|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea000018624b*\id |
|
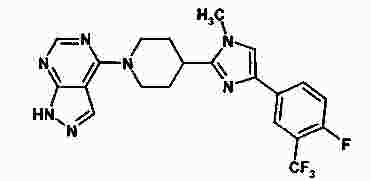
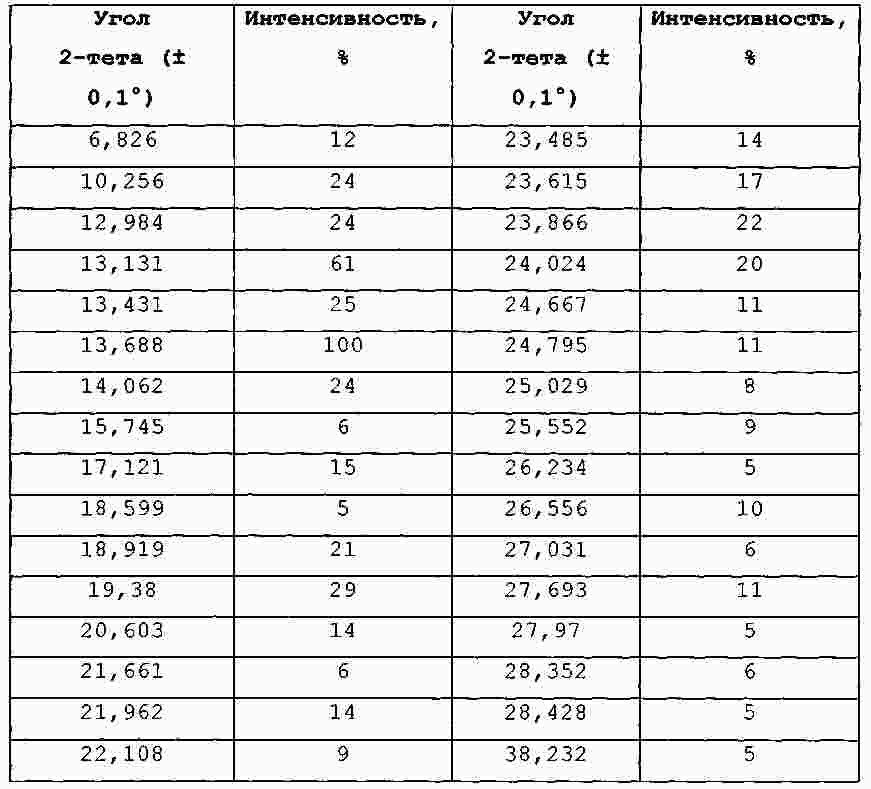
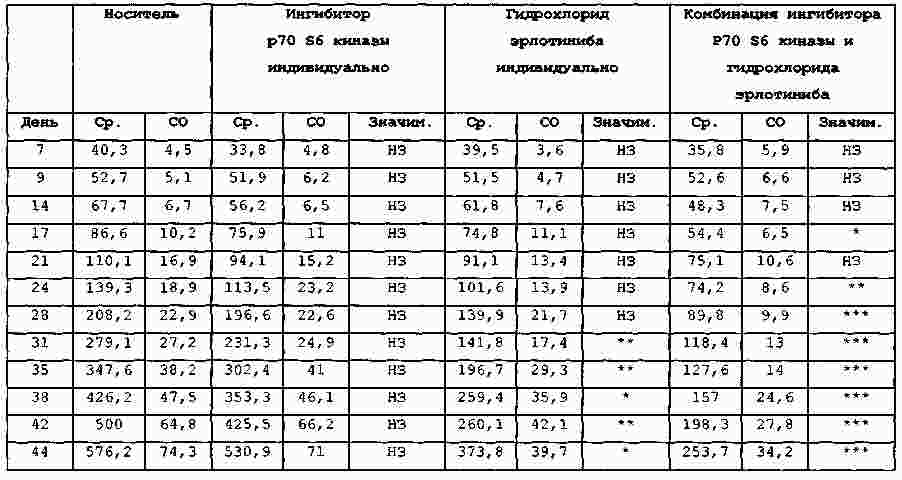
Термины запроса в документе Реферат В изобретении предложены продукт для комбинированной терапии рака, включающий соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин или его фармацевтически приемлемую соль и ингибитор EGFR, представляющий собой гидрохлорид эрлотиниба, для одновременного, раздельного или последовательного применения, и способ лечения мультиформной глиобластомы, аденокарциномы толстой кишки, немелкоклеточного рака легких, мелкоклеточного рака легких, резистентного к цисплатину мелкоклеточного рака легких, рака яичников, лейкемии, рака поджелудочной железы, рака простаты, карциномы молочной железы, почечно-клеточной карциномы, множественной миеломы, саркомы Капоши, лимфомы Ходжкина, лимфангиолейомиоматоза, неходжкинской лимфомы или саркомы. Формула [0001] Продукт, включающий соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин или его фармацевтически приемлемую соль и ингибитор EGFR (рецептора эпидермального фактора роста), представляющий собой гидрохлорид эрлотиниба, в виде комбинированного препарата для одновременного, раздельного или последовательного применения в терапии. [0002] Продукт, включающий соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин или его фармацевтически приемлемую соль и ингибитор EGFR, представляющий собой гидрохлорид эрлотиниба, в виде комбинированного препарата для одновременного, раздельного или последовательного применения для лечения мультиформной глиобластомы, аденокарциномы толстой кишки, немелкоклеточного рака легких, мелкоклеточного рака легких, резистентного к цисплатину мелкоклеточного рака легких, рака яичников, лейкемии, рака поджелудочной железы, рака простаты, карциномы молочной железы, почечно-клеточной карциномы, множественной миеломы, саркомы Капоши, лимфомы Ходжкина, лимфангиолейомиоматоза, неходжкинской лимфомы или саркомы. [0003] Продукт по п.2, отличающийся тем, что комбинированный препарат предназначен для одновременного, раздельного или последовательного применения для лечения немелкоклеточного рака легких. [0004] Продукт по любому из пп.1-3, отличающийся тем, что соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин или его фармацевтически приемлемая соль представляет собой п-толуолсульфонат 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1H-пиразоло[3,4-d]пиримидина. [0005] Продукт по любому из пп.1-4, отличающийся тем, что соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин или его фармацевтически приемлемую соль и ингибитор EGFR, представляющий собой гидрохлорид эрлотиниба, вводят перорально. [0006] Применение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидина или его фармацевтически приемлемой соли для введения одновременно, раздельно или последовательно в комбинации с ингибитором EGFR, представляющим собой гидрохлорид эрлотиниба, для лечения мультиформной глиобластомы, аденокарциномы толстой кишки, немелкоклеточного рака легких, мелкоклеточного рака легких, резистентного к цисплатину мелкоклеточного рака легких, рака яичников, лейкемии, рака поджелудочной железы, рака простаты, карциномы молочной железы, почечно-клеточной карциномы, множественной миеломы, саркомы Капоши, лимфомы Ходжкина, лимфангиолейомиоматоза, неходжкинской лимфомы или саркомы. [0007] Применение по п.6 для лечения немелкоклеточного рака легких. [0008] Применение по любому из пп.6, 7, отличающееся тем, что указанная соль представляет собой п-толуолсульфонат 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидина. [0009] Применение по любому из пп.6-8, отличающееся тем, что указанное соединение и ингибитор EGFR, представляющий собой гидрохлорид эрлотиниба, предназначены для перорального введения. [0010] Способ лечения ракового заболевания, выбранного из группы, состоящей из мультиформной глиобластомы, аденокарциномы толстой кишки, немелкоклеточного рака легких, мелкоклеточного рака легких, резистентного к цисплатину мелкоклеточного рака легких, рака яичников, лейкемии, рака поджелудочной железы, рака простаты, карциномы молочной железы, почечно-клеточной карциномы, множественной миеломы, саркомы Капоши, лимфомы Ходжкина, лимфангиолейомиоматоза, неходжкинской лимфомы или саркомы, включающий введение пациенту, который в этом нуждается, 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидина или его фармацевтически приемлемой соли и ингибитора EGFR, представляющего собой гидрохлорид эрлотиниба, в количествах, которые являются эффективными в комбинации. [0011] Способ по п.10, отличающийся тем, что раковое заболевание представляет собой немелкоклеточный рак легких. [0012] Способ по любому из пп.10, 11, отличающийся тем, что соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин или его фармацевтически