. В соответствии с биохимическими данными (смотри ниже), указывающими на то, что 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амид (соединение 80) имеет сравнимую активность в отношении Flk-1 и PDGFRp, было обнаружено также, что это соединение ингибирует PDGF-зависимое фосфорилирование рецепторов в клетках со значением IC50 приблизительно 0,03 мкМ. Способность 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амида (соединение 80) ингибировать c-kit, близкородственный RTK, который связывает фактор стволовых клеток (SCF), определяли, используя клетки MO7E, которые экспрессируют этот рецептор. В этих клетках 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламино-этил)амид (соединение 80) ингибировал SCF-зависимое c-kit фосфорилирование со значением IC50 0,010,1 мкМ. Это соединение также ингибировало SCF-стимулированнное c-kit фосфорилирование в бластах острой миелоидной лейкемии (AML), выделенных из периферической крови пациентов. В дополнение к испытанию на способность 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амида (соединение 80) ингибировать лиганд-зависимое фосфорилирование рецепторов в клетках, также исследовали in vitro его воздействие на лиганд-зависимый пролиферативный ответ клеток (см. табл. 4). В этих исследованиях в клетках, переведенных в пассивное состояние в результате сывороточного голодания в течение ночи, индуцировали синтез ДНК при добавлении подходящего митогенного лиганда. Как показано в табл. 3, 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламино-этил)амид (соединение 80) ингибировал PDGF-индуцированную пролиферацию клеток NIH-3T3, сверх-экспрессирующих PDGFRp или PDGFRa со значениями IC50 0,031 и 0,069 мкМ соответственно, и SCF-индуцированную пролиферацию клеток MO7E со значением IC50 0,007 мкМ. - 33 - 008137 Таблица 3 Биохимический Клеточная IC5o Рецептор Ki' (мкМ) Фосфорилирование рецептора (мкМ) Лиганд-зависимая пролиферация (мкМ) Flk-1/KDR 0,009 0,032 0,0043 PDGFRa 0,008 0,034 0,0314 PDGFRp 0,069й FGFR 0,83 0,7а c-kit 0,01-0,1b 0,007б ND = Не определен 1 Определен с использованием рекомбинантного фермента 2Определен с использованием сывороточно-голодавших клеток NIH-3T3, экспрессирующих Flk-1 3Определен с использованием сывороточно-голодавших HUVECs 4Определен с использованием сывороточно-голодавших клеток NIH-3T3, экспрессирующих PDGFR 5Определен с использованием сывороточно-голодавших клеток NIH-3T3, экспрессирующих PDGFR 6 Определен с использованием сывороточно-голодавших клеток MO7E Как показано в табл. 3, наблюдается общее соответствие между биохимическими и клеточными активностями 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амида (соединение 80), подтверждающее вывод, что это соединение проходит сквозь клеточные мембраны. Можно также сделать вывод, что клеточные ответы являются результатом активности соединения 80 в отношении указанной мишени. Наоборот, при испытании в присутствии полной среды роста in vitro требовались значительно более высокие концентрации 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламино-этил)амида (соединение 80) (более 10 мкМ) для ингибирования роста различных человеческих опухолевых клеток (см. табл. 4). Это указывает на то, что данное соединение не прямо ингибирует рост этих клеток в концентрациях, требуемых для ингибирования лиганд-зависимого фосфорилирования рецепторов и клеточной пролиферации. Таблица 4 Линия клеток Происхождение Ю5о (мкМ) LD50 (мкМ) НТ29 Карцинома прямой кишки А549 Карцинома легкого 9,5 NCI-H460 NSC карцинома легкого 8,9 SF767T Глиома 7,9 А431 Эпидермоидная карцинома 6,0 Кратко, результаты, представленные в табл. 4, были получены путем инкубирования клеток в течение 48 ч в полной среде для роста в присутствии серийных разведений 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламино-этил)амида. В конце периода роста определяли относительное количество клеток. Значения IC50 вычисляли как концентрацию соединения, которая ингибирует рост клеток на 50% относительно необработанных клеток. Значения