|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea200601895a*\id |
|
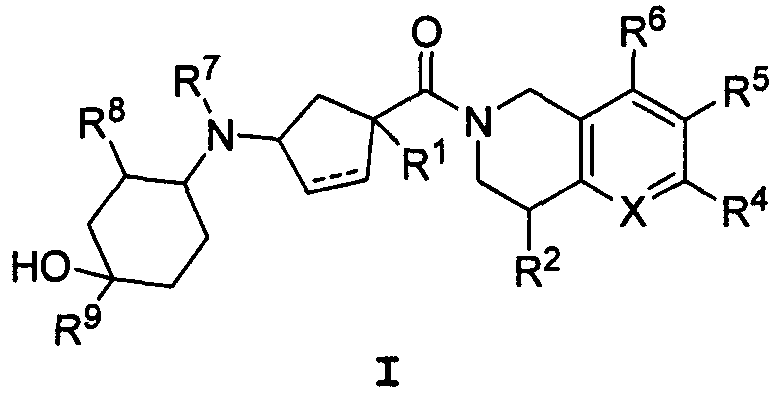
Термины запроса в документе Реферат Настоящее изобретение касается соединений формулы I которые являются модуляторами хемокиновых рецепторов. Соединения по изобретению и их композиции являются применимыми для лечения заболеваний, связанных с экспрессией и/или активностью хемокинового рецептора.
Полный текст патента (57) Реферат / Формула: которые являются модуляторами хемокиновых рецепторов. Соединения по изобретению и их композиции являются применимыми для лечения заболеваний, связанных с экспрессией и/или активностью хемокинового рецептора.
2420-139997ЕА/032 3-(4-ГЕТЕРОАРИЛЦИКЛОГЕКСИЛАМИНО)ЦИКЛОПЕНТАНКАРБОКСАМИДЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ ХЕМОКИНОВЫХ РЕЦЕПТОРОВ ОПИСАНИЕ Область техники, к которой относится изобретение Настоящее изобретение относится к соединениям, которые модулируют активность хемокиновых рецепторов, таких как CCR2. В некоторых вариантах осуществления соединения являются селективными по отношению к CCR2. Данные соединения могут применяться, например, для лечения заболеваний, ассоциированных с экспрессией или активностью хемокинового рецептора, таких как воспалительные заболевания, метаболические заболевания с ассоциированными характеристиками воспаления, иммунные заболевания и рак. Предпосылки создания изобретения Миграция и транспорт лейкоцитов из кровеносных сосудов в больные ткани вовлечены в инициацию нормальных воспалительных ответов для борьбы с заболеванием. Процесс, также известный как мобилизация лейкоцитов, имеет также отношение к наступлению и развитию воспалительных и аутоиммунных заболеваний. Полученная в результате этих заболеваний патология является производной от атаки защитных средств иммунной системы на заведомо нормальные ткани. Соответственно, предотвращение и блокировка мобилизации лейкоцитов для атаки на ткани при воспалительном заболевании, метаболическом заболевании, аутоиммунном заболевании и раке являлось бы высокоэффективным подходом к терапевтическому вмешательству. Различные классы лейкоцитарных клеток, которые вовлечены в клеточные иммунные ответы, включают моноциты, лимфоциты, нейтрофилы, эозинофилы, естественные клетки-киллеры, тучные клетки и базофилы. В большинстве случаев моноциты и лимфоциты являются классами лейкоцитов, которые инициируют, координируют и поддерживают хронические воспалительные ответы, и желательной является блокировка данных клеток от попадания в участки воспаления. Лимфоциты привлекают к участкам тканей моноциты, которые совместно с лимфоцитами являются ответственными за большинство действительных повреждений тканей, которые происходят при воспалительном заболевании. Известно, что инфильтрация лимфоцитов и/или моноцитов приводит к широкому ряду хронических, аутоиммунных заболеваний, а также отторжению пересаженных органов. Эти заболевания включают, но без ограничений, ревматоидный артрит, хронический контактный дерматит, астму, гипераллергические состояния, воспалительное заболевание кишечника, волчанку, системную красную волчанку, рассеянный склероз, атеросклероз, псориаз, саркоидоз, идиопатический фиброз легких, дерматомиозит, кожный пемфигоид и родственные заболевания (например, Pemphigus vulgaris, P. foliacious, P. erythematosis), гломерулонефриты, васкулиты, гепатит, диабет, отторжение аллотрансплантата и заболевание "трансплантат-против-хозяина". Полагают, что процесс, посредством которого лейкоциты покидают кровоток, аккумулируются в участках воспаления и дают начало заболеванию, имеет, по меньшей мере три стадии, которые были описаны как (1) роллинг, (2) активация/стойкая адгезия и (3) трансэндотелиальная миграция [Springer, Т. A., Nature 346:425-433 (1990); Lawrence and Springer, Cell 65:859-873 (1991); Butcher, E. C, Cell 67:1033-1036 (1991)]. Вторая стадия опосредована на молекулярном уровне хемоаттрактантными рецепторами. Хемоаттрактантные рецепторы на поверхности лейкоцитов связывают хемоаттрактантные цитокины, которые секретируются клетками на участке явного повреждения или инфекции. Связывание с рецептором активирует лейкоциты, увеличивает адгезивность адгезионных молекул, которые опосредуют трансэндотелиальную миграцию, и стимулирует направленную миграцию клеток по направлению к источнику хемоаттрактантного цитокина. Хемотаксические цитокины (лейкоцитарные хемоаттрактантные/активирующие факторы), также известные как хемокины, также известные как интеркрины и SIS-цитокины, представляют собой группу воспалительных/иммуномодуляторных полипептидных факторов с молекулярной массой 6-15 кДа, которые высвобождаются большим разнообразием клеток, таких как макрофаги, моноциты, эозинофилы, нейтрофилы, фибробласты, клетки сосудистого эндотелия, эпителиальные клетки, клетки гладкой мускулатуры и тучные клетки на участках воспаления (дан обзор в Luster, New Eng. J Med., 338, 436-445 (1998) и Rollins, Blood, 90, 909-928 (1997)). Хемокины также были описаны в Oppenheim, J. J. et al., Annu. Rev. Immunol., 9:617-648 (1991); Schall and Bacon, Curr. Opin. Immunol., 6:865-873 (1994); Baggiolini, M., et al., and Adv. Immunol., 55:97-179 (1994). Хемокины обладают способностью стимулировать направленную миграцию клеток - процесс, известный как хемотаксис. Хемокины могут быть сгруппированы в два основных подсемейства, на основании того, являются ли два аминоконцевых цистеиновых остатка непосредственно смежными (СС семейство) или разделенными одной аминокислотой (СХС семейство). Эти различия коррелируют с организацией двух подсемейств в два раздельных генных кластера. В пределах каждого генного кластера хемокины типично проявляют сходство последовательности в интервале между 25 и 60%. СХС-хемокины, такие как интерлейкин-8 (IL-8), нейтрофил-активирующий белок-2 (NAP-2) и белок стимуляторной активности роста меланомы (MGSA) являются хемотаксичными прежде всего для нейтрофилов и Т-лимфоцитов, в то время как СС-хемокины, такие как RANTES, MIP-la, М1Р-1р, моноцитарные хемотаксические белки (МСР-1, МСР-2, МСР-3, МСР-4 и МСР-5) и элтаксины (-1 и -2) являются хемотаксичными для, среди прочих клеточных типов, макрофагов, Т-лимфоцитов, эозинофилов, дендритных клеток и базофилов. Также существуют хемокины лимфотактин-1, лимфотактин-2 (оба С-хемокины) и фракталкин (СХХХС-хемокин), которые не попадают в оба из основных хемокиновых подсемейств. МСР-1 (также известный как MCAF (аббревиатура для хемотаксического и активирующего фактора макрофагов) или JE) представляет собой СС-хемокин, продуцируемый моноцитами/макрофагами клетками гладкой мускулатуры, фибробластами, и клетками сосудистого эндотелия, и вызывает клеточную миграцию и клеточную адгезию моноцитов (см., например Valente, A. J., et al., Biochemistry, 1988, 27, 4162; Matsushima, K., et al., J. Exp. Med., 1989, 169, 1485; Yoshimura, Т., et al., J. Immunol., 1989, 142, 1956; Rollins, B. J., et al., Proc. Natl. Acad. Sci. USA, 1988, 85, 3738; Rollins, B. J., et al., Blood, 1991, 78, 1112; Jiang, Y. , et al., J. Immunol., 1992, 148, 2423; Vaddi, K., et al., J. Immunol., 1994, 153, 4721), Т-лимфоциты памяти (см., например Carr, М. W., et al., Proc. Natl. Acad. Sci. USA, 1994, 91, 3652), T лимфоциты (см., например Loetscher, P., et al., FASEB J., 1994, 8, 1055) клетки естественных киллеров (см., например Loetscher, P., et al., J. Immunol., 1996, 156, 322; Allavena, P., et al., Eur. J. Immunol., 1994, 24, 3233), а также опосредование высвобождения гистамина базофилами (см., например Alam, R., et al., J. Clin. Invest., 1992, 89, 723; Bischoff, S. C, et al., J. Exp. Med., 1992, 175, 1271; Kuna, P., et al., J. Exp. Med., 1992, 175, 489). Кроме того, сообщается о высокой экспрессии МСР-1 при заболеваниях, где, как полагают, аккумуляция моноцитов/макрофагов и/или Т-клеток является важной при инициации или развитии заболеваний, таких как атеросклероз (см., например Hayes, I. М., et al., Arterioscler. Thromb. Vase. Biol., 1998, 18, 397; Takeya, M., et al., Hum. Pathol., 1993, 24, 534; Yla-Herttuala, S., et al., Proc. Natl. Acad. Sci. USA, 1991, 88, 5252; Nelken, N. A., J. Clin. Invest., 1991, 88, 1121), ревматоидный артрит (см., например, Koch, А. Е., et al., J. Clin. Invest., 1992, 90, 772; Akahoshi, Т., et al., Arthritis Rheum., 1993, 36, 762; Robinson, E., et al., Clin. Exp. Immunol., 101, 398), нефрит (см, например, Noris, M., et al., Lab. Invest., 1995, 73, 804; Wada, Т., at al., Kidney Int., 1996, 49, 761; Gesualdo, L., et al. , Kidney Int., 1997, 51, 155), нефропатия (см., например, Saitoh, A., et al., J. Clin. Lab. Anal., 1998, 12, 1; Yokoyama, H., et al., J. Leukoc. Biol., 1998, 63, 493), фиброз легких, саркоидоз легких (см., например, Sugiyama, Y., et al., Internal Medicine, 1997, 36, 856), астма, (см., например Karina, М., et al., J. Invest. Allergol. Clin. Immunol., 1997, 7, 254; Stephene, Т. H., Am. J. Respir. Crit. Care Med., 1997, 156, 1377; Sousa, A. R., et al., Am. J. Respir. Cell Mol. Biol., 1994, 10, 142), рассеянный склероз (см., например, McManus, С, et al., J. Neuroimmunol., 1998, 86, 20), псориаз (см., например, Gillitzer, R., et al., J. Invest. Dermatol., 1993, 101, 127), воспалительное заболевание кишечника (см., например, Grimm, М. С, et al., J. Leukoc. Biol., 1996, 59, 804; Reinecker, H. C, et al., Gastroenterology, 1995, 106, 40), миокардит (см., например, Seino, Y., et al., Цитокин, 1995, 7, 301), эндометриоз (см., например, Jolicoeur, С, et al., Am. J. Pathol., 1998, 152, 125), интраперитонеальная адгезия (см., например, Zeyneloglu, Н. В., et al., Human Reproduktion, 1998, 13, 1194), застойная сердечная недостаточность (см., например, Aurust, P., et al., Circulation, 1998, 97, 1136), хроническое заболевание печени (см., например, Marra, F., et al., Am. J. Pathol., 1998, 152, 423), вирусный менингит (см., например, Lahrtz, F., et al., Eur. J. Immunol., 1997, 27, 2484), болезнь Кавасаки (см., например, Wong, М. ; et al., J. Rheumatol., 1997, 24, 1179) и сепсис (см., например, Salkowski, С. A.; et al., Infect. Immun., 1998, 66, 3569). Кроме того, сообщается, что антитела против МСР-1 проявляют ингибиторный эффект или терапевтический эффект на животных моделях ревматоидного артрита (см., например, Schimmer, R. С, et al., J. Immunol., 1998, 160, 1466; Schrier, D. J., J. Leukoc. Biol., 1998, 63, 359; Ogata, H., et al., J. Pathol., 1997, 182, 106), рассеянного склероза (см., например, Karpus, W. J., et al., J. Leukoc. Biol., 1997, 62, 681), нефрита (см., например, Lloyd, С. M., et al., J. Exp. Med., 1997, 185, 1371; Wada, Т., et al., FASEB J., 1996, 10, 1418), астмы (см., например, Gonzalo, J.-A., et al., J. Exp. Med., 1998, 188, 157; Lukacs, N. W., J. Immunol., 1997, 158, 4398), атеросклероза (см., например, Guzman, L. A., et al., Circulation, 1993, 88 (suppl.), 1-371), гиперчувствительности отложенного типа (см., например, Rand, М. L., et al., Am. J. Pathol., 1996, 148, 855), легочной гипертензии (см., например, Kimura, Н., et al., Lab. Invest., 1998, 78, 571) и интраперитонеальной адгезии (см., например, Zeyneloglu, Н. В., et al., Am. J. Obstet. Gynecol., 1998, 179, 438). Также сообщается, что пептидный антагонист МСР-1, МСР-1(9-76) ингибирует артрит на мышиной модели (см., Gong, J.-H., J. Exp., 4ed., 1997, 186, 131), а также исследования на МСР-1-дефицитных мышах показали, что МСР-1 являются существенно необходимыми для мобилизации моноцитов in vivo (см., Lu, В., et al., J. Exp. Med., 1998, 187, 601; Gu, L., et al., Moll. Cell, 1998, 2, 275) . Хроническая обструктивная болезнь легких (ХОБЛ) находится в ряду наиболее частых причин смерти в западном обществе. Она определяется прогрессирующим ухудшением функции легкого, только частично обратимым бронходилятаторными лекарствами. ХОБЛ характеризуется хроническим воспалением в дыхательных путях или альвеолах, что отличается от того, что наблюдают при астме, включая увеличение числа нейтрофилов, макрофагов, Т-клеток CD8+ и/или тучных клеток в стенках воздушных путей, альвеолярных фрагментах и сосудистой гладкой мускулатуре. Полагают, что цитокины, ассоциированные с ХОБЛ, включают фактор некроза опухолей (TNF)-альфа, интерферон (IFN)-гамма, интерлейкин (IL)-1 бета, IL-6, IL-8 и МСР-1. Известно, что CCR2 является рецептором для МСР-1, и недавние данные подтверждают роль СР-1 и CCR2 в коррекции дыхательных путей и воспалении непосредственно или через макрофаги. Таким образом, антагонисты CCR2 представляют собой привлекательный подход к терапевтическому лечению ХОБЛ (De Boer, W. I., Chest, 2002, 121, 209S-218S). Литературные источники указывают, что хемокины, такие как МСР-1 и М1Р-1а, привлекают моноциты и лимфоциты к участкам заболевания и опосредуют их активацию и, таким образом, полагают, что они тесно вовлечены в инициацию, развитие и поддержание заболеваний со значительным участием моноцитов и лимфоцитов, таких как атеросклероз, диабет, рестеноз, ревматоидный артрит, псориаз, астма, язвенный колит, нефрит (нефропатия), рассеянный склероз, фиброз легких, миокардит, гепатит, панкреатит, саркоидоз, болезнь Крона, эндометриоз, конгестивная сердечная недостаточность, вирусный менингит, инфаркт мозга, нейропатия, болезнь Кавасаки и сепсис (см., например, Rovin, В. Н., et al. , Am. J. Kidney. Dis., 1998, 31, 1065; Lloyd, C, et al., Curr. Opin. Nephrol. Hypertens., 1998, 7, 281; Conti, P., et al., Allergy and Asthma Proc, 1998, 19, 121; Ransohoff, R. M., et al., Trends Neurosci., 1998, 21, 154; MacDermott, R. P., et al., Inflammatory Bowel Diseases, 1998, 4, 54). Хемокины связываются со специфичными рецепторами на поверхности клеток, принадлежащих к семейству связанных с G-белком семи трансмембранных доменных белков (обзор дан в Horuk, Trends Pharm. Sci., 15, 159-165 (1994)), которые именуют термином "хемокиновые рецепторы". При связывании с их родственными лигандами хемокиновые рецепторы передают внутриклеточный сигнал через ассоциированные тримерные G-белки, что приводит наряду с другими ответами, к быстрому увеличению концентрации внутриклеточного кальция, изменениям формы клеток, повышенной экспрессии молекул клеточной адгезии, дегрануляции и стимуляции клеточной миграции. Гены, кодирующие рецепторы специфичных хемокинов, были клонированы, и известно, что эти рецепторы представляют собой связанные с G-белком семь трансмембранных доменных белков, присутствующие на различных популяциях лейкоцитов. До настоящего времени идентифицированы, по меньшей мере, шесть СХС-хемокиновых рецепторов (CXCR1-CXCR6) и девять СС-хемокиновых рецепторов (CCR1-CCR8 и CCR10). Например, IL-8 является лигандом для CXCR1 и CXCR2, М1Р-1сс является лигандом для CCR1 и CCR5, а МСР-1 является лигандом для CCR2A и CCR2B (для ссылки, см., например, Holmes, W. Е., et al., Science 1991, 253, 1278-1280; Murphy P. M., et al., Science, 253, 12801283; Neote, K. et al, Cell, 1993, 72, 415-425; Charo, I. F. , et al., Proc. Natl. Acad. Sci. USA, 1994, 91, 2752-2756; Yamagami, S., et al., Biochem. Biophys. Res. Commun., 1994, 202, 1156-1162; Combadier, C, et al., The Journal of Biological Chemistry, 1995, 270, 16491-16494, Power, C. A., et al., J. Biol. Chem., 1995, 270, 19495-19500; Samson, M. , et al., Biochemistry, 1996, 35, 3362-3367; Murphy, P. M., Annual Review of Immunology, 1994, 12, 592-633). Сообщалось, что воспаление легкого и образование гранулемы подавляются у CCR1-дефицитных мышей (см. Gao, J.-L., et al., J. Exp. Med., 1997, 185, 1959; Gerard, C, et al., J. Clin. Invest., 1997, 100, 2022), и что мобилизация макрофагов и образование атеросклеротического повреждения уменьшается у CCR2-дефицитных мышей (см. Boring, L., et al., Nature, 1998, 394, 894; Kuziel, W. A., et al., Proc. Natl. Acad. Sci., USA, 1997, 94, 12053; Kurihara, Т., et al., J. Exp. Med., 1997, 186, 1757; Boring, L., et al., J. Clin. Invest., 1997, 100, 2552). Хемокиновые рецепторы также известны как сорецепторы для вхождения вируса, приводящего к вирусным инфекциям, таким как, например, ВИЧ-инфекция. Обратная транскрипция и процессинг белка являются классическими стадиями жизненного цикла вируса, который планируется блокировать антиретровирусными терапевтическими средствами. Несмотря на то, что многие новые лекарства, которые как полагают, блокируют вхождение вируса, остаются обещающими, в настоящее время не существует средства, к которому ВИЧ-1 не был бы способен приобрести резистентность. Для генерации генетического разнообразия, которое образует основу для резистентности, требуются множественные циклы репликации вируса. Комбинационная терапия, в которой репликация максимально подавляется, остается краеугольным камнем лечения ингибиторами вхождения вируса, как и другими средствам. Полагают, что нацеливание множественных стадий в пределах процесса вхождения вируса имеет потенциал для синергизма (Starr-Spires et al., Clin. Lab. Med., 2002, 22(3), 681). Вхождение ВИЧ-1 в клетки CD4(+) требует последовательных взаимодействий гликопротеинов оболочки вируса с CD4 и сорецепторам, таким как хемокиновые рецепторы CCR5 и CXCR4. Приемлемый подход для блокировки этого процесса состоит в применении низкомолекулярных антагонистов функции сорецептора. Молекула ТАК-779 представляет собой один такой антагонист CCR5, который действует для предотвращения инфекции ВИЧ-1. ТАК-77 9 ингибирует репликацию ВИЧ-1 на стадию мембранного слияния посредством блокировки взаимодействия гликопротеина поверхности др120 с CCR5. Участок связывания для ТАК-77 9 на CCR5 расположен вблизи от внеклеточной поверхности рецептора внутри полости, образованной между трансмембранными спиралями 1, 2, 3 и 7 (Dragic et al., Proc. Natl. Acad. Sci. USA, 2000, 97(10), 5639). Полагают, что хемокиновые рецепторы CXCR4 и CCR5 могут быть использованы в качестве сорецепторов Т клеточно-тропными (Х4) и макрофаг-тропными (R5) штаммами ВИЧ-1, соответственно, для вхождения в их клетки-хозяева. Размножение штаммов R5 ВИЧ-1 на лимфоцитах CD4 и макрофагах требует экспрессии сорецептора CCR5 на клеточной поверхности. Лица с недостатком CCR5 (CCR5 Дельта 32 гомозиготный генотип) являются фенотипически нормальными и резистентными к инфекции ВИЧ-1. Вхождение вируса может ингибироваться природным лигандом для CXCR4 (СХС-хемокин SDF-1) и CCR5 (СС-хемокины RANTES, М1Р-1альфа и М1Р-1бета). Первое непептидное соединение, которое взаимодействует с CCR5, а не с CXCR4, представляет собой производное четвертичного аммония, названное ТАК-779, которое также имеет высокую, но изменчивую активность против ВИЧ (De Clercq et al., Antivir. Chem. Chemother. 2001, 12 Suppl. 