приемлемая соль представляет собой п-толуолсульфонат 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидина. [0013] Способ по любому из пп.10, 12, отличающийся тем, что соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин или его фармацевтически приемлемую соль и ингибитор EGFR, представляющий собой гидрохлорид эрлотиниба, вводят перорально. Полный текст патента Уровень техники Путь "фосфотидилинозитол-3-киназа (PI3K)/АКТ/мишень рапамицина у млекопитающих" (mTOR) включает ряд сигнальных точек, которые играют важную роль в контроле роста и выживания клеток. Киназа Р70 S6 представляет собой серин-треонинпротеинкиназу, которая является эффектором следующей за ним части сигнального пути PI3K/AKT/mTOR. Киназа Р70 S6 фосфорилирует рибосомальный белок S6 в клетках и регулирует биогенез рибосом, рост клеток и протекание клеточного цикла в ответ на митогенное стимулирование. Киназа Р70 S6, как правило, активирована во многих солидных опухолях. Ингибиторы киназы р70 S6, применяемые для лечения указанных опухолей, описаны в WO 2006/046024 и WO 2008/075109. Рецептор эпидермального фактора роста (EGFR) представляет собой трансмембранный гликопротеин, который принадлежит к семейству структурно родственных рецепторных тирозинкиназ. EGFR включается в путь PI3K/AKT/mTOR на уровне поверхности клеток. Полагают, что EGFR играет важную роль во множестве путей трансдукции сигнала и, по всей видимости, в онкогенезе и росте опухоли. EGFR и его лиганды сверхэкспрессируются или вовлечены в аутокринные петлеобразные циклы роста опухолей различных типов. В EP 0817775 описаны серии производных 4-(замещенного фениламино)хиназолина, которые обладают ингибирующей активностью по отношению к EGFR и подходят для лечения рака. Существует необходимость в усовершенствованной терапии для лечения раковых заболеваний. Кроме того, существует необходимость в терапии, обладающей значительно большей эффективностью по сравнению с существующей терапией. Предпочтительные комбинированные терапии согласно настоящему изобретению демонстрируют повышенную эффективность по сравнению с лечением лишь одним терапевтическим агентом. Более предпочтительные комбинированные терапии согласно настоящему изобретению демонстрируют повышенную эффективность при введении каждого из терапевтических агентов в дозе, меньшей оптимальной. В настоящем изобретении предложен продукт, включающий соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин или его фармацевтически приемлемую соль и ингибитор EGFR в виде комбинированного препарата для одновременного, раздельного или последовательного применения в терапии. Также в настоящем изобретении предложен продукт, включающий соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин или его фармацевтически приемлемую соль и ингибитор EGFR в виде комбинированного препарата для одновременного, раздельного или последовательного применения для лечения мультиформной глиобластомы, аденокарциномы толстой кишки, немелкоклеточного рака легких, мелкоклеточного рака легких, резистентного к цисплатину мелкоклеточного рака легких, рака яичников, лейкемии, рака поджелудочной железы, рака простаты, карциномы молочной железы, почечно-клеточной карциномы, множественной миеломы, саркомы Капоши, лимфомы Ходжкина, лимфангиолейомиоматоза, неходжкинской лимфомы или саркомы. Также в настоящем изобретении предложено применение соединения 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин или его фармацевтически приемлемая соль для применения одновременно, раздельно или последовательно в комбинации с ингибитором EGFR для лечения мультиформной глиобластомы, аденокарциномы толстой кишки, немелкоклеточного рака легких, мелкоклеточного рака легких, резистентного к цисплатину мелкоклеточного рака легких, рака яичников, лейкемии, рака поджелудочной железы, рака простаты, карциномы молочной железы, почечно-клеточной карциномы, множественной миеломы, саркомы Капоши, лимфомы Ходжкина, лимфангиолейомиоматоза, неходжкинской лимфомы или саркомы. В настоящем изобретении также предложен способ лечения ракового заболевания, выбранного из группы, состоящей из мультиформной глиобластомы, аденокарциномы толстой кишки, немелкоклеточного рака легких, мелкоклеточного рака легких, резистентного к цисплатину мелкоклеточного рака легких, рака яичников, лейкемии, рака поджелудочной железы, рака простаты, карциномы молочной железы, почечно-клеточной карциномы, множественной миеломы, саркомы Капоши, лимфомы Ходжкина, лимфангиолейомиоматоза, неходжкинской лимфомы или саркомы, включающий введение пациенту, который в этом нуждается, соединения 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидина или его фармацевтически приемлемой соли и ингибитора EGFR в количествах, которые являются эффективными в комбинации. Соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин представляет собой ингибитор киназы р70 S6. Соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин