|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea000006198b*\id |
|
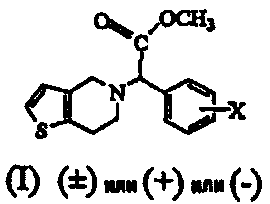
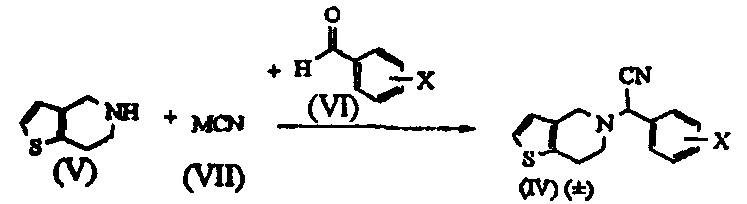
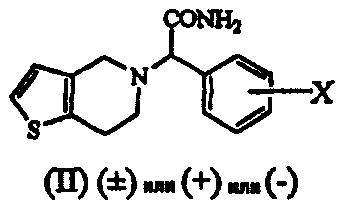
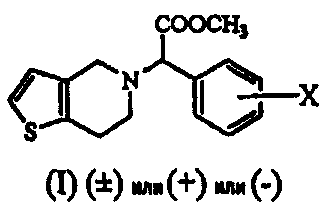
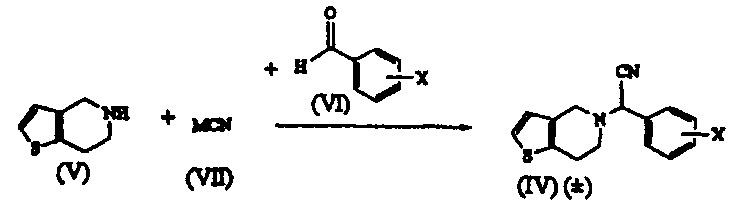
Термины запроса в документе Реферат 1. Способ получения соединения формулы (I) где Х обозначает атом водорода, фтора, хлора, брома или йода, предпочтительно 2-хлор, который включает: i) взаимодействие соединения формулы (V) или его соли с цианидом общей формулы (VII), где М обозначает щелочной металл, триметилсилил, Cu или водород, и последующее добавление соединения общей формулы (VI), где Х определен ранее, с получением рацемического соединения общей формулы (IV), где Х определен ранее; ii) взаимодействие соединения общей формулы (IV) в (+) форме или в любой из его оптически активных (+) или (-) форм с кислотными или щелочными реагентами с получением соединения формулы (II) или его соли с сохранением конфигурации; iii) взаимодействие соединения общей формулы (II) или в (+) форме, или в любой его оптически активной (+) или (-) форме с кислотными реагентами в присутствии метанола с получением соединения формулы (I) или его соли с сохранением конфигурации; iv) наконец, разделение (+) соединения формулы (I) или его соли на его оптические изомеры. 2. Способ по п.1, в котором необязательно после разделения рацемического соединения формулы (IV) на его оптические изомеры или же трансформирования рацемического соединения или его оптических изомеров на его соли имеет место высвобождение соли из соединения. 3. Способ по п.1, в котором необязательно после разделения рацемического соединения формулы (II) на его оптические изомеры или же трансформирования рацемического соединения или его оптических изомеров на его соли имеет место высвобождение соли из соединения. 4. Способ по п.1, в котором соединение формулы (II), (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамид, или его оптически активные изомеры, или его соли, где Х обозначает атом галогена, такой как хлор-, бром-, иодзаместитель, более предпочтительно 2-хлор, получают по способу, включающему i) обработку соединения формулы (IV), или его соли, или любой его оптически активной формы щелочными реагентами, когда исходный материал является рацемическим или кислотным катализатором, когда исходный материал является оптически активным, в присутствии подходящего растворителя; ii) образование соли рацемического соединения формулы (II), которую разделяют на ее соответствующие оптически чистые (+) и (-) формы. 5. Способ по п.4, который включает получение соединения формулы (II), отличающийся тем, что используемый щелочной реагент представляет собой гидроксид лития, гидроксид калия, гидроксид натрия и трет-бутилат калия. 6. Способ по п.4, который включает получение соединения формулы (II), где используемый кислотный реагент выбирают из группы, состоящей из уксусной кислоты, п-толуолсульфоновой кислоты, трифторуксусной кислоты, хлоруксусной кислоты, перхлорной кислоты, муравьиной кислоты или минеральных кислот, таких как безводные спиртовые галогенидводороды или водная хлористо-водородная кислота, серная кислота, HBr или их смеси. 7. Способ по п.4, который включает получение соединения формулы (II), где используемый растворитель представляет собой воду, (С 1 -С 4 )спирт, ацетон, уксусную кислоту, диметилформамид, ТГФ, ДМСО, диоксан, ДМЭ и т.п. или их смеси, предпочтительно смесь воды, метанола или трет-BuOH. 8. Способ по п.4, в котором соединение формулы (II), (+)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено-[3,2-с]пирид-5-ил)ацетамид, разделяют на его (+) и (-) формы с использованием 1-(R)- или 1-(S)-камфорсульфоновой-10-кислоты, винной кислоты, в особенности 1-(R)- или 1-(S)-камфорсульфоновой-10-кислоты, в присутствии растворителя, такого как вода, ацетон, этилацетат или их смесь, и используемая соль может находиться в молярном соотношении 0,5-1,1 эквивалентов. 9. Способ по п.4, в котором соединение формулы (II), (+)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено-[3,2-с]пирид-5-ил)ацетамид, превращают в соответствующую соль, причем заявленная соль представляет собой D-камфорсульфонат, L-камфорсульфонат, D-винную кислоту, L-винную кислоту или кислый сульфат. 10. Способ по п.1, в котором указанное соединение формулы (I) представляет собой метил(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетат, или его оптически активные изомеры, или его соли, где Х обозначает атом галогена, такой как хлор-, бром-, иодзаместитель, более предпочтительно 2-хлор, причем указанный способ включает i) обработку соединения формулы (II), или его соли, или любой его оптически активной формы кислотными реагентами в присутствии подходящего растворителя; ii) образование соли рацемического соединения формулы (I), которую разделяют на ее соответствующие оптически чистые (+) и (-) формы. 11. Способ по п.10, в котором указанный используемый кислотный реагент выбирают из концентрированной серной кислоты в количестве от 1 до 50 эквивалентов, предпочтительно добавление проводят при 0-5шС или при температуре флегмы используемого растворителя. 12. Способ по п.10, в котором используемый растворитель представляет собой этанол, который берут в количестве от 3 до 30 объемов, вместе с другими сорастворителями, такими как толуол, ДМСО, ксилол и т.п., или их смесями. 13. Способ по п.10, в котором продолжительность реакции составляет от 4 ч до 4 дней. 14. Способ по п.10, в котором указанную реакцию проводят при температуре от 40 до 140шС. 15. Способ по п.10, в котором соединение формулы (I) разделяют с использованием 1-(R)- или 1-(S)-камфорсульфоновой-10-кислоты, (R)- или (S)-винной кислоты, в особенности 1-(R)- или 1-(S)-камфорсульфоновой-10-кислоты, в присутствии растворителя, такого как вода, ацетон, этилацетат или их смесь. 16. Способ по п.15, в котором смесь, содержащую варьирующие соотношения двух энантиомеров 17. Способ получения соединения формулы (IV) и его солей где Х обозначает атом галогена, такой как хлор-, бром-, иодзаместитель, в особенности Х обозначает 2-хлор, а также его солей, который включает: i) взаимодействие соединения общей формулы (V) или его кислотно-аддитивной соли с цианидом общей формулы (VII), в которой М означает щелочной металл, ТМС, Cu или водород, с последующим взаимодействием с галогенбензальдегидом формулы (VI), где Х обозначает атом галогена, продукт которого разделяют на его оптически активные изомеры, и, при необходимости, высвобождение из их солей или превращение в их соли; или ii) взаимодействие галогенбензальдегида общей формулы (VI), в которой Х обозначает атом галогена, с цианидом общей формулы (VII), где М определен ранее, с последующей реакцией in situ с соединением общей формулы (V) или его кислотно-аддитивной солью; или iii) взаимодействие галогенбензальдегида общей формулы (VI), в которой Х обозначает атом галогена, с кислым сульфитом общей формулы (VIII), где М определен ранее, и затем с цианидом общей формулы (VII), где М определен ранее, с последующей реакцией in situ с соединением общей формулы (V) или его кислотно-аддитивной солью; и, наконец, разделение полученного таким образом соединения формулы (IV) или его соли на оптически чистую (+) и (-) форму соединения формулы (IV) или его соли. 18. Способ по п.17, в котором указанное взаимодействие проводят при температуре от 0 до 100шС, а в случае использования газообразного HCN температуру реакции понижают дополнительно до -30шС. 19. Способ по п.17, в котором указанное взаимодействие проводят в водной или неводной среде, содержащей кислоты, такие как уксусная кислота, пропионовая кислота, метанол и HCl/MeOH. 20. Способ по п.17, в котором используют кислотный катализатор, такой как сухая хлористо-водородная кислота, серная кислота, п-толуолсульфоновая кислота или уксусная кислота. 21. Способ по п.17, в котором соединение формулы (IV), (+)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрил, разделяют на его (-) и (+) формы с использованием 1-(R)- или 1-(S)-камфорсульфоновой-10-кислоты, винной кислоты, в особенности 1-(R)- или 1-(S)-камфорсульфоновой-10-кислоты, в присутствии ацетона, этилацетата или их смеси и воды. 22. Способ по п.17, в котором соединение формулы (IV), (+)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрил, в (+), или (-), или (+) формах превращают в соответствующую соль, причем заявленная соль представляет собой D-камфорсульфонат, L-камфорсульфонат, D-винную кислоту, L-винную кислоту или кислый сульфат. 23. Способ по п.1, в котором рацемизацию (-) изомера соединения формулы (II) проводят в смеси, которая может содержать (+) стереоизомер, для образования (+) соединения с использованием основания, такого как ЛДА, KОН, NaOH, K-t-BuOH, NaOMe, NaH, KH, в подходящем растворителе. 24. Способ по п.1, в котором рацемизацию (-) изомера соединения формулы (I) проводят в смеси, которая может содержать (+) стереоизомер, для образования (+) соединения с использованием основания, такого как ЛДА, NaOMe, NaH, KH, в подходящем растворителе. 25. Способ по п.16, в котором разделение проводят в присутствии ацетона в качестве растворителя, взятого в количестве от 5 до 10 объемов, который может содержать воду вплоть до 5%. 26. Способ по п.16, в котором указанный подходящий хиральный агент используют в молярном соотношении 1:1. 27. Способ по п.26, в котором подходящая температура составляет от 0шС до температуры флегмы растворителя.