1, 19. SCH-C (SCH 351125) представляет собой еще один низкомолекулярный ингибитор вхождения ВИЧ-1 через сорецептор CCR5. SCH-C, соединение оксим-пиперидина, представляет собой специфичный антагонист CCR5, как определено при анализах связывания с множеством рецепторов и сигнальной трансдукции. Это соединение специфически ингибирует ВИЧ-1-инфекцию, опосредованную CCR5 в клетках U-87 астроглиомы, но не обладает никаким эффектом на инфекцию СХС^4-экспресситующих клеток. (Strizki et al, Proc. Natl. Acad. Sci. USA, 2001, 98(22), 12718 or Tremblay et al., Antimicrobial Agents and Chemotherapy, 2002, 46(5), 1336). AD101, химически родственный с SCH-C, также ингибирует вхождение вируса иммунодефицита человека типа 1 (ВИЧ-1) через человеческие CCR5. Было обнаружено, что AD101 ингибирует вхождение ВИЧ-1 через CCR5 макаки-резуса, в то время как SCH-C так не действует. Среди восьми остатков, которые различаются при сравнении версий сорецептора человека и макаки, только один, а именно, метионин-198, отвечает за нечувствительность CCR5 макаки к ингибированию посредством SCH-C. Положение 198 находится в CCR5 трансмембранной (ТМ) спирали 5 и не расположено в пределах ранее определенного участка связывания для AD101 и SCH-C, который включает остатки в ТМ спиралях 1, 2, 3 и 7. На основании исследований аминокислотных замещений в CCR5, предположили, что область CCR5 рядом с остатком 198 может оказывать влияние на конформационное состояние этого рецептора. (Billick et al., 2004, J. Virol., 78(8), 4134). Идентификация соединений, которые модулируют активность хемокиновых рецепторов, представляет желательный подход к созданию лекарств для необходимой разработки фармакологических средств для лечения заболеваний, ассоциированных с активностью хемокинового рецептора. Соединения по настоящему изобретению помогают реализовывать эти и другие потребности. Краткое содержание сущности изобретения Настоящее изобретение предоставляет соединения формулы I: О R6 или его фармацевтически приемлемые соли или пролекарства, где составляющие элементы даны в настоящем описании. Настоящее изобретение дополнительно предоставляет композиции, содержащие соединение формулы I и фармацевтически приемлемый носитель. Настоящее изобретение дополнительно предоставляет способы модуляции активности хемокинового рецептора, включающие в себя контакт указанного рецептора с соединением формулы I. Настоящее изобретение дополнительно предоставляет способы лечения заболевания, ассоциированного с экспрессией или активностью хемокинового рецептора у пациента, включающие в себя введение пациенту терапевтически эффективного количества соединения формулы I. Настоящее изобретение дополнительно предоставляет применение соединения формулы I в терапии. Настоящее изобретение дополнительно предоставляет применение соединения формулы I для получения лекарственного средства для применения в терапии. Подробное описание изобретения Соединения Настоящее изобретение предоставляет соединения формулы I: О R6 или его фармацевтически приемлемые соли или пролекарства, в которых: пунктирная линия означает необязательную связь; X представляет собой N, N0 или CR3; R1 представляет собой Ci_6 алкил, (Со-6 алкил) -О- (Ci-б алкил) , (Со-б алкил) -S- (Ci-б алкил), (С0-б алкил) - (С3_7 циклоалкил) - (Со-6 алкил), ОН, C02R10, гетероциклил, CN, NR10R12, NS02R10, NCOR10, NC02R10, NCOR10, CRuC02R10, CRuOCOR10 или фенил; R2 представляет собой H, ОН, галоген, Ci-з алкил, NR10R12, C02R10, CONR10R12, NR10COR1:L; OCONR10R12, NR10CONR10R12, гетероциклил, CN, NR10-SO2-NR10R12, NR10-SO2-R12, SO2-NR10R12 или оксо; где указанный Ci-з алкил является необязательно замещенным 1-6 заместителями, выбранными из F и ОН; R3 представляет собой Н, ОН, галоген, Ci-б алкил, Ci-б алкокси, NR10Rn, NR10CO2Rn; NR^CONR^R11, NR10SO2NR10Ru, NR10-S02-R11, гетероциклил, CN, CONR10R12, C02R10, N02, SR10, SOR10, S02R10; или S02-NR10Rn; R4 представляет собой H, Ci_6 алкил, CF3, 0CF3, CI, F, Br или фенил; R5 представляет собой Ci_6 алкил, Ci-6 алкокси, CO- (Ci-б алкил) , Ci-б тиоалкокси, пиридил, F, CI, Br, C4-6 циклоалкил, C4-6 цикл оал кил окси, фенил, фенилокси, Сз-б циклоалкил, С3_б циклоалкилокси, гетероциклил, CN, или CO2R10; где указанный Ci-б алкил является необязательно замещенным одним или несколькими ОН или F; где указанный Ci_6 алкокси, СО- (Ci-б алкил) или Ci_6 тиоалкокси являются необязательно замещенными одним или несколькими F; где указанный пиридил, фенил или фенилокси являются необязательно замещенными одним или несколькими заместителями, выбранными из галогена, CF3, Ci_4 алкил и C02R10; где указанный Сз-б циклоалкил или Сз-б циклоалкилокси являются необязательно замещенными одним или несколькими F; R6 представляет собой Н, CF3, Ci_6 алкил, F, С1 или Вг; R7 представляет собой Н или Ci-б алкил, необязательно замещенные 1-3 заместителями, выбранными из галогена, ОН, С02Н, C02-(Ci_6 алкил) или Ci-з алкокси; R8 представляет собой Н, Ci-б алкил, F, Ci-з алкокси, Ci_3 галогеналкокси, С3-6 циклоалкил, Сз_б циклоалкилокси, ОН, C02R10, OCOR10; где указанный Ci-е алкил является необязательно замещенным одним или несколькими заместителями, выбранными из F, Ci-з алкокси, ОН или C02R10; или R7 и R8 вместе образуют мостиковую группу С2.4 алкилен или -(Со-2 алкил)-О-(Ci-з алкил)- с образованием 5-7 членного цикла; R9 представляет собой гетероциклил, необязательно замещенный 1-4 заместителями, выбранными из Ci-б алкила, С2_б алкенила, С2_6 алкинила, галогена, С1-4 галогеналкила, CN, N02, OR13, SR13, C(0)R14, С (0) OR13, C(0)NR15R16, NR15R16, NR15CONHR16, NR15C(0)R14, NR15C(0)OR13, S(0)R14, S(0)2R14, S(0)NR:L5R16 или S02NR15R16; R10 представляет собой H, Ci-б алкил, бензил, фенил или С3-6 циклоалкил, где указанные Ci-6 алкил, бензил, фенил или С3-6 циклоалкил являются необязательно замещенными 1-3 заместителями выбранными из галогена, ОН, С1-3 алкила, С1-3 алкокси, С02Н, С02-(Ci-б алкил) и CF3; R11 представляет собой Н, ОН, Ci-б алкил, Ci_6 алкокси, бензил, фенил или Сз_б циклоалкил, где указанные Ci-б алкил, бензил, фенил или Сз-б циклоалкил являются необязательно замещенными 1-3 заместителями, выбранными из галогена, ОН, Ci-з алкила, Ci-3 алкокси, С02Н, С02- (Ci-б алкил) и CF3; R12 представляет собой Н, Ci-б алкил, бензил, фенил или С3_6 циклоалкил, где указанные Ci-e алкил, бензил, фенил или С3-б циклоалкил являются необязательно замещенными 1-3 заместителями, выбранными из галогена, ОН, Ci_3 алкила, Сх_3 алкокси, С02Н, С02- (Ci-б алкил) и CF3; R13 и R14 представляют собой каждый независимо, Н, Ci-б алкил, Ci-б галогеналкил, С2_б алкенил, С2-б алкинил, арил, циклоалкил, арилалкил или циклоалкилалкил; R15 и R16 представляют собой каждый, независимо Н, С]_б алкил, Ci_6 галогеналкил, С2-б алкенил, С2_б алкинил, арил, циклоалкил, арилалкил или циклоалкилалкил; или R15 и R16 вместе с атомом N, к которому они присоединены, образуют 4-6 членную гетероциклильную группу. В некоторых вариантах осуществления X представляет собой N или N0. В некоторых вариантах осуществления X представляет собой CR3. В некоторых вариантах осуществления R1 представляет собой Ci-б алкил. В некоторых вариантах осуществления R1 представляет собой 2-пропил. В некоторых вариантах осуществления R2 представляет собой Н, ОН, галоген или Ci_3 алкил. В некоторых вариантах осуществления R2 представляет собой В некоторых вариантах осуществления R3 представляет собой Н, ОН, галоген или Ci_6 алкил. В некоторых вариантах осуществления R3 представляет собой В некоторых вариантах осуществления R4 представляет собой В некоторых вариантах осуществления R5 представляет собой С]_б алкил, замещенный 1-4 F. В некоторых вариантах осуществления R5 представляет собой CF3, В некоторых вариантах осуществления R6 представляет собой В некоторых вариантах осуществления R7 представляет собой В некоторых вариантах осуществления R8 представляет собой В некоторых вариантах осуществления R9 представляет собой гетероарил, необязательно замещенный 1-4 заместителями, выбранными из Ci~6 алкила, С2-б алкенила, С2_б алкинила, галогена, Ci-4 галогеналкила, CN, N02, OR13, SR13, C(0)R14, С (0) OR13, C(0)NR15R16, NR15R16, NR15CONHR16, NR15C(0)R14, NR15C (0) OR13, S(0)R14, S(0)2R14, S(0)NR15R16 или S02NR15R16. В некоторых вариантах осуществления R9 представляет собой гетероарил, где гетероарил представляет собой 5- или 6-членный гетероарил, необязательно замещенный 1-4 заместителями, выбранными из Ci_6 алкила, С2-б алкенила, С2_б алкинила, галогена, d-4 галогеналкила, CN, N02, OR13, SR13, C(0)R14, С (0) OR13, C(0)NR15R16, NR15R16, NR15CONHR16, NR15C(0)R14, NR15C (0) OR13, S(0)R14, S(0)2R14, S(0)NR15R16 или S02NR15R16. В некоторых вариантах осуществления R9 представляет собой гетероарил, где гетероарил представляет собой пиридил, пиримидинил, пиразинил, пиридазинил или триазинил, каждый необязательно замещенный 1-4 заместителями, выбранными из Ci-б алкила, С2-б алкенила, С2-б алкинила, галогена, С1-4 галогеналкила, CN, N02, OR13, SR13, C(0)R14, С (0) OR13, С (О) NR15R16, NR15R16, NR15CONHR16, NR15C(0)R14, NR15C (0) OR13, S(0)R14, S(0)2R14, S (0)NR15R16 или S02NR15R16. В некоторых вариантах осуществления R9 представляет собой гетероарил, где гетероарил представляет собой тиенил, фуранил, тиазолил, оксазолил или имидазолил, каждый необязательно замещенный 1-4 заместителями, выбранными из Ci-6 алкила, С2-б алкенила, С2_б алкинила, галогена, С1-4 галогеналкила, CN, N02, OR13, SR13, C(0)R14, С (О) OR13, C(0)NR15R16, NR15R16, NR15CONHR16, NR15C(0)R14, NR15C (0) OR13, S(0)R14, S(0)2R14, S(0)NR15R16 или S02NR15R16. В некоторых вариантах осуществления R9 представляет собой гетероарил, где указанный гетероарил представляет собой тиазолил, оксазолил, пиримидинил или пиридил, каждый необязательно замещенный 1-3 заместителями из группы F, Cl, Вг, I, метил, этил, метокси, этокси или трифторметил. В некоторых вариантах осуществления соединения имеют формулу II: В некоторых вариантах осуществления соединения имеют формулу Ша, ШЬ или Шс: В различных местах настоящего описания заместители соединений раскрыты в виде групп или интервалов. Конкретно подразумевают, что данное изобретение включает каждую индивидуальную подкомбинацию элементов таких групп и интервалов. Например, конкретно подразумевают, что термин "Ci-б алкил" индивидуально раскрывает метил, этил, Сз-алкил, С4-алкил, Сб-алкил и Сб-алкил. Для соединений по изобретению, в которых переменный элемент появляется более одного раза, каждый переменных! элемент может представлять собой различный фрагмент, выбранный из группы Маркуша, определяющей данный переменный элемент. Например, там, где описана структура, имеющая две группы R, которые одновременно присутствуют в одном и том же соединении; две группы R могут представлять собой различные фрагменты, выбранные из группы Маркуша, определенной для R. Дополнительно следует учитывать, что определенные признаки изобретения, которые, для ясности, описаны в контексте отдельных вариантов осуществления, могут также быть предложены в сочетании с единственным вариантом осуществления. Напротив, различные признаки изобретения, которые для краткости описаны в контексте единственного варианта осуществления, могут также быть предложены отдельно или в любой подходящей субкомбинации. При использовании в данном описании подразумевают, что термин "алкил" относится к насыщенной углеводородной группе, которая является прямоцепочечной или разветвленной. Примерные алкильные группы включают метил (Me), этил (Et), пропил (например, н-пропил и изопропил), бутил (например, н-бутил, изобутил, трет-бутил), пентил (например, н-пентил, изопентил, неопентил), и т.п. Алкильная группа может содержать от 1 до приблизительно 20, от 2 до приблизительно 20, от 1 до приблизительно 10, от 1 до приблизительно 8, от 1 до приблизительно б, от 1 до приблизительно 4 или от 1 до приблизительно 3 атомов углерода. Как используется в данном описании, термин "алкенил" относится к алкильной группе, имеющей один или более двойных углерод-углеродных связей. Примерные алкенильные группы включают этенил, пропенил, циклогексенил и т.п. Как используется в данном описании, термин "алкинил" относится к алкильной группе, имеющей один или более тройных углерод-углеродных связей. Примерные алкинильные группы включают этинил, пропинил и т.п. Как используется в данном описании, термин "галогеналкил" относится к алкильной группе, имеющей один или несколько заместителей - галогенов. Примерные галогеналкильные группы включают CF3, C2F5, CHF2, СС13, СНС12/ С2С15 и т.п. Как используется в данном описании, термин "арил" относится к моноциклическим или полициклическим (например, имеющим 2, 3 или 4 сопряженных цикла) ароматическим углеводородам, таким как, например, фенил, нафтил, антраценил, фенантренил, инданил, инденил и т.п. В некоторых вариантах осуществления арильные группы имеют от 6 до приблизительно 20 атомов углерода. Как используется в данном описании, термин "циклоалкил" относится к неароматическим карбоциклам, включающим циклизованные алкильные, алкенильные и алкинильные группы. Циклоалкильные группы могут включать моно- или полициклические (например, имеющие 2, 3 или 4 сопряженных цикла) циклические системы, а также спироциклические системы. Примерные циклоалкильные группы включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклопентенил, циклогексенил, циклогексадиенил, циклогептатриенил, норборнил, норпинил, норкарнил, адамантил и т.п. Также включены в определение циклоалкила фрагменты, которые имеют один или более ароматических циклов, сконденсированных (т.е. имеющих общую связь) с циклоалкильным циклом, например, бензопроизводные пентана, пентена, гексана и т.п. В некоторых вариантах осуществления, циклоалкильные группы могут иметь от приблизительно 3 до приблизительно 10 или приблизительно от 3 до приблизительно 7 циклообразующих атомов углерода. Как используется в данном описании, термин "гетероциклил" или "гетероцикл" относится к насыщенному или ненасыщенному циклическому углеводороду, где один или более из циклообразующих атомов углерода циклического углеводорода заменены на гетероатом, такой как О, S или N. Гетероциклильные группы могут быть ароматическими (например, "гетероарил") или неароматическими (например, "гетероциклоалкил"). Гетероциклильные группы могут также соответствовать гидрированным и частично гидрированным гетероарильным группам. Гетероциклильные группы могут включать моно- или полициклические (например, имеющие 2, 3 или 4 сопряженных цикла) циклические системы. Гетероциклильные группы могут характеризоваться, как имеющие 3-14 или 3-7 циклообразующих атомов. В некоторых вариантах осуществления гетероциклильные группы могут содержать, в дополнение по меньшей мере к одному гетероатому, от приблизительно 1 до приблизительно 13, от приблизительно 2 до приблизительно 10 или от приблизительно 2 до приблизительно 7 атомов углерода и могут быть присоединены через атом углерода или гетероатом. В дополнительных вариантах осуществления, гетероатом может быть окислен (например, иметь оксо- или сульфидо заместитель) или атом азота может быть кватернизован. Примеры гетероциклильных групп включают морфолино, тиоморфолино, пиперазинил, тетрагидрофуранил, тетрагидротиенил, 2,3-дигидробензофурил, 1,3-бензодиоксол, бензо-1,4-диоксан, пиперидинил, пирролидинил, изоксазолидинил, изотиазолидинил, пиразолидинил, оксазолидинил, тиазолидинил, имидазолидинил и т.п, а также любую из групп, перечисленных ниже для "гетероарила" и "гетероциклоалкила." Дополнительные примеры гетероциклов включают пиримидинил, фенантридинил, фенантролинил, феназинил, фенотиазинил, феноксатиинил, феноксазинйл, фталазинил, пиперазинил, пиперидинил, 3,б-дигидропиридил, 1,2,3,б-тетрагидропиридил, 1,2,5,б-тетрагидропиридил, пиперидонил, 4-пиперидонил, пиперонил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридооксазол, пиридоимидазол, пиридотиазол, пиридинил, пиридил, пиримидинил, пирролидинил, пирролинил, 2Н-.пирролил, пирролил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, тетразолил, 6Н-1,2,5-тиадиазинил, 1, 2, 3-тиадиазолил, 1,2,4-тиадиазолил, 1, 2, 5-тиадиазолил, 1, 3, 4-тиадиазолил, тиантренил, тиазолил, тиенил, тиенотиазолил, тиенооксазолил, тиеноимидазолил, тиофенил, триазинил, 1,2,-триазолил, 1,2,4-триазолил, 1, 2,5-триазолил, 1,3,4-триазолил, ксантенил, октагидро-изохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1, 2, 5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, оксазолидинил, хиназолинил, хинолинил, 4Н-ХИНОЛИЗИНИЛ, хиноксалинил, хинуклидинил, акридинил, азоцинил, бензимидазолил, бензофуранил, бензотиофуранил, бензотиофенил, бензоксазолил, бензтиазолил, бензтриазолил, бензтетразолил, бензизоксазолил, бензизотиазолил, бензимидазолинил, метилендиоксифенил, морфолинил, нафтиридинил, дека-гидрохинолинил, 2Н,6Н-1,5,2-дитиазинил, дигидрофуро[2,3-Ь]тетрагидрофуран, фуранил, фуразанил, карбазолил, 4аН-карбазолил, карболинил, хроманил, хроменил, циннолинил, имидазолидинил, имидазолинил, имидазолил, 1Н-индазолил, индолелил, индолинил, индолизинил, индолил, ЗН-индолил, изобензофуранил, изохроманил, изоиндазолил, изоиндолинил, изоиндолил, изохинолинил, изотиазолил и изоксазолил. Дополнительные примеры гетероциклов включают азетидин-1-ил, 2.5- дигидро-1Н-пиррол-1-ил, пипериндин-1-ил, пиперазин-1-ил, пирролидин-1-ил, изохинол-2-ил, пиридин-1-ил, 3.6- дигидропиридин-1-ил, 2,З-дигидроиндол-1-ил, 1,3,4,9-тетрагидрокарболин-2-ил, тиено[2,3-е]пиридин-6-ил, 3,4,10,10а-тетрагидро-1Н-пиразино[1,2-а]индол-2-ил, 1,2,4,4а,5,б-гексагидро-пиразино[1,2-а]хинолин-3-ил, пиразино[1,2-а]хинолин-3-ил, диазепан-1-ил, 1,4,5,б-тетрагидро-2Н-бензо[f]изохинолин-3-ил, 1,4,4а,5,6,10Ь-гексагидро-2Н- бензо[f]изохинолин-3-ил, 3,За,8,8а-тетрагидро-1Н-2-аза- циклопента[а]инден-2-ил и 2,3,4,7-тетрагидро-1Н-азепин-1-ил, азепан-1-ил. Как используется в данном описании, термин "гетероарильные" группы относится к ароматическому гетероциклу, имеющему, по меньшей мере, один гетероатом в качестве элемента цикла, такой как сера, кислород или азот. Гетероарильные группы включают моноциклические и полициклические (например, имеющие 2, 3 или 4 сопряженных цикла) системы. Примеры гетероарильных групп включают без ограничений, пиридил, пиримидинил, пиразинил, пиридазинил, триазинил, фурил (фуранил), хинолил, изохинолил, тиенил, имидазолил, тиазолил, индолил, пиррил, оксазолил, бензофурил, бензотиенил, бензтиазолил, изоксазолил, пиразолил, триазолил, тетразолил, индазолил, 1,2,4-тиадиазолил, изотиазолил, бензотиенил, пуринил, карбазолил, бензимидазолил, индолинил и т.п. В некоторых вариантах осуществления, гетероарильная группа имеет от 1 до приблизительно 20 атомов углерода, и в дополнительных вариантах осуществления от приблизительно 3 до приблизительно 20 атомов углерода. В некоторых вариантах осуществления, гетероарильная группа содержит от 3 до приблизительно 14, от 3 до приблизительно 7, или от 5 до 6 циклообразующих атомов. В некоторых вариантах осуществления, гетероарильная группа имеет от 1 до приблизительно 4, от 1 до приблизительно 3, или от 1 до 2 гетероатомов. Как используется в данном описании, термин "гетероциклоалкил" относится к неароматическим гетероциклам, включающим циклизованные алкильные, алкенильные и алкинильные группы, где один или более из циклообразующих атомов углерода заменены на гетероатом, такой как атом О, N, или S. Примерные "гетероциклоалкильные" группы включают морфолино, тиоморфолино, пиперазинил, тетрагидрофуранил, тетрагидротиенил, 2,3-дигидробензофурил, 1,3-бензодиоксол, бензо-1,4-диоксан, пиперидинил, пирролидинил, изоксазолидинил, изотиазолидинил, пиразолидинил, оксазолидинил, тиазолидинил, имидазолидинил и т.п. Также включены в определение гетероциклоалкила фрагменты, которые имеют один или более ароматических циклов, конденсированных (т.е. имеющих общую связь) с неароматическим гетероциклическим циклом, например, фталимидил, нафталимидил и бензопроизводные гетероциклов, таких как индоленовая и изоиндоленовая группы. В некоторых вариантах осуществления, гетероциклоалкильная группа имеет от 1 до приблизительно 20 атомов углерода, и в дополнительных вариантах осуществления от приблизительно 3 до приблизительно 20 атомов углерода. В некоторых вариантах осуществления, гетероциклоалкильная группа содержит от 3 до приблизительно 14, от 3 до приблизительно 7, или от 5 до б циклообразующих атомов. В некоторых вариантах осуществления, гетероциклоалкильная группа имеет от 1 до приблизительно 4, от 1 до приблизительно 3, или от 1 до 2 гетероатомов. В некоторых вариантах осуществления, гетероциклоалкильная группа содержит от 0 до 3 двойных связей. В некоторых вариантах осуществления, гетероциклоалкильная группа содержит от 0 до 2 тройных связей. Как используются в данном описании, термины "гало" или "галоген" включают фтор, хлор, бром и йод. Как используется в данном описании, термин "алкокси" относится к -О-алкильной группе. Примерные алкоксигруппы включают метокси, этокси, пропокси (например, н-пропокси и изопропокси), трет-бутокси и т.