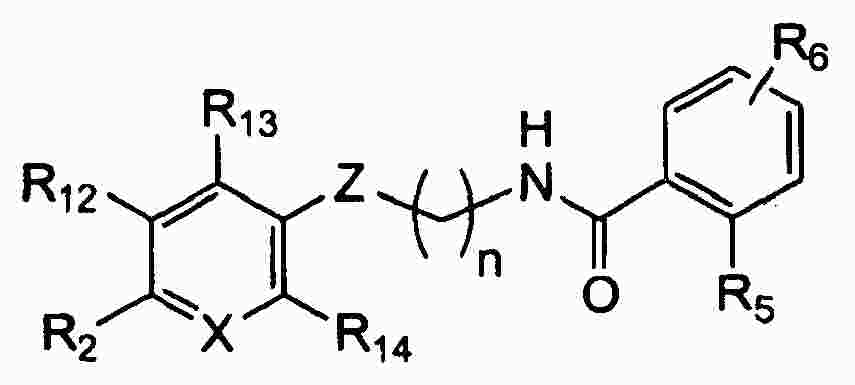
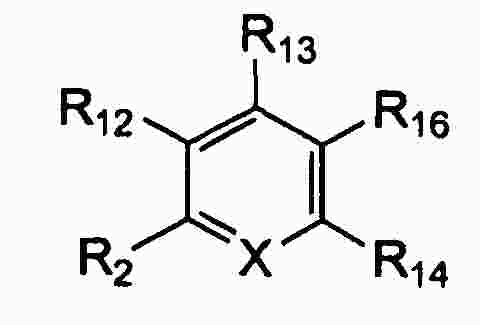
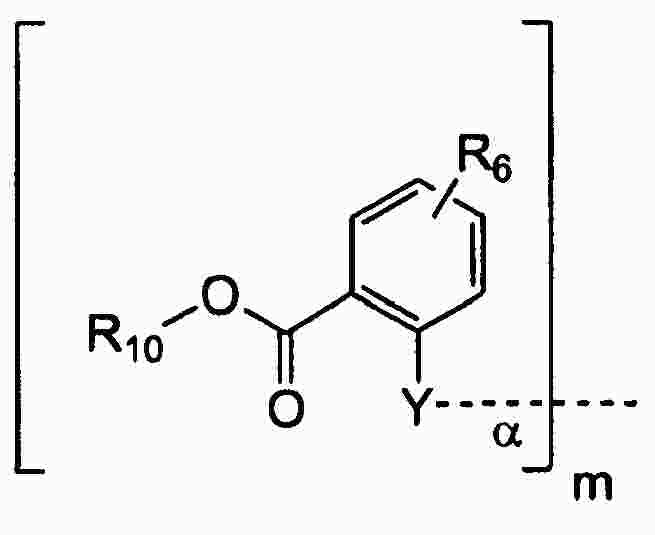
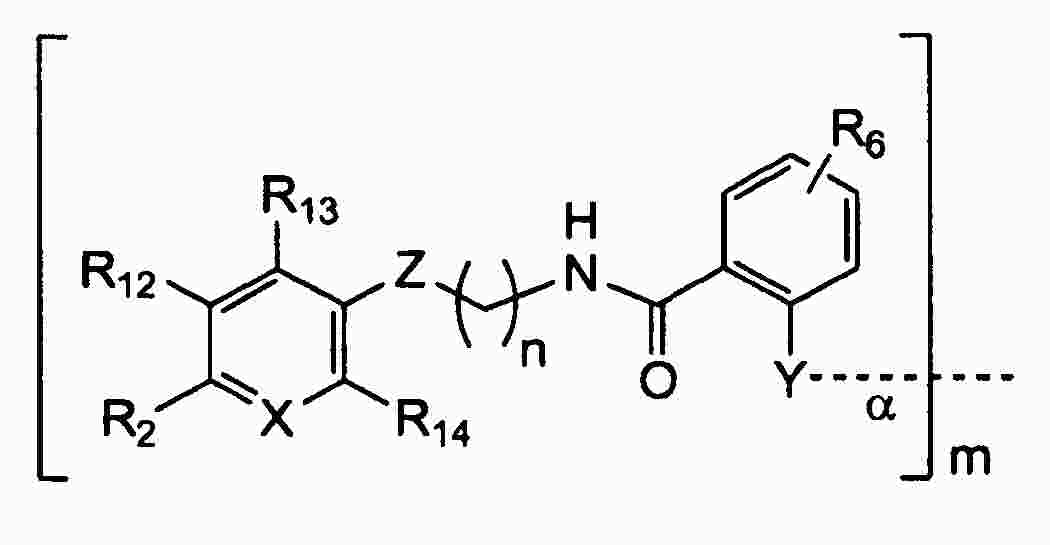
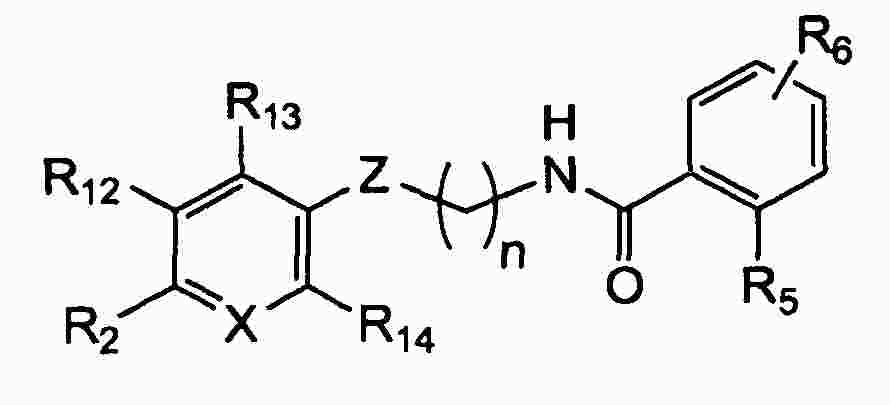
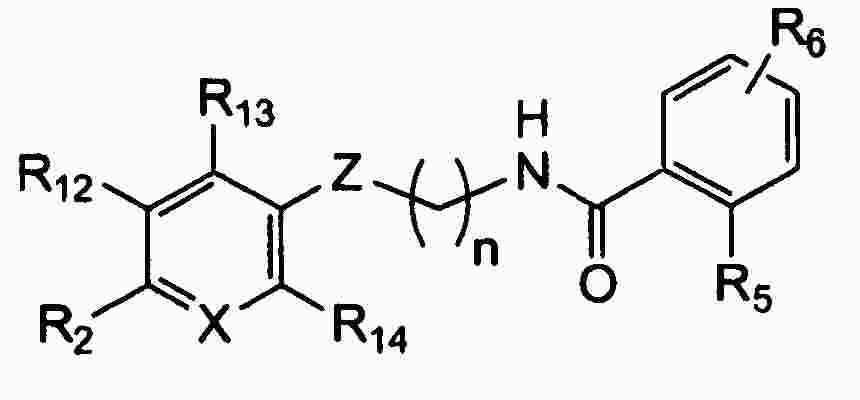
