|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea000018637b*\id |
|
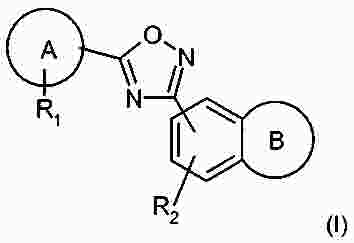
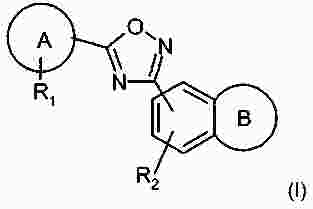
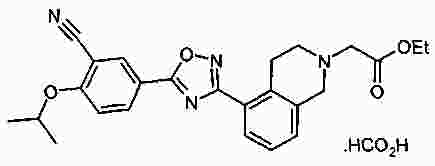
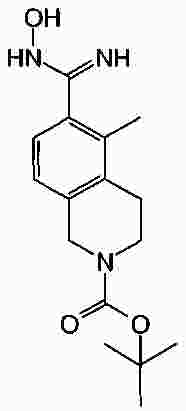
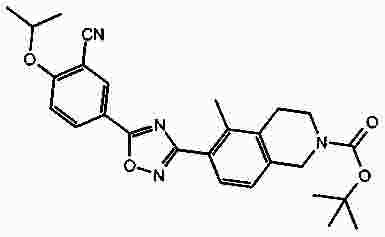
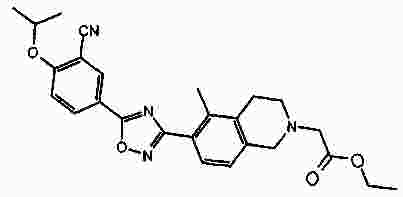
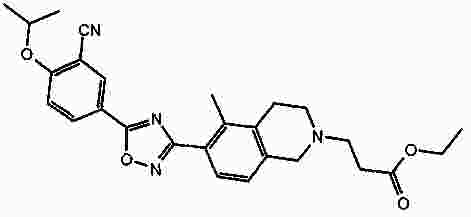
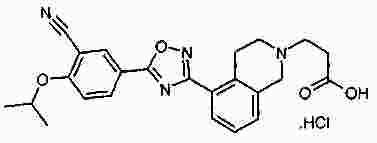
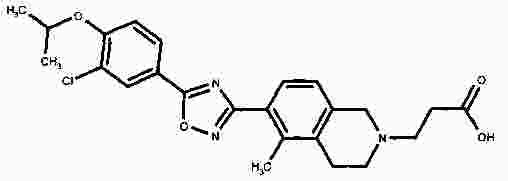
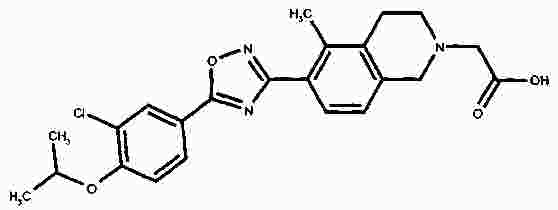
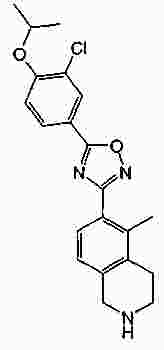
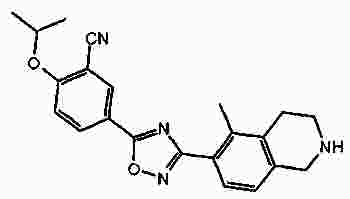
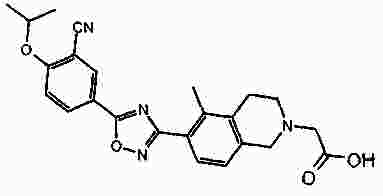
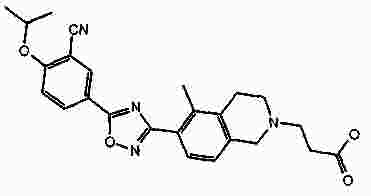
Термины запроса в документе Реферат В патенте раскрыты соединения на основе оксадиазола формулы (I) активные в отношении сфингозин-1-фосфата (S1P), которые могут быть использованы, в частности, для лечения рассеянного склероза. А обозначает фенил; В обозначает Формула [0001] Соединение формулы (I) или его фармацевтически приемлемая соль [0002] Соединение, выбранное из следующих соединений: [0003] Применение соединения по любому из пп.1-2 для лечения состояний или нарушений, опосредуемых рецептором S1P1, причем состояние или нарушение представляет собой рассеянный склероз. [0004] Применение соединения по любому из пп.1-2 для получения лекарственного средства для использования в лечении состояний или нарушений, опосредуемых рецептором S1P1, причем состояние или нарушение представляет собой рассеянный склероз. [0005] Способ лечения состояний или нарушений у млекопитающих, включая человека, которые могут быть опосредованы рецептором S1P1, который включает введение пациенту терапевтически безопасного и эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли, причем состояние представляет собой рассеянный склероз. Полный текст патента Изобретение относится к новым производным оксадиазола, имеющим фармакологическую активность, к способам их получения, к содержащим их фармацевтическим композициям и к их применению в лечении различных нарушений. Сфингозин-1-фосфат (S1P) представляет собой биоактивный липидный медиатор, образованный в результате фосфорилирования сфингозина сфингозинкиназами, и обнаруживается в высоких уровнях в крови. Он продуцируется и секретируется множеством типов клеток, включая клетки гематопоэтического происхождения, такие как тромбоциты и тучные клетки (Okamoto et al. 1998 J Biol Chem 273 (42): 27104; Sanchez and HIa 2004, J Cell Biochem 92:913). Он осуществляет разнообразные биологические действия, включая регуляцию пролиферации, дифференцировки, подвижности клеток, васкуляризации и активации воспалительных клеток и тромбоцитов (Pyne and Pyne 2 000, Biochem J. 349: 385). Были описаны пять подтипов SlP-чувствительных рецепторов: S1P1 (Edg-1), S1P2 (Edg-5), S1P3 (Edg-3), S1P4 (Edg-6) и S1P5 (Edg-8), образующих часть семейства рецепторов конъюгированного с G-белком гена дифференцировки эндотелия (Chun et al 2002 Pharmacological Reviews 54:265, Sanchez and HIa 2004 J Cellular Biochemistry, 92:913). Эти 5 рецепторов демонстрируют дифференциальную экспрессию мРНК, причем S1P1-3 широко экспрессируется, S1P4 экспрессируется на лимфоидных и гематопоэтических тканях и S1P5 - прежде всего в мозге и в меньшей степени в селезенке. Они сигнализируют через различные субпопуляции G-белков, промотируя различные биологические реакции (Kluk and HIa 2002 Biochem et Biophysica Acta 1582:72, Sanchez and HIa 2004, J Cellular Biochem 92:913). Предложенные роли для рецептора S1P1 включают трафик лимфоцитов, индукцию/супрессию цитокинов и эффекты в отношении эндотелиальных клеток (Rosen and Goetzl 2005 Nat Rev Immunol. 5:560). Агонисты рецептора S1P1 использовались во множестве аутоиммунных и трансплантационных моделей животных, включая модели MS Экспериментального Аутоиммунного Энцефаломелита (ЕАЕ), для уменьшения серьезности вызванного заболевания (Brinkman et al 2003 JBC 277:21453; Fujino et al 2003 J Pharmacol Exp Ther 305:70; Webb et al 2004 J Neuroimmunol 153:108; Rausch et al 2004 J Magn Reson Imaging 20:16). Эта активность, как сообщалось, опосредуется эффектом агонистов S1P1 на циркуляцию лимфоцитов через лимфатическую систему. Лечение агонистами S1P1 приводит к секвестрации лимфоцитов в пределах вторичных лимфоидных органов, таких как лимфатические узлы, вызывая обратимую периферическую лимфопению на моделях животных (Chiba et al 1998, J Immunology 160:5037, Forrest et al 2004 J Pharmacol Exp Ther 309:758; Sanna et al 2004 JBC 279:13839). Опубликованные данные относительно агонистов позволяют предположить, что лечение соединением вызывает исчезновение рецепторов S1P1 с поверхности клетки в результате интернализации (Graler and Goetzl 2004 FASEB J 18:551; Matloubian et al 2004 Nature 427:355; Jo et al 2005 Chem Biol 12:703), и именно это сокращение рецепторов S1P1 на иммунных клетках вносит свой вклад в сокращение движения Т-лимфоцитов из лимфатических узлов назад в кровоток. Делеция гена S1P1 вызывает эмбриональную летальность. Эксперименты по исследованию роли рецептора S1P1 в миграции и трафике лимфоцитов включали адоптивный перенос меченых S1P1-дефицитных Т-лимфоцитов в организм облученных мышей дикого типа. Эти клетки показали сниженный выход из вторичных лимфоидных органов (Matloubian et al 2004 Nature 427:355). S1P1 также отводили роль в модуляции слияния эндотелиальных клеток (Allende et al. 2003 102:3665, Blood Singelton et al 2005 FASEB J 19:1646). Относительно этого эндотелиального действия сообщалось, что агонисты S1P1 имели эффект на изолированные лимфатические узлы, что может вносить вклад в роль в коррекции иммунных нарушений. Агонисты S1P1 вызывают закрытие эндотелиальных стромальных "ворот" лимфатических пазух, которые дренируют лимфатические узлы и предотвращают выход лимфоцитов (Wei et al 2005, Nat. Immunology 6:1228). Было показано, что иммуносупрессивное соединение FTY720 (JP11080026-A) снижает количество циркулирующих лимфоцитов у животных и человека, имеет модулирующую заболевания активность при иммунных нарушениях на моделях животных и уменьшает скорость ремиссии в рецидивирующе-ремиттирующем рассеянном склерозе (Brinkman et al 2002 JBC 277:21453, Mandala et al 2002 Science 296:346, Fujino et al 2003 J Pharmacology and Experimental Therapeutics 305:45658, Brinkman et al. 2004 American J Transplantation 4:1019, Webb et al 2004 J Neuroimmunology 153:108, Morris et al 2005 EurJ Immunol 35:3570, Chiba 2005 Pharmacology and Therapeutics 108:308, Kahan et al 2003, Transplantation 76:1079, Kappos et al 2006 New Eng J Medicine 335:1124). Это соединение является пролекарством, которое фосфорилируется in vivo сфингозинкиназами с образованием молекулы, которая имеет агонистическую активность в отношении рецепторов S1P1, S1P3, S1P4 и S1P5. Клинические исследования показали, что лечение с использованием FTY720 приводит к брадикардии в первые 24 ч лечения (Kappos et al. 2006 New Eng J Medicine 335:1124). Ha основании ряда экспериментов на основе клеток и на животных считается, что брадикардия является следствием агонизма к рецептору S1P3. Эти эксперименты включают использование S1P3 нокаут-животных, которые, в отличие от мышей дикого типа, не демонстрируют брадикардию после введения FTY720 и использования S1P1-селективных соединений (Hale et al. 2004 Bioorganic & Medicinal Chemistry Letters 14:3501, Sanna et al 2004 JBC 279:13839, Koyrakh et al. 2005 American J Transplantation 5:529). Следовательно, существует потребность в соединениях-агонистах рецептора S1P1 с селективностью по S1P3, в отношении которых можно было бы ожидать сниженную тенденцию к индукции брадикардии. В следующих заявках на патент описаны производные оксадиазола как агонисты S1P1: WO 03/105771, WO 05/058848, WO 