представляет собой основание и соответственно реагирует с любыми органическими или неорганическими кислотами с образованием фармацевтически приемлемых солей. Термин "фармацевтически приемлемая соль", используемый в настоящей заявке, относится к солям соединения 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидина, которые являются, по существу, нетоксичными для живых организмов. Указанные соли включают фармацевтически приемлемые соли, перечисленные в Journal of Pharmaceutical Science, 66, 2-19 (1977), которые известны специалистам в данной области техники. Предпочтительными являются тозилатная (также известная как п-толуолсульфонатная) и гидрохлоридная соли. Особенно предпочтительной является тозилатная соль. "Ингибитор EGFR" означает любое соединение, пептид или антитело, которое представляет собой ингибитор EGFR. Предпочтительные ингибиторы EGFR включают эрлотиниб, цетуксимаб (Erbitux ®; описан в ЕР 0359282), панитумумаб (Vectibix ®, описан в ЕР 0359282) и гефитиниб (Iressa ®, описан в ЕР 0566226). Особенно предпочтительный ингибитор EGFR представляет собой эрлотиниб, N-(3-этиниллфенил)-6,7-бис-(2-метоксиэтокси)-4-хиназолинамин и, в частности, гидрохлорид эрлотиниба (Tarceva ®). Ингибитор EGFR эрлотиниб и способы его получения описаны в ЕР 0817775. Термин "комбинированная терапия" относится к лечению, включающему введение соединения 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидина или его фармацевтически приемлемой соли и ингибитора EGFR ("терапевтические агенты") в комбинации. Терапевтические агенты могут быть введены одновременно, раздельно или последовательно. Термин "лечащий" или "лечение" включает замедление, прерывание, подавление, контроль, остановку, снижение или обращение прогрессирования или степени тяжести симптома, нарушения, состояния или болезни. Термин "количества, которые являются эффективными в комбинации" означает количество соединения 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидина или его фармацевтически приемлемой соли и количество ингибитора EGFR, которые являются эффективными для лечения нарушений, описанных в настоящей заявке, при введении в комбинации. Количество каждого из фармацевтических агентов, которое является эффективным в комбинации, может быть равным количеству, которое является эффективным при введении терапевтического агента в отдельности, или может быть меньше количества, эффективного при введении терапевтического агента в отдельности (т.е. может представлять собой дозу, меньшую оптимальной). Комбинированную терапию, описанную в настоящем изобретении, можно применять для лечения пролиферативных нарушений, таких как рак, и для подавления ангиогенеза у млекопитающих. Во всех вариантах реализации настоящего изобретения предпочтительно рак, подвергаемый лечению, выбран из мультиформной глиобластомы, аденокарциномы толстой кишки, немелкоклеточного рака легких, мелкоклеточного рака легких, резистентного к цисплатину мелкоклеточного рака легких, рака яичников, лейкемии, рака поджелудочной железы, рака простаты, карциномы молочной железы, почечно-клеточной карциномы, множественной миеломы, саркомы Капоши, лимфомы Ходжкина, лимфангиолейомиоматоза, неходжкинской лимфомы или саркомы. Особенно предпочтительно рак, который подвергают лечению, представляет собой почечно-клеточную карциному или мультиформную глиобластому. Предпочтительно млекопитающее, нуждающееся в лечении, представляет собой человека. В альтернативном варианте реализации настоящего изобретения ингибитор EGFR можно применять одновременно, раздельно или последовательно в комбинации с соединением 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидином или его фармацевтически приемлемой солью для лечения рака, в частности раковых заболеваний, описанных выше. Соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин или его фармацевтически приемлемую соль можно применять для получения лекарственного средства, подходящего для применения в комбинированной терапии для лечения рака, в частности раковых заболеваний, описанных выше, при этом указанное лекарственное средство предназначено для введения в комбинации с ингибитором EGFR. В другом альтернативном варианте реализации настоящего изобретения ингибитор EGFR можно применять для получения лекарственного средства, подходящего для применения в комбинированной терапии для лечения рака, в частности раковых заболеваний, описанных выше, где указанное лекарственное средство предназначено для введения в комбинации с 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидином или его фармацевтически приемлемой солью. В другом альтернативном варианте реализации предложен фармацевтический состав, содержащий соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин или его фармацевтически приемлемую соль и ингибитор EGFR в комбинации с фармацевтически приемлемым носителем, разбавителем или