|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea200601849a*\id |
|
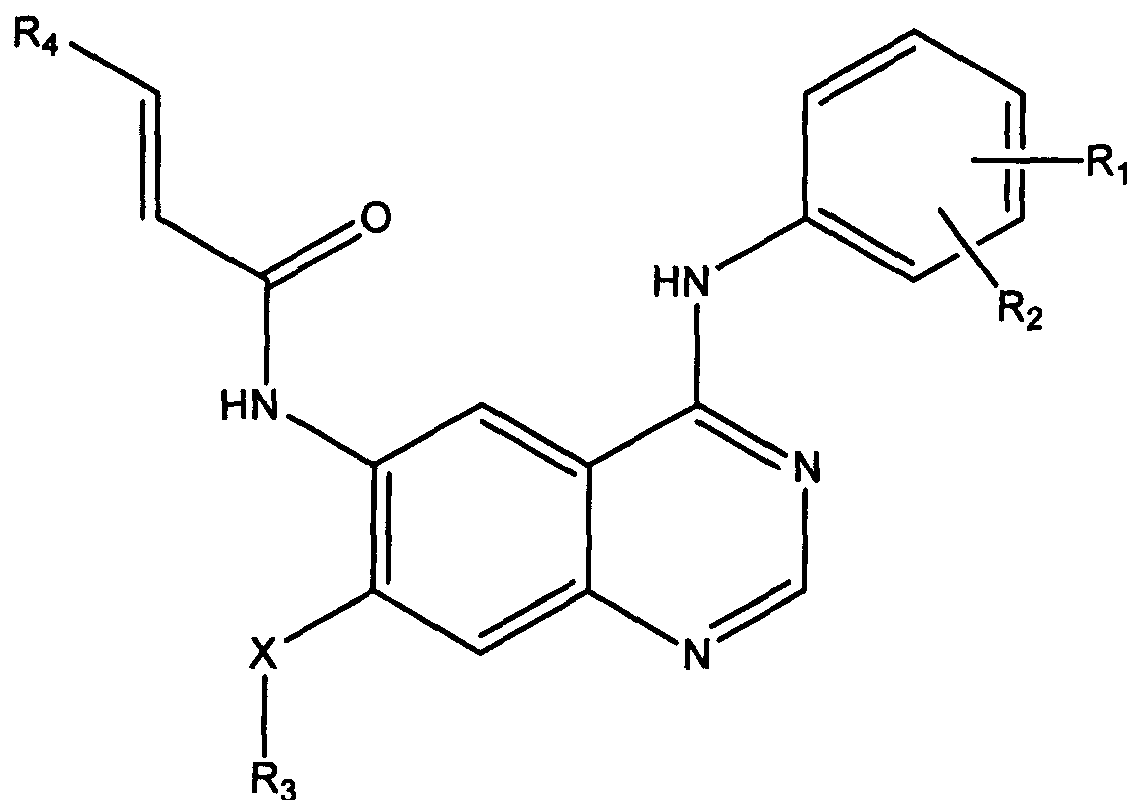
Термины запроса в документе Реферат В данном изобретении предложены хиназолиновые соединения формулы где R 1 представляет собой галогено; R 2 представляет собой H или галогено; R 3 представляет собой а) C 1 -C 3 алкил, возможно замещенный галогено; или б) -(CH 2 ) n -морфолино, -(CH 2 ) n -пиперидин, -(CH 2 ) n -пиперазин, -(CH 2 ) n -пиперазин-N(C 1 -C 3 алкил), -(CH 2 ) n -пирролидин или -(CH 2 ) n -имидазол; n равно от 1 до 4; R 4 представляет собой -(CH 2 ) m -Het; Het представляет собой морфолин, пиперидин, пиперазин, пиперазин-N(C 1 -C 3 алкил), имидазол, пирролидин, азепан, 3,4-дигидро-2H-пиридин или 3,6-дигидро-2H-пиридин, каждый из которых возможно замещен алкилом, галогено, OH, NH 2 , группой NH(C 1 -C 3 алкил) или N(C 1 -C 3 алкил) 2 ; m равно 1-3; и X представляет собой O, S или NH; или их фармацевтически приемлемая соль, а также способы и промежуточные соединения для их получения, полезные фармацевтические композиции и способы использования этих соединений в лечении пролиферативных заболеваний.
Полный текст патента (57) Реферат / Формула: где R 1 представляет собой галогено; R 2 представляет собой H или галогено; R 3 представляет собой а) C 1 -C 3 алкил, возможно замещенный галогено; или б) -(CH 2 ) n -морфолино, -(CH 2 ) n -пиперидин, -(CH 2 ) n -пиперазин, -(CH 2 ) n -пиперазин-N(C 1 -C 3 алкил), -(CH 2 ) n -пирролидин или -(CH 2 ) n -имидазол; n равно от 1 до 4; R 4 представляет собой -(CH 2 ) m -Het; Het представляет собой морфолин, пиперидин, пиперазин, пиперазин-N(C 1 -C 3 алкил), имидазол, пирролидин, азепан, 3,4-дигидро-2H-пиридин или 3,6-дигидро-2H-пиридин, каждый из которых возможно замещен алкилом, галогено, OH, NH 2 , группой NH(C 1 -C 3 алкил) или N(C 1 -C 3 алкил) 2 ; m равно 1-3; и X представляет собой O, S или NH; или их фармацевтически приемлемая соль, а также способы и промежуточные соединения для их получения, полезные фармацевтические композиции и способы использования этих соединений в лечении пролиферативных заболеваний.
PCT/IB2005/001139 МПК7: А61К 31/505, C07D 239/94, А61Р 43/00, 35/00 4-ФЕНИЛАМИНОХИНАЗОЛИН-6-ИЛ-АМИДЫ ССЫЛКИ НА РОДСТВЕННЫЕ ЗАЯВКИ Сделана ссылка на заявку США № 60/568872, поданную 6 мая 2004 года, описание которой включено в данное описание во всей его полноте. ОБЛАСТЬ ИЗОБРЕТЕНИЯ Данное изобретение относится к новым соединениям, которые действуют в качестве ингибиторов тирозинкиназ и применимы в способах лечения, предупреждения или ингибирования пролиферативных заболеваний, включая рак, атеросклероз, рестеноз, эндометриоз и псориаз. Более конкретно, данное изобретение относится к новым 4-анилино-6-замещенным-алкеноиламинохиназолиновым соединениям, полезным в лечении таких расстройств. ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ Замещенные 4-фениламинохиназолин-6-ил-амиды, полезные в лечении рака, известны из уровня техники, включая соединения из патента США № 5457105 (Barker), патента США № 5760041 (Wissner et al.), патента США № 5770599 (Gibson), патента США № 5929080 (Frost), патента США № 5955464 (Barker), патента США № 6251912 (Wissner et al.), патента США № 6344455 (Bridges et al.), патента США № 6344459 (Bridges et al.), патента США № 6414148 (Thomas et al.), патента США № 5770599 (Gibson et al.), заявки на патент США 2002/0173509 (Himmelsbach et al.) и патента США № 6323209 (Frost). Тем не менее сохраняется потребность в новых и эффективных соединениях для лечения пролиферативных расстройств. КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ Настоящее изобретение включает хиназолиновые соединения формулы где: Ri выбран из F, Br, CI или I; R2 выбран из Н, F, Br, CI или I; R3 выбран из: а) С1-С3 прямого или разветвленного алкила, возможно замещенного галогеном; или б) групп -(СН2)п-морфолино, -(СН2)п-пиперидин, -(СН2)п-пиперазин, -(СН2)п-пиперазин-М(СгСзалкил), -(СНг)п-пирролидин или -(СНг)п-имидазол; п представляет собой целое число от 1 до 4; R4 представляет собой -(CH2)m-Het; Het представляет собой гетероциклическую группировку, выбранную из группы состоящей из морфолина, пиперидина, пиперазина, группы пиперазин-М(С-|-Сзалкил), имидазола, пирролидина, азепана, 3,4-дигидро-2Н-пиридина или 3,6-дигидро-2Н-пиридина, где каждая гетероциклическая группировка возможно замещена 1-3 группами, выбранными из Ci-Сзалкила, галогена, ОН, NH2, групп МН(СгС3алкил) или М(СгСзалкил)2; m представляет собой целое число от 1 до 3; и X представляет собой О, S или NH; или их фармацевтически приемлемую соль. Данное изобретение также включает способы применения соединений по изобретению для лечения, ингибирования, предупреждения или контролирования развития пролиферативных заболеваний, включая рак, рестеноз, псориаз, атеросклероз или эндометриоз, каждый из которых включает введение фармацевтически или терапевтически эффективного количества соединения млекопитающему, нуждающемуся в этом. Данное изобретение также охватывает фармацевтические композиции, содержащие фармацевтически эффективное количество соединения по данному изобретению и один или более чем один фармацевтически приемлемый эксципиент и/или носитель. Данное изобретение также включает пути синтеза и промежуточные соединения, пригодные для получения соединений по изобретению. ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ Одна группа соединений по изобретению включает описанные выше соединения, где представляет собой галоген, a R2 представляет собой водород. Другая группа включает соединения, где Ri представляет собой фтор, a R2 представляет собой другой галоген. Следующая группа включает соединения, в которых Ri представляет собой 4-фторо, a R2 представляет собой 3-хлоро. Другая группа соединений по данному изобретению включает соединения формулы II: где: R3 выбран из: а) С1-С3 прямого или разветвленного алкила, возможно замещенного галогеном; или б) групп -(СН2)ч-морфолино, -(СН2)д-пиперидин, -(СН2^-пиперазин, -(СН2)д-пиперазин-М(С1-С3алкил), -(СН2)ч-пирролидин или -(СН2)д-имидазол; q представляет собой целое число от 1 до 2; R4 представляет собой -(CH2)m-Het; Het представляет собой группу пиперидин, пиперазин, пиперазин-г^Сг Сзалкил), имидазол, пирролидин, азепан или дигидропиридин, возможно замещенную 1 или 2 группами, выбранными из галогена или СгС3алкила; m представляет собой целое число от 1 до 3; и X представляет собой О, S или NH; или их фармацевтически приемлемую соль. Еще одна группа соединений по изобретению включает соединения формулы III: где: R3 представляет собой прямой или разветвленный СгСзалкил, возможно замещенный галогеном; Rs и R6 независимо выбраны из Н, Ci-Сзалкила, F, Br, I или CI; X представляет собой О, S или NH; и каждая из пунктирных линий, обозначенная как а и Ь, означает возможную двойную связь, при условии, что только одна двойная связь а или b существует в одном соединении; или его фармацевтически приемлемую соль. Подгруппа каждой группы описанных здесь соединений включает соединения, в которых X представляет собой О. Другие подгруппы включают соединения, в которых X представляет собой NH или S. Другая подгруппа каждой группы включает соединения, в которых X представляет собой О, и R3 представляет собой прямой или разветвленный Ci-Сзалкил, возможно замещенный 1-3 атомами галогенов. Другая подгруппа включает соединения, в которых R3 представляет собой полифторированный С2-С3алкил, такой как 1,1,2,2-тетрафторэтильная или 2,2,3,3,3-пентафторпропильная группы, или перфторированный Сг-Сзалкил, такой как пентафторэтильная или гептафторпропильная группа. Следует понимать, что прямые или разветвленные СгСзалкильные группы, определенные как R3, в указанных здесь группах могут быть галогенированы одной или более группами галогено, включая пергалогенирование, то есть присутствие максимального числа атомов галогенов, допускаемого валентными ограничениями данной алкильной группы (то есть R3 представляет собой трифторметил, пентафторэтил, гептафторпропил и так далее). Соединения по данному изобретению могут быть использованы для ингибирования активности тирозинкиназ, включая, в частности, егЬВ1, егЬВ2 и егЬВ4. Соединения по данному изобретению могут быть использованы в способах лечения, ингибирования, предупреждения или контролирования развития пролиферативных заболеваний, включая рак, рестеноз, псориаз, атеросклероз или эндометриоз. Клеточно-пролиферативные расстройства, которые можно лечить данными способами, включают злокачественные новообразования, скелетные нарушения, ангиогенные пролиферативные расстройства или пролиферативные расстройства кровеносных сосудов, фиброзные нарушения и мезангиально-клеточные пролиферативные нарушения. Фиброзные пролиферативные нарушения, то есть аномальное формирование внеклеточных матриксов, которые можно лечить посредством данных соединений и способов, включают атеросклероз, цирроз печени и мезангиально-клеточные пролиферативные нарушения (включая почечные заболевания человека, такие как гломерулонефрит, диабетическая нефропатия, злокачественный нефросклероз, синдромы тромботической микроангиопатии, отторжение трансплантатов и гломерулопатии). Каждый из описанных здесь способов включает введение нуждающемуся в этом млекопитающему фармацевтически или терапевтически эффективного количества соединения по данному изобретению или его фармацевтически приемлемой солевой формы. В данном изобретении также предложен способ лечения или ингибирования поликистозного заболевания почек у млекопитающего, включающий введение млекопитающему с поликистозным заболеванием почек фармацевтически эффективного количества соединения по данному изобретению. Этот способ применим к поликистозному заболеванию почек как аутосомально-рецессивной, так и аутосомально-доминантной формы. Кроме того, в данном изобретении предложен способ лечения или ингибирования полипов толстой кишки у млекопитающего, включающий введение млекопитающему с поликистозным заболеванием почек фармацевтически эффективного количества соединения по данному изобретению. Следует понимать, что способы ингибирования полипов толстой кишки включают способы, уменьшающие скорость роста полипов толстой кишки. Способы лечения или ингибирования полипов толстой кишки у млекопитающих также могут включать совместное введение или циклические схемы с использованием дополнительных фармацевтически эффективных агентов, таких как ингибиторы СОХ-2 (циклооксигеназы-2), включая целекоксиб; рофекоксиб; вальдекоксиб; лумиракоксиб (также известный как СОХ-189); LAS-34475; UR-8880; 2-(3,4-дифторфенил)-4-(3-гидрокси-3-метилбутокси)-5-[4-метилсульфонил)фенил]-3(2Н)-пиридазинон (АВТ-963); 3-[(3-хлорфенил)[4-(метилсульфонил)фенил]метилен]дигидро-2(ЗН)фуранон (BMS-347070); тилакоксиб; соединение 4-[5-(2,4-дифторфенил)-4,5-дигидро-3-(трифторметил)-1Н-пиразол-1-ил]-бензолсульфонамид (также известный как Е 6087); CS-502 [регистрационный номер службы Chemical Abstracts (Chemical Abstracts Service Registry Number ("CAS Per №")) 176429-82-6]; (6aR,10aR)-3-(1,1-flHMeTnnren^)-6а,7,10,10а-тетрагидро-1-гидрокси-6,6-диметил-6Н-дибензо[Ь,с1]пиран-9-карбоновая кислота ("CT-3"); CV-247; 2(5Н)-фуранон, 5,5-диметил-3-(1-метилэтокси)-4-[4-(метилсульфонил)фенил]- ("DFP"); карпрофен; деракоксиб; эторикоксиб (товарный знак ARCOXIA(r) от MERCK & CO., Inc., Whitehouse Station, New Jersey); GW-406381; аспирин; тиракоксиб; мелоксикам; нимесулид; 2-(ацетилокси)бензойной кислоты 3-[(нитроокси)метил]фениловый эфир ("NCX-4016"); парекоксиб (товарный знак согласно заявке, поданной на DYNASTAT(r) G. D. Searle & Co., Skokie, Illinois); ^ацетил-1_-треонил-!.-пролил-1_-аргинил-0-пролил-Ь-глутаминил-Ь-серил-Ьгистидил-иаспарагинил-Ьа-аспартилглицил-!--а-аспартил-1-фенилаланил-иа-глутамил-1-а-глутамил-1.