06/047195, WO 06/100633, WO 06/115188, WO 06/131336, WO 07/024922 и WO 07/1 16866. В следующих заявках на патент описаны производные тетрагидроизохинолинил-оксадиазола как агонисты рецептора S1P: WO 06/064757, WO 06/001463, WO 04/1 13330. В настоящее время был обнаружен структурно новый класс соединений, которые представляют собой агонисты рецептора S1P1. Изобретение поэтому относится к соединениям формулы (I) или к их фармацевтически приемлемым солям: А обозначает фенил; R 1 обозначает до двух заместителей, независимо выбранных из хлора, изопропокси и циано; R 2 обозначает водород или метил; В обозначает: R 3 обозначает водород, метил или (СН 2 ) 1-3 CO 2 H; R 4 обозначает водород. В одном варианте осуществления изобретения, А обозначает фенил; и/или R 1 обозначает до двух заместителей, независимо выбранных из хлора, изопропокси и циано; и/или R 2 обозначает водород; и/или В обозначает (а); и/или R 3 обозначает водород, метил или (СН 2 ) 1-3 CO 2 H; и/или R 4 обозначает водород. А обозначает фенил; R 1 обозначает до двух заместителей, независимо выбранных из хлора, изопропокси и циано; R 2 обозначает водород или метил; В обозначает (а); R 3 обозначает водород, метил или (СН 2 ) 1-3 СО 2 Н; R 4 обозначает водород. В одном варианте осуществления R 1 обозначает два заместителя, один из которых представляет собой изопропокси, а другой выбран из хлора или циано. В другом варианте осуществления R 1 обозначает два заместителя, выбранных из хлора, изопропокси и циано. В другом варианте осуществления R 1 обозначает хлор и изопропокси. В другом варианте осуществления R 1 обозначает хлор в положении 3 и изопропокси в положении 4, когда А обозначает фенил. В другом варианте осуществления R 1 обозначает изопропокси и циано. В другом варианте осуществления R 1 обозначает циано в положении 3 и изопропокси в положении 4, когда А обозначает фенил. В некоторых из соединений формулы (I), в зависимости от природы заместителя, имеются хиральные атомы углерода, и поэтому соединения формулы (I) могут существовать как стереоизомеры. Изобретение охватывает все оптические изомеры, такие как стереоизомерные формы соединений формулы (I), включая энантиомеры, диастереоизомеры и их смеси, такие как рацематы. Различные стереоизомерные формы могут быть разделены или отделены одна от другой обычными способами, или любой данный изомер может быть получен обычным стереоселективным или асимметричным синтезом. Некоторые из соединений, описанных здесь, могут существовать в различных таутомерных формах, и следует понимать, что изобретение охватывает все такие таутомерные формы. Следует понимать, что некоторые соединения по изобретению содержат как кислотные, так и основные группы и могут поэтому существовать как цвиттерионы при некоторых значениях рН. Подходящими соединениями по изобретению являются: 5-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1,2,3,4-тетрагидроизохинолин; 2-[(1-метилэтил)окси]-5-[3-(1,2,3,4-тетрагидро-5-изохинолинил)-1,2,4-оксадиазол-5-ил]бензонитрил; [5-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-3,4-дигидро-2(1Н)-изохинолинил]уксусная кислота; 3-[5-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-3,4-дигидро-2(1Н)-изохинолинил]пропановая кислота; 4-[5-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-3,4-дигидро-2(1Н)-изохинолинил]бутановая кислота; 2-[(1-метилэтил)окси]-5-[3-(2-метил-1,2,3,4-тетрагидро-5-изохинолинил)-1,2,4-оксадиазол-5-ил] бензонитрил; [8-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-3,4-дигидро-2Н-хромен-4-ил] уксусная кислота; 3-[6-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-3,4-дигидро-2(1Н)-изохинолинил]пропановая кислота; [6-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-3,4-дигидро-2(1Н)-изохинолинил]уксусная кислота; 6-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-1,2,3,4-тетрагидроизохинолин; 2-[(1-метилэтил)окси]-5-[3-(5-метил-1,2,3,4-тетрагидро-6-изохинолинил)-1,2,4-оксадиазол-5-ил]бензонитрил; [6-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-3,4-дигидро-2(1Н)-изохинолинил]уксусная кислота; 3-[6-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-3,4-дигидро-2(1Н)-изохинолинил]пропановая кислота или их фармацевтически приемлемые соли. Фармацевтически приемлемые производные соединений формулы (I) включают любую фармацевтически приемлемую соль, сложный эфир или соль такого сложного эфира соединения формулы (I), которая, при введении реципиенту, способна приводить (прямо или косвенно) к соединению формулы (I) или его активному метаболиту или остатку. Соединения формулы (I) могут образовывать соли. Следует понимать, что для использования в медицине соли соединений формулы (I) должны быть фармацевтически приемлемыми. Подходящие фармацевтически приемлемые соли являются очевидными для специалиста и включают описанные в J. Pharm. ScL, 1977, 66, 1-19, такие как соли присоединения с кислотой, образованные с неорганическими кислотами, например, хлористоводородную, бромистоводородную, серную, азотную или фосфорную кислоту; и органические кислоты, например, янтарную, малеиновую, уксусную, фумаровую, лимонную, винную, бензойную, п-толуолсульфоновую, метансульфоновую или нафталинсульфоновую кислоту. Некоторые из соединений формулы (I) могут образовывать соли присоединения с кислотой с одним или более эквивалентами кислоты. Настоящее изобретение включает все возможные стехиометрические и нестехиометрические формы. Соли могут также быть получены из фармацевтически приемлемых оснований, включая неорганические основания и органические основания. Соли, полученные из неорганических оснований, включают соли алюминия, аммония, кальция, меди, трехвалентного железа, двухвалентного железа, лития, магния, трехвалентного марганца, двухвалентного марганца, калия, натрия, цинка и т.п. Соли, полученные из фармацевтически приемлемых органических оснований, включают соли первичных, вторичных и третичных аминов; замещенные амины, включая природные замещенные амины; и циклические амины. Частные случаи фармацевтически приемлемых органических оснований включают аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этил-морфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трис(гидроксиметил)аминометан (TRIS, трометамол) и т.п. Соли можно также образовать из катионообменных смол, например, полиаминных смол. Когда соединение согласно настоящему изобретению является основным, соли могут быть получены из фармацевтически приемлемых кислот, включая неорганические и органические кислоты. Такие кислоты включают уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, этандисульфоновую, фумаровую, глюконовую, глутаминовую, бромисто-водородную, хлористо-водородную, изэтиновую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, муциновую, памовую, пантотеновую, фосфорную, пропионовую, янтарную, серную, винную, п-толуолсульфоновую кислоту и т.