LD50 вычисляли как концентрацию соединения, которая вызывает 50% сокращение количества клеток относительно количества клеток в начале эксперимента. Более релевантным, основанным на клетках анализом для оценки антиангиогенного потенциала 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амида (соединение 80) является in vitro анализ митогенеза с использованием эндотели-альных клеток пупочной вены человека (HUVECs) в качестве модельной системы пролиферации эндоте-лиальных клеток, критической для ангиогенного процесса. В этом анализе митогенный ответ, измеряемый как увеличение синтеза ДНК, индуцируется в сывороточно-голодавших HUVECs при добавлении VEGF или FGF. В этих клетках 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амид (соединение 80) ингибирует VEGF- и FGF-индуцированный митогенный ответ дозозависимым образом со значениями IC50 0,004 и 0,7 мкМ соответственно, когда соединение присутствовало на всем протяжении 48-часового анализа. Кратко, приведенные выше результаты были получены с использованием сывороточно-голодавших HUVECs, которые инкубировали с митогенными концентрациями VEGF (100 нг/мл) или FGF (30 нг/мл) - 34 - 008137 в присутствии серийных разведений 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амида (соединение 80) в течение 24 ч. Митоген-ный ответ в течение следующих 24 ч в присутствии лиганда и ингибитора количественно определяли путем измерения синтеза ДНК на основании включения бромдезоксиуридина в клеточную ДНК. В отдельных экспериментах соединение 80 ингибировало VEGF-зависимое фосфорилирование ERK 1/2 (р42/44МАР киназы), ранней по нисходящей мишени Flk-1/KDR, дозозависимым способом. Также было показано, что ингибирующее действие соединения 80 в этой системе длится долго с ингибировани-ем VEGF-зависимого фосфорилирования ERK 1/2 в течение 48 ч после удаления 5-(5-фтор-2-оксо-1,2-дигидроиндол-3 -илиденметил)-2,4-диметил-1Н-пиррол-3 -карбоновой кислоты (2-диэтиламино-этил)амида (соединение 80) из среды после короткого (2 ч) воздействия микромолярных концентраций этого соединения. VEGF признан важным фактором выживания эндотелиальных клеток. Так как 5-(5-фтор-2-оксо-1,2-дигидроиндол-3 -илиденметил)-2,4-диметил-1Н-пиррол-3 -карбоновой кислоты (2-диэтиламиноэтил)амид (соединение 80) ингибирует VEGF-зависимый митогенный ответ HUVECs, исследовали воздействие данного соединения на выживание HUVEC. В этих экспериментах в качестве показателей апоптоза использовали расщепление субстрата каспазы 3 поли-АЭР-рибозилполимеразы (PARP). HUVECs, культивированные в отсутствие сыворотки в течение 24 ч, имели значительные уровни расщепления PARP, определенные путем аккумуляции 23 кДа фрагмента PARP расщепления. Этому в значительной степени мешало добавление VEGF к клеточной среде, указывая на то, что VEGF действует как фактор выживания в данном анализе. Было показано, что 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амид (соединение 80) ингибирует KDR сигнализацию. Следовательно, 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амид (соединение 80) ингибирует VEGF-опосредованное HUVEC выживание дозозависимым образом. Таким образом, эти данные показывают, что соединение 80 индуцирует апоптоз в эндотелиальных клетках в культуре в присутствии VEGF. В. In vivo исследования эффективности 1. Эффективность против установленных опухолевых ксенотрансплантатов In vivo эффективность 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амида (соединение 80) исследовали в моделях подкожного (SC) ксенотрансплантата с использованием клеток человеческой опухоли, имплантированных в заднюю боковую область бестимусных мышей. После имплантации опухолям давали возможность развиться до размера 100-550 мм3 до начала перорального лечения данным соединением. Eжедневное пероральное введение соединения 80 вызывало дозозависимое ингибирование роста опухоли А431, когда лечение начинали после того, как опухоли вырастали до размера 400 мм3. Статистически значимое (Р <0,05) ингибирование роста опухоли наблюдалось при дозах 40 