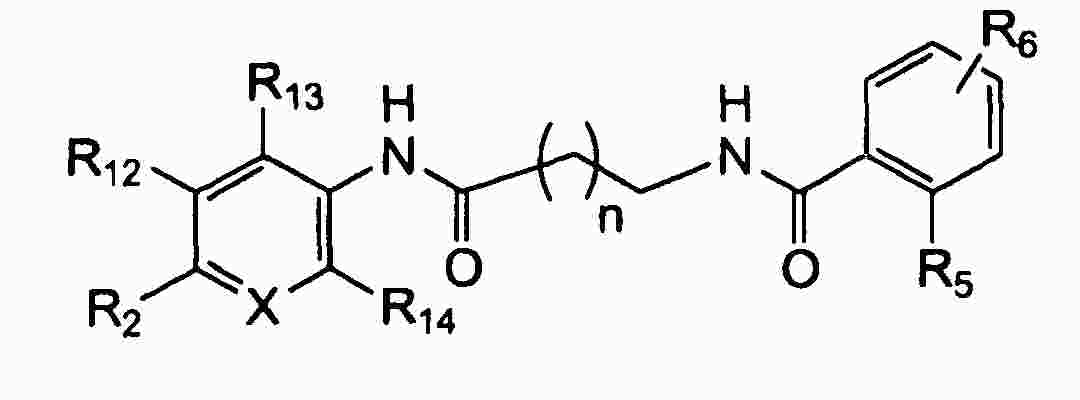
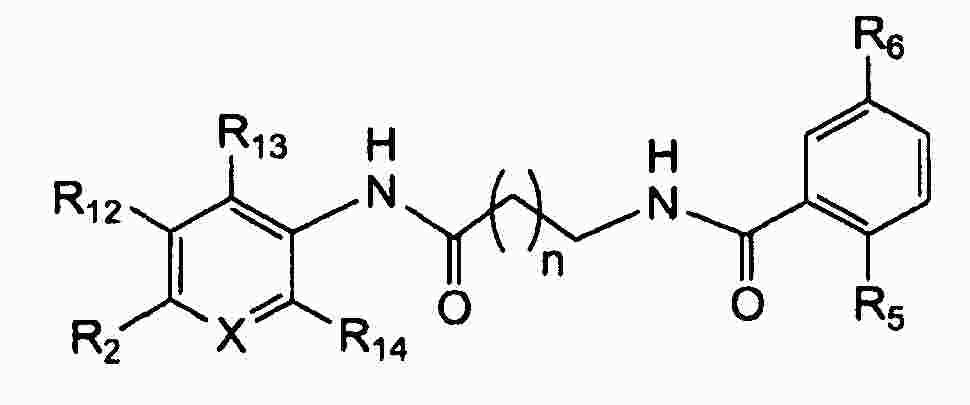
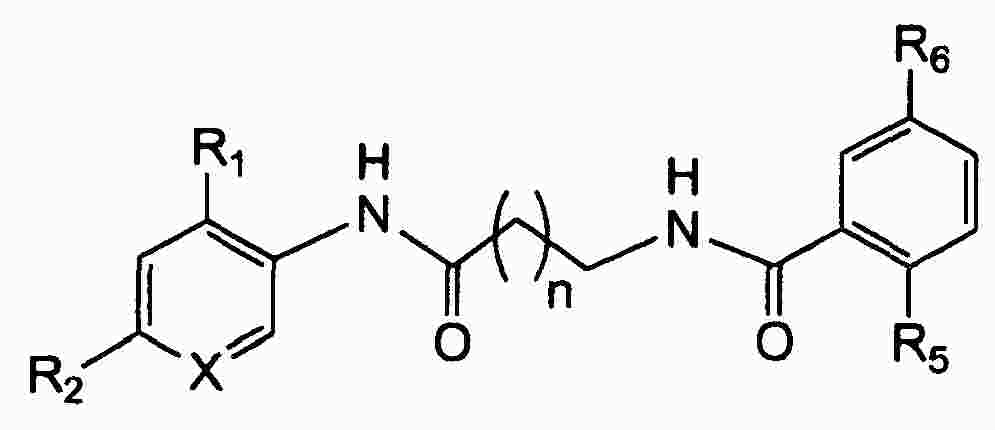
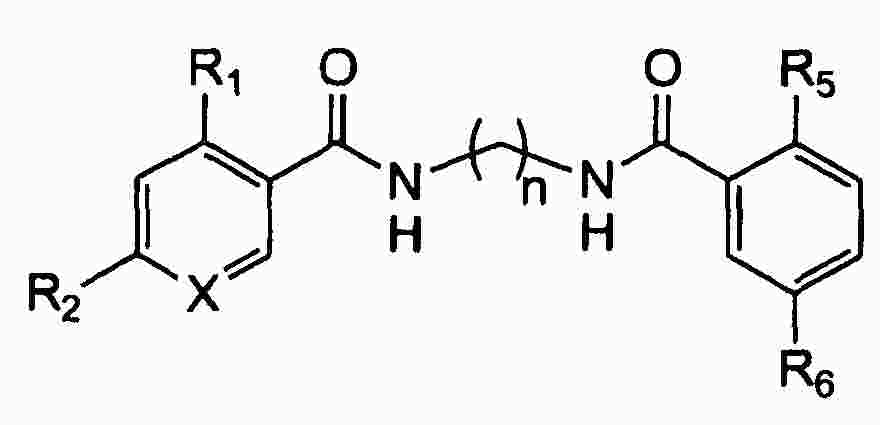
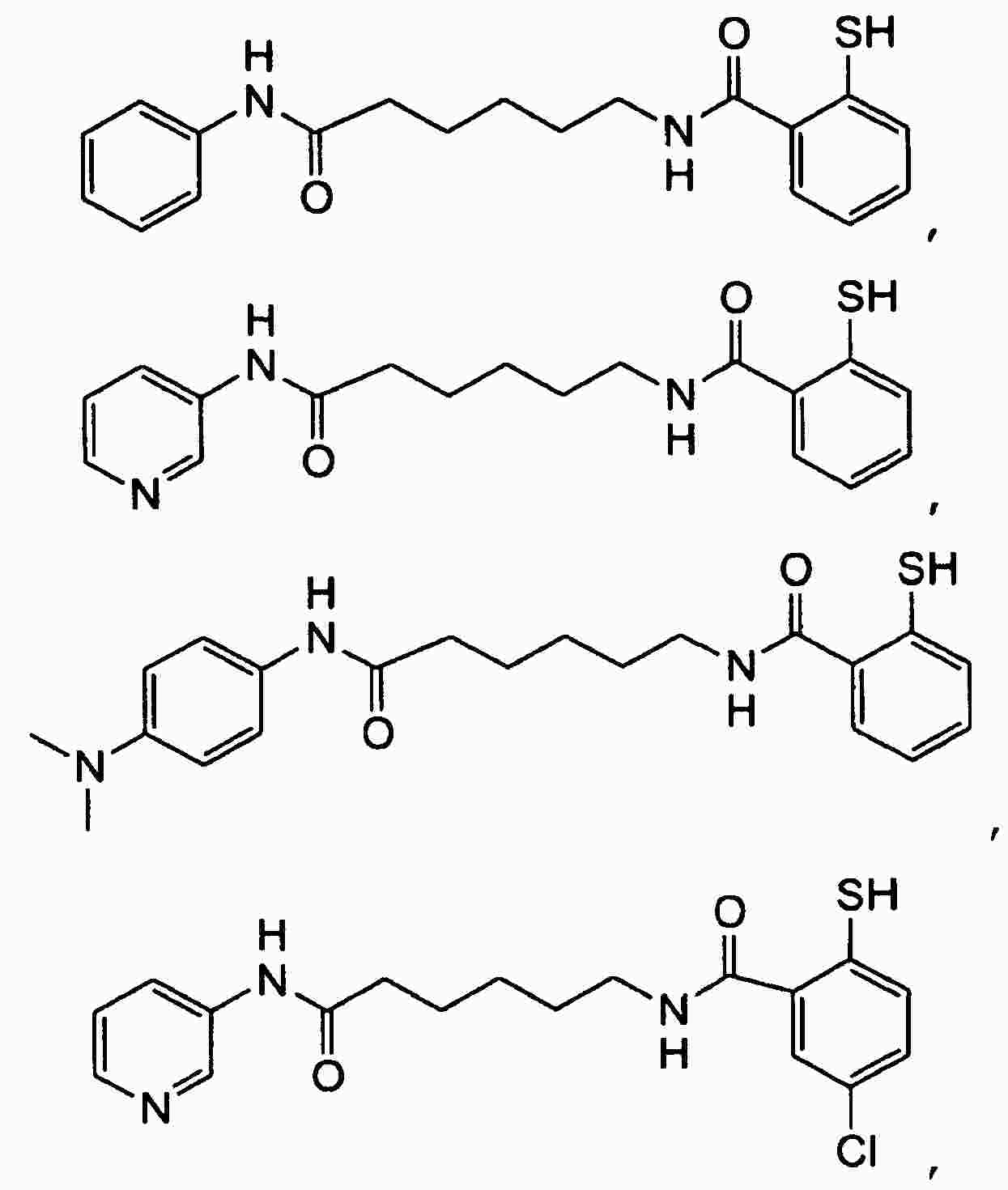
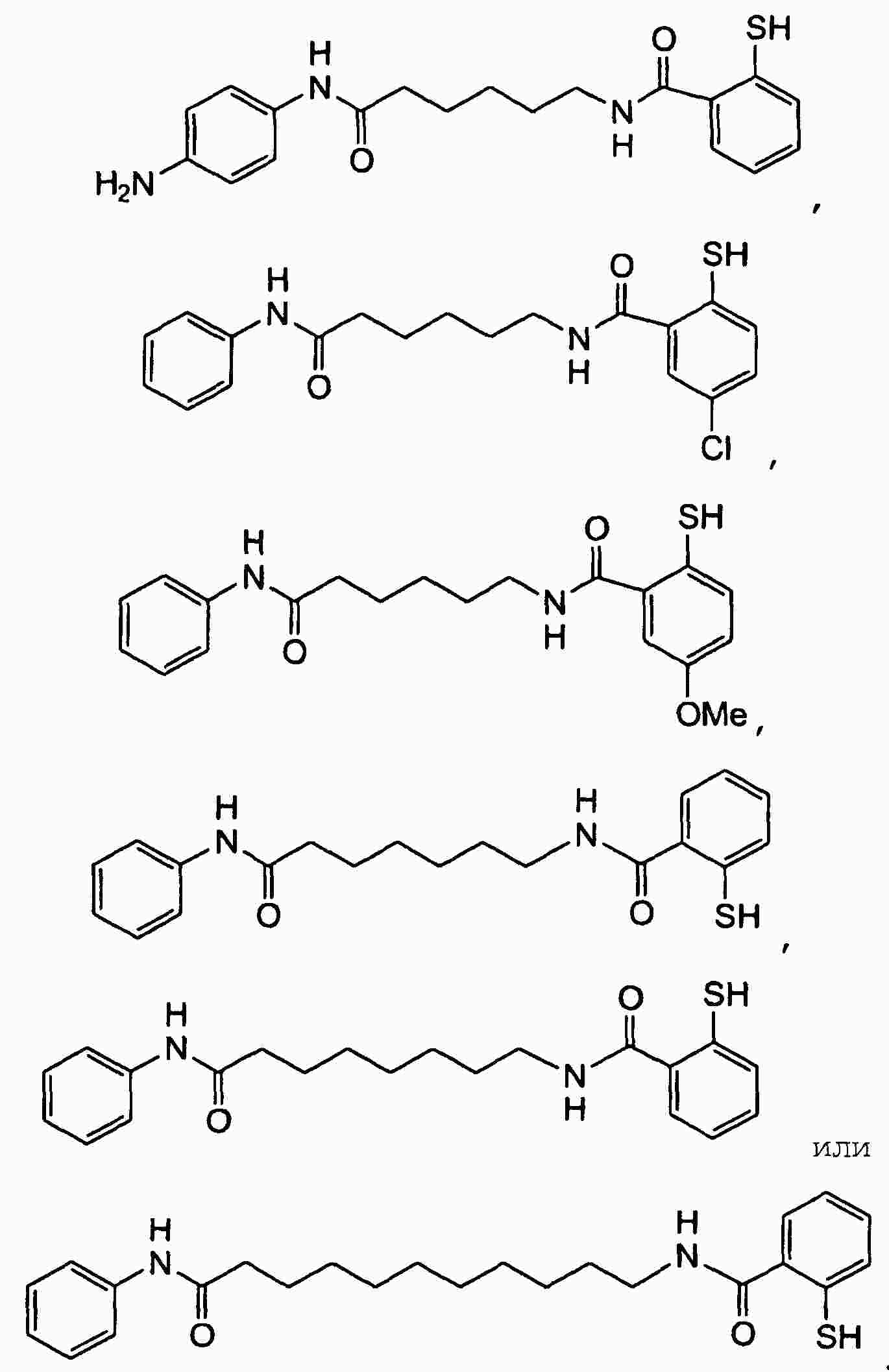