-изолейцил-1_-пролил-1_-а-глутамил-Ьа-глутамил-Ьтирозил-Ьлейцил-Ьглутамин (также известный как Р54, CAS Per. № 130996-28-0); рофекоксиб (товарный знак VIOXX(r) от MERCK & CO., Inc., Whitehouse Station, New Jersey); RevlMiD; 2,6-бис(1,1-диметилэтил)-4-[(Е)-(2-этил-1,1-диоксо-5-изотиазолидинилиден)метил]фенол ("S-2474"); 5(R)-тио-6-сульфонамид-3(2Н)-бензофуранон ("SVT-2016") и ^[3-(формиламино)-4-оксо-6-фенокси-4Н-1-бензопиран-7-ил]-метансульфонамид ("T-614") или их фармацевтически приемлемой соли. Данное изобретение также относится к способу лечения аномального клеточного роста у млекопитающего, в том числе человека, включающему введение указанному млекопитающему такого количества соединения формулы 1, как оно определено выше, или его фармацевтически приемлемой соли, сольвата или пролекарства, которое является эффективным в лечении аномального клеточного роста. В одном воплощении данного способа аномальный клеточный рост представляет собой рак, включая, без ограничения ими, рак легкого, рак кости, рак поджелудочной железы, рак кожи, рак головы или шеи, кожную или внутриглазную меланому, рак матки, рак яичников, рак прямой кишки, рак анальной области, рак желудка, рак толстой кишки, рак молочной железы, рак матки, карциному фаллопиевых труб, карциному эндометрия, цервикальную карциному, карциному влагалища, карциному вульвы, болезнь Ходжкина, рак пищевода, рак тонкого кишечника, рак эндокринной системы, рак щитовидной железы, рак паращитовидной железы, рак надпочечников, саркому мягких тканей, рак уретры, рак пениса, рак простаты, хроническую или острую лейкемию, лимфоцитарные лимфомы, рак мочевого пузыря, рак почки или мочеточника, почечноклеточную карциному, карциному почечной лоханки, новообразования в центральной нервной системе (ЦНС), первичную лимфому ЦНС, опухоли позвоночного столба, глиомы мозгового ствола, аденому гипофиза или комбинацию одного или более вышеуказанных злокачественных новообразований. В одном воплощении способ включает введение млекопитающему такого количества соединения формулы 1^ которое эффективно в лечении указанной раковой солидной опухоли. В одном предпочтительном воплощении солидная опухоль представляет собой рак молочной железы, легкого, толстой кишки, мозга, простаты, желудка, поджелудочной железы, яичника, кожи (меланома), эндокринной системы, матки, яичника и мочевого пузыря. В другом воплощении указанного способа указанный аномальный рост представляет собой доброкачественное пролиферативное заболевание, включая, без ограничения ими, псориаз, доброкачественную гипертрофию простаты или рестеноз. Данное изобретение также относится к способу лечения аномального клеточного роста у млекопитающего, включающему введение указанному млекопитающему соединения формулы 1 или его фармацевтически приемлемой соли, сольвата или пролекарства в количестве, которое эффективно в лечении аномального клеточного роста, в комбинации с противоопухолевым агентом, выбранным из группы, состоящей из ингибиторов митоза, алкилирующих агентов, антиметаболитов, интеркалирующих антибиотиков, ингибиторов факторов роста, ингибиторов клеточного цикла, ферментов, ингибиторов топоизомераз, модификаторов биологического ответа, антител, цитотоксических агентов, антигормонов и антиандрогенов. Данное изобретение также относится к фармацевтической композиции для лечения аномального клеточного роста у млекопитающего, включая человека, содержащей соединение формулы 1, как оно определено выше, или его фармацевтически приемлемую соль, сольват или пролекарство в количестве, которое является эффективным в лечении аномального клеточного роста, и фармацевтически приемлемый носитель. В одном воплощении указанной композиции указанный аномальный клеточный рост представляет собой рак, включая, без ограничения ими, рак легкого, рак кости, рак поджелудочной железы, рак кожи, рак головы или шеи, кожную или внутриглазную меланому, рак матки, рак яичников, рак прямой кишки, рак анальной области, рак желудка, рак толстой кишки, рак молочной железы, рак матки, карциному фаллопиевых труб, карциному эндометрия, цервикальную карциному, карциному влагалища, карциному вульвы, болезнь Ходжкина, рак пищевода, рак тонкого кишечника, рак эндокринной системы, рак щитовидной железы, рак паращитовидной железы, рак надпочечников, саркому мягких тканей, рак уретры, рак пениса, рак простаты, хроническую или острую лейкемию, лимфоцитарные лимфомы, рак мочевого пузыря, рак почки или мочеточника, почечноклеточную карциному, карциному почечной лоханки, новообразования центральной нервной системы (ЦНС), первичную ЦНС лимфому, опухоли позвоночного столба, глиому ствола мозга, аденому гипофиза или комбинацию одного или более вышеуказанных злокачественных новообразований, но не ограничиваясь ими. В другом предпочтительном воплощении указанной фармацевтической композиции указанный аномальный клеточный рост представляет собой псориаз, доброкачественную гипертрофию простаты или рестеноз. Данное изобретение также относится к способу лечения аномального клеточного роста у млекопитающего, который включает введение указанному млекопитающему соединения формулы 1 или его фармацевтически приемлемой соли, сольвата или пролекарства в количестве, которое является эффективным в лечении аномального клеточного роста, в комбинации с другим противоопухолевым агентом, выбранным из группы, состоящей из ингибиторов митоза, алкилирующих агентов, антиметаболитов, интеркалирующих антибиотиков, ингибиторов факторов роста, ингибиторов клеточного цикла, ферментов, ингибиторов топоизомераз, модификаторов биологического ответа, антител, цитотоксических агентов, антигормонов и антиандрогенов. Данное изобретение также охватывает фармацевтическую композицию для лечения аномального клеточного роста, которая включает соединение формулы 1, как оно определено выше, его фармацевтически приемлемую соль, сольват или пролекарство, которые эффективны в лечении аномального клеточного роста, и другой противоопухолевый агент, выбранный из группы, состоящей из ингибиторов митоза, алкилирующих агентов, антиметаболитов, интеркалирующих антибиотиков, ингибиторов факторов роста, ингибиторов клеточного цикла, ферментов, ингибиторов топоизомераз, модификаторов биологического ответа, антител, цитотоксических агентов, антигормонов и антиандрогенов. Данное изобретение также относится к способу лечения расстройства, ассоциированного с ангиогенезом, у млекопитающего, в том числе человека, включающему введение указанному млекопитающему такого количества соединения формулы 1, как оно определено выше, или его фармацевтически приемлемой соли, сольвата или пролекарства, которое эффективно в лечении указанного расстройства, в комбинации с одним или более противоопухолевыми агентами, перечисленными выше. Такие расстройства включают злокачественные опухоли, такие как меланома; глазные расстройства, такие как возрастная дегенерация желтого пятна, синдром предполагаемого гистоплазмоза глаз и ретинальная неоваскуляризация вследствие пролиферативной диабетической ретинопатии; ревматоидный артрит; расстройства потери кости, такие как остеопороз, болезнь Педжета, гуморальная гиперкальцемия при злокачественном развитии, гиперкальцемия вследствие метастазирующих в кости опухолей и остеопороз, индуцированный глюкокортикоидным лечением; коронарный рестеноз; и некоторые микробные инфекции, включая инфекции, ассоциированные с микробными патогенами, выбранными из аденовируса, гантавируса, Borrelia burgdorferi, Yersinia spp., Bordetella pertussis и Streptococcus группы A. Данное изобретение также относится к способу (и к фармацевтической композиции для) лечения аномального клеточного роста у млекопитающего, в которых используют некоторое количество соединения формулы 1 или его фармацевтически приемлемой соли, сольвата или пролекарства в комбинации с некоторым количеством одного или более веществ, выбранных из антиангиогенезных агентов, ингибиторов сигнальной трансдукции и антипролиферативных агентов, причем эти количества совместно эффективны в лечении указанного аномального клеточного роста. Антиангиогенезные агенты, такие как ингибиторы ММР-2 (матриксной металлопротеиназы 2), ингибиторы ММР-9 (матриксной металлопротеиназы 9) и ингибиторы СОХ-Н (циклооксигеназы II), могут быть использованы вместе с соединением формулы 1 в способах и фармацевтических композициях, описанных в данном описании изобретения. Примеры полезных ингибиторов СОХ-Н включают CELEBREX(tm) (целекоксиб), бекстра (валдекоксиб), паракоксиб, виокс (рофекоксиб) и аркоксиа (эторикоксиб). Примеры полезных ингибиторов матриксных металлопротеиназ описаны в WO 96/33172 (опубликованной 24 октября 1996 года), WO 96/27583 (опубликованной 7 марта 1996 года), заявке на европейский патент № 97304971.1 (поданной 8 июля 1997 года), заявке на европейский патент № 99308617.2 (поданной 29 октября 1999 года), WO 98/07697 (опубликованной 26 февраля 1998 года), WO 98/03516 (опубликованной 29 января 1998 года), WO 98/34918 (опубликованной 13 августа 1998 года), WO 98/34915 (опубликованной 13 августа 1998 года), WO 98/33768 (опубликованной 6 августа 1998 года), WO 98/30566 (опубликованной 16 июля 1998 года), публикации Европейского патента 606046 (опубликованной 13 июля 1994 года), публикации Европейского патента 931788 (опубликованной 28 июля 1999 года), WO 90/05719 (опубликованной 31 мая 1990 года), WO 99/52910 (опубликованной 21 октября 1999 года), WO 99/52889 (опубликованной 21 октября 1999 года), WO 99/29667 (опубликованной 17 июня 1999 года), международной заявке РСТ № PCT/IB98/01113 (поданной 21 июля 1998 года), заявке на европейский патент № 99302232.1 (поданной 25 марта 1999 года), заявке на патент Великобритании № 9912961.1 (поданной 3 июня 1999 года), предварительной заявке США № 60/148464 (поданной 12 августа 1999 года), патенте США 5863949 (выданном 26 января 1999 года), патенте США 5861510 (выданном 19 января 1999 года) и публикации европейского патента 780386 (опубликованной 25 июня 1997 года), все из которых включены в данное описание изобретения во всей своей полноте посредством ссылки. Предпочтительными ингибиторами ММР-2 и ММР-9 являются ингибиторы, имеющие незначительную активность в отношении ингибирования ММР-1 или не имеющие такой активности. Более предпочтительными являются ингибиторы, селективно ингибирующие ММР-2 и/или ММР-9 по сравнению с другими матриксными металлопротеиназами (то есть ММР-1, ММР-3, ММР-4, ММР-5, ММР-6, ММР-7, ММР-8, ММР-10, ММР-11, ММР-12 и ММР-13). Некоторыми конкретными примерами ингибиторов ММР, полезных в комбинации с соединениями по настоящему изобретению, являются AG-3340, RO 32-3555, RS 13-0830 и соединения, перечисленные в следующем списке: 3-[[4-(4-фторфенокси)бензолсульфонил]-(1-гидроксикарбамои-лциклопентил)амино]пропионовая кислота; гидроксиамид 3-э/сзо-3-[4-(4-фторфенокси)бензолсульфониламино]-8-окса-бицикло[3.2.1]октан-3-карбоновой кислоты; гидроксиамид (2R,3R)-1-[4-(2^op-4- фторбензилокси)бензолсульфонил]-3-гидрокси-3-метилпиперидин-2-карбоновой кислоты; гидроксиамид 4-[4-(4- фторфенокси)бензолсульфониламино]тетрагидропиран-4-карбоновой кислоты; 3-[[4-(4-фторфенокси)бензолсульфонил]-(1-гидроксикарбамоил-циклобутил)амино]пропионовая кислота; гидроксиамид 4-[4-(4-хлорфенокси)бензолсульфониламино]тетрагидропиран-4-карбоновой кислоты; гидроксиамид 3-[4-(4-хлорфенокси)бензолсульфониламино]тетрагидропиран-3-карбоновой кислоты; гидроксиамид (2R.3R) 1-[4-(4-фтор-2-метилбензилокси)бензолсульфонил]-3-гидрокси-3-метилпиперидин-2-карбоновой кислоты; 3-[[4-(4-фторфенокси)бензолсульфонил]-(1- гидроксикарбамоил-1-метилэтил)амино]пропионовая кислота; 3-[[4-(4-фторфенокси)бензолсульфонил]-(4-гидроксикарбамоил-тетрагидропиран-4-ил)амино]пропионовая кислота; гидроксиамид 3-экзо-3-[4-(4- хлорфенокси)бензолсульфониламино]-8-окса-бицикло[3.2.1]октан-3-карбоновой кислоты; гидроксиамид 3-энбо-3-[4-(4-фторфенокси)бензолсульфониламино]-8-окса-бицикло[3.2.1]октан-3-карбоновой кислоты; и гидроксиамид 3-[4-(4-фторфенокси)бензолсульфониламино]тетрагидрофуран-3-карбоновой кислоты; и фармацевтически приемлемые соли, сольваты и пролекарства указанных соединений. Ингибиторы VEGF (васкулярного эндотелиального фактора роста), например SU-11248, SU-5416 и SU-6668 (Sugen Inc. of South San Francisco, California, USA) также можно комбинировать с соединением формулы 1. Ингибиторы VEGF описаны, например, в WO 99/24440 (опубликованной 20 мая 1999 года), международной заявке PCT/IB99/00797 (поданной 3 мая 1999 года), в WO 95/21613 (опубликованной 17 августа 1995 года), WO 99/61422 (опубликованной 2 декабря 1999 года), патенте США 5834504 (выданном 10 ноября 1998 года), WO 98/50356 (опубликованной 12 ноября 1998 года), патенте США 5883113 (выданном 16 марта 1999 года), патенте США 5886020 (выданном 23 марта 1999 года), патенте США 5792783 (выданном 11 августа 1998 года), патенте США № 6653308 (выданном 25 ноября 2003 года), WO 99/10349 (опубликованной 4 марта 1999 года), WO 97/32856 (опубликованной 12 сентября 1997 года), WO 97/22596 (опубликованной 26 июня 1997 года), WO 98/54093 (опубликованной 3 декабря 1998 года), WO 98/02438 (опубликованной 22 января 1998 года), WO 99/16755 (опубликованной 8 апреля 1999 года) и WO 98/02437 (опубликованной 22 января 1998 года); все из которых включены в данное описание изобретения во всей своей полноте посредством ссылки. Другими примерами некоторых конкретных ингибиторов VEGF являются IM862 (Cytran Inc. of Kirkland, Washington, USA); авастин - анти-VEGF моноклональное антитело от Genentech, Inc. of South San Francisco, California; и ангиозим синтетический рибозим от Ribozyme (Boulder, Colorado) и Chiron (Emeryville, California). Ингибиторы егЬВ2-рецептора, такие как GW-282974 (Glaxo Wellcome pic), CP-724714 (Pfizer Inc.) и моноклональные антитела AR-209 (Aronex Pharmaceuticals Inc. of The Woodlands, Texas, USA) и 2B-1 (Chiron), можно вводить в комбинации с соединением формулы 1. Такие егЬВ2-ингибиторы включают герсептин, 2С4 и пертузумаб. Такие егЬВ2-ингибиторы включают ингибиторы, описанные в WO 98/02434 (опубликованной 22 января 1998 года), WO 99/35146 (опубликованной 15 июля 1999 года), WO 99/35132 (опубликованной 15 июля 1999 года), WO 98/02437 (опубликованной 22 января 1998 года), WO 97/13760 (опубликованной 17 апреля 1997 года), WO 95/19970 (опубликованной 27 июля 1995 года), патенте США 5587458 (выданном 24 декабря 1996 года) и патенте США 5877305 (выданном 2 марта 1999 года); все они включены в данное описание изобретения во всей своей полноте посредством ссылки. Ингибиторы егЬВ2-рецептора, полезные в настоящем изобретении, также описаны в предварительной заявке США № 60/117341, поданной 27 января 1999 года, и предварительной заявке США № 60/117346, поданной 27 января 1999 года; обе они включены в данное описание