Полный текст патента (57) Реферат / Формула: где Х обозначает атом водорода, фтора, хлора, брома или йода, предпочтительно 2-хлор, который включает: i) взаимодействие соединения формулы (V) или его соли с цианидом общей формулы (VII), где М обозначает щелочной металл, триметилсилил, Cu или водород, и последующее добавление соединения общей формулы (VI), где Х определен ранее, с получением рацемического соединения общей формулы (IV), где Х определен ранее; ii) взаимодействие соединения общей формулы (IV) в (+) форме или в любой из его оптически активных (+) или (-) форм с кислотными или щелочными реагентами с получением соединения формулы (II) или его соли с сохранением конфигурации; iii) взаимодействие соединения общей формулы (II) или в (+) форме, или в любой его оптически активной (+) или (-) форме с кислотными реагентами в присутствии метанола с получением соединения формулы (I) или его соли с сохранением конфигурации; iv) наконец, разделение (+) соединения формулы (I) или его соли на его оптические изомеры. 2. Способ по п.1, в котором необязательно после разделения рацемического соединения формулы (IV) на его оптические изомеры или же трансформирования рацемического соединения или его оптических изомеров на его соли имеет место высвобождение соли из соединения. 3. Способ по п.1, в котором необязательно после разделения рацемического соединения формулы (II) на его оптические изомеры или же трансформирования рацемического соединения или его оптических изомеров на его соли имеет место высвобождение соли из соединения. 4. Способ по п.1, в котором соединение формулы (II), (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамид, или его оптически активные изомеры, или его соли, где Х обозначает атом галогена, такой как хлор-, бром-, иодзаместитель, более предпочтительно 2-хлор, получают по способу, включающему i) обработку соединения формулы (IV), или его соли, или любой его оптически активной формы щелочными реагентами, когда исходный материал является рацемическим или кислотным катализатором, когда исходный материал является оптически активным, в присутствии подходящего растворителя; ii) образование соли рацемического соединения формулы (II), которую разделяют на ее соответствующие оптически чистые (+) и (-) формы. 5. Способ по п.4, который включает получение соединения формулы (II), отличающийся тем, что используемый щелочной реагент представляет собой гидроксид лития, гидроксид калия, гидроксид натрия и трет-бутилат калия. 6. Способ по п.4, который включает получение соединения формулы (II), где используемый кислотный реагент выбирают из группы, состоящей из уксусной кислоты, п-толуолсульфоновой кислоты, трифторуксусной кислоты, хлоруксусной кислоты, перхлорной кислоты, муравьиной кислоты или минеральных кислот, таких как безводные спиртовые галогенидводороды или водная хлористо-водородная кислота, серная кислота, HBr или их смеси. 7. Способ по п.4, который включает получение соединения формулы (II), где используемый растворитель представляет собой воду, (С 1 -С 4 )спирт, ацетон, уксусную кислоту, диметилформамид, ТГФ, ДМСО, диоксан, ДМЭ и т.п. или их смеси, предпочтительно смесь воды, метанола или трет-BuOH. 8. Способ по п.4, в котором соединение формулы (II), (+)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено-[3,2-с]пирид-5-ил)ацетамид, разделяют на его (+) и (-) формы с использованием 1-(R)- или 1-(S)-камфорсульфоновой-10-кислоты, винной кислоты, в особенности 1-(R)- или 1-(S)-камфорсульфоновой-10-кислоты, в присутствии растворителя, такого как вода, ацетон, этилацетат или их смесь, и используемая соль может находиться в молярном соотношении 0,5-1,1 эквивалентов. 9. Способ по п.4, в котором соединение формулы (II), (+)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено-[3,2-с]пирид-5-ил)ацетамид, превращают в соответствующую соль, причем заявленная соль представляет собой D-камфорсульфонат, L-камфорсульфонат, D-винную кислоту, L-винную кислоту или кислый сульфат. 10. Способ по п.1, в котором указанное соединение формулы (I) представляет собой метил(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетат, или его оптически активные изомеры, или его соли, где Х обозначает атом галогена, такой как хлор-, бром-, иодзаместитель, более предпочтительно 2-хлор, причем указанный способ включает i) обработку соединения формулы (II), или его соли, или любой его оптически активной формы кислотными реагентами в присутствии подходящего растворителя; ii) образование соли рацемического соединения формулы (I), которую разделяют на ее соответствующие оптически чистые (+) и (-) формы. 11. Способ по п.10, в котором указанный используемый кислотный реагент выбирают из концентрированной серной кислоты в количестве от 1 до 50 эквивалентов, предпочтительно добавление проводят при 0-5шС или при температуре флегмы используемого растворителя. 12. Способ по п.10, в котором используемый растворитель представляет собой этанол, который берут в количестве от 3 до 30 объемов, вместе с другими сорастворителями, такими как толуол, ДМСО, ксилол и т.п., или их смесями. 13. Способ по п.10, в котором продолжительность реакции составляет от 4 ч до 4 дней. 14. Способ по п.10, в котором указанную реакцию проводят при температуре от 40 до 140шС. 15. Способ по п.10, в котором соединение формулы (I) разделяют с использованием 1-(R)- или 1-(S)-камфорсульфоновой-10-кислоты, (R)- или (S)-винной кислоты, в особенности 1-(R)- или 1-(S)-камфорсульфоновой-10-кислоты, в присутствии растворителя, такого как вода, ацетон, этилацетат или их смесь. 16. Способ по п.15, в котором смесь, содержащую варьирующие соотношения двух энантиомеров 17. Способ получения соединения формулы (IV) и его солей где Х обозначает атом галогена, такой как хлор-, бром-, иодзаместитель, в особенности Х обозначает 2-хлор, а также его солей, который включает: i) взаимодействие соединения общей формулы (V) или его кислотно-аддитивной соли с цианидом общей формулы (VII), в которой М означает щелочной металл, ТМС, Cu или водород, с последующим взаимодействием с галогенбензальдегидом формулы (VI), где Х обозначает атом галогена, продукт которого разделяют на его оптически активные изомеры, и, при необходимости, высвобождение из их солей или превращение в их соли; или ii) взаимодействие галогенбензальдегида общей формулы (VI), в которой Х обозначает атом галогена, с цианидом общей формулы (VII), где М определен ранее, с последующей реакцией in situ с соединением общей формулы (V) или его кислотно-аддитивной солью; или iii) взаимодействие галогенбензальдегида общей формулы (VI), в которой Х обозначает атом галогена, с кислым сульфитом общей формулы (VIII), где М определен ранее, и затем с цианидом общей формулы (VII), где М определен ранее, с последующей реакцией in situ с соединением общей формулы (V) или его кислотно-аддитивной солью; и, наконец, разделение полученного таким образом соединения формулы (IV) или его соли на оптически чистую (+) и (-) форму соединения формулы (IV) или его соли. 18. Способ по п.17, в котором указанное взаимодействие проводят при температуре от 0 до 100шС, а в случае использования газообразного HCN температуру реакции понижают дополнительно до -30шС. 19. Способ по п.17, в котором указанное взаимодействие проводят в водной или неводной среде, содержащей кислоты, такие как уксусная кислота, пропионовая кислота, метанол и HCl/MeOH. 20. Способ по п.17, в котором используют кислотный катализатор, такой как сухая хлористо-водородная кислота, серная кислота, п-толуолсульфоновая кислота или уксусная кислота. 21. Способ по п.17, в котором соединение формулы (IV), (+)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрил, разделяют на его (-) и (+) формы с использованием 1-(R)- или 1-(S)-камфорсульфоновой-10-кислоты, винной кислоты, в особенности 1-(R)- или 1-(S)-камфорсульфоновой-10-кислоты, в присутствии ацетона, этилацетата или их смеси и воды. 22. Способ по п.17, в котором соединение формулы (IV), (+)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрил, в (+), или (-), или (+) формах превращают в соответствующую соль, причем заявленная соль представляет собой D-камфорсульфонат, L-камфорсульфонат, D-винную кислоту, L-винную кислоту или кислый сульфат. 23. Способ по п.1, в котором рацемизацию (-) изомера соединения формулы (II) проводят в смеси, которая может содержать (+) стереоизомер, для образования (+) соединения с использованием основания, такого как ЛДА, KОН, NaOH, K-t-BuOH, NaOMe, NaH, KH, в подходящем растворителе. 24. Способ по п.1, в котором рацемизацию (-) изомера соединения формулы (I) проводят в смеси, которая может содержать (+) стереоизомер, для образования (+) соединения с использованием основания, такого как ЛДА, NaOMe, NaH, KH, в подходящем растворителе. 25. Способ по п.16, в котором разделение проводят в присутствии ацетона в качестве растворителя, взятого в количестве от 5 до 10 объемов, который может содержать воду вплоть до 5%. 26. Способ по п.16, в котором указанный подходящий хиральный агент используют в молярном соотношении 1:1. 27. Способ по п.26, в котором подходящая температура составляет от 0шС до температуры флегмы растворителя.
006198 Область изобретения Настоящее изобретение относится к способу получения производных тиено[3,2-с]пиридина общей формулы (I) либо в рацемической, либо в оптически активной (+) или (-) формах и их солей, где Х обозначает заместитель в бензольном кольце и представляет собой атом водорода или галогена, такой как фтор, хлор, бром или йод. (1)(±)или(+)илн(-) Предпочтительно Х обозначает 2-хлор. Настоящее изобретение также описывает способ получения соединений общей формулы (II) или в рацемической, или в оптически активной (+) или (-) формах и их солей, где Х обозначает заместитель в бензольном кольце и представляет собой атом водорода или галогена, такой как фтор, хлор, бром или йод. (П)(±)или(+)или(.) Предпочтительно Х обозначает 2-хлор. Указанные соединения представляют собой полезные промежуточные соединения для получения соединения общей формулы (I). Соединения, представленные формулами (I) и (II), имеют один асимметричный атом углерода и, следовательно, для получения оптически активных соединений формулы (I) или (II) имеется возможность либо разделить рацемический промежуточный продукт/готовый продукт, либо использовать оптически активный промежуточный продукт. Предпосылки создания изобретения Производные тиено[3,2-с]пиридина, описанные в FR 2215948, FR 2530247 и FR 2612929, представляют собой фармакологически активные соединения и обладают значительными антиагрегирующими и антитромботическими свойствами. Одним таким примером является "Qopidogrel" (клопидогрел), сложный метиловый эфир (S)-(+)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пиридин-5-ил)уксусной кислоты, и его фармацевтически приемлемые соли. Позже было обнаружено, что биологическая активность присуща лишь ^)-(+)-стереоизомеру (патент США 4847265). Поскольку "клопидогрел-основание" представляет собой маслянистую жидкость, то для того, чтобы получить подходящую композицию, основание превращают в фармацевтически приемлемую соль. Приемлемые соли "клопидогрела-основания" могут быть получены с таурохолатом, гидробромидом и серной кислотой. Описание уровня техники Известные методы синтеза соединений общей формулы (I) (US 4529596, GB 0420706 и GB 0466569) включают использование производных а-галогенфенилуксусной кислоты, которые по своей природе вызывают слезоточивость и раздражение. Способы синтеза таких соединений включают множество стадий, а также имеют другие недостатки, связанные с используемыми химическими веществами/реагентами, которые обычно трудны для работы с ними, для масштабного производства и оказывают неблагоприятное воздействие как с точки зрения здоровья человека, так и с точки зрения окружающей среды. Кроме того, общий выход продукта в таких процессах варьирует от слабого до среднего. Имеющиеся в литературе различные другие стратегии синтеза включают использование дорогостоящих или опасных химических веществ, которые, тем не менее, не улучшают в значительной мере выход желательного продукта. В последнее время в качестве стандарта для изучения метаболизма был получен с использованием ортометаллирования/хлорирования производных бензойной кислоты радиоактивно меченный (бензол-U-13С), рацемический (±)-клопидогрел, с общим выходом 7% (Chem. Abst., 133:281711, 2000). Различные другие стратегии описаны в приведенных ниже документах: WO 98/51681, WO 98/51682, WO 98/51689, WO 99/18110, US 4876362, US 5036156, US 5132435, US 5139170, US 5204469 и US 6080875. В последнее время в заявке на патент (WO 99/65915) был описан новый полиморф бисульфата кло-пидогрела (обозначенный как форма II), который имеет точку плавления 176±3°С. Указывается также, что соединение, описанное в более раннем патенте США (US 4847265), имеет иную точку плавления, примерно 184±3°С (в настоящее время обозначенное как форма I). Было показано, что оба полиморфа имеют отличные и характерные ДРЛ и ИК спектры. Следовательно, настоящее изобретение относится также к обеспечению недорогого и коммерчески жизнеспособного способа получения соединений формулы (I) с хорошими выходами. - 1 - 006198 Цели изобретения Основной целью настоящего изобретения является обеспечение нового способа получения производных тиено[3,2-с]пиридина, представленных общей формулой (I), или в рацемической, или в оптически активной (+) или (-) формах и их солей, где Х обозначает атом водорода или атом галогена, такой как фтор, хлор, бром или йод. (I) (±)или(+)нлн(-) Другой целью настоящего изобретения является разработка нового коммерчески жизнеспособного способа получения производных тиено[3,2-с]пиридина, представленных общей формулой (I), или в рацемической, или в оптически активной (+) или (-) формах и их солей. Конкретной целью настоящего изобретения является разработка нового способа производства сложного метилового эфира (S)-(+)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пиридин-5-ил)уксусной кислоты в виде бисульфатной соли, то есть клопидогрела-бисульфата, где X обозначает 2-хлор-заместитель. сссо С1' (I) (+) Предпочтительной целью настоящего изобретения является обеспечение нового коммерчески жизнеспособного способа производства клопидогрела-бисульфата. Другой важной целью настоящего изобретения является разработка нового коммерчески жизнеспособного способа