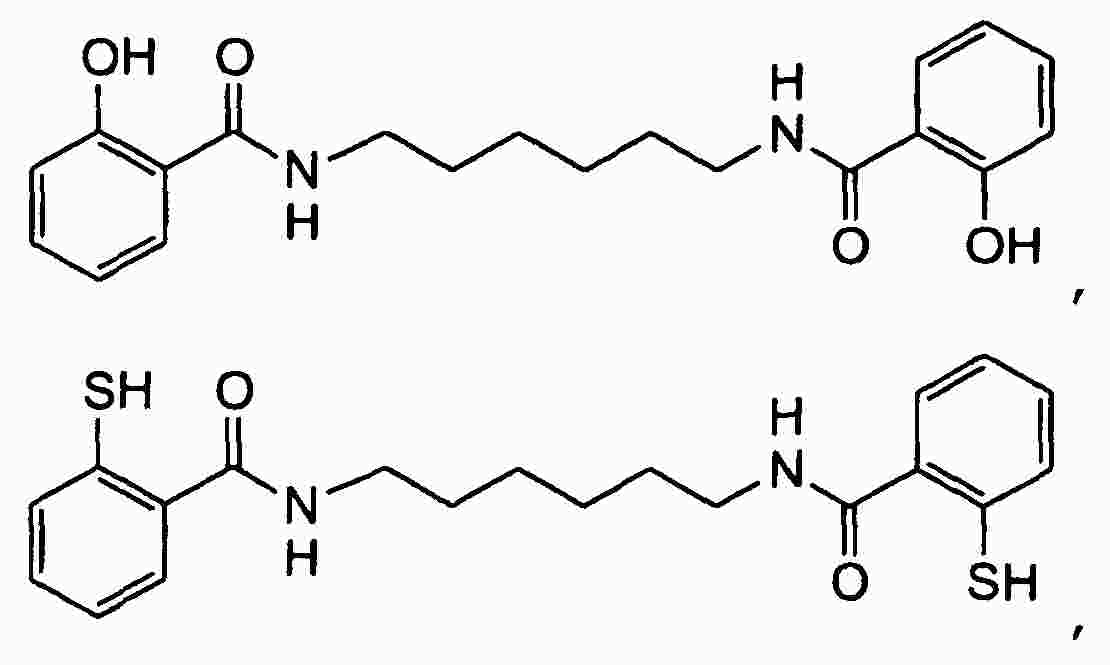
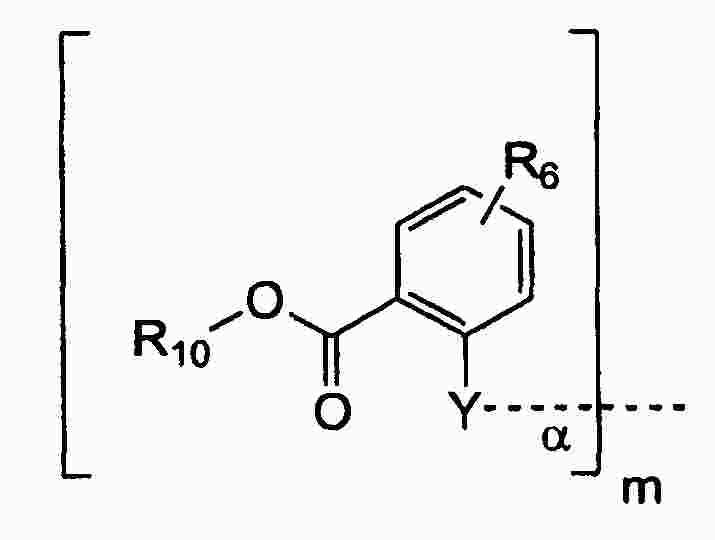
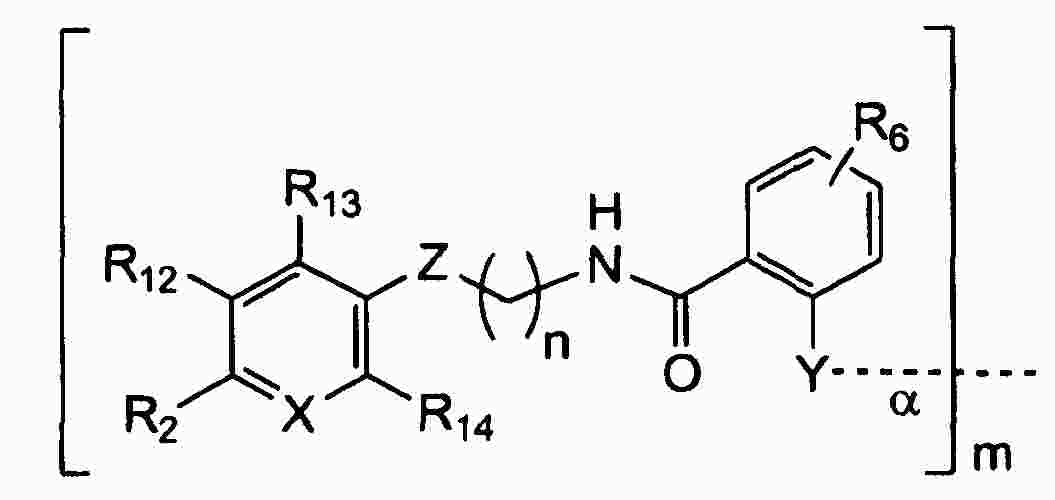
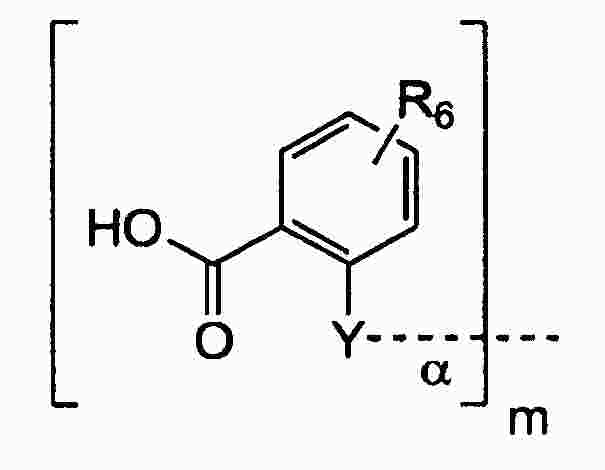
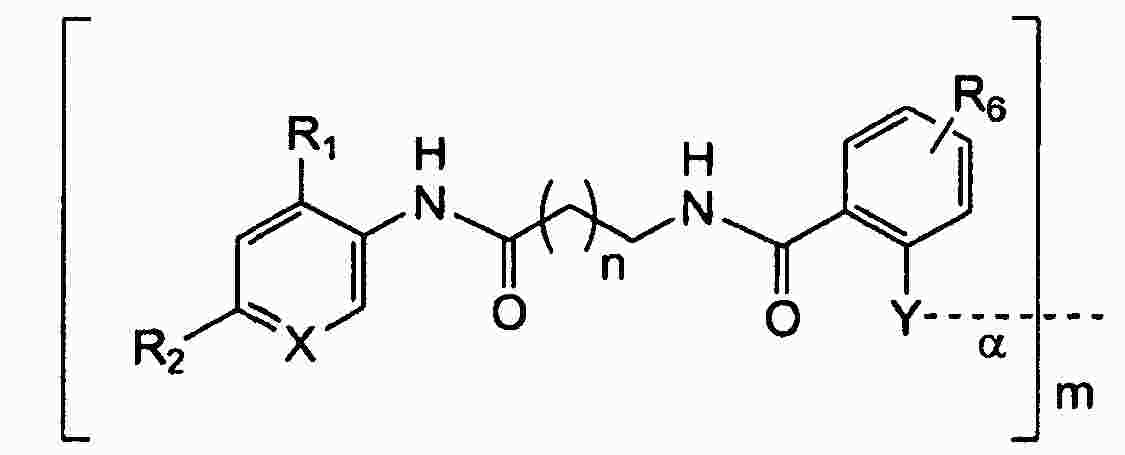
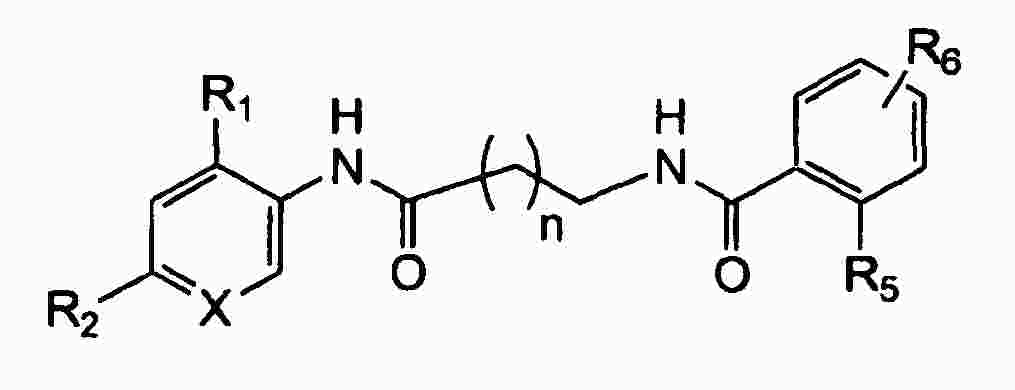