изобретения во всей своей полноте посредством ссылки. Другие ингибиторы егЬВ2-рецептора включают ТАК-165 (Takeda) и GW-572016 (Glaxo-Wellcome). Кроме того, показано, что различные другие соединения, такие как производные стирола, обладают ингибиторными свойствами в отношении тирозинкиназ, и некоторые ингибиторы тирозинкиназ были определены как ингибиторы егЬВ2-рецептора. Пять совсем недавних европейских патентных публикаций, а именно ЕР 0566226 А1 (опубликовано 20 октября 1993 года), ЕР 0602851 А1 (опубликовано 22 июня 1994 года), ЕР 0635507 А1 (опубликовано 25 января 1995 года), ЕР 0635498 А1 (опубликовано 25 января 1995 года) и ЕР 0520722 А1 (опубликовано 30 декабря 1992 года), относятся к некоторым бициклическим производным, в частности хиназолиновым производным, обладающим противораковыми свойствами, являющимися результатом их ингибиторных свойств в отношении тирозинкиназ. Кроме того, международная заявка на патент WO 92/20642 (опубликованная 26 ноября 1992 года) относится к некоторым бис-моно- и бициклическим арильным и гетероарильным соединениям в качестве ингибиторов тирозинкиназ, полезных в ингибировании аномальной клеточной пролиферации. Международные заявки на патент WO 96/16960 (опубликованная 6 июня 1996 года), WO 96/09294 (опубликованная 6 марта 1996 года), WO 97/30034 (опубликованная 21 августа 1997 года), WO 98/02434 (опубликованная 22 января 1998 года), WO 98/02437 (опубликованная 22 января 1998 года) и WO 98/02438 (опубликованная 22 января 1998 года) также относятся к замещенным бициклическим гетероароматическим производным в качестве ингибиторов тирозинкиназ, полезных для той же самой цели. Другими заявками на патент, относящимися к противораковым соединениям, являются международная заявка на патент WO 00/44728 (опубликованная 3 августа 2000 года), ЕР 1029853А1 (опубликованная 23 августа 2000 года) и WO 01/98277 (опубликованная 12 декабря 2001 года); все из которых включены в данное описание изобретения во всей своей полноте посредством ссылки. Другие антипролиферативные агенты, которые могут быть использованы с соединениями по настоящему изобретению, включают ингибиторы фермента фарнезилпротеинтрансферазы и ингибиторы рецепторной тирозинкиназы PDGFr, включая соединения, раскрытые и заявленные в следующих заявках на патент США: 09/221946 (поданной 28 декабря 1998 года); 09/454058 (поданной 2 декабря 1999 года); 09/501163 (поданной 9 февраля 2000 года); 09/539930 (поданной 31 марта 2000 года); 09/202796 (поданной 22 мая 1997 года); 09/384339 (поданной 26 августа 1999 года); и 09/383755 (поданной 26 августа 1999 года); и соединения, раскрытые и заявленные в следующих предварительных заявках на патент США: 60/168207 (поданной 30 ноября 1999 года); 60/170119 (поданной 10 декабря 1999 года); 60/177718 (поданной 21 января 2000 года); 60/168217 (поданной 30 ноября 1999 года); и 60/200834 (поданной 1 мая 2000 года). Каждая из приведенных выше заявок на патент и предварительных заявок на патент включена в данное описание изобретения во всей своей полноте посредством ссылки. Соединение формулы 1 также можно применять с другими агентами, полезными в лечении аномального клеточного роста или рака, включая, без ограничения ими, агенты, способные усиливать противоопухолевые иммунные ответы, такие как антитела к CTLA4 (цитотоксическому лимфоцитарному антигену 4) и другие агенты, способные блокировать CTLA4; и антипролиферативные агенты, такие как другие ингибиторы фарнезилпротеинтрансферазы, например ингибиторы фарнезилпротеинтрансферазы, описанные в ссылках, приведенных выше в разделе "Предшествующий уровень техники". Специфичные к CTLA4 антитела, которые можно применять в настоящем изобретении, включают антитела, описанные в предварительной заявке США 60/113647 (поданной 23 декабря 1998 года), которая включена в данное описание изобретения во всей своей полноте посредством ссылки. Соединение формулы I может быть применено в виде монотерапии или терапия может включать одно или более чем одно другое противоопухолевое вещество, выбранное, например, из ингибиторов митоза, например винбластина; алкилирующих агентов, например цисплатина, оксалиплатина, карбоплатина и циклофосфамида; антиметаболитов, например 5-фторурацила, капецитабина, цитозинарабинозида и гидроксимочевины, или, например, одного из предпочтительных антиметаболитов, раскрытых в заявке на европейский патент № 239362, такого как Л/-(5-[М-(3,4-дигидро-2-метил-4-оксохиназолин-6-илметил)-Л/-метиламино]-2-теноил)-1_-глутаминовая кислота; ингибиторов ростовых факторов; ингибиторов клеточного цикла; интеркалирующих антибиотиков, например адриамицина и блеомицина; ферментов, например интерферона; и антигормонов, например антиэстрогенов, таких как нолвадекс (тамоксифен), или, например, антиандрогенов, таких как казодекс (4'-циано-3-(4-фторфенилсульфонил)-2-гидрокси-2-метил-3'-(трифторметил)пропионанилид). Соединения по настоящему изобретению можно использовать сами по себе или в комбинации с одним или более чем одним противораковым агентом или агентом заместительной терапии. Например, соединения по настоящему изобретению можно применять с цитотоксическими агентами, например одним или более чем одним, выбранным из группы, состоящей из камптотецина, иринотекана HCI (камптосар), эдотекарина, SU-11248, эпирубицина (элленс), доцетаксела (таксотер), паклитаксела, ритуксимаба (ритуксан), бевацизумаба (авастин), иматиниба мезилата (гливак), эрбитукса, гефитиниба (пресса) и их комбинаций. Данное изобретение также включает применение соединений настоящего изобретения вместе с гормональной терапией, например эксеместаном (аромазин), лупроном, анастрозолом (аримидекс), тамоксифена цитратом (нолвадекс), трелстаром и их комбинациями. Кроме того, согласно изобретению предложено соединение по настоящему изобретению само по себе или в комбинации с одним или более чем одним продуктом заместительной терапии, например продуктом, выбранным из группы, состоящей из филграстима (нейпоген), ондансетрона (зофран), фрагмина, прокрита, алокси, эменда или их комбинаций. Такое совместное лечение может быть достигнуто посредством одновременного, последовательного или раздельного введения отдельных компонентов лечения. Соединения по изобретению можно применять с противоопухолевыми агентами, алкилирующими агентами, антиметаболитами, антибиотиками, противоопухолевыми агентами растительного происхождения, производными камптотецина, ингибиторами тирозинкиназ, антителами, интерферонами и/или модификаторами биологического ответа. В этом отношении следующее далее представляет собой неограничивающий список примеров дополнительных агентов, которые можно применять с соединениями по изобретению: • алкилирующие агенты включают, без ограничения ими, N-оксид азотистого иприта, циклофосфамид, ифосфамид, мелфалан, бусульфан, митобронитол, карбоквон, тиотепа, ранимустин, нимустин, темозоломид, AMD-473, алтретамин, АР-5280, апазиквон, бросталлицин, бендамустин, кармустин, эстрамустин, фотемустин, глуфосфамид, ифосфамид, KW-2170, мафосфамид и митолактол, но этим не ограничиваются; платина-координированные алкилирующие соединения включают цисплатин, карбоплатин, эптаплатин, лобаплатин, недаплатин, оксалиплатин или сатрплатин; • антиметаболиты включают, без ограничения ими, метотрексат, 6-меркаптопуринрибозид, меркаптопурин, 5-фторурацил (5-FU) сам по себе или в комбинации с лейковорином, тегафур, UFT, доксифлуридин, кармофур, цитарабин, цитарабина окфосфат, эноцитабин, S-1, гемцитабин, флударабин, 5-азацитидин, капецитабин, кладрибин, клофарабин, децитабин, эфлорнитин, этинилцитидин, цитозинарабинозид, гидроксимочевину, TS-1, мелфалан, неларабин, нолатрексед, окфосфат, динатрия преметрексед, пентостатин, пелитрексол, ралтитрексед, триапин, триметрексат, видарабин, винкристин, винорелбин; или например, один из предпочтительных антиметаболитов, раскрытых в заявке на европейский патент №239362, такой как N-(5-[N-(3,4-дигидро-2-метил-4-оксохиназолин-6-илметил)-г> 1-метиламино]-2-теноил)-1_-глутаминовая кислота; • антибиотики включают, но не ограничены ими: акларубицин, актиномицин D, амрубицин, аннамицин, блеомицин, даунорубицин, доксорубицин, элсамитруцин, эпирубицин, галарубицин, идарубицин, митомицин С, неморубицин, неокарзиностатин, пепломицин, пирарубицин, ребеккамицин, стималамер, стрептозоцин, валрубицин или зиностатин; • агенты для гормональной терапии, например эксеместан (аромазин), лупрон, анастрозол (аримидекс), доксеркальциферол, фадрозол, форместан, антиэстрогены, такие как тамоксифена цитрат (нолвадекс) и фулвестрант, трелстар, торемифен, ралоксифен, лазофоксифен, летрозол (фемара), или антиандрогены, такие как бикалутамид, флутамид, мифепристон, нилутамид, Casodex(r) (4'-циано-3-(4-фторфенилсульфонил)-2-гидрокси-2-метил-З'-(трифторметил)пропионанилид) и их комбинации; • противоопухолевые вещества растительного происхождения включают, например, вещества, выбранные из ингибиторов митоза, например винбластин, доцетаксел (таксотер) и паклитаксел; • цитотоксические ингибирующие топоизомеразу агенты включают один или более чем один агент, выбранный из группы, состоящей из акларубицина, амонафида, белотекана, камптотецина, 10-гидроксикамптотецина, 9-аминокамптотецина, дифломотекана, иринотекана HCI (камптосар), эдотекарина, эпирубицина (элленс), этопозида, эксатекана, гиматекана, луртотекана, митоксантрона, пирарубицина, пиксантрона, рубитекана, собузоксана, SN-38, тафлупозида и топотекана и их комбинаций; • иммунологические средства включают интерфероны и множество других агентов, повышающих иммунитет. Интерфероны включают интерферон-альфа, интерферон-альфа-2а, интерферон-альфа-2Ь, интерферон-бета, интерферон-гамма-1а или интерферон-гамма-п1. Другие агенты включают филграстим, лентинан, сизофилан, ТераЦис, убенимекс, WF-10, алдеслейкин, алемтузумаб, ВАМ-002, дакарбазин, даклизумаб, денилейкин, гемтузумаб, озогамицин, ибритумомаб, имиквимод, ленограстим, лентинан, меланомную вакцину (корикса), молграмостин, OncoVAX-CL, сарграмостим, тазонермин, теклейкин, тималазин, тозитумомаб, вирулизин, Z-100, эпратузумаб, митумомаб, ореговомаб, пемтумомаб, провенж; • модификаторы биологического ответа представляют собой агенты, которые модифицируют защитные механизмы живых организмов или биологические ответы, такие как выживаемость, рост или дифференцировка тканевых клеток, с целью придания им противоопухолевой активности. Такие агенты включают крестин, лентинан, сизофиран, пицибанил или убенимекс; • другие противораковые агенты включают алитретиноин, амплиген, атрасентан, бексаротен, бортезомиб, босентан, кальцитриол, эксисулинд, финастерид, фотемустин, ибандроновую кислоту, милтефозин, митоксантрон, I-аспарагиназу, прокарбазин, дакарбазин, гидроксикарбамид, пегаспаргазу, пентостатин, тазаротин, TLK-286, велкад, тарцева или третиноин; • другие антиангиогенные соединения включают ацитретин, фентретинид, талидомид, золедроновую кислоту, ангиостатин, аплидин, силенгтид, комбретастатин А-4, эндостатин, галофугинон, ребимастат, ремоваб, ревлимид, скваламин, украин и витаксин; • платина-координированные соединения включают, без ограничения ими, цисплатин, карбоплатин, недаплатин или оксалиплатин; • производные камптотецина включают, без ограничения ими, камптотецин, 10-гидроксикамптотецин, 9-аминокамптотецин, иринотекан, SN-38, эдотекарин и топотекан; • тирозинкиназные ингибиторы представляют собой пресса или SU5416; • антитела включают герцептин, эрбитукс, авастин или ритуксимаб; • интерфероны включают интерферон-альфа, интерферон-альфа-2а, интерферон-альфа-2Ь, интерферон-бета, интерферон-гамма-1а или интерферон-гамма-п1; • модификаторы биологического ответа представляют собой агенты, которые модифицируют защитные механизмы живых организмов или биологические ответы, такие как выживаемость, рост или дифференцировка тканевых клеток, с целью придания им противоопухолевой активности. Такие агенты включают крестин, лентинан, сизофиран, пицибанил или убенимекс. Другие противоопухолевые агенты включают митоксантрон, I-аспарагиназу, прокарбазин, дакарбазин, гидроксикарбамид, пентостатин или третиноин. "Аномальный клеточный рост", при использовании в данном описании изобретения, если не указано иное, относится к клеточному росту, который не зависит от нормальных регуляторных механизмов (например утрата контактного ингибирования). Он включает аномальный рост: (1) опухолевых клеток (опухоли), которые пролиферируют в результате экспрессии мутированной тирозинкиназы или сверхэкспрессии рецепторной тирозинкиназы; (2) доброкачественных или злокачественных клеток других пролиферативных заболеваний, при которых наблюдается аберрантная активация тирозинкиназы; (3) любых опухолей, которые пролиферируют посредством рецепторных тирозинкиназ; (4) любых опухолей, которые пролиферируют посредством аберрантной активации серин/треонинкиназ; и (5) доброкачественных или злокачественных клеток других пролиферативных заболеваний, при которых наблюдается аберрантная активация серин/треонинкиназ. В данном изобретении также предложен способ ингибирования тирозинкиназ у млекопитающего, включающий введение нуждающемуся в этом млекопитающему фармацевтически или терапевтически эффективного количества соединения по данному изобретению или его фармацевтически приемлемой солевой