получения полиморфной формы (I) клопидогрела, имеющей точку плавления 184±3°С. Еще одной целью настоящего изобретения является возвращение в цикл в рамках нового способа левовращающего изомера клопидогрела либо варьирующей смеси (+) и (-) стереоизомеров с целью получения (+)-клопидогрела-бисульфата. Другая цель настоящего изобретения включает обеспечение способа получения соединения (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида формулы (II) или в рацемической, или в оптически активных (+) или (-) формах и их солей. CONH2 (П)(±)или(+)или(-) Другой целью настоящего изобретения является разработка способа получения соединения формулы (II), где Х обозначает 2-хлор, в рацемической, а также оптически активной (+) или (-) формах, имеющих приемлемую химическую и хиральную чистоту, а также их солей. Правовращающий изомер соединения формулы (II) соответствующей чистоты или его соли представляют собой полезные промежуточные соединения для синтеза (+)-клопидогрела-бисульфата. Еще одной целью настоящего изобретения является разработка нового способа превращения (R)-(-)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида или его смеси с варьирующими незначительными количествами его оптического антипода почти в 1:1 смесь (+) и (-) изомера. Целью настоящего изобретения является также разработка способа получения соединения формулы (III), (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)уксусной кислоты, в рацемической (±) или либо в оптически активной (+), либо в (-) форме и их солей. Еще одной целью настоящего изобретения является разработка нового способа превращения (R)-(-)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)уксусной кислоты в смеси в (S)-(+) стереоизомер. Еще одной целью настоящего изобретения является разработка способа получения соединения формулы (IV), (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрила, и его солей. Еще одной целью настоящего изобретения является разработка нового способа превращения (R)-(-)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрила или его смеси с варьирующими незначительными количествами его оптического антипода почти в 1:1 смесь (+) и (-) изомера. Приведенный в настоящем описании способ обеспечивает простой и альтернативный метод получения соединений общей формулы (I), в особенности ^)-(+)-клопидогрела-бисульфата, полиморфной формы I. - 2 - 006198 Краткое описание сущности изобретения Приведенные выше и другие цели настоящего изобретения достигаются за счет способа согласно настоящему изобретению при использовании соединений формулы (II) CONH2 (П)(±)или(+)или(-) или их солей в или рацемической или оптически активной (+) или (-) формах, как показано на схеме 1. Настоящее изобретение необязательно предлагает способ разделения (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида на оптически активные (+) или (-) формы, которые могут использоваться для получения (+)-клопидогрела-бисульфата. Настоящее изобретение необязательно относится также к способу разделения (±)-2-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)уксусной кислоты на оптически активные (+) или (-) формы. Настоящее изобретение необязательно относится также к способу разделения (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрила на оптически активные (+) или (-) формы. Подробное описание изобретения Соответственно, настоящее изобретение относится к способу получения соединений формулы (I) или в рацемической, или в оптически активной (+) или (-) формах и их солей, где Х обозначает атом водорода или атом галогена, такой как фтор, хлор, бром или йод. Более конкретно, настоящее изобретение относится к способу получения клопидогрела-бисульфата. Способ получения соединений формулы (I) или их солей включает использование соединений формулы (II) CONHj <хго (П)(±)илм(+)нли(-) или их солей в или рацемической или оптически активной (+) или (-) формах, как показано на схеме 1. Схема 1 соон MeOH/SOCl,^/' V^O 'C^,J> (Ш)(±)^+)или(-) COOCH. Рацемизация кон, растворитель. CN " CONHj СОП> (П) (+),(+) или (-) )(+)или(+)или(-) Разделение ,/HCl/MeOH Разделение Ow ,ОСН5 *С СОШ, Рацемизация CONHj да <Д)(+) (П)(-) (l) Каждое промежуточное соединение, показанное на схеме 1, имеет один хиральный центр. В этой связи, для получения оптически активного продукта, такого как соединение, представленное формулой (I), в частности клопидогрела и его солей, можно использовать оптически активное промежуточное соединение, получаемое на первой стадии процесса. Настоящее изобретение предлагает способ получения соединений формулы (I) и их солей, как показано на схеме 1, который включает 1) получение соединения формулы (IV), (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрила, как показано на схеме 2, то есть посредством реакции Штреккера; - 3 - 006198 2) разделение, при необходимости, рацемической смеси соединения формулы (IV) на его оптически активные (+) и (-) стереоизомеры; и возвращение в рецикл нежелательного стереоизомера в процесс рацемизацией; 3) превращение соединения формулы (IV) или в рацемической, или в оптически активной (+) или (-) форме или его соли в соединение формулы (II), (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамид, или соответствующую оптически активную (+) или (-) форму на основе используемого исходного материала; 4) разделение, при необходимости, рацемического соединения формулы (II) на его оптически активные (+) и (-) стереоизомеры; и возвращение в рецикл нежелательного стереоизомера в процесс рацемизацией; 5) превращение соединения формулы (II) или в рацемической, или в оптически активной (+) и (-) форме или его соли или в оптически активное, или в рацемическое соединение формулы (I), сложный метиловый эфир (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетата, в рацемической или оптически активной (+) и (-) форме и его соль на основе используемого исходного материала; 6) дальнейшее разделение и/или превращение рацемического/оптически активного соединения формулы (I) в его фармацевтически приемлемые соли и/или высвобождение рацемического или оптически активного соединения формулы (I) из его солей. Альтернативно, любое из соединений формулы (IV) или (II) или в рацемической, или в оптически активной (+) или (-) форме может быть трансформировано в соответствующие соединения формулы (III), которые могут быть затем превращены в соответствующее соединение формулы (I). Соединение формулы (IV) в рацемической или оптически активных (+) или (-) формах может быть непосредственно превращено в соответствующее соединение формулы (I). Необязательно, в указанных выше способах могут использоваться подходящие кислотно-аддитивные соли промежуточных соединений формул II, III и IV. Подходящие кислоты, используемые в настоящем способе, могут быть выбраны из уксусной, бензойной, фумаровой, малеиновой, лимонной, винной, гентизиновой, метансульфоновой, этансульфоновой, бензолсульфоновой, п-толуолсульфоновой, камфорсульфоновой, хлористо-водородной, серной, бромисто-водородной кислот и т.п. Другой аспект настоящего изобретения относится к разработке способа получения нового промежуточного соединения формулы (IV) и его солей. аУ)(±)или(+)или(-) Еще один аспект способа согласно настоящему изобретению включает получение промежуточного соединения общей формулы (IV), приведенного на схеме 2, реакцией Штреккера, использующей вторичный амин (Organic Synthesis Collective Volume III, p.275). Схема 2 (IV) (t) Способ получения соединений формулы (IV) включает взаимодействие амина формулы (V) или его соли с производным цианида общей формулы (VII), где М обозначает или щелочной металл, такой как Na, К, Li, или Н, триметилсилил (ТМС) и т.п., с 2-хлорбензальдегидом формулы (VI). Синтез амина или его соли формулы (V) описан в работе FR 2608607. Указанная выше реакция может быть осуществлена различными способами. Несколько таких методик описано на схеме 2, как показано выше. Вначале, амин формулы (V) или его соль подвергают взаимодействию с цианидом (VII), где М определен ранее, с последующим добавлением 2-хлорбензальдегида (VI). Альтернативно, 2-хлорбензальдегид (VI) обрабатывают цианидом формулы (VII), где М определен ранее, и промежуточный циангидрин далее подвергают взаимодействию с амином формулы (V) или его - 4 - 006198 солью. По альтернативной методике, 2-хлорбензальдегид формулы (VI) добавляют к производному бисульфита (кислого сульфата) формулы (VIII), где М' обозначает Na, К, Li и т.п., с последующей реакцией с цианидом формулы (VII), где М определен ранее, и, наконец, с амином формулы (V) или его солью в реакции in situ. Независимо от вариаций в методике реакции, полученный выход полученного в результате промежуточного соединения (IV) является сопоставимым. Предпочтительный способ включает добавление 2-хлорбензальдегида формулы (VI) к производному бисульфита формулы (VIII). Образованную соль обрабатывают цианидом формулы (VII) и, наконец, амином формулы (V) или его солью в присутствии подходящих реагента и растворителей. Подходящие реагенты включают кислотные катализаторы, такие как ледяная уксусная кислота (Synthesis, 1989, 616-618), хлористо-водородная кислота, серная кислота, метансульфоновая кислота, трифторуксусная кислота, полифосфорная кислота и т.п. Подходящие растворители могут представлять собой гидрофильные растворители, протонные или апротонные, которые включают воду, ^1-^)^^^, тетрагидрофуран, диметилформамид, ДМСО, диок-сан, 1,2-диметоксиэтан, уксусную кислоту, пропионовую кислоту и т.п., или смесь таких растворителей. Предпочтительный растворитель представляет собой смесь растворителя и воды в варьирующих соотношениях. Более предпочтительная реакционная среда включает смесь, содержащую воду и (C1 -C4)спирт в соотношении, варьирующем от 1:1 до 1:10. Когда реакцию проводят в апротонном или гидрофобном растворителе, необходимы межфазный катализатор и двухфазная система растворителей. Соответствующий межфазный катализатор, используемый в таком случае, может представлять собой галогенид тетрабутиламмония, галогенид бензилтри-метиламмония и т.п. В указанную реакцию могут быть внесены соответствующие добавки. Такие подходящие добавки могут представлять собой цикло[^)-гистидин-^)-фенилаланин] и т.п. Температура реакции может варьировать в диапазоне от -30°С до температуры флегмы используе-мого(ых) растворителя(ей). Предпочтительная температура находится в диапазоне от 0 до 100°С и более предпочтительно от 40 до 80°С. Однако при использовании HCN(g) (схема 2, промежуточное соединение VII, М=Н) требуемая температура находится в диапазоне от примерно -30 до -10°С. Указанная реакция может быть проведена в отсутствие или в присутствии инертной атмосферы, такой как N2, Не или АГ. Длительность реакции может варьировать от 1 ч до 3 дней, более конкретно от 2 ч до 2 дней. Предпочтительно осуществлять реакцию соединения формулы (V), производного бисульфита (VIII) и производного цианида (VII) с 2-хлорбензальдегидом (VI) в соотношении предпочтительно от 1 до 1,2 эквивалентов. Получаемое при этом рацемическое цианосоединение (IV) может быть разделено на оптически активные (+) и (-) формы. Получаемое при этом цианосоединение (IV) может быть превращено в соответствующую кислоту формулы (III), амид формулы (II) или кислоту формулы (I), как показано на cхеме 1 (R.C. Larrock, in "Comprehensive Organic Transformations", John Wiley & Sons, Inc, 1999, 2nd Ed., 815-818 и содержащиеся в работе ссылки). Еще один аспект настоящего изобретения относится к превращению промежуточных соединений (II) и (III) в соединения формулы (I), как показано на cхеме 1. Каждый из (±), (+) или (-) изомеров промежуточных соединений формул (II) и (III) может быть превращен в соответствующий изомер соединений формулы (I). Предпочтительный путь получения соединения формулы (I) включает превращение любого из (±), (+) или (-) изомеров цианосоединения (IV) и его солей в амидное соединение формулы (II) в присутствии подходящих кислотных/основных реагентов в соответствующих растворителях. Последующее разделение амида на оптически активную (+) или (-) форму или ее соли и оптически активный амид затем превращается в оптически активный сложный эфир формулы (I) в присутствии подходящего катализатора и реагента. Реакция превращения цианосоединения формулы (IV) в амидное соединение формулы (II) может быть проведена в присутствии реагентов, которые включают кислоту или основание. Подходящими кислотами, которые могут использоваться, являются уксусная кислота, п-толуолсульфоновая кислота, трифторуксусная кислота, хлоруксусная кислота т. п. или безводные спиртовые или водные растворы минеральных кислот, таких как серная кислота, НС1, НВГ и т.п. Основание является предпочтительным, когда исходным материалом является рацемическая смесь. Подходящее основание, которое может использоваться в настоящем способе, представляет собой гидроксид лития, гидроксид натрия, гидроксид калия, трет-бутилат калия или их смеси, предпочтительно гидроксиды щелочных металлов. В указанных выше реакциях вместе с гидроксидами щелочных металлов может также использоваться избыток перекисей водорода или пероксидов металлов. Подходящий(е) растворитель(и) для указанной реакции мо-жет(гут) представлять собой водный(ые), полярный(ые) или протонный(ые) растворитель(и), такие как вода, (^-0^01^*1', ацетон, уксусная кислота, диметилформамид, ТГФ, ДМСО, диоксан, ДМЭ и т.п., или их смеси, предпочтительно растворитель состоит из воды, метанола или трет-ВиОН или их смеси в соотношении, варьирующем от 1:1 до 1:10. - 5 - 006198 Температура находится в диапазоне от 20 до 250°С, предпочтительно от 50 до 150°С. Используемые в указанном способе реагенты могут варьировать от 0,01 до 1,2 мол.экв. Реакция может быть проведена в отсутствие инертной атмосферы или в инертной атмосфере, такой как N2, Не или АГ. Реакцию в щелочных условиях предпочтительно проводить в инертной атмосфере. Длительность реакции может варьировать от 1/2 ч до 5 дней, предпочтительно от 2 ч до 2 дней. Амид формулы (II) или в рацемической, или в оптически активной (+) или (-) форме или его соль может быть превращен в соответствующий сложный метиловый эфир формулы (I) в присутствии по меньшей мере 1 эквивалента метанола и кислоты в подходящем растворителе. Подходящие кислоты, которые могут использоваться в данном способе, включают уксусную кислоту, полифосфорную кислоту, п-толуолсульфоновую кислоту, трифторуксусную кислоту, хлоруксусную кислоту или минеральные кислоты, которые включают серную кислоту, НС1, НВГ и т.