|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea200601247a*\id |
|
Термины запроса в документе Реферат Соединения формулы (I) в которой n представляет собой целое число от 1 до 6 включительно, R 1 и R 2 представляют собой атом водорода, C 1 -C 6 -алкильную группу или арил-C 1 -C 6 -алкильную группу, R 3 и R 4 представляют собой атом водорода или C 1 -C 6 -алкильную группу, R 5 и R 6 представляют собой атом водорода или C 1 -C 6 -алкил, галоген, гидрокси, C 1 -C 6 -алкокси, циано, нитро, C 2 -C 6 -ацил, C 1 -C 6 -алкоксикарбонил, C 1 -C 6 -тригалоалкил или C 1 -C 6 -тригалоалкокси группу или необязательно замещенную аминогруппу, R 7 представляет собой атом водорода, C 1 -C 6 -алкильную группу или арилалкильную группу. Лекарственные средства.
Полный текст патента (57) Реферат / Формула: в которой n представляет собой целое число от 1 до 6 включительно, R 1 и R 2 представляют собой атом водорода, C 1 -C 6 -алкильную группу или арил-C 1 -C 6 -алкильную группу, R 3 и R 4 представляют собой атом водорода или C 1 -C 6 -алкильную группу, R 5 и R 6 представляют собой атом водорода или C 1 -C 6 -алкил, галоген, гидрокси, C 1 -C 6 -алкокси, циано, нитро, C 2 -C 6 -ацил, C 1 -C 6 -алкоксикарбонил, C 1 -C 6 -тригалоалкил или C 1 -C 6 -тригалоалкокси группу или необязательно замещенную аминогруппу, R 7 представляет собой атом водорода, C 1 -C 6 -алкильную группу или арилалкильную группу. Лекарственные средства.
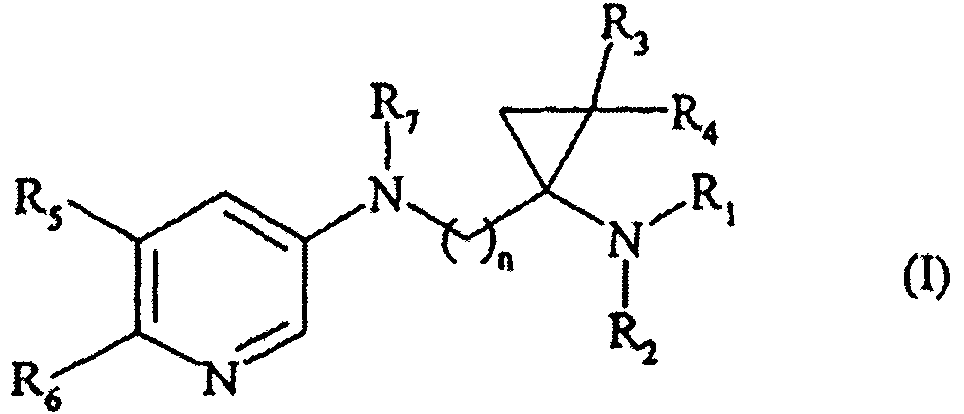
92260 Заявка № 200601247 Заявитель Ле Лаборатуар Сервье, FR НОВЫЕ МНОГОКРАТНО ЗАМЕЩЕННЫЕ СОЕДИНЕНИЯ 1,1-ПИРИДИЛАМИНОЦИКЛОПРОПАНАМИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, КОТОРЫЕ ИХ СОДЕРЖАТ Настоящее изобретение относится к новым многократно замещенным соединениям 1,1-пиридиламиноциклопропанамина, к способу их получения и к фармацевтическим композициям, которые их содержат. Соединения согласно настоящему изобретения являются чрезвычайно ценными в фармакологии, поскольку они специфически взаимодействуют с центральными никотиновыми рецепторами а4р2 типа, и применяются для лечения патологий нервной системы, связанных со старением головного мозга, а также для лечения нарушений настроения, боли и абстинентного табачного синдрома. Старение популяции в связи с повышением предполагаемой продолжительной жизни при рождении приводит к значительному повышению случаев патологий нервной системы, связанных со старением, в особенности болезни Альцгеймера. -2- Основными клиническими проявлениями старения головного мозга и в особенности нейропатологий, связанных со старением, являются недостаточности процессов памяти и познавательных способностей, что может приводить к слабоумию. В результате многочисленных исследований было 5 показано, что среди различных нейромедиаторов ацетилхолин играет важную роль в процессах памяти и что при определенных нейродегенеративных заболеваниях происходит значительное повреждение холинергических путей передачи сигналов в нейронах или при старении головного мозга происходит неадекватная активация. В связи с этим различные терапевтические подходы 10 направлены на предотвращение разрушения нейромедиатора путем ингибирования ацетилхолинэстеразы или обеспечение замены для недостающего нейромедиатора. В последнем случае были предложены холинергические агонисты мускаринового типа, которые специфичны к пост-синаптическим Ml рецепторам. 15 В последнее время было показано, что холинергические повреждения, связанные с болезнью Альцгеймера, поражают в большей степени нейроны, несущие никотиновые рецепторы, по сравнению с нейронами, несущими мускариновые рецепторы (Schroder и др., "Alzheimer disease : therapeutic strategies", Birkhauser Boston, 1994, 181-185). Кроме того, в различных исследованиях было показано, 20 что никотин обладает свойствами, улучшающими память (Prog. Neuropsychopharmacol., 1992, 16, 181-191) и что эти свойства оказывают влияние как на процессы памяти (Psychopharmacol., 1996, 123, 88-97), так и на улучшение внимания и бодроствования (Psychopharmacol., 1995, 118, 195-205). Кроме того, никотин обладает нейрозащитным действием по отношению к возбуждающе- 25 токсическим средствам, таким как глутамат (Brain Res., 1994, 644. 181-187). Все эти результаты с большой вероятностью могут быть связаны с эпидемиологическими исследованиями, в которых была показана низкая степень проявления болезни Альцгеймера и болезни Паркинсона у курильщиков. Кроме того, в некоторых исследованиях была показана ценность никотина для лечения 30 нарушений настроения, таких как депрессивные состояния, страх или шизофрения. В завершение, было показано, что никотин обладает обезболивающими свойствами. Все терапевтические свойства никотина, а также -3- свойства, описанные для других никотиновых средств, основываются на действии на центральные рецепторы, которые структурно и фармакологически отличаются от периферических рецепторов (мышц и ганглий). Центральные рецепторы а4р2 типа наиболее часто встречаются в центральной нервной 5 системе и вовлечены в большинство терапевтических действий никотина (Life Sci., 1995, 56, 545-570). В некоторых документах, таких как Synlett, 1999, 7, 1053-1054 ; J. Med. Chem, 1985, 28(12), 1953-1957 и 1980, 23(3), 339-341 ; 1970, 13.(5), 820-826 ; 1972, Ц(Ю), 1003-1006 ; J. Am. Chem. Soc, 1987, 109(13), 4036-4046, или нескольких 10 патентах или патентных заявках, таких как DE 36 08 727, ЕР 124 208 или WO 94/10158, описаны и заявлены соединения, содержащие 1,1- или 1,2-дизамещенную циклопропановую часть. Ни в одном из перечисленных документов не было описано или предположено, что эти соединения обладают фармакологическим действием, специфическим для никотиновых рецепторов, и, 15 в особенности, для центральных никотиновых рецепторов а4р2 типа, и это новое свойство соединений описано заявителем. В патентной заявке ЕР 1 170 281 описаны 1,1- и 1,2-дизамещенные циклопропановые соединения, которые являются никотиновыми лигандами. Следовательно, соединения согласно настоящему изобретению являются новыми 20 и представляют собой эффективные селективные никотиновые лиганды центрального рецептора подтипа а4р2. Поэтому они применяются для лечения недостаточности памяти, связанной со старением головного мозга и с нейродегенеративными заболеваниями, такими как болезнь Альцгеймера, болезнь Паркинсона, болезнь Пика, корсаковский психоз и деменция лобной 25 доли и подкорковая деменциям, а также для лечения нарушений настроения, синдрома Тоуретте, синдрома гиперактивности с недостаточностью внимания, абстинентного табачного синдрома и боли. В частности, настоящее изобретение относится к соединениям формулы (I): -4- в которой : п представляет собой целое число от 1 до 6 включительно, Ri и R2, которые могут быть одинаковыми или разными, каждый независимо 5 друг от друга представляет собой атом водорода, линейную или разветвленную Ci-Сб-алкильную группу или арил-С^-Сб-алкильную группу, где алкильная часть может быть линейной или разветвленной, R3 и R4, которые могут быть одинаковыми или разными, каждый независимо 10 друг от друга представляет собой атом водорода или линейную или разветвленную СрСб-алкильную группу, R5 и R6, которые могут быть одинаковыми или разными, каждый независимо друг от друга представляет собой атом водорода или линейный или разветвленный Ci-Сб-алкил, галоген, гидрокси, линейный или 15 разветвленный СрСб-алкокси, циано, нитро, линейный или разветвленный Сг-Сб-ацил, линейный или разветвленный С\-Сь-алкоксикарбонил, линейную или разветвленную Ci-Сб-тригалоалкильную или линейную или разветвленную Ci-Сб-тригалоалкокси группу или аминогруппу, необязательно замещенную 20 одной или двумя линейными или разветвленными СрСб-алкильными группами, R7 представляет собой атом водорода, линейную или разветвленную С\- Сб-алкильную группу или арил-С^Сб-алкильную группу, где алкильная часть может быть линейной или разветвленной, 25 где под арильной группой подразумевается фенильная, бифенильная, нафтильная, дигидронафтильная, тетрагидронафтильная, инданильная или инденильная группа, каждая из этих групп необязательно замещена одной или -5 - несколькими одинаковыми или разными группами, выбранными из атомов галогена, линейного или разветвленного Ci-Сб-алкила, гидрокси, циано, нитро, линейного или разветвленного Ci-Сб-алкокси, линейного или разветвленного Сг-С7-ацила, линейного или разветвленного Ci-Сб-алкоксикарбонила, линейной или 5 разветвленной Ci-Сб-тригалоалкильной и линейной или разветвленной С\-Сь-тригалоалкокси групп и аминогрупп, необязательно замещенных одной или двумя линейными или разветвленными СрСб-алкильными группами. Предпочтительными соединениями по изобретению являются те соединения, в которых п представляет собой целое число, равное 1. 