п. Фармацевтически приемлемые соли присоединения могут быть получены обычным образом реакцией с подходящей кислотой или производным кислоты. Фармацевтически приемлемые соли с основаниями могут быть получены обычным образом реакцией с подходящим неорганическим или органическим основанием. Соединения формулы (I) могут быть получены в кристаллической или некристаллической форме, и, в случае кристаллической формы, могут быть гидратированы или сольватированы. Это изобретение включает стехиометрические гидраты или сольваты, а также соединения, содержащие переменные количества воды и/или растворителя. В рамки изобретения входят все соли, сольваты, гидраты, комплексы, полиморфы, пролекарства, меченные радиоактивным изотопом производные, стереоизомеры и оптические изомеры соединений формулы (I). Потенциал и эффективность соединений по изобретению в отношении рецептора S1P1 могут быть определены тестом ГТФ γS, проводимом на клонированном рецепторе человека, как описано здесь. Соединения формулы (I) продемонстрировали агонистическую активность в отношении рецептора S1P1 при использовании функциональных тестов, описанных здесь. Соединения формулы (I) и их фармацевтически приемлемые соли могут поэтому быть использованы в лечении состояний или нарушений, опосредуемых рецептором S1P1. В частности, соединения формулы (I) и их фармацевтически приемлемые соли могут быть использованы в лечении рассеянного склероза. Следует понимать, что "лечение" в рамках изобретения включает профилактику, а также облегчение установленных симптомов. Таким образом, изобретение также относится к соединениям формулы (I) или к их фармацевтически приемлемым солям для применения в качестве терапевтических веществ, в частности, в лечении состояний или нарушений, опосредуемых рецептором S1P1. В частности, изобретение относится к соединению формулы (I) или к его фармацевтически приемлемой соли для применения в качестве терапевтического вещества в лечении рассеянного склероза. Изобретение также относится к способу лечения состояний или нарушений у млекопитающих, включая человека, которые могут быть опосредованы рецептором S1P1, который включает введение пациенту терапевтически безопасного и эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В частности, изобретение относится к способу лечения рассеянного склероза. Изобретение относится к способу лечения рассеянного склероза, который включает введение пациенту терапевтически безопасного и эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В другом аспекте изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли в получении лекарственного средства для использования в лечении состояний или нарушений, опосредуемых рецептором S1P1. В частности, изобретение относится к соединению формулы (I) или к его фармацевтически приемлемой соли для применения в получении лекарственного средства для использования в лечении рассеянного склероза. Все публикации, включая, но не ограничиваясь ими, патенты и заявки на патент, процитированные в этом описании, включены путем ссылки, как если бы каждая индивидуальная публикация была специально и индивидуально обозначена как включенная в настоящее описание путем ссылки. Следующие описания и примеры иллюстрируют получение соединений по изобретению. Аббревиатуры: г - граммы; мг - миллиграммы; мл - миллилитры; мкл - микролитры; MeCN - ацетонитрил; МеОН - метанол; EtOH - этанол; Et 2 O - простой диэтиловый эфир; EtOAc - этилацетат; DCM - дихлорметан; DIAD - диизопропил азодикарбоксилат; DIPEA - N,N-диизопропилэтиламин; DMAP - N,N-диметил-4-пиридинамин; DME - 1,2-бис(метилокси)этан; DMF - N,N-диметилформамид; DMSO - диметилсульфоксид; EDAC - N-(3-диметиламинопропил)-N'-этилкарбодиимид гидрохлорид; EDC - N-(3-диметиламинопропил)-N'-этилкарбодиимид гидрохлорид; EDCI - N-(3-диметиламинопропил)-N'-этилкарбодиимид гидрохлорид; HOBT/HOBt – гидроксибензотриазол; IPA - изопропиловый спирт; NCS - N-хлорсукцинимид; РуВОР - бензотриазол-1-ил-окситрипирролидинофосфоний гексафторфосфат; THF- тетрагидрофуран; dba - дибензилиден ацетон; RT- температура окружающей среды; °С - градусы Цельсия; М - моль; Н - протон; S - синглет; d - дублет; t - триплет; q - квартет; МГц - мегагерц; MeOD - меченый дейтерием метанол; LCMS - жидкостная хроматография с масс-спектрометрией; LC/MS - жидкостная хроматография с масс-спектрометрией; MS - масс-спектрометрия; ES - электрораспыление; МН + - масса ион+Н + ; MDAP - масс-направленная автоматизированная препаративная жидкостная хроматография; насыщ. - насыщенный; Boc - трет-бутилоксикарбонил; SCX или SCX-3 - экстракционная твердофазная (SPE) колонка с остатками бензолсульфоновой кислоты, иммобилизованными на твердой фазе (например, колонки IST Isolute ™). Способы, описанные ниже, приведены в иллюстративных целях, промежуточные соединения в описанных в примерах получениях, не обязательно были получены от конкретных описанных загрузок. Пример получения 1. 1,1-Диметилэтил-5-[(гидроксиамино)(имино)метил]-3,4-дигидро-2(1Н)-изохинолинкарбоксилат 1,2,3,4-тетрагидро-5-изохинолинкарбонитрил гидрохлорид (5,0 г/25,7 ммоль, от Fluorchem) разделяли между DCM (100 мл) и 2н. NaOH. Слой DCM собирали, высушивали (гидрофобная фритта) и упаривали. Свободное основание, 1,2,3,4-тетрагидро-5-изохинолинкарбонитрил, растворяли в сухом DCM (100 мл) и обрабатывали бис(1,1-диметилэтил)бикарбонатом (1,1 экв., 6,17 г). Реакционную смесь перемешивали при температуре окружающей среды под аргоном в течение 18 ч, промывали 2н. NaOH (100 мл), 2н. HCl (100 мл), высушивали (гидрофобная фритта) и упаривали, получая сырой 1,1-диметилэтил-5-циано-3,4-дигидро-2(1Н)-изохинолинкарбоксилат, который использовали без дальнейшей очистки (LCMS 100%, ЯМР смесь 2:1 с tBuOH). Изолированный выход 7,58 г. 1 Н ЯМР (400 МГц, CDCl 3 ) δ (среди прочего) 7,52(1Н, д), 7,35-7,16 (2Н, м), 4,60 (2Н, ушир.с), 3,71 (2Н, т), 3,04 (2Н, т), 1,5 (9Н, с); m/z (API-ES) 203 [М+Н-56] + . Сырой материал, полученный выше, гидроксиламин.HCl (14,31 г, 206 ммоль) и бикарбонат натрия (21,63 г, 257 ммоль) добавляли в круглодонную колбу на 500 мл, содержащую этанол (200 мл). Реакционную смесь нагревали при 65 °С в течение 24 ч. Охлажденную реакционную смесь упаривали и разделяли между DCM (2 ×100 мл) и водой (100 мл). Объединенные слои DCM собирали и высушивали. Анализ LC/MS показал, что материал имеет чистоту ~80%. Этот материал растворяли в этаноле (100 мл) и фильтровали, чтобы удалить нерастворенные примеси. Анализ LC/MS показал, что материал имеет чистоту ~92%. 1,1-диметилэтил-5-[(гидроксиамино)(имино)метил]-3,4-дигидро-2(1Н)-изохинолинкарбоксилат использовали без дальнейшей очистки. 1 Н ЯМР (400 МГц, CDCl 3 ) δ 7,82 (1Н, ушир.с), 7,36-7,16 (3H, м), 4,80 (2Н, ушир.с), 4,58 (2Н, ушир.с), 3,59 (2Н, ушир.т), 2,98 (2Н, т), 1,49 (9Н, с); m/z (API-ES) 292 [М+Н] + . Пример получения 2. 3-Хлор-4-[(1-метилэтил)окси]метилбензоат 3-Хлор-4-гидроксиметилбензоат (50 г, 0,27 моль), K 2 CO 3 (74 г, 0,54 моль) и йодопропан (29,5 мл, 0,23 моль) перемешивали при температуре окружающей среды в DMF (100 мл). Через 18 ч растворитель удаляли выпариванием под вакуумом, и остаток подвергали хроматографии на колонке с силикагелем 60 в смеси EtOAc/гексан (1:1), получая целевое соединение в форме масла (55 г, 90%). 1 Н ЯМР (400 МГц, CDCl 3 ) δ 8,05 (1Н, д), 7,89 (1Н, дд), 6,94 (1Н, д), 4,60-4,72 (1Н, м), 3,89 (3H, с), 1,41 (6Н, д); m/z (API-ES) 229 [М+Н] + . Пример получения 3. 1,1-Диметилэтил-5-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-3,4-дигидро-2(1Н)-изохинолинкарбоксилат 1,1-Диметилэтил-5-[(гидроксиамино)(имино)метил]-3,4-дигидро-2(1Н)-изохинолин карбоксилат (пример получения 1) (2 г, 6,86 ммоль) растворяли в тетрагидрофуране (THF)(100 мл) и перемешивали с гидридом натрия (60%-ая дисперсия, 0,302 г, 7,55 ммоль) под аргоном при температуре окружающей среды в течение 30 мин. Затем добавляли 3-хлор-4-[(1-метилэтил)окси]метилбензоат (пример получения 2) (2,355 г, 10,30 ммоль), и реакционную смесь нагревали при температуре кипения растворителя в течение 1,5 ч. Охлажденную реакционную смесь упаривали и разделяли между DCM (100 мл) и водой (100 мл). Водный слой промывали DCM (50 мл) и объединенные слои DCM высушивали (гидрофобная фритта) и упаривали. Сырой продукт очищали хроматографией через тонкий слой силикагеля, элюируя DCM, получая 1,1-диметилэтил-5-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-3,4-дигидро-2(1Н)-изохинолин карбоксилат (2,57 г, 5,47 ммоль, 80%-ый выход). 1 Н ЯМР (400 МГц, CDCl 3 ) δ 8,23 (1Н, с), 8,05 (1Н, д), 7,95 (1Н, д), 7,34 (1Н, т), 7,27 (1Н, д), 7,06 (1Н, д), 4,76-4,63 (1Н, м), 4,66 (2Н, ушир.