формы. Более конкретно, в данном изобретении дополнительно предложен способ необратимого ингибирования тирозинкиназ у млекопитающего. Раскрытые здесь способы также включают способы необратимого ингибирования тирозинкиназ, включая EGFR (рецептор эпидермального фактора роста), PDGFR (рецептор тромбоцитарного фактора роста), c-src, erbB1, егЬВ2 и егЬВ4. Данное изобретение также может быть охарактеризовано как включающее способ ингибирования секреции VEGF у млекопитающего. Дополнительный способ включает ингибирование фосфорилирования тирозина в егЬВЗ у млекопитающего. Раскрытые здесь соединения также полезны в качестве ингибиторов pan-erbB, то есть ингибиторов множественных егЬВ киназ при каждом введении. Специалисты в данной области могут с легкостью определить пациентов, нуждающихся в описанном здесь лечении. В частности, пациенты с более высоким риском развития рестеноза включают субъектов, которые подвергались процедурам ангиопластики, шунтирования или пересадок, либо были реципиентами других сосудистых процедур или травм. Субъекты с более высоким риском развития атеросклероза включают субъектов с ожирением, потребляющих пищу с высоким содержанием жиров, имеющих повышенные уровни холестерина, либо страдающих гипертензией. Раскрытые способы полезны в лечении млекопитающих, включая людей, животных-спутников человека, таких как собаки и кошки, и сельскохозяйственных животных, таких как лошади, овцы, свиньи, козы, крупный рогатый скот и так далее. Термин "рак или злокачественное новообразование" включает, без ограничения ими, следующие виды злокачественных новообразований: рак молочной железы; яичников; шейки; простаты; яичка; пищевода; глиобластому; нейробластому; рак желудка; кожи, кератоакантому; рак легкого, эпидермоидную карциному, крупноклеточную карциному, аденокарциному, мелкоклеточный рак легкого; немелкоклеточный рак легкого; рак кости; толстой кишки, аденокарциному, аденому; рак поджелудочной железы, аденокарциному, рак щитовидной железы, фолликулярную карциному, недифференцированную карциному, папиллярную карциному; семиному; меланому; саркому; карциному мочевого пузыря; карциному печени и желчевыводящих путей; карциному почки; миелоидные расстройства; лимфоидные расстройства, болезнь Ходжкина, волосковые клетки; рак ротовой полости и глотки (рта), рак губ, языка, глотки; рак тонкой кишки; рак ободочной и прямой кишок, толстой кишки, прямой кишки; рак головного мозга и центральной нервной системы; и лейкемию. Кроме того, соединения по данному изобретению могут быть использованы для лечения пациентов, нуждающихся в ингибировании секреции васкулярного эндотелиального фактора роста (VEGF). Пациенты, нуждающиеся в ингибировании секреции VEGF, включают пациентов, имеющих злокачественное новообразование, диабетическую ретинопатию, ревматоидный артрит, псориаз, рестеноз, атеросклероз, остеопороз, эндометриоз, субъетов, перенесших имплантацию эмбриона, или субъектов, страдающих другими заболеваниями, при которых играют роль ангиогенез и неоваскуляризация. Соединения по настоящему изобретению могут быть использованы в способах ингибирования фосфорилирования тирозина в erbB1, егЬВ2 и егЬВ4. Пациетами, нуждающимися в ингибировании фосфорилирования тирозина в erbB1, егЬВ2 и егЬВ4, являются пациенты, страдающие вышеупомянутыми заболеваниями в отношении ингибирования EGFR и ингибирования секреции VEGF, или имеющие риск данных заболеваний. Соединения по изобретению могут быть введены людям и животным перорально, ректально, парентерально (внутривенно, внутримышечно или подкожно), внутриполостным образом, внутривагинально, внутрибрюшинно, внутрь мочевого пузыря, местно (порошки, мази или капли), либо в виде буккального или назального спрея. Соединение может быть введено само по себе или как часть фармацевтически приемлемой композиции, которая включает фармацевтически приемлемые эксципиенты. Композиции, подходящие для парентеральной инъекции, могут содержать физиологически приемлемые стерильные водные и неводные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки для разведения с образованием стерильных растворов или дисперсий для инъекций. Примеры подходящих водных и неводных носителей, разбавителей, растворителей или наполнителей включают воду, этанол, полиолы (пропиленгликоль, полиэтиленгликоль, глицерин и подобное), их подходящие смеси, растительные масла (такие как оливковое масло) и инъекционные органические сложные эфиры, такие как этилолеат. Необходимую текучесть можно поддерживать, например, используя покрытие, например лецитин, путем поддерживания требуемого размера частиц в случае дисперсий, и путем использования поверхностно-активных веществ. Соединения по изобретению можно легко применять в водных препаратах. Например, 4-пиперидин-1-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амид имеет растворимость в воде примерно 10 мкг/мл при рН 6,3, и его растворимость возрастает при более низких значениях рН. Эти композиции могут также содержать адъюванты, такие как консерванты, увлажняющие агенты, эмульгирующие и диспергирующие агенты. Предупреждение действия микроорганизмов может быть достигнуто с помощью различных антибактериальных и противогрибковых агентов, например парабенов, хлорбутанола, фенола, сорбиновой кислоты и подобного. Также может быть желательным включение изотонических агентов, например Сахаров, хлорида натрия и тому подобного. Пролонгированной абсорбции инъекционной фармацевтической формы можно добиться путем использования агентов, замедляющих абсорбцию, например моностеарата алюминия и желатина. Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах активное соединение смешано по меньшей мере с одним инертным традиционным эксципиентом (или носителем), таким как цитрат натрия или дикальция фосфат, или (а) наполнителями или носителями, такими как, например, крахмалы, лактоза, сахароза, глюкоза, маннит и кремниевая кислота; (б) связывающими агентами, такими как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и аравийская камедь; (в) увлажняющими агентами, такими как, например, глицерин; (г) разрыхляющими агентами, такими как, например, агар-агар, карбонат кальция, картофельный крахмал или крахмал из тапиоки, альгиновая кислота, некоторые сложные силикаты и карбонат натрия; (д) замедлителями растворения, такими как, например, парафин; (е) усилителями абсорбции, такими как, например, четвертичные аммониевые соединения; (ж) увлажняющими агентами, такими как, например, цетиловый спирт и глицеринмоностеарат; (з) адсорбентами, такими как, например, каолин и бентонит; и (и) смазывающими веществами, такими как, например, тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия или их смеси. В случае капсул, таблеток и пилюль лекарственные формы также могут содержать буферные агенты. Твердые композиции аналогичного типа можно также использовать в качестве заполнителей мягких и твердых желатиновых капсул, используя такие эксципиенты, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и тому подобное. Такие твердые лекарственные формы как таблетки, драже, капсулы, пилюли и гранулы, могут быть изготовлены с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие покрытия, хорошо известные в данной области техники. Они могут содержать замутняющие агенты, а также могут иметь такой состав, чтобы высвобождать активное соединение в определенной части желудочно-кишечного тракта замедленным образом. Примерами композиций для заполнения, которые можно использовать, являются полимерные вещества и воски. Активное соединение также может находиться в микроинкапсулированной форме, если целесообразно, с одним или более чем одним вышеуказанным эксципиентом. Жидкие лекарственные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии,. сиропы и эликсиры. В дополнение к активному соединению жидкие лекарственные формы могут содержать инертные разбавители, обычно используемые в данной области, такие как вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как, например, этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла, в частности хлопковое масло, арахисовое масло, масло из зародышей кукурузы, оливковое масло, касторовое масло и кунжутное масло, глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбитана, или смеси этих веществ, и тому подобное. Помимо инертных разбавителей, композиция может также включать адъюванты, такие как увлажняющие агенты, эмульгирующие и суспендирующие агенты, подсластители, корригенты и отдушки. Суспензии в дополнение к активному соединению могут содержать суспендирующие агенты, такие как, например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сорбитановые сложные эфиры, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, или смеси этих веществ, и тому подобное. Композиции для ректального введения предпочтительно представляют собой суппозитории, которые можно приготовить путем смешивания соединений по настоящему изобретению с подходящими нераздражающими эксципиентами или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при обычных температурах, но жидкими при температуре тела и, следовательно, плавятся в прямой кишке или вагинальной полости и высвобождают активный компонент. Лекарственные формы для местного введения включают мази, порошки, спреи и ингалируемые формы. Активный компонент смешивают в стерильных условиях с физиологически приемлемым носителем и любыми консервантами, буферами или пропеллентами, как это может требоваться. Подразумевается, что офтальмические препараты, глазные мази, порошки и растворы также входят в объем данного изобретения. Данное изобретение также включает фармацевтически или терапевтически приемлемые соли, сложные эфиры, амиды и пролекарственные формы соединений по данному изобретению. Термины "фармацевтически приемлемые соли, сложные эфиры, амиды и пролекарства", при использовании в данном описании, относятся к тем карбоксилатным солям, солям присоединения аминокислот, сложным эфирам, амидам и пролекарствам соединений по настоящему изобретению, которые являются, в рамках компетентного медицинского суждения, пригодными для использования в контакте с тканями пациента без чрезмерной токсичности, раздражения, аллергического ответа и подобного, соответствуют разумному соотношению польза/риск и эффективны при предполагаемом использовании, а также, где это возможно, к цвиттерионным формам соединений по изобретению. Термин "соли" относится к относительно нетоксичным солям присоединения органических и неорганических кислот соединений по настоящему изобретению. Эти соли могут быть получены in situ в ходе заключительного выделения и очистки соединения или путем отдельного взаимодействия очищенного соединения в форме его свободного основания с подходящей органической или неорганической кислотой и выделения образованной таким образом соли. Репрезентативные соли включают гидробромид, гидрохлорид, сульфат, бисульфат, нитрат, ацетат, оксалат, валерат, олеат, пальмитат, стеарат, лаурат, борат, бензоат, лактат, фосфат, тозилат, цитрат, малеат, фумарат, сукцинат, тартрат, нафтилат, мезилат, глюкогептонат, лактобионат и лаурилсульфонат и тому подобное. Они могут включать катионы щелочных и щелочноземельных металлов, таких как натрий, литий, калий, кальций, магний и тому подобное, а также нетоксичные катионы аммония, четвертичного аммония и аминов, включая, без ограничения ими, аммоний, тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, триэтиламин, этиламин и тому подобное (смотри, например, статью S.M. Berge, et al., "Pharmaceutical Salts," J. Pharm. Sci., 1977;66:1-19, которая включена в данное описание изобретения посредством ссылки). Примеры фармацевтически приемлемых нетоксичных сложных эфиров соединений по данному изобретению включают СгСбалкиловые сложные эфиры, где алкильная группа имеет прямую или разветвленную цепь. Приемлемые сложные эфиры включают также Сб-С/Циклоалкиловые сложные эфиры, а также арилалкиловые сложные эфиры, такие как, но без ограничения ими, бензиловые сложные эфиры. Предпочтительными являются СгСдалкиловые сложные эфиры. Сложные эфиры соединений по настоящему изобретению могут быть получены традиционными способами. Примеры фармацевтически приемлемых нетоксичных амидов соединения по данному изобретению включают амиды, производные от аммиака, первичных СгСбалкиламинов и вторичных диСгСвалкиламинов, где алкильные группы имеют прямую или разветвленную цепь. В случае вторичных аминов такой амин может также находиться в форме 5- или 6-членного гетероцикла, содержащего один атом азота. Предпочтительными являются амиды, производные от аммиака, первичных Ci-Сзалкиламинов и вторичных диСгСгалкиламинов. Амиды соединения по изобретению могут быть получены традиционными способами. Термин "пролекарство" относится к соединениям, которые быстро трансформируются in vivo, например, в результате гидролиза в крови, с образованием родительского соединения вышеуказанных формул. Подробное обсуждение приведено в Т. Higuchi and V. Stella, "Pro-drugs as Novel Delivery Systems," Vol. 14 of the A.C.S. Symposium Series, и в Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987, оба их которых включены в описание посредством ссылки. Соединение по изобретению может быть введено пациенту в фармацевтически или терапевтически эффективных дозах в интервале от примерно 0,1 до примерно 