мг/кг/сутки (74% ингибирование) и 80 мг/кг/сутки (84% ингибирование) (см. табл. 6). В предварительных экспериментах более высокая (160 мг/кг/сутки) доза соединения не была более эффективной против установленных опухолей А431 по сравнению с дозой 80 мг/кг/сутки. Кроме того, мыши, которых лечили дозой 160 мг/кг/сутки, теряли вес, что указывает на то, что более высокие дозы также были не очень толерантными. Аналогичные результаты были получены в эксперименте, где опухолям А431 давали возможность достичь размера только 100 мм3 (см. табл. 4). В этом втором эксперименте полная регрессия опухолей произошла у шести из восьми животных при дозе 80 мг/кг/сутки в течение 21 суток. У этих шести животных опухоли не возобновляли рост в течение 110-дневного периода наблюдения после окончания лечения. У двух животных, у которых опухоли выросли до большого размера (2000-3000 мм3), эти опухоли регрессировали в ответ на второй раунд лечения соединением 80. Важно, что во всех экспериментах на эффективность 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амид (соединение 80) в количестве 80 мг/кг/сутки был в высокой степени толерантным, даже при непрерывном введении в течение более 100 суток. - 35 - 008137 Таблица 5 Начальный Соединение1 % ингибирования Р-значение объем опухоли (мг/кг/сутки) (сутки) (мм3) 400 84 (36) 0,001 74 (36) 0,003 51 (36) 0,130 100 93 (40) 0,002 75 (40) 0,015 61 (40) 0,059 Соединение 80. Кратко, результаты, представленные в табл. 5, были получены с использованием клеток А431 (0,5х106 клеток/мышь), которые имплантировали подкожно в заднюю боковую область бестимусных мышей. Eжедневное пероральное введение 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амида (соединение 80) в носителе на основе кремофора или носителя-контроля начинали, когда опухли достигали указанного среднего объема. Опухоли измеряли, используя штангенциркуль, и объем опухоли рассчитывали как произведение длина х ширина х высота. Р-значения вычисляли путем сравнения размера опухолей животных, которых лечили соединением 80 (n=8), с опухолями животных, которых лечили носителем (n=16), в последний день эксперимента, используя двусторонний t-критерий Стьюдента. Эффективность соединения 80 против установленных человеческих опухолей различного происхождения определяли, используя ксенотрансплантаты Colo205 (карцинома прямой кишки), SF763T (глиома) и NCl-H460 (немелкоклетоный рак легкого) (см. табл. 6). Эти эксперименты проводили с использованием 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амида (соединение 80), который вводили перорально в количестве 80 мг/кг/сутки - доза, которая была эффективной и в высокой степени толерантной. Таблица 6 Тип опухоли Начальный объем опухоли (мм3) % ингибирования (сутки) значение А4311 Эпидермоидная 100 93 (40) 0,002 А4311 Эпидермоидная 400 84 (36) 0,001 Со1о205 Толстой кишки 370 77 (54) 0,028 NCI-Н460 Легкого 300 61 (54) 0,003 SF763T Глиома 550 53 (30) 0,001 |-з-'-'-^ Данные из эксперимента, представленного в табл. 5 В вышеупомянутых экспериментах соединение 80 вводили один раз в сутки в количестве 80 мг/кг/сутки в носителе на основе кремофора сразу после того, как опухоли достигали указанного размера. Процент ингибирования по сравнению с контрольной группой, которую лечили носителем, вычисляли по окончании экспериментов. Р-значения вычисляли путем сравнения размеров опухолей животных, которых лечили соединением, с размерами опухолей тех животных, которых лечили носителем, используя двусторонний t-критерий Стьюдента. Хотя 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амид (соединение 80) ингибировал рост всех типов опухолей, приведенных в табл. 7, существовало различие в ответе разных моделей ксенотрансплантации. Конкретно, рост опухолей NCl-H640 и SF763T приостанавливался или значительно замедлялся, а опухоли СО1О205, такие как опухоли F431, регрессировали при лечении 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амидом. Чтобы дать молекулярное обоснование различия в ответе между моделями