с), 3,67 (2Н, ушир.т), 3,25 (2Н, ушир.т), 1,51 (9Н, с), 1,45 (6Н, д); m/z (API-ES) 414, 416 [М+Н-56] + . Пример получения 4. 1,1-Диметилэтил-5-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-3,4-дигидро-2(1Н)-изохинолинкарбоксилат К раствору 3-циано-4-[(1-метилэтил)окси]бензойной кислоты (может быть получена, как описано в WO2005/58848, 737 мг, 3,59 ммоль) и оксалилхлорида (360 мкл) в DCM (20 мл) добавляли DMF (20 мкл). Раствор перемешивали при температуре окружающей среды в течение 90 мин, затем концентрировали в вакууме и повторно растворяли в сухом DMF (6 мл). Отдельно 1,1-диметилэтил-5-[(гидроксиамино) (имино)метил]-3,4-дигидро-2(1Н)-изохинолинкарбоксилат (пример получения 1, 1,05 г, 3,60 ммоль), N,N-диметил-4-пиридинамин (DMAP) (20 мг) и N,N-диизопропилэтиламин (DIPEA) (1,31 мл, 7,50 ммоль) растворяли в DMF (10 мл). 5 мл первого раствора DMF добавляли ко второму раствору, и желтый раствор перемешивали при температуре окружающей среды в течение 1 ч и затем при 95 °С в течение 18 ч. Реакционную смесь концентрировали в вакууме, затем разделяли между DCM (50 мл) и насыщенным водным раствором NaHCO 3 (50 мл). Органические фракции промывали насыщенным водным раствором NaHCO 3 (50 мл), затем объединенные водные экстрагировали DCM (20 мл). Объединенные органические фракции концентрировали в вакууме, получая сырое коричневое масло. Флэш-хроматография (градиент МеОН [0,5-4%] в DCM) и концентрация в вакууме дала целевое соединение в форме твердого вещества светло-желтого цвета (512 мг, 37%). 1 Н ЯМР (400 МГц, CDCl 3 ) δ 8,42 (1Н, д), 8,34 (1Н, дд), 7,96 (1Н, д), 7,34 (1Н, т), 7,29 (1Н, д), 7,13 (1Н, д), 4,80 (1Н, септет), 4,67 (2Н, с), 3,68 (2Н, т), 3,24 (2Н, т), 1,51 (9Н, с), 1,48 (6Н, д); m/z (ES) 361 [М+Н-100] + . Пример получения 5. [5-(5-{3-Циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-3,4-дигидро-2(1Н)-изохинолинил]этилацетат соль муравьиной кислоты Суспензию 2-[(1-метилэтил)окси]-5-[3-(1,2,3,4-тетрагидро-5-изохинолинил)-1,2,4-оксадиазол-5-ил]бензонитрил (пример 3, 28,8 мг, 0,080 ммоль), этил бромацетат (35,5 мкл, 0,32 ммоль) и Cs 2 CO 3 (130 мг, 0,40 ммоль) в DMF (2 мл) перемешивали при температуре окружающей среды в течение 1 ч и затем нагревали при 60 °С в течение 5 мин в микроволновом реакторе. Суспензию добавляли в картридж SCX, и продукт элюировали метанольным раствором аммиаком. Концентрация в вакууме дала сырой продукт (43 мг), который был очищен MDAP, получая целевое соединение как непигментированная пленка (11,6 мг, 32%). 1 Н ЯМР (400 МГц, CDCl 3 ) δ 8,41 (1Н, д), 8,33 (1Н, дд), 8,09 (1Н, ушир.с), 8,01 (1Н, д), 7,34 (1Н, кажущийся т), 7,21 (1Н, д), 7,12 (1Н, д), 4,80 (1Н, септет), 4,24 (2Н, кв), 4,00 (2Н, с), 3,52 (2Н, с), 3,35 (2Н, т), 3,07 (2Н, т), 1,48 (6Н, д), 1,31 (3H, т); m/z (ES) 447 [М+Н] + . Пример получения 6. 3-[5-(5-{3-Циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-3,4-дигидро-2(1Н)-изохинолинил]метилпропаноат соль муравьиной кислоты Суспензию 2-[(1-метилэтил)окси]-5-[3-(1,2,3,4-тетрагидро-5-изохинолинил)-1,2,4-оксадиазол-5-ил] бензонитрила (пример 3, 28,8 мг, 0,080 ммоль), метил-3-бромпропионата (35 мкл, 0,32 ммоль) и Cs 2 CO 3 (130 мг, 0,40 ммоль) в DMF (2 мл) перемешивали при температуре окружающей среды в течение 1 ч и затем нагревали при 60 °С в течение 5 мин в микроволновом реакторе. Добавляли дополнительное количество метил-3-бромпропионата (35 мкл, 0,32 ммоль), и суспензию перемешивали при температуре окружающей среды в течение 3,5 ч и затем нагревали при увеличивающихся температурах в микроволновом реакторе, пока температура не достигла 150 °С за 30 мин. Суспензию фильтровали и концентрировали в вакууме, затем очищали MDAP, получая целевое соединение в форме желтой пленки (5,9 мг, 17%). 1 Н ЯМР (400 МГц, CDCl 3 ) δ 8,42(1Н, д), 8,33 (1Н, дд), 8,21 (1Н, ушир.с), 8,04 (1Н, д), 7,35 (1Н, т), 7,24 (1Н, д), 7,13 (1Н, д), 4,80 (1Н, септет), 3,99 (2Н, с), 3,72 (3H, с), 3,38 (2Н, т), 3,12-3,06 (4Н, м), 2,78 (2Н, т), 1,48 (6Н, д); m/z (ES) 447 [М+Н] + . Пример получения 7. 4-[5-(5-{3-Циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-3,4-дигидро-2(1Н)-изохинолинил]этилбутаноат соль муравьиной кислоты Суспензию 2-[(1-метилэтил)окси]-5-[3-(1,2,3,4-тетрагидро-5-изохинолинил)-1,2,4-оксадиазол-5-ил]бензонитрила (пример 3, 28,8 мг, 0,080 ммоль), этил-4-бромбутирата (45,8 мкл, 0,32 ммоль) и Cs 2 CO 3 (130 мг, 0,40 ммоль) в DMF (2 мл) перемешивали при температуре окружающей среды в течение 1 ч и затем нагревали при 60 °С в течение 5 мин в микроволновом реакторе. Добавляли дополнительное количество этил-4-бромбутирата (45,8 мкл, 0,32 ммоль), и суспензию перемешивали при температуре окружающей среды в течение 3,5 ч. Затем ее фильтровали, концентрировали в вакууме и очищали MDAP, получая целевое соединение в форме желтой пленки (8,0 мг, 21%). 1 Н ЯМР (400 МГц, CDCl 3 ) δ 8,42 (1Н, д), 8,34 (1Н, дд), 8,27 (ушир.с), 8,09 (1Н, д), 7,39 (1Н, т), 7,27 (1Н, д), 7,13 (1Н, д), 4,80 (1Н, септет), 4,19 (2Н, с), 4,13 (2Н, кв), 3,47 (2Н, т), 3,28 (2Н, т), 2,99 (2Н, т), 2,44 (2Н, т), 2,10 (2Н, квинтет), 1,48 (6Н, д), 1,25 (3H, т); m/z (ES) 475 [М+Н] + . Пример получения 8. 3-Хлор-4-[(1-метилэтил)окси]бензоилхлорид В круглодонную колбу загружали 3-хлор-4-[(1-метилэтил)окси]бензойную кислоту (от Paragos Product List, 10,2 г, 47,5 ммоль), дихлорметан (158 мл) и оксалилхлорид (8,29 мл, 95 ммоль). Реакционную смесь охлаждали до 0 °С в бане лед/вода, после чего добавляли N,N-диметилформамид (0,158 мл). Раствору давали нагреться до температуры окружающей среды в течение ночи. Растворитель упаривали, получая целевое соединение в форме твердого вещества кремового цвета (11,4 г). 1 Н ЯМР (Хлороформ-d) δ: 8,14 (д, 1Н), 8,00 (дд, 2,5 Гц, 1Н), 6,98 (д, 1Н), 4,73 (септет, 1Н), 1,44 (д, 6Н). Пример получения 9. 2-Метил-1-(метилокси)-3-[(Е)-2-нитроэтенил]бензол К раствору 2-метил-3-(метилокси)бензальдегида (от Allichem Product List; 20 г; 133 ммоль) в нитрометане (400 мл) добавляли ацетат аммония (6,16 г; 80 ммоль), и полученную оранжевую смесь перемешивали в течение 1 ч при 100 °С, затем охлаждали до температуры окружающей среды и концентрировали в вакууме. Остаток разделяли между этилацетатом ( ×2) и солевым раствором, и органические слои промывали солевым раствором, высушивали (MgSO 4 ) и концентрировали в вакууме. Растирание со смесью дихлорметан/простой эфир дало целевое соединение в форме твердого вещества желтого цвета (6,6 г). 1 Н ЯМР (CDCl 3 ) δ: 8,35 (д, 1Н), 7,48 (д, 1Н), 7,22 (т, 1Н), 7,11 (д, 1Н), 6,96 (д, 1Н), 3,86 (с, 3H), 2,33 (с, 3H). После упаривания маточных растворов дополнительное количество продукта (7,22 г) получали растиранием с простым диэтиловым эфиром. Маточные растворы снова упаривали, и остаток очищали хроматографией, элюируя этилацетатом (5-10%) в циклогексане, получая дополнительное количество продукта (3,45 г) после растирания с простым эфиром. Пример получения 10. {2-[2-метил-3-(метилокси)фенил]этил}амин К боргидриду лития (2М в THF; 2,0 мл; 4,0 ммоль) (мутный раствор) при температуре окружающей среды добавляли по каплям за 0,5 мин. хлортриметилсилан (1,02 мл; 8,00 ммоль). Образовывался осадок, и через приблизительно 3 мин 2-метил-1-(метилокси)-3-[(Е)-2-нитроэтенил]бензол (пример получения 9, 193 мг; 1,0 ммоль) в THF (4 мл) добавляли по каплям через шприц за 5 мин, удостоверяясь, что температура оставалась приблизительно 25 °С (использование ванны с холодной водой). Раствор перемешивали при температуре окружающей среды в течение ночи. Смесь охлаждали с помощью ванны со льдом, медленно добавляли метанол, и растворитель удаляли в вакууме. Остаток разделяли между 25% водным раствором гидроксида натрия и дихлорметаном. Водную фазу отделяли и экстрагировали три раза дихлорметаном, и объединенные органические слои упаривали. Очистка остатка твердофазной экстракцией (колонка SCX) с элюированием метанолом, затем 2н. аммиаком в метаноле, с последующем упариванием содержащей аммиак фракции дала целевое соединение (80 мг). 