п. Как используется в данном описании, термин "тиоалкокси" относится к -S-алкильной группе. Как используется здесь термин "галогеналкокси" относится к -О-галогеналкильной группе. Примерная галогеналкоксигруппа представляет собой OCF3. Как используется в данном описании, термин "циклоалкилокси" относится к -О-циклоалкилу. Как используется в данном описании, термин "аралкил" относится к алкильной группе, замещенной арильной группой. Как используется в данном описании, термин "циклоалкилалкил" относится к алкильной группе, замещенной циклоалкильной группой. Как используется в данном описании, термин "гетероциклилалкил" относится к алкильному фрагменту, замещенному гетерокарбоциклильной группой. Примерные гетероциклилалкильные группы включают "гетероарилалкил" (алкил, замещенный гетероарилом) и "гетероциклоалкилалкил" (алкил, замещенный гетероциклоалкилом). В некоторых вариантах осуществления, гетероциклилалкильные группы имеют от 3 до 24 атомов углерода в дополнение к, по меньшей мере, одному циклообразующему гетероатому. Как используется в данном описании, термин "оксо" относится к =0. Соединения, описанные в данном описании, могут быть асимметричными (например, имеющими один или более стереоцентров). Подразумевают, что рассматриваются все стереоизомеры, такие как энантиомеры и диастереомеры, если не указано иначе. Соединения настоящего изобретения, которые содержат асимметрически замещенные атомы углерода, могут быть выделены в оптически активной или рацемических формах. Способы получения оптически активных форм из оптически активных исходных веществ известны в данной области, как, например, посредством разделения рацемических смесей или посредством стереоселективного синтеза. Многие геометрические изомеры олефинов, C=N двойных связей, и т.п. могут также присутствовать в соединениях, описанных в данном документе, и все такие стабильные изомеры рассматриваются в настоящем изобретении. Цис- и транс- геометрические изомеры соединений настоящего изобретения описаны и могут быть выделены в виде смеси изомеров или как разделенные изомерные формы. Разделение рацемических смесей соединений может осуществляться посредством любого из многочисленных способов, известных в данной области. Примерный способ включает фракционную перекристаллизацию с использованием "хирального разделяющего агента", который является оптически активной, солеобразующей органической кислотой. Подходящими разделяющими средствами для способов фракционной перекристаллизации являются, например, такие оптически активные кислоты, как D- и L-формы винной кислоты, диацетилвинной кислоты, дибензоилвинной кислоты, миндальной кислоты, яблочной кислоты, молочной кислоты или различные оптически активные камфорсульфоновые кислоты, такие как (3-камфорсульфоновая кислота. Другие разделяющие средства, пригодные для способов фракционной кристаллизации, включают стереоизомерно чистые формы ог-метилбензиламина (например, 5- и й-формы, или диастереомерно чистые формы), 2-фенилглицинол, норэфедрин, эфедрин, Н-метилэфедрин, циклогексилэтиламин, 1,2-диаминоциклогексан и т.п. Разделение рацемических смесей может также осуществляться элюированием колонки, заполненной оптически активным разделяющим агентом (например, динитробензоилфенилглицином). Подходящая композиция элюирующего растворителя может быть определена специалистом в данной области. Соединения по изобретению также включают таутомерные формы, такие как кето-енольные таутомеры. Соединения по изобретению могут также включать все изотопы атомов, встречающихся в промежуточных соединениях или конечных соединениях. Изотопы включают такие атомы, имеющие одинаковый атомный номер, но различные массовые числа. Например, изотопы водорода включают тритий и дейтерий. Фраза "фармацевтически приемлемые" используется в данном описании для обозначения тех соединений, веществ, композиций, и/или дозированных форм, которые, в пределах обоснованного медицинского заключения, являются пригодными для применения в контакте с тканями людей и животных без избыточных токсичности раздражения, аллергической реакции или других проблем или осложнений, соответствующих рациональному соотношению благоприятное воздействие/риск. Настоящее изобретение также включает фармацевтически приемлемые соли соединений, описанных в данном документе. Как используется в данном описании, термин "фармацевтически приемлемые соли" относится к производным раскрытых соединений, где родительское соединение модифицируют преобразованием существующего фрагмента кислоты или основания в их солевую форму. Примеры фармацевтически приемлемых солей включают, но без ограничений, минеральные или органические кислотные соли основных остатков, таких как амины; щелочные или органические соли кислотных остатков, таких как карбоновые кислоты; и т.п. Фармацевтически приемлемые соли настоящего изобретения включают общепринятые нетоксичные соли или четвертичные аммониевые соли родительского соединения образованные, например, из нетоксичных неорганических или органических кислот. Фармацевтически приемлемые соли настоящего изобретения могут быть синтезированы из родительского соединения, которое содержит основный или кислотный фрагмент, общепринятыми химическими способами. В целом, такие соли могут быть получены взаимодействием свободных кислотных или основных форм этих соединений со стехиометрическим количеством соответствующих основания или кислоты в воде или органическом растворителе, или в их смеси; обычно являются предпочтительными неводные среды, такие как эфир, этил ацетат, этанол, изопропанол или ацетонитрил. Перечни подходящих солей можно найти в работах Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, Pa., 1985, p.1418 и Journal of Pharmaceutical Science, 66, 2 (1977), каждая из которых включена в данное описание во всей полноте в качестве ссылки. Настоящее изобретение также включает пролекарства соединений, описанных в данном документе. Как он используется в данном описании, термин "пролекарства" относится к любым ковалентно связанным носителям, которые высвобождают активное родительское лекарство, при введении млекопитающему. Пролекарства могут быть получены модификацией функциональных группы, присутствующей в соединениях, таким образом, что данные модификации расщепляются, либо при рутинных манипуляциях, либо in vivo, до родительских соединений. Пролекарства включают соединения, в которых гидроксильные, амино, сульфгидрильные или карбоксильные группы связываются с любой группой так, что при введении млекопитающему, расщепляются с образованием свободной гидроксильной, амино, сульфгидрильной или карбоксильной группы соответственно. Примеры пролекарств включают, но без ограничения указанными, ацетатные, формиатные и бензоатные производные спиртовой и аминной функциональных групп в соединениях по изобретению. Получение и применение пролекарств обсуждается Т. Higuchi и V. Stella, "Pro-drugs as Novel Delivery Systems," Vol.14 of the A.C.S. Symposium Series, и в Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association и Pergamon Press, 1987, которые обе включены в данное описание в качестве ссылки во всей полноте. Синтез Соединения по изобретению, включая их соли, гидраты и сольваты, могут быть получены с использованием известных методов органического синтеза и могут быть синтезированы в соответствии с любым из многочисленных возможных путей синтеза. Реакции для получения соединений по изобретению могут осуществляться в подходящих растворителях, которые могут быть легко выбраны специалистом, квалифицированным в области органического синтеза. Подходящие растворители могут являться по существу нереакционноспособными по отношению к исходным веществам (реагентам), промежуточным соединениям или продуктам при температурах, при которых осуществляются реакции, например, температурах, которые могут находиться в интервале от температуры замерзания растворителя до температуры кипения растворителя. Данная реакция может осуществляться в одном растворителе или в смеси более чем одного растворителя. В зависимости от конкретной стадии реакции, могут быть выбраны подходящие растворители для конкретной стадии реакции. Получение соединения по изобретению может включать защиту различных химических групп и снятие защитных групп. Необходимость в защите и снятии защитных групп и выбор соответствующих защитных групп могут быть легко определены специалистом в данной области. Химию защитных групп можно найти, например, в работе T.W. Green и P.G.M. Wuts, Protective Groups in Organic Synyhesis, 3rd. Ed., Wiley & Sons, Inc., New York (1999), которая включена в данное описание во всей полноте в качестве ссылки. Мониторинг реакций можно проводить в соответствии с любым подходящим способом, известным в данной области. Например, образование продукта можно регистрировать спектроскопическими средствами, такими как спектроскопия ядерного магнитного резонанса (например, 1Н или 13С), инфракрасная спектроскопия, спектрофотометрия (например, УФ-видимая) или масс-спектрометрия или хроматографией, такой как высокоэффективная жидкостная хроматография (ВЭЖХ) или тонкослойная хроматография. Примерные пути синтеза соединений по изобретению предоставлены на схемах 1-6 ниже, где составляющие элементы изображенных формул определены в данном документе. Промежуточные соединения формулы 1-5 могут быть получены с использованием методики, описанной на схеме 1. Доступная для приобретения карбоновая кислота 1-1 может быть преобразована в сложный эфир, такой как метиловый эфир, обработкой смесью йодометан/карбонат калия в ДМФА. Полученный в результате сложный эфир 1-2 может быть подвергнут алкилированию галогенидом, таким как йодид (R1!) с использованием основания, такого как гексаметилдисилазид лития (LHMDS), с получением алкилированного продукта 1-3 в виде смеси цис- и транс-диастереомеров (отношение 4:1). Минорный транс-диастереомер может быть удален кристаллизацией с последующим гидролизом сложного эфира до кислоты. Полученную в результате энантиомерно кислоту 1-4 подвергают гидрированию с использованием катализатора, такого как Pd-C с получением насыщенной карбоновой кислоты 1-5. Схема 1 1-5 Промежуточные соединения формулы 2-5 могут быть получены с использованием методик, представленных на схеме 2. Последовательное восстановление доступного для приобретения 2-1 (например, 4-трифторметилфенилацетонитрила) в амин гидрированием с использованием катализатора, такого как никель Ренея, ацилирование амина трифторуксусным ангидридом может приводить к получению амида 2-3. Полученный в результате амид может быть обработан формальдегидом в присутствии сильной кислоты, такой как серная кислота с получением циклизованного производного тетрагидроизохинолина 2-4. Удаление трифторацетильной группы обработкой карбонатом калия в воде и этаноле дает промежуточное соединение 2-5. Схема 2 2-5 Соединения формулы 3-9 (например, 5-аза- тетрагидроизохинолин) могут быть получены в соответствии со схемой 3. Доступное для приобретения промежуточное соединение формулы 3-1 (например, 5-трифторметилпиридин-2-ол) может быть бромирован обработкой бромом в уксусной кислоте с образованием промежуточных соединений формулы 3-2. Литиирование промежуточных соединений формулы 3-2 с использованием алкиллития, такого как н-бутиллитий или трег-бутиллитий, с последующим гашением ДМФА может приводить к получению альдегида 3-3. Альдегид может быть преобразован в цианогруппу обработкой гидроксиламином в присутствии формиата натрия/муравьиной кислоты с получением промежуточных соединений 3-4. Последующая конверсия гидроксила в хлор (3-5), замена хлора трет-бутилметилмалонатом дает сложный диэфир З-б. Гидрирование в присутствии катализатора, такого как никель Ренея, восстанавливает цианогруппу до амина, который циклизуется с метиловым сложным эфиром с образованием лактама. Обработка лактама сильной кислотой, такой как ТФУК (трифторуксусная кислота), удаляет трет-бутоксикарбонильный фрагмент. Полученный в результате лактам 3-8 восстанавливают до амина с использованием борана, который очищают посредством защиты амина группой Вое. Удаление Вое с использованием кислоты, такой как НС1 в диоксане, позволяет получить продукт 3-9. Схема 3 HO' "N' "R4 3-1 Br2/NaOAc AcOH BrvV NaH/ТГФ Ji Л. , н-бутиллитий/ДМФА НО N R4 НО N R4 3-2 3'3 NaHC02/HC02H HONH2 трет-бутилметилмалонат ^ NaH/ТГФ r5 фосфорилхлорид ^уД^^ * JUL ХИНОЛИН Н2/Ренея Ni EtOH ТФУ/?Н2С!2 HN 3-8 1.ВНЗ/ТГФ 2. (Boc)20 3. HCI N' "R4 3-5 o^oX 3-7 Re 3-9 Промежуточное соединение карбоновой кислоты формулы 1-5 может сочетаться с амином формулы 4-1 с использованием стандартного средства образования амида, такого как РуВгор или ВОР. Когда X=N, полученный в результате амид 4-2 может быть окислен с использованием окислителя, такого как тСРВА с получением N-оксида 4-4. Удаление группы Вое с 4-2 и 4-4 с использованием кислоты, такой как НС1 в диоксане, приводит к получению свободных аминов 4-3 и 4-5 соответственно. Схема 4 BocHN рос агент сочетания 4-1 BocHN BocHN "-" R2 6 HCI HCI 4-3 R2 4-5 R2 О Циклогексаноновые производные формулы 5-5 могут быть получены с использованием последовательности, изображенной на схеме 5. Гетероцикл (R9-X; где X является Н или галогеном) может быть литиирован обработкой бутиллитием, и полученный в результате анион может быть погашен моноэтиленкеталем 1,4-циклогексанона (5-2) с получением спирта 5-3. Обработка 5-3 водной кислотой, такой как НС1 в воде, преобразует кеталь в кетон. Алкилированием полученного в результате кетона 5-4 обработкой LDA с последующим гашением алкилгалогенидом, таким как R8I, получают циклогексаноновые производные формулы 5-5. Заместители на гетероцикле могут присутствовать перед литиированием, или гетероцикл может быть дериватизирован на последующих стадиях в соответствии с рутинными способами, такими как дополнительные реакции литиирования, с образованием соединений по изобретению. Схема 5 R9-X буллитий 5-1 о Х=Н, галогенид НО R9 - RSjOO 5-2 HCI/H20 5-3 LDA/RBI О - 5-4 5-5 Соединения по изобретению могут быть собраны, как показано на схеме 6. Восстановительным аминированием кетона формулы 5-5 амином формулы 4-3 с использованием восстановителя, такого как триацетоксиборгидрид натрия получают соединения формулы 6-1. Связывающий амин 6-1 может быть необязательно алкилирован, ацилирован, защищен и т.п в соответствии с рутинными способами с образованием производных формулы 6-2. Схема 6 В некоторых вариантах осуществления, соединения по изобретению могут модулировать активность одного или более хемокиновых рецепторов. Термин "модулировать" означает способность увеличивать или уменьшать активность рецептора. Соответственно, соединения по изобретению могут применяться в способах модуляции хемокинового рецептора посредством контакта рецептора с любым одним или несколькими из соединений или композиций, описанных в данном документе. В некоторых вариантах осуществления соединения настоящего изобретения могут действовать как ингибиторы хемокиновых рецепторов. В дополнительных вариантах осуществления соединения по изобретению могут применяться для модуляции активности хемокинового рецептора у лица, нуждающегося в модуляции рецептора посредством введения модулирующего количества соединения формулы I. Хемокиновые рецепторы, с которыми связываются настоящие соединения и/или которые подвергают модулированию, включают любой хемокиновый рецептор. В некоторых вариантах осуществления, хемокиновый рецептор принадлежит к СС-семейству хемокиновоых рецепторов, включающему например, CCRl, CCR2, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8 и CCR10. В некоторых вариантах осуществления, хемокиновый рецептор представляет собой CCR2. Соединения по изобретению могут быть селективными. Под "селективными" подразумевают, что соединение связывается с хемокиновым рецептором или ингибирует хемокиновый рецептор с большей аффинностью или активностью, соответственно, по сравнению, по меньшей мере, с одним другим хемокиновым рецептором. Соединения по изобретению могут являться селективными ингибиторами или лигандами CCR2, что означает, что соединения по изобретению могут связываться с CCR2 или ингибировать его с большей аффинностью или активностью, соответственно, чем для еще одного хемокинового рецептора, такого как, по меньшей мере, один из CCR1, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8 и CCR10. В некоторых вариантах осуществления соединения по изобретению имеют селективность связывания или ингибирования, более высокую для CCR2 по сравнению с CCR5. В некоторых вариантах осуществления соединения по изобретению имеют селективность связывания или ингибирования, более высокую для CCR2 по сравнению с CCR1. В некоторых вариантах осуществления соединения по изобретению имеют селективность связывания или ингибирования, более высокую для CCR2 по сравнению с любыми другими CCR. Селективность может быть, по меньшей мере, приблизительно 10-кратной, по меньшей мере, приблизительно 20-кратной, по меньшей мере, приблизительно 50-кратной, по меньшей мере, приблизительно 100-кратной, по меньшей мере, приблизительно 200-кратной, по меньшей мере, приблизительно 500-кратной или, по меньшей мере, приблизительно 1000-кратной. Сродство связывания и активность ингибирования могут быть измерены в соответствии с рутинными способами в данной области, такими как в соответствии с анализами, представленными в данном описании. Настоящее изобретение дополнительно предоставляет способы лечения заболевания или нарушения, ассоциированных с хемокиновым рецептором, у отдельного лица (например, пациента) посредством введения лицу, нуждающемуся в таком лечении, терапевтически эффективного количества или дозы соединения настоящего изобретения или его фармацевтической композиции. Заболевание, ассоциированное с хемокиновым рецептором, может включать любое заболевание, нарушение или состояние, которое непосредственно или опосредованно связано с экспрессией или активностью хемокина или хемокинового рецептора. Заболевание, ассоциированное с хемокиновым рецептором или хемокином, может также включать любое заболевание, нарушение или состояние, которое может быть предотвращено, облегчено или излечено посредством модуляции активности хемокинового рецептора. Примеры заболеваний, нарушений и состояний, ассоциированных с хемокинами или хемокиновым рецептором, включают воспаление и воспалительные заболевания, метаболические заболевания, иммунные нарушения и рак. В некоторых вариантах осуществления, заболевание, ассоциированное с хемокиновым рецептором, представляет собой вирусную инфекцию, такую как ВИЧ-инфекция. Примеры воспалительных заболеваний включают заболевания, имеющие воспалительный компонент, такие как астма, сезонный и круглогодичный аллергический ринит, синусит, конъюнктивит, возрастную пятнистую дегенерацию, пищевую аллергию, скомброидное отравление, псориаз, крапивницу, зуд, экзему, воспалительное заболевание кишечника, тромбическое заболевание, воспаление среднего уха, цирроз печени, заболевание сердца, болезнь Альцгеймера, сепсис, рестеноз, атеросклероз, диабет типа II, метаболический синдром, рассеянный склероз, болезнь Крона, язвенный колит, заболевания легких, связанные с гиперчувствительностью, индуцированный лекарствами фиброз легких, хроническая обструктивная болезнь легких (ХОБЛ), ревматоидный артрит, нефрит, язвенный колит, атопический дерматит, инсульт, острое повреждение нерва, саркоидоз, гепатит, эндометриоз, невропатическая боль, пневмония с гиперчувствительностью, эозинофильная пневмония, гиперчувствительность отложенного типа, интерстициальная болезнь легких (ILD) (например, идиопатический фиброз легких, или ILD, ассоциированную с ревматоидным артритом, системную красную волчанку, анкилозирующий спондилит, системный склероз, синдром Сегрена, полимиозит или дерматомиозит) и т.