1000 мг в сутки. Для нормального взрослого человека с массой тела примерно 70 кг достаточной является дозировка в интервале от примерно 0,01 до примерно 100 мг на килограмм массы тела в сутки. Конкретные применяемые дозировки могут, однако, варьироваться. Например, дозировка может зависеть от ряда факторов, включая требования пациента, тяжесть подлежащего лечению состояния и фармакологическую активность используемого соединения. Методология определения оптимальных дозировок для конкретного пациента хорошо известна специалистам в данной области. Одна схема введения людям включает введение соединения по данному изобретению, такого как 4-пиперидин-1-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амид или его фармацевтически приемлемая соль, сложный эфир или амид, в интервале дозирования от примерно 500 мг до примерно 1000 мг в сутки в единой или разделенных дозах. Фармацевтически полезные композиции для применения в этой схеме могут содержать отдельные лекарственные формы, содержащие 100 мг, 200 мг, 250 мг, 500 мг или 1000 мг активного соединения и один или более чем один фармацевтически приемлемый носитель или эксципиент. Соединения по изобретению могут быть приготовлены в виде препарата традиционными способами с получением традиционных лекарственных форм для доставки млекопитающим различными путями, включая пероральный, парентеральный (то есть подкожный, внутривенный и внутримышечный), чрескожный, например кожный пластырь с замедленным высвобождением или крем, а также посредством устройств для замедленного высвобождения, таких как осмотические насосы, суппозитории и буккальные накладки. Следующие неограничивающие примеры дополнительно показывают, как можно легко приготовить в виде препаратов соединения по данному изобретению. Препарат в виде таблетки 100 мг На На 10000 таблетку таблеток (г) (г) 0,10 4-пиперидин-1-илбут-2-еновой кислоты [4-(3-хлор- 1000 4-фторфениламино)-7-метоксихиназолин-6-ил]амид На 10000 таблетку таблеток (г) (г) 0,080 Лактоза 800 0,010 Кукурузный крахмал (для смеси) 100 0,008 Кукурузный крахмал (для клейстера) 0,148 1480 0,002 Стеарат магния (1%) 0,150 1500 Активный агент по данному изобретению, лактозу и кукурузный крахмал (для смеси) смешивают до однородного состояния. Кукурузный крахмал (для пасты) суспендируют в 600 мл воды и нагревают при перемешивании с образованием клейстера. Этот клейстер используют для гранулирования смешанных порошков. Влажные гранулы пропускают через ручное сито № 8 и сушат при 80°С. Сухие гранулы затем пропускают через сито № 16. В смесь вводят смазывающее вещество, 1% стеарата магния, и ее прессуют в таблетки в обычном таблеточном прессе. Эти таблетки пригодны для лечения злокачественных новообразований, таких как рак молочной железы, простаты, легкого, яичников, толстой кишки, поджелудочной железы, меланома, рак пищевода, рак мозга, саркома Капоши и лимфомы. Приготовление пероральной суспензии Ингредиент Кол-во 4-пиперидин-1-илбут-2-еновой кислоты [4-(3-хлор-4- 500 мг фторфениламино)-7-метоксихиназолин-6-ил]амид Раствор сорбита (70% N.F. (Национальный формуляр, США)) 40 мл Бензоат натрия 150 мг Сахарин 10 мг Вишневый корригент 50 мг Дистиллированная вода, сколько требуется до 100 мл Раствор сорбита добавляют к 40 мл дистиллированной воды и суспендируют в нем пиридопиримидин. Добавляют и растворяют сахарин, бензоат натрия и корригент. Объем доводят до 100 мл дистиллированной водой. Каждый миллилитр сиропа содержит 5 мг соединения по изобретению. Приготовление парентерального раствора В растворе 700 мл пропиленгликоля и 200 мл воды для инъекций при перемешивании суспендируют 20,0 г 4-пиперидин-1-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида. После завершения суспендирования, с помощью соляной кислоты доводят рН до 5,5, а объем доводят до 1000 мл водой для инъекций. Препарат стерилизуют, заполняют в ампулы на 5,0 мл, каждая из которых содержит 2,0 мл (что представляет собой 40 мг соединения по изобретению), и запаивают их в атмосфере азота. Суппозитории Смесь 400 мг 4-пиперидин-1-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида и 600 мг теобромового масла перемешивают при 60°С до однородного состояния. Смесь охлаждают и оставляют затвердевать в конической форме с получением суппозиториев массой 1 г. Препарат с замедленным высвобождением Пятьсот миллиграмм 4-пиперидин-1-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида превращают в гидрохлоридную соль и помещают в осмотический насос Oros для контролируемого высвобождения в лечении атеросклероза. Препарат в виде кожного пластыря Сто миллиграмм 4-пиперидин-1-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида смешивают со 100 мг пропиленгликоля монолаурата в полидиметилсилоксановом адгезиве. Смесь наслаивают на гибкую пленку, образованную адгезивным препаратом полибутена, полиизобутилена и пропиленгликоля монолаурата. Слои размещают между 2 слоями полиуретановой пленки. К адгезивной поверхности прикрепляют открепляемую накладку, которую удаляют перед наложением на поверхность кожи. Пропиленгликоля монолаурат служит в качестве агента, усиливающего проницаемость. Соединение по настоящему изобретению может существовать в несольватированной, а также в сольватированных формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и тому подобное. В общем случае для целей настоящего изобретения сольватированные формы считаются эквивалентными несольватированным формам. Предполагается, что рассматриваемое соединение получено либо синтетическим путем, либо биологическим путем, например посредством метаболизма. Фармацевтически или терапевтически эффективное количество соединения следует понимать как количество, достаточное для ингибирования активности белков и механизмов фосфорилирования, описанных в данном описании, у млекопитающего в степени, которая лимитирует, ингибирует или предупреждает прогресс и развитие пролиферативного заболевания или другого рассматриваемого расстройства, опосредованного тирозинкиназами. Фармацевтически или терапевтически эффективное количество в контексте лечения, ингибирования, предупреждения или регулирования развития клеточно-пролиферативного расстройства также можно понимать как количество, достаточное, чтобы вызвать гибель клеток, ингибировать рост клеток, вызывающих данное расстройство, устранить дискомфорт, вызванный расстройством, или продлить жизнь пациента, страдающего этим расстройством. Неограничивающие примеры соединений, входящих в объем данного изобретения, включают следующие соединения: 4-пиперидин-1-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амид; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метилсульфанилхиназолин-6-ил]амид; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метиламинохиназолин-6-ил]амид; 4-пиперидин-1-илбут-2-еновой кислоты [4-(5-хлор-4-фторфениламино)-7-изопропоксихиназолин-6-ил]амид; 4-пиперидин-1-илбут-2-еновой кислоты [4-(3-бромфениламино)-7-метоксихиназолин-6-ил]амид; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-этоксихиназолин-6-ил]амид; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-пропоксихиназолин-6-ил]амид; 4-(4-фторпиперидин-1-ил)бут-2-еновой кислоты [4-(3-хлор-4- фторфен и л ам и но)-7-метоксихи назол и н-6-и л]ам ид; 4-(3-фторпиперидин-1-ил)бут-2-еновой кислоты [4-(3-хлор-4- фторфениламино)-7-метоксихиназолин-6-ил]амид; 4-(2-фторпиперидин-1-ил)бут-2-еновой кислоты [4-(3-хлор-4- фторфениламино)-7-метоксихиназолин-6-ил]амид; 4-морфолин-4-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихи назол и н-6-и л ]ам ид; 4-азепан-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амид; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-трифторметоксихиназолин-6-ил]амид; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-фторметоксихиназолин-6-ил]амид; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-фторэтоксихиназолин-6-ил]амид; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-(2-фторэтилсульфанил)хиназолин-6-ил]амид; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-трифторэтоксихиназолин-6-ил]амид; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-дифторэтоксихиназолин-б-ил]амид; 4-пиперидин-1-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-(3-морфолин-4-илпропокси)хиназолин-6-ил]амид; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-(2-пиперидин-1-илэтокси)хиназолин-6-ил]амид; 4-(3,4-дигидро-2Н-пиридин-1-ил)бут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амид; [4-(3-хлор-4-[4-(3-хлор-4-[4-(3-хлор-4-[4-(3-хлор-4-[4-(3-хлор-4-[4-(3-хлор-4-[4-(3-хлор-4- 4-(3,4-дигидро-2Н-пиридин-1 -ил)бут-2-еновой кислоты фторфениламино)-7-этоксихиназолин-6-ил]амид; 4-(3,4-дигидро-2Н-пиридин-1 -ил)бут-2-еновой кислоты фторфениламино)-7-метилсульфанилхиназолин-6-ил]амид; 4-(3,4-дигидро-2Н-пиридин-1 -ил)бут-2-еновой кислоты фторфениламино)-7-фторэтоксихиназолин-6-ил]амид; 4-(3,6-дигидро-2Н-пиридин-1 -ил)бут-2-еновой кислоты фторфениламино)-7-метоксихиназолин-6-ил]амид; 4-(3,6-дигидро-2Н-пиридин-1-ил)бут-2-еновой кислоты фторфениламино)-7-метоксихиназолин-6-ил]амид; 4-(3,б-дигидро-2Н-пиридин-1 -ил)бут-2-еновой кислоты фторфениламино)-7-фторэтоксихиназолин-6-ил]амид; 4-(3,6-дигидро-2Н-пиридин-1 -ил)бут-2-еновой кислоты фторфениламино)-7-метилсульфанилхиназолин-6-ил]амид; 4-пиперазин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амид; 4-(4-метилпиперазин-1-ил)бут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амид; 4-имидазол-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амид; 4- пирролидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амид; 5- пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амид; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метилсульфанилхиназолин-6-ил]амид; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метиламинохиназолин-6-ил]амид; 5-пиперидин-1-илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-изопропоксихиназолин-6-ил]амид; 5-пиперидин-1-илпент-2-еновой кислоты [4-(3-бромфениламино)-7-метоксихиназолин-6-ил]амид; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-этоксихиназолин-6-ил]амид; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-пропоксихиназолин-6-ил]амид; 5-(4-фторпиперидин-1-ил)пент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амид; 5-(3-фторпиперидин-1-ил)пент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амид; 5-(2-фторпиперидин-1-ил)пент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амид; 5-морфолин-4-илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амид; 5-азепан-1 -ил пент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-б-ил]амид; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-трифторметоксихиназолин-6-ил]амид; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-фторметоксихиназолин-б-ил]амид; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-фторэтоксихиназолин-6-ил]амид; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-(2-фторэтилсульфанил)хиназолин-6-ил]амид; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-трифторэтоксихиназолин-6-ил]амид; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-дифторэтоксихиназолин-6-ил]амид; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-(3-морфолин-4-илпропокси)хиназолин-6-ил]амид; 5- пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-(2-пиперидин-1-илэтокси)хиназолин-6-ил]амид; 6- пиперидин-1 -илгекс-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амид; 6-пиперидин-1 -илгекс-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метилсульфанилхиназолин-6-ил]амид; 6-пиперидин-1 -илгекс-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метиламинохиназолин-б-ил]амид; 6-пиперидин-1 -илгекс-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-этоксихиназолин-6-ил]амид и 6-пиперидин-1-илгекс-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-фторэтоксихи назол ин-б-и л ]а мид или их фармацевтически приемлемую солевую форму. Соединения по данному изобретению могут быть получены с использованием способов и веществ, известных из уровня техники. Соединения по данному изобретению, где X представляет собой кислород, могут быть получены, как показано на следующей ниже Схеме 1, где анилиновая группа по положению 4 представляет собой 4-фтор-З-хлоранилиновую группу. Схема 1 4-хлор-7-фтор-6-нитрохиназолин (7) может быть получен способами, аналогичными описанным в J.Med. Chem. 1996, 39, 918-928. В общем случае, 2-амино-4-фторбензойная кислота (1) может быть подвергнута взаимодействию с формамидином (2) и уксусной кислотой (3) в присутствии 2-метоксиэтанола с получением 7-фтор-ЗН-хиназолин-4-она (4). 7-фтор-ЗН-хиназолин-4-он (4) затем может быть подвергнут нитрованию до 7-фтор-6-нитро-ЗН-хиназолин-4-она (5), который может быть обработан тионилхлоридом с получением 4-хлор-6-нитро-7-фтор-ЗН-хиназолина (6). 4-Хлор-хиназолиновое соединение (6) можно объединить с замещенным желательным образом анилином, представленным выше 4-фтор-З-хлоранилином, в присутствии третичного амина и изопропанола с получением 4-анилино-6-нитро-7-фторхиназолина (7). 