1 Н ЯМР (CDCl 3 ) δ: 7,10 (т, 1Н), 6,78 (д, 1Н), 6,73 (д, 1Н), 3,81 (с, 3H), 2,91 (т, 2Н), 2,77 (т, 2Н), 2,18 (с, 3H), 1,76 (ушир.с, 2Н). Пример получения 11. {2-[2-Метил-3-(метилокси)фенил]этил}формамид {2-[2-Метил-3-(метилокси)фенил]этил}амин (пример получения 10; 1,75 г; 10,6 ммоль) нагревали с обратным холодильником с этилформиатом (97%; 15 мл) в течение 15 ч. Удаление растворителя дало сырой продукт (приблизительно 2 г), который очищали хроматографией на силикагеле, элюируя градиентом 13-63% этилацетата в циклогексане, получая целевое соединение (1,57 г). MS m/z 194 [МН + ] Пример получения 12. 5-Метил-6-(метилокси)-3,4-дигидро-2(1Н)-изохинолинкарбальдегид К раствору {2-[2-метил-3-(метилокси)фенил]этил}формамида (пример получения 11; 1,93 г; 10,0 ммоль) в муравьиной кислоте (20 мл) добавляли параформальдегид (0,315 г; 10,5 ммоль), и полученную смесь нагревали с обратным холодильником (100 °С) в течение 20 мин. Смесь быстро охлаждали ледяной холодной водой до температуры окружающей среды и растворитель удаляли. Очистка хроматографией на силикагеле с элюированием градиентом метанола в дихлорметане дала целевое соединение в форме твердого вещества белого цвета (1,64 г), который использовали без дальнейшей очистки. MS m/z 206 [МН + ]. Пример получения 13. 5-Метил-1,2,3,4-тетрагидро-6-изохинолинол К раствору 5-метил-6-(метилокси)-3,4-дигидро-2(1Н)-изохинолинкарбальдегида (пример получения 12; 1,49 г; 7,26 ммоль) в дихлорметане (25 мл) при 0 °С под азотом медленно добавляли трибромид бора (9,09 г; 36,3 ммоль). После 85 мин при 0 °С медленно добавляли метанол (25 мл), и полученную смесь оставляли при температуре окружающей среды в течение трех дней. Удаление растворителя и растирание со смесью метанол/дихлорметан дало белый твердый остаток; маточные растворы концентрировали, и остаток загружали на экстракционный твердофазный картридж SCX (20 г) и элюировали метанолом, затем 2н. аммиаком в метаноле, элюируя продукт. После выпаривания растворителя целевое соединение (780 m мг) получали в форме твердого вещества светло-желтого цвета. 1 Н ЯМР (ДМСО-d 6 ) δ: 8,91 (ушир.с, 1Н), 6,62 (д, 1Н), 6,56 (д, 1Н), 3,70 (с, 2Н), 2,92 (т, 2Н), 2,48 (т, 2Н), 1,96 (с, 3H) Пример получения 14. 1,1-Диметилэтил-6-гидрокси-5-метил-3,4-дигидро-2(1Н)-изохинолинкарбоксилат К суспензии 5-метил-1,2,3,4-тетрагидро-6-изохинолинола (пример получения 13; 163 мг; 1,0 ммоль) в дихлорметане (5 мл) при 0 °С добавляли триэтиламин (0,21 мл; 1,50 ммоль), затем бис(1,1-диметилэтил)бикарбонат (240 мг; 1,10 ммоль). Растворитель упаривали, и остаток разделяли между этилацетатом и солевым раствором. Водный слой затем экстрагировали этилацетатом, и объединенные органические слои высушивали (MgSO 4 ) и упаривали, получая желтое масло. Очистка флэш-хроматографией с элюированием с градиентом 5-25% этилацетата в циклогексане дала целевое соединение в форме белой пены (232 мг). MS m/z 262 [М-Н]. Пример получения 15. 1,1-Диметилэтил-5-метил-6-{[(трифторметил)сульфонил]окси}-3,4-дигидро-2(1Н)-изохинолинкарбоксилат К раствору 1,1-диметилэтил-6-гидрокси-5-метил-3,4-дигидро-2(1Н)-изохинолинкарбоксилат (пример получения 14; 263 мг; 1,00 ммоль) в дихлорметане (5 мл) при -30 °С под азотом добавляли пиридин (0,162 мл; 2,00 ммоль) с последующим медленным добавлением трифторметансульфонового ангидрида (0,186 мл; 1,10 ммоль). После 0,5 ч при температуре от -30 до -20 °С растворитель удаляли, и остаток собирали этилацетатом. Раствор промывали 1н. соляной кислотой, затем насыщенным раствором бикарбоната натрия, высушивали (MgSO 4 ) и концентрировали в вакууме, получая целевое соединение в форме светло-желтого масла (368 мг). MS m/z 394 [М-Н] Пример получения 16. 1,1-Диметилэтил-6-циано-5-метил-3,4-дигидро-2(1Н)-изохинолинкарбоксилат Раствор 1,1-диметилэтил-5-метил-6-{[(трифторметил)сульфонил]окси}-3,4-дигидро-2(1Н)-изохинолинкарбоксилата (пример получения 15; 395 мг; 1,00 ммоль) в DMF (4 мл) дегазировали под высоким вакуумом при перемешивании в течение 15 минут, после чего добавляли цианид цинка (153 мг; 1,30 ммоль) и тетракис(трифенилфосфин)палладий (0) (116 мг; 0,10 ммоль) под азотом. Полученную темно-желтую смесь перемешивали при 100 °С в течение 6 ч. Растворитель удаляли, остаток растворяли в этилацетате, и раствор промывали насыщенным бикарбонатом натрия. Водный слой затем экстрагировали этилацетатом, и объединенные органические слои промывали солевым раствором, высушивали (MgSO 4 ) и упаривали, получая сырой продукт. Очистка флэш-хроматографией на силикагеле с элюированием с градиентом 5-25% этилацетата в циклогексане дала целевое соединение (214 мг) в форме твердого вещества белого цвета. 1 H ЯМР (CDCl 3 ) δ: 7,44 (д, 1Н), 7,04 (д, 1Н), 4,60 (с, 2Н), 3,69 (т, 2Н), 2,75 (т, 2Н), 2,46 (с, 3H), 1,49 (с, 9Н). Пример получения 17. 1,1-Диметилэтил-6-[(гидроксиамино)(имино)метил]-5-метил-3,4-дигидро-2(1Н)-изохинолинкарбоксилат 1, 1-диметилэтил-6-циано-5-метил-3,4-дигидро-2(1Н)-изохинолинкарбоксилат (пример получения 16; 404 мг; 1,48 ммоль) перемешивали в течение ночи при 80 °С с гидрохлоридом гидроксиламина (619 мг; 8,90 ммоль) и бикарбонатом натрия (748 мг; 8,9 ммоль) в этаноле (10 мл). Добавляли дополнительное количество бикарбоната натрия (400 мг) и гидрохлорида гидроксиламина (300 мг), и нагревание продолжали в течение приблизительно 8 ч. Смесь охлаждали до комнатной температуры, и полученное твердое вещество удаляли, промывали этанолом и высушивали под высоким вакуумом, получая целевое соединение в форме твердого вещества белого цвета (431 мг). 1 Н ЯМР (ДМСО-d 6 ) δ: 7,06 (д, 1Н), 6,99 (д, 1Н), 4,48 (ушир.с, 2Н), 3,58 (т, 2Н), 2,67 (т, 2Н), 2,19 (с, 3H), 1,42 (с, 9Н) (обмениваемые не указаны; не видны четко). Пример получения 18. 1,1-Диметилэтил-6-[{[({3-хлор-4-[(1-метилэтил)окси]фенил}карбонил)окси] амино}(имино)метил]-5-метил-3,4-дигидро-2(1Н)-изохинолинкарбоксилат К раствору 1,1-диметилэтил-6-[(гидроксиамино) (имино)метил]-5-метил-3,4-дигидро-2(1Н)-изохинолинкарбоксилата (пример получения 17; 435 мг; 1,42 ммоль) в дихлорметане (10 мл) добавляли триэтиламин (0,30 мл; 2,14 ммоль), затем 3-хлор-4-[(1-метилэтил)окси]бензоилхлорид (пример получения 8; 365 мг; 1,57 ммоль) в дихлорметане (5 мл). Через 30 мин реакционную смесь концентрировали, и остаток разделяли между этилацетатом и насыщенным бикарбонатом натрия. Водный слой затем экстрагировали этилацетатом, и объединенные органические слои промывали солевым раствором, высушивали (MgSO 4 ) и концентрировали в вакууме. Очистка флэш-хроматографией на силикагеле с элюированием с градиентом 10-50% этилацетата в циклогексане дала целевое соединение в форме белой пены (413 мг). MS m/z 500 [М-Н]. Пример получения 19. 1,1-Диметилэтил-6-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-3,4-дигидро-2(1Н)-изохинолинкарбоксилат Раствор 1,1-диметилэтил-6-[{[({3-хлор-4-[(1-метилэтил)окси]фенил}карбонил)окси]амино}(имино) метил]-5-метил-3,4-дигидро-2(1Н)-изохинолинкарбоксилата (Пример получения 18; 413 мг/ 0,823 ммоль) в 1,4-диоксане (15 мл) перемешивали при 110 °С в течение приблизительно 18 ч. Смесь охлаждали до температуры окружающей среды и растворитель удаляли. Очистка флэш-хроматографией на силикагеле с элюированием 5-25% этилацетата в циклогексане, затем 13-63% этилацетатом в циклогексане, дала целевое соединение в форме белой пены (220 мг). 1 Н ЯМР (Хлороформ-d) δ: 8,24 (д, 1Н), 8,05 (дд, 1Н), 7,76 (д, 1Н), 7,10 (д, 1Н), 7,06 (д, 1Н), 4,72 (септет, 1Н), 4,63 (с, 2Н), 3,71 (т, 2Н), 2,84 (т, 2Н), 2,52 (с, 3H), 1,51 (с, 9Н), 1,45 (д, 6Н). Пример получения 20. 