наполнителем. Соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин или его фармацевтическую соль и ингибитор EGFR можно вводить различными способами. Указанные вещества можно вводить одинаковыми способами или различными способами. Предпочтительно соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]-пиримидин или его фармацевтически приемлемую соль или ингибитор EGFR вводят орально. Более предпочтительно оба указанных вещества вводят перорально. Оптимальные режимы дозировки каждого из терапевтических агентов, применяемых в комбинированной терапии согласно настоящему изобретению, можно варьировать в зависимости от, например, способа введения, болезни, подвергаемой лечению, и применяемого ингибитора EGFR. Например, дозировка соединения 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидина или его фармацевтически приемлемой соли может находиться в диапазоне от 100 до 2000 мг в день. Предпочтительные дозировки находятся в диапазоне от 600 до 1600 мг в день. В предпочтительном варианте реализации соединения вводят дважды в день и каждая доза находится в диапазоне от 300 до 800 мг. Дозировка ингибитора EGFR гидрохлорида эрлотиниба может находиться в диапазоне от 10 до 450 мг в день. Предпочтительные дозировки гидрохлорида эрлотиниба составляют 150 или 100 мг в день. Комбинированную терапию можно проводить в течение единственного установленного периода времени, например 6 месяцев. Комбинированную терапию можно проводить в циклическом режиме, в котором присутствуют периоды альтернативного лечения и отсутствия лечения. Предпочтительно комбинированную терапию проводят непрерывно (исключая прогрессирование болезни или проявление неприемлемой токсичности). В одном из вариантов реализации соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин или его фармацевтически приемлемую соль и ингибитор EGFR вводят раздельно. При раздельном введении соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин или его фармацевтически приемлемую соль и ингибитор EGFR можно вводить согласно различным режимам дозировки и при помощи различных способов введения. В другом варианте реализации соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин или его фармацевтически приемлемую соль и ингибитор EGFR вводят последовательно. В указанном варианте реализации каждый из терапевтических агентов можно вводить первым. Предпочтительно первым вводят ингибитор EGFR, a затем вводят соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин или его фармацевтически приемлемую соль. Предпочтительно время между введением дозы одного из терапевтических агентов и введением дозы другого терапевтического агента составляет менее чем 8 ч, более предпочтительно менее чем 4 ч, еще более предпочтительно менее чем 1 ч. В другом варианте реализации соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин или его фармацевтически приемлемую соль и ингибитор EGFR вводят одновременно. В указанном варианте реализации агенты можно вводить в одном и том же составе или одновременно при помощи различных способов введения. Терапевтический агент, представляющий собой тозилат 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидина, предпочтительно вводят перорально. Также предпочтительно, что в течение курса лечения в день вводят две дозы тозилата 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидина, и каждая из доз находится в диапазоне от 300 до 800 мг. В одном из вариантов реализации терапевтические агенты, применяемые в комбинированной терапии, представляют собой тозилат 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидина и гидрохлорид эрлотиниба. Предпочтительно, что тозилат 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидина вводят перорально согласно предпочтительному режиму дозировки, описанному выше. В указанном варианте реализации предпочтительно, что гидрохлорид эрлотиниба также вводят перорально. Также предпочтительно, что в день вводят одну дозу эрлотиниба, и каждая из доз составляет 100 или 150 мг. В указанном варианте реализации предпочтительно, что комбинированную терапию проводят непрерывно. Соединение 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин и его фармацевтически приемлемые соли могут быть получены согласно способам, описанным ниже. Получение промежуточного 4-хлор-1Н-пиразоло [3,4-d]пиримидина. К раствору аллопуринола (20 г, 146,94 ммоль) в толуоле (205,71 мл) добавляли фосфорилхлорид (68,27 мл, 734,68 ммоль) и диизопропилэтиламин (56,38 мл, 323,26 ммоль) и нагревали смесь при 80 °С в течение 2 ч. Удаляли растворитель в вакууме до половины объема и добавляли смесь в 2 М раствор двухосновного фосфата калия (734,68 мл, 1,47 моль) в воде при 4 °C. Перемешивали смесь в течение ночи при комнатной температуре (КТ). Отфильтровывали осадок через фильтр Celite ® и затем промывали EtOAc. Отделяли фильтрат, промывали водный слой дополнительным количеством EtOAc, объединяли органические