10 Заместители Rj и R2, которые являются предпочтительными согласно настоящему изобретению, представляют собой атом водорода и линейную или разветвленную СрСб-алкильную группу. Более предпочтительно, заместители R\ и R2, которые являются предпочтительными согласно настоящему изобретению, представляют собой 15 атом водорода и метальную группу. Заместители R3 и R4, которые являются предпочтительными согласно настоящему изобретению, представляют собой атом водорода. Заместители R5 и К^, которые являются предпочтительными согласно настоящему изобретению, представляют собой атом водорода, линейную или 20 разветвленную Ci-Сб-алкильную группу и атом галогена. Благоприятно, предпочтительными соединениями по изобретению являются соединения, в которых R5 представляет собой атом водорода и представляет собой линейную или разветвленную СрСб-алкильную группу или атом галогена. Еще более благоприятно, предпочтительными соединениями по изобретению 25 являются соединения, в которых R5 представляет собой атом водорода и R6 представляет собой метильную группу или атом галогена. -6- Заместитель R7 в соответствии с изобретением предпочтительно представляет собой атом водорода или линейную или разветвленную СрСб-алкильную группу. Благоприятно, заместитель R7 в соответствии с изобретением предпочтительно 5 представляет собой атом водорода или метальную группу. Наиболее благоприятно, предпочтительными соединениями по изобретению являются: N- {[ 1 -(метиламино)циклопропил]метил } пиридин-3 -амин N-MCTHJI-N- {[ 1 -(метиламино)циклопропил]метил} пиридин-3-амин 10 N- [(1 -аминоциклопропил)мети л] пиридин-3 -амин N-{[1 -(диметиламино)циклопропил] метил} пиридин-3 -амин N-[( 1 -аминоцикл опропил)метил] -//-метилпиридин-З -амин 7^-{[1-(диметиламино)циклопропил]метил}-Лг-метилпиридин-3-амин 6-хлор-Л^-{[1-(метиламино)циклопропил]метил}пиридин-3-амин 15 6-хлор-Л^-метил-Л^-{[1-(метиламино)циклопропил]метил}пиридин-3-амин 6-бром-Л^-{[1-(метиламино)циклопропил]метил}пиридин-3-амин 6-бром-Л^-метил-М-{[1-(метиламино)циклопропи л] метил }пиридин-3-амин б-метил-N- {[ 1 -(метиламино)циклопропил]метил} пиридин-3-амин Л^,6-диметил-Л^- {[ 1 -(метиламино)циклопропил] метил} пиридин-3-амин 20 Среди фармацевтически приемлемых кислот можно отметить, но не ограничиваясь только ими, соляную кислоту, бромистоводородную кислоту, серную кислоту, фосфоновую кислоту, уксусную кислоту, трифторуксусную кислоту, молочную кислоту, пировиноградную кислоту, малоновую кислоту, янтарную кислоту, глутаровую кислоту, фумаровую кислоту, винную кислоту, 25 малеиновую кислоту, лимонную кислоту, аскорбиновую кислоту, щавелевую кислоту, метансульфоновую кислоту, камфорную кислоту и т.д. Среди фармацевтически приемлемых оснований можно отметить, но не ограничиваясь только ими, гидроксид натрия, гидроксид калия, триэтиламин, /и/?е/и-бутиламин и т.д. -7- Энантиомеры, диастереоизомеры, а также соли присоединения с фармацевтически приемлемой кислотой предпочтительных соединений составляют неотъемлемую часть изобретения. Настоящее изобретение также относится к способу получения соединений 5 формулы (I), который характеризуется тем, что в качестве исходного вещества применяют соединение формулы (II): в которой R-з, R4 и п имеют значения, указанные для формулы (I), соединения формулы (II) подвергают реакции с дифенилфосфорилазидом в 10 присутствии триэтиленамина в толуоле, с последующим добавлением трет-бутанола, получая соединения формулы (III) : в которой R3, R4 и п имеют значения, указанные выше, соединения формулы (III) подвергают реакции с соединением формулы (IV): в которой Hal представляет собой атом галогена и R'i представляет собой группу, выбранную из линейного или разветвленного Ci-Сб-алкила и арил-Ср Сб-алкила, где алкильная часть может быть линейной или разветвленной, в присутствии основания в безводном растворителе, получая соединения формулы R'l-Hal (IV) 20 (V): Н3СООС , N-COOtBu (V) в которой R3, R4, п и R'i имеют значения, указанные выше, -8- соединения формул (III) и (V) составляют соединения формулы (VI): HjCOOC (VI) Vi N-COOtBu R, в которой R3, R4 и n имеют значения, указанные выше, и Rj имеет значения, указанные для формулы (I), соединения формулы (VI) помещают в присутствие восстановителя, получая соединения формулы (VII): (VII) -COOtBu в которой R3, R4, п и Rj имеют значения, указанные выше, соединения формулы (VII) подвергают воздействию окислителя, обычного для органического синтеза, получая соединения формулы (VIII) : ОНО (vni) Vi N-COOtBu в которой R3, R4, п и Rj имеют значения, указанные выше, соединения формулы (VIII) подвергают реакции с соединением формулы (IX) : (IX) в которой R5 и Re имеют значения, указанные для формулы (I), в присутствии триацетоксиборогидрида натрия, получая соединения формулы (X) : в которой R3, R4, R5, Яб, п и Rj имеют значения, указанные выше, соединения формулы (X): - или помещают в присутствие кислоты в диоксане, получая соединения формулы (I/а), частный случай соединений формулы (I): в которой Rj, R3, R4, R5, R6 и п имеют значения, указанные выше, соединения формулы (I/а) : - или помещают в присутствие водного раствора формальдегида и муравьиной кислоты, получая соединения формулы (I/b), частный случай соединений формулы (I): в которой Rj, R3, R4, R5, Re и п имеют значения, указанные выше, - или подвергают реакции с соединением формулы (XI) : R*2 - Hal (XI) в которой Hal имеет значения, указанные выше, и R'2 представляет собой группу, выбранную из линейного или разветвленного Ci-Сб-алкила и арил-Ci-Сб-алкила, где алкильная часть может быть линейной или разветвленной, в -10- присутствии основания в безводном растворителе, получая соединения формулы (1/с), частный случай соединений формулы (I) : в которой R), R3, R4, R5, R6, п и R'2 имеют значения, указанные выше, 5 - или, если Rj представляет собой бензильную группу, помещают в присутствие соляной кислоты и палладия на угле, получая соединения формулы (I/d), частный случай соединений формулы (I): в которой R3, R4, R5, R6 и п имеют значения, указанные выше, 10 соединения формулы (I/d) обрабатывают муравьиной кислотой и водным раствором формальдегида, получая соединения формулы (1/е), частный случай соединений формулы (I) : в которой R3, R4, R5, R6 и п имеют значения, указанные выше, 15 соединения формул (I/b), (I/c), (I/d) и (1/е) составляют соединения формулы (I/f), частный случай соединений формулы (I) : -11 - (I/O в которой Rj, R2, R3, R4> R5, Re и п имеют значения, указанные выше, соединения формулы (I/f) подвергают реакции с соединением формулы (XII): R'7-СООН (XII), 5 в которой R7 представляет собой группу, выбранную из линейного или разветвленного Ci-Cs-алкила и арил-С^Сб-алкила, где алкильная часть может быть линейной или разветвленной, в присутствии связующего вещества, с последующим восстановлением амида, получая соединения формулы (I/g), частный случай соединений формулы (I) : в которой Ri, R2, R3, R4, R5, R6 и п имеют значения, указанные выше, и RM7 представляет собой группу, выбранную из линейного или разветвленного Cj-Сб-алкила и арил-СрСб-алкила, где алкильная часть может быть линейной или разветвленной, (I/g) - или помещают в присутствие карбонилдиимидазола и муравьиной кислоты в диметилформамиде, получая соединения формулы (XIII) : N-COOtBu (XIII) в которой Rj, R3, R4, R5, R6, и п имеют значения, указанные выше, - 12- соединения формулы (XIII) подвергают реакции с боран-метилсульфидным комплексом в тетрагидрофуране, получая соединения формулы (XIV) : (XIV) в которой Rj, R3, R4, R5, Яб и п имеют значения, указанные выше, соединения формулы (XIV) подвергают таким же условиям реакции, как и для соединений формулы (X), воздействию кислоты в диоксане, получая соединения формулы (I/h), частный случай соединений формулы (I): в которой R], R3, R4, R5, Re и п имеют значения, указанные выше, соединения формулы (I/h), если Rj представляет собой бензильную группу, подвергают таким же условиям реакции, как и для соединений формулы (1/а), получая соединения формулы (I/i), частные случаи соединений формулы (I): в которой R3, R4, R5, R$ и п имеют значения, указанные выше, соединения формулы (I/i) подвергают таким же условиям реакции, как и для соединений формулы (I/d), получая соединения формулы (I/j), частные случаи соединений формулы (I): -13 - ]JI-CH3 сн3 (I/j) в которой R3, R4, R5, Rg и п имеют значения, указанные выше, совокупность соединений формул (I/а) - (I/j), представляющее собой совокупность соединений по изобретению, очищают, если это является 5 подходящим, в соответствии с обычными методиками очистки, могут быть разделены на их разные изомеры в соответствии с обычный методикой разделения, и превращены, если это является подходящим, в их соли присоединения с фармацевтически приемлемой кислотой или основанием. Соединения формул (И), (IV), (IX), (XI) и (XII) или являются коммерческими 10 продуктами, или их получают согласно обычным способам органического синтеза, которые хорошо известны специалисту в данной области. Как правило, под изомерами соединений по изобретению подразумевают оптические изомеры, такие как энантиомеры и диастереоизомеры. Более предпочтительно, чистые энантиомерные формы соединений по изобретению 15 могут быть разделены, используя в качестве исходных веществ смеси энантиомеров, которые подвергают реакции с рацемат-разделяющим средством, которое может высвобождаться, где указанное средство само находится в виде чистого энантиомера, что обеспечивает получение соответствующих диастереоизомеров. Затем диастереоизомеры разделяют согласно методикам 20 разделения, которые хорошо известны специалисту в данной области, таким как кристаллизация или хроматография, и после этого разделяющее средство удаляют с помощью обычных методик органической химии, что обеспечивает получение чистого энантиомера. Соединения по изобретению, которые находятся в виде смеси 25 диастереоизомеров, выделяют в чистом виде с помощью обычных методик разделения, таких как хроматография. -14- В определенных предпочтительных случаях, способ получения соединений по изобретению может приводить к преимущественному образованию одного энантиомера или диастереоизомера по сравнению с другими. Благодаря их фармакологическим свойствам в качестве никотиновых лигандов и 5 их избирательности по отношению к рецептору подтипа а4р2, соединения согласно настоящему