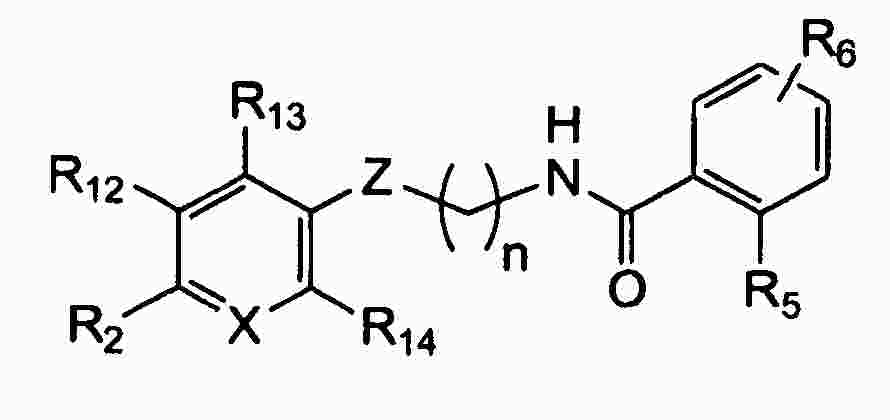
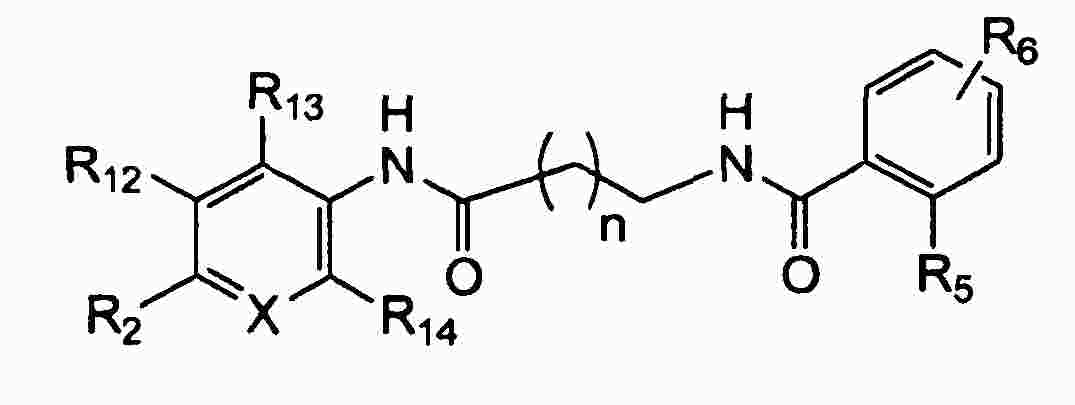
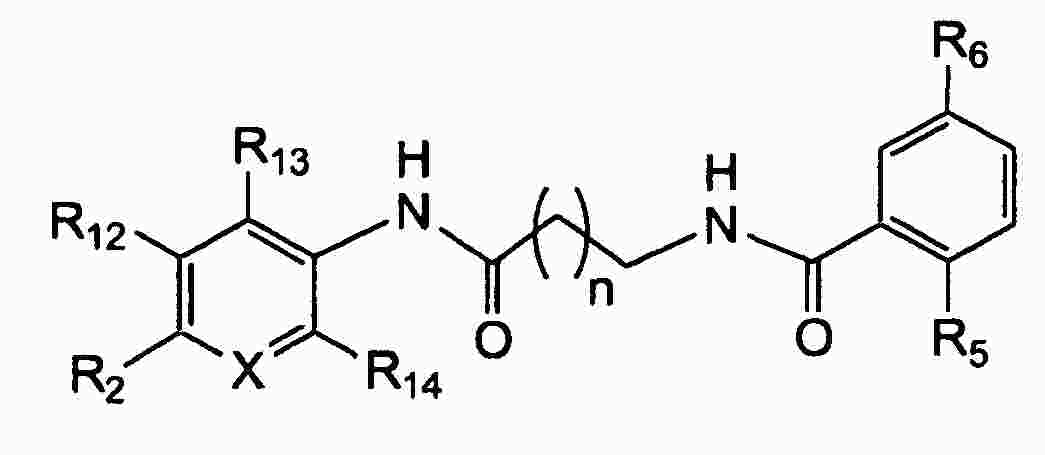
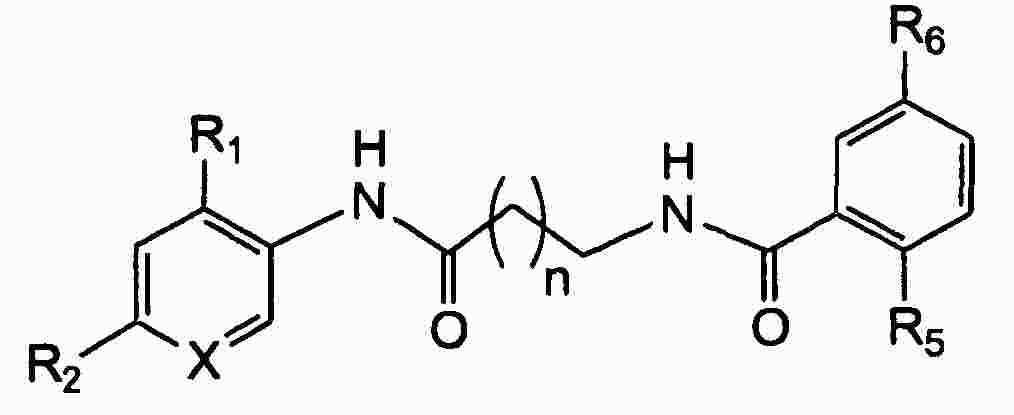
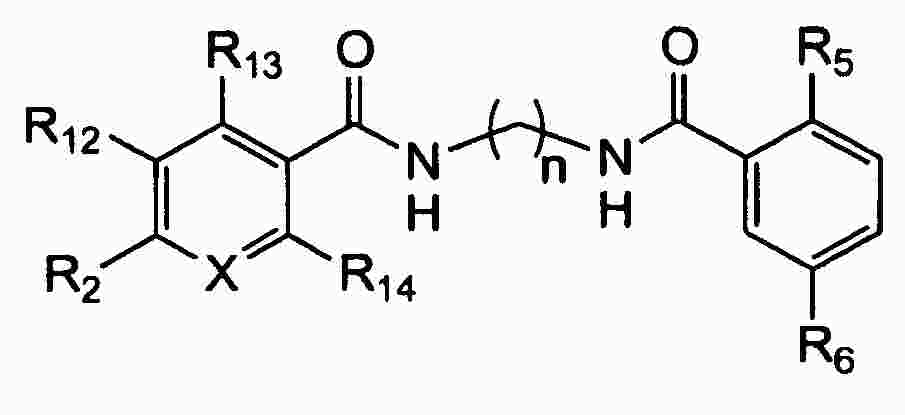