ксенотрансплантации, исследовали опухоли SF763. Поэтому опухоли SF763, которые были менее чувствительными к лечению 5-(5-фтор-2-оксо-1,2-дигидроиндол-3-илиденметил)-2,4-диметил-1 Н-пиррол-3-карбоновой кислоты (2 - 36 - 008137 диэтиламиноэтил)амидом, оценивали на молекулярном уровне с использованием иммуногистологиче-ских методов определения эффекта лечения этим соединением. Эти исследования вначале проводили на опухолях SF763T, так как опухоли этого типа сильно васкуляризированы микрососудами, которые интенсивно экспрессируют маркер эндотелиальных клеток CD31, и, следовательно, хорошо подходят для исследований плотности микрососудов (MVD) в опухолях. Иммуногистологическая оценка опухолей SF763 показала, что опухоли животных, которых лечили, имели пониженную MVD по сравнению с контролями, которые лечили носителем, что согласуется с антиангиогенным механизмом действия соединения 80; MVD составила 24,2±4,1 у животных, которых лечили соединением 80, по сравнению с 39,3±5,7 у животных, которых лечили только носителем. Как и ожидалось от остановки связанного с этим роста опухоли, ярко выраженное ингибирование пролиферации опухолевых клеток было явным в опухолях, которые лечили соединением 80. Эти опухоли имели половинное значение митотического индекса от опухолей, которые лечили носителем (данные не приведены). Воздействие соединения 80 на MVD и пролиферацию опухолевых клеток указывает на то, что соединение обладает сильным антиангиогенным и противоопухолевым эффектами, даже в условиях, когда опухоли не регрессируют. Способность соединения 80 ингибировать фосфорилирование PDGFR и последующую сигнализацию in vivo оценивали также на опухолях SF763, которые экспрессировали высокие уровни PDGFR8. Лечение опухолей SF763 соединением 80 сильно ингибировало фосфорилирование тирозина PDGFRP в установленных опухолях SF763T. Соединение 80 также снижало уровни фосфорилированной (активированной) фосфолипазы С гамма (PLC-y), непосредственного следующего по нисходящей индикатора активации PDGFR. Эти данные показывают, что пероральное введение соединения 80 оказывает прямое воздействие на активность мишени (PDGFR) в опухолях in vivo. На основании того, что способность соединения 80 ингибировать VEGF-зависимую сигнализацию в HUVECs in vitro была продолжительной (см. выше), эффективность соединения оценивали, когда это соединение вводили в модель опухоли Со1о205 не часто. Как показано в табл. 7, 80 мг/кг (91% ингибиро-вание) и 40 мг/кг/ (84% ингибирование) были эффективными при введении ежедневно, но не при введении два раза в неделю. Наоборот, более высокая доза соединения 80 (160 мг/кг) не ингибирует (52% ин-гибирование) рост укрепившихся опухолей Colo205 при введении два раза в неделю, что позволяет предположить, что это соединение может продемонстрировать эффективность при нечастом введении в более высоких дозах. Следует отметить, что режимы дозирования может определить специалист в данной области техники без большего, чем необходимо экспериментирования. Таблица 7 Доза (мг/кг) Частота % ингибирования Р-значение 160 Два раза в неделю 0,085 Один раз в неделю Ежедневно 0,039 Два раза в неделю Один раз в неделю Ежедневно 0,028 Два раза в неделю NS: не значимо (Р> 0,05) Кратко, результаты, представленные в табл. 7, были получены с использованием клеток Со1о205 (0,5x106 клеток/мышь), которые имплантировали подкожно в заднюю боковую область бестимусных мышей. Пероральное введение соединения 80 в соответствии с указанной схемой начинали, когда опухоли достигали размера 400 мм3. Опухоли измеряли штангенциркулем и объем опухолей вычисляли как произведение длина х ширина х высота. Р-значения вычисляли путем сравнения размера опухолей у животных, которых лечили соединением 80, с опухолями животных, которых лечили носителем, в последний день эксперимента, используя двусторонний t-критерий Стьюдента. 