п. Примеры иммунных нарушений включают ревматоидный артрит, псориатический артрит, системную красную волчанку, тяжелую псевдопаралитическую миастению, юношеский диабет; гломерулонефрит, аутоиммунный тиреоидит, отторжение пересаженного органа, включающее отторжение аллотрансплантата и заболевание трансплантат против хозяина. Примеры раковых заболеваний включают разновидности рака, такие как рак молочной железы, рак яичника, множественная миелома и т.п., которые характеризуются инфильтрацией макрофагов (например, макрофагов, ассоциированных с опухолью или TAMs) в опухоли или больные ткани. Как используется в данном описании, термин "контакт" относится к привнесению вместе указанных фрагментов в системе in vitro или системе in vivo. Например, "контакт" хемокинового рецептора с соединением изобретения включает введение соединения по настоящему изобретению лицу или пациенту, такому как человек, имеющему хемокиновый рецептор, а также, например, введение соединения по изобретению в образец, содержащий клеточный или очищенный препарат, содержащий хемокиновый рецептор. Как используется в данном описании, термин "индивид" или "пациент", используемый взаимозаменяемо, относится к любому животному, включая людей, предпочтительно мышам, крысам, другим грызунам, кроликам, собакам, кошкам, свиньям, крупному рогатому скоту, овцам, лошадям или приматам и, наиболее предпочтительно людям. Как используется в данном описании, фраза "терапевтически эффективное количество" относится к количеству активного соединения или фармацевтического средства, которое вызывает биологический или медицинский ответ, который считается значимым в ткани, системе, животном, субъекте или человеке с точки зрения исследователя, ветеринара, врача или другого клинициста, что включает один или несколько из следующих случаев: (1) профилактика заболевания; например, профилактика заболевания, состояния или нарушения у субъекта, который может быть предрасположен к заболеванию, состоянию или нарушению, но еще не сталкивался с заболеванием и не проявлял патологию и симптоматологию заболевания (неограничивающие примеры представляют собой профилактику заболеваний легких, связанных с гиперчувствительностью, индуцированного лекарствами фиброза легких, хронической обструктивной болезни легких (ХОБЛ), заболевания трансплантат против хозяина и/или отторжения аллотрансплантата после трансплантации, вирусной инфекции, резистентности к инсулину, атеросклероза или профилактику аллергических реакций, таких как атопический дерматит, гиперчувствительность отложенного типа или сезонный или круглогодичный аллергический ринит); (2) ингибирование заболевания и его развития/ например, ингибирование заболевания, состояния или нарушения у субъекта, который сталкивается или проявляет патологию или симптоматологию заболевания, состояния или нарушения (т.е. прекращение дальнейшего развития патологии и/или симптоматологии), такое как ингибирование воспалительного или аутоиммунного ответа при заболеваниях легких, связанных с гиперчувствительностью, индуцированном лекарствами фиброзе легких, хронической обструктивной болезни легких (ХОБЛ), ревматоидном артрите, волчанке или псориазе или ингибирование развития атеросклеротических бляшек, болезни Альцгеймера, пятнистой дегенерации или развитии резистентности к инсулину до диабетического состояния, или ингибирование опухолевого роста, или стабилизация вирусной нагрузки в случае вирусной инфекции; и (3) облегчение симптомов заболевания; например, облегчение симптомов заболевания, состояния или нарушения у субъекта, который сталкивается или проявляет патологию или симптоматологию заболевания, состояния или нарушения (т.е. обращение патологии и/или симптоматологии), такое как уменьшение аутоиммунного ответа при заболеваниях легких, связанных с гиперчувствительностью, индуцированном лекарствами фиброзе легких, хронической обструктивной болезни легких (ХОБЛ), ревматоидном артрите, волчанке или псориазе, или уменьшение опухоли, ассоциированной с раком, или снижение вирусной нагрузки в случае вирусной инфекции. Одно или более дополнительных фармацевтических средств таких как, например, противовирусные средства, антитела, противовоспалительные средства, стимуляторы секреции инсулина и сенсибилизаторы, средства, модулирующие сывороточные липиды и переносчики липидов и/или иммуносуппрессоры могут применяться в сочетании с соединениями настоящего изобретения для лечения заболевания, нарушения или состояний, ассоциированных с хемокиновыми рецепторами. Средства могут быть объединены с настоящими соединениями в виде разовой или непрерывной лекарственной формы или средства могут вводиться одновременно или последовательно в виде раздельных лекарственных форм. Подходящие противовирусные средства, предполагаемые для применения в сочетании с соединениями настоящего изобретения, могут включать в себя нуклеозидные и нуклеотидные ингибиторы обратной транскриптазы (NRTIs), ненуклеозидные ингибиторы обратной транскриптазы (NNRTIs), ингибиторы протеаз, ингибиторы входа, ингибиторы слияния, ингибиторы созревания и другие противовирусные лекарства. Примеры подходящих NRTIs включают зидовудин (AZT); диданозин (ddl); зальцитабин (ddC)/ ставудин (d4T); ламивудин (ЗТС); абакавир (1592U89); адефовир дипивоксил [bis(РОМ)-РМЕА]; лобукавир (BMS-180194); ВСН-10652; эмитрицитабин [(-)-FTC]; 6eTa-L-FD4 (также называемый 6eTa-L-D4C и именуемый бета-Ь-2', 3'-диклеокси-5-фтор-цитиден); DAPD, ((-)-бета-0-2,б,-диамино-пурин диоксолан); и лоденозин (FddA). Типичные NNRTIs включают невирапин (BI-RG-587); делавирадин (ВНАР, U-90152); эфавиренц (DMP-266); PNU-142721; AG-1549; МКС-442 (1-(этокси-метил)-5-(1-метилэтил)-6- (фенилметил)-(2,4(1Н,ЗН)пиримидил недион); и (+)-каланолид А (NSC-675451) и В. Типичные подходящие ингибиторы протеаз включают саквинавир (Ro 31-8959); ритонавир (АВТРЕТ-538); индинавир (МК-639); нелфинавир (AG-1343); ампренавир (141W94); лазинавир (BMS-234475); DMP-450; BMS-2322623; АВТРЕТ-378; и AG-1 549. Другие противовирусные средства включают гидроксимочевину, рибавирин, IL-2, IL-12, пентафузид, энфувиртид, С-34, циклотриазадисульфонамид CADA, РА-4 57 и Yissura Project No.11607. В некоторых вариантах осуществления, противовоспалительные или анальтезирующие средства, предполагаемые для применения в сочетании с соединениями настоящего изобретения, могут включать, например, агонист опиатов, ингибитор липоксигеназ, такой как ингибитор 5-липоксигеназы, ингибитор циклооксигеназы, такой как ингибитор циклооксигеназы-2, ингибитор интерлейкинов, такой как ингибитор интерлейкина-1, ингибитор ФНО, такой как инфликсимаб, этанерцепт или адалимумаб, антагонист NNMA, ингибитор оксида азота или ингибитор синтеза оксида азота, нестероидное противовоспалительное средство или цитокин-супрессорное противовоспалительное средство, например, такие как ацетаминофен, аспирин, кодеин, фентанил, ибупрофен, индометацин, кетодлолак, морфин, напроксен, фенацетин, пироксикам, стероидный анальгетик, суфентанил, сунлиндак, тенидап и т.п. Сходным образом, растворимые соединения могут вводиться вместе с болеутоляющим средством; потенцирующим средством, таким как кофеин, Н2-антагонист, симетикон, гидроксид алюминия или магния; противоотечным средством, таким как фенилэфрин, фенилпропаноламин, псевдоэфедрин, оксиметазолин, эпинефрин, нафазолин, ксилометазолин, пропилгексэдфин или лево-дезоксиэфедрин; средство против кашля, такое как кодеин, гидрокодон, карамифен, карбетапентан или декстраметорфан; диуретик; и седативное или неседативное антигистаминное средство. В некоторых вариантах осуществления, фармацевтические средства, предполагаемые для применения в сочетании с соединениями по настоящему изобретению могут включать, но не ограничиваясь ими (а) антагонисты VLA-4, такие как антагонисты, описанные в патенте США 5510332, W095/15973, W096/01644, W096/06108, W096/20216, W096/229661, W096/31206, W096/4078, W097/030941, W097/022897, WO 98/426567, W098/53814, W098/53817, W098/538185, W098/54207 и W098/58902; (Ь) стероиды, такие как беклометазон, метилпреднизолон, бетаметазон, преднизон, дексаметазон и гидрокортизон; (с) иммуносуппрессоры, такие как циклоспорин, такролимус, рапарницин и другие иммуносуппрессоры типа FK506; (d) антигистамины (антагонисты HI-гистамина), такие как бромфенирамин, хлорфенирамин, дексахлорфенирамин, трипролидин, клемастин, дифенгидрамин, дифенилпиралин, трипеленнамин, гидроксизин, метдилазин, прометазин, тримепразин, азатадин, ципрогептадин, антазолин, фенирамина пириларнин, астернизол, терфенадин, лоратадин, цетиризин, фексфенадин, десеарбоэтоксилоратадин и т.п; (е) нестероидные антиастматики, такие как тербуталин, метапротеренол, фенотерол, изоэтаиин, албутерол, битолтерол, пирбутерол, теофиллин, кромолин натрия, атропин, бромид ипратропия, антагонисты лейкотриенов (например, зафирлукаст, монтелукаст, пранлукаст, иралукаст, побилукаст, SKB-106203), ингибиторы биосинтеза лейкотриенов (например, зилейтон, BAY-1005); (f) нестероидные противовоспалительные средства (NSAIDs), такие как производные пропионовой кислоты (например, алминопрофен, беноксапрофен, буклоксиновая кислота, карпрофен, фенбуфен, фенопрофен, флупрофен, флурбипрофен, ибупрофен, индопрофен, кетопрофен, миропрофен, напроксен, оксапрозин, пирпрофен, пранопрофен, супрофен, тиапрофеновая кислота и тиоксапрофен), производные уксусной кислоты (например, индометацин, ацернетацин, алклофенак, клидинак, диклофенак, фенклофенак, фенклозиновая кислота, фентиазак, фурофенак, ибуфенак, изоксепак, окспинак, сулиндак, тиопинак, толметин, зидометацин и зомерипак), производные фенарниновой кислоты (флуфенапниновая кислота, меклофенаминовая кислота, рнефенаминовая кислота, нифлуминовая кислота и толфвенарниновая кислота), производные бифенилкарбоновой кислоты (дифлунизал и флуфенизал), оксикарны (изоксикарн, пироксикам, судоксикам и теноксикан), салицилаты (ацетилсалициловая кислота, сульфасалазин) и пиразолоны (апазон, безпиперилон, фепразон, мофебутазон, оксифенбутазон, фенилбутазон); (д) ингибиторы циклооксигеназы-2 (СОХ-2); (h) ингибиторы фосфодиэстеразы типа IV (PDE-IV); (i) другие антагонисты хемокиновых рецепторов, особенно, CXCR-4, CCR1, CCR2, CCR3 и CCR5; (j) средства понижения холестерина, такие как ингибиторы HMG-CoA редуктазы (ловастатин, сирривастатин и правастатин, флувастатин, аторвастатин и другие статины), секвестранты (холестирамин и колестипол), никотиновая кислота, производные фенофибриновой кислоты (гемфиброзил, клофибрат, фенофибрат и бензафибрат) и пробукол; (к) противовоспалительные биологические средства, такие как средства анти-ФИО-терапии, антитела против IL-1 рецептора, CTLA-4Ig, против CD20 и против-VLA4; (1) противодиабетические средства, такие как инсулин, сульфонилмочевины, бигуаниды (метформин), ингибиторы U.-глюкозидазы (акарбозы) и орлитазоны (троглитазон и пиоглитазон); (т) препараты бета интерферона (интерферон бета-1о., интерферон бета-1 Р) ; (п) другие соединения, такие как аминосалициловые кислоты, антиметаболиты, такие как азатиоприн и 6-меркаптопурин и цитотоксические противораковые химиотерапевтические средства. Массовое отношение соединения настоящего изобретения ко второму активному ингредиенту может изменяться и будет зависеть от эффективной дозы каждого ингредиента. Например, антагонист CCR2 может применяться в сочетании с противовоспалительным средством при лечении воспаления, метаболического заболевания, аутоиммунного заболевания, рака или вирусной инфекции для улучшения ответа на лечение по сравнению с ответом на терапевтическое средство в отдельности, без обострения его токсических эффектов. Дополнительные или синергетические эффекты являются желательными результатами сочетания антагониста CCR2 настоящего изобретения с дополнительным средством. Фармацевтические готовые формы и дозированные формы При использовании в качестве фармацевтических средств соединения формулы I могут вводиться в виде фармацевтических композиций. Эти композиции могут быть получены путем, хорошо известным в области фармацевтики, и могут вводиться различными путями, в зависимости от того, является ли желательным местное или системное лечение, и от области, подлежащей лечению. Введение может быть местным (включая офтальмологическое и на слизистые мембраны, включая интраназальную, вагинальную и ректальную доставку), легочным (например, ингаляцией или инсуффляцией порошков или аэрозолей, включая распылитель; интратрахеальным, интраназальным, эпидермальным и чрезкожным), пероральным или парентеральным. Парентеральное введение включает внутривенное, внутриартериальное, подкожное, внутрибрюшинное, внутримышечное или инъекционное или инфузионное; или внутричерепное, например, интратекальное или внутрижелудочковое введение. Парентеральное введение может осуществляться в виде разовой болюсной дозы или, например, посредством непрерывного перфузионного насоса. Фармацевтические композиции и готовые формы для местного введения могут включать чрезкожные пластыри, мази, лосьоны, кремы, гели, капли, суппозитории, спреи, жидкости и порошки. Общепринятые фармацевтические носители, водные, порошковые или масляные основы, загустители и т.п. могут быть необходимыми или желательными. Могут также применяться презервативы с покрытием, перчатки и т.п. Данное изобретение также включает фармацевтические композиции, которые содержат в качестве активного ингредиента, одно или более из соединений формулы I, приведенной выше, в сочетании с одним или более фармацевтически приемлемых носителей. При создании композиций изобретения активный ингредиент типовым образом смешивают с эксципиентом, разбавляют эксципиентом или помещают внутрь такого носителя в виде, например, капсулы, саше, бумажки или другого контейнера. Когда эксципиент служит в качестве разбавителя, он может представлять собой твердое, полутвердое или жидкое вещество, которое действует в качестве переносчика, носителя или среды для активного ингредиента. Таким образом, композиции могут быть представлены в виде таблеток, пилюль, порошков, лепешек, саше, облаток, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозолей (в виде твердого вещества или в жидкой среде), мазей, содержащих, например, вплоть до 10% масс, активного соединения, мягких и твердых желатиновых капсул, суппозиториев, стерильных растворов для инъекций и стерильно упакованных порошков. При получении готовой формы активное соединение может быть размолото с получением соответствующего размера частиц перед объединением с другими ингредиентами. Если активное соединение является по существу нерастворимым, оно может быть размолото до размера частиц менее, чем 200 меш. Если активное соединение является по существу водорастворимым, размер частиц может быть отрегулирован размолом с получением по существу однородного распределения в готовой форме, например, приблизительно 40 меш. Некоторые примеры подходящих эксципиентов включают лактозу, декстрозу, сахарозу, сорбит, маннит, крахмалы, камедь акации, фосфат кальция, альгинаты, трагакант, желатин, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, воду, сироп и метилцеллюлозу. Готовые формы могут дополнительно включать: смазывающие средства, такие как тальк, стеарат магния и минеральное масло; увлажняющие средства; эмульгирующие и суспендирующие средства; консерванты, такие как метил- и пропилгидроксибензоаты; подсластители; и отдушки. Композиции изобретения могут быть сформированы таким образом, чтобы получить быстрое, замедленное или отложенное высвобождение активного ингредиента после введения пациенту с использованием методик, известных в данной области. Композиции могут быть сформированы в единичную дозированную лекарственную форму, причем каждая дозировка содержит приблизительно от 5 до приблизительно 1000 мг (1 г), более обычно, приблизительно от 100 до приблизительно 500 мг активного ингредиента. Термин "единичные дозированные формы" относится к физически дискретным единицам, пригодным в качестве разовых дозировок для людей и других млекопитающих, причем каждая единица содержит заранее определенное количество активного вещества для получения желательного терапевтического эффекта, в сочетании с подходящим фармацевтическим эксципиентом. В некоторых вариантах осуществления соединения или композиции изобретения содержат от приблизительно 5 до приблизительно 50 мг активного ингредиента. Рядовой специалист в данной области оценит, что эти варианты осуществления относятся к соединениям или композициям, содержащим от приблизительно 5 до приблизительно 10, от приблизительно 10 до приблизительно 15, от приблизительно 15 до приблизительно 20, от приблизительно 20 до приблизительно 25, от приблизительно 25 до приблизительно 30, от приблизительно 30 до приблизительно 35, от приблизительно 35 до приблизительно 40, от приблизительно 4 0 до приблизительно 45, или от приблизительно 4 5 до приблизительно 50 мг активного ингредиента. В некоторых вариантах осуществления, соединения или композиции изобретения содержат от приблизительно 50 до приблизительно 500 мг активного ингредиента. Рядовой специалист в данной области оценит, что эти варианты осуществления относятся к соединениям или композициям, содержащим от приблизительно 50 до приблизительно 75, от приблизительно 75 до приблизительно 100, от приблизительно 100 до приблизительно 125, от приблизительно 125 до приблизительно 150, от приблизительно 150 до приблизительно 175, от приблизительно 175 до приблизительно 200, от приблизительно 200 до приблизительно 225, от приблизительно 225 до приблизительно 250, от приблизительно 250 до приблизительно 275, от приблизительно 275 до приблизительно 300, от приблизительно 300 до приблизительно 325, от приблизительно 325 до приблизительно 350, от приблизительно 350 до приблизительно 375, от приблизительно 375 до приблизительно 400, от приблизительно 4 00 до приблизительно 425, от приблизительно 425 до приблизительно 4 50, от приблизительно 450 до приблизительно 475, или от приблизительно 475 до приблизительно 500 мг активного ингредиента. В некоторых вариантах осуществления, соединения или композиции изобретения содержат от приблизительно 500 до приблизительно 1000 мг активного ингредиента. Рядовой специалист в данной области оценит, что эти варианты осуществления относятся к соединениям или композициям, содержащим от приблизительно 500 до приблизительно 550, от приблизительно 550 до приблизительно 600, от приблизительно 600 до приблизительно 650, от приблизительно 650 до приблизительно 700, от приблизительно 700 до приблизительно 750, от приблизительно 750 до приблизительно 800, от приблизительно 800 до приблизительно 850, от приблизительно 850 до приблизительно 900, от приблизительно 900 до приблизительно 950, или от приблизительно 950 до приблизительно 1000 мг активного ингредиента. Активное соединение может быть эффективным в широком диапазоне дозировки и, в целом, вводится в фармацевтически эффективном количестве. Следует понимать, однако, что количество соединения, вводимое в действительности, будет определяться лечащим врачом, в соответствии с учитываемыми обстоятельствами, включающими состояние, подлежащее лечению, выбранный путь введения, действительное вводимое соединение, возраст, массу тела и ответ индивидуального пациента, тяжесть симптомов пациента и т.