изобретению применяются для лечения недостаточности памяти, связанной со старением головного мозга и с нейродегенеративными заболеваниями, такими как болезнь Альцгеймера, болезнь Паркинсона, болезнь Пика, корсаковский психоз и деменция лобной доли и подкорковая деменциям, а 10 также для лечения нарушений настроения, синдрома Тоуретте, синдрома гиперактивности с недостаточностью внимания, абстинентного табачного синдрома и боли. Настоящее изобретение также относится к фармацевтическим композициям, которые содержат в качестве активного компонента по меньшей мере одно 15 соединение формулы (I), его изомер или его соль присоединения с фармацевтически приемлемой кислотой или основанием, отдельно или в сочетании с одним или несколькими фармацевтически приемлемыми, инертными, нетоксичными наполнителями или носителями. Фармацевтические композиции в соответствии с изобретением для 20 парентерального введения включают, главным образом, стерильные водные или неводные растворы, дисперсии, суспензии и эмульсии, а также стерильные порошки для восстановленных растворов для инъекций или дисперсий. Фармацевтические композиции согласно изобретению для перорального введения в виде твердых форм включают, в особенности, таблетки или драже, 25 подъязычные таблетки, саше, желатиновые капсулы и гранулы, а для перорального, назального, трансбуккального или внутриглазного введения в виде жидких форм включают, в особенности, эмульсии, растворы, суспензии, капли, сиропы и аэрозоли. Фармацевтические композиции для ректального или вагинального введения 30 предпочтительно представляют собой суппозитории, а композиции для -15- чрескожного введения включают, в особенности, порошки, аэрозоли, кремы, мази, гели и пластыри. Фармацевтические композиции, указанные выше, приведены только с целью иллюстрации изобретения и никоим образом его не ограничивают. 5 Среди фармацевтически приемлемых, инертных, нетоксичных, наполнителей или носителей можно отметить, но только с целью иллюстрации и никоим образом не ограничиваясь ими, разбавители, растворители, консерванты, смачивающие вещества, эмульгирующие средства, диспергирующие средства, связывающие вещества, агенты, вызывающие набухание, дезинтеграторы, 10 вещества для замедленного высвобождения, замасливатели, абсорбенты, суспендирующие вещества, красители, ароматизаторы и т.д. Полезная дозировка изменяется в зависимости от возраста и веса пациента, пути введения и применяемой фармацевтической композиции, природы и тяжести расстройства, и применения любых сопутствующих видов лечения. Дозировка 15 находится в диапазоне от 1 мг до 500 мг в сутки на одно или больше введений. Примеры, представленные ниже, приведены только с целью иллюстрации и никоим образом не ограничивают изобретение. Используемые исходные материалы являются известными продуктами или продуктами, которые получают в соответствии с известными способами. Разные 20 примеры получения приводят к синтезу промежуточных продуктов, которые пригодны для получения соединений согласно изобретению. Структуры соединений, описанных в примерах и в получениях, были определены согласно обычным спектрофотометрическим способам (инфракрасная спектрометрия, ядерный магнитный резонанс, масс-25 спектрометрия, и т.д). Точки плавления определяли при помощи нагревающей пластинки Кофлера или нагревающей пластинки под микроскопом. Если соединение находится в виде соли, то приведенная точка плавления и элементный микроанализ соответствует солевой форме продукта. -16- Получение 1 : трет-Бутнл 1-(формил)циклопропил(метил)карбамат Этап 1: Метил 1-[(трет-бутоксикарбонил)амино]циклопропанкарбоксилат Раствор 80 г 1-(метоксикарбонил)циклопропанкарбоновой кислоты и 78 мл триэтиламина в 550 мл толуола, к которому добавляли 152 г 5 дифенилфосфорилазида, нагревали до 80°С. После прекращения выделения газа, температуру доводили до 50°С и добавляли 61 г /и/?е/и-бутанола. После осуществления реакции в течение 7 часов при 80°С, смесь концентрировали. Остаток ресуспендировали в простом эфире, промывали насыщенным раствором ИагСОз, затем 1 н. раствором соляной кислоты, и после этого раствором 10 ИаНСОз. После высушивания и упаривания органической фазы, остаток ресуспендировали в 300 мл циклогексана и затем концентрировали насухо. Полученный остаток растирали в порошок в пентане, фильтровали и затем высушивали, выделяя ожидаемый продукт. Этап 2 : Метил 1-Ктует-15 бутоксикарбонил)(метил)аминоЫиклопропанкарбоксилат 24,1 г гидрида натрия порциями добавляли к раствору, охлажденному до 5°С, 99,7 г соединения, полученного на вышеописанном этапе 1, в 1,7 л безводного диметилформамида. Через 15 минут при 5°С и после этого 3 часа при температуре окружающей среды, по каплям добавляли 38,2 мл метилйодида. 20 После осуществления реакции в течение 20 часов смесь упаривали. Остаток ресуспендировали в простом эфире и затем обрабатывали обычным способом. При осуществлении хроматографии на силикагеле (дихлорметан) выделяли ожидаемый продукт. Этап 3 : трет-Бутил 1-(гидроксиметил)циклопропил(метил)карбамат 25 Раствор 100 мл 2М борогидрида лития в тетрагидрофуране добавляли к раствору 23 г соединения, полученного на вышеописанном этапе 2, в 100 мл тетрагидрофурана. После перемешивания в течение 20 часов при температуре окружающей среды, затем 8 часов при температуре флегмы, реакционную смесь охлаждали до 0°С, гидролизивали, разводили простым эфиром, разделяли, -17- высушивали и концентрировали. При хроматографировании остатка на силикагеле (дихлорметан/тетрагидрофуран : 95/5) выделяли ожидаемый продукт. Этап 4 : трет-Бутил 1-формилииклопропил(метил)карбамат 33,5 г диметилсульфоксида добавляли при -60°С, в течение 20 минут, к раствору, 5 содержащему 25,8 г оксалилхлорида в 430 мл дихлорметана. После перемешивания в течение 20 минут при -60°С, добавляли смесь, содержащую 34,3 г соединения с вышеописанного этапа 3, в 100 мл дихлорметана, при -60°С в течение 1 часа. После перемешивания в течение 30 минут при -60°С, вливали 81 мл триэтиламина в течение 20 минут при -60°С и затем температуре снова 10 позволяли повышаться до 20°С. Вливали в 60 мл воды, осуществляли разделение и водную фазу несколько раз экстрагировали дихлорметаном. Объединенные дихлорметановые фазы промывали насыщенным раствором хлорида натрия и высушивали над сульфатом натрия и затем концентрировали насухо. При осуществлении хроматографии на силикагеле (дихлорметан/тетрагидрофуран : 15 97/3) получали 31,2 г ожидаемого продукта. Получение 2 : трет-Бутил бензил{1-[(пиридин-3- иламино)метил]циклопропил}-карбамат Этап 1: Метил 1-[бензил(трет- бутоксикарбонил)амино]ииклопропанкарбоксилат 20 21,5 г соединения с этапа 1 получения 1 и 216 мл диметилформамида вносили в трехгорлую колбу. При 20°С добавляли 4,8 г 60% гидрида натрия в масле. Перемешивали в течение 2 часов при температуре окружающей среды. В течение 20 минут вливали в 18 мл бензилбромида и перемешивали в течение 20 часов при температуре окружающей среды. Нагревали в течение 1 часа при 60°С, 25 затем концентрировали насухо. Остаток ресуспендировали в простом эфире, и промывали 10% раствором карбоната натрия и затем 10% раствором хлорида лития. Высушивали над сульфатом натрия и концентрировали насухо. При осуществлении хроматографии на силикагеле (дихлорметан/циклогексан : 85/15, затем чистый дихлорметан) получали 21,3 г ожидаемого продукта в виде смолы. 30 Этап 2: трет-Бутил бензилЦ-гидроксиметил)циклопропил]карбамат - 18- В течение 20 минут, 70 мл 2М борогидрида лития в ТГФ вливали при 20°С в смесь 21,2 г соединения, полученного на вышеописанном этапе 1, и 100 мл тетрагидрофурана. Перемешивали в течение 20 часов при 20°С и затем в течение 1 часа при температуре флегмы. Охлаждали до 5°С, затем проводили гидролиз с 5 помощью 24 мл воды и после этого 20 мл 10% водного раствора карбоната натрия. Добавляли 500 мл простого эфира, осуществляли разделение и водную фазу экстрагировали простым эфиром. Объединенные эфирные фазы высушивали над сульфатом натрия и концентрировали насухо. Получали 19,3 г требуемого продукта. 10 Точка плавления: 68°С Этап 3 : трет-Бутил бензил(1-формилииклопропил)карбамат В течение 20 минут, 10,3 мл диметилсульфоксида вливали при -60°С в смесь 7,94 мл оксалилхлорида и 145 мл дихлорметана. Смесь перемешивали в течение 20 минут при -60°С. Вливали в 19,3 г соединения, полученного на 15 вышеописанном этапе 2, в течение 1 часа и перемешивали в течение 30 минут при -60°С. Вливали в 27,3 мл триэтиламина и перемешивали в течение 20 минут при -60°С. Температуре позволяли повышаться до 20°С и вливали в 50,6 мл воды. После перемешивания в течение 10 минут при 20°С, осуществляли разделение и водную фазу экстрагировали дихлорметаном. Объединенные 20 органические фазы высушивали над сульфатом натрия и концентрировали насухо. При осуществлении хроматографии на силикагеле (дихлорметан) получали 16,45 г ожидаемого продукта. Точка плавления: 67°С Этап 4 : трет-Бутил бензил{1-1(пиридин-3-25 иламино)метил1ииклопропил}карбамат 38,8 г продукта, полученного на вышеописанном этапе 3, 14,7 г 3-аминопиридина и 282 мл ЗА молекулярного сита добавляли к 2,5 л дихлорметана. Перемешивали в течение 2 часов при 20°С и затем добавляли 149 г триацетоксиборогидрида натрия. Перемешивали в течение 4 дней при 20°С. 30 Фильтровали и фильтрат промывали 10% водным раствором карбоната натрия, высушивали над сульфатом натрия и концентрировали насухо. При -19- осуществлении хроматографии на силикагеле (дихлорметан/тетрагидрофуран : 90/10) получали 36,2 г ожидаемого продукта в виде смолы. Пример 