2. Эффективность соединения 80 в модели диссеминированного заболевания В дополнение к поддержанию непрерывного роста солидных первичных опухолей, ангиогенез также является существенным компонентом, поддерживающим развитие диссеминированного заболевания - 37 - 008137 в результате метастазирования из первичной опухоли. Воздействие соединения 80 на развитие диссеми-нированного заболевания исследовали в мышиной модели колонизации меланомы легкого B16-F1. В этой модели клетки B16-F1, инокулированные внутривенно через хвостовую вену бестимусных мышей, колонизируют легкие и образуют опухоли. Как показано в табл. 7, пероральное введение соединения 80 в количестве 80 мг/кг/сутки эффективно снижает содержание клеток B16-F1 в легком, как это оценено путем измерения общей массы легкого. Эти данные показывают, что соединение 80 может ингибировать диссеминированное заболевание in vivo. Таблица 8 Масса легкого (г) % ингибирования Р-значение Носитель 0,83±0,07 Соединение1 0,41±0,04 <0,001 Соединение 80 Кратко, результаты, представленные в табл. 8, были получены с использованием бестимусных мышей, которым инокулировали опухолевые клетки B16-F1 (5x105 клеток/мышь) через хвостовую вену. Мышей лечили ежедневно перорально вводимым соединением 80 в количестве 80 мг/кг/сутки (n=10) или носителем (n=18) в течение 24 суток после инокуляции опухолевых клеток. В конце периода лечения мышей умерщвляли и их легкие извлекали и взвешивали. Процент ингибирования вычисляли путем сравнения массы легких тех животных, которых лечили соединением 80, с массой легких животных, которых лечили только носителем. Р-значения определяли, используя двусторонний t-критерий Стьюдента. II. Ингибиторы СОХ-2 Способность ингибиторов СОХ-2 замедлять рост опухолей испытывали, используя легочную модель Льюиса, описанную ниже. Мышам делали подкожные инъекции в левую лапу (1х106 опухолевых клеток, суспендированных в 30% Матригель) и объем опухоли оценивали, используя плетизмометр, два раза в неделю в течение 30-60 дней. Кровь брали дважды в течение эксперимента по 24-часовому протоколу для оценки концентрации в плазме и общего воздействия анализом AUC (площади под кривой). Данные выражают как среднее ± SEM (стандартная ошибка среднего). Тесты Стьюдента и Манн-Витнея применяли для оценки различий между средними значениями, используя пакет программного обеспечения InStat. Целекоксиб, даваемый с пищей в дозах между 160-3200 м.д. (миллионные доли), удерживал рост этих опухолей. Ингибирующее воздействие целекоксиба было дозозависимым и находилось в пределах от 4 до 85% по сравнению с контрольными опухолями. Анализ легочного метастазирования выполняли у всех животных путем подсчета метастазов в стереомикроскопе и путем гистохимического анализа соседних участков легкого. Целекок-сиб не воздействует на легочное метастазирование в более низкой дозе 160 м.д., однако поверхностное метастазирование снижались более чем на 50%, когда его вводили в дозах 480-3200 м.д. Кроме того, гис-топатологический анализ выявил, что целекоксиб дозозависимым образом уменьшает размер метастатических поражений в легком. 2. Модель НТ-20 Мышам делали подкожные инъекции в левую лапу (1 х106 опухолевых клеток, суспендированных в 30% Матригель), и объем опухоли оценивали, используя плетизмометр, дважды в неделю в течение 3060 дней. Имплантация раковых клеток толстой кишки человека (НТ-29) голым мышам порождает опухоли, которые достигают 0,6-2 мл за 30-50 суток. Кровь берут два раза в ходе эксперимента по 24-часовому протоколу для оценки концентрации в плазме и общего воздействия анализом AUC. Данные выражают как среднее ± SEM. Тесты Стьюдента и Манн-Витнея использовали для оценки различий между средними значениями, используя пакет программного обеспечения InStat. А. Мышей, инъецированных раковыми клетками НТ-29, лечили цитоксином внутрибрюшинно в дозах 50 мг/кг на день 5, 7 и 9 в присутствии или в отсутствие целекоксиба в пище. Эффективность обоих агентов оценивали путем измерения объема опухоли. Лечение с использованием родственного целекок-сибу ингибитора СОХ-2 (SC-58236) уменьшало объем опухоли на 89%. В таком же анализе индометацин, который вводили в почти максимально толерантной дозе 2 