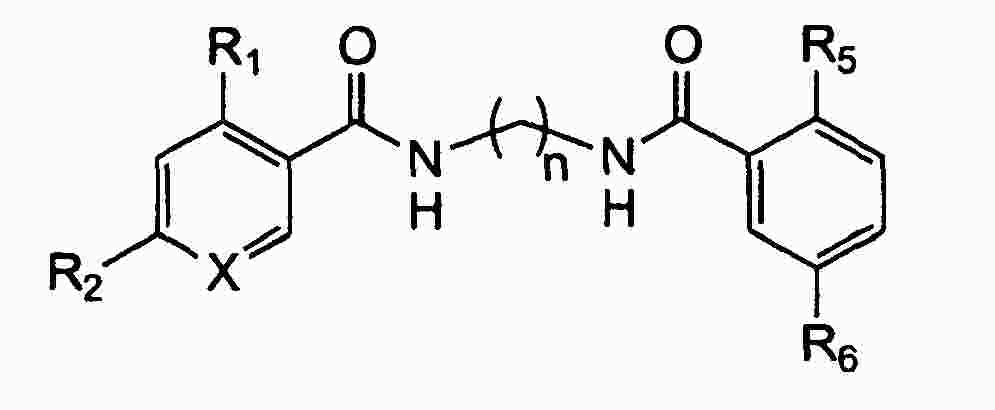
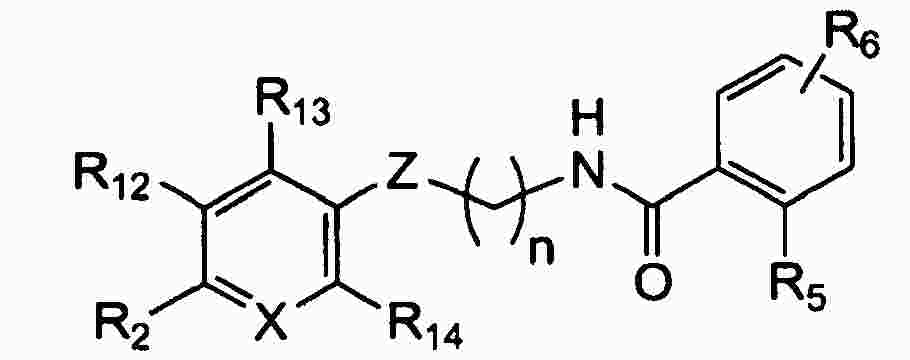
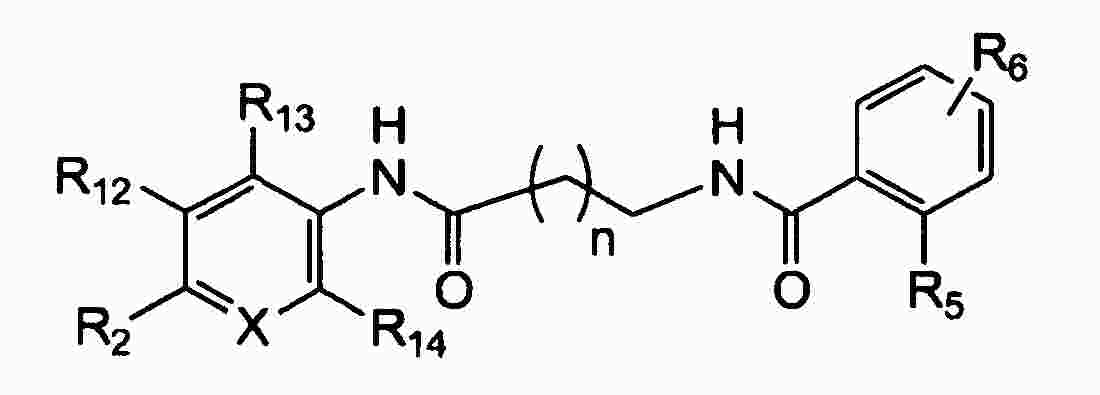
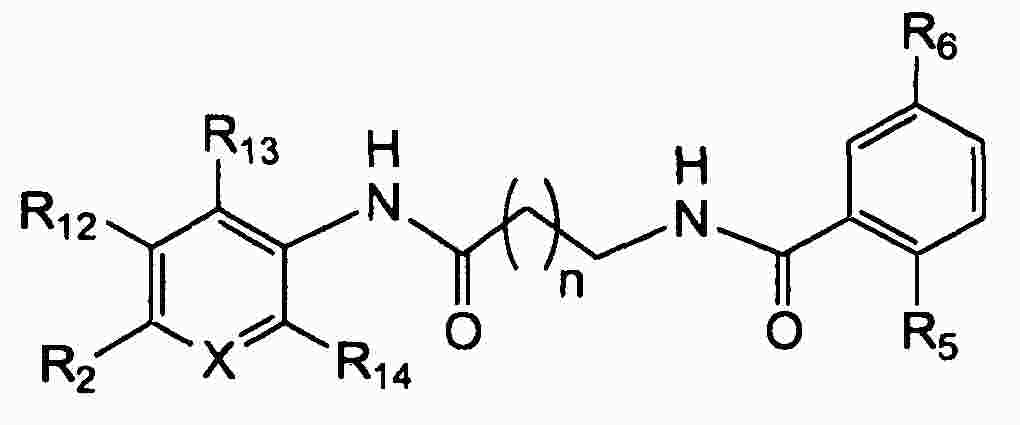
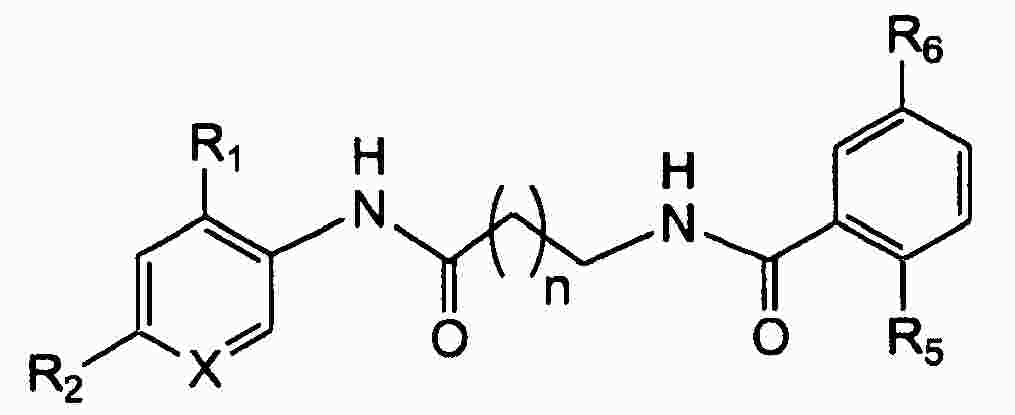
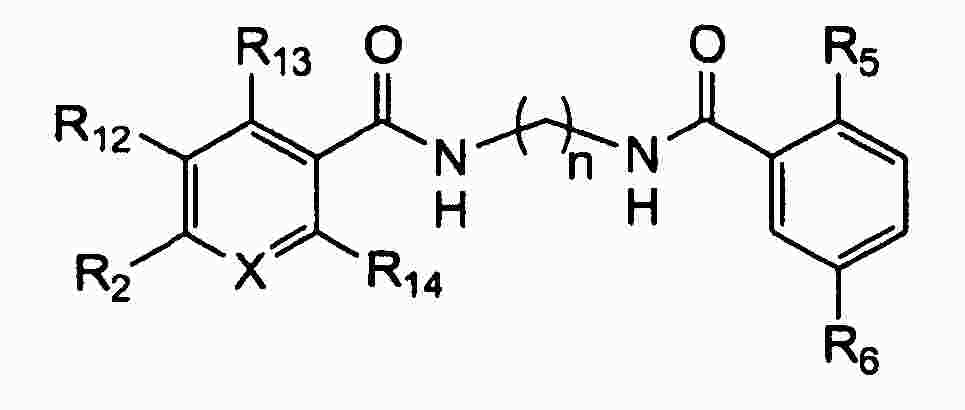