3-[6-(5-{3-Хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-3,4-дигидро-2(1Н)-изохинолинил]этилпропаноат К суспензии 6-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-1,2,3,4-тетрагидроизохинолина трифторацетата (пример 10; 90 мг; 0,18 ммоль) в ацетонитриле (3 мл) добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU; 0,16 мл; 1,09 ммоль). К раствору добавляли этилакрилат (0,03 мл; 0,27 ммоль), и полученную смесь перемешивали при 70 °С в течение 0,5 ч. Смесь охлаждали до температуры окружающей среды и растворитель удаляли. Остаток разделяли между этилацетатом и солевым раствором, и водный слой затем экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором, высушивали (MgSO 4 ) и концентрировали в вакууме, получая целевое соединение (90 мг). MS m/z 484 [МН + ]. Пример получения 21. [6-(5-{3-Хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-3,4-дигидро-2(1Н)-изохинолинил]этилацетат К суспензии 6-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-1,2,3,4-тетрагидроизохинолин трифторацетата (пример 10; 90 мг; 0,18 ммоль) в DMF (3 мл) под азотом при температуре окружающей среды добавляли карбонат цезия (177 мг; 0,54 ммоль), затем этилбромацетат (0,026 мл; 0,23 ммоль). Через 35 мин смесь разбавляли этилацетатом, и осадок удаляли. Раствор промывали водой, и водный слой затем экстрагировали этилацетатом. Органический слой промывали солевым раствором, высушивали над MgSO 4 и концентрировали в вакууме, получая целевое соединение (75 мг). MS m/z 470 [МН + ] Пример получения 22. 1,1-Диметилэтил-6-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-3,4-дигидро-2 (1Н)-изохинолинкарбоксилат Оксалилхлорид (571 мг, 4,5 ммоль) добавляли к раствору 3-циано-4-[(1-метилэтил)окси]бензойной кислоты (от АК Scientific Product Catalog; 800 мг, 3,90 ммоль) в дихлорметане (15 мл) с последующим добавлением DMF (каталитическое количество). Реакционную смесь перемешивали в течение 45 мин, добавляли дополнительное количество оксалилхлорида (0,1 мл) и DMF (1 капля), и перемешивание продолжали в течение 20 мин. Растворитель упаривали, остаток упаривали совместно с толуолом, затем высушивали под высоким вакуумом в течение 30 мин. Сырой хлорангидрид кислоты растворяли в ацетонитриле (10 мл) и добавляли к суспензии 1,1-диметилэтил 6-[(гидроксиамино)(имино)метил]-5-метил-3,4-дигидро-2(1Н)-изохинолинкарбоксилата (пример получения 17; 916 мг, 3.00 ммоль) и триэтиламина (607 мг, 6,00 ммоль) в ацетонитриле (15 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 40 мин, затем нагревали с обратным холодильником в течение 24 ч. Реакционную смесь охлаждали, и растворитель упаривали. Остаток разделяли между этилацетатом и насыщенным раствором бикарбоната натрия. Органическую фазу отделяли, промывали солевым раствором, высушивали (MgSO 4 ) и упаривали, получая сырой продукт (1,3 г). Остаток очищали хроматографией, элюируя с 8-38% этилацетата в циклогексане, получая целевое соединение (486 мг). 1 Н ЯМР (Хлороформ-d) δ: 8,42 (д, 1Н), 8,33 (дд, 1Н), 7,76 (д, 1Н), 7,12 (д, 1Н), 7,1 1 (д, 1Н), 4,80 (септет, 1Н), 4,64 (с, 2Н), 3,72 (т, 2Н), 2,84 (т, 2Н), 2,52 (с, 3H), 1,51 (с, 9Н), 1,48 (д, 6Н). Пример получения 23. [6-(5-{3-Циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-3,4-дигидро-2(1Н)-изохинолинил]этилацетат Смесь соли трифторуксусной кислоты с 2-[(1-метилэтил)окси]-5-[3-(5-метил-1,2,3,4-тетрагидро-6-изохинолинил)-1,2,4-оксадиазол-5-ил]бензонитрилом (пример 11; 100 мг, 0,20 ммоль), этилбромацетата (44 мг, 30 мкл, 0,26 ммоль) и карбоната цезия (200 мг, 0,61 ммоль) в сухом DMF (3 мл) перемешивали при температуре окружающей среды в течение 1 ч. Реакционную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (3 ×5 мл). Объединенные экстракты высушивали и упаривали. Очистка хроматографией на силикагеле с элюированием с 10%-ым этилацетатом в циклогексане дала целевое соединение в форме бесцветного масла (85 мг). MS m/z 461 [МН] + . Пример получения 24. 3-[6-(5-{3-Циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-3,4-дигидро-2(1Н)-изохинолинил]этилпропаноат Смесь соли трифторуксусной кислоты с 2-[(1-метилэтил)окси]-5-[3-(5-метил-1,2,3,4-тетрагидро-6-изохинолинил)-1,2,4-оксадиазол-5-ил]бензонитрилом (пример 11; 100 мг, 0,20 ммоль), этилакрилата (31 мг, 33 мкл, 0,31 ммоль) и 1,8-диазабицикло[5.4.0]ундец-7-ена (DBU; 187 мг, 185 мкл, 1,23 ммоль) в ацетонитриле (3 мл) перемешивали при температуре окружающей среды в течение 3 ч. Растворитель упаривали, и остаток растворяли в этилацетате (10 мл); раствор промывали водой (5 мл), высушивали и упаривали. Очистка остатка хроматографией на силикагеле с элюированием с 10%-ым этилацетатом в циклогексане дала целевое соединение в форме бесцветного масла (86 мг). MS m/z 475 [МН] + . Пример 1. 5-(5-{3-Хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1,2,3,4-тетрагидроизохинолин гидрохлорид 1,1-диметилэтил-5-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-3,4-дигидро-2(1Н)-изохинолинкарбоксилат (пример получения 3) (2,57 г, 5,47 ммоль) перемешивали в 4М HCl в 1,4-диоксане (100 мл). Через 1 ч реакционная смесь стала мутной и перемешивание продолжали в течение еще 16 ч. Упаривание дало 5-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-1,2,3,4-тетрагидроизохинолин гидрохлорид (2.23 г, 5,49 ммоль, 100%-ый выход). 1 Н ЯМР (400 МГц, d 6 -ДМСО) δ 9,43 (2Н, с), 8,19 (1Н, с), 8,11 (1Н, дд), 8,03-8,00 (1Н, м), 7,51-7,45 (3H, м), 4,89 (1Н, септет), 4,38 (2Н, с), 3,43 (2Н, т), 3,35-3,32 (2Н, м), 1,37 (6Н, д); m/z (API-ES) 370, 372 [М+Н] + . Пример 2. 2-[(1-Метилэтил)окси]-5-[3-(1,2,3,4-тетрагидро-5-изохинолинил)-1,2,4-оксадиазол-5-ил]бензонитрил гидрохлорид Смесь 1,1-диметилэтил-5-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-3,4-дигидро-2(1Н)-изохинолинкарбоксилата (пример получения 4; 53,7 мг, 0,117 ммоль) и 4М HCl в диоксане (2 мл) перемешивали при температуре окружающей среды в течение 64 ч. Смесь концентрировали в вакууме, остаток растирали с Et 2 O и высушивали в вакууме, получая твердое вещество светло-коричневого цвета (35,1 мг, 76%). 1 Н ЯМР (400 МГц, d 6 -ДМСО) δ 9,52 (2Н, ушир.с), 8,52 (1Н, д), 8,40 (1Н, дд), 8,02 (1Н, дд), 7,57, (1Н, д), 7,51-7,48 (2Н, м), 4,99 (1Н, септет), 4,38 (2Н, с), 3,39-3,34 (4Н, м), 1,39 (6Н, д); m/z (ES) 361 [М+Н] + . Пример 3. 2-[(1-Метилэтил)окси]-5-[3-(1,2,3,4-тетрагидро-5-изохинолинил)-1,2,4-оксадиазол-5-ил]бензонитрил Смесь 1,1-диметилэтил-5-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-3,4-дигидро-2(1Н)-изохинолинкарбоксилата (пример получения 4, 512 мг, 1,11 ммоль) и 4М HCl в диоксане (20 мл) перемешивали при температуре окружающей среды в течение 18 ч. Смесь концентрировали в вакууме, получая твердое вещество светло-желтого цвета, которое повторно растворяли в МеОН. Этот раствор загружали в картридж SCX-3 (10 г), и продукт элюировали 1%-ым NH 3 в МеОН. Концентрация дала желтое масло, которое затвердевало при отстаивании в течение ночи (366 мг, 91%). 1 Н ЯМР (400 МГц, d 6 -ДМСО) δ 8,38 (1Н, д), 8,32 (1Н, дд), 7,94 (1Н, д), 7,32-7,17 (2Н, м), 7,13 (1Н, м), 4,80 (1Н, септет), 4,10 (2Н, с), 3,20-3,14 (4Н, м), 1,48 (6Н, д); m/z (ES) 361 [М+Н] + . Пример 4. Гидрохлорид [5-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-3,4-дигидро-2(1Н)-изохинолинил]уксусной кислоты Раствор соли муравьиной кислоты с [5-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-3,4-дигидро-2(1Н)-изохинолинил]этилацетатом (пример получения 5, 11,6 мг, 0,026 ммоль) и LiOH (0,3 мг) в смеси THF-MeOH-вода (300 мкл, 300 мкл, 200 мкл) нагревали при 100 °С в течение 3 мин в микроволновом реакторе. Добавляли 2М водный раствор HCl (1 мл), затем раствор экстрагировали DCM (2 ×2 мл). Объединенные органические фазы концентрировали в вакууме, получая целевое соединение в форме твердого вещества белого цвета (10,1 мг, 85%). 