мг/кг/сутки с питьевой водой, ингибировал образование опухоли на 77%. Более того, селективный ингибитор СОХ-2 полностью ингибировал образование легочных метастазов, тогда как неселективный NSAID (нестероидное противовоспалительное средство) индометацин был неэффективным. Результаты этих исследований показывают, что целекок-сиб, вводимый с пищей имеющим опухоль мышам, может задерживать рост опухолей и метастазирова-ние при введении в качестве единственного терапевтического средства. Кроме того, положительный результат наблюдался, когда целекоксиб вводят в комбинации с цитотоксическим агентом, таким как цик-лофосфамид. Б. Во втором анализе мышей, которым инъекцировали клетки рака толстой кишки НТ-29, лечили целекоксибом (10, 40 или 160 м.д.) в пище, начиная с дня 10. Наблюдали приблизительно дозозависимый эффект (табл. 9). - 38 - 008137 Таблица 9. Ингибирование целекоксибом карциномы толстой кишки человека НТ-29 Дни Носитель 10 м. д. 40 м. д. 160 м. д. 0,114 0,124 0,125 0,120 0,25 0,25 0,19 0,14 Л AR П 07 \i,t- i \J,i-1 0,79 0,57 0,4 0,3 1,38 0,89 0,68 0,49 1,9 1.49 1,04 0,8 Объем (мл) III. In vivo анализы с использованием протеинкиназ в комбинации с селективным ингибитором циклооксигеназы-2 для лечения рака. Способность ингибитора протеинкиназы сдерживать рост опухолей в комбинации с селективным ингибитором СОХ-2 можно испытывать в модели ксенотрансплантата 1483, описанной ниже. Ксенотрансплантат 1483 представляет собой животный ксенотрансплантат, который моделирует эпителиальный рак человека, экспрессирующий циклооксигеназу-2 (СОХ-2) в опухолевых клетках и в сосудистой сети. Модель ксенотрансплантата человеческой опухоли сквамозно-клеточной карциномы головы и шеи голых мышей (клеточная линия 1483), которая экспрессирует СОХ-2 в опухолевых клетках и в сосудистой сети, аналогична человеческому эпителиальному раку. Авторы полагают, что эта модель представляет собой человеческий эпителиальный рак и должна быть хорошей моделью для корреляции эффективности противораковых лекарственных средств, включая СОХ-2 ингибиторы, с эффективностью у людей. Материалы и методы: Культура клеток: Клетки 1483 сквамозно-клеточной карциномы шеи и головы человека (HNSCC) хранят в замороженных виалах, содержащих 3х106 клеток, 90% сыворотку пода коровы (FBS) и 10% диметилсульфоксид (ДМСО). Берут замороженный виал и быстро размораживают при 37°С и помещают во флакон Т-162 см2 (Corning), содержащий среду D-MEM/F12 (GibcoBRL) с 15 мМ HEPES буфером, L-глутамином, пири-доксина гидрохлоридом и 10% FBS. Клетки выращивают в инкубаторе с 5% CO2 и температурой 37°С. Среду меняют каждый день и клетки пассируют при конфлюэнтности 80-90%. Для пассирования клеток флакон промывают 10 мл фосфатного буферного солевого раствора (PBS), отсасывают и добавляют 2 мл трипсин/EDTA (0,25%/1 мМ, GibcoBRL), помещают обратно в инкубатор, через 5 мин клетки отсоединяются. Добавляют 8 мл указанной выше среды к промывке флакона и переносят в стерильную центрифужную пробирку емкостью 50 мл. Добавляют 30 мл среды, смешивают, подсчитывают клетки, используя гемацитометр, и пассируют клетки в Т-162 см2, вмещающий 3-4х106 клеток. Животная модель 1483: Меняют среду за 24 ч до сбора клеток 1483 перед инъекцированием голым мышам. Трипсинизиру-ют клетки 1483 как описано выше в разделе "Культура клеток". Подсчитывают клетки и определяют количество клеток. Центрифугируют клетки при 1000 об./мин в течение 5 мин при комнатной температуре. Ресуспендируют гранулы клеток и объединяют их (если центрифужных пробирок емкостью 50 мл несколько) в одну центрифужную пробирку емкостью 50 мл с буферным солевым раствором Хэнкса (HBSS, GibcoBRL) и центрифугируют как раньше. Следует иметь приблизительно на 25% клеток больше, чем реально требуется для инъекции, чтобы иметь лишние клетки. Если делают инъекции 72 мышам и имеется 100х106 клеток, все эти клетки готовят для инъекции мышам. Инъецируют клетки 1483 в количестве 1 х106 клеток в 0,03 мл/мышь. Инъецируют клетки с 30% Матригелем (Collaborative Biomedical