п. Для получения твердых композиций, таких как таблетки, основной активный ингредиент смешивают с фармацевтическим эксципиентом для образования твердой композиции предварительной готовой формы, содержащей гомогенную смесь соединения настоящего изобретения. Ссылка на эти композиции предварительной готовой формы как гомогенные означает, что активный ингредиент обычно равномерно распределен по всей композиции, таким образом, чтобы композиция могла быть легко подразделена на равноэффективные единичные дозированные формы, такие как таблетки, пилюли и капсулы. Эта твердая предварительная готовая форма далее подразделяется на единичные дозированные формы типа, описанного выше, содержащие, например, от 0,1 до приблизительно 1000 мг активного ингредиента настоящего изобретения. Таблетки или пилюли настоящего изобретения могут быть покрыты или иным образом составлены с получением дозированной формы, обеспечивающей преимущество пролонгированного действия. Например, таблетка или пилюля может содержать внутренний компонент дозировки и внешний компонент дозировки, причем последний находится в виде конверта для предыдущего. Два компонента могут быть разделены энтеросолюбильным слоем, который служит для сопротивления разрушению в желудке и позволяет внутреннему компоненту проходить в двенадцатиперстную кишку или иметь задержанное высвобождение. Для таких энтерасолюбильных слоев или покрытий могут применяться разнообразные вещества, причем такие вещества включают полимерные кислоты и смеси полимерных кислот с такими веществами как шеллак, цетиловый спирт и ацетат целлюлозы. Жидкие формы, в которые соединения и композиции настоящего изобретения могут включаться для перорального введения или посредством инъекции, включают водные растворы, подходящим образом ароматизированные сиропы, водные или масляные суспензии и ароматизированные эмульсии со съедобными маслами, такими как хлопковое масло, кунжутное масло, кокосовое масло или арахисовое масло, а также эликсиры и сходные фармацевтические переносчики. Композиции для ингаляции или инсуффляции включают растворы и суспензии в фармацевтически приемлемых, водных или органических растворителях или их смесях, и порошки. Жидкие или твердые композиции могут содержать подходящие фармацевтически приемлемые эксципиенты, как описано выше. В некоторых вариантах осуществления, композиции вводятся посредством перорального или назального респираторного пути для местного или системного эффекта. Композиции могут быть распылены при использовании инертных газов. Распыленные растворы могут вдыхаться непосредственно из распыляющего устройства, или распыляющее устройство может присоединяться к надеваемым на лицо маскам или дыхательному прибору с попеременным положительным давлением. Композиции в виде раствора, суспензии или порошка могут вводиться перорально или назально из устройств, которые доставляют готовую форму соответствующим образом. Количество соединения или композиции, вводимое пациенту будет изменяться в зависимости от того, что вводится, цели введения, как, например, профилактика или терапия, состояния пациента, способа введения и т.п. В терапевтических приложениях, композиции могут вводиться пациенту, уже страдающему от заболевания в количестве, достаточном для излечения или, по меньшей мере, частичного прекращения симптомов заболевания и его осложнений. Эффективные дозы будут зависеть от состояния излечиваемого заболевания, а также от заключения лечащего клинициста в зависимости от таких факторов, как тяжесть заболевания, возраст, масса тела и общее состояние пациента и т.п. Композиции, вводимые пациенту могут быть представлены в виде фармацевтических композиций, описанных выше. Эти композиции могут быть стерилизованы общепринятыми методами стерилизации или могут быть стерильно отфильтрованы. Водные растворы могут быть упакованы для применения как таковые или лиофилизированы, причем лиофилизированный препарат объединяют со стерильным водным носителем перед введением. рН препаратов соединения типично будет иметь значения между 3 и 11, более предпочтительно от 5 до 9 и, наиболее предпочтительно, от 7 до 8. Следует понимать, что применение некоторых из вышеуказанных эксципиентов, носителей или стабилизаторов будет приводить в результате к образованию фармацевтических солей. Терапевтическая дозировка соединений настоящего изобретения может изменяться в соответствии, например, с конкретным применением, для которого проводится лечение, путем введения соединения, здоровьем и состоянием пациента и заключением лечащего врача. Пропорция или концентрация соединения по изобретению в фармацевтической композиции может изменяться в зависимости от ряда факторов, включающих дозировку, химические характеристики (например, гидрофобность) и путь введения. Например, соединения по изобретению могут быть предоставлены в водном физиологическом буферном растворе, содержащем приблизительно от 0,1 до приблизительно 10% м/об соединения для парентерального введения. Некоторые типичные интервалы доз составляют от приблизительно 1 мкг/кг до приблизительно 1 г/кг массы тела в день. В некоторых вариантах осуществления, интервал дозы составляет от приблизительно 0,01 мг/кг до приблизительно 100 мг/кг массы тела в день. Дозировка, вероятно, зависит от таких переменных, как тип и степень развития заболевания или нарушения, общее состояние здоровья конкретного пациента, относительная биологическая эффективность выбранного соединения, эксципиент готовой формы и ее путь введения. Эффективные дозы могут быть экстраполированы на основании кривых доза-ответ, полученных в результате исследований на тест-системах in vitro или животных моделях. Соединения по изобретению могут также быть сформулированы в сочетании с одним или несколькими дополнительными активными ингредиентами, которые могут включать, любое фармацевтическое средство, такое как антитела, иммуносуппрессоры, противовоспалительные средства, химиотерапевтические средства, средства, снижающие уровень липидов, средства, повышающие уровень липопротеинов высокой плотности (HDL), средства стимуляции секреции инсулина или сенсибилизаторы, лекарства, применяемые для лечения ревматоидного артрита и т.п. Режим лечения ревматоидного артрита (РА) Пациенты с ревматоидным артритом (РА), проходящие агрессивный курс лечения средствами модификации заболевания (метотрексат, антималярийные препараты, золото, пеницилламин, сульфасалазин, дапсон, лефлунамид или биологическое средство) могут достигать меняющихся степеней контроля заболевания, включая полные ремиссии. Эти клинические ответы ассоциированы с улучшением в стандартизованной балльной оценке активности заболевания, конкретно критериев ACR, которые включают: боль, функцию, количество болезненных суставов, количество опухших суставов, общая оценка пациента, общая оценка врача, лабораторные измерения воспаления (CRP и ESR) и радиологическая оценка структурного повреждения сустава. Требуют введения лекарства, модифицирующие протекающее заболевание (DMARDs), для поддержания оптимального благоприятного эффекта. Хроническое дозирование этих средств связано со значительной токсичностью и истощением защитных сил организма хозяина. Дополнительно пациенты часто становятся невосприимчивыми к конкретной терапии, и требуется альтернативный режим. По этим причинам новая, эффективная терапия, которая позволяет отменить стандартные DMARDs, была бы клинически важным достижением. Пациенты со значительным ответом на средства анти-ФНО-терапии (инфликсимаб, этанерцепт, адалимумаб), средства анти- IL-1-терапии (кинарет) или другие модифицирующие заболевание антиревматические лекарства (DMARDs) , включая, но не ограничиваясь указанным, метотрексат, циклоспорин, соли золота, антималярийные препараты, пеницилламин или лефлунамид, которые достигли клинической ремиссии заболевания, могут проходить курс лечения веществом, которое ингибирует экспрессию и/или активность CCR2, включая, например, нуклеиновые кислоты (например, антисмысловые или сиРНК молекулы), белки (например, антитела против CCR2), низкомолекулярные ингибиторы (например, соединения, раскрытые в данном описании, и другие ингибиторы хемокинового рецептора, известные в данной области). В некоторых вариантах осуществления вещество, которое ингибирует экспрессию и/или активность CCR2, представляет собой низкомолекулярный ингибитор (или антагонист) CCR2. Антагонист CCR2 может дозироваться перорально q.d. или b.i.d при дозе, не превышающей приблизительно 500 мг в день. Пациентам может быть отменена их проводимая терапия или уменьшена ее дозировка, и лечение может поддерживаться антагонистом CCR2. Лечение пациентов сочетанием антагониста CCR2 и их текущей терапии может осуществляться, например, в течение приблизительно от одного до приблизительно двух дней, перед прерыванием или снижением дозы DMARD и продолжением лечения антагонистом CCR2. Имеются многочисленные преимущества замены традиционных DMARDS антагонистами CCR2. Традиционные DMARDs обладают серьезными кумулятивными дозо-ограничивающими побочными эффектами, наиболее частым из которых является повреждение печени, а также иммуносуппрессорные действия. Ожидают, что антагонизм CCR2 будет обладать улучшенным продолжительным профилем безопасности и не будет иметь сходных иммуносуппрессорных свойств, ассоциированных с традиционными DMARDs. Дополнительно, время полужизни биологических средств типично составляет дни или недели, что является проблемой при рассмотрении неблагоприятных реакций. Ожидается, что время полужизни перорально биодоступного антагониста CCR2 будет составлять порядок часов, так что риск от продолжительного воздействия лекарства после неблагоприятного события является очень минимальным, по сравнению с биологическими средствами. Также применимые в настоящее время биологические средства (инфликсимаб, этанерцепт, адалимумаб, кинарет) обычно дают либо внутривенно (в.в.), либо подкожно (п.к.), что требует введения доктором или само-инъекцию пациентом. Это приводит к возможности реакции от инфузии или реакциям на участке инъекции. Этих эффектов можно избежать, используя перорально вводимый антагонист CCR2. Режим лечения диабета и резистентности к инсулину Диабет типа 2 является одной из ведущих причин заболеваемости и смертности в западных странах. У преобладающего большинства пациентов, заболевание характеризуется дисфунцией панкреатических бета-клеток, сопровождаемой резистентностью к инсулину в печени и в периферических тканях. На основании первичных механизмов, которые ассоциированы с заболеванием, доступны два общих класса пероральных терапий для лечения диабета типа 2: стимуляторы секреции инсулина (сульфонилмочевины, такие как глибурид) и сенсибилизаторы инсулина (метформин и тиазолидиндионы, такие как розиглитазон). Было показано, что комбинационная терапия, которая направлена на оба механизма, управляет метаболическими дефектами этого заболевания и во многих случаях можно показать, что она снижает необходимость во введении экзогенного инсулина. Однако со временем, резистентность к инсулину часто прогрессирует, приводя к необходимости дальнейшего дополнения инсулина. Кроме того, было продемонстрировано, что преддиабетическое состояние, на которое ссылаются как на метаболический синдром, характеризуется нарушенной переносимостью глюкозы, особенно в сочетании с ожирением. У большинства пациентов, у которых развивается диабет типа 2, начинается развитие резистентности к инсулину, причем у этих пациентов имеет место гипергликемия, когда они не могут далее выносить степень гиперинсулинемии, необходимый для предотвращения потери гомеостаза глюкозы. Наступление компонента резистентности к инсулину является в высокой степени предсказательным в отношении наступления заболевания и ассоциирован с увеличением риска развития диабета типа 2, гипертензии и коронарной болезни сердца. Одним из основных коррелятов нарушенной переносимости глюкозы и развития от инсулин-резистентного состояния до диабета типа 2 является наличие центрального ожирения. Большинство пациентов с диабетом типа 2 являются тучными, и само ожирение ассоциировано с резистентностью к инсулину. Ясно, что центральное ожирение является основным фактором риска для развития резистентности к инсулину, ведущей к диабету типа 2, с предположением, что сигналы от висцерального жира вносят вклад в развитие резистентности к инсулину и прогрессирование к заболеванию. В дополнение к секретируемым белковым факторам ожирение индуцирует клеточный воспалительный ответ, при котором макрофаги, производимые костным мозгом, накапливаются в жировых депо, становясь макрофагами жировой ткани. Макрофаги жировой ткани накапливаются пропорционально измерениям ожирения. Макрофаги, инфильтрованные тканью, являются источником многих воспалительных цитокинов, для которых была продемонстрирована способность индуцировать резистентность к инсулину в адипоцитах. Жировая ткань продуцирует МСР-1 пропорционально ожирению, что позволяет предположить, что его активность посредством передачи сигналов через CCR2 также может играть важную роль в накоплении макрофагов в жировой ткани. Неизвестно, является ли взаимодействие MCP-1/CCR2 непосредственно ответственным за мобилизацию моноцитов к жировой ткан, будет ли сниженная мобилизация макрофагов к жировой ткани у людей непосредственно приводить со сниженной выработке провоспалительных молекул и связана ли выработка провоспалительных молекул непосредственно с резистентностью к инсулину. Пациенты, у которых наблюдают резистентность к инсулину либо преддиабетическую (нормогликемическую), либо диабетическую (гипергликемическую), могут проходить курс лечения веществом, которое ингибирует экспрессию и/или активность CCR2, включая, например, нуклеиновые кислоты (например, антисмысловые или молекулы малых интерферирующих PHK-siRNA), белки (например, антитела против CCR2), низкомолекулярные ингибиторы (например, соединения, раскрытые в данном описании, и другие ингибиторы хемокинового рецептора, известные в данной области). В некоторых вариантах осуществления, вещество, которое ингибирует экспрессию и/или активность CCR2 представляет собой низкомолекулярный ингибитор (или антагонист) CCR2. Антагонист CCR2 может дозироваться перорально q.d. или b.i.d при дозе, не превышающей приблизительно 500 мг в день. Пациентам может быть отменена их проводимая терапия или уменьшена ее дозировка, и лечение может поддерживаться антагонистом CCR2. Альтернативно, лечение антагонистом CCR2 может применяться для дополнения текущей терапии, для усиления ее эффективности или для предотвращения развития в дальнейшую инсулиновую зависимость. Имеются многочисленные преимущества замены или дополнения традиционных средств антагонистами CCR2. Такие средства могут быть применимыми, например, для предотвращения развития от преддиабетического, инсулинрезистентного состояния до диабетического состояния. Такие средства могут снижать или заменять необходимость в применении сенсибилизаторов инсулина, с их присущими токсическими свойствами. Такие средства могут также снижать необходимость в дополнении экзогенного инсулина или пролонгировать период до наступления этой потребности. Режим лечения атеросклероза Атеросклероз представляет собой состояние, характеризуемое отложением жировых веществ в стенках артерий. Бляшка охватывает такие отложения жировых веществ, холестерина, продуктов жизнедеятельности клеток, кальций и другие вещества, которые встраиваются во внутреннюю выстилку артерии. Бляшки могут вырастать достаточно крупными, чтобы значительно снижать кровоток через артерию. Однако более значительное повреждение происходит, когда бляшка становится неустойчивой и разрывается. Бляшки, которые разрываются, приводят к образованию тромбов, которые могут блокировать кровоток или разрушаться и перемещаться к другим частям тела. Если тромб блокирует кровеносный сосуд, который обеспечивает снабжение сердца, он вызывает сердечный приступ. Если он блокирует кровеносный сосуд, который обеспечивает питание мозга, это вызывает инсульт. Атеросклероз является медленным, комплексным заболеванием, которое типично начинается в детстве и часто прогрессирует по мере того, как люди становятся старше. Высокий уровень холестерина в крови является основным фактором риска для коронарной болезни сердца. На основании того, что холестерин является первичной композицией бляшки, развитие образования бляшки управляется снижением циркулирующего холестерина или повышением уровня холестерин-переносящих липопротеинов высокой плотности (HDL). Циркулирующий холестерин может быть снижен, например, ингибированием его синтеза в печени с использованием или снижением поступления из пищи. Такие медикаменты, которые действуют через этот механизм, могут включать лекарственные средства, которые применяют, чтобы понизить высокие уровни холестерина: поглотители желчных кислот, ингибиторы синтеза липопротеинов, ингибиторы синтеза холестерина и производные фибриновой кислоты. Уровни циркулирующих HDL могут дополнительно быть увеличены, посредством введения, например, пробукола или высоких доз ниацина. Было показано, что терапия, которая направлена на множественные механизмы, замедляет развитие заболевания и прогрессирование к разрыву бляшки. Атеросклероз в типичном случае сопровождается клеточным воспалительным ответом, при котором макрофаги, производимые костным мозгом, накапливаются в жировых полосках вдоль стенки сосуда, становясь пенистыми клетками. Пенистые клетки являются источником многих воспалительных цитокинов, для которых была продемонстрирована способность индуцировать развитие бляшки, и ферментов, которые могут стимулировать дестабилизацию бляшки. Атеросклеротическая ткань также продуцирует МСР-1, что позволяет предположить, что его активность посредством подачи сигналов через CCR2 также может играть важную роль в накоплении макрофагов в виде пенистых клеток в бляшках. Продемонстрировано, что CCR2-/- мыши имеют значительно пониженный уровень макрофагов в жировых полосках, генерируемый в качестве результата высокожировой диеты или генетического изменения в метаболизме липидов. Пациенты, у которых наблюдают высокий циркулирующий холестерин, низкие HDL, или повышенный циркулирующий CRP, или у которых визуализируют присутствие бляшки в стенке сосуда или с любыми другими очевидными признаками наличия атеросклероза, могут проходить курс лечения веществом, которое ингибирует экспрессию и/или активность CCR2, включая, например, нуклеиновые кислоты (например, молекулы антисмысловых или малых интерферирующих РНК), белки (например, антитела против CCR2), низкомолекулярные ингибиторы (например, соединения, раскрытые в данном описании, и другие ингибиторы хемокинового рецептора, известные в данной области). В некоторых вариантах осуществления, вещество, которое ингибирует экспрессию и/или активность CCR2, представляет собой низкомолекулярный ингибитор (или антагонист) CCR2. Антагонист CCR2 может дозироваться перорально q.d. или b.i.d при дозе, не превышающей приблизительно 500 мг в день. Пациентам может быть отменена проводимая им терапия или уменьшена ее дозировка, и лечение может поддерживаться антагонистом CCR2. Альтернативно, лечение антагонистом CCR2 может применяться для дополнения текущей терапии с целью усиления ее эффективности для, например, предотвращения развития бляшки, стабилизации уже сформированной бляшки или индуцирования регрессии бляшки. Имеются многочисленные преимущества замены или дополнения традиционных средств антагонистами CCR2. Такие средства могут быть применимыми, например, для предотвращения развития бляшки до стадии нестабильности с ассоциированным риском разрыва бляшки. Такие средства могут снижать или заменять необходимость в применении лекарств, модифицирующих холестерин, или лекарств повышающих HDL, с присущими им токсическими свойствами, включающими, но, не ограничиваясь ими, гиперемию, повреждение печени и повреждение мыщц, такое как миопатия. Такие средства могут также снижать необходимость в хирургическом вмешательстве для открытия стенки сосуда или пролонгировать период до этого вмешательства или до тех пор, когда потребуется применение антикоагулянтов для ограничения повреждения вследствие возможного разрыва бляшки. Меченые соединения и способы аналитического тестирования Еще один аспект настоящего изобретения относится к соединениям формулы I, меченным флуоресцентным красителем, спиновой меткой, тяжелым металлом или радиоактивным изотопом, которые могут быть полезны не только при визуализации, но также в аналитических тестах, как in vitro так и in vivo, для локализации и количественной оценки хемокинового рецептора в образцах тканей, включая человеческие, и для идентификации лигандов хемокинового рецептора посредством ингибирования связывания меченого соединения. Соответственно, настоящее изобретение включает аналитические тесты хемокинового рецептора, которые содержат такие меченые соединения. Настоящее изобретение дополнительно включает изотопно-меченые соединения формулы I. "Изотопно" или "радиоактивно-меченое" соединение представляет собой соединение по изобретению, где один или более атомов замещены или заменены атомом, имеющим атомную массу или массовое число, отличные от атомной массы или массового числа, типично встречающихся в природе (т.