слои, сушили MgSO 4 , фильтровали и концентрировали в вакууме с получением вышеназванного соединения (16 г, выход 70,45%) в виде желтого твердого вещества. МС (APCI): m/z=155,1 [M+H]. Получение гидрохлорида 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидина. К раствору 4-фтор-3-(трифторметил)фенацилбромида (60,00 г, 1,00 экв., 210,50 ммоль) в этилацетате (450 мл, 4,60 моль) добавляли метенамин (1,10 экв., 231,55 ммоль, 32,46 г). Реакционную смесь перемешивали при КТ (комнатной температуре) в течение ночи. Растворитель удаляли в вакууме и растирали твердое вещество в метил-трет-бутиловом эфире (МТБЭ). Фильтровали и сушили при пониженном давлении. Добавляли этанол (450 мл, 7,73 моль), затем хлороводород (150 мл, 8,30 экв., 1,75 моль) и перемешивали смесь при КТ в течение ночи. Растворитель удаляли в вакууме и сушили твердое вещество в вакууме при 50 °C в течение недели с получением гидрохлорида 2-амино-1-(4-фтор-3-трифторметилфенил)этанона (54,23 г, выход 100%) в виде белого твердого вещества. К раствору моно-трет-бутилового эфира пиперидин-1,4-дикарбоновой кислоты (1,20 экв., 252,61 ммоль, 57,92 г) в тетрагидрофуране (ТГФ) (400 мл) добавляли N-метилморфолин (3 экв., 631,52 ммоль, 69,66 мл). Охлаждали смесь до -10 °C на бане сухой лед/ацетон. Поддерживая температуру, равную ниже -5 °C, по каплям добавляли изобутилхлорформиат (1,1 экв., 2 31,56 ммоль, 30,26 мл). Через 30 мин при -5-10 °C добавляли гидрохлорид 2-амино-1-(4-фтор-3-трифторметилфенил)этанона (54,23 г, 1,00 экв., 210,51 ммоль), суспендированный в ТГФ (300 мл), и перемешивали смесь в бане при -5 °C в течение 20 мин. Перемешивали в течение 1 ч при КТ. Добавляли воду и EtOAc, затем промывали органический слой водой и насыщенным водным раствором хлорида натрия. Сушили MgSO 4 , фильтровали и удаляли растворитель в вакууме. Суспендировали неочищенное вещество в МТБЭ и перемешивали в течение 2 ч. Фильтровали твердое вещество в вакууме с получением трет-бутилового эфира 1-[2-(4-фтор-3-трифторметилфенил)-2-оксоэтилкарбамоил]пиперидин-4-карбоновой кислоты (64,44 г, выход 70,79%). К раствору трет-бутилового эфира 1-[2-(4-фтор-3-трифторметилфенил)-2-оксоэтилкарбамоил]пиперидин-4-карбоновой кислоты (29,4 г, 1,00 экв., 67,99 ммоль) в 1-бутаноле (150 мл, 1,64 моль) добавляли ацетат аммония (15 экв., 1,02 моль, 7 8,61 г), затем добавляли триэтиламин (1 экв., 67,99 ммоль, 9,48 мл). Перемешивали смесь при 160 °C в запаянной трубке в течение 3 ч. Добавляли EtOAc и воду, затем дополнительно промывали органический слой водой и насыщенным водным раствором хлорида натрия и концентрировали в вакууме. Растирали твердое вещество в МТБЭ, фильтровали и сушили при пониженном давлении с получением трет-бутилового эфира 4-[4-(4-фтор-3-трифторметилфенил)-1Н-имидазол-2-ил]пиперидин-1-карбоновой кислоты (18,23 г, 44,10 ммоль, выход 64,86%) в виде белого твердого вещества. К раствору гидроксида калия (1,5 экв., 58,16 ммоль, 3,2 6 г) в 200 мл диметилсульфоксида (ДМСО) добавляли трет-бутиловый эфир 4-[4-(4-фтор-3-трифторметилфенил)-1Н-имидазол-2-ил]пиперидин-1-карбоновой кислоты (16,03 г, 1,00 экв., 38,77 ммоль) в 40 мл ДМСО. Через 5 мин при КТ добавляли одну порцию метилйодида (1,1 экв., 42,65 ммоль, 2,66 мл). Перемешивали при КТ в течение 2 ч, затем добавляли смесь в ледяную воду. Фильтровали твердое вещество, промывали водой и сушили при пониженном давлении. Твердое вещество растирали в горячем гептане, фильтровали и сушили при пониженном давлении с получением трет-бутилового эфира 4-[4-(4-фтор-3-фторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-карбоновой кислоты (8,7 г, выход 52,4 9%) в виде белого твердого вещества. К раствору трет-бутилового эфира 4-[4-(4-фтор-3-фторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-карбоновой кислоты (8,7 г, 1,00 экв., 20,35 ммоль) в дихлорметане (101,77 мл) при КТ добавляли хлорид водорода (4,00 экв., 81,41 ммоль, 20,35 мл). Раствор перемешивали при КТ в течение 1 ч. Удаляли растворитель при пониженном давлении и растворяли неочищенное вещество в изопропиловом спирте (101,77 мл). Добавляли 4-хлор-1Н-пиразоло[3,4-d]пиримидин (1,65 экв., 33,58 ммоль, 5,19 г) и триэтиламин (10 экв., 203,54 ммоль, 28,37 мл). Перемешивали смесь при кипячении с обратным холодильником в течение 1 ч. Удаляли растворитель при пониженном давлении, растирали неочищенное вещество в воде и оставляли на ночь. Твердое вещество фильтровали и растирали в горячем ацетонитриле, фильтровали и сушили в вакууме. Получали 4-{4-[4-(4-фтор-3-трифторметилфенил)-3-метил-1H-имидазол-2-ил]пиперидин-1-ил}-1H-пиразоло[3,4-d]пиримидин (8,42 г, 18,86 ммоль, выход 92,66%) в виде светло-желтого твердого вещества. К суспензии 4-{4-[4-(4-фтор-3-трифторметилфенил)-3-метил-1H-имидазол-2-ил]пиперидин-1-ил}-1H-пиразоло[3,4-d]пиримидина (7,5 г, 1,0 0 экв., 16,84 ммоль) в дихлорметане (50 мл) добавляли хлороводород (1,1 экв., 18,52 ммоль, 4,63 мл) и перемешивали смесь в течение 1 ч при КТ. Удаляли растворитель в вакууме и растирали неочищенное вещество в МТБЭ в течение 1 ч. Фильтровали твердое вещество и сушили в вакууме в течение ночи с получением гидрохлорида 4-{4-[4-(4-фтор-3-трифторметилфенил)-3-метил-1H-имидазол-2-ил]пиперидин-1-ил}-1H-пиразоло[3,4-d]пиримидина (7,99 г, 16,58 ммоль, выход 98,47%) в виде белого твердого вещества. 