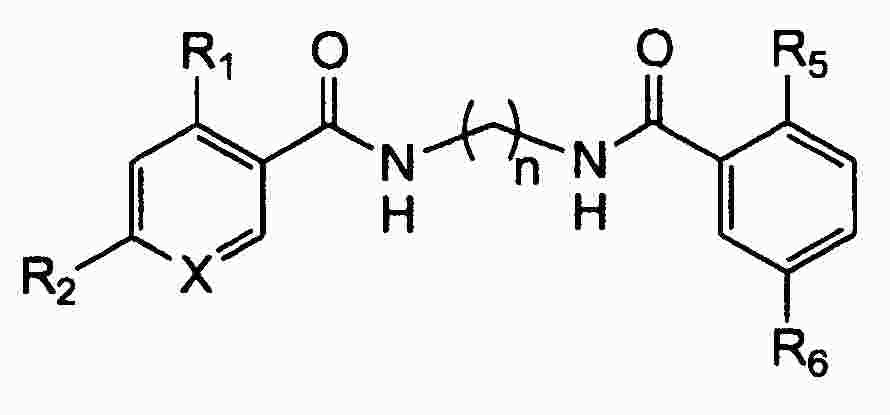
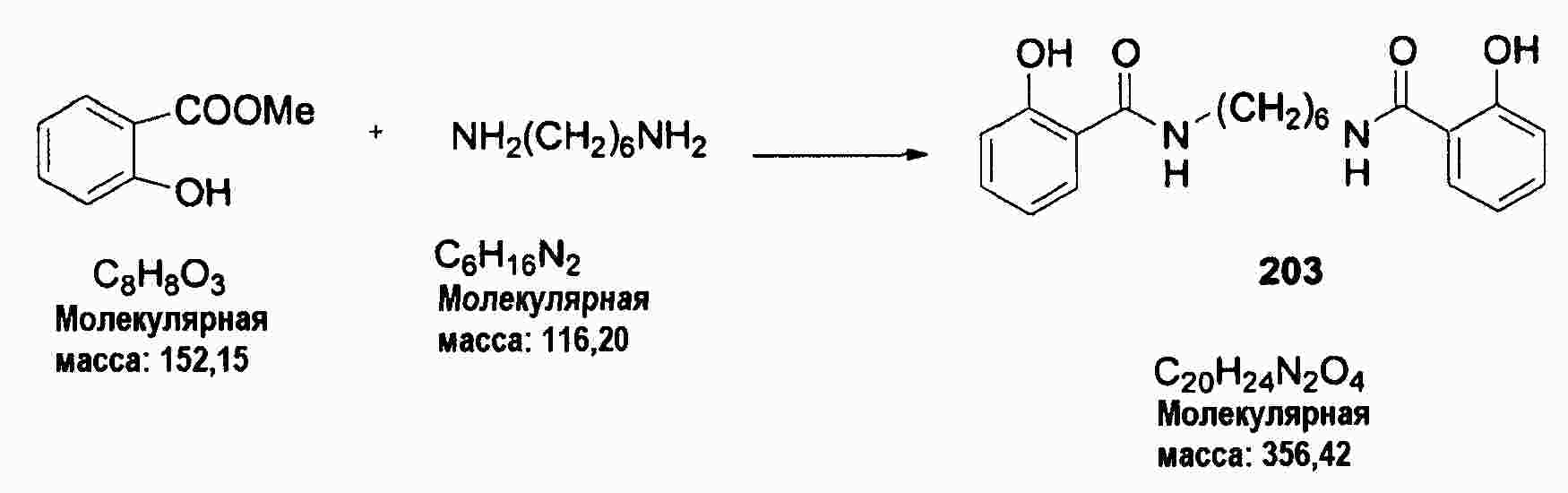
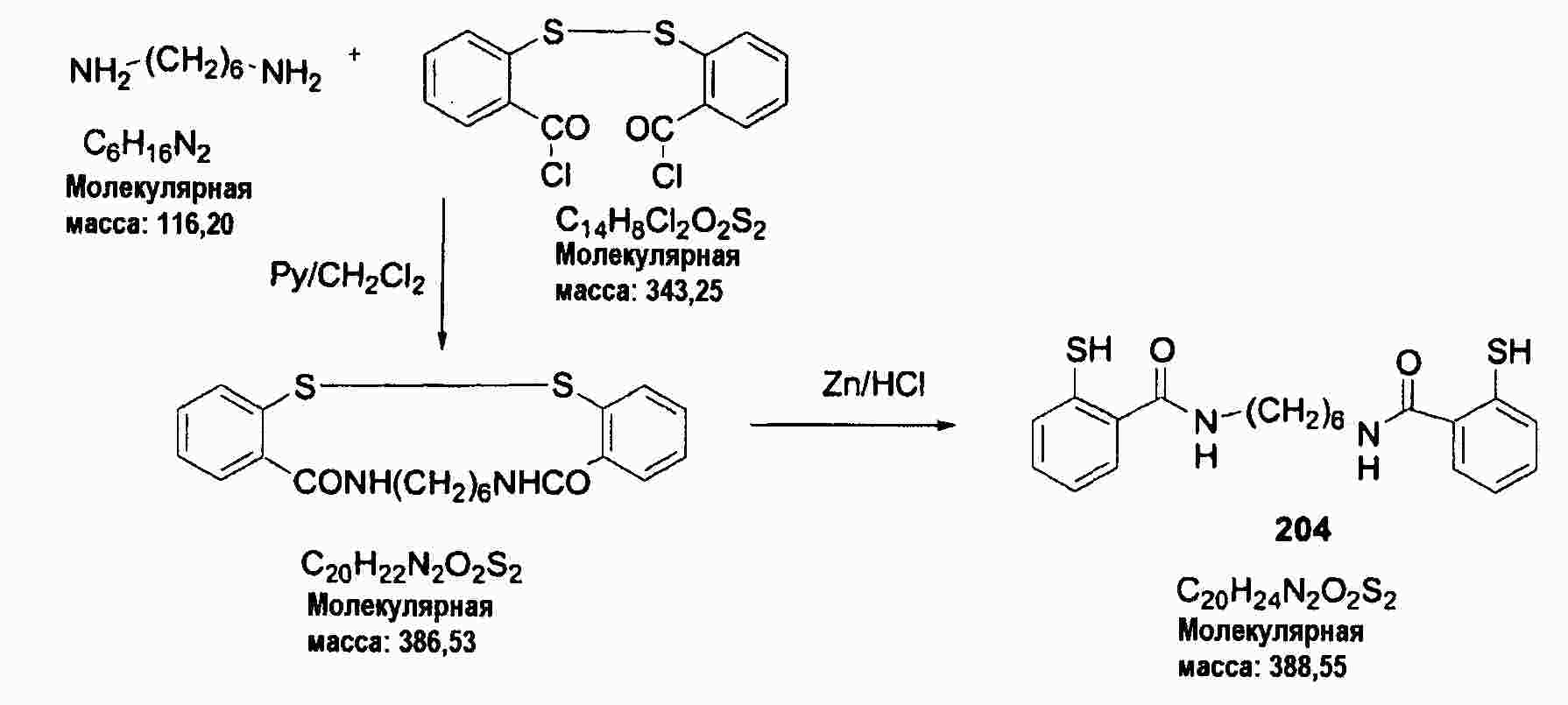
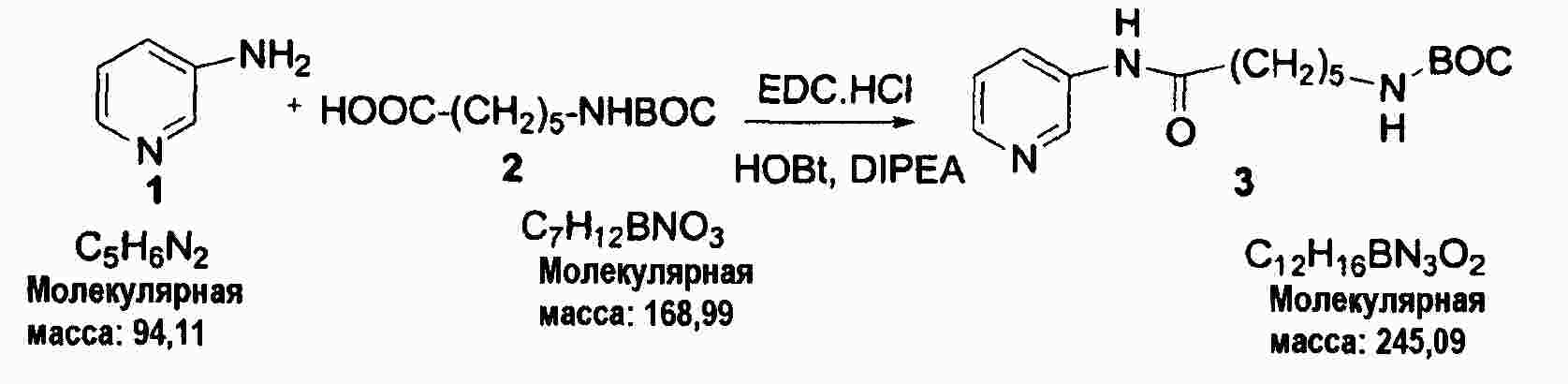
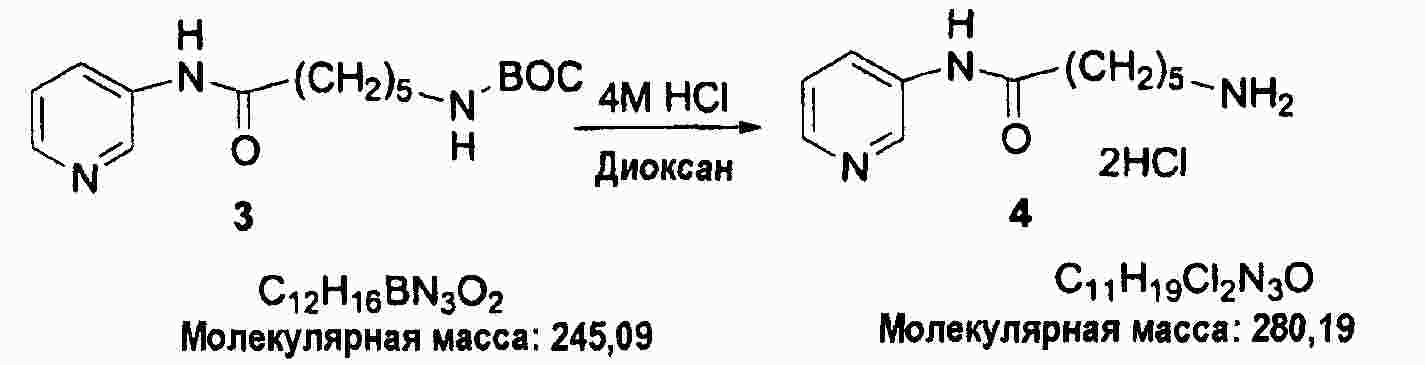
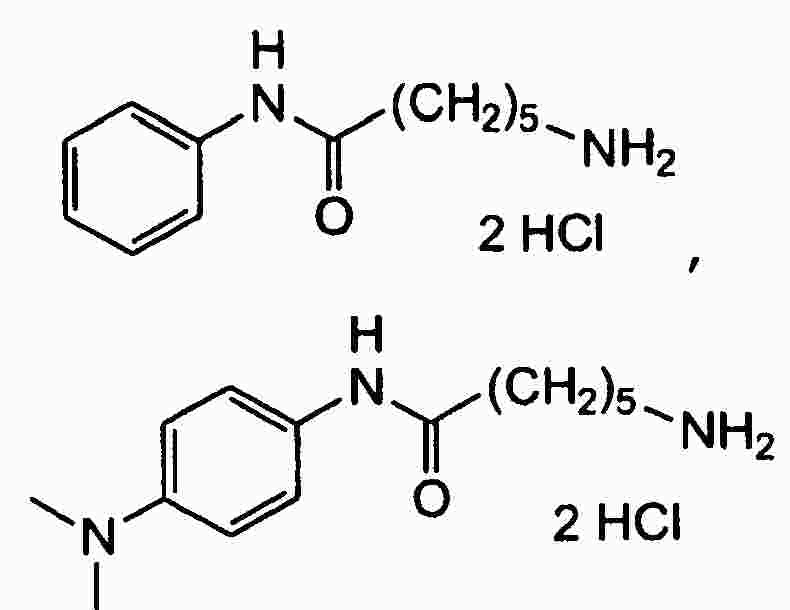