4-Анилино-6-нитро-7-фторхиназолин (7) можно подвергнуть взаимодействию со спиртом формулы R3OH, где R3 является таким, как определено выше, с получением 7-алкоксилированного соединения (8). Восстановление 6-нитросоединения (8) дает 6-амино-аналог (9). Соединение (9) с аминогруппой по положению 6 можно подвергнуть взаимодействию с галогеноалкеноилхлоридои (12), таким как 4-бромбут-2-еноилхлорид, 5-бромпент-2-еноилхлорид, 4-хлорбут-2-еноилхлорид или 5-хлорпент-2-еноилхлорид, с получением [4-анилино]-7-алкоксилированный-хиназолин-6-ил-амида алкеновой кислоты (13). Галогеноалкеноилхлоридные агенты, пригодные для этой схемы, могут быть получены способами, известными из уровня техники, такими как обработка походящей галогеноалкеновой кислоты, представленной эфиром бромалкеновой кислоты (10), первичным спиртом, с получением соответствующей галогеноалкеновой кислоты (11), которая в свою очередь может быть обработана оксалилхлоридом с получением желаемого галогеноалкеноилхлорида (12). И, наконец, хиназолин-6-алкановокислотное соединение (13) может быть подвергнуто взаимодействию с циклическим амином, таким как пиперидин, пиперазин и так далее, с получением желаемого конечного соединения (14). Следует понимать, что соединения, имеющие алкоксигруппы по положению 7, могут быть получены, как выше, с использованием спирта формулы R3OH, где R3 представляет собой алкильную группу, как она определена выше, известного из уровня техники, включая, но не ограничиваясь ими, метанол, этанол, пропанол, изопропанол, фторметанол, хлорметанол, дифторметанол, дихлорметанол, трифторметанол, трихлорметанол, 1-фторэтанол, 2-фторэтанол, 2-хлорэтанол, 2-иодэтанол, 2-бромэтанол, 1,1-дифторэтанол, 2,2-дифторэтанол, 2,2-дихлорэтанол, 1,2,2-трифторэтанол, 2,2,2-трифторэтанол, 1,1,2,2-тетрафторэтанол, пентафторэтанол, З-фтор-1-пропанол, 2,3-дифтор-1 -пропанол, 3,3-дифтор-1-пропанол, 2,3,3-трифтор-1-пропанол, 3,3,3-трифтор-1-пропанол, 1,1,3-трифтор-1-пропанол, 1,2,2,3-тетрафтор-1-пропанол, 2,3,3,3-тетрафторпропанол, 2,2,3,3,3-пентафтор-1-пропанол, 1,2,3,3.З-пентафтор-1-пропанол, 1,1,2,3,3,3-гексафтор-1-пропанол, гептафтор-1-пропанол, 2-фтор-2-пропанол, 1,1-дифтор-2-пропанол, 1,3-дифтор-2-пропанол, 1-фтор-2-пропанол, 1,1,1-трифтор-2-пропанол, 1,1,3,3-тетрафтор-2-пропанол, 1,1,3,3,3-пентафтор-2-пропанол, 1,1,2,3,3-гексафтор-2-пропанол, 1,1,1,3,3,3-гексафтор-2-пропанол, 1,1,1,2,3,3,3-гептафтор-2-пропанол и так далее. В объем данного изобретения входят также полезные промежуточные соединения формулы: где: Y представляет собой N02, NH2 или группировку галогено-(СН2)т-СН=СН-C(0)-NH2-; галогено представляет собой F, CI, Вг или I; m представляет собой целое число от 1 до 3; 1Чз выбран из: а) моно-, ди- или тригалогенированной метильной группы; б) прямого или разветвленного С2-Сзалкила, возможно замещенного галогено; или в) групп -(СН2)п-морфолино, -(СН2)п-пиперидин, -(СН2)п-пиперазин, -(СН2)п-пиперазин-г> 1(С1-Сзалкил), -(СН2)п-пирролидин или -(СН2)п-имидазол. Эти соединения, в частности, включают соединения указанной выше формулы, где R3 представляет собой группу этан, пропан, изопропан, фторметан, хлорметан, дифторметан, дихлорметан, трифторметан, трихлорметан, 1-фторэтан, 2-фторэтан, 2-хлорэтан, 2-иодэтан, 2-бромэтан, 1,1-дифторэтан, 2,2-дифторэтан, 2,2-дихлорэтан, 1,2,2-трифторэтан, 2,2,2-трифторэтан, 1,1,2,2-тетрафторэтан, пентафторэтан, З-фтор-1-пропан, 2,3-дифтор-1 -пропан, 3,3-дифтор-1-пропан, 2,3,3-трифтор-1-пропан, 3,3,3-трифтор- 1- пропан, 1,1,3-трифтор-1-пропан, 1,2,2,3-тетрафтор-1-пропан, 2,3,3,3-тетрафторпропан, 2,2,3,3,3-пентафтор-1-пропан, 1,2,3,3,3-пентафтор-1-пропан, 1,1,2,3,3,3-гексафтор-1-пропан, гептафтор-1-пропан, 2-фтор-2-пропан, 1,1-дифтор-2-пропан, 1,3-дифтор-2-пропан, 1-фтор-2-пропан, 1,1,1-трифтор-2-пропан, 1,1,3,3-тетрафтор-2-пропан, 1,1,3,3,3-пентафтор-2-пропан, 1,1,2,3,3-гексафтор-2-пропан, 1,1,1,3,3,3-гексафтор-2-пропан или 1,1,1,2,3,3,3-гептафтор- 2- пропан. Соединения по данному изобретению, где X представляет собой серу, могут быть получены, как показано на следующей ниже Схеме 1, путем замены спирта R3OH, взаимодействующего с (3-хлор-4-фторфенил)-(7-галогено-6-нитро-3,4-дигидрохинолин-4-ил)амином (Соединение 7), на соответствующий алкилтиол формулы R3SH, где R3 является таким, как определено здесь выше. Полезные алкилтиоловые соединения формулы R3SH включают, без ограничения ими, метантиол, этантиол, 1-пропантиол, 2-пропантиол, фторметантиол, 2-фторэтантиол, 2,2-дифторэтантиол, 2,2,2-трифторэтантиол и так далее. Аналогично, соединения по данному изобретению, где X представляет собой -NH-, могут быть получены, как показано на следующей ниже Схеме 1, путем замены спирта R3OH, взаимодействующего с (3-хлор-4-фторфенил)-(7-галогено-6-нитро-3,4-дигидрохинолин-4-ил)амином (Соединение 7), на соответствующий алкиламин формулы R3NH, где R3 является таким, как определено здесь выше. Пригодные алкиламины включают, без ограничения ими, метиламин, этиламин, пропиламин, изопропиламин, 1-фторметиламин, 1,1-дифторметиламин, 1,1,1-трифторметиламин, 2-фторэтиламин, 2,2-дифторэтиламин, 2,2,2-трифторэтиламин, 3-фторпропиламин, 3,3-дифторпропиламин, 3,3,3-трифторпропиламин, 2,3,3-тетрафторпропиламин, 2,2,3,3,3-пентафторпропиламин, 1,1,2,2,3,3,3-гептафторпропиламин и так далее. Пиперидиновые соединения, пригодные для получения групп R4 в соединениях по данному изобретению, включают, без ограничения ими, пиперидин, 2-фторпиперидин, 3-фторпиперидин, 4-фторпиперидин, 4-бромпиперидин, 4-хлорпиперидин, 2-гидроксипиперидин, 3-гидроксипиперидин, 2-метилпиперидин, 3-метилпиперидин, 4-метилпиперидин, 4-этилпиперидин, 4-пропилпиперидин, 2-аминопиперидин, 3-аминопиперидин, 4-аминопиперидин, 2-метилпиперидин, 2,3-диметилпиперидин, 3,3-диметилпиперидин, 2,4-диметилпиперидин, 2,5-диметилпиперидин, 2,6-диметилпеперидин, 3,5-диметилпиперидин, 2-метил-5-этилпиперидин, З-метил-4-гидроксипиперидин, 2,6-диметил-4-гидроксипиперидин, 2,5-диметил-4-гидроксипиперидин, 2,3-диметил-4-гидроксипиперидин, 3,3-дифторпиперидин, 4,4-дифторпиперидин, 4,4-дигидроксипиперидин, 2,4,6-триметилпиперидин и так далее. Пример 1 4-Пиперидин-1-илбут-2-еновой кислоты Г4-(3-хлор-4-фторфениламино)-7-(2-фторэтокси)хиназолин-6-ил1амид 7-фтор-6-нитро-4-хлорхиназолин (14,73 г, 65 ммоль) объединяли с 3-хлор-4-фторанилином (9,49 г, 65 ммоль) и триэтиламином (10 мл, 72 ммоль) в 150 мл изопропанола. Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов с получением суспензии желтого цвета. Твердую фазу собирали фильтрацией, промывали изопропанолом и затем водой. Твердое вещество сушили в вакуумной печи при 40°С в течение ночи с получением 19,83 г (91%) продукта в виде твердого оранжевого вещества. MS (APCI, m/z, М+1) (масс-спектрометрия, химическая ионизация при атмосферном давлении): 337,0 К раствору 2-фторэтанола (5,19 г, 80 ммоль) в 200 мл THF (тетрагидрофуран) порциями добавляли NaH (60% в минеральном масле, 3,55 г, 88 ммоль). Реакционную смесь перемешивали в течение 60 минут при комнатной температуре. К реакционной смеси добавляли 7-фтор-6-нитро-4-(3-хлор-4-фторанилин)хиназолин (18,11 г, 54 ммоль) в виде твердого вещества, промывая THF. Реакционную смесь нагревали до 65°С в течение 26 часов. Реакционную смесь охлаждали до комнатной температуры и гасили водой. В вакууме удаляли THF. Полученный остаток быстро обрабатывали ультразвуком в воде, затем твердую фазу собирали фильтрацией. Твердое вещество растирали с МеОН, отфильтровывали и сушили при 40°С в вакуумной печи в течение ночи с получением 12,63 г продукта. Дополнительное количество продукта получали путем концентрирования досуха МеОН фильтрата и хроматографии с элюированием смесью 50% EtOAc/гексан. Выделенное вещество растирали с МеОН (2Х), отфильтровывали и сушили, получая 3,90 г Общий выход: 16,53 г, 81% MS (APCI, m/z, М+1): 381,0 7-(2-Фторэтокси)-6-нитро-4-(3-хлор-4-фторанилин)хиназолин (0,845 г, 2,2 ммоль) в 50 мл THF гидрировали с никелем Ренея (0,5 г) в качестве катализатора в течение 15 часов. Катализатор отфильтровывали, и фильтрат выпаривали с получением 0,77 г продукта (99%). MS (APCI, m/z, М+1): 351,2 Метил-4-бромкротонат (85%, 20 мл, 144 ммоль) гидролизовали с помощью Ва(ОН)2 в смеси ЕЮН/Н20, как описано в J.Med.Chem. 2001, 44(17), 2729-2734. MS (APCI, m/z, M-1): 163,0 К раствору 4-бромкротоновой кислоты (4,17 г, 25 ммоль) в CH2CI2 (20 мл) добавляли оксалилхлорид (33 мл, 38 ммоль) и несколько капель DMF (диметилформамида). Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. Растворитель и избыток реагента удаляли в вакууме. Полученный остаток растворяли в 10 мл THF и добавляли к охлажденной до 0°С смеси 6-амино-7-(2-фторэтокси)-4-(3-хлор-4-фторанилин)хиназолина (5,28 г, 15 ммоль) и триэтиламина (5,2 мл, 37 ммоль). Реакционную смесь перемешивали при 0°С в течение 1 часа. К реакционной смеси добавляли воду, и в вакууме удаляли THF. Продукт экстрагировали в CH2CI2 (400 мл). Органический слой сушили над MgS04, фильтровали и концентрировали. Неочищенное вещество хроматографировали на силикагеле, элюируя смесью 0-4% MeOH/CH2CI2. Отделяли выделенную золотистую пену. Выход: 4,58 г, 61% MS (APCI, m/z, М-1): 497,1 К раствору вышеуказанного соединения (3,35 г, 6,7 ммоль) и TEA (триэтаноламин) (2,80 мл, 20 ммоль) в 10 мл DMA (N.N-диметилацетамид) при 0°С добавляли пиперидин (0,75 мл, 6,7 ммоль). Реакционную смесь перемешивали при 0°С в течение 17 часов. К реакционной смеси добавляли воду до тех пор, пока явно не образовывался осадок. Реакционную смесь обрабатывали ультразвуком в течение 40 минут, и жидкость декантировали. Остаток растворяли в CH2CI2, сушили над MgS04, фильтровали и концентрировали. Вещество хроматографировали на силикагеле, элюируя смесью 4-10% MeOH/CH2CI2. Выделенный остаток растирали с ацетонитрилом (2Х) и собирали посредством фильтрации. Обнаруженные примеси: продукт присоединения пиперидина по Михаэлю (Michael) (2,2% при первом растирании ацетонитрила). Из ацетонитрильных фильтратов можно получить дополнительное вещество. Выход: 0,95 г, 27% MS (APCI, m/z, М+1): 502,3 Пример 2 4-Пиперидин-1 -илбут-2-еновой кислоты И-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амид (путь синтеза № 1) Указанное в заголовке соединение и другие 7-метокси-аналоги по данному изобретению могут быть получены, как описано в Примере 1, путем замены 2-фторзтанола, используемого в Примере 1, стехиометрическим количеством метанола. Пример 3 М-Г4-(3-Хлор-4-фторфениламино)-7-метоксихиназолин-6-ил1-3-пиперидин-1-илакриламид (путь синтеза № 2) Альтернативный путь синтеза для соединений по данному изобретению включает получение цепи заместителя по положению 6 в виде Het-алкеноилхлорида, как показано на Схеме 2 ниже. Схема 2 12 3 4 SOCI2 Следует понимать, что другие соединения в рамках данного изобретения могут быть получены с использованием Het-бутеноилгалогенидной, Het-пентеноилгалогенидной и Het-гексеноилгалогенидной групп формулы: где R* является таким, как описано в данном описании, a halo представляет собой F, CI, Вг или I, предпочтительно CI или Вг. Одна конкретная группа этих Het-алкеноилгалогенидов включает соединения, в которых halo представляет собой CI или Br, R4 представляет собой -(CH2)m-Het, m является целым числом от 1 до 3, и Het представляет собой пиперидиновую или замещенную пиперидиновую группировки, описанные выше. Пример 4 4-Пиперидин-1 -илбут-2-еновой кислоты Г4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил1амид (путь синтеза № 3) Схема 3 ^^^СООН н м 2-метокси Н (Г 1 " +2 \ м этанол FY^N'" HN03/H2S04 , niM о 77,86% г N 81,36% 1 2 3 О CI ли г N 5 " ОН "°ч V с О N №Ренея,Н2 L,> ^/^CI СНзОМа 02f^AN _J N _и Ji Л J THF, 97,6% N^^^N - 92,1% . НзСсЛ^гГ XT J 8 НзССГ^^ГГ ]Yf THF TV^N HgCO^^V 57% H3CcA^V 10 18 О 0NH 0 86% w 3 70% 11 12 13 14 CI OCH3 I /0- / -/ 3 AcOH,NaCNBH3 Q.