1 Н-ЯМР (300 МГц, ДМСО): δ 14,01-13,99 (м, 1Н), 8,57-8,54 (м, 2Н), 8,26-8,19 (м, 3H), 7,72-7,63 (м, 1Н), 5,23-5,20 (м, 2Н), 3,89 (с, 3H), 3,41 (м, 2Н), 2,15-2,07 (м, 3H), 1,10 (с, 2Н). Получение п-толуолсульфоната 4-{4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1H-имидазол-2-ил]пиперидин-1-ил}-1H-пиразоло[3,4-d]пиримидина. Охлаждали раствор 4-фтор-3-(трифторметил)фенацилбромида (93% чистота, определена при помощи ВЭЖХ, 1000 г, 3,51 моль) и ТГФ (5 л) до <5 °C в ледяной бане. По каплям в течение 1 ч при <5 °C добавляли раствор азида натрия (239 г, 3,68 моль, 1,05 экв.) в воде (800 мл). После перемешивания при <5 °C в течение 1 ч отделяли и отбрасывали водный слой. Остающийся холодным органический слой медленно добавляли в течение 3 ч к раствору трифенилфосфина (920,2 г, 3,51 моль, 1,0 экв.), моногидрата п-толуолсульфоновой кислоты (1335 г, 7,02 моль, 2,0 экв.) и ТГФ (5 л). Поддерживали температуру <15 °C в течение указанного добавления и в ходе добавления осаждались твердые вещества. Перемешивали реакционную смесь при <20 °C в течение 2 ч, затем фильтровали твердое вещество, промывали ТГФ (3 ×2 л) и сушили при 50 °C в вакууме с получением 1167,4 г (85%, 92% с поправкой на чистоту исходного вещества) п-толуолсульфоната 2-амино-1-(4-фтор-3-трифторметилфенил)этанона в виде белого кристаллического твердого вещества. Смешивали п-толуолсульфонат 2-амино-1-(4-фтор-3-трифторметилфенил)этанона (1133 г, 2,88 моль), 1-(трет-бутоксикарбонил)пиперидин-4-карбоновую кислоту (795 г, 3,47 моль, 1,20 экв.), ТГФ (3450 мл) и этилацетат (7500 мл) с получением жидкой белой суспензии. Охлаждали суспензию до <5 °C в ледяной бане и добавляли ангидрид 2-пропанфосфоновой кислоты (Т 3 Р) (50% раствор в EtOAc) (2385 г, 3,75 моль, 1,3 экв.). Затем в течение 1 ч добавляли N-метилморфолин (795 мл, 7,21 моль, 2,5 экв.), поддерживая температуру <10 °C. Нагревали полученную суспензию до температуры окружающей среды и перемешивали в течение 2 ч. Реакцию гасили путем добавления воды. Отделяли органическую фазу, затем промывали водным NaHCO 3 , водным NaCl. Органическую фазу нагревали до 50 °C в роторном испарителе и добавляли н-гептан. Отгоняли растворитель в вакууме до конечного объема суспензии, составляющего примерно 5 л. Охлаждали суспензию до КТ и фильтровали твердые вещества, промывали н-гептаном (2 ×1 л) и затем сушили в вакуумируемом сушильном шкафу при 50 °C в течение ночи с получением трет-бутилового эфира 1-[2-(4-фтор-3-трифторметилфенил)-2-оксоэтилкарбамоил]пиперидин-4-карбоновой кислоты (1124,8 г, 90%) в виде белого твердого вещества. Смешивали трет-бутиловый эфир 1-[2-(4-фтор-3-трифторметилфенил)-2-оксоэтилкарбамоил]пиперидин-4-карбоновой кислоты (100 г, 231 ммоль), ацетат аммония (178,3 г, 2,31 моль, 10 экв.) и метанол (1000 мл). Реактор, применяемый для указанного превращения, представляет собой спиральную трубку из нержавеющей стали с внутренним диаметром 1/16'' (общий внутренний объем трубки в печи составляет 541 моль). Нагревали реактор в печи до 140 °C. Контролировали давление реакционной смеси в указанной трубке, равное 250 psig (1,72 МПа) при помощи регулятора, что обеспечивает нагревание раствора выше его обычной температуры кипения. Непрерывно откачивали раствор, полученный выше, из нагретой трубки под давлением со скоростью 6,01 мл/мин (достигая общего времени выдерживания в нагретой трубке, равного 90 мин). После выхода раствора из сушильного шкафа охлаждали раствор до 20 °C в теплообменнике типа "труба в трубе". После того как весь раствор проходит через реактор (общее время процесса составляет 8 ч), концентрировали полученный оранжевый раствор в вакууме при 30 °C до достижения общего объема, равного 600 мл. Добавляли ацетонитрил (200 мл) и нагревали раствор до 50 °C. По каплям добавляли воду (700 мл) с затравкой в течение 2 ч для кристаллизации продукта. Охлаждали полученную суспензию до 20 °C и отфильтровывали твердое вещество, затем промывали 20% раствором МеОН в воде (2 ×200 мл). Полученное твердое вещество сушили в вакууме при 50 °C. Повторно суспендировали твердое вещество в ацетонитриле (200 мл) при 50 °C. Охлаждали суспензию до температуры окружающей среды, отфильтровывали твердое вещество и промывали ацетонитрилом (100 мл) с получением трет-бутилового эфира 4-[4-(4-фтор-3-трифторметилфенил)-1Н-имидазол-2-ил]пиперидин-1-карбоновой кислоты (54,43 г, 132 ммоль, 57%) в виде беловатого твердого вещества. Растворяли трет-бутиловый эфир 4-[4-(4-фтор-3-трифторметилфенил)-1Н-имидазол-2-ил]пиперидин-1-карбоновой