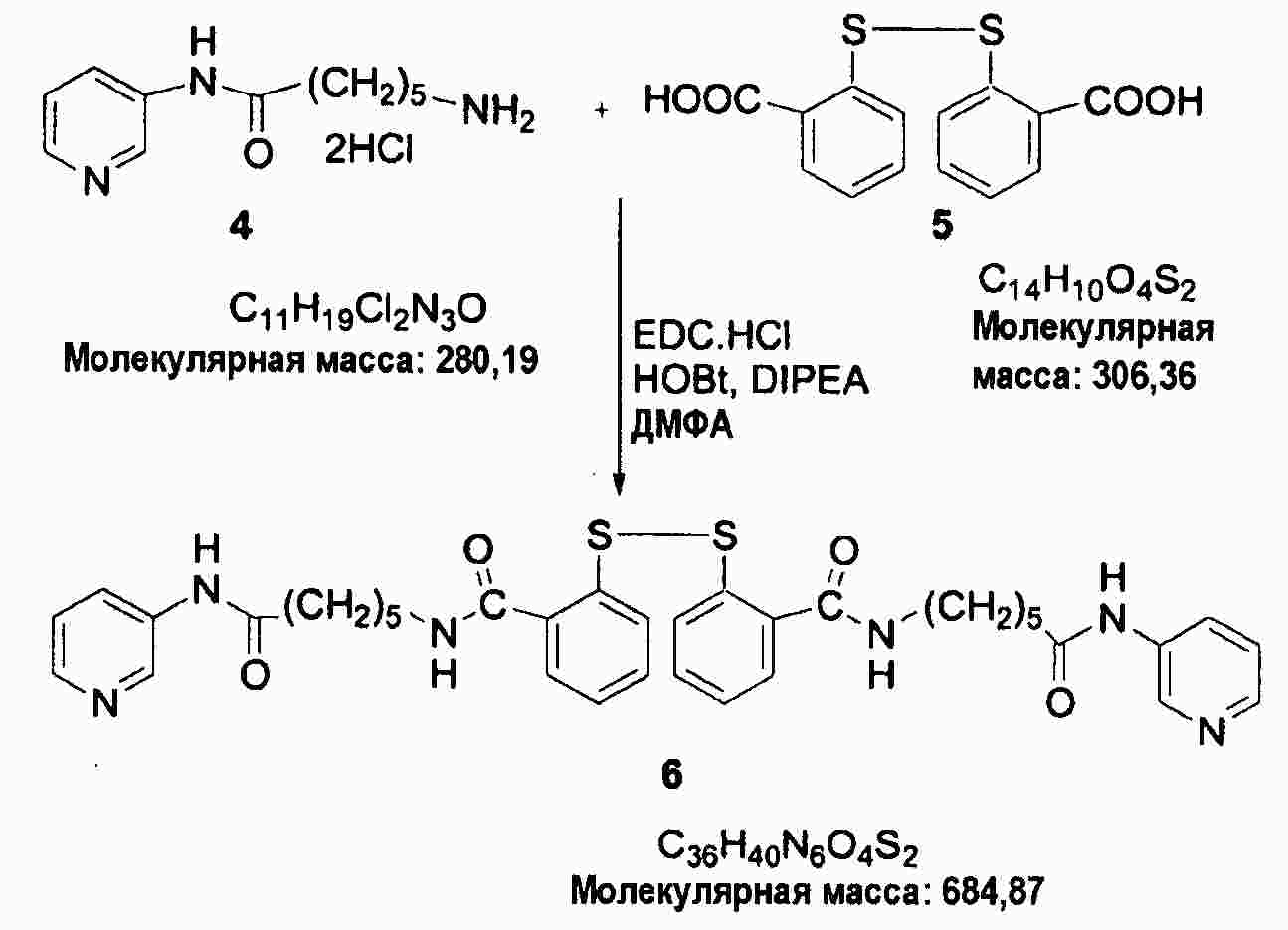
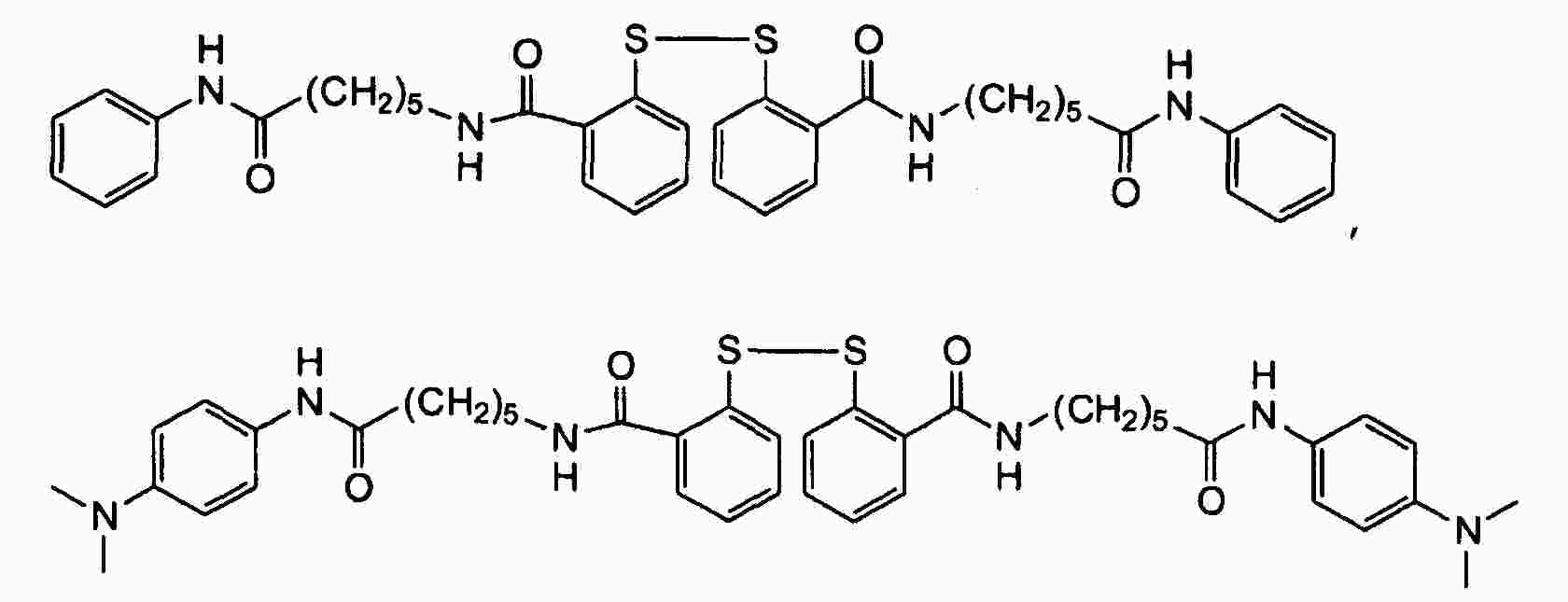
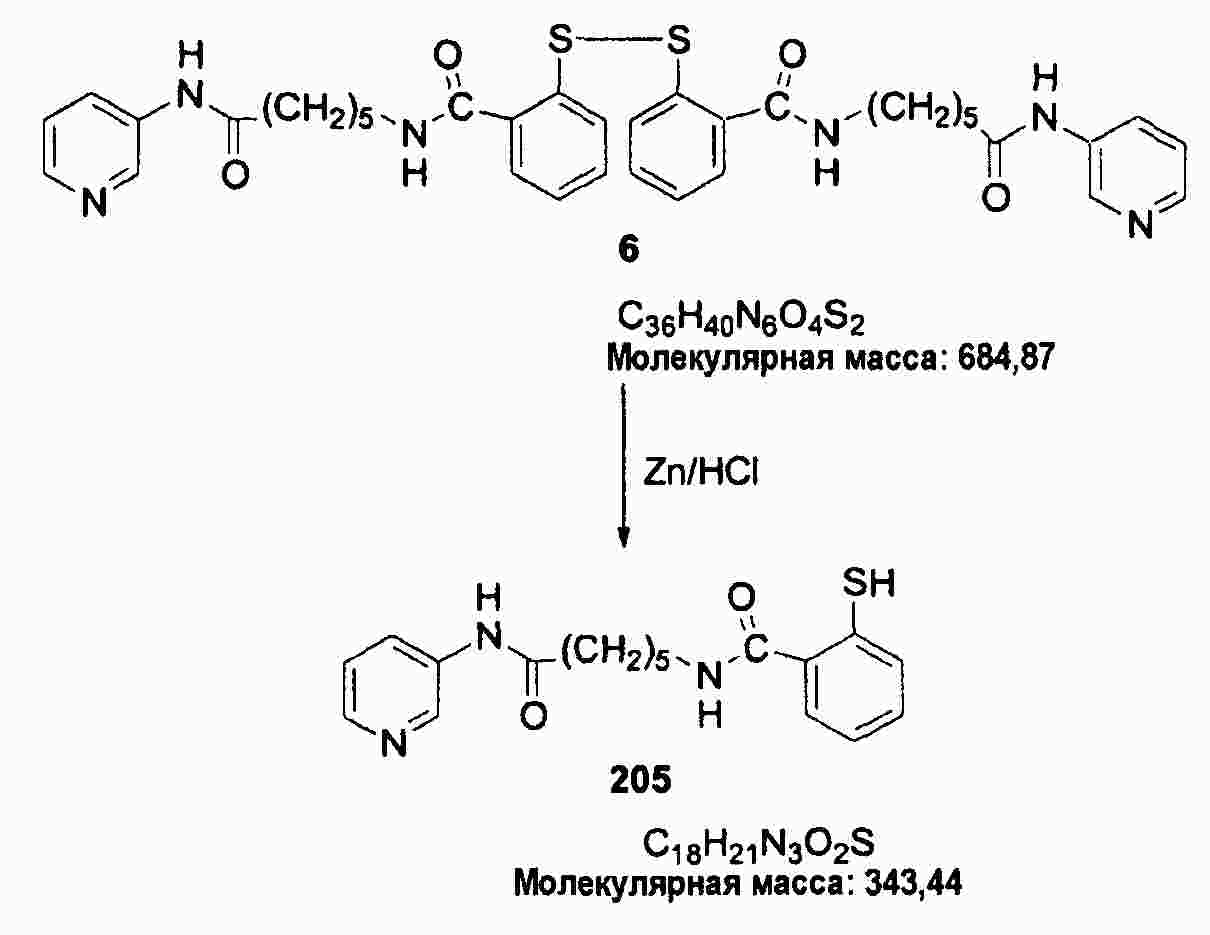
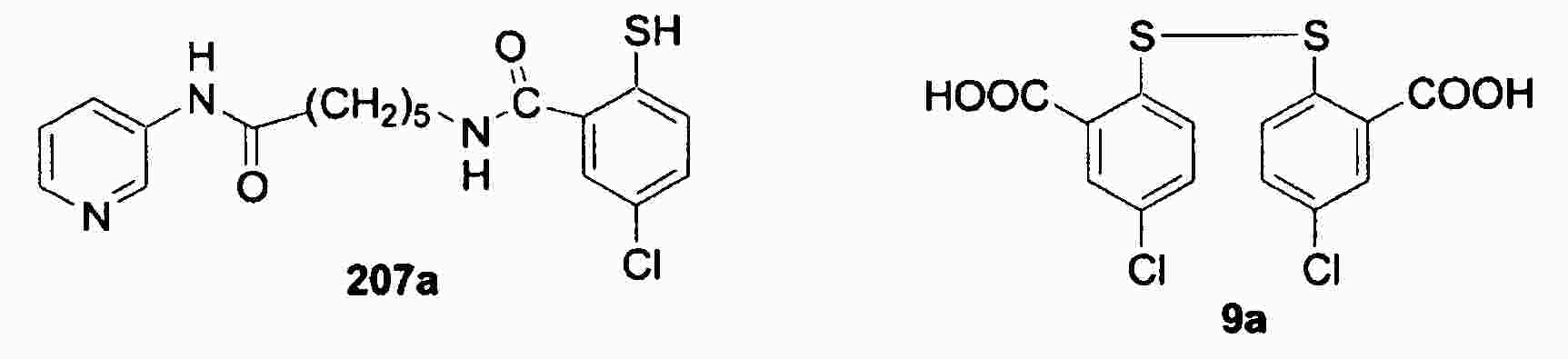
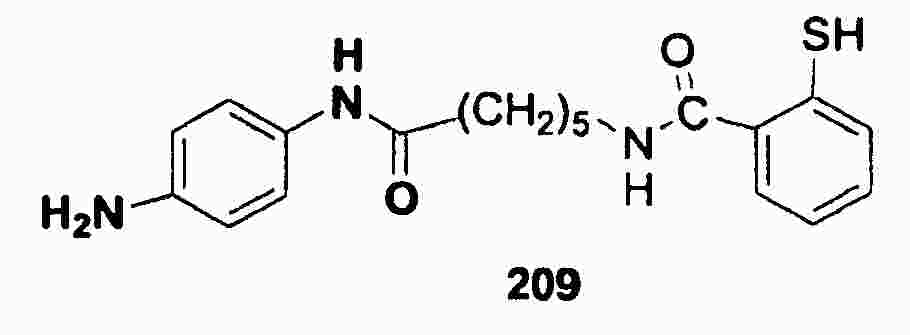
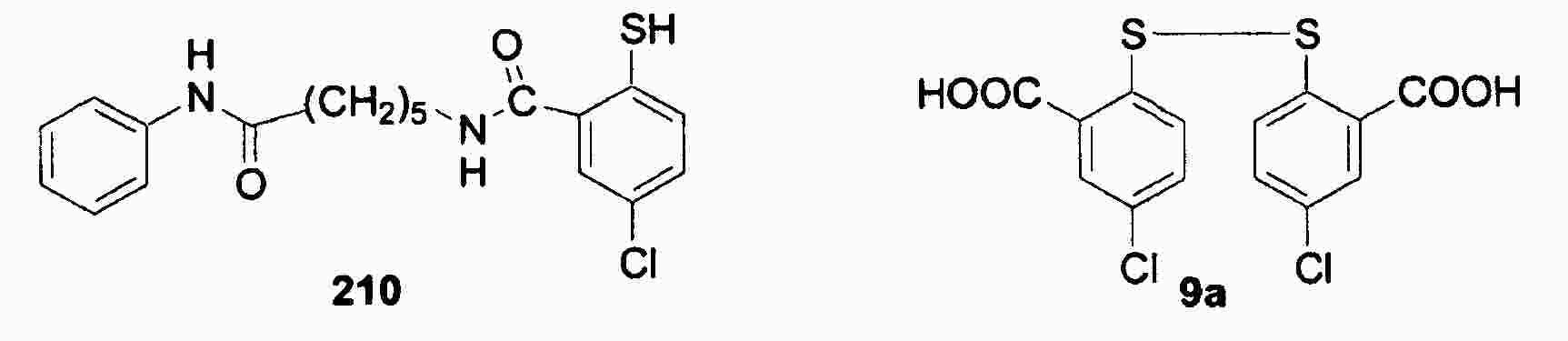