1 Н ЯМР (400 МГц, d 4 -МеОН) δ 8,48 (1Н, д), 8,44 (1Н, дд), 8,21 (1Н, д), 7,44 (1Н, т), 7,47 (2Н, кажущийся д), 4,97 (1Н, септет), 4,70 (2Н, с), 4,32 (2Н, с), 3,67 (2Н, т), 3,57 (2Н, т), 1,47 (6Н, д); m/z (ES) 419 [М+Н] + . Пример 5. Гидрохлорид 3-[5-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-3,4-дигидро-2(1Н)-изохинолинил]пропионовой кислоты Раствор соли муравьиной кислоты с 3-[5-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-3,4-дигидро-2(1Н)-изохинолинил]метилпропаноатом (пример получения 6, 5,9 мг, 0,0132 ммоль) и LiOH (0,15 мг) в смеси THF-MeOH-вода (300 мкл, 300 мкл, 100 мкл) нагревали при 100 °С в течение 3 мин в микроволновом реакторе. Добавляли последовательно 2М водный раствор HCl (1 мл) и DCM (2 мл), в результате чего образовывался осадок. Фильтрация дала целевое соединение в форме твердого вещества белого цвета (6,5 мг, колич.). 1 Н ЯМР (400 МГц, d 6 -ДМСО) δ 12,71 (1Н, ушир.с), 11,41 (1Н, ушир.с), 8,53 (1Н, д), 8,41 (1Н, дд), 8,05 (1Н, д), 7,57 (1Н, д), 7,53 (1Н, т), 7,46 (1Н, д), 4,99 (1Н, септет), 4,53 (2Н, ушир.с), 3,46-3,43 (4Н, м), 3,65 (2Н, ушир.с), 2,98 (2Н, т), 1,39 (6Н, d); m/z (ES) 433 [М+Н] + . Пример 6. Гидрохлорид 4-[5-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-3,4-дигидро-2(1Н)-изохинолинил]бутановой кислоты Целевое соединение получали подобно примеру 4, заменяя [5-(5-{3-циано-4-[(1-метилэтил)окси] фенил}-1,2,4-оксадиазол-3-ил)-3,4-дигидро-2(1Н)-изохинолинил]этилацетат солью муравьиной кислоты с 4-[5-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-3,4-дигидро-2(1Н)-изохинолинил]этилбутаноатом (пример получения 7, 8,0 мг). Целевое соединение получали в форме твердого вещества белого цвета (10,1 мг). 1 Н ЯМР (400 МГц, d 4 -МеОН) δ 8,36 (1Н, д), 8,33 (1Н, дд), 8,23 (1Н, д), 7,43 (1Н, т), 7,37 (1Н, д), 7,35 (1Н, д), 4,85 (1Н, септет), 4,52 (2Н, ушир.с), 3,63-3,54 (4Н, ушир.м), 3,30 (2Н, кажущийся дд), 2,43 (2Н, т), 2,06 (2Н, квинтет), 1,36 (6Н, д); m/z (ES) 447 [М+Н] + . Пример 7. 2-[(1-метилэтил)окси]-5-[3-(2-метил-1,2,3,4-тетрагидро-5-изохинолинил)-1,2,4-оксадиазол-5-ил]бензонитрил К раствору 2-[(1-метилэтил)окси]-5-[3-(1,2,3,4-тетрагидро-5-изохинолинил)-1,2,4-оксадиазол-5-ил]бензонитрила (пример 3, 25,0 мг, 0,069 ммоль) и формальдегида (37% вес./об. в Н 2 О, 8,0 мкл, 0,10 ммоль) в DCM (5 мл) добавляли NaBH(OAc) 3 (29 мг, 0,14 ммоль), и полученный раствор перемешивали при температуре окружающей среды в течение 20 мин. Добавляли солевой раствор (5 мл), и водный слой экстрагировали DCM (5 мл). Объединенные органические фазы концентрировали в вакууме, получая твердое вещество белого цвета. Это твердое вещество повторно растворяли в смеси ДМСО-МеОН, добавляли в картридж SCX и промывали МеОН. Продукт элюировали NH 3 в МеОН; концентрация в вакууме дала твердое вещество белого цвета (20,1 мг, 78%). 1 Н ЯМР (400 МГц, CDCl 3 ) δ 8,41 (1Н, д), 8,32 (1Н, дд), 7,98 (1Н, д), 7,29 (1Н, т), 7,20 (1Н, д), 7,12 (1Н, д), 4,79 (1Н, септет), 3,67 (2Н, с), 3,30 (2Н, т), 2,75 (2Н, т), 2,49 (3H, с), 1,47 (6Н, д). Пример 8. 3-[6-(5-{3-Хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-3,4-дигидро-2(1Н)-изохинолинил]пропановая кислота К раствору 3-[6-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-3,4-дигидро-2(1Н)-изохинолинил]этилпропаноата (пример получения 20; 90 мг; 0,186 ммоль) в этаноле (2 мл) и THF (2 мл) при температуре окружающей среды добавляли 2н. гидроксид натрия (0,186 мл; 0,372 ммоль), и полученную смесь перемешивали при температуре окружающей среды приблизительно в течение 2,5 ч. Удаление большей части растворителей, разбавление приблизительно 1 мл воды с последующим добавлением уксусной кислоты {приблизительно 300 мкл) дало осадок. Смесь экстрагировали дихлорметаном, получая, после удаления растворителя и растирания с простым диэтиловым эфиром, целевое соединение в форме твердого вещества белого цвета (40 мг). 1 Н ЯМР (ДМСО-d 6 ) δ: 8,15 (д, 1Н), 8,09 (дд, 1Н), 7,64 (д, 1Н), 7,43 (д, 1Н), 7,09 (д, 1Н), 4,88 (септет, 1Н), 3,64 (с, 2Н), 2,76 (ушир.с, 4Н), 2,72 (т, 2Н), 2,44 (т, 2Н), 2,41 (с, 3H), 1,36 (д, 6Н). MS m/z 456 [МН + ]. Пример 9. [6-(5-{3-Хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-3,4-дигидро-2(1Н)-изохинолинил]уксусная кислота К раствору [6-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-3,4-дигидро-2(1Н)-изохинолинил]этилацетата (пример получения 21; 75 мг; 0,16 ммоль) в этаноле (2 мл) и THF (2 мл) при температуре окружающей среды добавляли 2н. гидроксид натрия, и полученную смесь перемешивали при температуре окружающей среды в течение 40 мин. Растворители удаляли, и остаток растворяли в воде (приблизительно 1 мл). Добавляли уксусную кислоту (1 мл), все растворители удаляли, и остаток упаривали совместно с толуолом, получая твердое вещество светло-желтого цвета. Это твердое вещество растирали с простым диэтиловым эфиром, получая целевое соединение, которое отфильтровывали в форме твердого вещества светло-желтого цвета (50 мг). MS m/z 442 [МН + ]. 1 Н ЯМР (ДМСО-d 6 ) δ: 8,16 (ушир.с, 1Н), 8,10 (д, 1Н), 7,62 (д, 1Н), 7,44 (д, 1Н), 7,05 (д, 1Н), 4,88 (септет, 1Н), 3,70 (ушир.с, 2Н), 2,91 (ушир.с, 2Н), 2,77 (ушир.с, 4Н), 2,42 (ушир.с, 3H), 1,36 (д, 6Н). Пример 10. 6-(5-{3-Хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-1,2,3,4-тетрагидроизохинолин трифторацетат К раствору 1,1-диметилэтил-6-(5-{3-хлор-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-3,4-дигидро-2(1Н)-изохинолинкарбоксилата (пример получения 19; 210 мг; 0.434 ммоль) в дихлорметане (3 мл) при 0 °С добавляли по каплям трифторуксусную кислоту (3 мл). Полученную смесь перемешивали при 0 °С в течение 1 ч, получая светло-желтый раствор. Удаление растворителя и выпаривание совместно с толуолом дало твердое вещество белого цвета, которое растирали с простым диэтиловым эфиром, получая целевое соединение в форме твердого вещества белого цвета (206 мг). 1 Н ЯМР (ДМСО-d 6 ) δ: 9,08 (ушир.с, 2Н), 8,17 (д, 1Н), 8,10 (дд, 1Н), 7,78 (д, 1Н), 7,45 (д, 1Н), 7,28 (д, 1Н), 4,84-4,94 (м, 1Н), 4,38 (ушир.с, 2Н), 3,45-3,52 (м, 2Н), 2,98 (т, 2Н), 2,46 (с, 3H), 1,36 (д, 6Н). MS m/z 384 [МН + ]. Пример 11. Соль трифторуксусной кислоты и 2-[(1-метилэтил)окси]-5-[3-(5-метил-1,2,3,4-тетрагидро-6-изохинолинил)-1,2,4-оксадиазол-5-ил]бензонитрила Трифторуксусную кислоту (3 мл) добавляли к охлажденному льдом раствору 1,1-диметилэтил-6-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-3,4-дигидро-2(1Н)-изохинолинкарбоксилата (Пример получения 22; 486 мг, 1,02 ммоль) в дихлорметане (3 мл). Реакционную смесь перемешивали при 0 °С в течение 30 мин. Растворитель упаривали, и остаток совместно упаривали из толуола ( ×2). Растирание остатка с простым диэтиловым эфиром дало целевое соединение в форме бесцветного твердого вещества, которое отфильтровывали и высушивали (485 мг). 1 Н ЯМР (400 МГц, CDCl 3 ) δ: 1,48 (6Н, д), 2,54 (3H, с), 3,09 (2Н, м), 3,5 (2Н, затененный остаточным растворителем), 4,36 (2Н, с), 4,80 (1Н, м), 7,08-7,15 (2Н, м), 7,85 (1Н, д), 8,33 (1Н, д), 8,42 (1Н, с), 10,20 (2Н, ушир.с). MS m/z 375 [МН] + . Пример 12. [6-(5-{3-Циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-3,4-дигидро-2(1Н)-изохинолинил]уксусная кислота 2М гидроксида натрия (2 мл) добавляли к суспензии [6-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-3,4-дигидро-2(1Н)-изохинолинил]этилацетата (пример получения 23; 80 мг, 0,17 ммоль) в этаноле (2 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч. Растворитель упаривали, и остаток разбавляли водой (5 мл). Раствор подкисляли уксусной кислотой и экстрагировали этилацетатом (3 ×5 мл). Водную фазу упаривали, и остаток очищали MDAP, получая целевое соединение в форме бесцветного твердого вещества (7 мг). 