Products) и 70% HBSS. Ресуспен-дируют объединенные гранулы с 2,1 мл (70%) холодного HBSS, затем добавляют 0,9 мл (30%) размороженного разжиженного холодного Матригеля и хорошо смешивают на льду. Хранят этот клеточный препарат на льду все время до инъекции мышам. В этих исследованиях используют самцов голых мышей в возрасте 4-6 недель (Harlen). Анестезируют мышей, используя СО2/О2 газ, а затем делают им инъекцию в середину правой задней лапы, используя туберкулиновый шприц на 0,5 см3 (Beckerson & Dickerson). Мышей взвешивают для определения массы тела на день инъекции (день 0) - исходной массы на начало исследования. Взвешивают мышей в день 7 и измеряют правую заднюю лапу для определения объема опухоли на лапе, используя плетизмометр (Stoelting Co.). Плетизмометр представляет собой прибор для измерения объема лапы путем вытеснения воды. Измеряют несколько левых неинъецированных лап и среднее для фонового измерения для вычитания из правой лапы, имеющей опухоль. Животным дают тестируемые соединения в жевательной пище, когда размер опухолей достигает - 39 - 008137 100-200 мкл, и продолжают давать пищу с соединением в течение всего исследования. Некоторым мышам дают только ингибитор протеинкиназы или селективный ингибитор циклооксигеназы-2. Некоторым мышам дают и ингибитор протеинкиназы, и селективный ингибитор циклооксигеназы-2 ежедневно в соответствующей дозе, определенной по результатам in vitro анализа. Взвешивают и измеряют мышей на протяжении всего исследования в дни 7, 10, 14, 17, 21, 24 и 28. Лечение животных соединением можно начать в день 0 (профилактическое) или сразу после того, как только образуется опухоль, приблизительно в день 7 (терапевтическое). Примерно на день 30 мыши, которым давали носитель (контроль) будут иметь большие опухоли (примерно 1,0-1,5 мл) и начнут терять вес, и в это время животных, которым давали носитель, умерщвляют. Если в подвергнутых лечению группах ингибирование заметно, и их состояние здоровья хорошее, умерщвляют половину этой группы лечения, а половину подвергнутой лечению группы оставляют в живых для определения задержки роста опухоли. Специалисту в данной области очевидно, что данное изобретение пригодно для выполнения задач и достижения целей и получения преимуществ, упомянутых, так же как и неотъемлемых, в данном описании изобретения. Молекулярные комплексы и способы, процедуры, обработки, молекулы, конкретные соединения, описанные в данной заявке, на данный момент являются иллюстративными и не предназначены ограничивать объем изобретения. Изменения и другие применения, которые будут иметь место у специалистов в данной области, которые охвачены замыслом данного изобретения, определены объемом формулы изобретения. Специалисту в данной области очевидно, что можно вносить модификации в данное изобретение, раскрытое в данной заявке, не выходя за рамки объема и замысла данного изобретения. Все патенты и публикации, упомянутые в материалах заявки, характеризуют уровень техники в области, к которой относится данное изобретение. Все патенты и публикации включены в данное описание изобретения ссылкой в такой же степени, как если бы каждая отдельная публикация была отдельно и конкретно указана, как включенная в данное описание изобретения ссылкой. Способ лечения или предупреждения рака, при котором млекопитающему, нуждающемуся в таком лечении, вводят терапевтически эффективное количество ингибитора протеинкиназы в комбинации с селективным ингибитором циклооксигеназы-2, где указанный ингибитор протеинкиназы представляет собой 5-[фтор-2-оксо-1,2-дигидроиндол-3-илиденметил]-2,4-диметил-1Н-пиррол-3-карбоновой кислоты (2-диэтиламиноэтил)амид формулы или его фармацевтически приемлемую соль, и указанный селективный ингибитор циклооксигеназы-2 представляет собой 4-[5-(4-метилфенил)-3-(трифторметил)-1Н-пиразол-1-ил]фенилсульфонамид (целе-коксиб) формулы ФОРМУЛА ИЗОБРЕТЕНИЯ или его фармацевтически приемлемую соль. Евразийская патентная организация, ЕАПВ Россия, 109012, Москва, Малый Черкасский пер., 2/6 - 40 - |