п., которые могут находиться в различных формах, таких как кислоты, растворенные в спирте, безводные кислоты, растворенные в спирте или насыщенные спиртом, и используемый спирт может представлять собой метанол. Предпочтительной кислотой является концентрированная серная кислота в соотношении от 1 до 50 эквивалентов. Подходящие растворители для указанной выше трансформации могут представлять полярный или протонный растворитель, такой как гидрофильные растворители, включающие метанол, ацетон, уксусную кислоту, ТГФ, ДМСО, диоксан, ДМЭ и т. п., или их смеси. Предпочтительный растворитель состоит из метанола по меньшей мере в 1 эквиваленте, но может иметься и в большом избытке, так что он действует в этом случае как растворитель. Иногда можно также использовать инертные сорастворители, такие как толуол, ксилол и т.п. Температурный диапазон составляет от 20 до 250°С, предпочтительно от 50 до 150°С. Реакция может быть проведена в отсутствие инертной атмосферы или в инертной атмосфере, такой как N2, Не или АГ. Длительность реакции может варьировать от 3 ч до 5 дней, предпочтительно от 4 ч до 2 дней. Можно осуществить превращение соединения формулы (IV) в его рацемической или оптически активной (+) или (-) форме или его соли в соответствующее производное уксусной кислоты формулы (III) в присутствии подходящего растворителя и реагента. Подходящие(й) растворители(ь) могут(жет) представлять собой водные или спиртовые растворители. Подходящие реагенты для указанной выше реакции включают кислоты и основания. Можно также превратить цианосоединение формулы (IV) или в рацемической, или в оптически активной (+) или (-) форме или его соли непосредственно в сложный метиловый эфир формулы (I) в присутствии по меньшей мере 1 эквивалента кислоты и по меньшей мере 1 эквивалента метанола в подходящих растворителях по известным в литературе методикам. Кислота формулы (III) или в рацемической или в оптически активной (+) или (-) форме или ее соли могут быть превращены в соответствующий сложный метиловый эфир формулы (I) в присутствии подходящего реагента в соответствующих растворителях и в присутствии по меньшей мере 1 эквивалента метанола. Подходящий реагент, который может использоваться в настоящей реакции, включает тионилхло-рид, хлорангидриды кислот, такие как пивалоилхлорид, алкилформиаты, такие как этил- или метилхлор-формиаты, и другие такие реагенты, которые активируют СООН группу, в соотношении эквивалентов 1:1. Растворитель, подходящий для указанных выше трансформаций, может представлять собой полярный или протонный растворитель, такой как метанол, ацетон, диметилформамид, ТГФ, ДМСО, дихлор-метан, дихлорэтан, диоксан, ДМЭ и т. п., или их смеси. Предпочтительный растворитель состоит из метанола, взятого по меньшей мере в 1 эквиваленте, но может присутствовать и в большом избытке, так что при этом он действует как растворитель. Температурный диапазон составляет от 20 до 250°С, предпочтительно от 50 до 150°С. Реагенты, используемые в указанном выше способе, могут составлять от 0,01 молей до эквимоляр-ных количеств. Реакция может быть проведена в отсутствие инертной атмосферы или в инертной атмосфере, такой как N2, He или АГ. Длительность реакции может варьировать от 3 ч до 5 дней, предпочтительно от 3 ч до 2 дней. Указанный способ получения соединений общей формулы (I), показанный на схеме 1, имеет следующие преимущества: 1) требует меньшего числа стадий для получения соединений формулы (I); 2) используемые реагенты/химические соединения легко доступны; 3) на различных стадиях используются более мягкие реакционные условия; 4) возможно получать хиральные/оптически активные промежуточные соединения на каждой из стадий (I, II, III, IV или V); 5) возможно рацемизировать нежелательные изомеры, что повышает эффективность процесса и снижает загрязнение окружающей среды; 6) указанные выше факторы способствуют повышению рентабельности описанного способа. Соединения формул (I), (II), (III) и (IV) могут быть разделены различными способами с получением оптически активных соединений формул (I), (II), (III) и (IV), которые могут дать клопидогрел с желательной стереохимической конфигурацией (R.A. Sheldon, in "Chirotechnology", Marcel Dekker, Inc. NY, - 6 - 006198 Basel, 1993, 173-204 и содержащиеся в работе ссылки; А.К Collins, G.N. Sheldrack and J. Crosby, in "Chi-rality in Industry II", John Wiley & Sons, Inc., 1997, 81-98 и содержащиеся в работе ссылки, E.L. Eliel and S.H. Wilen, in "Stereochemistry of Organic Compound", John Wiley & Sons, Inc, 1999, 297-464 и содержащиеся в работе ссылки). Способ разделения включает растворение рацемической смеси (формул I, II, III или IV) в соответствующем растворителе и добавление подходящего хирального реагента. Среда может необязательно содержать воду в количестве примерно <5%. Подходящие растворители выбирают на основе того, что диастереомерная соль осаждается отлично от других форм. Отделение диастереомерной соли может происходить или спонтанно, или при добавлении сорастворителя, или путем высаливания или выпаривания растворителя, или при добавлении сорастворителя. Альтернативно, отделение может быть просто проведено путем перемешивания при подходящей температуре в растворителе(ях) до тех пор, пока одна из солей предпочтительно не выпадет в осадок. Очистка диастереомерной соли возможна при кипячении с обратным холодильником в подходящем растворителе. Свободное основание высвобождается из его соли при использовании подходящего основного реагента. Диастереомерную соль растворяют или суспендируют в смеси воды и органического растворителя и нейтрализуют основанием при перемешивании. Свободное основание получают после отделения водного слоя и выпаривания органического растворителя. Используемые в процессе разделения растворители могут включать растворители или их смеси, такие как (^-00^^^ (^-^кетен, диметилформамид, этилацетат, метилацетат, метилэтилкетон, ацето-нитрил, пропионитрил, ТГФ, диоксан и т.п., используемый растворитель необязательно может содержать воду вплоть до 5%, но присутствие воды или ее содержание не является решающим фактором. Подходящий температурный диапазон для разделения включает температуру от 0°С до температуры флегмы используемого растворителя, предпочтительно от 0 до 80°С. Кислотные хиральные реагенты, которые могут использоваться для образования диастереомерной соли, включают винную кислоту, миндальную кислоту, камфорсульфоновую кислоту, молочную кислоту, малеиновую кислоту, молочную кислоту, аминокислоты и т.п. При повторной кристаллизации из подходящего растворителя осажденная соль обогащается солью правовращающего изомера желательного диастереомера, что дает продукт с постоянным оптическим вращением. Подходящий основной реагент для гидролиза диастереомерной соли включает карбонат натрия, карбонат калия, бикарбонат натрия и бикарбонат калия в водной среде при температурах, варьирующих от 5 до 25°С. Наконец, желательная соль соединения формулы (II), (III) или (IV) или фармацевтически приемлемая соль соединения формулы (I) могут быть образованы из соответствующего стереоизомера и подходящей кислоты. Оптически чистое (S)-(+) соединение формулы (I) превращают в его бисульфатную соль с помощью 70-98% серной кислоты в соответствующем растворителе и при подходящей температуре, что приводит к получению требуемого (+)-клопидогрела-бисульфата, полиморфа I. Альтернативно, образованные диастереомеры могут быть отделены с помощью традиционных методик очистки, таких как фракционная кристаллизация, колоночная хроматография и т. п., с последующим расщеплением соли и с получением продукта желательной стереохимии. Предпочтительно использовать такой хиральный реагент, который может селективно образовывать диастереомер либо с R, либо с S стереоизомером промежуточного соединения (I), (II), (III) или (IV). Используемый хиральный реагент может присутствовать в соотношении от 0,5 до 1,1 молей. Определение энантиомерной чистоты (+)-правовращающего и (-)-левовращающего энантиомеров может быть проведено с помощью протонной ЯМР спектроскопии при добавлении хиральных редкоземельных реагентов (сдвиговых реагентов) или с помощью ВЭЖХ с использованием хиральной стационарной фазы, а также путем измерения оптического вращения. Абсолютная стереохимия диастереомерной соли (II), (III) или (IV) соединений может быть определена с помощью традиционных методик, таких как рентгеновская кристаллография. Абсолютная стереохимия хиральных соединений может быть также определена при сравнении ее с эталонными стандартами, известными в литературе. Фармацевтически приемлемые соли минеральных и органических кислот оптически активных энантиомеров клопидогрела получают с использованием различных кислых солей, что составляет часть настоящего изобретения, однако, они не ограничиваются сульфатами водорода, гидрогалогенидами, тау-рохолатами и т.п. Более конкретно, способ настоящего изобретения приводит к получению клопидогрела-бисульфата с точкой плавления 184±3°С, которая является характеристикой формы I клопидогрела-бисульфата. Альтернативно, форма II клопидогрела-бисульфата может быть также получена по известному способу (WO 99/65915, FR 98 07464). Способ согласно настоящему изобретению также включает способ возвращения в рецикл нежелательного стереоизомера через процесс рацемизации. Условия для проведения рацемизации всех промежуточных соединений общей формулы (II), (III) или (IV), а также конечного продукта I включают наличие аналогичного растворителя и катализатора в эквимолярных количествах. Подходящий катализатор пред- 7 006198 ставляет собой, в основном, основание, такое как ЛДА (лития диизопропиламид), КОН, NaOH, K+-t-BuO-, NaOMe, NaH, KH и т.п. Подходящий для использования в ходе разделения растворитель может включать растворители или их смеси, такие как (C1-C4)спирт, (C1-C4)кетон, этилацетат, метилацетат, метилэтилке-тон, ТГФ, диоксан и т.п., при этом используемый растворитель необязательно может содержать воду вплоть до 5%. Подходящий температурный диапазон для разделения включает температуру от 0°С до температуры флегмы используемого растворителя, предпочтительно от 0 до 80°С. Описанный в настоящем изобретении способ иллюстрируется приведенными ниже примерами. Указанные примеры даны только для иллюстрации и никоим образом не должны рассматриваться как ограничивающие область настоящего изобретения. Пример 1. (±)-(2-Хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрил (IV). К раствору 8,98 г (86,33 ммоль) бисульфита натрия в воде (27 мл) добавляют 12,4 г (86,33 ммоль) о-хлорбензальдегида, что приводит к образованию белого осадка. К указанному осадку добавляют 15 г (0,107 моль) 6,7-дигидро-4Н-тиено[3,2-с]пиридина и затем добавляют 4,4 г (89,7 ммоль) NaCN (растворенного в 15 мл воды). Реакционную смесь нагревают при температуре 40-50°С в течение 6 ч и затем гасят реакцию выливанием в воду (50 мл). Смесь экстрагируют 2х100 мл этилацетата. Органический слой сушат над безводным сульфатом натрия и растворитель удаляют при пониженном давлении. Выход указанного в заголовке продукта составляет 24 г (97%). Полученный продукт характеризуют с использованием ИК спектра, масс-спектра, 13С-ЯМР и 1H-ЯМР, значения которых приведены ниже. ИК спектр (см-1): 2227 (w, CN группа). Масс-спектр (m/z): 289,1 (М+Н)+. 13С-ЯМР ^СЬ): 5 136,46, 132,78, 132,38, 130,69, 130,46, 130,38, 129,90, 126,73, 124,96, 123,01, 115,09, 59,12, 49,30, 47,66, 25,47. ^-ЯМР (CDCl3): 5 7,2-7,7 (4Н, м), 7,0 (1Н, д), 6,69 (1Н, д), 5,32 (1Н, с), 3,78 (1Н, д), 3,65 (1Н, д), 2,83,0 (4Н, м). Пример 2. (±)-(2-Хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрил (IV). 140,5 г (1 моль) о-хлорбензальдегида и 65 г (1,01 моль) KCN добавляют к 3,5 л метанола. К реакционной смеси добавляют 139,05 г (1 моль) 6,7-дигидро-4Н-тиено[3,2-с]пиридина и 190 мл ледяной уксусной кислоты, все нагревают до 60°С в течение 20 ч при перемешивании. Через 8 ч начинает появляться осадок, затем реакционную смесь выливают в воду и экстрагируют этилацетатом (2х25 мл). Растворитель удаляют при пониженном давлении и осадок очищают, как описано в примере 1. Выход продукта составляет 187 г (65%), и его точка плавления равна 123-124°С. Полученный продукт описывают с использованием ИК спектра, масс-спектра, 13С-ЯМР и ^-ЯМР, и оказалось, что он идентичен продукту, полученному в примере 1. Пример 3. (±)-(2-Хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрил (IV). 27,6 г (266 ммоль) бисульфита натрия растворяют в 100 мл воды и после растворения в 100 мл метанола добавляют 38,2 г (271 ммоль) о-хлорбензальдегида. Образуется густая белая суспензия, которую нагревают при 60°С в течение 1 ч, затем добавляют к 36,97 г (266 ммоль) 6,7-дигидро-4Н-тиено[3,2-с]пиридина при 40°С и все перемешивают в течение 2 ч. К указанной реакционной смеси осторожно добавляют 17,29 г (266 ммоль) KCN, растворенного в 50 мл воды, и продолжают нагревание при 40°С в течение 5-6 ч с получением белого осадка. Реакционную смесь обрабатывают, как описано в примере 1, достигая выхода продукта 54,6 г (72%). Полученный продукт характеризуют с использованием ИК спектра, масс-спектра, 13С-ЯМР и 1H-ЯМР, и оказалось, что он идентичен продукту, полученному в примере 1. Пример 4. (±)-(2-Хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрил (IV). Добавляют 4,5 г (25,64 ммоль) гидрохлорида 6,7-дигидро-4Н-тиено[3,2-с]-5-пиридиния (при 10°С) к раствору 1,95 г (30 ммоль) цианида калия в 2 мл ледяной воды и затем добавляют по каплям 5 мл концентрированной хлористо-водородной кислоты при 0°С. После добавления Hd добавляют по каплям 3,3 г (23,47 ммоль) о-хлорбензальдегида, растворенного в 50 мл метанола. Затем реакционную смесь выдерживают при комнатной температуре в течение 3 дней и далее при 50°С в течение 3 ч. Значение рН доводят до 7,5-8,0 путем добавления по каплям NH4OH и экстрагируют продукт этилацетатом (2х50 мл). Растворитель сушат над сульфатом натрия и выпаривают при пониженном давлении. Количество полученного продукта составляет 1,67 г (18%), точка плавления равна 122-124°С. Полученный продукт описывают с использованием ИК спектра, масс-спектра, 13С-ЯМР и ^-ЯМР, и оказалось, что он идентичен продукту, полученному в примере 1. Пример 5. (±)-(2-Хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрил (IV). 