1 : ]\-{[1-(Метиламино)цнклопропил]метил}пиридин-3-амин дигидрохлорид 5 Этап 1: трет-Бутил метил{1-[(пиридин-3- иламино)метил1ииклопропил}карбамат 15,9 г триацетоксиборогидрида натрия добавляли при 20°С, в атмосфере азота, к смеси, содержащей 3,0 г продукта из получения 1, 300 мл дихлорметана, 1,56 г 3-аминопиридина и 30 мл ЗА молекулярного сита. Реакционную смесь Ю перемешивали в течение 2 дней при 20°С и фильтровали. Фильтрат промывали 10% раствором карбоната натрия, высушивали над сульфатом натрия и концентрировали насухо. При осуществлении хроматографии на силикагеле (толуол/этанол: 95/5) получали 3,0 г ожидаемого продукта. Этап 2 : N-{11-(Метиламино) циклопу опил]метил}пиу ид ин-3-амин 15 дигидрохлорид При 20°С, 14 мл 4 н. соляной кислоты в диоксане вливали в раствор, содержащий 1,0 г продукта, полученного на вышеописанном этапе 1, в 50 мл диоксана. Перемешивали в течение 20 часов при температуре окружающей среды. Добавляли простой эфир, и нерастворимое вещество отфильтровали с 20 отсасыванием и растворяли в этаноле. Концентрировали насухо и остаток растирали в порошок в простом эфире. Кристаллы отфильтровали с отсасыванием и высушивали в вакууме при 30°С. Получали 0,78 г ожидаемого продукта. Масс-спектрометрия (ESI) : m/z = 178,1 Th ([М+Н]+) 25 Точка плавления: 190-195°С Ппимеп 2 : ^Метил-^{[1-(метиламино)циклопропил]метил}пиридин-3-амин дигидрохлорид -20- Этап 1: трет-Бутил (1-{1формил(пиридин-3- ил)амино]метил}циклопропил(метил)карбамат Раствор 23,8 г карбонилдиимидазола и 30 мл ДМФА вливали при 5°С в раствор, содержащий 6,14 г муравьиной кислоты и 30 мл ДМФА. Перемешивали в 5 течение 1 часа при 5°С и затем в течение 3 часов при 20°С. Охлаждали до 5°С и затем вливали в смесь, содержащую 7,4 г продукта, полученного на этапе 1, примера 1 и 75 мл ДМФА. Перемешивали в течение 20 часов при 20°С и концентрировали насухо под давлением 1 торр. Остаток ресуспендировали в дихлорметане, промывали 10% водным раствором карбоната натрия, 10 высушивали над сульфатом натрия и затем концентрировали насухо. При осуществлении хроматографии на силикагеле (толуол/этанол : 95/5) получали 6,2 г ожидаемого продукта. Этап 2: М-Метил-М-{[1-(метиламино)ииклопропил1метил}пиридин-3-амин дигидрохлорид 15 При 0°С, 1,6 мл ЮМ боран-метилсульфидного комплекса вливали в смесь, содержащую 2,0 г соединения, полученного на вышеописанном этапе 1, и 20 мл тетрагидрофурана. Температуре позволяли повышаться до 40°С и затем нагревали в колбе с обратным холодильником в течение 3 часов. Охлаждали до 0°С и затем вливали в 3 мл метанола. Перемешивали в течение 1 час и затем 20 медленно выливали в солянокислый метанол до установления значения рН < 2. Наблюдали выделение газа. После прекращения выделения, нагревали в колбе с обратным холодильником в течение 3 часов. Концентрировали насухо. Остаток ресуспендировали в дихлорметане и затем промывали 1 н. раствором гидроксида натрия. Высушивали над сульфатом натрия и концентрировали насухо. Остаток 25 хроматографировали на силикагеле (дихлорметан/метанол/гидроксид аммония 15 н. : 95/5/0,5). Получали 0,7 г основания, которое растворяли в простом эфире. Добавляли эфирный хлористый водород до достижения кислого значения рН. Наблюдали выпадение осадка. Простой эфир отделяли и смолу растирали в порошок с чистым простым эфиром. Наблюдали кристаллизацию. Кристаллы 30 отфильтровали с отсасыванием и высушивали при 40°С под давлением 1 торр. Получали 0,8 г ожидаемого продукта. Масс-спектрометрия (ESI) : m/z =192,1 Th ([М+Н]+) -21 - Точка плавления: 180-184°С Пример 3 : ]Ч-{[1-Бензиламино)циклопропил]метил}пиридин-3-амин 50 мл 4М соляной кислоты в диоксане добавляли при 20°С к смеси, содержащей 5 7,15 г соединения из получения 2, 80 мл диоксана и 80 мл метанола. Перемешивали в течение 3 дней при 20°С. Концентрировали насухо, добавляли метанол, снова осуществляли концентрирование, и дополнительно два раза повторяли добавление и отгонку метанола. Остаток ресуспендировали в 300 мл метанола и добавляли 80 мл силикагеля. Концентрировали насухо (образование 10 пасты). При осуществлении хроматографии на силикагеле (СНгС^/метанол : 80/20) получали 6,6 г ожидаемого продукта в виде смолы. Пример 4: 1Ч-[(1-Аминоциклопропил)метил]пиридин-3-амин дигидрохлорид 4,0 г соединения из примера 3 растворяли в 200 мл этанола и добавляли 1,0 мл 15 11,8 н. соляной кислоты. Концентрировали насухо и остаток растворяли в 200 мл этанола в колбе с обратным холодильником. После охлаждения добавляли 200 мл циклогексена и затем, в атмосфере азота, 1,2 г 10% палладия на угле. Нагревали в колбе с обратным холодильником в течение 20 часов, затем фильтровали и концентрировали насухо. Остаток ресуспендировали в 200 мл 20 этанола и 5 мл воды и снова концентрировали. Остаток растворяли в метаноле и добавляли 32 мл силикагеля (образование пасты). Концентрировали насухо. Хроматографировали на 550 мл силикагеля (дихлорметан/метанол : 80/20). Получали соединение, которое ресуспендировали в 12 мл 35% раствора гидроксида натрия. Несколько раз экстрагировали простым эфиром, и 25 объединенные эфирные фазы высушивали над сульфатом натрия и концентрировали насухо. Остаток растворяли в этаноле, добавляли солянокислый этанол до достижения рН 1 и концентрировали насухо. Остаток растворяли в изопропаноле при нагревании, и охлаждали, что вызывало кристаллизацию. Кристаллы отфильтровали с отсасыванием и высушивали при 30 50°С под давлением 0,5 торр. Получали 1,6 г ожидаемого продукта. Масс-спектрометрия (EI) : m/z = 163,1 Th (М+) -22- Точка плавления: 195-199°С Пример 5: ГЧ-{[1-(Диметиламино)циклопропил]метил}пиридин-3-амин дигидрохлорид 1,26 г основания соединения из примера 4 растворяли в 25,2 мл муравьиной 5 кислоты и 25,2 мл 37% формальдегида. Нагревали в течение 5 часов при 70°С. Концентрировали насухо и остаток ресуспендировали в 20 мл воды и снова концентрировали. Остаток ресуспендировали в 15 мл 35% раствора гидроксида натрия и экстрагировали простым эфиром, и объединенные эфирные фазы высушивали над сульфатом натрия и концентрировали насухо. Остаток 10 хроматографировали на 230 мл силикагеля (дихлорметан/метанол : 95/5) и получали 1,17 г остатка, который растворяли в 25 мл изопропанола. Добавляли 3 мл 4М соляной кислоты в диоксане и затем разводили с помощью 25 мл простого эфира. Кристаллы отфильтровали с отсасыванием и высушивали и получали 1,25 г ожидаемого продукта. 15 Масс-спектрометрия (ESI) : m/z =192,1 Th ([М+Н]+) Точка плавления: 208-210°С Пример 6: ^{[1-(Бензиламино)циклопропил]метил}-^метилпиридин-3-амин Этап 1: трет-Бутил бензил(1-{(формил(пиридин-3-20 ил)амино1метил}ииклопропил)карбамат Соединение получали в соответствии с методикой с этапа 1 примера 2, заменяя соединение с этапа 1 примера 1 соединением из получения 2. Этап 2: М-{[1-(бензиламино)циклопропил]метил}-М-метилпиридин-3-амин Соединение получали в соответствии с методикой с этапа 2 примера 2, 25 используя соединение с вышеописанного этапа 1. Пример 7 : 1Ч-{(1-Аминоциклопропил)метил]-ГЧ-метилпиридин-3-амин дигидрохлорид -23- 5,5 мл 11,8 н. соляной кислоты добавляли к смеси, содержащей 17,7 г соединения из примера 6 и 900 мл этанола. Смесь подогревали для получения раствора. Добавляли 900 мл циклогексена и затем, в атмосфере азота, 6 г 10% палладия на угле. Нагревали в колбе с обратным холодильником в течение 20 5 часов. Катализатор отфильтровали и фильтрат концентрировали насухо. Остаток ресуспендировали в 60 мл 35% раствора гидроксида натрия и осуществляли экстракцию большим количеством простого эфира. Объединенные эфирные фазы высушивали над сульфатом натрия и концентрировали насухо. Остаток хроматографировали на силикагеле (дихлорметан/метанол : 93/7). Получали 10 основание требуемого продукта, которое превращали в соль путем добавления соляной кислоты в небольшом избытке в этаноле. Разводили простым эфиром. Кристаллы отфильтровали с отсасыванием и высушивали при 40°С под давлением 1 торр. Получали 2,48 г ожидаемого продукта. Масс-спектрометрия (ESI): m/z = 178,1 Th ([М+Н]+) 15 Точка плавления: 218-222°С Пример 8 : ]Ч-{[1-(Диметиламино)циклопропил]метил}-1Ч-метилпиридин-3-амин дигидрохлорид Соединение получали в соответствии методикой из примера 5, заменяя 20 соединение из примера 4 соединением из примера 7. Масс-спектрометрия (ESI): m/z = 206,2 Th ([М+Н]+) Точка плавления: 198-201°С Пример 9 : 6-Хлор-М-{[1-(метиламиноциклопропил]метил}пиридин-3-амин 25 дигидрохлорид Этап 1: трет-Бутил (1-{[(6-хлорпиридин-3- ил)амино]метил\ииклопропил)метилкарбамат 10 мл уксусной кислоты добавляли к смеси, содержащей 10,8 г соединения из получения 1, 100 мл метанола и 6,9 г 5-амино-2-хлорпиридина. Перемешивали в 30 течение 30 минут при температуре окружающей среды. Охлаждали до 5°С и -24- порциями добавляли 4,4 г цианоборогидрида натрия. Перемешивали в течение 20 часов при 20°С. Добавляли 10 мл воды и концентрировали насухо. Остаток ресуспендировали в дихлорметане и водном растворе карбоната калия. Осуществляли разделение и водную фазу несколько раз экстрагировали дихлорметаном. Объединенные органические фазы высушивали над сульфатом натрия и концентрировали насухо. При осуществлении хроматографии на силикагеле (дихлорметан/тетрагидрофуран : 98/2) получали 9,9 г ожидаемого продукта. Этап 2: 6-Хлор-И-{(1-(метиламиноииклопропил1метил}пиридин-3-амин дигидрохлорид Соединение получали в соответствии с методикой с этапа 2 примера 1, используя соединение с вышеописанного этапа 1. Масс-спектрометрия (ESI): m/z = 212,1 Th ([М+Н]+) Точка плавления: 196-202°С Пример 10 : 