е. природных). Подходящие радионуклиды, которые могут быть включены в соединения настоящего изобретения, включают, но, не ограничиваясь ими, 2Н (также пишут как D для дейтерия) , 3Н (также пишут как Т для трития) , ПС, 13С, 14С, 13N, 15N, 150, 170, 180, 18F, 35S, 36С1, 82Вг, 75Вг, 76Вг, 77Вг, 1231 , 1241, 1251 и 1311. Радионуклид, который включают в растворимые меченные радиоактивным изотопом соединения будет зависеть от конкретного применения такого меченного радиоактивным изотопом соединения. Например, для мечения in vitro хемокинового рецептора и тестов на конкурентное связывание, соединения, которые включают 3Н, 14 ЙР IPS 1*31 С, Br, I, I, "S в целом, будут наиболее применимыми. Для применений, связанных с радиоактивной визуализацией, ПС, 18F, Х2Ъ1 , 1231 , 1241 , 1311 , 75Вг, 7бВг или 77Вг в целом, будут наиболее применимыми. Следует понимать, что "радиоактивно-меченое" или "меченое соединение" представляет собой соединение, в которое включен, по меньшей мере, один радионуклид. В некоторых вариантах осуществления радионуклид выбирают из группы, состоящей из 3Н, 14С, 125I, 35S и 82Вг. Синтетические способы для введения радиоактивных изотопов в органические соединения являются применимыми к соединениям по изобретению и хорошо известны в данной области. Меченое радиоактивным изотопом соединение по изобретению может применяться в скрининговом аналитическом тесте для идентификации/оценки соединения. В общих чертах, вновь синтезированное или идентифицированное соединение (т.е. тестируемое соединение) может быть оценено с точки зрения его способности снижать связывание меченного радиоактивным изотопом соединения по изобретению с хемокиновым рецептором. Соответственно, способность тестируемого соединения конкурировать с меченным радиоактивным изотопом соединением за связывание с хемокиновым рецептором непосредственно коррелирует с его сродством связывания. Наборы Настоящее изобретение также включает фармацевтические наборы, применимые, например, при лечении или предотвращении хемокин-ассоциированного заболевания, которые включают один или более контейнеров, содержащих фармацевтическую композицию, включающую в себя терапевтически эффективное количество соединения формулы I. Такие наборы могут дополнительно включать, если требуется, один или более из различных общепринятых фармацевтических компонентов набора, такие как, например, контейнеры с одним или более фармацевтически приемлемыми носителями, дополнительные контейнеры и т.д., как будет легко видно для специалистов в данной области. Инструкции, либо в виде вкладок или как этикетки, указывающие количества вводимых компонентов, инструкции по введению и/или инструкции по смешиванию компонентов могут также быть включены в набор. Изобретение будет описано более детально посредством конкретных примеров. Следующие примеры предлагаются для иллюстративных целей, и подразумевается, что они не ограничивают изобретение никаким образом. Квалифицированные специалисты в данной области легко распознают разнообразие некритических параметров, которые могут изменяться или модифицироваться с получением по существу тех же результатов. ПРИМЕРЫ Пример 1 4-[((1R,3S)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}циклопентил)амино]-1- пиридин-2-илциклогексанол Стадия А-1 BocHN Метил (1R,4S)-4-[ (трет-бутоксикарбонил)амино]циклопент-2-ен-1-карбоксилат К раствору {1R,45)-4-[(трет- бутоксикарбонил )амино]циклопент-2-ен-1-карбоновой кислоты (10,0 г, 44 ммоль) в ДМФА (25 мл) добавляют карбонат калия (6,33 г, 45,8 ммоль) с последующим добавлением метилйодида (4,0 мл, 64 ммоль). После перемешивания при комнатной температуре в течение ночи, реакционную смесь разбавляют EtOAc. Раствор промывают водой четыре раза и насыщенным солевым раствором один раз, сушат (MgS04) и концентрируют. Остаток сушат в условиях высокого вакуума в течение ночи с получением соединения заголовка (11 г, 99%). МС рассчитано для C12H19NO4: (М+Н)+ 242; найдено 142,1 (М-Вос+Н)+. ХН ЯМР (CDC13) 5 5, 86 (м, 2Н) , 4,90 (м, 1Н), 4,80 (м, 1Н) , 3,72 (с, ЗН) , 3,50 (м, 1Н) , 2,51 (м, 1Н), 1,86 (м, 1Н), 1,42 (с, 9Н). Стадия А-2 BocHN Метил (IS,4S)-4-[(трет-бутоксикарбонил)амино]-1- изопропилциклопент-2-ен-1-карбоксилат К 1,00М раствору гексаметилдисилазида лития в ТГФ (202 мл) при -78°С добавляют раствор метил (1.R, 4S)-4-[(трет-бутоксикарбонил) ймино] циклопент-2-ен-1-карбоксилата (22,10 г, 91,59 ммоль) в ТГФ (36,2 мл) в течение 10 мин. Раствор перемешивают при -78°С в течение 30 мин перед добавлением изопропилйодида (10,0 мл, 100 ммоль) одной частью. Смесь далее переносят в морозильную камеру, работающую при -24°С, и выдерживают в течение ночи. Реакцию гасят водным раствором хлорида аммония и полученный в результате раствор экстрагируют эфиром три раза. Эфирные слои сушат над сульфатом натрия и упаривают в вакууме. Остаток очищают флэш-хроматографией на оксиде кремния с элюированием смесью 10% этилацетат/гексан с получением соединения, указанного в заголовке (20,2 г). МС рассчитано для Ci5H25N04: (М+Н)+ 284; найдено 184,2 (М-Вос+Н)+. Стадия А-3 BocHN (1S,4S) -4-[ (трет-бутоксикарбонил) амино]-1-изопропилциклопент-2-ен-1-карбоновая кислота К раствору метил (IS,4S)-4-[(трет-бутоксикарбонил)амино]-1-изопропилциклопент-2-ен-1-карбоксилата (18,42 г, 65 ммоль) в ТГФ (500 мл), метаноле (500 мл) и воде (100 мл) добавляют моногидрат гидроксида лития (5,00 г, 119 ммоль). Смесь нагревают при кипячении с обратным холодильником в течение ночи. Через 18 часов, ТСХ показывает наличие лишь следового количества исходного вещества. Органические растворители удаляют в вакууме и водный слой экстрагируют эфиром (200 мл) для удаления непрореагировавшего исходного вещества. Водный слой подкисляют концентрированной НС1 до рН=4 при охлаждении в бане со льдом. Полученный в результате раствор экстрагируют метиленхлоридом три раза. Экстракты сушат над MgS04 и концентрируют с получением твердого вещества (17 г). Твердое вещество растворяют в горячем этилацетате (22 мл) и к раствору добавляют гексаны (550 мл) . Раствор медленно охлаждают до комнатной температуры перед переносом в морозильную камеру, работающую в интервале температур от -22 до -24°С. Через два дня кристаллы удаляют и жидкость упаривают в вакууме с получением требуемого продукта в виде белого пенистого твердого вещества (9,78 г, 56%). МС рассчитано для Ci4H23N04: (М+Н)+ 270; найдено 170,1 (М-Вос+Н) +. Стадия А-4 BocHN-^jf?^OH (1S,3R)-3-[(трет-бутоксикарбонил)амино]-1-изопропилциклопентанкарбоновая кислота К раствору (IS, 4S) -А-[(трет-бутоксикарбонил)амино]-1-изопропилциклопент-2-ен-1-карбоновой кислоты (9,78 г, 36,3 ммоль) в этаноле (250 мл) добавляют 10% палладия на углероде (550 мг) . Смесь встряхивают над водородом при 55 фунт/кв.дюйм в течение ночи и фильтруют через целит. Фильтрат упаривают в ваккуме с получением соединения заголовка (9,45 г, 96%). МС рассчитано для Ci4H25N04: (М+Н)+ 272; найдено 172,1 (М-Вос+Н)+. Стадия В-1 мн2 F3C 2-[4-(Трифторметил)фенил]этанамин [4-(Трифторметил)фенил]ацетонитрил (10,0 г, 54 ммоль) растворяют в 2,00М раствор аммиака в метаноле (100 мл) в колбе Парра. К нему добавляют никель Ренея (приблизительно 1 г) . Смесь встряхивают над водородом (50 фунт/кв.дюйм) в течение 20 ч и фильтруют через целит, который далее промывают метиленхлоридом несколько раз. Фильтрат концентрируют с получением соединения заголовка в виде твердого вещества. МС рассчитано для C9Hi0F3N: (М+Н)+ 190; найдено 173,1 (M+H-NH3)+. Стадия В-2 .jCtV 2,2,2-трифтор-Ы- {2-[4 - (трифторметил)фенил]этил}ацетамид К раствору 2-[4-(трифторметил)фенил]этанамина (9,7 г, 44 ммоль) и N,N-диизопропилэтиламина (13 мл, 77 ммоль) в метиленхлориде (80 мл), охлажденному в бане со льдом, медленно добавляют трифторуксусный ангидрид (9,05 мл, 64 ммоль) через шприц. После перемешивания в бане со льдом в течение 10 мин баню со льдом удаляют и перемешивание продолжают в течение дополнительных 30 мин. Реакцию гасят добавлением воды и полученный в результате раствор экстрагируют метиленхлоридом дважды. Объединенные экстракты промывают 1н НС1 и насыщенным солевым раствором, сушат (MgS04) , фильтруют и концентрируют с получением неочищенного продукта (14,2 г). Кристаллизацией из EtOAc и гексанов получают соединение заголовка (8,5 г, 68%) в виде белых игл. МС рассчитано для CiiH9F6NO: (М+Н)+ 28 6; найдено 2-(трифторацетил)- 7-(трифторметил)-1,2,3,4-тетрагидроизохинолин 2,2,2-трифтор-Ы-{2-[4-(трифторметил)фенил]этил}ацетамид (4,00 г, 14 ммоль) и параформальдегид (0,63 г) объединяют в колбе и растворяют в уксусной кислоте (11 мл). Медленно добавляют серную кислоту (11 мл). Смесь переходит из мутного раствора в прозрачный и наблюдают экзотермическую реакцию. Через 4 0 мин колбу погружают в баню со льдом. Реакцию гасят холодной водой, и полученный в результате раствор экстрагируют EtOAc три раза. Объединенные экстракты промывают водой, насыщенным раствором бикарбоната натрия и насыщенным солевым раствором, сушат (MgS04) , фильтруют и концентрируют с получением желтого масла (4,1 г, 83%). МС рассчитано для Ci2H9F6NO: (М+Н)+ 298; найдено 298, 0. Стадия В-4 286,0. Стадия В-3 7- (трифторметил)-1,2,3,4-тетрагидроизохинолин 2-(трифторацетил)-7-(трифторметил)-1,2,3,4-тетрагидроизохинолин (4,1 г, 12 ммоль) растворяют в этаноле (16,0 мл). Добавляют карбонат калия (4,0 г, 29 ммоль) и воду (4 мл) и смесь кипятят с обратным холодильником в течение 2 часов. После охлаждения, раствор разбавляют водой и экстрагируют дихлорметаном четыре раза. Объединенные экстракты сушат (MgS04) , фильтруют и концентрируют. Очисткой хроматографией высокого давления на силикагеле с элюированием в градиенте от 100% А до 20% В (A=l%NH4OH/5%MeOH/EtOAc; B=l%NH4OH/MeOH) в течение 13 мин получают соединение заголовка (1,4 г, 59%). МС рассчитано для Ci0Hi0F3N: (М+Н)+ 202; найдено 202,0. Стадия С-1 Трет-бутил ((1R,3S)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}циклопентил)карбамат К раствору 7-(трифторметил)-1,2,3,4-тетрагидроизохинолина (1,7 г, 8,45 ммоль) Стадии В-4, (IS,3R)-3-[(трет- бутоксикарбонил) амино]-1-изопропилциклопентанкарбоновой кислоты (2,75 г, 10,14 ммоль) Стадии А-4, 4-диметиламинопиридина (0,70 г, 5,73 ммоль) и N, N-диизопропилэтиламина (7,0 мл, 40,2 ммоль) в метиленхлориде (30,0 мл) добавляют гексафторфосфат бромотрис(пирролидино)фосфония (5,119 г, 10,98 ммоль). После перемешивания в течение 36 часов при комнатной температуре, раствор концентрируют в ваккуме. Остаток очищают колоночной хроматографией на оксиде кремния с элюированием смесью 20% этилацетат/гексаны с получением 2,1 г (55%) требуемого продукта. МС рассчитано для С24НззГ3Ы2Оз: (М+Н) + 455; найдено 355 (М-Вос+Н)+. Стадия С-2 (1R,3S)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2 (1Н)-ил]карбонил}циклопентанамин Трет-бутил ( (1R, 35)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}циклопентил)карбамат (0,80 г, 1,76 ммоль) растворяют в 4М растворе НС1 в 1,4-диоксане (10 мл). После перемешивания в течение 2 часов, раствор упаривают в ваккуме с получением требуемого продукта (0,64 г, 94%) в виде НС1 соли. МС рассчитано для C19H25F3N2O: (М+Н)+ 355; найдено 355,2. Стадия D-1 8-пиридин-2-ил-1, 4-диоксаспиро[4.5]декан-8-ол К раствору 2-бромпиридина (14 г, 88,6 ммоль) в безводном эфире (300 мл), охлажденному при -78°С медленно добавляют раствор 2,5М н-бутиллития (36 мл). После добавления перемешивание продолжают при -78°С в течение 1 часа. К раствору медленно добавляют раствор 1,4-циклогександиона моноэтиленкеталя (15 г, 96 ммоль) в безводном эфире (300 мл) . По завершении добавления смеси дают нагреться до 0°С, и перемешивание продолжают в течение 1 часа. Реакцию гасят добавлением водного раствора (100 мл) хлорида аммония (4,5 г). Органическую фазу отделяют и водную фазу экстрагируют метиленхлоридом 4 раза. Объединенные органические фазы сушат MgS04 и концентрируют. Кристаллизацией из EtOAc получают 7 г требуемого продукта. Маточный раствор жидкость очищают на силикагеле с элюированием 10% MeOH/EtOAc с получением еще 3 г требуемого продукта. МС рассчитано для C13H17NO3: (М+Н)+ 236; найдено 236,0. Стадия D-2 сЯо 4-гидрокси-4-(пиридин-2-ил)циклогексанон Вышеуказанный продукт растворяют в ТГФ (30 мл) и Зн растворе НС1 в воде (30 мл) . Смесь перемешивают при 50°С в течение 3 часов. После охлаждения до комнатной температуры, к раствору добавляют NaHC03 при перемешивании до окончания выделения пузырьков. Органическую фазу отделяют и водный слой экстрагируют EtOAc три раза. Объединенные органические слои сушат над MgS04 и концентрируют. Остаток растирают в EtOAc с получением 5,5 г соединения заголовка. МС рассчитано для (М+Н)+ 192; найдено 192,0. Стадия D-3 4- [ ((1R,3S)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)ил]карбонил}циклопентил)амино]-1 -пиридин-2-илциклогексанол К раствору 4-гидрокси-4-пиридин-2-илциклогексанона (42,3 мг, 0,221 ммоль) Стадии D-2 и (1R,35)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)- ил]карбонил}циклопентанамина (103 мг, 0,221 ммоль) Стадии С-2 в СН2С12 (8 мл) добавляют триацетоксиборгидрид натрия (200 мг, 1,0 ммоль). После перемешивания при комнатной температуре в течение ночи, реакцию гасят водным раствором NaOH. Раствор экстрагируют СН2С12. Органический слой концентрируют в вакууме. Остаток очищают посредством ВЭЖХ с получением двух диастереомеров. Изомер 1: ЖХМС рассчитано для СзоНз8ЕзМ302: (М+Н) + 530; найдено 530,1. Изомер 2: МС найдено 530,1. Пример 2 4-[((1R, 3S)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)ил]карбонил}циклопентил)амино]-1-(1,3- оксазол-2-ил)циклогексанол Стадия А 4-гидрокси-4-(1,З-оксазол-2-ил)циклогексанон К раствору 1,3-оксазола (2,0 мл, 30,41 ммоль) в ТГФ (20 мл) добавляют 1,0М раствор борана в ТГФ (30,4 мл) при комнатной температуре. Смесь перемешивают в течение 1 часа перед охлаждением до -78°С. К вышеуказанному раствору добавляют 1,6М раствор н-бутиллития в гексане (19 мл). После перемешивания при -78°С в течение одного часа, добавляют раствор 1,4-диокса-спиро[4.5]декан-8-она (5,22 г, 33,45 ммоль) в ТГФ (10 мл). После перемешивания при -78°С в течение 5 часов, реакцию гасят добавлением ЗМ раствора НС1 в воде (40 мл) . Полученный в результате раствор перемешивают при комнатной температуре в течение ночи, нейтрализуют карбонатом калия и экстрагируют EtOAc три раза. Объединенные экстракты сушат над MgS04 и концентрируют. Очисткой флэш-хроматографией на оксиде кремния с элюированием EtOAc получают требуемый продукт (3,9 г, 70%). ЖХ-МС рассчитано для C9HnN03: (М+Н)+ 182; найдено 182,0. 4-[((1R,3S)-З-изопропил-3-{[7- (трифторметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}циклопентил)амино]-1-(1,3-оксазол-2-ил)циклогексанол К раствору {1R,35)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}циклопентанамина (113,0 мг, 0,3188 ммоль) Стадии C-2 примера 1 в ТГФ (1 мл) добавляют 4-гидрокси-4-(1,З-оксазол-2-ил)циклогексанон (80,3 мг, 0,443 ммоль) с последующим добавлением триэтилэтиламина (0,5 мл, 3,6 ммоль) и, наконец, триацетоксиборгидрида натрия (135 мг, 0,638 ммоль). После перемешивания в течение ночи реакцию гасят добавлением раствора NaOH. ¦ Полученный в результате раствор экстрагируют метиленхлоридом. Экстракт сушат Стадия В над MgS04 и упаривают в ваккуме. Остаток очищают флэш-хроматографией с элюированием EtOAc/l%NH40H с получением двух изомеров. Изомер 1: 73 мг. МС рассчитано для C28H36F3N3O3: (М+Н) + 520/ найдено 520,1. Изомер 2: 56 мг. МС найдено 520,1. Пример 3 4-[((1R,3S)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}циклопентил)амино]-1-пиримидин-2-илциклогексанол Стадия А 8-Пиримидин-2-ил-1, 4-диокса-спиро[4. 5]декан-8-ол К раствору 2-бромпиримидина (0,20 г, 1,258 ммоль) в безводном метиленхлориде (3,0 мл) по каплям добавляют 1,6М н-бутиллития в гексане (0,86 мл) при -78°С. Реакционную смесь перемешивают в течение 29 мин при -78°С и по каплям добавляют 1,4-диокса-спиро[4.5]декан-8-он (0,196 г, 1,26 ммоль) в CH2CI2 (3 мл) . Реакционную смесь перемешивают при -7 8°С в течение 50 мин и гасят водным раствором NH4C1. После нагрева до комнатной температуры, смесь экстрагируют СН2С1г три раза. Объединенные экстракты сушат над MgS04, фильтруют и концентрируют в ваккуме с получением 0,50 г неочищенного продукта. Очисткой колоночной хроматографией на силикагеле с элюированием 0 -> 50% EtOAc в гексанах получают 0,159 г (54%) требуемого продукта в виде светло-коричневого желтого твердого вещества. МС (М+Н)+ 237,2. Стадия В 4-гидрокси-4-пиримидин-2-илциклогексанон К продукту со Стадии А (190 ммоль, 44 г) в ТГФ (200 мл) добавляют водный раствор НС1 (300 ммоль, 100 мл) . Реакционную смесь перемешивают в течение 2 дней и экстрагируют, используя диэтиловый эфир. Водный слой далее нейтрализуют, используя водный раствор NaOH (50%) до получения рН 11 и экстрагируют, используя EtOAc (6x300 мл). Органические слои объединяют и сушат над MgS04 и концентрируют в ваккуме. Остаток очищают флэш-хроматографией на оксиде кремния с получением требуемого кетона (18 г, 49%). МС (М+Н)+ 193,1. Стадия С 4-[((1R,3S)-З-изопропил-3-{ [7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}циклопентил) амино]-1-пиримидин-2-илциклогексанол К раствору 4-гидрокси-4-пиримидин-2-илциклогексанона (59,8 мг, 0,31 ммоль) и (1R,35)-З-изопропил-3-{[7- (трифторметил)-3,4-дигидроизохинолин-2(1Н)- ил]карбонил}циклопентанамина (110 мг, 0,31 ммоль) Стадии С-2 Примера 1 в СН2С12 (10 мл) добавляют триацетоксиборгидрид натрия (131 мг, 0,62 ммоль). После перемешивания в течение ночи добавляют еще триацетоксиборгидрида натрия для доведения количества эквивалентов восстановителя до 5 эквивалентов. После перемешивания в течение еще 5 часов реакцию гасят водным раствором NaOH. Полученный в результате раствор экстрагируют EtOAc три раза. Объединенные экстракты сушат над MgS04 и концентрируют. Очисткой флэш-хроматографией с последующей ВЭЖХ получают два изомера. Изомер 1 и изомер 2: МС рассчитано для C29H37F3N402: (М+Н)+ 531; найдено 531,1. CF3 Пример 4 4-[((1R,3S)-3-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}циклопентил)амино]-1-(1,3- тиазол-2-ил)циклогексанол Стадия А 8- (1,З-тиазол-2-ил)-1, 4-диоксаспиро[4.5]декан-8-ол 1,6М раствор н-бутиллития в гексанах (8,1 мл, 12,92 ммоль) добавляют к раствору тиазола (1,0 г, 11,75 ммоль) в ТГФ (10 мл) при -7 8°С при перемешивании под N2. После перемешивания при -78°С в течение 1 часа, раствор 1,4-циклогександиона моноэтиленкеталя (1,84 г, 11,75 ммоль) в ТГФ (10 мл) добавляют к раствору через шприц и перемешивание продолжают в течение 3 часов при -78°С. Добавляют воду (5 мл), и реакционную смесь нагревают до комнатной температуры и экстрагируют, используя EtOAc три раза. Объединенные органические слои сушат (MgS04) , фильтруют, концентрируют в ваккуме и хроматографируют с получением 2,53 г (89%) требуемого соединения. МС (М+Н)+ = 242,2. 