кислоты (80,02 г, 183,69 ммоль) в ДМСО (1060 мл). Добавляли одну порцию KOH (18,47 г, 279,82 ммоль, 1,5 экв.). Добавляли метилйодид (27,74 г, 193,48 ммоль, 1,05 экв.) в течение 30 мин при 25 °C. Перемешивали раствор при 25 °C в течение 1 ч. В течение 5 мин к раствору добавляли смесь затравочных кристаллов трет-бутилового эфира 4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-карбоновой кислоты (0,17 г) и воды (80 мл). Перемешивали полученную жидкую суспензию при 25 °C в течение 30 мин. Дополнительно добавляли воду (240,73 мл) в течение 30 мин при 25 °C. Отфильтровывали твердое вещество и промывали 20% ДМСО в воде (2 ×120 мл), затем водой (120 мл). Твердое вещество сушили в вакууме при 60 °C. Растворяли полученные твердые вещества в этаноле (480 мл) при 50 °C. Добавляли воду (240 мл) в течение 5 мин. Затем в течение 30 мин добавляли затравку трет-бутилового эфира 4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-карбоновой кислоты (0,038 г) и дополнительное количество воды (240 мл). Охлаждали полученную суспензию до 25 °C в течение 2 ч. Фильтровали твердые вещества и промывали осадок 20% раствором EtOH в воде. Осуществляли сушку твердого вещества в вакууме при 60 °C с получением трет-бутилового эфира 4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-карбоновой кислоты (72,36 г, 92%) в виде белого твердого вещества. Готовили раствор безводной HCl путем медленного добавления ацетилхлорида (193,14 мл, 2,71 моль, 4,00 экв.) к метанолу (1160 мл) в течение 45 мин при <5 °C. Полученный раствор добавляли в отдельную колбу, содержащую раствор трет-бутилового эфира 4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-карбоновой кислоты (290 г, 678,46 ммоль) в метаноле (2320 мл) в течение 90 мин при 20 °C. Перемешивали реакционную смесь при 20 °C в течение ночи. Концентрировали реакционную смесь в вакууме при 30 °C. Добавляли ДМСО (1080 мл, 15,20 моль, 1,08 л, 1,19 кг) и продолжали перегонку до достижения внутренней температуры, равной 50 °C, при давлении, равном 20 мм рт. ст. (2,67 кПа). Добавляли ДМСО до достижения общего объема, равного 2030 мл. Затем добавляли триэтиламин (473 мл, 3,39 моль, 5 экв.) через капельную воронку в течение 30 мин. Помещали твердый 4-хлор-1Н-пиразоло[3,4-d]пиримидин (110,29 г, 713,58 ммоль, 1,05 экв.) равными порциями равномерно в течение 30 мин. Полученную суспензию перемешивали при 20 °C в течение ночи. Нагревали суспензию до 80 °C. Добавляли воду (229 мл) с получением прозрачного раствора. Добавляли в реакционную смесь затравку и медленно добавляли дополнительное количество воды (1273 мл) в течение 4 ч до полной кристаллизации продукта. Охлаждали суспензию до 50 °C и отфильтровывали твердое вещество. Промывали осадок 30% раствором воды в ДМСО (2 ×290 мл), затем водой (290 мл). Сушили твердые вещества в вакууме при 60 °C с получением 4-{4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил}-1Н-пиразоло[3,4-d]пиримидина (301 г, 99%) в виде беловатого твердого вещества. Растворяли 4-{4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил}-1Н-пиразоло[3,4-d]пиримидин (20 г, 44,9 ммоль) в смеси 20:1 H 2 O:ацетон (360 мл). К реакционной смеси добавляли раствор моногидрата п-толуолсульфоновой кислоты (10,25 г, 53,9 ммоль, 1,2 экв.) в смеси 20:1 H 2 O:ацетон (40 мл) в течение 20 мин при 20 °C. Реакционную смесь нагревали до 55 °C, выдерживали в течение 1 ч, затем охлаждали до 25 °C в течение 1 ч. Твердое вещество отфильтровывали и промывали осадок водой (40 мл). Сушка в вакууме при 50 °C позволила получить п-толуолсульфонат 4-{4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил}-1Н-пиразоло[3,4-d]пиримидина (23,9 г, 86%) в виде белого твердого вещества. Получение кристаллического п-толуолсульфоната 4-{4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил}-1Н-пиразоло[3,4-d]пиримидина. В круглодонную колбу вместимостью 1 л, снабженную подвесной мешалкой, помещали 60,12 г 4-{4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил}-1Н-пиразоло[3,4-d]пиримидина (полученного в соответствии с любым из представленных выше примеров получения без конечной стадии получения соли), затем 250 мл 5% водного раствора МеОН. Полученную суспензию перемешивали и добавляли моногидрат п-толуолсульфоновой кислоты (26,88 г), затем промывали оставшимися 50 мл 5% водного раствора МеОН. Полученную суспензию перемешивали и охлаждали кристаллы до 5 °C. Через 1 ч при 5 °C перемешивание останавливали и фильтровали суспензию через воронку Бюхнера. Ополаскивали колбу 75 мл холодного 5% водного раствора МеОН и применяли указанный промывочный раствор для промывки осадка на фильтре. Твердые вещества переносили на тарелку для взвешивания и сушили при 50 °C в вакууме в течение дня и ночи с медленным откачиванием воздуха. Конечная масса составляла 71,44 г. Проводили анализ методом порошковой рентгеновской дифракции при помощи дифракторметра D4 