6-Хлор-К-метил-К-{[1-(метиламино)циклопропил)метилпиридин-3-амин дигидрохлорид Этап 1: трет-Бутил (1-{[(6-хлорпиридин-3- ил)(формил)амино1метил}ииклопропил)метилкарбамат Соединение получали в соответствии с методикой с этапа 1 примера 2, используя соединение с этапа 1 примера 9. Этап 2 : трет-Бутил (1-{[(6-хлорпиридин-3- ил)(метил)амино]метил}циклопропил)метилкарбамат При 0°С, 2,2 мл ЮМ боран-метилсульфидного комплекса вливали в смесь, содержащую 3,0 г продукта, полученного на вышеописанном этапе 1, и 30 мл тетрагидрофурана. Охлаждение прекращали, и, если температура стабилизировалась, нагревали в колбе с обратным холодильником в течение 3 часов. После охлаждения, вливали в солянокислый метанол до получения значения рН 2. Перемешивали в течение 1 часа при 20°С, затем нагревали в -25- колбе с обратным холодильником в течение 1 часа. Концентрировали насухо и остаток ресуспендировали в смеси дихлорметана и 4 н. раствора гидроксида натрия. Осуществляли разделение и водную фазу экстрагировали дихлорметаном. Объединенные органические фазы высушивали над сульфатом натрия и концентрировали насухо. Остаток хроматографировали на 200 г силикагеля (дихлорметан/тетрагидрофуран : 95/5). Получали 1,6 г ожидаемого продукта. Этап 3: 6-Хлор-И-метил-М-{[1-метиламино)циклопропил1метилпиридин-3-амин дигидрохлорид Продукт получали в соответствии с методикой с этапа 2 примера 1, используя продукт, полученный на вышеуказанном этапе 2. Масс-спектрометрия (ESI) : m/z = 226,1 Th ([М+Н]+) Точка плавления: 133-136°С Пример 11 : 6-Бром-]Ч-{[1-(метиламиноциклопропил]метил}пиридин-3-амин фумарат Этап 1: трет-Бутил (1-{[(6-бромпиридин-3- ил)амино]метил}ииклопропил)метилкарбамат Продукт получали в соответствии с методикой с этапа 1 примера 9, используя 5-амино-2-бромпиридин вместо 5-амино-2-хлорпиридина. Этап 2: б-Бром-N-ifl-(метилам ино) и иклопропил1метил}пир ид ин-3-амин фумарат 3,5 мл трифторуксусной кислоты добавляли к смеси, содержащей 2 г продукта, полученного на вышеописанном этапе 1, и 20 мл дихлорметана. Перемешивали в течение 20 часов при 20°С. Добавляли 10% водный раствор карбоната натрия до достижения рН > 9. Добавляли дихлорметан. Осуществляли разделение и органическую фазу высушивали над сульфатом натрия и концентрировали насухо. Остаток ресуспендировали в 5 мл этанола. Добавляли 0,6 г фумаровой кислоты, растворенной в 10 мл этанола. Наблюдали кристаллизацию. Кристаллы отфильтровали с отсасыванием, промывали этанолом, затем простым эфиром и -26- высушивали при 50°С под давлением 1 торр. Получали 1,6 г ожидаемого продукта. Масс-спектрометрия (ESI) : m/z = 256,0 Th ([М+Н]+) Точка плавления: 165-169°С 5 Пример 12 : 6-Бром-1Ч-метил-1Ч-{[1-(метиламино)циклопропил)метил}пиридин-3-амин фумарат Этап 1: трет-Бутил (1-{[(6-бромпиридин-3- ил)(формил)аминоъметил}циклопропил)метилкарбамат 10 Продукт получали в соответствии с методикой с этапа 1 примера 2, используя соединение с этапа 1 примера 11. Этап 2 : трет-Бутил (1-{[(6-бромпиридин-3- ил)(метил)аминоШетил}циклопропил)метилкарбамат Продукт получали в соответствии с методикой с этапа 2 примера 10, используя 15 продукт с вышеописанного этапа 1. Этап 3 : 6-Бром-М-метил-№Ш-(метиламино)ииклопропил1метил}пиридин-3-амин фумарат Продукт получали в соответствии с методикой с этапа 2 примера 11, используя продукт, полученный на вышеуказанном этапе 2. 20 Масс-спектрометрия (ESI) : m/z = 270,1 Th ([М+Н]+) Точка плавления: 146-150°С Пример 13 : 6-Метил-1Ч-{[1-(метиламино)циклопропил]метил}пиридин-3-амин дигидрохлорид 25 Этап 1: трет-Бутил метил(1-{[(6-метилпиридин-3-ил)амино1метил1ииклопропил)карбамат Соединение получали в соответствии с методикой с этапа 1 примера 9, используя 5-амино-2-метилпиридин вместо 5-амино-2-хлорпиридина. -27- Этап 2: 6-Метил-N-dl-(метиламине) ииклопропил1метил}пиридин-3-амин дигидрохлорид Соединение получали в соответствии с методикой с этапа 2 примера 9, используя продукт с вышеописанного этапа 1. 5 Масс-спектрометрия (ESI) : m/z = 192,2 Th ([М+Н]+) Точка плавления: 230-232°С Пример 14 : 1Ч,6-Диметил-М-{[1-(метиламино)циклопропил]метил}пиридин-3-амин дигидрохлорид Ю Этап 1: трет-Бутил (1-(1Формил(6-метилпиридин-3-иламиноШетил}ииклопропил)метилкарбамат Соединение получали в соответствии с методикой с этапа 1 примера 2, используя соединение с этапа 1 примера 13. Этап 2 : трет-Бутил метил(1-Пметил(6-метилпиридин-3-15 ил)амино1метил}ииклопропил)карбамат Соединение получали в соответствии с методикой с этапа 2 примера 2, используя продукт с вышеописанного этапа 1. Этап 3: N,6-Ииметил^-{(!-(метиламине)ииклопропил]метил}пиридин-3-амин дигидрохлорид 20 Соединение получали в соответствии с методикой с этапа 2 примера 1, используя продукт, полученный на вышеуказанном этапе 2. Масс-спектрометрия (ESI) : m/z = 206,2 Th ([М+Н]+) Точка плавления: 112-115°С Пример 15 : 25 6-Хлор-1Ч-{[1-(диметиламино)циклопропил]метил}пиридин-3-амин дигидрохлорид Соединение получали в соответствии методикой из примера 5, заменяя соединение из примера 4 соединением из примера 9. -28- Масс-спектрометрия (EI) : m/z 225,1 Th (M+) Точка плавления: 158-160 °C Пример 16 : 6-Хлор-Лг-{[1-(диметиламино)циклопропил]метил}-Лг-метилпиридин-3-амин 5 дигидрохлорид Этап 1: (6-Хлорпиридин-3-ил)({1- (формил (метил) амино/ииклопропилЫетил) формам ид В течение 15 минут, 1,85 мл муравьиной кислоты добавляли к 3,7 мл уксусного ангидрида при 5°С, и затем смесь нагревали в течение 2 часов при 55°С. После 10 возвращения до температуры окружающей среды, добавляли 7,4 мл тетрагидрофурана и затем реакционную смесь охлаждали до -20°С. В течение 30 минут, вливали в раствор 1,57 г основания соединения из примера 9 в 18:5 мл тетрагидрофурана. Температуру поддерживали равной -20°С в течение одного часа и затем равной 0°С в течение 20 часов. Осуществляли концентрирование, и 15 остаток ресуспендировали в 10% водном растворе карбоната натрия и экстрагировали дихлорметаном. Дихлорметан высушивали над сульфатом натрия и концентрировали насухо. При осуществлении хроматографии на силикагеле (дихлорметан/метанол: 97,5/2,5) получали 1,73 г ожидаемого продукта. 20 Этап 2: 6-Хлор-^{[1-(диметиламино)ииклопроп ил fметил }-N-метилпиридин-3-амин дигидрохлорид 1,73 г соединения, полученного на вышеописанном этапе 1, растворяли в 48 мл тетрагидрофурана и затем добавляли 3,6 мл ЮМ боран-метилсульфидного комплекса. Перемешивали в течение 20 часов при 20°С, затем нагревали в колбе 25 с обратным холодильником в течение 3 часов. Охлаждали до 5°С и вливали в 6,5 мл метанола; перемешивали в течение одного часа и концентрировали насухо. Остаток ресуспендировали в дихлорметане и промывали 10% водным раствором гидрокарбоната натрия. Органическую фазу высушивали над сульфатом натрия и концентрировали и при хроматографировании остатка на силикагеле 30 (дихлорметан/метанол : 97,5/2,5) получали основание ожидаемого продукта. 0,7 г этого основания растворяли в 5 мл изопропанола и добавляли раствор соляной -29- кислоты в простом эфире, получая, после отфильтровывания осадка и высушивания, 0,85 г ожидаемого соединения. Масс-спектрометрия (ESI) : m/z 240,1 Th ([М+Н]+) Точка плавления: 163-166 °С 5 Пример 17 : ]Ч-Бензнл-6-хлор-1Ч-{[1-(метиламино)циклопропил]метил}пиридин-3-амин дигидрохлорид Этап 1: трет-Бутил (1-{[бензоил(6-хлорпиридин-3- ил)амино1метил}циклопропил)метилкарбамат 10 10,17 г соединения, полученного на этапе 1 примера 9, растворяли в 200 мл тетрагидрофурана, добавляли 4,88 мл триэтиламина, охлаждали до 5°С и, в течение 30 минут, вливали в 5,05 г бензоилхлорида. Перемешивали в течение одного часа при 5°С и затем в течение одного часа при температуре окружающей среды, и после этого нагревали в колбе с обратным холодильником в течение 2 15 часов. Концентрировали насухо и остаток ресуспендировали в дихлорметане и промывали 50% водным раствором карбоната калия. Органическую фазу высушивали над сульфатом натрия и концентрировали, и при хроматографировании остатка на силикагеле (дихлорметан/тетрагидрофуран : 96/4) получали 13,9 г ожидаемого продукта. 20 Этап 2 : трет-Бутил (1-{[бензил(6-хлорпиридин-3- ил)амино1метил}ииклопропил)метилкарбамат 13,8 г соединения, полученного на вышеописанном этапе 1, растворяли в 120 мл тетрагидрофурана, охлаждали до 5°С и затем добавляли 8,3 мл ЮМ боран-метилсульфидного комплекса. После возвращения до температуры окружающей 25 среды, нагревали в колбе с обратным холодильником в течение 3 часов. Охлаждали до 5°С и вливали в 15,1 мл метанола; перемешивали в течение одного часа, затем подкисляли до рН 3, используя раствор соляной кислоты в метаноле. Нагревали в колбе с обратным холодильником в течение одного часа, затем концентрировали насухо. Остаток ресуспендировали в дихлорметане и 30 промывали 10% водным раствором карбоната натрия. Органическую фазу -30- высушивали над сульфатом натрия и концентрировали, и при хроматографировании остатка на силикагеле (дихлорметан до дихлорметан/бутанон : 90/10) получали 6,7 г ожидаемого соединения. Этап 3 : М-Бензил-6-хлоу-№{[1-(метиламино)циклопуопил1метил}пиуидин-5 3-амин дигидрохлорид Соединение получали в соответствии с методикой с этапа 2 примера 1, заменяя соединение с этапа 1 примера 1 соединением с вышеописанного этапа 2. Масс-спектрометрия (ESI) : m/z 302,1 Th ([М+Н] ) Точка плавления: 156-157 °С 10 Пример 18 : 7У-Бензил-6-хлор-Лг-{[1-(диметиламино)циклопропил]метил}пиридин-3-амин гидрохлорид 3,1 г основания соединения из примера 17 растворяли в 60 мл муравьиной кислоты. Добавляли 60 мл 37% водного раствора формальдегида и смесь 15 нагревали в течение 4 часов при 70°С. Концентрировали насухо, и остаток ресуспендировали в 50% водном растворе карбоната калия и экстрагировали дихлорметаном. Органическую фазу высушивали над сульфатом натрия, концентрировали, и остаток хроматографировали на силикагеле (дихлорметан/ацетон : 96/4). Выделяли фракцию, содержащую 0,48 г основания 20 ожидаемого соединения, из которой, после растворения в 5 мл этанола и добавления раствора соляной кислоты в простом эфире, получали, после отфильтровывания осадка и высушивания, 0,4 г ожидаемого соединения. Масс-спектрометрия (ESI) : m/z 316,16 Th ([М+Н]+) Точка плавления: 190-192°С Фармакологические исследования соединений по изобретению Пример А : Вытеснение связывания [ 1]-а-бунгаротоксина на никотиновых рецепторах электрического органа электрического ската -31 - Целью этого исследования, проведенного в соответствии со способом, описанным в J. Pharmacol. Exp. Ther., 1994, 271; 624-631, являлась оценка сродства соединений согласно настоящему изобретению к никотиновым рецепторам "мышечного" типа. 5 Мембраны (1-5 мкг/мл) электрического органа электрического ската инкубировали (1 час, 22°С) в присутствии серийных разведений (0,01-10 мкМ) каждого соединения по изобретению (разведения, начиная от 10 мМ маточного 125 раствора в ДМСО) в присутствии [ 1]-ос-бунгаротоксина (С.