1 Н ЯМР (400 МГц, d 4 MeOH) δ: 1,46 (6Н, д), 2,55 (3H, с), 3,19 (2Н, т), 3,35 (2Н, с), 3,67 (2Н, т), 3,78 (2Н, с), 4,53 (2Н, с), 4,95 (1Н, м) (частично затененный водным пиком), 7,20 (1Н, д), 7,54 (1Н, д), 7,85 (1Н, d), 8,50-8,60 (2Н, м). MS m/z 433 [МН] + . Пример 13. 3-[6-(5-{3-Циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-3,4-дигидро-2(1Н)-изохинолинил]пропановой кислоты натриевая соль 2М гидроксида натрия (2 мл) добавляли к раствору 3-[6-(5-{3-циано-4-[(1-метилэтил)окси]фенил}-1,2,4-оксадиазол-3-ил)-5-метил-3,4-дигидро-2(1Н)-изохинолинил]этилпропаноата (пример получения 24; 80 мг, 0,17 ммоль) в этаноле (2 мл) при 60 °С. Реакционную смесь перемешивали при 60 °С в течение 2 часов, охлаждали до температуры окружающей среды и разбавляли водой (2 мл). Твердое вещество отфильтровывали, промывали небольшим количеством воды и высушивали, получая целевое соединение в форме бесцветного твердого вещества (55 мг). 1 Н ЯМР (400 МГц, d 6 ДМСО) δ: 1,39 (6Н, д), 2,08 (2Н, т), 2,44 (3H, с), 2,59-2,78 (6Н, м), 3,56 (2Н, с), 4,98 (1Н, м), 7,09 (1Н, д), 7,55 (1Н, д), 7,65 (1Н, д), 8,40 (1Н, дд), 8,50 (1Н, с). MS m/z 447 [МН] + . Для мембранных препаратов все стадии осуществляли при 4 °С. Клетки гепатомы крысы, стабильно экспрессирующие рецептор S1P1 человека, или клетки базофильного лейкоза крысы (RBL), стабильно экспрессирующие рецептор S1P3 человека, выращивали до 80% слияния, после чего собирали в 10 мл фосфатного буферного солевого раствора (PBS) и центрифугировали при 1200 об/мин в течение 5 мин. После удаления супернатанта осадок повторно суспендировали, и клетки гомогенизировали в стеклянной мешалке Waring 2 циклами по 15 с в 200 мл буфера (50 мМ HEPES, 1 мМ лейпептина, 25 мкг/мл бацитрацина, 1 мМ EDTA, 1 мМ PMSF, 2 мкМ пепстатина А). Мешалку погружали в лед на 5 мин после первого цикла и на 10-40 мин после заключительного цикла, чтобы дать пене рассеяться. Материал затем центрифугировали при 500 g в течение 20 мин, и супернатант центрифугировали в течение 36 мин при 48000 g. Осадок повторно суспендировали в том же самом буфере, как описано выше, но без PMSF и пепстатина А. Материал затем пропускали через иглу диаметром 0,6 мм, собирали до необходимого объема, (обычно ×4 объем от первоначального осадка клеток), разделяли на аликвоты и сохраняли замороженным при -80 °С. Все стадии осуществляли при 4 °С. Клетки гомогенизировали в стеклянной мешалке Waring в ходе 2 циклов по 15 секунд в 200 мл буфера (50 мМ HEPES, 1 мМ лейпептина, 25 мкг/мл бацитрацина, 1 мМ EDTA, 1 мМ PMSF, 2 мкМ пепстатина А). Мешалку погружали в лед на 5 мин после первого цикла и на 10-40 мин после заключительного цикла, чтобы дать пене рассеяться. Материал затем центрифугировали при 500 g в течение 20 мин, и супернатант центрифугировали в течение 36 минут при 48000 g. Осадок повторно суспендировали в том же самом буфере, как описано выше, но без PMSF и пепстатина А. Материал затем пропускали через иглу диаметром 0,6 мм, собирали до необходимого объема, (обычно ×4 объем от первоначального осадка клеток), разделяли на аликвоты и сохраняли замороженным при -80 °С. Мембраны гепатомы крысы с S1P1 человека (1,5 мкг/лунка) налепляли на покрытые агглютинином проростков пшеницы (WGA) гранулы для теста сцинтилляционной близости (SPA) (0,125 мг/лунка) в тестовом буфере (HEPES 20 мМ, MgCl 2 10 мМ, NaCl 100 мМ и рН, доведенный до 7,4 с использованием КОН 5М, также добавляли ГДФ 10 мкМ FAC (конечная тестовая концентрация) и сапонин 90 мкг/мл FAC). После 30 мин предварительного соединения на льду гранулы и мембранную суспензию распределяли в белые полипропиленовые 384-луночные планшеты Greiner LV (5 мкл/лунка), содержащие 0,1 мкл соединения. Затем в планшеты с агонистом добавляли 5 мкл/лунка [ 35 S]-ГТФ γS (0,5 нМ конечная концентрация меченого лиганда), составленного в тестовом буфере. Конечную тестовую смесь (10,1 мкл) затем центрифугировали при 1000 об./мин. в течение 5 мин, затем немедленно считывали на приборе Viewlux. Все тестируемые соединения растворяли в ДМСО в концентрации 10 мМ и готовили в 100%-ом ДМСО, используя стадию разведения 1 в 4, чтобы получить 11-точечные кривые доза-ответ. Разведения переносили на тестовые планшеты, обеспечивая постоянную концентрацию ДМСО в планшетах для всех тестов. Все данные подвергали нормализации до средних значений из 16 верхних и 16 нижних контрольных лунок на каждом планшете. Затем использовали построение кривой по 4 параметрам. Мембраны RH7777, экспрессирующие S1P1 (1.5 мкг/лунка), гомогенизировали, пропуская через иглу калибра 23G. Их налепляли на покрытые WGA гранулы для SPA (0,125 мг/лунка) в тестовом буфере (HEPES 20 мМ, MgCl 2 10 мМ, NaCl 100 мМ и рН, доведенный до 7,4 с использованием KOH 5М). Также добавляли ГДФ 10 мкМ FAC и сапонин 90 мкг/мл FAC. После 30 мин предварительного соединения на льду гранулы и мембранную суспензию распределяли в белые полипропиленовые 384-луночные планшеты Greiner LV (5 мкл/лунка), содержащие 0,1 мкл соединения. Затем в планшеты добавляли 5 мкл/лунка [ 35 S]-ГТФ γS (0,5 нМ для S1P1 или 0,3 нМ для S1P3, конечная концентрация меченого лиганда), составленного в тестовом буфере. Конечную тестовую смесь (10,1 мкл) затем герметизировали, центрифугировали, затем немедленно считывали на приборе Viewlux. Проиллюстрированные в примерах соединения по изобретению имели рЕС50> 5. Примеры 1-7, 8, 9, 11-13 имели рЕС50> 7. Примеры 2, 4, 6 и 8 имели рЕС50> 8. Мембраны клеток базофильного лейкоза крысы с S1P3 (RBL-2Н3) (1,5 мкг/лунка) налепляли на покрытые WGA гранулы для SPA (0,125 мг/лунка) в тестовом буфере (HEPES 20 мМ, MgCl 2 3 мМ, NaCl 100 мМ и рН, доведенный до 7,4 с использованием КОН 5М), также добавляли ГДФ 10 мкМ FAC (конечная тестовая концентрация) и сапонин 90 мкг/мл FAC). После 30 мин предварительного соединения на льду гранулы и мембранную суспензию распределяли в белые полипропиленовые 384-луночные планшеты Greiner LV (5 мкл/лунка), содержащие 0,1 мкл соединения. Затем в планшеты с агонистом добавляли 5 мкл/лунка [ 35 3]-ГТФ γS (0,5 нМ конечная концентрация меченого лиганда), составленного в тестовом буфере. Конечную тестовую смесь (10.1 мкл) затем центрифугировали при 1000 об./мин в течение 5 мин, затем немедленно считывали на приборе Viewlux. Все тестируемые соединения растворяли в ДМСО в концентрации 10 мМ и готовили в 100%-ом ДМСО, используя стадию разведения 1 в 4, чтобы получить 11-точечные кривые доза-ответ. Разведения переносили на тестовые планшеты, обеспечивая постоянную концентрацию ДМСО в планшетах для всех тестов. Все данные подвергали нормализации до средних значений из 16 верхних и 16 нижних контрольных лунок на каждом планшете. Затем использовали построение кривой по 4 параметрам. Мембраны RBL, экспрессирующие S1P3 (1.5 мкг/лунка) гомогенизировали, пропуская через иглу калибра 23G. Их налепляли на покрытые WGA гранулы для SPA (0,125 мг/лунка) в тестовом буфере (HEPES 20 мМ, MgCl 2 10 мМ, NaCl 100 мМ и рН, доведенный до 7,4 с использованием KOH 5М). Также добавляли ГДФ 10 мкМ FAC и сапонин 90 мкг/мл FAC. После 30 мин предварительного соединения на льду гранулы и мембранную суспензию распределяли в белые полипропиленовые 384-луночные планшеты Greiner LV (5 мкл/лунка), содержащие 0,1 мкл соединения. Затем в планшеты добавляли 5 мкл/лунка [ 35 S]-ГТФ γS (0,5 нМ для S1P1 или 0,3 нМ для S1P3, конечная концентрация меченого лиганда), составленного в тестовом буфере. Конечную тестовую смесь (10,1 мкл) затем герметизировали, центрифугировали, затем немедленно считывали на приборе Viewlux. Проиллюстрированные в примерах соединения, проверенные в этом тесте, имели рЕС50 <6, многие имели рЕС50 <5. Примеры 1, 4, 8-10, 12 и 13 имели рЕС50 <5. |