^-L H2N-^JKF + OHC-(JKOCH3 ^ XXl!wCI 15 16 ^ З-Хлор-4-фторфениламин 15 (50,31, 345,6 ммоль) и 3,4-диметокси-бензальдегид 16 (57,43 г, 345,6 ммоль) смешивали в 500 мл IPA (изопропиловый спирт) и охлаждали в ледяной воде. Добавляли ледяную уксусную кислоту (20,76 г, 345,6 моль) и затем одной порцией цианоборгидрид натрия. Реакционную смесь перемешивали при комнатной температуре (КТ) в течение 24 часов. После завершения реакции при КТ по каплям добавляли 250 мл 10%-ного NaOH. Смесь перемешивали в течение Уг часа. Суспензию затем отфильтровывали и промывали IPA, а затем сушили в вакууме. Масса 88,75 г (17, 87%). Соединения 6 (3 г, 13,18 ммоль) и 17 (3,9 г, 13,18 ммоль) объединяли в CH3CN (25 мл) и нагревали в течение 1 часа. Масс-спектроскопия не выявила присутствия исходного вещества. Добавляли насыщенный К2СОз, и реакционную смесь экстрагировали ЗХ ЕЮАс. Органические слои объединяли, промывали рассолом и концентрировали в вакууме с получением 6,48 г соединения 7 (78,4%). Соединение 7 (72,76 г, 149,4 ммоль) добавляли к холодному раствору NaOMe в 1,5 л сухого МеОН под N2. Охлаждающую баню удаляли, и смесь нагревали до кипения с обратным холодильником и перемешивали в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры и гасили водой до тех пор, пока продукт не выпадал в осадок. Твердую фазу отфильтровывали и промывали водой и гексанами. Продукт суспендировали в кипящем с обратным холодильником ЕЮАс и отфильтровывали горячим с получением 68,75 г твердого желтого вещества 8 (73%). Соединение 8 (63,62 г, 127,5 моль) гидрировали, используя никель Ренея в качестве катализатора, с получением 43,82 г соединения 9 (100%). К суспензии соединения 13 (10,5 г, 51,2 ммоль) в 200 мл дихлорметана, содержащей 8 капель DMF, медленно добавляли оксалилхлорид (6,5 г, 51,18 ммоль). После того, как реакционная смесь становилась гомогенной, растворитель удаляли, а оставшееся твердое вещество светло-желтого цвета суспендировали в 200 мл DMAC (N.N-диметилацетамид), и постепенно добавляли соединение 9 (20 г, 42,65 ммоль) в виде твердого вещества. Реакционную смесь перемешивали в течение 15 минут и медленно выливали в 1 н. NaOH. Смесь экстрагировали ЗХ ЕЮАс. Объединенные органические слои промывали рассолом, фильтровали и концентрировали в вакууме с получением 28,4 г (100%) соединения 10. Соединение 10 (13,07 г, 21,08 ммоль) растворяли в трифторуксусной кислоте (TFA) (74 г, 649 ммоль) и нагревали до 30°С в течение 24 часов. Реакционную смесь охлаждали до КТ и постепенно выливали в охлажденный 1 н. раствор NaOH в рассоле. Образовывался осадок, его отфильтровывали и ЗХ промывали водой, а затем сушили. В результате перекристаллизации из толуола получали чистый ^[4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]-3-пиперидин-1-илакриламид (9,90 г, 89%). Из вышеприведенных примеров очевидно, что данное изобретение включает полезные промежуточные соединения формулы: где Y представляет собой NH2, N02 или группировку R4-(CH2)m-CH=CH-C(0)-NH2-; a m является целым числом от 1 до 3; R3 выбран из: а) прямого или разветвленного СгСзалкила, возможно замещенного галогеном; или б) групп -(СН2)п-морфолино, -(СН2)п-пиперидин, -(СН2)п-пиперазин, -(СН2)п-пиперазин-^СгСзалкил), -(СН2)п-пирролидин или -(СН2)п-имидазол; п является целым числом от 1 до 4; R4 представляет собой -(CH2)m-Het; Het представляет собой гетероциклическую группировку, выбранную из группы, состоящей из морфолина, пиперидина, пиперазина, группы пиперазин-^СгСзалкил), имидазола, пирролидина, азепана, 3,4-дигидро-2Н-пиридина или 3,6-дигидро-2Н-пиридина, где каждая гетероциклическая группировка возможно замещена группами в количестве от 1 до 3, выбранными из d-Сзалкила, галогена, ОН, NH2, групп NH(C1-Cзaлкил) или ^С1-С3алкил)2; m является целым числом от 1 до 3; и X представляет собой О, S или NH. Пример 5 4-Пиперидин-1 -илбут-2-еновой кислоты Г4-(3-хлор-4-Фторфениламино)-7-метоксихиназолин-6-ил1амид (путь синтеза № 4) Схема 4 (3-Хлор-4-фторфенил-(7-метокси-6-нитрохиназолин-4-ил)амин (1) (17,26 г, 0,0495 ммоль) суспендировали в 350 мл уксусного ангидрида в атмосфере азота, нагревали до 90°С и выдерживали при этой температуре в течение 24 часов, затем постепенно охлаждали до КТ. Образовывалась бледноокрашенная суспензия. Охлаждали до 0°С в течение 1 часа. Твердые вещества отфильтровывали, и колбу и осадок на фильтре промывали 2x50 мл IPA. Продукт, М-(3-хлор-4-фторфенил)-М-(7-метокси-6-нитрохиназолин-4-ил)ацетамид (2), сушили в вакуумной печи при 60°С в течение 24 часов. Масса: 17,97 г (92,4%). HPLC (высокоэффективная жидкостная хроматография): 99,45%, rt (время удерживания)=13,705 мин. Ni Ренея (5,0 г) суспендировали в МеОН, затем в THF для удаления воды. М-(3-хлор-4-фторфенил)-М-(7-метокси-б-нитрохиназолин-4-ил)ацетамид (2) (19,2 г, 49 ммоль) суспендировали в THF (500 мл) и загружали в реактор. Реакционную смесь нагревали до 60°С и создавали давление водорода 60 фунтов на кв. дюйм (413,7 кПа). Через почти 17 часов загружали дополнительно 10,0 г катализатора, и реакция завершалась в течение 38 часов. Реакционную смесь фильтровали и промывали THF. Твердые вещества концентрировали в вакуумном испарителе, и растворитель заменяли на гексаны. При добавлении гексанов в осадок выпадало бледно-желтое твердое вещество. Растворитель удаляли под вакуумом для отгонки оставшегося THF. Фильтровали и промывали большими количествами гексанов. Продукт, Ы-(6-амино-7-метоксихиназолин-4-ил)-г> 1-(3-хлор-4-фторфенил)ацетамид (3), сушили в вакуумной печи при 70°С в течение 24 часов. Масса составляет 16,75 г (94,49%). HPLC: tm(100%). К раствору DMF (60 мг), 4-пиперидин-1-илбут-2-еновой кислоты в 40 мл DCM (дихлорметан) при комнатной температуре добавляли оксалилхлорид, и реакционную смесь перемешивали в течение одного часа. Растворитель упаривали под вакуумом, и полученное твердое вещество суспендировали в 150 мл DMAC. К реакционной смеси добавляли N-(6-aMHHO-7-метоксихиназолин-4-ил)-М-(3-хлор-4-фторфенил)ацетамид (3) в виде твердого вещества. Реакция завершалась через 45 минут. Затем смесь по каплям добавляли к 300 мл 2 н. NaOH, и водный слой экстрагировали ЕЮАс. Объединенный органический слой концентрировали до 100 мл и перемешивали в течение 2 суток при комнатной температуре. Добавляли 300 мл этилового эфира и 100 мл 2 н. NaOH, и выпавшее в осадок твердое вещество собирали фильтрацией. Конечный продукт перекристаллизовывали из этиленхлорида с получением 5,5 г чистого продукта. Как отмечалось выше, перед использованием катализатор, никеля Ренея, может быть обработан спиртом, таким как метанол или этанол, а затем промыт THF. Дополнительные катализаторы для использования в данной реакции включают платину на углероде или палладий на углероде, предпочтительно в присутствии 1-4 эквивалентов уксусной кислоты. Следует понимать, что удаление ацетильной группы у соединения (5) с получением соединения (6), выше, может быть осуществлено способами, известными из уровня техники, включая как щелочные, так и кислые условия. Удаление в кислых условиях может быть осуществлено с использованием, среди прочих, кислот, известных из уровня техники, уксусной кислоты или метансульфоновой кислоты. В объем данного изобретения входят полезные промежуточные соединения формулы: где Y представляет собой NH2, NO2 или группировку R4-(CH2)m-CH=CH-C(0)-NH2-; и m является целым числом от 1 до 3; R3 выбран из: а) прямого или разветвленного d-Сзалкила, возможно замещенного галогеном; или б) групп -(СН2)п-морфолино, -(СН2)п-пиперидин, -(СН2)п-пиперазин, -(СН2)п-пиперазин-г> 1(С1-Сзалкил), -(СН2)п-пирролидин или -(СН2)п-имидазол; п является целым числом от 1 до 4; R4 представляет собой (CH2)m-Het; Het представляет собой гетероциклическую группировку, выбранную из группы, состоящей из морфолина, пиперидина, пиперазина, группы пиперазин-ЫССгСзалкил), имидазола, пирролидина, азепана, 3,4-дигидро-2Н-пиридина или 3,6-дигидро-2Н-пиридина, где каждая гетероциклическая группировка возможно замещена 1-3 группами, выбранными из С1-С3алкила, галогена, ОН, NH2, групп МН(С1-С3алкил) или 1Ч(С-|-Сзалкил)2; m является целым числом от 1 до 3;и X представляет собой О, S или NH. Примеры таких соединений включают М-(3-хлор-4-фторфенил)-М-(7-метокси-б-нитрохиназолин-4-ил)ацетамид, М-(6-амино-7-метоксихиназолин-4-ил)-М-(3-хлор-4-фторфенил)ацетамид и М-{4-[ацетил-(3-хлор-4- фторфенил)амино]-7-метоксихиназолин-6-ил}-3-пиперидин-1-илакриламид. Пример 6 4-Пиперидин-1 -илбут-2-еновой кислоты Г4-(3-хлор-4-ФторФениламино)-7-(2,2-дифторэтокси)хиназолин-6-ил]амид 0,6 г 60%-ного NaH добавляли порциями к раствору 1,23 г 2,2-дифторэтанола в 20 мл THF и перемешивали при комнатной температуре в течение 15 минут. Добавляли 2,02 г (3-хлор-4-фторфенил)-(7-фтор-6-нитрохиназолин-4-ил)амина в виде твердого вещества, и смесь нагревали до 65°С в течение 1 часа, затем охлаждали до комнатной температуры. Добавляли воду, и под вакуумом удаляли THF. Смесь обрабатывали ультразвуком, и полученный твердые вещества собирали фильтрацией и сушили под вакуумом в течение ночи с получением 2,93 г неочищенного (3-хлор-4-фторфенил)-[7-(2,2-дифторэтокси)-6-нитрохиназолин-4-ил]амина. Неочищенный (3-хлор-4-фторфенил)-[7-(2,2-дифторэтокси)-6-нитрохиназолин-4-ил]амин растворяли в THF и восстанавливали с помощью никеля Ренея в качестве катализатора с получением М4-(3-хлор-4-фторфенил)-7-(2,2-дифторэтокси)хиназолин-4,6-диамина. 0,45 г бромбут-2-еновой кислоты растворяли в 10 мл CH2CI2 вместе с 2 каплями DMF. При комнатной температуре добавляли 0,47 мл оксалилхлорида и перемешивали в течение ночи. Смесь упаривали досуха с получением 4-бромбут-2-еноилхлорида. 0,5 г М4-(3-хлор-4-фторфенил)-7-(2,2-дифторэтокси)хиназолин-4,6-диамина растворяли в 10 мл THF и 1,2 мл 1М,1М-диизопропилэтиламина (DIEA) и добавляли 0,48 г 4-бромбут-2-еноилхлорида, смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли 0,27 мл пиперидина и перемешивали при комнатной температуре в течение ночи. Добавляли дополнительно 0,7 мл пиперидина, и смесь нагревали до 70°С. Через 3 часа реакционную смесь выливали в воду, твердую фазу экстрагировали этилацетатом, промывали водой и рассолом, сушили над NaSC^ и подвергали флэш-хроматографии с 0-4%-ным раствором метанола в хлороформе с получением 0,2 г 4-пиперидин-1-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-(2,2-дифторэтокси)хиназолин-6-ил]амида. MS (масс-спектрометрия) (М+Н)+ 520. Пример 7 4-Пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-(3-морфолин-4-илпропокси)хиназолин-6-ил1амид Стадия 1: 4-пиперидин-1-илбут-2-еновой кислоты метиловый эфир Метил-4-бромкротонат (2 г, 11,2 ммоль) растворяли в дихлорметане (20 мл) и охлаждали до 0°С. Медленно добавляли пиперидин (1,11 мл, 11,2 ммоль). Смесь перемешивали при 0°С в течение 1 часа. Растворитель удаляли в вакууме. Неочищенное вещество использовали как таковое. MS m/z 184 (М+1). Стадия 2: 4-пиперидин-1-илбут-2-еновая кислота -HCI 4-пиперидин-1-илбут-2-еновой кислоты метиловый эфир (2,05 г, 11,2 ммоль) и концентрированную соляную кислоту (10 мл) объединяли в диоксанах (30 мл) и нагревали до кипения с обратным холодильником в течение ночи. Смесь концентрировали в вакууме. Остаток кристаллизовали из IPA с получением нужного продукта (390 мг, 17%). 400 МГц 1Н-ЯМР (DMSO-d6) 5 6.80 (dt, 1Н, J = 15,6, 7,1 Гц), 6.14 (d, 1Н, J = 15,6 Гц), 3.85 (d, 1Н, J = 7,1 Гц), 2.89 (m, 4Н), 1.54 (m, 6Н). MS m/z 170 (М+1). Стадия 3: 4-пиперидин-1-илбут-2-еноилхлорид HCI-соль 4-пиперидин-1-илбут-2-еновой кислоты (250 мг, 1,48 ммоль) растворяли в дихлорметане (15 мл). Добавляли диметилформамид (3 капли). Медленно добавляли оксалилхлорид (155 мкл, 1,77 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Смесь концентрировали при пониженном давлении, и остаток использовали как таковой. Стадия 4: 4-пиперидин-1-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-(3-морфолин-4-илпропокси)хиназолин-6-ил1амид М*4*-(3-хлор-4-фторфенил)-7-(3-морфолин-4-илпропокси)хиназолин-4,6-диамин (510 мг, 1,18 ммоль) и DIPEA (620 мкл, 3,55 ммоль) объединяли в тетрагидрофуране (10 мл) и охлаждали до 0°С. Добавляли 4-пиперидин-1-илбут-2-еноилхлорид (278 мг, 1,48 ммоль), и реакционную смесь перемешивали при 0°С в течение 2 часов. Смесь гасили этилацетатом, сушили с помощью MgSC> 4 и концентрировали. Остаток очищали, используя хроматографию на диоксиде кремния, элюируя 15%-20%-ным МеОН в CH2CI2, с получением нужного продукта (20 мг). 400 МГц 1H-flMP(DMSO-d6) 8 8.82 (s, 1Н), 8.50 (s, 1Н), 8.11 (dd, 1Н, J = 6,9, 2,6 Гц), 7.77 (m, 1Н), 7.40 (t, 1Н, J = 9,0 Гц), 7.25 (s, 1Н), 6.76 (m, 1Н), 6.53 (m, 1Н), 4.23 (t, 2Н, J = 6,0 Гц), 3.55 (m, 4Н), 3.08 (m, 2Н), 2.40 (m, ЮН), 1.96 (m, 2Н), 1.29 (m, 6Н). MS m/z 584 (М+1). Пример 8 4-(3-Фторпиперидин-1-ил)бут-2-еновой_кислоты_Г4-(3-хлор-4- Фторфениламино)-7-метоксихиназолин-6-ил1амид 99 мг 3-фторпиперидина гидрохлорида, 300 мг 4-хлорбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида и 0,37 мл DIEA растворяли в 5 мл THF и перемешивали при 70°С в течение ночи. Затем смесь разбавляли этилацетатом, промывали водой и рассолом и сушили над Na2S04. Полученные твердые вещества подвергали флэш-хроматографии с 0-4%-ным раствором метанола в хлороформе с получением 275 мг 4-(3-фторпиперидин-1 -ил)бут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихи назол и н-6-и л]амида. (М+Н)+ 488. Пример 9 4-(4-Фторпиперидин-1-ил)бут-2-еновой_кислоты_Г4-(3-хлор-4- фторфениламино)-7-метоксихиназолин-6-ил1амид 131 мг 4-фторпиперидина гидробромида добавляли к 300 мг 4-хлорбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида и 0,37 мл DIEA растворяли в 5 мл THF, перемешивали при 70°С в течение ночи. Затем смесь разбавляли этилацетатом, промывали водой и рассолом и сушили над Na2S04. Полученное твердое вещество подвергали флэш-хроматографии с 0-4%-ным раствором метанола в хлороформе с получением 189,4 мг 4-(4-фторпиперидин-1 -ил)бут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида. (М+Н)+ 488. Пример 10 4-азепан-1-илбут-2-еновой кислоты Г4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил1амид (3-Хлор-4-фторфенил)-(7-фтор-6-нитрохиназолин-4-ил)амин суспендировали в 100 мл метанола, добавляли 2 мл 50%-ного NaOH в воде, и смесь нагревали при 70°С в течение 2 часов. Смесь затем выливали в воду и интенсивно перемешивали в течение 30 минут, затем фильтровали и промывали водой, сушили под вакуумом при 60°С в течение ночи с получением 7,2 г (3-хлор-4-фторфенил)-(7-метокси-6-нитрохиназолин-4-ил)амина. 