А. : 7,4 ТБк/ммоль: 0,2 нМ) в буфере Кребса (Трис-НС1 50 мМ, КС1 5 мМ, MgCl2 1 мМ, СаС12 2 мМ, 10 NaCl 100 мМ, рН 7,4) с 0,01% БСА; конечный объем: 500 мкл. Неспецифическое связывание определяли путем инкубирования мембран в присутствии ос-бунгаротоксина (1 мкМ). В результате проведенного исследования было установлено, что вплоть до концентрации 10 мкМ все соединения согласно настоящему изобретению не 15 обладают существенным действием на никотиновые рецепторы "мышечного" типа (Kj> 10"5M). Пример В : Вытеснение связывания [3Н]-эпибатидина на никотиновых рецепторах IMR32 клеток Целью этого исследования, проведенного в соответствии с методикой, 20 описанной в Molec. Pharmacol., 1995, 48 ; 280-287, являлось определение сродства соединений согласно настоящему изобретению к рецепторам "ганглионарного" типа (American Soc. Neuroscience, 2000, 26, 138). Мембраны (250 мкг/мл) клеток нейробластомы IMR-32 инкубировали (2 часа, 20°С) в присутствии серийных разведений (0,01-10 мкМ) каждого соединения по 25 изобретению (разведения, начиная от 10 мМ маточного раствора в ДМСО) и (±)-[ Н]-эпибатидина (С.А. : 2464 ГБк/ммоль: 1,5 нМ) в фосфатном буфере (NaH2P04 20 мМ, рН 7,4); конечный объем: 250 мкл. Неспецифическое связывание определяли путем инкубирования мембран в присутствии 300 мкМ (-)никотина. 30 В результате проведенного исследования было установлено, что вплоть до концентрации 10 мкМ все соединения согласно настоящему изобретению не -32- обладают существенным действием на никотиновые рецепторы "ганглионарного" типа (К; > 10~5М). Пример С: Вытеснение связывания [ Н]-оксотреморина-М на мускариновых рецепторах головного мозга крыс 5 Целью этого исследования, проведенного в соответствии со способом, описанным в Naumyn-Schmiederberg's Arch. Pharmacol., 2001, 363, 429-438, являлось определение сродства соединений согласно настоящему изобретению к мускариновым рецепторам. Мембраны (250 мкг/мл) головного мозга крыс инкубировали (2 часа, 20°С) в Ю присутствии серийных разведений (0,01-10 мкМ) каждого соединения по изобретению (разведения, начиная от 10 мМ маточного раствора в ДМСО) и [ Н]-оксотреморина-М (С.А.: 3174 ТБк/ммоль: 2 нМ) в фосфатном буфере (NaH2P04 20 мМ, рН 7,4); конечный объем: 250 мкл. Специфическое связывание определяли путем инкубирования мембран в присутствии атропина (1 мкМ). 15 Сродство соединений согласно настоящему изобретению к мускариновым рецепторам характеризовали путем определения Kj. В результате проведенного исследования было установлено, что вплоть до концентрации 10 мкМ, все соединения согласно настоящему изобретению не обладают сродством к мускариновым рецепторам (К; > 10 5М). 125 20 Пример D: Вытеснение связывания [ 1]-а-бунгаротоксина на никотиновых рецепторах "а7 типа" головного мозга крыс Целью этого исследования, проведенного в соответствии со способом, описанным в Molec. Pharmacol., 1986, 30 ; 427-436, являлось определение 25 сродства соединений согласно настоящему изобретению к центральным никотиновым рецепторам а7 типа. Мембраны (1000 мкг/мл) головного мозга инкубировали (5 часов, 37°С) в присутствии серийных разведений (0,01-10 мкМ) каждого соединения согласно настоящему изобретению (разведения, начиная от 10 мМ маточного раствора в 125 30 ДМСО) и [ 1]-а-бунгаротоксина (С.А. : 7,4 ТБк/ммоль: 1 нМ) в буфере Кребса -33- (Трис-HCl 50 мМ, КС1 5 мМ, MgCl2 1 мМ, СаС12 2 мМ, NaCl 100 мМ, рН 7,4) с 0,05% БСА; конечный объем: 500 мкл. Неспецифическое связывание определяли путем инкубирования мембран в присутствии а-бунгаротоксина (1 мкМ). Сродство соединений согласно настоящему изобретению к никотиновым 5 рецепторам а7 типа характеризовали путем определения Kj. В результате проведенного исследования было установлено, что вплоть до концентрации 10 мкМ, большинство соединений согласно настоящему изобретению не обладает сродством к центральным никотиновым рецепторам а7 типа. Определенные соединения по изобретению обладают К; порядка 10 мкМ, 10 такое как в примере 2, которое имеет Kj 8,3 х 10 "6М. Пример Е : Вытеснение связывания [ Н]-цитизина на никотиновых рецепторах "сс4р2 типа" головного мозга крыс Целью этого исследования, проведенного в соответствии с методикой, описанной в Molec. Pharmacol., 1990, 39 ; 9-12, являлось определение сродства 15 соединений согласно настоящему изобретению к центральным никотиновым рецепторам а4р2 типа. Мембраны (250 мкг/мл) головного мозга крыс инкубировали (2 часа, 20°С) в присутствии серийных разведений (0,01-10 мкМ) каждого соединения согласно настоящему изобретению (разведения, начиная от 10 мМ маточного раствора в 20 ДМСО) и [3Н]-цитизина (С.А. : 1184 ГБк/ммоль : 2 нМ) в фосфатном буфере (NaH2P04 20 мМ, рН 7,4); конечный объем: 250 мкл. Неспецифическое связывание определяли путем инкубирования мембран в присутствии 10 мкМ (-)никотина. Сродство соединений согласно настоящему изобретению к центральным никотиновым рецепторам а4р2 типа характеризовали путем 25 определения Kj. Полученные результаты свидетельствуют о том, что соединения согласно настоящему изобретению обладают сильным сродством к центральным никотиновым рецепторам ос4р2 типа. Так, соединения из примеров 1, 2 и 10 -8 -8 -8 имеют значения К, 3,4 х 10 " М, 1,5 х 10 ~ М и 1,6 х 10 " М, соответственно. -34- Эти результаты, а также результаты, полученные в примерах А - D, указывают на то, что соединения согласно настоящему изобретению являются сильнодействующими центральными никотиновыми лигандами, которые специфичны к рецепторам а4(52 типа. Пример F: Измерение в условиях in vivo высвобождения ацетилхолина с помощью интракортикального микродиализа крыс Wistar, находящихся в сознании Системное введение никотина и никотиновых агонистов вызывает повышение в условиях in vivo ацетилхолина в различных участках головного мозга (Neurochem. Res., 1996, 21, 1181-1186 ; Eur. J. Pharmacol., 1998, 35_L 181-188; Br. J. Pharmacol., 1999, 127, 1486-1494). Микродиализный зонд имплантировали в серединную префронтальную кору самцов крыс Wistar. Через шесть или семь дней после имплантации зонды перфузировали раствором Рингера (NaCl 147 мМ, КС1 2,7 мМ, СаС12 1,2 мМ, MgCl2 1 мМ, неостигмин 20 нМ) со скоростью потока 1 мкл/мин, при этом животные могли свободно двигаться. Через 2 часа в виварии внутрибрюшинно вводили исследуемый продукт. Контрольной группе животных вводили растворитель, используемый для продукта. Затем каждые 30 минут в течение 4 часов собирали диализаты (30 мкл) для измерения корковых внесинаптических концентраций ацетилхолина с помощью ВЭЖХ с амперометрическим определением. Результаты выражали в пг ацетилхолина/диализат, и сравнивали между разными группами с помощью дисперсионного анализа, используя 2 фактора (обработка х время), и измерение повторяли через определенное время. Полученные результаты показали, что соединения согласно настоящему изобретению повышали в условиях in vivo кортикальное высвобождение ацетилхолина в зависимости от дозы для доз в интервале от 0,3 до 3 мг/кг ВБ, что указывает на а4р2-агонистический характер. Так, соединения из примеров 1 и 2, через один час после введения в дозе 3 мг/кг ВБ, вызывают повышение на +70% и +80% соответственно, высвобождение ацетилхолина в префронтальной коре крыс Wistar, находящихся в сознании. -35- Пример G: Сокращения брюшных мышц, вызванные фенил-и- бензохиноном (PBQ) у NMRI мышей Внутрибрюшинное введение спиртового раствора PBQ вызывает абдоминальные судороги у мышей (Ргос. Soc. Exp. Biol., 1957, 95, 729-731). Судороги 5 характеризуются неоднократными сокращениями брюшных мышц, сопровождаемыми разгибаниями задних конечностей. Большинство обезболивающих средств противодействует таким брюшным судорогам (Brit. J. Pharmacol. Chem., 1968, 32, 295-310). В период времени t=0 мин животных взвешивали и ВБ вводили исследуемое соединение. Контрольной группе 10 животных вводили растворитель, используемый для продукта. В период времени t=30 мин, ВБ вводили спиртовой раствор PBQ (0,2%) в количестве 0,25 мл/мышь. Сразу после введения PBQ животных помещали в цилиндры из органического стекла (длина 19,5 см; внутренний диаметр 5 см). Наблюдали за реакцией животных в течение периода времени от t=35 мин до t=45 мин, при 15 этом исследователь записывал общее число брюшных судорог на животное. Результаты