6,25 г (44,46 ммоль) о-хлорбензальдегида растворяют в 60 мл толуола. В ту же самую колбу вносят 10 мл ледяной уксусной кислоты и 1,24 г (4,343 ммоль) цикло[^)-гистидин-^)-фенилаланин] и температуру понижают до -25°С. Затем добавляют 7 г (50,35 ммоль) 6,7-дигидро-4Н-тиено[3,2-с]пиридина, и через реакционную колбу пропускают газ HCN (со скоростью 30 пузырьков/мин) при -25°С в течение 6 ч, и затем перемешивают все при 31°С в течение 2 дней. Растворитель удаляют в вакууме и остаток очищают, - 8 - 006198 как указано в примере 1. Количество полученного продукта составляет 5,5 г (43%), точка плавления равна 124-125°С. Полученный продукт описывают с использованием ИК спектра, масс-спектра, 13С-ЯМР и ^-ЯМР, и оказалось, что он идентичен продукту, полученному в примере 1. Пример 6. (±)-(2-Хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрил (IV). По методике примера 5 получают аддукт между 104,3 г (1 моль) бисульфита натрия и 144,39 г (1,02 моль) о-хлорбензальдегида, к которому при температуре 31°С добавляют 150 г (1,078 моль) 6,7-дигидро-4Н-тиено[3,2-с] пиридина и перемешивают все в течение 1 ч. Добавляют по каплям 102 г TMC-CN и температуру поддерживают при 31°С в течение 6 ч, что приводит к появлению белого продукта, который выделяют и очищают по методике примера 1. Выход полученного продукта составляет 30 г (10%), точка плавления примерно равна 123-124°С. Полученный продукт описывают с использованием ИК спектра, масс-спектра, 13С-ЯМР и ^-ЯМР, и оказалось, что он идентичен продукту, полученному в примере 1. Пример 7. (±)-(2-Хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрил (IV). К 19,5 г (250 ммоль) KCN, растворенного в воде (20 мл), добавляют 43 г (250 ммоль) 6,7-дигидро-4Н-тиено[3,2-с]пиридина и затем добавляют по каплям 50 мл концентрированной хлористо-водородной кислоты. После завершения добавления добавляют по каплям раствор 33 г (230 ммоль) о-хлорбензальде-гида в 100 мл метанола и все перемешивают в течение 8 ч при 31°С. Значение рН реакционной смеси доводят до 7,5-8,0 с помощью NH4OH и продукт экстрагируют этилацетатом (2х500 мл) и промывают водой (2х500 мл), солевым раствором (500 мл) и сушат над сульфатом натрия. Растворитель выпаривают при пониженном давлении с получением 50 г (74%) продукта, точка плавления которого равна примерно 123-125°С. 13 1 Полученный продукт описывают с использованием ИК спектра, масс-спектра, 13С-ЯМР и ^-ЯМР, и оказалось, что он идентичен продукту, полученному в примере 1. Пример 8. (±)-(2-Хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрил (IV). К раствору 35,5 г (342 ммоль) бисульфита натрия в 35 мл воды добавляют по каплям 49,1 г (349 ммоль) о-хлорбензальдегида, и при этом сразу же образуется аддукт. К нему добавляют 50 г (284,9 ммоль) гидрохлорида 6,7-дигидро-4Н-тиено[3,2-с]пиридина и кипятят с обратным холодильником в течение 5 ч. Обычная процедура обработки и очистка дают 40 г (40%) продукта. Полученный продукт описывают с использованием ИК спектра, масс-спектра, 13С-ЯМР и ^-ЯМР, и оказалось, что он идентичен продукту, полученному в примере 1. Пример 9. (±)-(2-Хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрил (IV). 123,83 г (880 ммоль) о-хлорбензальдегида и 44 г (897 ммоль) цианида натрия добавляют к смеси 100 мл метанола и воды (1:1). К указанной смеси добавляют 150 г (1070 ммоль) 6,7-дигидро-4Н-тиено[3,2-с] пиридина и затем добавляют 10 мл концентрированной хлористо-водородной кислоты и перемешивают все при 31°С в течение 2 дней. Значение рН реакционной смеси доводят до 7,5-8,0 с помощью NH4OH и продукт экстрагируют этилацетатом (2х50 мл), промывают водой (2х50 мл), солевым раствором (50 мл), сушат над сульфатом натрия и выделяют, как указано в примере 1. Количество образованного белого твердого продукта составляет 33 г (13%), который характеризуют обычным образом. Пример 10. (±)-(2-Хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамид (II). 48 г (0,116 моль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрила суспендируют в 240 мл t-BuOH и в виде одной порции при перемешивании добавляют 18,26 г (0,332 моль) КОН. Реакционную смесь кипятят с обратным холодильником при температуре 80-82°С в течение 3 ч, затем охлаждают до 30°С, добавляют 240 мл воды и перемешивают в течение 20 мин. Отделяют нижний водный слой и медленно, в течение 15 мин добавляют 720 мл свежей охлажденной воды (5-10°С). Продукт экстрагируют этилацетатом (2х50 мл), промывают водой (2х50 мл), солевым раствором (50 мл), сушат над сульфатом натрия и далее выделяют путем выпаривания растворителя при пониженном давлении. При обработке гексаном получают твердое вещество. Выход продукта составляет 48 г (94%). Полученный продукт характеризуют с использованием ИК спектра, масс-спектра, 13С-ЯМР и 1H-ЯМР, значения которых приведены ниже. ИК спектр (см-1): 1656 (s, C=O группа), 2333,7 (N-H). Масс-спектр (m/z): 307,2 (М+Н)+. 13С-ЯМР (CDCl3): 5 173,82, 135,32, 133,42, 133, 130,27, 129,99, 129,4, 126,98, 125,18, 122,98, 69,12, 50,77, 49,10, 25,82. ^-ЯМР ^СЬ): 5 7,4-7,5 (4Н, м), 7,24 (1Н, д), 7,0 (1Н, с), 6,66 (1Н, д), 6,0 (1Н, с), 4,88 (1Н, д), 3,61 (2Н, кв), 2,88 (4Н, м). Точка плавления: 125-127°С. Пример 11. (±)-(2-Хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамид (II). По методике примеров 1-9 получают 100 мг (0,3466 ммоль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрила формулы (IV), растворяют его в 5 мл Hd и 1 мл трифторуксусной кислоты, добавляют 5 мл трет-бутанола и кипятят с обратным холодильником в течение 4 ч. После за - 9 - 006198 вершения реакции продукт выделяют, как описано в примере 11. Выход продукта составляет 40 мг (38%). 13 1 Полученный продукт описывают с использованием ИК спектра, масс-спектра, 13С-ЯМР и 1Н-ЯМР, и оказалось, что он идентичен продукту, полученному в примере 10. Пример 12. (±)-(2-Хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамид (II). К 100 мг (0,346 ммоль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрила формулы (IV) (приготовленного по методике примеров 1-9), добавляют 5 мл муравьиной кислоты и 5 мл концентрированной хлористо-водородной кислоты и реакционную смесь перемешивают в течение 48 ч при 25-30°С и затем кипятят с обратным холодильником при температуре приблизительно 100°С в течение 6 ч и перемешивают в течение 8 дней при 25-30°С. После завершения реакции реакционную смесь обрабатывают, как указано в примере 10. Выход продукта составляет 50 г (47%). Полученный продукт описывают с использованием ИК спектра, масс-спектра, 13С-ЯМР и ^-ЯМР. Указанный продукт идентичен продукту примера 10. Пример 13. (±)-(2-Хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамид (II). 200 мг (0,694 ммоль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрила формулы (IV) (приготовленного по методике примеров 1-9) добавляют к 5 мл НВг и 5 мл Н2О и перемешивают в течение 72 ч при комнатной температуре. Затем реакционную смесь кипятят с обратным холодильником при температуре 100°С в течение 11 ч и продукт выделяют, как указано в примере 10. Выход продукта составляет 50 г (47%). Полученный продукт описывают с использованием ИК спектра, масс-спектра, 13С-ЯМР и ^-ЯМР, и оказалось, что он идентичен продукту, полученному в примере 10. Пример 14. (±)-(2-Хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамид (II). 1 г (3,47 ммоль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрила растворяют в 5 мл H2SO4 (50%). К реакционной смеси добавляют 0,405 г безводного NaCl и реакционную смесь кипятят с обратным холодильником в течение 2-3 ч. В конце реакции продукт выделяют, как указано в примере 10. Выход продукта составляет 600 мг (57%). Полученный продукт описывают с использованием ИК спектра, масс-спектра, 13С-ЯМР и ^-ЯМР, и оказалось, что он идентичен продукту, полученному в примере 10. Пример 15. (±)-(2-Хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамид (II). 1 г (3,47 ммоль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрила растворяют в 5 г (5 ммоль) НСЮ4 и к реакционной смеси добавляют 10 мл H2O. Реакционную смесь кипятят с обратным холодильником при температуре 105°С в течение 7 ч и затем перемешивают при комнатной температуре в течение 12 ч. Значение рН поднимают до 10-12 с использованием 10% (вес/объем) раствора NaOH и затем экстрагируют 50 мл дихлорметана. Отделяют органический слой, промывают его водой и выпаривают при пониженном давлении. Остаток обрабатывают гексаном с получением 50 мг (47% выход) твердого вещества с точкой плавления 125-127°С. Полученный продукт описывают с использованием ИК спектра, масс-спектра, 13С-ЯМР и ^-ЯМР, и оказалось, что он идентичен продукту, полученному в примере 10. Пример 16. (±)-(2-Хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамид (II). 1 г (3,47 ммоль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрила суспендируют в 10 мл t-BuOH и при перемешивании вносят 277 мг (6,925 ммоль) дробленого NaOH. Реакционную смесь кипятят с обратным холодильником при температуре 80-82°С в течение 4 ч и затем охлаждают до комнатной температуры. Продукт выделяют путем экстракции этилацетатом. Органический экстракт выпаривают, и оставшийся после обработки гексаном масляный материал представляет собой 400 мг твердого вещества (38%). Полученный продукт описывают с использованием ИК спектра, масс-спектра, 13С-ЯМР и ^-ЯМР, и оказалось, что он идентичен продукту, полученному в примере 10. Пример 17. (±)-(2-Хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамид (II). 3 г (0,01 ммоль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрила суспендируют в 50 мл ацетона. К указанному раствору добавляют 1 г (0,023 ммоль) NaOH, растворенного в 10 мл воды, затем добавляют в виде одной порции 5 мл H2O2 (0,05 моль, 30%, вес/объем) и кипятят все с обратным холодильником в течение 3 ч. Реакционную смесь охлаждают до комнатной температуры и снова добавляют 5 мл H2O2 (0,05 моль, 30%, вес/объем) и перемешивают в течение 12 ч. Продукт выделяют при добавлении избытка воды и экстрагируют 1 л этилацетата. Органический экстракт выпаривают с получением 2,8 г неочищенного масляного материала, который после обработки гексаном дает 1 г твердого вещества (31%). Полученный продукт описывают с использованием ИК спектра, масс-спектра, 13С-ЯМР и ^-ЯМР, и оказалось, что он идентичен продукту, полученному в примере 10. Пример 18. (±)-(2-Хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамид (II). К 10 г (0,0346 ммоль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрила, суспендированного в 30 мл изопропилового спирта, медленно добавляют 3,9 г (0,0589 моль) дробленого КОН (85%) и смесь нагревают и добавляют 120 мл воды. Значение рН водного слоя доводят до 7 с по- 10 006198 мощью разбавленной хлористо-водородной кислоты. Отфильтровывают белый твердый осадок и промывают его 100 мл воды. Выход продукта составляет 9 г (85%), и точка плавления равна 122°С. Полученный продукт описывают с использованием ИК спектра, масс-спектра, 13С-ЯМР и ^-ЯМР, и оказалось, что он идентичен продукту, полученному в примере 10. Пример 19. Бисульфатная соль (II) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)аце-тамида. 2 г (6,48 ммоль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида формулы (II) растворяют в 10 мл ацетона. К реакционной смеси добавляют 1 мл серной кислоты и все перемешивают в течение 0,5 ч. Затем добавляют 5 мл диэтилового эфира и все перемешивают в течение ночи при комнатной температуре с получением соли. Соль (2 г, 76%) отделяют фильтрованием и промывают ацетоном. Точка плавления: 199,1°С. ИК спектр: 1682,8, 3116 (см-1). Пример 20. (±)-(2-Хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)уксусная кислота (III). К 100 мг (0,3466 ммоль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрила, растворенного в 2 мл трет-бутанола, добавляют 1,5 мл Hd. Реакционную смесь кипятят с обратным холодильником при температуре 100°С в течение 9 ч. В конце, после завершения реакции, добавляют воду и значение рН доводят до 4 с помощью 10% раствора КОН. Продукт экстрагируют 5 мл дихлорметана и обрабатывают, как указано в предыдущем примере. Выход составляет 40 мг (38%). Полученный продукт характеризуют с использованием ИК спектра, масс-спектра, 13С-ЯМР и 1H-ЯМР, значения которых приведены ниже. ИК спектр (см-1): 1637,5 (s, группа), 3399,37 (O-H). Масс пики (m/z): 308,1 (М+Н)+. ^-ЯМР (5 м.д.): 5 7,22-7,89 (4Н, м), 7,11-7,12 (1Н, д), 6,61-6,63 (1Н, д), 3,57-3,67 (2Н, д), 4,13 (2Н, с), 3,32-3,42 (2Н, с). Пример 21. (±)-(2-Хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)уксусная кислота (III). 5 г (17,33 ммоль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрила добавляют к 100 мл Hd и смесь перемешивают в течение 2 дней и затем кипятят с обратным холодильником в течение 15 ч. В конце реакции реакционную смесь выливают в воду и значение рН доводят до 4 с помощью 10% раствора КОН. Продукт экстрагируют 2 мл дихлорметана, промывают водой и органический слой выпаривают с получением остатка. Обычная очистка остатка дает 2 г твердого материала (38%). Полученный продукт характеризуют с использованием ИК спектра, масс-спектра, 13С-ЯМР и 1H-ЯМР. Указанный продукт оказывается идентичным продукту, полученному в примере 20. Пример 22. (±)-Метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетат (I). Смешивают 10 г (32,62 ммоль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетами-да (полученного по методике примера 15) с 19,8 г (161,7 ммоль) ДМФДМА (диметилформамида диме-тилацеталь) в 100 мл метанола. Смесь кипятят с обратным холодильником при температуре 70°С в течение 14 ч. Затем реакцию гасят добавлением в смесь воды и экстрагируют ее этилацетатом. Органический экстракт выпаривают при пониженном давлении с получением 5 г масляного продукта (48%). Указанное масло используют без дальнейшей обработки для получения солей сложного эфира (I). Пример 23. (±)-Метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетат (I). 15 г (0,0490 моль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида растворяют в 105 мл метанола при перемешивании. К указанному раствору добавляют при комнатной температуре по каплям в течение 1,5 ч 45 мл (0,823 моль) концентрированной серной кислоты (98%). Затем реакционную смесь кипятят с обратным холодильником при температуре 80°С в течение 26 ч, после чего отгоняют метанол. К образовавшемуся остатку добавляют при перемешивании 200 мл этилацетата при температуре от 0 до 5°С. После добавления 99 г (1,764 моль) КОН, растворенного в 300 мл воды, добавляют к реакционной смеси и перемешивают все в течение 0,5 ч. В итоге реакционную смесь фильтруют и оставляют выстаиваться. Органический слой отделяют и сушат над безводным Na2SO4. Растворитель выпаривают с получением масляного продукта. Выход составляет 10 г (64%). Пример 24. Бисульфатная соль (±)-метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил) ацетата (I). 