4-гидрокси-4 -(1,3-тиазол-2-ил)циклогексанон Раствор 8-(1,З-тиазол-2-ил)-1,4-диоксаспиро[4.5]декан-вола (1,0 г, 4,14 ммоль) в 20 мл смеси ТГФ/Зн НС1 (1:1) перемешивают в течение 1 часа при 50°С. После охлаждения до комнатной температуры, Смесь обрабатывают Na2C03 до рН 8 и экстрагируют EtOAc три раза. Объединенные органические слои промывают насыщенным раствором NaCl, сушат (MgS04) , и концентрируют с получением 0,80 г (98%) соединения заголовка. Стадия В N НО МС (М+Н)+ = 198,2. Стадия С CF3 4-[( (1R, 3S) -З-изопропил-3- { [ 7- (трифторметил) -3,4-дигидроизохинолин-2(1Н)-ил]карбонил}циклопентил)амино]-1-(1,3-тиазол-2-ил)циклогексанол К раствору трифторацетатной соли (1R,3S)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н) - ил]карбонил}циклопентанамина (46,8 мг, 0,132 ммоль) и 4-гидрокси-4-(1,З-тиазол-2-ил)циклогексанона (30,4 мг, 0,154 ммоль) в дихлорметане (10 мл) добавляют триэтиламин (23,6 мкл, 0,169 ммоль) с последующим добавлением триацетоксиборгидрида натрия (56 мг, 0,2 6 ммоль) при комнатной температуре. После перемешивания в течение ночи смесь разбавляют дихлорметаном и нейтрализуют ЫаНСОз. Органический слой промывают насыщенным солевым раствором, сушат над Na2S04 и концентрируют. Остаток очищают флэш-хроматографией на оксиде кремния с получением двух изомеров. Изомер 1 и изомер 2: ЖХ-МС рассчитано для C28H36F3N3O2 S: (М+Н)+ 536; найдено 536,2. 4-[((1R,3S)-З-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)амино]-1-(1,3-тиазол- Пример 5 2-ил)циклогексанол Стадия А-1 З-бром-5-(трифторметил)пиридин-2-ол к раствору 5-(трифторметил)пиридин-2-ола (10,52 г, 62 ммоль) и ацетата натрия (5,29 г, 64 ммоль) в ледяной уксусной кислоте (38 мл) добавляют бром (3,36 мл, 65 ммоль) при комнатной температуре. Белый мутный раствор медленно переходит в прозрачный коричневый раствор, который нагревают при 80°С в течение 2,5 час. Смеси дают нагреться охладиться до комнатной температуры и далее упаривают при пониженном давлении. Остаток нейтрализуют насыщенным раствором NaHC03 до рН=8. Полученный в результате раствор экстрагируют EtOAc три раза. Объединенные экстракты сушат над MgS04, фильтруют, упаривают в ваккуме с получением 15,1 г (99,8%) неочищенного продукта (15,1 г, 98,8%) в виде белого твердого вещества. ЖХ-МС рассчитано для C6H3BrF3NO: (М+Н)+ 241,9; найдено 241, 9/243, 9. Стадия А-2 2- гидрокси-5-(трифторметил)никотинальдегид 3- бром-5-(трифторметил)пиридин-2-ол (8,20 г, 31,2 ммоль) добавляют небольшими частями к суспензии гидрида натрия (0,8575 г, 33,94 ммоль) в безводном ТГФ (76 мл) при комнатной температуре. После полного добавления, реакционную смесь охлаждают до -78°С и обрабатывают 1,7М раствором трет-бутиллития в пентане (40,0 мл), который добавляют по каплям через шприц в течение периода, равного 15 мин. После перемешивания в течение 5 мин, медленно добавляют безводный ДМФА (8,16 мл, 105 ммоль), поддерживая при этом температуру ниже -50°С. Реакционную смесь далее перемешивают в течение ночи, давая ей нагреться до комнатной температуры. Полученную в результате светло-коричневую смесь гасят добавлением насыщенного раствора NH4C1. рН раствора регулируют до 9-10 добавлением водного раствора NaHC03. Полученный в результате раствор экстрагируют EtOAc четыре раза. Объединенные экстракты сушат (MgS04) , фильтруют и концентрируют с получением коричневого твердого неочищенного продукта (7,33 г, > 100% выход неочищенного продукта). МС рассчитано для C7H4F3NO2 (М+Н)+ 192; найдено 192,1. Стадия А-3 2-Гидрокси-5- (трифторметил)никотинонитрил Смесь 2-гидрокси-5-(трифторметил)никотинальдегида (3,80 г, 17,9 ммоль) (приблизительно 90% чистота), формиата натрия (1,46 г, 20,8 ммоль), гидрохлорида гидроксиламина (1,47 г, 20,8 ммоль) в муравьиной кислоте (36,6 мл) перемешивают при комнатной температуре в течение 2 часов (мутный коричневый раствор) и далее нагревают при кипячении с обратным холодильником в течение ночи (первоначально прозрачный коричневый раствор, далее снова мутный). После охлаждения до комнатной температуры реакционную смесь гасят водой и экстрагируют EtOAc три раза. Объединенные органические слои промывают водой, сушат над MgS04, фильтруют и концентрируют в ваккуме с получением 3,03 г (90%) требуемого неочищенного продукта (приблизительно 75% чистоты) в виде коричневого твердого вещества. МС рассчитано для C7H3F3N2O: (М+Н)+ 18 9; найдено 189,0. Стадия А-4 2-хлор-5-(трифторметил)никотинонитрил К смеси фосфорилхлорида (1,28 мл, 13,6 ммоль) и хинолина (трифторметил)никотинонитрил (2,93 г, 11,7 ммоль) (неочищенный, 75% чистоты). Полученную в результате смесь нагревают при кипячении с обратным холодильником в течение 4 часов. После охлаждения до 100°С, медленно добавляют воду (7,0 мл). Смесь далее охлаждают до комнатной температуры и осторожно нейтрализуют насыщенным раствором NaHC03. Полученный в результате раствор экстрагируют EtOAc три раза и органические слои объединяют, сушат над MgS04, фильтруют, упаривают в (0,834 мл, 6,92 ммоль) добавляют 2-гидрокси-5- ваккуме. Неочищенный продукт (2,35 г) очищают флэш-хроматографией (15:85 EtOAc/гексаны) с получением 1,81 г (75%) требуемого соединения в виде темно-коричневого твердого вещества (> 85% чистоты). Стадия А-5 Трет-бутил метил [З-циано-5-(трифторметил)пиридин-2-ил]малонат К суспензии гидрида натрия (0,752 г, 29,8 ммоль) в ТГФ (18 мл) под азотом добавляют по каплям раствор трет-бутилметилмалоната (3,18 мл, 17,9 ммоль) в безводном ТГФ (15 мл) шприцем в течение периода 15 мин. Реакционную смесь перемешивают в течение 30 мин перед медленным добавлением раствора 2-хлор-5-(трифторметил)никотинонитрила (4,0 г, 14,5 ммоль) в ТГФ (30 мл). После перемешивания при комнатной температуре в течение ночи, реакционную смесь гасят водным раствором NH4C1. ТГФ удаляют в ваккуме и водный раствор экстрагируют EtOAc три раза. Объединенные органические слои сушат над MgS04, фильтруют, упаривают в ваккуме. Неочищенный коричневый продукт (7,1 г) очищают флэш-хроматографией (10:90 EtOAc/гексаны) с получением 4,20 г (84%) требуемого продукта в виде желтого масла. ЖХ-МС рассчитано для C15H15F3N2O4: (М+Н)+ 34 5; найдено 245, 0 (M-C02tBu+H+l)+. Стадия А-6 Грет-бутил 7-оксо-З- (трифторметил) -5, 6, 78-тетрагидро-1, 6-нафтиридин-8-карбоксилат К раствору трег-бутилметил [З-циано-5- (трифторметил)пиридин-2-ил]малоната (4,02 г, 11,7 ммоль) в этаноле (60 мл) добавляют взвесь никеля Ренея (0,60 г, 10 ммоль). Смесь переносят в аппарат Парра и гидрируют в атмосфере водорода при 4 0 фунт/кв.дюйм в течение ночи. Суспензию фильтруют через целит и фильтрат упаривают в ваккуме с получением 3,68 г (99,6%) требуемого продукта в виде желтого масла. ЖХ-МС рассчитано для C14H15F3N2O3: (М+Н)+ 317; найдено 217,1 (M-C02tBu+H+l)+. Стадия А-7 3- (трифторметил)-5,8-дигидро-1, 6-нафтиридин-7(6Н)-он К раствору трет-бутил 7-оксо-З-(трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридин-8-карбоксилата (3,60 г, 11,4 ммоль) в метиленхлориде (14 мл) добавляют трифторуксусную кислоту (6,75 мл) и полученную в результате смесь перемешивают при комнатной температуре в течение 0,5 часов. Раствор упаривают при пониженном давлении и остаток растворяют в CH2CI2. Смесь нейтрализуют медленным добавлением насыщенного раствора NaHC03 и органический слой удаляют. Водный слой экстрагируют CH2CI2 четыре раза, и объединенные органические слои сушат над MgSCU, фильтруют, упаривают в ваккуме с получением 2,45 г (100%) требуемого продукта в виде желтого твердого вещества. ЖХ-МС рассчитано для C9H7F3N2O: (М+Н)+ 217; найдено 217,0. Стадия А-8 3-(трифторметил)-5, 6, 7,8-тетрагидро-1, 6-нафтиридин К желтой суспензии 3-(трифторметил)-5,8-дигидро-1, 6-нафтиридин-7(6Н)-она (2,08 г, 9,62 ммоль) в ТГФ (14 мл) добавляют медленно 1,0М раствор борана в ТГФ (48,5 мл) и полученный в результате прозрачный желтый раствор перемешивают при комнатной температуре в атмосфере N2 в течение ночи. После реакции в течение ночи, обнаруживают два пика. Основной пик (> 80%) представляет собой борановый комплекс, минорный пик является пиком требуемого продукта ( <15%). Для разрушения боранового комплекса, мутную реакционную смесь обрабатывают добавлением по каплям 6М НС1 (12 мл). Генерируются большие количества пузырьков и тепла. Полученный в результате слегка мутный желтый раствор перемешивают при комнатной температуре в течение ночи. После упаривания растворителей желтый неочищенный продукт растворяют в 25 мл ДМСО и медленно обрабатывают ТФУК (4 мл) с получением желто-коричневого прозрачного раствора, который очищают препаратом ВЭЖХ с получением приблизительно 2,60 г (63%) требуемого светло-желтого липкого продукта в виде ди-ТФУК-соли. ЖХ-МС 203,0 (М+Н)+. Для дальнейшей очистки продукта, трифторуксусной кислоты (ТФУК) соль, полученную выше, нейтрализуют обработкой раствором NaOH. К полученному в результате свободному основанию (270 мг, 1,3 ммоль) в дихлорметане (10 мл) добавляют ди-трет-бутилдикарбонат (580 мг, 2,7 ммоль) с последующим добавлением диизопропилэтиламина (520 мг, 4,0 ммоль). После перемешивания в течение ночи при комнатной температуре раствор разбавляют дихлорметаном, промывают насыщенным раствором ЫаНСОз, водой и насыщенным солевым раствором, сушат над Na2S04, фильтруют, и концентрируют. Остаток очищают флэш-хроматографией на оксиде кремния с элюированием 50% этилацетата в гексанах с получением Вос-защищенного продукта. Продукт обрабатывают 4М раствором НС1 в 1,4-диоксане (10 мл). После перемешивания при комнатной температуре в течение 2 часов раствор упаривают в ваккуме. Остаток обрабатывают эфиром с получением требуемого продукта в виде белого твердого вещества (159 мг) . МС рассчитано для C9H9F3N2: (М+Н)+ 203; найдено 203, 0. Стадия В-1 / ^ N Трет-бутил ((1R,3S)-З-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)карбамат тетрагидро-1,6-нафтиридина (159 мг, 0,574 ммоль) Стадии А-1, (15, 3R)-3-[(трет-бутоксикарбонил)амино]-1- изопропилциклопентанкарбоновой кислоты (0,21 г, 0,79 ммоль) Стадии А-4 в Примере 1 в дихлорметане (10 мл) добавляют К раствору дигидрохлорида 3- (трифторметил)-5,6, 7, 8- 4-диметиламинопиридин (38 мг, 0,32 ммоль) и диизопропилэтиламин (180 мг, 1,4 ммоль) с последующим добавлением гексафторфосфата бромотрис(пирролидино)фосфония (270 мг, 0,57 ммоль). После перемешивания в течение ночи при комнатной температуре раствор разбавляют дихлорметаном, промывают насыщенным раствором NaHC03, водой и насыщенным солевым раствором, сушат над Na2S04, фильтруют, упаривают в ваккуме. Остаток очищают флэш-хроматографией на оксиде кремния с элюированием 30% этилацетата в гексанах с получением 300 мг требуемого продукта. МС рассчитано для C23H32F3N3O3: (М+Н)+ 456; найдено 356,2 (М-Вос+Н)+. Стадия В-2 (1R, 3S) -З-изопропил-3- { [3- (трифторметил) -7, 8-дигидро-1, 6-нафтиридин-б(5Н)-ил]карбонил}циклопентанамин Грет-бутил ( (1R, 3S) -З-изопропил-3-{ [3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)карбамат (300 мг, 0,6 ммоль) растворяют в 4М растворе НС1 в 1,4-диоксане (20 мл). После перемешивания при комнатной температуре в течение 2 часов, раствор концентрируют. Остаток обрабатывают эфиром с получением 250 мг желтого твердого вещества. МС рассчитано для Ci8H24F3N30: (М+Н)+ 356; найдено 356, 1. Стадия С 4-[ ((1R,3S)-З-изопропил-3-{[3-(трифторметил)-1,8-дигидро-1, 6-нафтиридин-б(5Н)-ил]карбонил}циклопентил)амино]-1- (1,3-тиазол-2-ил)циклогексанол К раствору (1R,3S)-З-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентанамина (41 мг, 0,12 ммоль) Стадии В-2, 4-гидрокси-4-(1,З-тиазол-2-ил)циклогексанона (46 мг, 0,23 ммоль) Примера 4, и триэтиламина (0,064 мл, 0,46 ммоль) в метиленхлориде (10 мл) добавляют триацетоксиборгидрид натрия (73 мг, 0,35 ммоль) при комнатной температуре. После перемешивания при комнатной температуре в течение ночи смесь разбавляют метиленхлоридом и нейтрализуют ЫаНСОз. Органический слой промывают водой и насыщенным солевым раствором, сушат над Na2S04, фильтруют, упаривают в ваккуме. Остаток очищают флэш-хроматографией на оксиде кремния с элюированием от 0% до 5% триэтиламина в этилацетате с получением двух изомеров (15 мг и 13 мг) . МС рассчитано для C27H35F3N402 S: (М+Н)+ 537; найдено 537,2. Пример 6 4- [ ( (1JR, 35) -З-изопропил-3-{ [7- (трифторметил) -3, 4- дигидроизохинолин-2(1Н)-ил]карбонил}циклопентил)амино]-1-(4- метил-1,З-тиазол-2-ил)циклогексанол Соединение заголовка получают по методике, сходной с описанной для Примера 4, исходя из 4-метил-1,3-тиазола. МС рассчитано для C29H38F3N302 S: (М+Н)+ 550; найдено 550,2. Пример 7 4-[((1R,35)-З-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)амино]-1-(4-метил-1,3- тиазол-2-ил)циклогексанол Соединение заголовка получают по методике, аналогичной описанной для Примера 5, исходя из 4-метил-1,3-тиазола. МС рассчитано для C28H37F3N4O2 S: (М+Н)+ 551; найдено 551,3. Пример 8 4-[((1R,3S)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}циклопентил)амино]-1-(5-метил-1,З-тиазол-2-ил)циклогексанол Стадия А ГрОО 8-(5-метил-1,З-тиазол-2-ил)-1,4-диоксаспиро[4.5]декан-8-ол 1,6М раствор н-бутиллития в гексанах (5,70 мл, 9,12 ммоль) добавляют к 8-(1,З-тиазол-2-ил)-1,4-диоксаспиро[4.5]декан-8-олу (1,00 г, 4,14 ммоль) в ТГФ (10 мл) при -7 8°С с перемешиванием в атмосфере N2. После перемешивания при -78°С в течение 1 часа, метилйодид (0,71 мл, 9,12 ммоль) добавляют к раствору шприцем при -7 8°С. Реакционной смеси дают медленно нагреться до комнатной температуры и перемешивают в течение ночи. Добавляют воду и EtOAc. Водный слой экстрагируют EtOAc три раза. Объединенные органические слои промывают насыщенным раствором NaCl, сушат (MgS04) , концентрируют и подвергают флэш-хроматографии, используя 20% EtOAc/гексаны, с получением 0,77 г (71%) соединения заголовка. МС (М+Н)+ = 256,1. Стадия В 4-Гидрокси-4-(5-мвтил-1,З-тиазол-2-ил)циклогексанон Раствор 8- (5-метил-1,З-тиазол-2-ил)-1,4- диоксаспиро [4.5] декан-8-ола (1,0 г, 4,14 ммоль) в 20 мл смеси ТГФ/Зн НС1 (1:1) перемешивают в течение 1 часа при 50°С. После охлаждения до комнатной температуры, смесь обрабатывают Ыа2СОз до рН 8 и экстрагируют EtOAc три раза. Объединенные органические слои промывают насыщенным раствором NaCl, сушат (MgS04) и концентрируют с получением 0,82 г (99%) требуемого продукта. МС (М+Н)+ = 212,2. Стадия С 4- [((1R,3S)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2 (1Н)-ил]карбонил}циклопентил)амино]-1-(5-метил-1,З-тиазол-2-ил)циклогексанол К раствору ТФУК-соли (1R,35)-З-изопропил-3-{[7- (трифторметил)-3,4-дигидроизохинолин-2(1Н)- ил]карбонил}циклопентанамина (40 мг, 0,1 ммоль) Стадии С-2 из Примера 1 и 4-гидрокси-4-(5-метил-1,З-тиазол-2- ил)циклогексанона (32,6 мг, 0,15 ммоль) в дихлорметане (10 мл) добавляют триэтиламин (31 мкл, 0,22 ммоль) с последующим добавлением триацетоксиборгидрида натрия (56 мг, 0,26 ммоль) при комнатной температуре. После перемешивания в течение ночи смесь разбавляют метиленхлоридом и нейтрализуют NaHCCb. Органический слой сушат над Na2S04 и концентрируют. Остаток очищают флэш-хроматографией на оксиде кремния с элюированием 5% МеОН/СН2С1г с получением двух изомеров (13 мг и 10 мг) . ЖХ-МС рассчитано для С29Нз8ГзЫ302 S: (М+Н)+ 550; найдено 550,2. Пример 9 4-[((1R,3S)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}циклопентил)амино]-1-(1,3- тиазол-5-ил)циклогексанол Стадия А 8-(1,З-тиазол-5-ил)-1,4-диоксаспиро[4,5]декан-8-ол Раствор 2-триметилсилилтиазола (2,5 г, 15,89 ммоль) в ТГФ (20 мл) добавляют к 1,6М раствору н-бутиллития в гексанах (11,9 мл, 19,07 ммоль) при -78°С с перемешиванием в атмосфере N2. После перемешивания при -78°С в течение 0,5 часа раствор 1,4-циклогександиона моноэтиленкеталя (2,48 г, 15,89 ммоль) в ТГФ (20 мл) добавляют к раствору шприцем и перемешивание продолжают в течение 1 часа при -78°С. Добавляют воду (5 мл) и EtOAc, и реакционную смесь нагревают до комнатной температуры и экстрагируют EtOAc три раза. Объединенные органические слои сушат (MgS04) , фильтруют, и кристаллизуют из EtOAc с получением 3,4 г (90%) требуемого продукта. МС (М+Н)+ = 242,1. 