выражали в процентах ингибирования количества брюшных судорог, измеренных у контрольных животных, для активной дозы исследуемого соединения. Полученные результаты свидетельствуют об ингибировании порядка -80% для 20 активных доз 10 мг/кг ВБ, что подтверждает наличие обезболивающих свойств соединений по изобретению. Так, соединения из примеров 1, 2, 9 и 10, при введении в дозе 10 мг/кг ВБ, уменьшают на -50%, -76%, -52% и -69%, соответственно, количество брюшных судорог, вызываемых введением мышам PBQ. 25 Пример Н : Социальное распознавание у крыс Wistar Тест на социальное распознавание впервые был описан в 1982 году (J. Сотр. Physiol., 1982, 96, 1000-1006), и впоследствии был предложен различными авторами (Psychopharmacology, 1987, 9J., 363-368 ; Psychopharmacology, 1989, 97, 262-268) для исследования действия новых соединений на процессы памяти и 30 познавательные способности. Тест основывается на естественном выражении обонятельной памяти у крыс и их естественной склонности забывать и -36- предоставлять возможность оценивать запоминание путем распознавания молодого родственного животного взрослой крысой. Молодую крысу (21 день), выбранную случайно, помещали на 5 минут в клетку, в которой жила взрослая крыса. При помощи видеоустройства экспериментатор наблюдал за социальным 5 распознавательным поведением взрослой крысы и определял его общую продолжительность. После этого молодую крысу удаляли из клетки взрослой крысы и помещали в ее собственную клетку до второго введения. Взрослой крысе внутрибрюшинно вводили исследуемое соединение и через 2 часа снова вносили в контакт (5 минут) с молодой крысой. Затем снова наблюдали за 10 социальным распознавательным поведением и оценивали его продолжительность. Критерием оценки являлось разница (Т2-Т1), выраженная в секундах, между временем "распознавания" для двух встреч. Полученные результаты свидетельствуют о наличии разницы (Т2-Т1) в интервале от -19 с до -36 с для доз в интервале от 1 до 3 мг/кг ВБ, что указывает 15 на то, что соединения по изобретению значительно усиливают запоминание, даже в низких дозах. Так, соединения из примеров 1, 2 и 4 в дозе 3 мг/кг ВБ вызывают разницу (Т2-Т1) 36, 29 и 19 с соответственно. Пример Т : Распознавание предметов у крыс Wistar Исследование распознавания предметов у крыс Wistar (Behav. Brain Res., 1988, 20 3_i, 47-59) основывается на самопроизвольной исследовательской активности животного и характеризует эпизодическую память у людей. Это исследование памяти чувствительно к старению (Eur. J. Pharmacol., 1997, 325, 173-180), а также к нарушению функции холинергической системы (Pharm. Biochem. Behav. 1996, 53(2), 277-283) и оно основывается на различиях в исследовании 2 25 объектов достаточно сходного размера, один из которых является обычным, а другой - новым. Перед исследованием животные привыкали к окружающей среде (отделенной от объекта). В течение первого сеанса животных помещали (на 3 минуты) в отделенное пространство, в котором находилось 2 идентичных объекта. Измеряли продолжительность исследования каждого объекта. В течение 30 второго сеанса (3 минуты), который осуществляли через 24 часа, один из двух объектов заменяли новым. Измеряли продолжительность исследования каждого объекта. Критерием оценки являлась разница дельта, выраженная в секундах, -37- между временем исследования нового объекта и обычного (сходного) объекта в течение второго сеанса. Контрольные животные, которым предварительно перорально за 60 минут до каждого сеанса вводили носитель, исследуют обычный объект и новый объект одинаково, что свидетельствует о том, что 5 объект, представленный ранее, забывается. Животные, которым вводили соединение, улучшающее процессы памяти и познавательные способности, предпочтительно исследуют новый объект, что указывает на то, что объект, представленный ранее, запоминается. Полученные результаты свидетельствуют о наличие определенной разницы 10 дельта порядка 9 с для доз в интервале от 0,01 до 0,3 мг/кг ПО, что указывает на значительное усиление запоминания соединениями согласно изобретению, даже при введении в очень низких дозах. Так, соединения из примера 1 и 2 в дозе 0,3 мг/кг ПО вызывают разницу дельта 9 и 4 с соответственно. Пример J: Фармацевтические композиции для 1000 таблеток, 15 каждая из которых содержит 10 мг активного компонента Соединение из примера 1............. Гидроксипропилметилцеллюлоза Пшеничный крахмал.................... 15 г Лактоза 90 г Стеарат магния 2 г -38- НОВАЯ РЕДАКЦИЯ ФОРМУЛЫ ИЗОБРЕТЕНИЯ 1. Соединение, формулы (I): в которой : п представляет собой целое число от 1 до 6 включительно, Ri и R.2, которые могут быть одинаковыми или разными, каждый независимо друг от друга представляет собой атом водорода, линейную или разветвленную Ci-Сб-алкильную группу или арил-СрСб-алкильную группу, где алкильная часть может быть линейной или разветвленной, R.3 и R.4, которые могут быть одинаковыми или разными, каждый независимо друг от друга представляет собой атом водорода или линейную или разветвленную СрСб-алкильную группу, R.5 и R.6, которые могут быть одинаковыми или разными, каждый независимо друг от друга представляет собой атом водорода или линейный или разветвленный Ci-Сб-алкил, галоген, гидрокси, линейный или разветвленный Ci-Сб-алкокси, циано, нитро, линейный или разветвленный Сг-Сб-ацил, линейный или разветвленный Ci-Сб-алкоксикарбонил, линейную или разветвленную C\-CQ-тригалоалкильную или линейную или разветвленную СрСб-тригалоалкокси группу или аминогруппу, необязательно замещенную одной или двумя линейными или разветвленными СрСб-алкильными группами, R.7 представляет собой атом водорода, линейную или разветвленную Cj- Сб-алкильную группу или арил-С^Сб-алкильную группу, где алкильная часть может быть линейной или разветвленной, -39- где под арильной группой подразумевается фенильная, бифенильная, нафтильная, дигидронафтильная, тетрагидронафтильная, инданильная или инденильная группа, каждая из этих групп необязательно замещена одной или несколькими одинаковыми или разными группами, выбранными из атомов галогена, линейного или разветвленного Cj-Сб-алкила, гидрокси, циано, нитро, линейного или разветвленного СрСб-алкокси, линейного или разветвленного С2-С7-ацила, линейного или разветвленного Ci-Сб-алкоксикарбонила, линейной или разветвленной Ci-Сб-тригалоалкильной и линейной или разветвленной Cj-Сб-тригалоалкокси групп и аминогрупп, необязательно замещенных одной или двумя линейными или разветвленными СрСб-алкильными группами, их энантиомерами, диастереоизомерами, а также добавками солей, полученных с использованием фармацевтически приемлемых кислот или оснований. 2. Соединения формулы (I) в соответствии с п. 1, отличающиеся тем, что п представляет собой целое число, равное 1, их энантиомеры, диастереоизомеры, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием. 3. Соединения формулы (I) в соответствии с п. 1, отличающиеся тем, что R\ и R.2, которые могут быть одинаковыми или разными, каждый независимо друг от друга представляет собой атом водорода или линейную или разветвленную С\-Сб-алкильную группу, их энантиомеры, диастереоизомеры, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием. 4. Соединения формулы (I) в соответствии с п. 1, отличающиеся тем, что R3 и R4 представляют собой атом водорода, их энантиомеры, диастереоизомеры, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием. 5. Соединения формулы (I) в соответствии с п. 1, отличающиеся тем, что R5 и R6, которые могут быть одинаковыми или разными, каждый независимо друг от друга представляет собой атом водорода или линейную или разветвленную С]- -40- Сб-алкильную группу, их энантиомеры, диастереоизомеры, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием. 6. Соединения формулы (I) в соответствии с п. 1, отличающиеся тем, что R7 представляет собой атом водорода или линейную или разветвленную СрСб-алкильную группу, их энантиомеры, диастереоизомеры, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием. 7. Соединения формулы (I) в соответствии с п. 1, которые представляют собой: N-{[1 -(метил амино)цикл опропил] метил } пиридин-3 -амин N-uemn-N- {[ 1 -(метиламино)циклопропил ] метил} пиридин-3 -амин N-[(1 -аминоциклопропил)метил] пиридин-3 -амин N-{[1 -(диметиламино)цикл опропил] метил} пиридин-3 -амин N- [(1 -аминоциклопропил)метил] -М-метилпиридин-З -амин N- {[ 1 -(диметиламино)циклопропил] метил} -N-метилпиридин-З -амин 6-хлор-Л^-{[1-(метиламино)циклопропил]метил}пиридин-3-амин б-хлор-Л^-метил-Т/- {[ 1 -(метиламино)цикл опропил]метил} пиридин-3 -амин б-бром-iV- {[ 1 -(метиламино)циклопропил]метил} пиридин-3 -амин 6-бром-^У-метил-^/- {[ 1 -(метиламино)циклопропил]метил} пиридин-3 -амин 6-метил-ЛЦ[1-(метиламино)циклопропил]метил}пиридин-3-амин Л^,6-диметил-Л^-{[1-(метиламино)циклопропил]метил}пиридин-3-амин, их энантиомеры, диастереоизомеры, а также их соли присоединения с фармацевтически приемлемой кислотой или основанием. 