10 г (±)-метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетата, полученного по методике примера 22, растворяют в 100 мл охлажденного льдом ацетона и добавляют 2 мл концентрированной серной кислоты при температуре от 0 до 5°С. Образованный кристаллический продукт от белого до не совсем белого цвета отделяют фильтрованием и промывают 20 мл ацетона. Полученный продукт сушат в вакуумной печи при 50°С. Выход указанного в заголовке продукта составляет 1,2 г (56%). Пример 25. (±)-Метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетат (I). 2 г (0,00652 моль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида и 2 мл (0,0308 моль) метансульфоновой кислоты и 20 мл метанола смешивают и раствор кипятят с обратным холодильником при температуре 85°С в течение 12 ч. Избыток растворителя удаляют при пониженном давлении. Значение рН доводят до примерно 8 водным раствором бикарбоната натрия при 0°С и продукт экстрагируют 70 мл этилацетата. Объединенные органические экстракты сушат над безводным Na2SO4 и - 11 - 006198 концентрируют. Полученный остаток очищают колоночной хроматографией с использованием смеси гексана:этилацетата в качестве элюента. Полученный таким образом продукт концентрируют, хранят в атмосфере азота и выдерживают в холодильнике перед превращением в соль. Выход указанного в заголовке продукта составляет 0,419 г (20%). Пример 26. (±)-Метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетат (I). 1 г (0,00326 моль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида растворяют в 20 мл метанола и кипятят с обратным холодильником при температуре 85°С. В ходе кипячения с обратным холодильником добавляют по каплям в течение 1 ч 10 мл полифосфорной кислоты и кипячение с обратным холодильником продолжают в течение 6 ч. Избыток растворителя удаляют при пониженном давлении. К остатку добавляют при 0°С 50 мл этилацетата и реакционную смесь подщелачивают водным NaHCO3 до рН 9. Из двух разделившихся фаз отделяют органический слой и сушат его над безводным Na2SO4 и концентрируют. Полученный остаток очищают колоночной хроматографией с использованием смеси гексана: этилацетата (9:1) в качестве элюента. Полученный продукт хранят в атмосфере азота и выдерживают в холодильнике перед превращением в соль. Выход указанного в заголовке продукта составляет 310 мг (30%). Пример 27. (±)-Метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетат (I). 1 г (0,00326 моль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида добавляют при 0°С к 2 мл толуола при перемешивании с последующим добавлением по каплям 1 мл тетрахлорида титана и затем реакционную смесь перемешивают при 0°С в течение 1 ч. После этого добавляют 18 мл метанола и далее реакционную смесь перемешивают в течение 36 ч при 29°С и затем нагревают с обратным холодильником в течение 3 ч. Растворитель отгоняют при пониженном давлении и остаток добавляют при 0°С к водному раствору карбоната натрия. Продукт экстрагируют 20 мл этилацетата и органический слой отделяют, сушат над безводным Na2SO4, концентрируют и очищают колоночной хроматографией с использованием смеси гексана: этилацетата в качестве элюента. Полученный продукт хранят в атмосфере азота и выдерживают в холодильнике перед превращением в соль. Выход указанного в заголовке продукта составляет 0,157 г (12-15%). Пример 28. (±)-Метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетат (I). К 5 г (16,31 ммоль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида добавляют при перемешивании 25 мл РОС13. Содержимое кипятят с обратным холодильником до полного потребления амида (примерно в течение 4 ч). Затем добавляют 20 мл метанола и 5 мл концентрированной H2SO4 и перемешивают все при комнатной температуре в течение 1 ч. После этого реакционную смесь кипятят с обратным холодильником в течение 1 ч. Реакционную смесь гасят добавлением водного Na2CO3 при 0°С и экстрагируют 200 мл этилацетата. Органический слой отделяют и сушат над безводным Na2SO4, концентрируют и очищают колоночной хроматографией с использованием смеси гекса-на: этилацетата в качестве элюента. Полученный продукт хранят в атмосфере азота и выдерживают в холодильнике перед превращением в соль. Выход указанного в заголовке продукта составляет 0,943 г (18%). Пример 29. (±)-Метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетат (I). 1 г (0,00326 моль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида растворяют 10 мл метанола. Реакционную смесь насыщают НС1 при 0°С, перемешивают при комнатной температуре в течение 4 ч и затем кипятят с обратным холодильником в течение 6 ч. Растворитель удаляют при пониженном давлении. К остатку добавляют 10 мл этилацетата и водный раствор NH033 доводят до рН 9 (при 0°С). Органический слой отделяют, сушат над безводным Na2SO4 и выпаривают при пониженном давлении. Остаток далее очищают колоночной хроматографией с использованием смеси гекса-на: этилацетата в качестве элюента. Полученный продукт хранят в атмосфере азота и выдерживают в холодильнике перед превращением в соль. Выход указанного в заголовке продукта составляет 0,188 г (18%). Пример 30. (±)-Метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетат (I). К 1 г (0,00326 моль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида добавляют 3 г (24,2 ммоль) ДМФДМА (диметилформамида диметилацеталь) и 10 мл метанола. Реакционную смесь кипятят с обратным холодильником при 70°С в течение 14 ч и затем выливают в воду и экстрагируют этилацетатом. Объединенные органические слои сушат над безводным Na2SO4 и выпаривают при пониженном давлении. Остаток далее очищают колоночной хроматографией с использованием смеси гексана:этилацетата в качестве элюента (9:1). Выход указанного в заголовке продукта составляет 500 мг (48%). Пример 31. (S)-(+)-(2-Хлорфенил)-(6,7-дигидро-4H-тиено[3,2-с]пирид-5-ил)ацетамид (II). а) Раствор 5 г (16,31 ммоль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида (полученного по методике примеров 10-15) и 4,15 г (16,2 ммоль) моногидрата (^)-(+)-камфор-10-сульфоновой кислоты в 100 мл ацетона перемешивают при комнатной температуре в течение 20 ч. Затем указанный раствор выдерживают в течение 1 недели в морозилке. Появляется несколько кристаллов, последующее концентрирование по объему путем выпаривания растворителя при пониженном давлении и повторная перекристаллизация в морозилке в течение нескольких дней дают 3,3 г (75% выход) (S)-2-(2 - 12 - 006198 хлорфенил)-(4,5,6,7-тетрагидротиено[3,2-с]пирид-5-ил)ацетамида соли (+)-камфорсульфоновой кислоты. Указанную соль далее очищают перекристаллизацией в ацетоне до достижения постоянного значения удельного оптического вращения. Полученный продукт соответствующим образом сушат. Ниже приведены типичные физико-химические характеристики полученного продукта. Диапазон плавления: 223-225°С (разложение). УОВ: +51° (С=1, МеОН). b) 3,3 г (S)-(+)-(2-хлорфенил)-(6,7-дигидро-4H-тиено[3,2-с]пирид-5-ил)ацетамида соли 10-Б-кам-форсульфоновой кислоты добавляют к 20 мл насыщенного водного раствора Na2CO3 и продукт экстрагируют 20 мл этилацетатата. Отделяют органический слой, сушат его над Na2SO4 и концентрируют при пониженном давлении. Полученный продукт представляет собой неочищенное масло, которое при очистке дает 1,6 г белых кристаллов (64%). Полученный продукт характеризуют по различным физико-химическим характеристикам, включая ИК спектр, масс-спектр, 13С-ЯМР и ^-ЯМР, значения которых приведены ниже. Диапазон плавления: 153-154°С. УОВ: +41° (С=1, МеОН). Оптическая чистота: 99,5% - по данным ВЭЖХ с использованием хиральной колонки. ИК спектр (см-1): 1656 (s, группа), 2333,7 (m, C-N), 3357,9 (s, N-h str.) Macc-пики (m/z): 307,2 (M+H)+. 13С-ЯМР (CDCl3): 5 173,25, 134,84, 132,87, 132,65, 129,98, 129,56, 129,0, 126,72, 124,87, 122,64, 68,65, 50,43, 48,73, 25,28. ^-ЯМР (CDCl3): 5 7,4-7,5 (4H, м), 7,07 (1H, д), 7,06 (1H, д), 6,66 (1Н, д), 6,5 (1Н, с), 4,9 (1Н, с), 3,6 (2Н, кв.), 2,88 (4Н, м). ВЭЖХ чистота: 99,85%. Пример 32. (S)-(+)-(2-Хлорфенил)-(6,7-дигидро-4H-тиено[3,2-с]пирид-5-ил)ацетамид (II). a) 2 г (6,5 ммоль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида добавляют к 30 мл ацетона. К указанному раствору при температуре 15-20°С в течение 4 ч добавляют 0,82 г (3,28 ммоль) моногидрата (^)-(+)-камфор-10-сульфоновой кислоты в 10 мл ацетона. Реакционную смесь перемешивают еще в течение 5 мин, при этом образуется несколько кристаллов. Растворитель отгоняют при пониженном давлении и затем реакционную смесь выдерживают в холодных условиях при температуре ниже 8°С. Образованный осадок отфильтровывают и промывают растворителем. Выход указанного в заголовке продукта составляет 1,2 (60%) в диапазоне температуры плавления=223-225°С и [a]D=+51° (С=1, СН3ОН). b) К суспензии 1,1 г (+)-(S)-(2-хлорфенил)-(6,7-дигидро-4H-тиено[3,2-с]пирид-5-ил)ацетамида соли (^)-(+)-камфор-10-сульфоновой кислоты в 50 мл воды добавляют 50 мл насыщенного водного раствора Na2CO3. Реакционную смесь перемешивают в течение некоторого времени и затем добавляют 100 мл этилацетата. Отделяют органический слой и отгоняют с получением указанного в заголовке продукта. Количество полученного продукта составляет 600 мг (60% относительно присутствующего в рацемической смеси правовращающего изомера). Полученный продукт характеризуют по различным физико-химическим характеристикам, данные которых приведены ниже. Диапазон плавления: 149-153°С. [CX]D: +38° (С=1, СН3ОН) при % ее=97,24%. Пример 33. (S)-(+)-(2-Хлорфенил)-(6,7-дигидро-4H-тиено[3,2-с]пирид-5-ил)ацетамид (II). a) 5 г (16,3 ммоль) (±)-(2-хлорфенил)-(6,7-дигидро-4H-тиено[3,2-с]пирид-5-ил)ацетамидa добавляют к 60 мл этилацетата. 2 г (8,6 ммоль) моногидрата (^)-(+)-камфор-10-сульфоновой кислоты растворяют в минимальном количестве воды и в один прием добавляют к указанному выше раствору и перемешивают при 35-40°С в течение 1 ч. За короткий промежуток времени образуется соль, которую отделяют, промывают 50 мл ацетона и сушат. Количество полученного продукта составляет 1,81 г (36%). Полученный продукт характеризуют по различным физико-химическим характеристикам, данные которых приведены ниже. Диапазон плавления: 223-225°С. [a]D: +52,12° (С=1, СН3ОН). b) К суспензии 1,8 г (S)-(+)-(2-хлорфенил)-(6,7-дигидро-4H-тиено[3,2-с]пирид-5-ил)ацетамида соли (1S)-(+)-камфор-10-сульфоновой кислоты в 100 мл воды добавляют 50 мл водного раствора бикарбоната натрия. После перемешивания смеси добавляют 150 мл этилацетата. Объединенные органические слои отгоняют с получением указанного в заголовке продукта. Количество полученного продукта составляет 1 г (56%). Полученный продукт характеризуют по различным физико-химическим характеристикам, данные которых аналогичны таковым в примере 31 и приведены ниже. Диапазон плавления: 153-154°С. [a]D: +42° (С=1, СН3ОН) и % ее=100% (по данным ВЭЖХ на хиральной колонке). - 13 - 006198 Пример 34. (Я)-(-)-(2-Хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамид (II). a) 5 г (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида (I) (0,016 моль) добавляют к 60 мл этилацетата и затем в один прием добавляют к раствору 2 г (0,0086 моль) моногидрата (1R)-(-)-камфор-10-сульфоновой кислоты (КСК) в 5 мл воды. Реакционную смесь перемешивают при температуре окружающей среды в течение 1 ч. Далее реакционную смесь хранят в морозилке в течение 1 недели, при этом появляется небольшое количество кристаллов. После выпаривания растворителя при пониженном давлении и хранения на холоде осаждается соль, которую фильтруют и промывают 50 мл ацетона. Количество полученного продукта составляет 1,7 г (39%). Полученный продукт характеризуют по различным физико-химическим характеристикам (R)-(-)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида соли (Ж)-(-)-камфор-10-сульфоновой кислоты, значения которых приведены ниже. Диапазон плавления (-)-КСК-(-)ацетамида: 219-220°С. [OC]D (-)-КСК-(-)ацетамида: -52,12° (С=1, СН3ОН). b) К суспензии 1,6 г (R)-(-)-(2-хлорфенил)-(6,7-дигидро-4H-тиено[3,2-с]пирид-5-ил)ацетамида соли (Ж)-(-)-камфор-10-сульфоновой кислоты в 100 мл воды добавляют 50 мл раствора в воде бикарбоната натрия. После перемешивания смеси добавляют 150 мл этилацетата. Органический слой экстрагируют и объединенные органические слои отгоняют с получением указанного в заголовке продукта. Количество полученного продукта составляет 900 мг (36%). Полученный продукт характеризуют по различным физико-химическим характеристикам, значения которых приведены ниже. Диапазон плавления: 145-149°С. [O]D ацетамида: -36,49° (С=1, СН3ОН). Пример 35. Бисульфат ^)-(+)-метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетата (I). 