7,1 г (3-хлор-4-фторфенил)-(7-метокси-6-нитрохиназолин-4-ил)амина восстанавливали в THF с помощью никеля Ренея в качестве катализатора, затем фильтровали и выпаривали с получением 6,4 г М4-(3-хлор-4-фторфенил)-7-метоксихиназолин-4,6-диамина (выход 99%). Этот продукт подвергали взаимодействию с 4-хлорбут-2-еноилхлоридом, как описано на Схеме 1, с получением 4-хлорбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихи назол и н-6-и л ]а м ида. 300 г 4-хлорбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида и 78 мг азепана растворяли в 5 мл THF и продували азотом. Добавляли 0,25 мл DIEA, и смесь перемешивали при 70°С в течение 2 суток. Смесь затем разбавляли 20 мл этилацетата, промывали водой и рассолом и сушили над Na2S04. Полученное твердое вещество подвергали флэш-хроматографии с 0-4%-ным раствором метанола в хлороформе. Продукт растворяли в CH2CI2 и обрабатывали избытком HCI и эфира, затем выпаривали до сухости с получением 115 мг 4-азепан-1-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида (выход 33%). (М+Н)+ 484. Пример 11 - Анализ erbB-киназы на основе ELISA (твердофазного иммуноферментного анализа) ErbB1, егЬВ2 и егЬВ4 цитоплазменные слитые белки получали путем клонирования последовательности егЬВ1 (от Met-668 до А1а1211), егЬВ2 (от Пе-675 до Val-1256) и последовательности егЬВ4 (от Gly-259 до Gly-690) в бакуловирусном векторе pFastBac с помощью полимеразной цепной реакции (Polymerase Chain Reaction, PCR). Белки экспрессировали в инфицированных бакуловирусом клетках насекомых Sf9 в виде слитых с глутатион-S-трансферазой (GST) белков. Белки очищали аффинной хроматографией с помощью глутатионсефарозных гранул. Ингибирование erbB-тирозинкиназной активности оценивали, используя анализ рецепторных тирозинкиназ на основе ELISA. Киназные реакционные смеси (50 мМ HEPES, рН 7,4, 125 мМ NaCI, 10 мМ MgCI2, 100 мкМ ортованадата натрия, 2 мМ дитиотреитола, 20 мкМ АТР (аденозинтрифосфат), тестируемое соединение или контроль-носитель и 1-5 нМ GST-erbB на реакционный объем 50 мкл) вносили в 96-луночные планшеты, покрытые 0,25 мг/мл poly-Glu-Tyr (Sigma). Реакционные смеси инкубировали в течение 6 минут при комнатной температуре при встряхивании. Киназные реакции останавливали путем удаления реакционной смеси, затем лунки промывали промывочным буфером, содержащим 3% бычьего сывороточного альбумина (BSA) и 0,1% Твин 20 в забуференном фосфатом физиологическом растворе (PBS). Фосфорилированные тирозиновые остатки детектировали путем добавления 0,2 мкг/мл антифосфотирозинового антитела (Oncogene Ab-4; 50 мкл/лунку), конъюгированного с пероксидазой хрена (HRP) на 25 минут при встряхивании при комнатной температуре. Антитело удаляли, и планшеты промывали (3% BSA и 0,1% Твин 20 в PBS). Добавляли HRP субстрат 3,3',5,5'-тетраметилбензиден (SureBlueTMB, Kirkegaard & Perry Labs) (50 мкл на лунку) и инкубировали в течение 10-20 минут при встряхивании при комнатной температуре. ТМВ реакцию останавливали добавлением 50 мкл останавливающего раствора (0,09 н. H2SO4). Сигнал оценивали количественно путем измерения поглощения при 450 нм. Значения Ю5о (50%-ная ингибирующая концентрация) для тестируемых соединений определяли, используя Microsoft Excel. Соединение ErbB1 ICsn (нМ) ErbB2 Юм (нМ) ErbB4 ICm (nM) Пример № 1 6,44 77,5 Пример №2 6,9 16,7 83,67 Пример №6 9,87 244,39 154 Пример №7 11,35 84,05 61,5 Пример №8 45,34 212,11 233,8 Пример №9 18,08 247,12 147 Пример № 10 12,13 85,98 41,33 ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы: где: Ri выбран из F, Br, CI или I; R2 выбран из Н, F, Вг, CI или I; R3 выбран из: а) прямого или разветвленного Ci-Сзалкила, возможно замещенного одним или более чем одним галогеном; или б) групп -(СНг)п-морфолино, -(СН2)п-пиперидин, -(СН2)п-пиперазин, -(СН2)п-пиперазин-М(С1-Сзалкил), -(СН2)п-пирролидин или -(СН2)п-имидазол; п представляет собой целое число от 1 до 4; R4 представляет собой -(CH2)m-Het; Het представляет собой гетероциклическую группировку, выбранную из группы морфолин, пиперидин, пиперазин, пиперазин-М(С1-С3алкил), имидазол, пирролидин, азепан, 3,4-дигидро-2Н-пиридин или 3,6-дигидро-2Н-пиридин, где каждая гетероциклическая группировка возможно замещена 1-3 группами, выбранными из Ci-Сзалкила, галогена, ОН, NH2, групп МН(СгС3алкил) или М(С1-Сзалкил)2; m представляет собой целое число от 1 до 3; и X представляет собой О, S или NH; или его фармацевтически приемлемая соль. 2. Соединение по п. 1 формулы: где: R3 выбран из: а) прямого или разветвленного СгСзалкила, возможно замещенного одним или более чем одним галогеном; или б) групп -(СН2)ч-морфолино, -(СН2)ч-пиперидин, -(СН2)ч-пиперазин, -(СН2)ч-пиперазин-М(С1-С3алкил), -(СН2) <,-пирролидин или -(СН2)р-имидазол; q представляет собой целое число от 1 до 2; R4 представляет собой -(CH2)m-Het; Het представляет собой группу пиперидин, пиперазин, nnnepa3HH-N(Ci-Сзалкил), имидазол, пирролидин, азепан или дигидропиридин, возможно замещенную 1 или 2 группами, выбранными из галогена или СгСзалкила; m представляет собой целое число от 1 до 3; и X представляет собой О, S или NH; или его фармацевтически приемлемая соль. Соединение по любому из пп.1-2 формулы: где: R3 представляет собой прямой или разветвленный C-i-Сзалкил, возможно замещенный одним или более чем одним галогеном; R5 и Re независимо выбраны из Н, СгСзалкила, F, Вг, I или CI; X представляет собой О, S или NH; и каждая из пунктирных линий, обозначенных как а и Ь, означает возможную двойную связь, при условии, что только одна двойная связь а или b присутствует в одном соединении; или его фармацевтически приемлемая соль. 4. Соединение по любому из пп.1-3, где X представляет собой О, и R3 представляет собой прямой или разветвленный С-рСзалкил, возможно замещенный 1-3 атомами галогенов. 5. Соединение, выбранное из группы, состоящей из: 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7- метоксихиназолин-6-ил]амида; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метилсульфанилхиназолин-6-ил]амида; 4-пиперидин-1-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метиламинохиназолин-6-ил]амида; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-изопропоксихиназолин-6-ил]амида; 4-пиперидин-1-илбут-2-еновой кислоты [4-(3-бромфениламино)-7-метоксихиназолин-6-ил]амида; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-этоксихиназолин-6-ил]амида; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-пропоксихиназолин-6-ил]амида; 4-(4-фторпиперидин-1-ил)бут-2-еновой кислоты [4-(3-хлор-4- фторфениламино)-7-метоксихиназолин-6-ил]амида; 4-(3-фторпиперидин-1-ил)бут-2-еновой кислоты [4-(3-хлор-4- фторфениламино)-7-метоксихиназолин-6-ил]амида; 4-(2-фторпиперидин-1-ил)бут-2-еновой кислоты [4-(3-хлор-4- фторфениламино)-7-метоксихиназолин-б-ил]амида; 4-морфолин-4-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида; 4-азепан-1-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида; 4-пиперидин-1-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-трифторметоксихиназолин-6-ил]амида; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-фторметоксихиназолин-6-ил]амида; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-фторэтоксихиназолин-6-ил]амида; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-(2-фторэтилсульфанил)хиназолин-6-ил]амида; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-трифторэтоксихиназолин-б-ил]амида; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-дифторэтоксихиназолин-6-ил]амида; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-(3-морфолин-4-илпропокси)хиназолин-6-ил]амида; 4-пиперидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-(2-пиперидин-1-илэтокси)хиназолин-6-ил]амида; 4-(3,4-дигидро-2Н-пиридин-1 -ил)бут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида; 4-(3,6-дигидро-2Н-пиридин-1 -ил)бут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-б-ил]амида; 4-пиперазин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида; 4-(4-метилпиперазин-1-ил)бут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида; 4-имидазол-1-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихи назол и н-6-и л]а м ида; 4- пирролидин-1 -илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида; 5- пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-мети лсул ьфа н и лхи назол и н-6-и л]ам ида; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метиламинохиназолин-6-ил]амида; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-изопропоксихиназолин-6-ил]амида; 5-пиперидин-1-илпент-2-еновой кислоты [4-(3-бромфениламино)-7-метоксихиназолин-6-ил]амида; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-этоксихиназолин-6-ил]амида; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-пропоксихиназолин-6-ил]амида; 5-(4-фторпиперидин-1-ил)пент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида; 5-(3-фторпиперидин-1-ил)пент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида; 5-(2-фторпиперидин-1-ил)пент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида; 5-морфолин-4-илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида; 5-азепан-1 -ил пент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-трифторметоксихи назол и н-6-ил]ам ида; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-фторметоксихиназолин-6-ил]амида; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-фторэтоксихиназолин-6-ил]амида; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-(2-фторэтилсульфанил)хиназолин-6-ил]амида; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-трифторэтоксихиназолин-6-ил]амида; 5-пиперидин-1-илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-дифторэтоксихиназолин-6-ил]амида; 5-пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-(3-морфолин-4-илпропокси)хиназолин-6-ил]амида; 5- пиперидин-1 -илпент-2-еновой кислоты [4-(3-хлор-4-фторфениламино)~ 7-(2-пиперидин-1-илэтокси)хиназолин-6-ил]амида; 6- пиперидин-1 -илгекс-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида; 6-пиперидин-1 -илгекс-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метилсульфанилхиназолин-6-ил]амида; 6-пиперидин-1 -илгекс-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-мети л а м и нохи назол и н-б-и л]ам ида; 6-пиперидин-1-илгекс-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-этоксихиназолин-6-ил]амида или 6-пиперидин-1-илгекс-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-фторэтоксихиназолин-6-ил]амида или их фармацевтически приемлемой солевой формы. 6. Фармацевтическая композиция, содержащая фармацевтически эффективное количество соединения по п.1 и фармацевтически приемлемый носитель. 7. Фармацевтическая композиция, содержащая фармацевтически эффективное количество 4-пиперидин-1-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида или его фармацевтически приемлемой солевой, сложноэфирной или амидной формы и фармацевтически приемлемый носитель. 8. Способ лечения пролиферативных расстройств у млекопитающего, включающий введение нуждающемуся в этом млекопитающему фармацевтически эффективного количества соединения по п.1 или его фармацевтически приемлемой солевой, сложноэфирной или амидной формы. 9. Способ лечения пролиферативных расстройств у млекопитающего, включающий введение нуждающемуся в этом млекопитающему фармацевтически эффективного количества 4-пиперидин-1-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида или его фармацевтически приемлемой солевой, сложноэфирной или амидной формы. 10. Способ лечения рака молочной железы у млекопитающего, включающий введение нуждающемуся в этом млекопитающему фармацевтически эффективного количества 4-пиперидин-1-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида или его фармацевтически приемлемой солевой, сложноэфирной или амидной формы. 11. Способ лечения рака ободочной кишки у млекопитающего, включающий введение нуждающемуся в этом млекопитающему фармацевтически эффективного количества 4-пиперидин-1-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида или его фармацевтически приемлемой солевой, сложноэфирной или амидной формы. 12. Способ лечения немел коклеточного рака легкого у млекопитающего, включающий введение нуждающемуся в этом млекопитающему фармацевтически эффективного количества 4-пиперидин-1-илбут-2-еновой кислоты [4-(3-хлор-4-фторфениламино)-7-метоксихиназолин-6-ил]амида или его фармацевтически приемлемой солевой, сложноэфирной или амидной формы. 13. Соединение формулы: halo где R4 является таким, как определено в п.1, a halo представляет собой F, CI, Вг или I. 14. Соединение формулы: где: Y представляет собой NH2, N02 или группировку R4-(CH2)m-CH=CH-C(0)-NH2-; и m представляет собой целое число от 1 до 3; R3 выбран из: а) прямого или разветвленного Ci-Сзалкила, возможно замещенного галогеном; или б) групп -(СН2)п-морфолино, -(СН2)п-пиперидин, -(СН2)п-пиперазин, -(СН2)п-пиперазин-М(СгСзалкил), -(СН2)п-пирролидин или -(СНУп-имидазол; п представляет собой целое число от 1 до 4; R4 представляет собой -(CH2)m-Het; Het представляет собой гетероциклическую группировку, выбранную из группы, состоящей из морфолина, пиперидина, пиперазина, группы пиперазин-М(СгСзалкил), имидазола, пирролидина, азепана, 3,4-дигидро-2Н-пиридина или 3,6-дигидро-2Н-пиридина, где каждая гетероциклическая группировка возможно замещена группами в количестве от 1 до 3, выбранными из СгС3алкила, галогена, ОН, NH2, групп МН(СгС3алкил) или М(СгС3алкил)2; m представляет собой целое число от 1 до 3; и X представляет собой О, S или NH. 15. Соединение формулы: где: Y представляет собой N02, NH2 или группировку галогено-(СН2)т-СН=СН-C(0)-NH2-; галогено представляет собой F, CI, Вг или I; m представляет собой целое число от 1 до 3; R3 выбран из: а) моно-, ди- или тригалогенированной метильной группы; б) прямого или разветвленного Сг-Сзалкила, возможно замещенного галогено; или в) групп -(СН2)п-морфолино, -(СН2)п-пиперидин, -(СН2)п-пиперазин, -(СН2)п-пиперазин-М(С1-Сзалкил), -(СНг)п-пирролидин или -(СН2)п-имидазол. |