4-гидрокси-4-(1,З-тиазол-5-ил)циклогексанон К раствору 8-(1, З-тиазол-5-ил)-1,4-диоксаспиро[4.5]декан-8-ола (0,95 г, 4,14 ммоль) в ТГФ (20 мл) добавляют водный раствор Зн НС1 (10 мл). Смесь перемешивают в течение 1 часа при 50°С. После охлаждения до комнатной температуры раствор обрабатывают Na2C03 до рН 8 и экстрагируют EtOAc три раза. Объединенные органические слои промывают насыщенным раствором NaCl, раствор сушат (MgS04) и концентрируют с получением 0,78 г (98%) требуемого продукта. МС (М+Н)+ = 198,2. Стадия С 4-[((1R,3S) -З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}циклопентил)амино]-1-(1,3-тиазол-5-ил)циклогексанол Стадия В раствору ТФУК-соли (1R, 3S)-З-изопропил-3-{ [7- (трифторметил)-3,4-дигидроизохинолин-2(1Н)- ил]карбонил}циклопентанамина (30,0 мг, 0,0846 ммоль) Стадии С-2 Примера 1 и 4-гидрокси-4-(1,З-тиазол-5-ил)циклогексанона (23,4 мг, 0,119 ммоль) в дихлорметане (8 мл) добавляют триэтиламин (0,0236 мл, 0,169 ммоль) с последующим добавлением триацетоксиборгидрида натрия (42 мг, 0,20 ммоль) при комнатной температуре. После перемешивания в течение ночи, смесь разбавляют дихлорметаном и нейтрализуют насыщенным раствором ЫаНСОз. Органический слой промывают насыщенным солевым раствором, сушат над Na2S04 и концентрируют. Остаток очищают флэш-хроматографией на силикагеле с элюированием 5% МеОН/СН2С1г с получением двух изомеров. ЖХ-МС для изомеров: рассчитано для C28H36F3N3O2 S (М+Н)+ 536; найдено 536,2. Пример 10 4-[((1Д,35)-З-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)амино]-1-(1,3-тиазол- 5-ил)циклогексанол К раствору (1.R, 3S) -З-изопропил-3-{ [3- (трифторметил) -7, 8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентанамина (41 мг, 0,12 ммоль) Стадии В-2 Примера 5, 4-гидрокси-4-(1,3-тиазол-5-ил)циклогексанона (46 мг, 0,23 ммоль) и триэтиламина (0,064 мл, 0,46 ммоль) в метиленхлориде (10 мл) добавляют триацетоксиборгидрид натрия (73 мг, 0,35 ммоль) при комнатной температуре. Смесь перемешивают в течение ночи, разбавляют CH2CI2 и нейтрализуют насыщенным ЫаНСОз. Органический слой промывают водой и насыщенным солевым раствором, сушат над Na2S04, фильтруют и концентрируют. Остаток очищают флэш-хроматографией на силикагеле с элюированием от 0% до 5% триэтиламина в этилацетате с получением двух изомеров. ЖХ-МС для двух изомеров: рассчитано для С^Н^Гз^Ог S: (М+Н)+ 537; найдено 537,1. Пример 11 4-[((1R,3S)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}циклопентил)амино]-1-(2-метил-1,З-тиазол-5-ил)циклогексанол Стадия А 4-Гидрокси-(2-метил-1,З-тиазол-5-ил)циклогексанон К раствору 1,3-тиазола (1 г, 11,76 ммоль) в ТГФ (20 мл) при -78°С добавляют 1,6М раствор н-бутиллития (9 мл). После перемешивания при -7 8°С в течение 1 часа, добавляют йодометан (1,7 г, 11,76 ммоль). Перемешивание продолжают при -78°С в течение 3 часов перед добавлением 1,6М раствора н-бутиллития (9 мл). После перемешивания при -78°С в течение еще одного часа добавляют 1,4-циклогександион моноэтиленкеталя (1,8 г, 11,76 ммоль) в ТГФ (5 мл). Реакционную смесь перемешивают в течение еще 3 часов и гасят добавлением EtOAc и насыщенным солевым раствором. Органическую фазу отделяют, промывают насыщенным солевым раствором, сушат над MgS04 и концентрируют. Флэш-хроматографией на силикагеле с элюированием от 20% до 50% EtOAc/гексанах получают кеталь в виде масла. Масло растворяют в ТГФ (5 мл) и 5% водном НС1 (10 мл) . После перемешивания при комнатной температуре в течение ночи раствор нейтрализуют Ыа2СОз и экстрагируют EtOAc три раза. Экстракты сушат над MgS04 и концентрируют с получением требуемого продукта (0,8 г, 30%). МС рассчитано для CioHi3N02S: (М+Н)+ 212; найдено 212,0. Стадия В 4-[((1R,3S)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(IE)-ил]карбонил}циклопентил)амино]-1-(2-метил-1,З-тиазол-5-ил)циклогексанол К раствору ТФУК-соли (1R,3S)-З-изопропил-3-{[7- (трифторметил)-3,4-дигидроизохинолин-2(1Н)- ил]карбонил}циклопентанамина (30,0 мг, 0,0846 ммоль) со Стадии С-2 Примера 1 и 4-гидрокси-4-(2-метил-1,З-тиазол-5-ил)циклогексанона (25,1 мг, 0,119 ммоль) в дихлорметане (8 мл) добавляют триэтиламин (0,0236 мл, 0,169 ммоль) с последующим добавлением триацетоксиборгидрида натрия (42 мг, 0,20 ммоль) при комнатной температуре. После перемешивания в течение ночи смесь гасят добавлением насыщенного раствора NaHC03. Полученный в результате раствор экстрагируют СН2С12 три раза. Объединенные экстракты промывают насыщенным солевым раствором, сушат над Na2S04 и концентрируют. Остаток очищают флэш-хроматографией на силикагеле с элюированием 5% МеОН/СН2С12 с получением двух изомеров (15 и 12 мг) . ЖХ-МС для двух изомеров: рассчитано для C29H38F3N302 S (М+Н)+ 550; найдено 550, 2. Пример 12 4-[({1R,35)-З-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)амино]-1-(2-метил-1,3- тиазол-5-ил)циклогексанол К раствору (1R,35) -З-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентанамина (4 мг, 0,12 ммоль) Стадии В-2 Примера 5, 4-гидрокси-4-(2-метил-1,З-тиазол-5-ил)циклогексанона (49 мг, 0,23 ммоль) и триэтиламина (0,064 мл, 0,46 ммоль) в метиленхлориде (10 мл) добавляют триацетоксиборгидрид натрия (73 мг, 0,35 ммоль) при комнатной температуре. Смесь перемешивают при комнатной температуре в течение ночи и гасят добавлением насыщенного раствора NaHC03. Полученный в результате раствор экстрагируют CH2CI2 три раза. Объединенные экстракты промывают водой и насыщенным солевым раствором, сушат над Na2S04, фильтруют, упаривают в ваккуме. Остаток очищают флэш-хроматографией на силикагеле с элюированием от 0% до 5% триэтиламина в этилацетате с получением двух изомеров. ЖХ-МС для двух изомеров: рассчитано для C28H37F3N4O2 S (М+Н)+ 551; найдено 551,3. Пример А In vitro анализы CCR2 Функциональная способность новых соединений по изобретению являться антагонистами хемокинового рецептора (например, CCR2) может быть определена с применением подходящего скрининга (например, высокопроизводительного анализа). Например, средство может быть протестировано в анализе на внеклеточное подкисление, анализ потока кальция, анализ связывания лигандов, анализ фосфорилирования, анализ интернализации рецептора или анализ хемотаксиса (см., например, Hesselgesser et al., J Biol. Chem. 273(25):15687-15692 (1998); WO 00/05265 и WO 98/02151). В подходящем анализе используют белок CCR2, который может быть выделен или рекомбинантно модифицирован, который имеет, по меньшей мере, одно свойство, одну активность или одну функциональную характеристику белка CCR2 млекопитающих. Специфическим свойством может являться свойство связывания (например, с лигандом или ингибитором), сигнальная активность (например, активация G-белка млекопитающих, индукция быстрого и временного увеличения концентрации цитозольного свободного кальция [Ca++]i, индукция специфического фосфорилирования белка), функция клеточного ответа (например, стимуляция хемотаксиса или высвобождения воспалительного медиатора лейкоцитами) и т.п. В примере анализа связывания композиция, содержащая белок CCR2 или его вариант, поддерживается в условиях, пригодных для связывания. Рецептор CCR2 контактирует с соединением, подлежащим тестированию, и связывание обнаруживают или измеряют. В примере анализа на клеточной основе применяют клетки, которые выделяют из периферической крови человека и структурно экспрессируют белок CCR2. Альтернативно, клетка, которая устойчиво или время от времени трансфицируется вектором или кассетой экспрессии, имеющими последовательность нуклеиновых кислот, которая кодирует рецептор CCR2, может рассматриваться в качестве источника белка CCR2. Клетки поддерживаются в условиях, соответствующих условиям экспрессии рецептора, и контактируют со средством в условиях, соответствующих связыванию. Связывание может быть обнаружено с использованием стандартных методов. Например, степень связывания может определяться относительно подходящего контроля. Также вместо целых клеток может применяться клеточная фракция, такая как мембранная фракция, содержащая рецептор. Обнаружение связывания или образования комплекса в аналитическом тесте может быть детектировано непосредственно или опосредованно. Например, средство может быть мечено подходящей меткой (например, флуоресцентной меткой, меткой, изотопной меткой, ферментной меткой и т.п.), и связывание может определяться посредством детектирования метки. Специфичное и/или конкурентное связывание может быть оценено в исследованиях по конкуренции или вытеснению с использованием немеченого средства или лиганда в качестве конкурента. CCR2-aнтaгoниcтичecкaя активность соединений по изобретению может быть представлена в виде концентрации ингибитора, требующейся для 50% ингибирования (IC50 значения) специфического связывания в анализах связывания с рецептором с использованием 1251-меченого МСР-1, в качестве лиганда, и моноядерных клеток периферической крови (PBMCs), полученных из нормальной человеческой цельной крови центрифугированием в градиенте плотности. Специфическое связывание предпочтительно определяют как общее связывание (например, общие овм (отсчеты в минуту) на фильтрах за вычетом неспецифического связывания. Неспецифическое связывание определяют как количество овм, все еще обнаруживаемое в присутствии избытка немеченого конкурента (например, МСР-1). Пример В Анализ связывания PBMCs человека используют для тестирования соединений по изобретению в аналитическом тесте на связывание. Например, от 200000 до 500000 клеток инкубируют с 0,1-0,2 нМ 1251-меченного МСР-1, с немеченным конкурентом (10 нМ МСР-1) или без него или различными концентрациями тестируемых соединений. 1251-меченый МСР-1 получают подходящими способами или покупают у коммерческих поставщиков (Perkin Elmer, Boston MA) . Реакции связывания проводят в 50-250 мкл буфера для связывания, состоящего из 1М HEPES рН 7,2 и 0,1% BSA (бычий сывороточный альбумин) в течение 30 мин при комнатной температуре. Реакции связывания останавливают сбором мембран посредством быстрой фильтрации через стекловолоконные фильтры (Perkin Elmer), которые предварительно смачивают в 0,3% полиэтиленимине или забуференном фосфатом физиологическом растворе (PBS). Фильтры споласкивают приблизительно 600 мкл буфера для связывания, содержащего 0,5М NaCl или PBS, далее сушат, и количество связанной радиоактивности определяют подсчетом на гамма-счетчике (Perkin Elmer). В соответствии с этой методикой анализа связывания соединения настоящего изобретения имеют IC50 значения менее, чем приблизительно 3000 нМ. Пример С Анализ хемотаксиса Способность соединений по изобретению к антагонизму функции CCR2 определяют в аналитическом тесте на хемотаксис лейкоцитов, используя моноядерные клетки периферической крови человека, в модифицированной камере Бойдена (Neuro Probe). 500000 клеток в свободной от сыворотки среде DMEM (In Vitrogen) инкубируют с ингибиторами или без них и нагревают до 37°С. Камеру для хемотаксиса (Neuro Probe) также предварительно нагревают. 4 00 мкл нагретого 10 нМ МСР-1 добавляют на дно камеры во все лунки, за исключением отрицательного контроля, в который добавляют DMEM. Мембранный фильтр 8 микрон (Neuro Probe) помещают на верхнюю часть и крышку камеры закрывают. Клетки далее добавляют в отверстия крышки камеры, которые связаны с лунками камеры ниже фильтрующей мембраны. Всю камеру инкубируют при 37°С, 5% С02 в течение 30 минут. Клетки далее аспирируют, крышку камеры открывают и фильтр плавно удаляют. Верхнюю часть фильтра промывают 3 раза PBS, а нижнюю часть оставляют незатронутой. Фильтр далее сушат воздухом и окрашивают красителем Wright Geimsa (Sigma). Фильтры подсчитывают под микроскопом. Лунки отрицательного контроля служат в качестве исходного фона и их вычитают из всех значений. Антагонистическую активность определяют, сравнивая количество клеток, которые мигрируют ко дну камеры в лунки, содержащие антагонист, с количеством клеток, которые мигрируют ко дну камеры в контрольные лунки с МСР-1. В соответствии с данным аналитическим тестом на хемотаксис соединения по изобретению имеют значения 1С5о менее, чем приблизительно 3000 нМ. Различные модификации изобретения в дополнение к описанным в данном документе, будут очевидными для квалифицированных специалистов в данной области из вышеизложенного описания. Подразумевают, что такие модификации также попадают в объем прилагаемой формулы изобретения. Каждая ссылка, включающая патенты, патентные заявки и публикации, цитируемая в настоящей заявке, включена в данное описание в качестве ссылки во всей своей полноте. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы I: О R6 или его фармацевтически приемлемая соль или пролекарство, где: пунктирная линия означает необязательную связь; X представляет собой N, N0 или CR3; R1 представляет собой Ci-б алкил, (Со-е алкил) -0- (Ci_6 алкил), (С0-б алкил) -S- (Ci-б алкил), (Со-6 алкил) - (С3-7 циклоалкил)- (Со-6 алкил), ОН, C02R10, гетероциклил, CN, NR10R12, NS02R10, NCOR10, NC02R10, NCOR10, CRnC02R10, CRnOCOR10 или фенил; R2 представляет собой H, ОН, галоген, Ci-з алкил, NR10R12, C02R10, CONR10R12, NR^COR11; OCONR10R12, NR10CONR10R12, гетероциклил, CN, NR10~SO2-NR10R12, NR10-SO2-R12, SO2-NR10R12 или оксо; где указанный Ci-з алкил является необязательно замещенным 1-6 заместителями, выбранными из F и ОН; R3 представляет собой Н, ОН, галоген, Ci-б алкил, Ci-б алкокси, NR10R11, NR10CO2Ru; NR10CONR10RU, NR10SO2NR10Ru, NR10-SO2-R11, гетероциклил, CN, CONR10R12, C02R10, N02, SR10, SOR10, S02R10; или SO2-NR10Rn; R4 представляет собой H, Ci_6 алкил, CF3, 0CF3, CI, F, Br или фенил; R5 представляет собой Ci-б алкил, Ci-б алкокси, C0-(Ci-6 алкил) , Ci_6 тиоалкокси, пиридил, F, CI, Br, C4-6 циклоалкил, С4_б циклоалкилокси, фенил, фенилокси, С3-6 циклоалкил, С3-6 циклоалкилокси, гетероциклил, CN, или C02R10; где указанный Ci-б алкил является необязательно замещенным одним или несколькими ОН или F; где указанный Ci-6 алкокси, СО-(Ci_6 алкил) или Ci-б тиоалкокси являются необязательно замещенными одним или несколькими F; где указанный пиридил, фенил или фенилокси являются необязательно замещенными одним или несколькими заместителями, выбранными из галогена, CF3, Ci-4 алкила и C02R10; где указанный С3-6 циклоалкил или С3-6 циклоалкилокси являются необязательно замещенными одним или несколькими F; R6 представляет собой Н, CF3, Ci_6 алкил, F, С1 или Вг; R7 представляет собой Н или Ci-б алкил, необязательно замещенные 1-3 заместителями, выбранными из галогена, ОН, СОгН, СОг- (Ci-б алкил) или Ci-з алкокси; R8 представляет собой Н, Ci-б алкил, F, Ci-з алкокси, С1-3 галогеналкокси, С3_б циклоалкил, С3_б циклоалкилокси, ОН, CO2R10, OCOR10; где указанный Ci-б алкил является необязательно замещенным одним или несколькими заместителями, выбранными из F, Ci-з алкокси, ОН или CO2R10; или R7 и R8 вместе образуют мостиковую группу С2-4 алкилен или -(Со-2 алкил)-О-(Ci-з алкил)- с образованием 5-7 членного цикла; R9 представляет собой гетероциклил, необязательно замещенный 1-4 заместителями, выбранными из Ci-б алкила, С2-6 алкенила, С2-6 алкинила, галогена, С1-4 галогеналкила, CN, NO2, OR13, SR13, C(0)R14, С (О) OR13, C(0)NR15R16, NR15R16, NR15CONHR16, NR15C(0)R14, NR15C (0)0R13, S(0)R14, S(0)2R14, S(0)NRL5R16 или S02NR15R16; R10 представляет собой H, Ci-б алкил, бензил, фенил или С3-6 циклоалкил, где указанные Ci-e алкил, бензил, фенил или С3-6 циклоалкил являются необязательно замещенными 1-3 заместителями выбранными из галогена, ОН, Ci_3 алкила, Ci_3 алкокси, СОгН, С0г~ (Ci_6 алкила) и CF3; R11 представляет собой Н, ОН, Ci_6 алкил, Ci-e алкокси, бензил, фенил или Сз-б циклоалкил, где указанные Ci-б алкил, бензил, фенил или С3-6 циклоалкил являются необязательно замещенными 1-3 заместителями, выбранными из галогена, ОН, Ci_3 алкила, С1-3 алкокси, СОгН, СО2- (Ci-e алкила) и CF3; R12 представляет собой Н, Ci-б алкил, бензил, фенил или Сз-б циклоалкил, где указанные Ci-б алкил, бензил, фенил или С3-6 циклоалкил являются необязательно замещенными 1-3 заместителями, выбранными из галогена, ОН, Ci-з алкила, С1-3 алкокси, СОгН, СО2-(Ci_6 алкила) и CF3; R13 и R14 представляют собой, каждый независимо, Н, Ci_6 алкил, Ci-б галогеналкил, С2-6 алкенил, С2-6 алкинил, арил, циклоалкил, арилалкил или циклоалкилалкил; R15 и R16 представляют собой каждый, независимо Н, Ci_6 алкил, Ci-б галогеналкил, С2_б алкенил, С2-б алкинил, арил, циклоалкил, арилалкил или циклоалкилалкил; или R15 и R16 вместе с атомом N, к которому они присоединены, образуют 4-6 членную гетероциклильную группу. Соединение по п.1, где X представляет собой N или N0. Соединение по п.1, где X представляет собой CR3. Соединение по п.1, где R1 представляет собой Ci-б алкил. Соединение по п.1, где R1 представляет собой 2-пропил. Соединение по п.1 , где R2 представляет собой Н, ОН галоген или Ci-з алкил. Соединение по п.1, где R2 представляет собой Н. Соединение по п.1 , где R3 представляет собой Н, ОН галоген или Ci-б алкил. Соединение по п.1, где R3 представляет собой Н. 10. Соединение по п.1, где R 4 представляет собой Н. 11. Соединение по п.1, где R 5 представляет собой Ci-б алкил замещенный 1-4 F. 12. Соединение по п.1, где R5 представляет собой CF3. 13. Соединение по п.1, где R6 представляет собой Н. 14. Соединение по п.1 , где R7 представляет собой Н. 15. Соединение по п.1 , где R8 представляет собой Н. 16. Соединение по п.1, где R9 представляет собой гетероарил, необязательно замещенный 1-4 заместителями, выбранными из С]_б алкила, С2_б алкенила, С2-б алкинила, галогена, Ci-4 галогеналкила, CN, N02, OR13, SR13, C(0)R14, C(0)0R13, C(0)NR15R16, NR15R16, NR15CONHR16, NR15C(0)R14, NR15C (0) OR13, S(0)R14, S(0)2R14, S(0)NR15R16 или S02NR15R16. 17. Соединение по п.16, где указанный гетероарил представляет собой 5- или 6-членный гетероарил, необязательно замещенный 1-4 заместителями, выбранными из Ci-б алкила, С2_б алкенила, С2_б алкинила, галогена, Ci_4 галогеналкила, CN, N02, OR13, SR13, C(0)R14, С (О) OR13, C(0)NR15R16, NR15R16, NR15CONHR16, NR15C(0)R14, NR15C(0)OR13, S(0)R14, S(0)2R14, S(0)NR15R16 или S02NR15R16. 18. Соединение по п.16 где указанный гетероарил представляет собой пиридил, пиримидинил, пиразинил, пиридазинил или триазинил, каждый необязательно замещенный 1-4 заместителями, выбранными из Ci-б алкила, С2-б алкенила, С2-б алкинила, галогена, Ci_4 галогеналкила, CN, N02, OR13, SR13, C(0)R14, С (0) OR13, C(0)NR15R16, NR15R16, NR15CONHR16, NR15C(0)R14, NR15C(0)OR13, S(0)R14, S(0)2R14, S (0) NR15R16 или S02NR15R16. 19. Соединение по п.16 где указанный гетероарил представляет собой тиенил, фуранил, тиазолил, оксазолил или имидазолил, каждый необязательно замещенный 1-4 заместителями, выбранными из Ci-б алкила, С2_б алкенила, С2-б алкинила, галогена, Сх-4 галогеналкила, CN, N02, OR13, SR13, C(0)R14, С (0) OR13, C(0)NR15R16, NR15R16, NR15CONHR16, NR15C(0)R14, NR15C (0) OR13, S(0)R14, S(0)2R14, S (0)NR15R16 или S02NR15R16. 20. Соединение по п.16 где указанный гетероарил представляет собой тиазолил, оксазолил, пиримидинил или пиридил, каждый необязательно замещенный 1-3 группами F, С1, Br, I, метил, этил, метокси, этокси или трифторметил. 21. Соединение по п.1, имеющее формулу II: 22. Соединение по п.1, имеющее формулу IIla, II lb или Шс: 87 О 23. Соединение по п.1 выбранное из следующих соединений: 4-[((1R,3S)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}циклопентил)амино]-1-пиридин-2-илциклогексанол; 4-[((1R,3S)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}циклопентил)амино]-1-(1,3-оксазол-2-ил)циклогексанол; 4-[((1R,3S)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}циклопентил)амино]-1-пиримидин-2-илциклогексанол; 4-[((1R,3S)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}циклопентил)амино]-1-(1,3-тиазол-2-ил)циклогексанол; 4-[((1R,3S)-З-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)амино]-1-(1,3-тиазол-2-ил)циклогексанол; 4-[((1R,35)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}циклопентил)амино]-1-(4-метил-1,З-тиазол-2-ил)циклогексанол; 4-[((1R,35)-З-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)амино]-1-(4-метил-1,З-тиазол-2-ил)циклогексанол; 4-[((1R,3S)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}циклопентил)амино]-1-(5-метил-1,З-тиазол-2-ил)циклогексанол; 4-[((1R,3S)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}циклопентил)амино]-1-(1,3-тиазол-5-ил)циклогексанол; 4-[([1R,3S)-З-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,б-нафтиридин-б(5Н)-ил]карбонил}циклопентил)амино]-1-(1,3-тиазол-5-ил)циклогексанол; 4-[((1R, 3S)-З-изопропил-3-{[7-(трифторметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}циклопентил)амино]-1-(2-метил-1,З-тиазол-5-ил)циклогексанол; и 4- [ ( (If?, 3S) -З-изопропил-3-{ [3- (трифторметил) -7, 8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)амино]-1-(2-метил-1,З-тиазол-5-ил)циклогексанол; или его фармацевтически приемлемая соль. 24. Композиция, содержащая соединение по любому из п.п.1-23 и фармацевтически приемлемый носитель. 25. Способ модуляции активности хемокинового рецептора, включающий в себя контакт указанного рецептора с соединением по любому из п.п.1-23. 26. Способ по п.25, где указанный рецептор представляет собой CCR2. 27. Способ по п.25, где указанная модуляция соответствует ингибированию. 28. Способ лечения заболевания, связанного с экспрессией или активностью хемокинового рецептора у пациента, включающий в себя введение указанному пациенту терапевтически эффективного количества соединения по любому из п.п.1-23. 29. Способ по п.28, где указанное заболевание представляет собой воспалительное заболевание или иммунное нарушение. 30. Способ по п.28, где указанное заболевание представляет собой ревматоидный артрит, атеросклероз, волчанку, рассеянный склероз, невропатическую боль, отторжение трансплантата, диабет, или ожирение. 31. Способ по п. 28 где указанное заболевание представляет собой рак. 32. Способ по п.28, дополнительно включающий в себя введение противовоспалительного средства. По доверенности |