15 г (+)-(S)-(2-хлорфенил)-(6,7-дигидро-4H-тиено[3,2-с]пирид-5-ил)ацетамида (0,0489 моль) превращают в соответствующий сложный эфир, как указано в примере 23. Сложный эфир может быть превращен в свою бисульфатную соль по методике, описанной в примере 24. Количество полученного продукта составляет 7 г (44,5%) и его температура плавления=184-185°С. Точка плавления, ИК спектр и ДРЛ полученного продукта соответствуют параметрам продукта, полученного в ЕР 281459 и US 4847265, обозначаемого в настоящее время как форма I полиморфа клопи-догрела-бисульфата (WO 99/65915). Полученный продукт характеризуют по различным физико-химическим характеристикам, как показано ниже. [a]D: +55° (С=1, СН3ОН). Диапазон плавления: 185°С±2°С. ИК спектр: 2980, 1755, 1224, 1175 и 840, с соответствующим % от процента. Коэффициент пропускания: примерно 45, 16, 19, 15, 45. ДРЛ определяют, сверяя с картиной ДРЛ формы I, приведенной в WO 99/65915. Пример 36. Бисульфат да-(-)-метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетата (I). С использованием 5 г (R)-(-)-(2-хлорфенил)-(6,7-дигидро-4H-тиено[3,2-с]пирид-5-ил)ацетамида, полученного в примере 34, можно осуществить превращение в сложный эфир, как показано в примере 23. Затем сложный эфир может быть превращен в бисульфатную соль, как показано в примере 24. Количество полученного продукта составляет 3,01 г (44%). Полученный продукт характеризуют по различным физико-химическим характеристикам/показателям чистоты, как показано ниже. [a]D: -52° (С=1, СН3ОН). Пример 37. Бисульфат ^)-(+)-метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетат (I). a) 10 г (0,0185 моль) (+)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида соли (^)-(+)-камфор-10-сульфоновой кислоты растворяют в 40 мл метанола при 0°С. К раствору по каплям в течение 1,5 ч добавляют 15 мл (0,28 моль) концентрированной H2SO4. Постепенно повышают температуру реакционной смеси и кипятят с обратным холодильником в течение 26 ч. В конце кипячения растворитель отгоняют при пониженном давлении. Образовавшийся осадок смешивают со 120 мл этилацетата и значение рН доводят до 9-10 с помощью водного раствора карбоната натрия для экстракции. Органический слой отделяют, сушат над безводным Na2SO4 и выпаривают в вакууме. Полученный продукт очищают колоночной хроматографией с использованием смеси гексана: этилацетата в качестве элюента (9:1). Объединенный элюат выпаривают при пониженном давлении с получением 5,76 г (97%) продукта. b) 2 г ^)-(+)-метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетата превращают в бисульфатную соль по методике примера 24. Получают 2,2 г (84%) продукта, который идентичен продукту, полученному в примере 35. - 14 - 006198 Пример 38. Бисульфатная соль полиморфа I (+)-метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с] пирид-5-ил)ацетата (I). Добавляют 52,5 мл охлажденного льдом ацетона к 17,5 г (+)-метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетата при перемешивании при 0°С. Добавляют по каплям 3,6 мл концентрированной H2SO4 и температуру поддерживают ниже 5°С. Далее добавляют 20 мл ацетона и реакционную смесь продолжают перемешивать еще в течение 4 ч при комнатной температуре. Осадок отделяют (17 г, 74%), сушат в вакууме при температуре, не превышающей 50°С. Полученный продукт характеризуют по различным физико-химическим характеристикам и выявляют, что он идентичен продукту, полученному в примере 35, как показано ниже. Удельное оптическое вращение (УОВ): +54° (С=1, СН3ОН). Точка плавления: 185°С±2°С. ИК и ДРЛ определяют, сравнивая с соответствующими литературными данными. Пример 39. Бисульфатная соль полиморфа I (+)-метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с] пирид-5-ил)ацетата (I). К 2,1 г (+)-метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетата добавляют 7,6 мл ацетона с получением прозрачного раствора. К указанному раствору медленно добавляют 0,887 г H2SO4 (80%) и температуру поддерживают около 20°С в атмосфере азота. Далее реакционную смесь охлаждают до -20°С в течение 2 ч, после чего температуру доводят до комнатной (20°С). Реакционную смесь перемешивают при температуре 20-25°С. Осадок отделяют (600 мг), сушат в вакууме при температуре, не превышающей 50°С. Полученный продукт характеризуют по различным физико-химическим характеристикам и выявляют, что он идентичен продукту, полученному в примере 35, как показано ниже: УОВ (aD): +54,03° (С=1,89, МеОН). Точка плавления: 185°С±1°С. Хиральная чистота: 99,63% (ее). ИК и ДРЛ определяют, сравнивая с соответствующими литературными данными. Пример 40. Бисульфатная соль полиморфа I (+)-метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с] пирид-5-ил)ацетата (I). К 2 г (+)-метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетата добавляют 5 мл ацетона и перемешивают при температуре 25-30°С. Температуру реакционной смеси повышают с 25 до 65°С и держат на уровне 65°С в течение 5 мин. При температуре 50-52°С добавляют 0,676 г концентрированной H2SO4. Реакционную смесь охлаждают от 52 до 5°С, вносят дополнительное количество ацетона и перемешивают еще в течение 5 мин. Далее реакционную смесь перемешивают при температуре 25-30°С в течение 12 ч, отфильтровывают полученный густой осадок, промывают его 5 мл ацетона и сушат полученный остаток в вакуумной печи. Выход указанного в заголовке продукта составляет 1,27 г (47%). Полученный продукт характеризуют по различным физико-химическим характеристикам и выявляют, что он идентичен продукту, полученному в примере 35, как показано ниже: УОВ (aD): +54,03° (С=1,89, МеОН). Точка плавления: 185°С±1°С. Хиральная чистота: 99,80% (ее). ИК и ДРЛ определяют, сравнивая с соответствующими литературными данными. Пример 41. Бисульфатная соль полиморфа I (+)-метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с] пирид-5-ил)ацетата (I). К 1,98 г (+)-метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетата добавляют 5 мл ацетона и перемешивают при температуре 25-30°С. Температуру реакционной смеси повышают с 25°С до 50-52°С и добавляют в виде одной порции 0,7 г концентрированной H2SO4 (95%) при постоянном перемешивании, затем реакционную смесь резко охлаждают до 0 - -5°С в течение 10 мин. Далее реакционную смесь перемешивают при температуре 25-30°С в течение 12 ч, отфильтровывают полученный густой осадок, промывают его 5 мл ацетона и полученный остаток (1,6 г, 62%) сушат в вакуумной печи. Полученный продукт характеризуют по различным физико-химическим характеристикам и выявляют, что он идентичен продукту, полученному в примере 35, как показано ниже. УОВ (aD): +55,96° (С=1,89, МеОН). Точка плавления: 185°С±1°С. Хиральная чистота: 99,85% (ее). ИК и ДРЛ определяют, сравнивая с соответствующими литературными данными. Пример 42. Рацемизация (R)-(-)-(2-хлорфенил)-(6,7-дигидро-4H-тиено[3,2-с]пирид-5-ил)ацетамида (II). 20 г смеси левовращающего (R)-(-)-(2-хлорфенил)-(6,7-дигидро-4H-тиено[3,2-с]пирид-5-ил)ацетамида формулы (II) и 11 г трет-бутилата калия в 100 мл трет-бутанола перемешивают при температуре 25 -30°С в течение 30 мин. После завершения реакции реакционную смесь выливают в 750 мл охлажденной воды с получением желтого осадка. Полученный твердый материал отфильтровывают и растворяют в метилендихло-риде. Органический слой промывают 2х100 мл ДМ воды (2х100 мл) и концентрируют с получением 18 г со - 15 - 006198 ответствующего рацемического амида, т.е. (±)-2-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)аце-тамида (II). Удельное оптическое вращение рацемического амида (II) составляет ±1° (С=1, СН3ОН). Пример 43. Рацемизация (R)-(-)-(2-хлорфенил)-(6,7-дигидро-4H-тиено[3,2-с]пирид-5-ил)ацетамида (II). 1 г смеси левовращающего (R)-(-)-(2-хлорфенил)-(6,7-дигидро-4H-тиено[3,2-с]пирид-5-ил)ацетамида формулы (II) и 1 г трет-бутилата калия в 20 мл ДМСО перемешивают при температуре 50-60°С в течение 3 ч. После завершения реакции реакционную смесь выливают в 150 мл охлажденной воды с получением желтого осадка. Указанный твердый материал отфильтровывают и растворяют в метилендихлориде. Органический слой промывают 2х25 мл ДМ воды (2х25 мл) и концентрируют с получением 0,8 г соответствующего рацемического амида, т.е. (±)-2-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида. Удельное оптическое вращение рацемического амида (II) составляет ±0,5° (C=1, СН3ОН). Пример 44. Рацемизация (R)-(-)-(2-хлорфенил)-(6,7-дигидро-4H-тиено[3,2-с]пирид-5-ил)ацетамида (II). 1 г смеси левовращающего (R)-(-)-(2-хлорфенил)-(6,7-дигидро-4H-тиено[3,2-с]пирид-5-ил)ацетамида формулы (II) и 250 мг гидрида натрия в сухом тетрагидрофуране перемешивают при температуре 25 -30°С в течение 3 ч. После завершения реакции реакционную смесь медленно выливают в 150 мл охлажденной воды с получением желтого осадка. Указанный твердый материал отфильтровывают и растворяют в метиленди-хлориде. Органический слой промывают 2х25 мл ДМ воды (2х25 мл) и концентрируют с получением 0,95 г соответствующего рацемического амида, т. е. (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида (II). Пример 45. Рацемизация (R)-(-)-(2-хлорфенил)-(6,7-дигидро-4H-тиено[3,2-с]пирид-5-ил)ацетамида (II). 1 г смеси левовращающего (R)-(-)-(2-хлорфенил)-(6,7-дигидро-4H-тиено[3,2-с]пирид-5-ил)ацетамида формулы (II) и 21 г трет-бутилата калия в 100 мл ДМСО перемешивают при температуре 50-60°С в течение 3 ч. После завершения реакции реакционную смесь выливают в 750 мл охлажденной воды с получением желтого осадка. Указанный твердый материал отфильтровывают и растворяют в метилендихлориде. Органический слой промывают 2х100 мл ДМ воды (2х100 мл) и концентрируют с получением 0,8 г соответствующего рацемического амида, т.е. (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида. Пример 46. ^)-(+)-Метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетат (I). 5 г (+)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида растворяют в 35 мл метанола и раствор охлаждают до 0 - -5°С. Медленно, в течение 1 ч, добавляют 15 мл (0,28 моль) концентрированной H2SO4 (98%) и поддерживают температуру на уровне -5°С. После завершения добавления реакционную смесь перемешивают при комнатной температуре в течение 30 мин. Раствор кипятят с обратным холодильником при 60°С в течение 36 ч. Реакционную смесь охлаждают до комнатной температуры и отгоняют растворитель при пониженном давлении. Остаток переносят в 200 мл охлажденной воды и охлаждают до -5°С. Значение рН реакционной смеси доводят до 9-10 с помощью водного раствора карбоната натрия. Остаток экстрагируют 100 мл этилацетата. Органический слой сушат над безводным Na2SO4 и выпаривают в вакууме. Объединенный элюат выпаривают при пониженном давлении с получением 3,2 г неочищенного продукта, и проводят очистку колоночной хроматографией с использованием смеси гексан: этилацетат (9:1) в качестве элюента. Пример 47. ^)-(+)-Метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетат (I). 5 г (+)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида растворяют в 45 мл метанола и затем добавляют 15 мл толуола. Быстро добавляют 5 мл (0,28 моль) концентрированной H2SO4 и затем медленно добавляют еще 10 мл концентрированной H2SO4, поддерживая температуру на уровне 90°С. После этого реакционную смесь кипятят с обратным холодильником при 90°С в течение 13 ч. Растворитель отгоняют при пониженном давлении. К остатку добавляют 50 мл воды и рН реакционной смеси доводят до значения 9-10 с помощью водного карбоната натрия. Остаток экстрагируют 100 мл этил-ацетата. Органический слой сушат над безводным Na2SO4 и выпаривают в вакууме. Объединенный элю-ат выпаривают при пониженном давлении с получением 2,8 г неочищенного продукта и проводят очистку колоночной хроматографией с использованием смеси гексан:этилацетат (9:1) в качестве элюента. Пример 48. ^)-(+)-Метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетат (I). 5 г (S)-(+)-(2-хлорфенил)-(6,7-дигидро-4H-тиено[3,2-с]пирид-5-ил)ацетамида растворяют в 50 мл метанола и раствор охлаждают до -15°С. В течение 1 ч добавляют по каплям 15 мл (0,28 моль) концентрированной H2SO4 (98%), поддерживая температуру на уровне -15°С. После завершения добавления постепенно повышают температуру реакционной смеси, перемешивают при 31°С в течение 30 мин и затем кипятят с обратным холодильником при 70°С в течение 21 ч. Растворитель отгоняют при пониженном давлении. К остатку добавляют 50 мл воды и перемешивают в течение 30 мин с последующим охлаждением до -5°С. Значение рН реакционной смеси доводят до 9-10 с помощью водного раствора карбоната натрия. Остаток экстрагируют 100 мл этилацетата. Органический слой сушат над безводным Na2SO4 и выпаривают в вакууме. Выход составляет 3,8 г неочищенного продукта, который очищают колоночной хроматографией с использованием смеси гексан:этилацетат (9:1) в качестве элюента. Пример 49. ^)-(+)-Метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетат (I). 5 г (+)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида растворяют в 20 мл метанола и раствор нагревают до 65-70°С. Медленно добавляют в течение 1 ч 15 мл (0,28 моль) охлажденной - 16 - 006198 концентрированной H2SO4 (98%, -15°С), поддерживая температуру на уровне -15°С. После завершения добавления реакционную смесь нагревают при 70°С в течение 16 ч. Растворитель отгоняют при пониженном давлении и к остатку добавляют 50 мл воды и перемешивают все в течение 30 мин с последующим охлаждением до -5°С. Значение рН реакционной смеси доводят до 9-10 с помощью водного раствора карбоната натрия. Остаток экстрагируют 100 мл этилацетата. Органический слой сушат над безводным Na2SO4 и выпаривают в вакууме. Объединенный элюат выпаривают при пониженном давлении с получением 2,8 г неочищенного продукта, который очищают колоночной хроматографией с использованием смеси гексан: этилацетат (9:1) в качестве элюента с получением 2 г указанного соединения (I) и 1 г исходного материала (S)-(+)-(II). Пример 50. (S)-(+)-(2-Хлорфенил)-(6,7-дигидро-4H-тиено[3,2-с]пирид-5-ил)ацетамид (II). a) Раствор 5 г (16,31 ммоль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамида (полученный по методике примеров 10-15) и 4,15 г (16,2 ммоль) моногидрата (^)-(+)-камфор-10-сульфоновой кислоты в 100 мл ацетона выдерживают в теплом состоянии в течение 20 ч с последующим охлаждением до температуры ниже 10°С. Появляется несколько кристаллов, маточную жидкость концентрируют и повторяют перекристаллизацию, после чего хранят в холодных условиях в течение нескольких дней с получением 3,1 г (70% выход) ^)-2-(2-хлорфенил)-(4,5,6,7-тетрагидротиено[3,2-с]пирид-5-ил)ацетамида соли (+)-камфорсульфоновой кислоты. Указанную соль далее очищают перекристаллизацией в ацетоне, достигая постоянного удельного оптического вращения. Полученный продукт соответствующим образом сушат. Указанный продукт имеет следующие типичные физико-химические характеристики. Диапазон плавления: 223-225°С (разложение). УОВ: +52° (С=1, МеОН). b) 3,1 г (S)-(+)-(2-хлорфенил)-(6,7-дигидро-4H-тиено[3,2-с]пирид-5-ил)ацетамида соли 10^-кам-форсульфоновой кислоты добавляют к 20 мл насыщенного раствора Na2CO3 и продукт экстрагируют 20 мл этилацетата. Органический слой отделяют, сушат над Na2SO4 и концентрируют при пониженном давлении. Полученный продукт представляет собой неочищенное масло, которое при очистке дает 1,5 г белых кристаллов (60%). Пример 51. Хиральное удаление (-)-стереоизомера из смеси, содержащей избыток (+)-метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетата (I). 