Endeaver, снабженного источником CuKa излучения ( λ=1,54056 Å), с рабочими характеристиками 40 кВ и 50 мА. Образец сканировали в диапазоне 2 θ от 4 до 40 ° с шагом 0,009 2 θ и скоростью сканирования ≥1,5 с на шаг. Определение эффективности в организме. Самкам мышей СВ-17 (Fox Chase SCID) Model #CB17SC-M подкожно имплантировали в бок клетки человеческой немелкоклеточной карциномы легких А549 в 0,2 мл матригеля. Примерно через 1 неделю после имплантирования, когда размер опухоли достигает примерно 100 мг, мышей случайным образом разделяли на группы по 10 мышей и вводили им перорально один раз в день 2,5 мг/кг 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидина (ингибитор р70 S6 киназы, в составе 35% ПЭГ300/10% ГПБЦД/10% ПС80 в Н 2 О, концентрация ингибитора составляет 0,31 мг/мл) или 20 мг/кг гидрохлорида эрлотиниба (в составе NaCMC Tween 80, концентрация гидрохлорида эрлотиниба составляет 2,5 мг/мл) или комбинацию (2,5 мг/кг ингибитора р70 S6 киназы и 20 мг/кг гидрохлорида эрлотиниба). Группа носителей состоит из 2 носителей (NaCMC Tween 80 и 35% ПЭГ300/10% ГПБЦД/10% ПС80 в H 2 O) в комбинации. Лечение продолжали в течение 38 дней. Объемы опухоли измеряли с применением стандартных процедур дважды в неделю и публиковали результаты. Размер опухоли и массу тела записывали и анализировали дважды в неделю. Объем опухоли оценивали при помощи формулы: v=l ×w 2 ×0,536, где l - больший из измеренных диаметров, a w - меньший из перпендикулярных диаметров. Для анализа применяли программное обеспечение SAS версии 8.2 (SAS Institutes Inc, Cary, NC) для определения логарифма объема опухоли с применением модели повторяющихся измерений ANOVA с пространственной ковариационной структурой. Для каждого измеренного периода времени группы, содержащие лекарственные средства, сравнивали с контрольными группами, содержащими носитель. Значения объемов опухолей даны в виде "значение ± стандартная ошибка" для каждой из групп, содержащей лекарственное средство, определенные при помощи повторяющихся измерений ANOVA для каждой группы (SAS for Mixed Models, 2 nd Ed., Littell et al., SAS Institutes Inc., Cary, NC). Данные зависимости объема опухоли от времени для каждого животного применяли для вычисления площади под кривой (AUC) с применением правила трапеции. Указанные AUC анализировали при помощи 2-факторной ANOVA модели с применением логарифмического преобразования для сглаживания различий между группами. Два фактора представляли собой дозировку 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидина и дозировку гидрохлорида эрлотиниба. Влияние взаимодействия между представленными факторами применяли для исследования отклонений от аддитивности (An Introduction to Statistical Methods and Data Analysis, R. Lyman Ott, 1993, Duxbury Press, Belmont, CA). Результаты указанного исследования представлены ниже в таблице, n (число мышей) составляет 9 для всех данных, за исключением данных, полученных для дня 17 в группе, получавшей носители, и данных, полученных для дня 38 в группе, получавшей гидрохлорид эрлотиниба, где число мышей равно 8. Данные в столбце, озаглавленном "Значим." отражали, является ли различие по сравнению с группой, получавшей носитель, статистически значимым: НЗ=незначительно (р> 0,05), *=0,01 <р ≤0,05, **=0,001 <р ≤0,01, ***=р ≤0,001. AUC логарифмического объема опухоли для комбинации (3,58) статистически отличается (р <0,0008) от данных для носителя (3,91), тогда как значения AUC логарифмического объема опухоли для групп, получавших лекарственные средства, ингибитором р70 S6 киназы, введенным индивидуально (3,85), и гидрохлоридом эрлотиниба, введенным индивидуально (3,76), незначительно отличались от данных, полученных для носителя. Группы, получавшие лекарственные средства: ингибитор р70 S6 киназы индивидуально и гидрохлорид эрлотиниба индивидуально также значительно отличались от группы с комбинацией лекарственных средств при попарном сравнении значений AUC логарифмического объема опухоли. При комбинировании с ингибитором EGFR, гидрохлоридом эрлотиниба, 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин проявляет способность к ингибированию роста немелкоклеточных раковых опухолей А549 в легких. В указанном исследовании 4-[4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил]-1Н-пиразоло[3,4-d]пиримидин вводили в дозировке 2,5 мг/кг ежедневно в течение 35 дней, а гидрохлорид эрлотиниба вводили в дозировке 20 мг/кг ежедневно в течение 35 дней. Указанные дозировки были меньше оптимальных дозировок для каждого из агентов, при этом комбинация проявляла повышенную эффективность по сравнению с группами, получавшими ингибитор киназы р70 S6 и гидрохлорид эрлотиниба в отдельности. Комбинированная терапия не привела к какой-либо выраженной токсичности. Представленные результаты показывают, что модулирование находящихся выше и ниже частей пути PI3K обеспечивает повышенную эффективность. |