8. Способ получения соединений формулы (I) в соответствии с п. 1, который характеризуется тем, что в качестве исходного вещества применяют соединение формулы (И): в которой R3, R4 и п имеют значения, указанные для формулы (I), соединения формулы (И) подвергают реакции с дифенилфосфорилазидом в присутствии триэтиленамина в толуоле, с последующим добавлением трет-бутанола, получая соединения формулы (III) : -41 - %.i N-COOtBu H в которой R3, R4 и п имеют значения, указанные выше, соединения формулы (III) подвергают реакции с соединением формулы (IV) : R'i - Hal (IV) в которой Hal представляет собой атом галогена и R'i представляет собой группу, выбранную из линейного или разветвленного Ci-Сб-алкила и арил-Ср Сб-алкила, где алкильная часть может быть линейной или разветвленной, в присутствии основания в безводном растворителе, получая соединения формулы (V): н3соое (V) в которой R3, R4, п и R'i имеют значения, указанные выше, соединения формул (III) и (V) составляют соединения формулы (VI): Х-1 y-cootBu в которой R3, R4 и п имеют значения, указанные выше, и Ri имеет значения, указанные для формулы (I), соединения формулы (VI) помещают в присутствие восстановителя, получая соединения формулы (VII): (VII) -42- в которой R.3, R.4, п и Ri имеют значения, указанные выше, соединения формулы (VII) подвергают воздействию окислителя, обычного для органического синтеза, получая соединения формулы (VIII): онс (VIII) Vi N-COOtBu в которой R3, R4, п и R\ имеют значения, указанные выше, соединения формулы (VIII) подвергают реакции с соединением формулы (IX): в которой R5 и Rg имеют значения, указанные для формулы (I), в присутствии триацетоксиборогидрида натрия, получая соединения формулы (X) : (X) N- COOtBu в которой R3, R4, R5, Rg, п и Rj имеют значения, указанные выше, соединения формулы (X): - или помещают в присутствие кислоты в диоксане, получая соединения формулы (I/а), частный случай соединений формулы (I): т> кг (1/а) в которой Ri, R3, R4, R5, R6 и п имеют значения, указанные выше, соединения формулы (1/а): -43 - - или помещают в присутствие водного раствора формальдегида и муравьиной кислоты, получая соединения формулы (I/b), частный случай соединений формулы (I): в которой Rj, R3, R4, R5, R6 и п имеют значения, указанные выше, - или подвергают реакции с соединением формулы (XI): R'2 - Hal (XI) в которой Hal имеет значения, указанные выше, и R'2 представляет собой группу, выбранную из линейного или разветвленного Ci-Сб-алкила и арил-Cj-Сб-алкила, где алкильная часть может быть линейной или разветвленной, в присутствии основания в безводном растворителе, получая соединения формулы (1/с), частный случай соединений формулы (I): в которой Ri, R3, R4, R5, Re, п и R'2 имеют значения, указанные выше, - или, если Ri представляет собой бензильную группу, помещают в присутствие соляной кислоты и палладия на угле, получая соединения формулы (I/d), частный случай соединений формулы (I): -44в которой R3, R4, R5, Rg и п имеют значения, указанные выше, соединения формулы (I/d) обрабатывают муравьиной кислотой и водным раствором формальдегида, получая соединения формулы (1/е), частный случай соединений формулы (I) : в которой R3, R4, R5, Re и п имеют значения, указанные выше, соединения формул (I/b), (I/c), (I/d) и (1/е) составляют соединения формулы (I/f), частный случай соединений формулы (I) : в которой Rj, R2, R3, R4> &5> Re и п имеют значения, указанные выше, соединения формулы (I/f) подвергают реакции с соединением формулы (XII): R'7 - СООН (XII), в которой R*7 представляет собой группу, выбранную из линейного или разветвленного Ci-Cs-алкила и арил-С^Сз-алкила, где алкильная часть может быть линейной или разветвленной, в присутствии связующего вещества, с последующим восстановлением амида, получая соединения формулы (I/g), частный случай соединений формулы (I) : -45 - в которой Ri, R.2, R-з, R4> Rs> Кб и п имеют значения, указанные выше, и R"7 представляет собой группу, выбранную из линейного или разветвленного Cj-Сб-алкила и арил-СуСб-алкила, где алкильная часть может быть линейной или разветвленной, - или помещают в присутствие карбонилдиимидазола и муравьиной кислоты в диметилформамиде, получая соединения формулы (XIII) : в которой Rj, R3, R4, R5, Re и п имеют значения, указанные выше, соединения формулы (XIII) подвергают реакции с боран-метилсульфидным комплексом в тетрагидрофуране, получая соединения формулы (XIV): в которой Ri, R3, R4, R5, R6 и п имеют значения, указанные выше, соединения формулы (XIV) подвергают таким же условиям реакции, как и для соединений формулы (X), воздействию кислоты в диоксане, получая соединения формулы (I/h), частный случай соединений формулы (I): N-COOtBu (ХШ) (XIV) в которой Ri, R3, R4, R5, R6 и п имеют значения, указанные выше, -46- соединения формулы (I/h), если Rj представляет собой бензильную группу, подвергают таким же условиям реакции, как и для соединений формулы (1/а), получая соединения формулы (I/i), частные случаи соединений формулы (I): 5 в которой R3, R4, R5, Re и п имеют значения, указанные выше, соединения формулы (I/i) подвергают таким же условиям реакции, как и для соединений формулы (I/d), получая соединения формулы (I/j), частные случаи соединений формулы (I): 10 в которой R3, R4, R5, R6 и п имеют значения, указанные выше, совокупность соединений формул (1/а) - (I/j), представляющее собой совокупность соединений по изобретению, очищают, если это является подходящим, в соответствии с обычными методиками очистки, могут быть разделены на их разные изомеры в соответствии с обычный методикой 15 разделения, и превращены, если это является подходящим, в их соли присоединения с фармацевтически приемлемой кислотой или основанием. 9. Фармацевтические композиции, которые содержат в качестве активного компонента по меньшей мере одно соединение в соответствии с любым из п.п. 17, отдельно или в сочетании с одним или несколькими фармацевтически 20 приемлемыми, инертными, нетоксичными наполнителями или носителями. (I/j) -47- 10. Фармацевтические композиции в соответствии с п. 9, которые содержат по меньшей мере один активный компонент в соответствии с любым из п.п. 1-7 для применения в качестве специфического никотинового лиганда а402 рецепторов. 11. Фармацевтические композиции в соответствии с п. 9, которые содержат по 5 меньшей мере один активный компонент в соответствии с любым из п.п. 1-7 для применения для лечения недостаточности памяти, связанной со старением головного мозга и с нейродегенеративными заболеваниями, а также для лечения нарушений настроения, синдрома Тоуретте, синдрома гиперактивности с недостаточностью внимания, абстинентного табачного синдрома и боли. Ю 12. Фармацевтические композиции в соответствии с п. 9, которые содержат по меньшей мере один активный компонент в соответствии с любым из п.п. 1-7 для применения для лечения недостаточности памяти, связанной с болезнью Альцгеймера, болезнью Паркинсона, болезнью Пика, корсаковским психозом или деменциями лобной доли и подкорковыми деменциями. ЕВРАЗИЙСКОЕ ПАТЕНТНОЕ ВЕДОМСТВО ЕАПВ/ОП-2 ОТЧЕТ О ПАТЕНТНОМ ПОИСКЕ (статья 15(3) ЕАПК и правило 42 Патентной инструкции к ЕАПК) Номер евразийской заявки: 200601247 Дата подачи: 27 июля 2006 (27.07.2006) | Дата испраш иваемого приоритета: 28 июля 2005 (28.07.2005) Название изобретения: Новые многократно замещенные соединения 1,1 -пиридилоксициклопропанамина, способ их получения и фармацевтические композиции, которые их содержат Заявитель: ЛЕ ЛАБОРАТУАР СЕРВЬЕ 1_| Некоторые пункты формулы не подлежат поиску (см. раздел I дополнительного листа) Единство изобретения не соблюдено (см. раздел II дополнительного листа) А. КЛАССИФИКАЦИЯ ПРЕДМЕТА ИЗОБРЕТЕНИЯ: C07D 213/74 (2006.01) А61К31/4406 (2006.01) А61Р 25/16 (2006.01) А61Р25/28 (2006.01) Согласно Международной патентной классификации (МПК) или национальной классификации и МПК Б. ОБЛАСТЬ ПОИСКА: Минимум просмотренной документации (система классификации и индексы МПК) C07D 213/74, C07D 213/65, А61К 31/44, А61К 31/4406, А61Р 25/00, А61Р 25/16, А61Р 25/28 Другая проверенная документация в той мере, в какой она включена в область поиска: В. ДОКУМЕНТЫ, СЧИТАЮЩИЕСЯ РЕЛЕВАНТНЫМИ Категория* Ссылки на документы с указанием, где это возможно, релевантных частей Относится к пункту № ЕР 1170281 Al (LES LABORATOIRES SERVIER) 09.01.2002, примеры 17-20, 23-24, 29,35, п.п. 1, 12, 15-17 формулы 1-12 GUANDALINIL. et al. "Synthesis and pharmacological evaluation of some (pyridyl)cyclopropylmethyl amines and their methiodides as nicotinic receptor ligands", FARMACO, 2002 Jun., 57 (6), p. 487-496 (реферат) [он-лайн] [ Найдено 2006-11-29] Найдено из базы данных PubMed, PMID: 12088064 1-12 RU 2095350 С1 (ЦЕНТР ПО ХИМИИ ЛЕКАРСТВЕННЫХ СРЕДСТВ (ЦХЛС-ВНИХФИ)) 10.11.1997, реферат 1-7 RU 2203889 С2 (НОВАРТИС АГ и др.) 10.05.2003, реферат 1-7 ^последующие документы указаны в продолжении графы В ^| данные о патентах-аналогах указаны в приложении * Особые категории ссылочных документов: "А" документ, определяющий общий уровень техники "Е" более ранний документ, но опубликованный на дату подачи евразийской заявки или после нее "О" документ, относящийся к устному раскрытию, экспонированию и т.д. "Р" документ, опубликованный до даты подачи евразийской заявки, но после даты испрашиваемого приоритета "D" документ, приведенный в евразийской заявке "Т" более поздний документ, опубликованный после даты приоритета и приведенный для понимания изобретения "X" документ, имеющий наиболее близкое отношение к предмету поиска, порочащий новизну или изобретательский уровень, взятый в отдельности "Y" документ, имеющий наиболее близкое отношение к предмету поиска, порочащий изобретательский уровень в сочетании с другими документами той же категории " &" документ, являющийся патентом-аналогом "L" документ, приведенный в других целях Дата действительного завершения патентного поиска: 30 ноября 2006 (30.11.2006) Наименование и адрес Международного поискового органа: Федеральный институт промышленной собственности РФ, 123995,Москва, Г-59, ГСП-5, Бережковская наб., 30-1. Факс: 243-3337, телетайп: 114818 ПОДАЧА Уполномоченное лицо: Т. Владимирова |