2 г (0,0173 моль) метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетата (в котором величина ее составляет 80%) растворяют в 10 мл ацетона и реакционную смесь перемешивают в течение 10 мин, после чего кипятят с обратным холодильником. К реакционной смеси добавляют 1,49 г гидрата (^)-(+)-камфор-10-сульфоновой кислоты в 0,8 мл воды и затем 1 мл ацетона. Затем всю реакционную смесь кипятят с обратным холодильником в течение 1 ч и постепенно охлаждают. Указанную смесь далее перемешивают в течение ночи при комнатной температуре. Прозрачный раствор охлаждают до температуры 0 - -5°С, получая при этом осадок. Полученную соль добавляют к этилацетату и воде и далее подщелачивают с использованием NaHCO3, органический слой промывают водой, концентрируют при пониженном давлении с получением 0,386 г свободного основания с хиральной чистотой, равной 99,85% (+)-изомера (ее=99,7%). Пример 52. Хиральное удаление (-)-стереоизомера из смеси, содержащей избыток (+)-метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетата (I). 2 г (0,0173 моль) метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетата (в котором величина ее составляет 90%) растворяют в 10 мл ацетона и реакционную смесь перемешивают в течение 10 мин, после чего кипятят с обратным холодильником. К реакционной смеси добавляют 1,49 г гидрата (^)-(+)-камфор-10-сульфоновой кислоты в 0,8 мл воды и затем добавляют 1 мл ацетона. Затем всю реакционную смесь кипятят с обратным холодильником в течение 1 ч и постепенно охлаждают. Указанную смесь перемешивают в течение ночи при комнатной температуре. Прозрачный раствор охлаждают до температуры 0 - -5°С, получая при этом осадок. Полученную соль добавляют к этилацетату и воде и подщелачивают с использованием NaHCO3. Органический слой промывают водой, концентрируют при пониженном давлении с получением 0,386 г свободного основания с хиральной чистотой, равной 99,85% (+)-изомера (ее=99,7%). Пример 53. Хиральное удаление (-)-стереоизомера из смеси, содержащей избыток (+)-метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетата (I). 2 г (0,0173 моль) метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетата (в котором величина ее составляет 95%) растворяют в 10 мл ацетона и реакционную смесь перемешивают в течение 10 мин, после чего кипятят с обратным холодильником. К реакционной смеси добавляют 1,49 г гидрата (^)-(+)-камфор-10-сульфоновой кислоты в 0,8 мл воды и затем добавляют 1 мл ацетона. Затем всю реакционную смесь кипятят с обратным холодильником в течение 1 ч и постепенно охлаждают. Указанную смесь затем перемешивают в течение ночи при комнатной температуре. Прозрачный раствор охлаждают до температуры 0 - -5°С, получая при этом осадок. Полученную соль добавляют к этилацетату и воде и затем подщелачивают с использованием NaHCO3. Органический слой промывают водой, концентрируют - 17 - 006198 под пониженным давлением с получением 0,386 г свободного основания с хиральной чистотой, равной 99,85% (+)-изомера (ее=99,7%). Пример 54. Разделение (±)-метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетата (I). 33 г (0,1 моль) (±)-метил (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетата растворяют в 200 мл ацетона, реакционную смесь нагревают до 60-70°С и кипятят с обратным холодильником в течение 15 мин. К реакционной смеси добавляют 25,6 г гидрата (^)-(+)-камфор-10-сульфоновой кислоты, растворенной в воде, при этом начинается образование соли при температуре 60-70°С. После завершения образования соли реакционную смесь постепенно охлаждают до комнатной температуры и затем до температуры 0-5°С. Диастереомерную соль отделяют фильтрованием, промывают ацетоном и сушат. Выход диастереомерного продукта составляет 20,5 г (70%). Получение (-) изомера указанного в заголовке соединения проводят с использованием разбавленного раствора бикарбоната натрия с последующей экстракцией этилацетатом, удалением растворителя, получая в итоге 10,9 г (66%) указанного в заголовке соединения. Пример 55. Разделение (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрила (IV). a) К 5 г (0,0173 моль) (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрила, растворенного в 100 мл ацетона, добавляют в один прием 4,35 г (0,0174 моль) гидрата (^)-(+)-камфор-10-сульфоновой кислоты, растворенной в 5 мл воды, поддерживая в ходе добавления температуру 60-62°С. Смесь перемешивают при 60°С в течение 60 ч. Перемешивание продолжают в течение ночи и затем выдерживают в холодильнике в течение 1 дня. Осажденную диастереомерную соль затем отфильтровывают с получением 730 мг продукта. b) К 730 мг (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрила соли (1S)-(+)-камфор-10-сульфоновой кислоты в 20 мл воды добавляют 10 мл (5%) раствора Na2CO3 (рН 10). После перемешивания смеси к органическому слою добавляют 100 мл этилацетата. Затем органический слой отделяют и отгоняют с получением продукта. Выход продукта составляет 400 мг (16%). Удельное оптическое вращение (aD): +7,5787° (С=1, ДМФ). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы (I) (c) (±)".(+)".(-) где Х обозначает атом водорода, фтора, хлора, брома или йода, предпочтительно 2-хлор, который включает: i) взаимодействие соединения формулы (V) или его соли 20я- -- W (VID с цианидом общей формулы (VII), где М обозначает щелочной металл, триметилсилил, Cu или водород, и последующее добавление соединения общей формулы (VI), где Х определен ранее, с получением рацемического соединения общей формулы (IV), где Х определен ранее; ii) взаимодействие соединения общей формулы (IV) в (±) форме или в любой из его оптически активных (+) или (-) форм | CONH, ссп> -• <хго- 0V)Cb).(+)M(.) <Д> "".(+)-Н с кислотными или щелочными реагентами с получением соединения формулы (II) или его соли с сохранением конфигурации; iii) взаимодействие соединения общей формулы (II) или в (±) форме, или в любой его оптически активной (+) или (-) форме 9°**" соосн, I - UAXJHj <хго-ссп> ОТ) (±)".(+)".(-) " а) (±)""(+)",(.) с кислотными реагентами в присутствии метанола с получением соединения формулы (I) или его соли с сохранением конфигурации; iv) наконец, разделение (±) соединения формулы (I) или его соли на его оптические изомеры. - 18 - 006198 2. Способ по п.1, в котором необязательно после разделения рацемического соединения формулы (IV) на его оптические изомеры или же трансформирования рацемического соединения или его оптических изомеров на его соли имеет место высвобождение соли из соединения. 3. Способ по п.1, в котором необязательно после разделения рацемического соединения формулы (II) на его оптические изомеры или же трансформирования рацемического соединения или его оптических изомеров на его соли имеет место высвобождение соли из соединения. 4. Способ по п.1, в котором соединение формулы (II), (2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетамид, или его оптически активные изомеры, или его соли, где Х обозначает атом галогена, такой как хлор-, бром-, иодзаместитель, более предпочтительно 2-хлор, получают по способу, включающему i) обработку соединения формулы (IV), или его соли, или любой его оптически активной формы щелочными реагентами, когда исходный материал является рацемическим или кислотным катализатором, когда исходный материал является оптически активным, в присутствии подходящего растворителя; ii) образование соли рацемического соединения формулы (II), которую разделяют на ее соответствующие оптически чистые (+) и (-) формы. 5. Способ по п.4, который включает получение соединения формулы (II), отличающийся тем, что используемый щелочной реагент представляет собой гидроксид лития, гидроксид калия, гидроксид натрия и трет-бутилат калия. 6. Способ по п.4, который включает получение соединения формулы (II), где используемый кислотный реагент выбирают из группы, состоящей из уксусной кислоты, п-толуолсульфоновой кислоты, трифторуксусной кислоты, хлоруксусной кислоты, перхлорной кислоты, муравьиной кислоты или минеральных кислот, таких как безводные спиртовые галогенидводороды или водная хлористо-водородная кислота, серная кислота, HBr или их смеси. 7. Способ по п.4, который включает получение соединения формулы (II), где используемый растворитель представляет собой воду, (С1-С4)спирт, ацетон, уксусную кислоту, диметилформамид, ТГФ, ДМСО, диоксан, ДМЭ и т.п. или их смеси, предпочтительно смесь воды, метанола или трет-BuOH. 8. Способ по п.4, в котором соединение формулы (II), (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено-[3,2-с]пирид-5-ил)ацетамид, разделяют на его (+) и (-) формы с использованием 1-(R)- или 1-(S)-камфорсульфоновой-10-кислоты, винной кислоты, в особенности 1-(R)- или 1-^)-камфорсульфоновой-10-кислоты, в присутствии растворителя, такого как вода, ацетон, этилацетат или их смесь, и используемая соль может находиться в молярном соотношении 0,5-1,1 эквивалентов. 9. Способ по п.4, в котором соединение формулы (II), (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено-[3,2-с]пирид-5-ил)ацетамид, превращают в соответствующую соль, причем заявленная соль представляет собой D-камфорсульфонат, L-камфорсульфонат, D-винную кислоту, L-винную кислоту или кислый сульфат. 10. Способ по п.1, в котором указанное соединение формулы (I) представляет собой метил(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетат, или его оптически активные изомеры, или его соли, где Х обозначает атом галогена, такой как хлор-, бром-, иодзаместитель, более предпочтительно 2-хлор, причем указанный способ включает сосен, СОХ-* (Г)(±)".(+)".(-) i) обработку соединения формулы (II), или его соли, или любой его оптически активной формы кислотными реагентами в присутствии подходящего растворителя; ii) образование соли рацемического соединения формулы (I), которую разделяют на ее соответствующие оптически чистые (+) и (-) формы. 11. Способ по п.10, в котором указанный используемый кислотный реагент выбирают из концентрированной серной кислоты в количестве от 1 до 50 эквивалентов, предпочтительно добавление проводят при 0-5°С или при температуре флегмы используемого растворителя. 12. Способ по п.10, в котором используемый растворитель представляет собой этанол, который берут в количестве от 3 до 30 объемов, вместе с другими сорастворителями, такими как толуол, ДМСО, ксилол и т. п., или их смесями. 13. Способ по п.10, в котором продолжительность реакции составляет от 4 ч до 4 дней. 14. Способ по п.10, в котором указанную реакцию проводят при температуре от 40 до 140°С. 15. Способ по п.10, в котором соединение формулы (I) разделяют с использованием 1-(R)- или 1-^)-камфорсульфоновой-10-кислоты, (R)- или ^)-винной кислоты, в особенности 1-(R)- или 1-(S) - 19 - 006198 камфорсульфоновой-10-кислоты, в присутствии растворителя, такого как вода, ацетон, этилацетат или их смесь. 16. Способ по п.15, в котором смесь, содержащую варьирующие соотношения двух энантиомеров (-)-I и хирально обогащают до (+)-(I)-стереоизомера или подвергают хиральному удалению (-)-(I)-энантиомера из варьирующей по соотношению смеси (-)-I и (+)-!-стереоизомеров. 17. Способ получения соединения формулы (IV) и его солей СО" + ~ со\> т ОШ) дух*) где Х обозначает атом галогена, такой как хлор-, бром-, иодзаместитель, в особенности Х обозначает 2-хлор, а также его солей, который включает: i) взаимодействие соединения общей формулы (V) или его кислотно-аддитивной соли с цианидом общей формулы (VII), в которой М означает щелочной металл, ТМС, Cu или водород, с последующим взаимодействием с галогенбензальдегидом формулы (VI), где Х обозначает атом галогена, продукт которого разделяют на его оптически активные изомеры, и, при необходимости, высвобождение из их солей или превращение в их соли; или ii) взаимодействие галогенбензальдегида общей формулы (VI), в которой Х обозначает атом галогена, с цианидом общей формулы (VII), где М определен ранее, с последующей реакцией in situ с соединением общей формулы (V) или его кислотно-аддитивной солью; или iii) взаимодействие галогенбензальдегида общей формулы (VI), в которой Х обозначает атом галогена, с кислым сульфитом общей формулы (VIII), где М определен ранее, и затем с цианидом общей формулы (VII), где М определен ранее, с последующей реакцией in situ с соединением общей формулы (V) или его кислотно-аддитивной солью; и, наконец, разделение полученного таким образом соединения формулы (IV) или его соли на оптически чистую (+) и (-) форму соединения формулы (IV) или его соли. 18. Способ по п.17, в котором указанное взаимодействие проводят при температуре от 0 до 100°С, а в случае использования газообразного HCN температуру реакции понижают дополнительно до -30°С. 19. Способ по п.17, в котором указанное взаимодействие проводят в водной или неводной среде, содержащей кислоты, такие как уксусная кислота, пропионовая кислота, метанол и HCl/MeOH. 20. Способ по п.17, в котором используют кислотный катализатор, такой как сухая хлористоводородная кислота, серная кислота, п-толуолсульфоновая кислота или уксусная кислота. 21. Способ по п.17, в котором соединение формулы (IV), (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрил, разделяют на его (-) и (+) формы с использованием 1-(R)- или 1-(S)-камфорсульфоновой-10-кислоты, винной кислоты, в особенности 1-(R)- или 1-^)-камфорсульфоновой-10-кислоты, в присутствии ацетона, этилацетата или их смеси и воды. 22. Способ по п.17, в котором соединение формулы (IV), (±)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено[3,2-с]пирид-5-ил)ацетонитрил, в (±), или (-), или (+) формах превращают в соответствующую соль, причем заявленная соль представляет собой D-камфорсульфонат, L-камфорсульфонат, D-винную кислоту, L-винную кислоту или кислый сульфат. 23. Способ по п.1, в котором рацемизацию (-) изомера соединения формулы (II) проводят в смеси, которая может содержать (+) стереоизомер, для образования (±) соединения с использованием основания, такого как ЛДА, KОН, NaOH, K-t-BuOH, NaOMe, NaH, KH, в подходящем растворителе. 24. Способ по п.1, в котором рацемизацию (-) изомера соединения формулы (I) проводят в смеси, которая может содержать (+) стереоизомер, для образования (±) соединения с использованием основания, такого как ЛДА, NaOMe, NaH, KH, в подходящем растворителе. 25. Способ по п.16, в котором разделение проводят в присутствии ацетона в качестве растворителя, взятого в количестве от 5 до 10 объемов, который может содержать воду вплоть до 5%. 26. Способ по п.16, в котором указанный подходящий хиральный агент используют в молярном соотношении 1:1. 27. Способ по п.26, в котором подходящая температура составляет от 0°С до температуры флегмы растворителя. Евразийская патентная организация, ЕАПВ Россия, 109012, Москва, Малый Черкасский пер., 2/6 - 20 - |