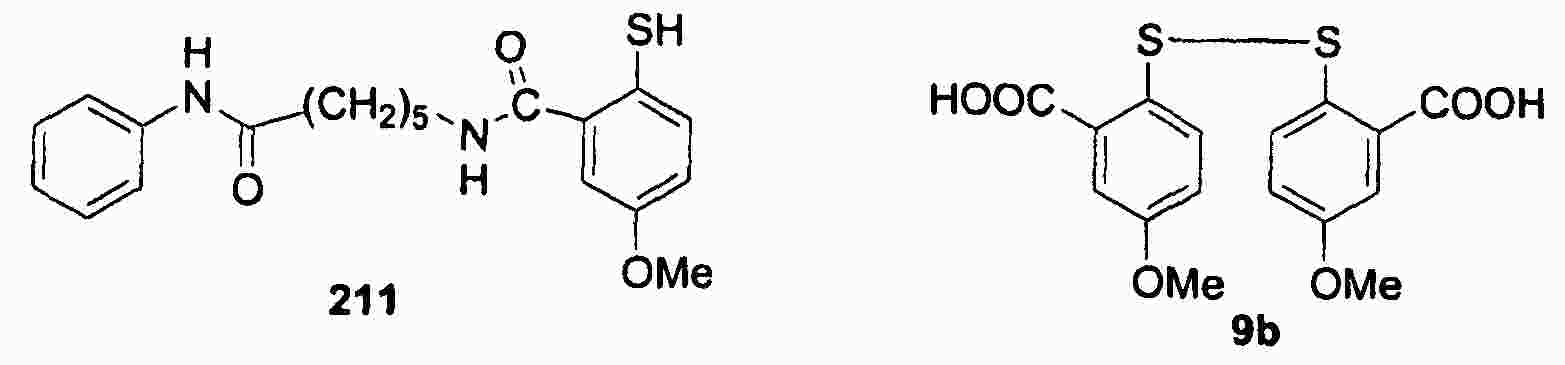
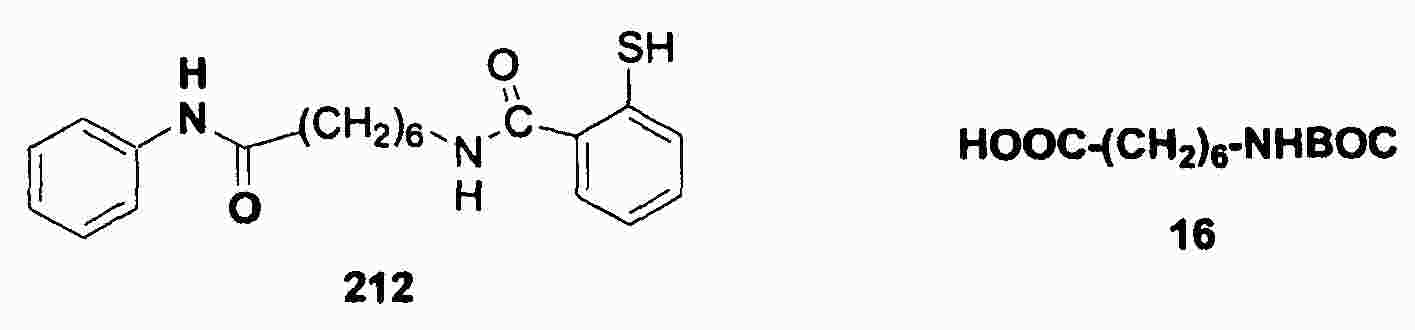
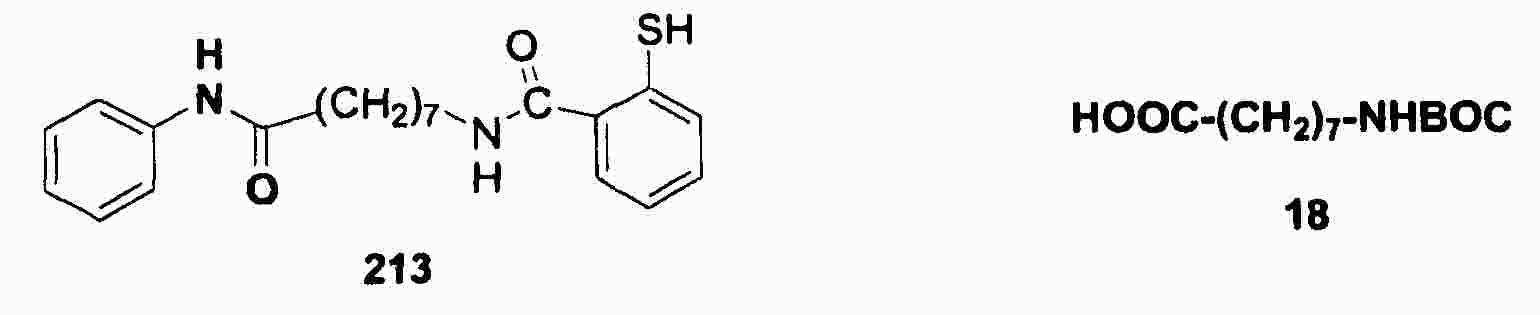
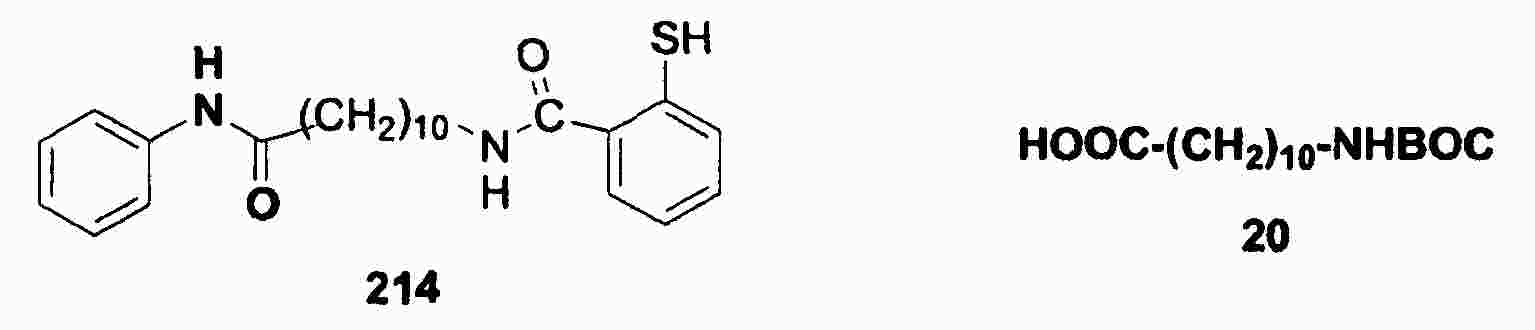
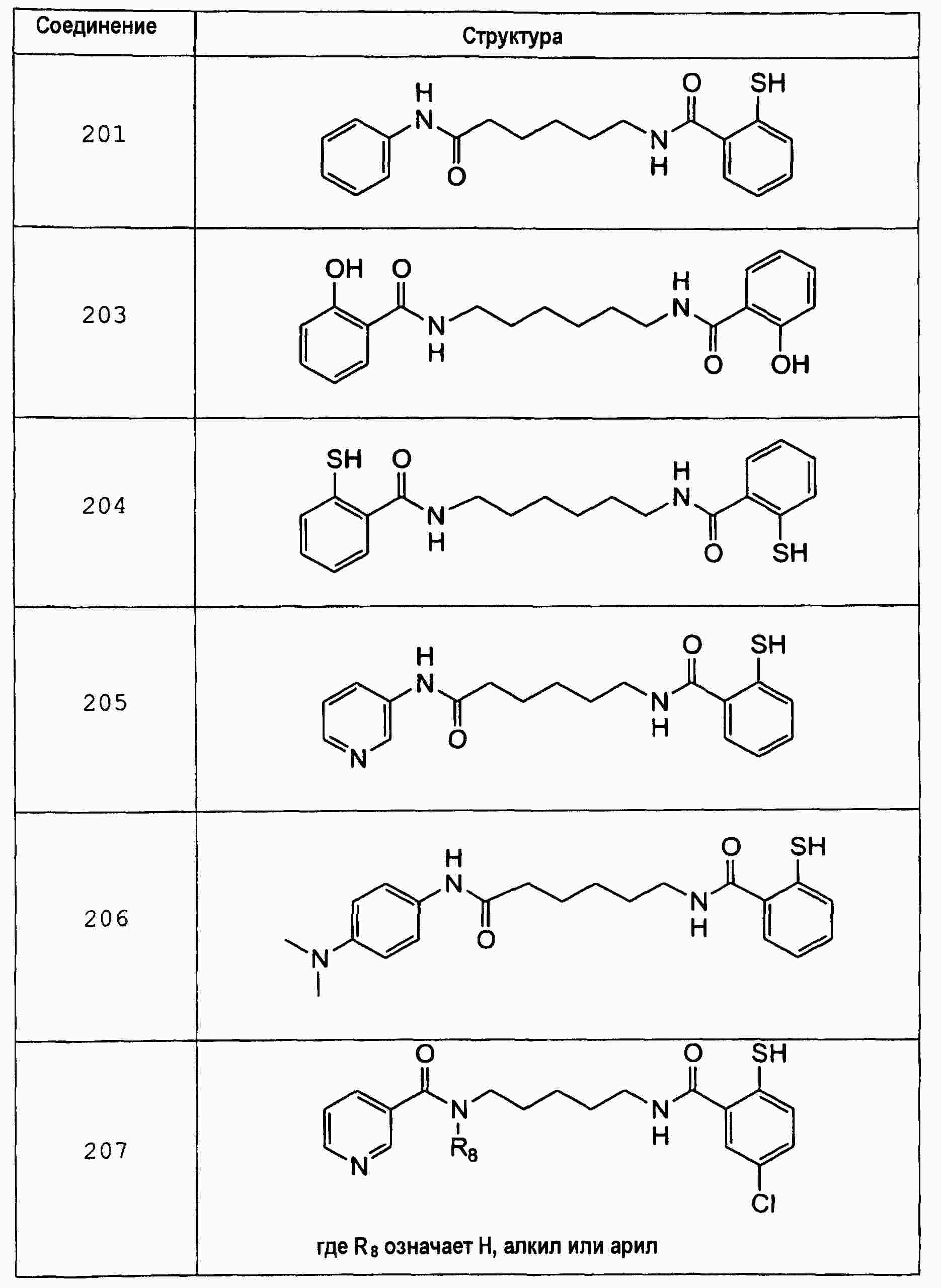
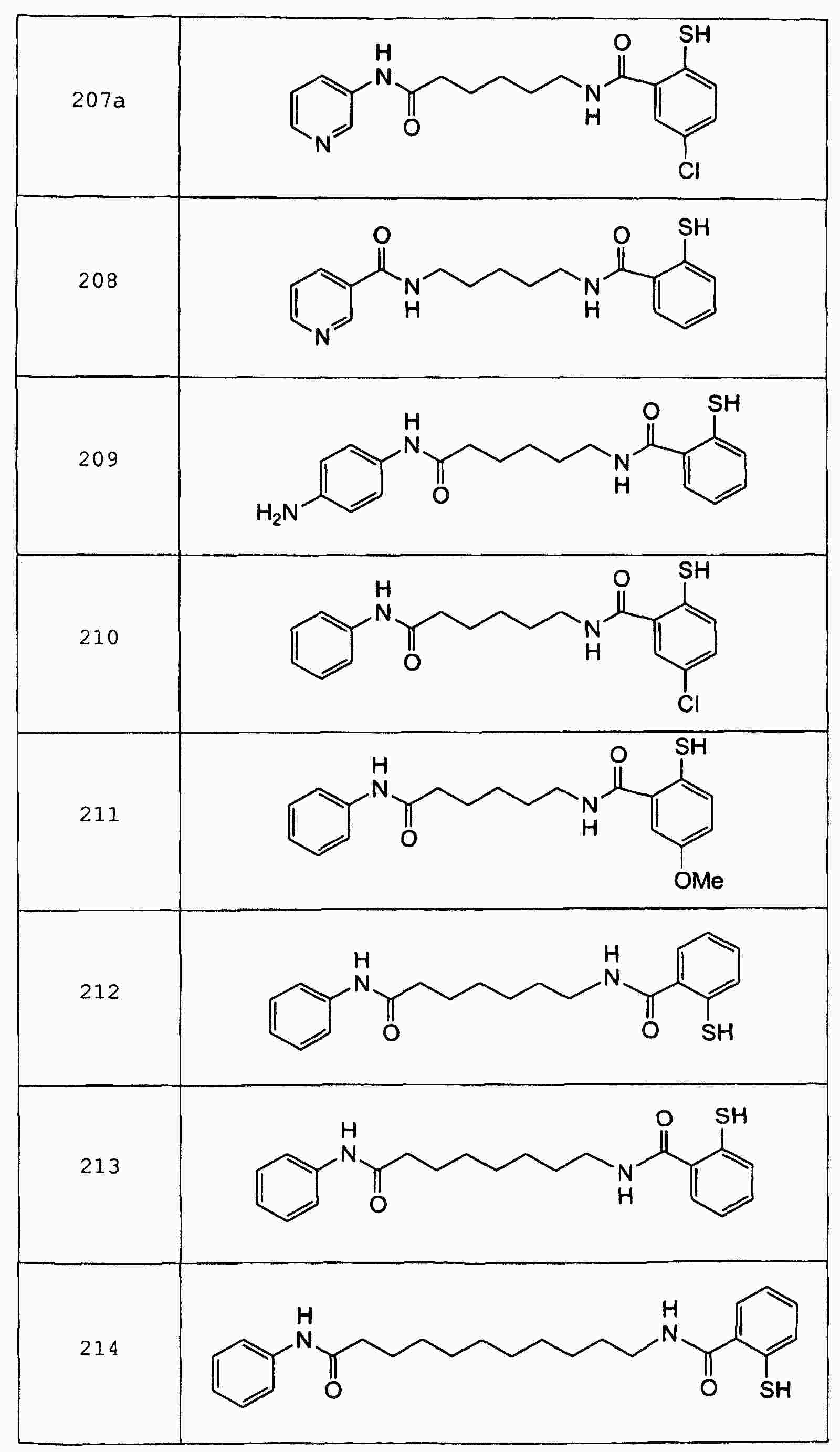
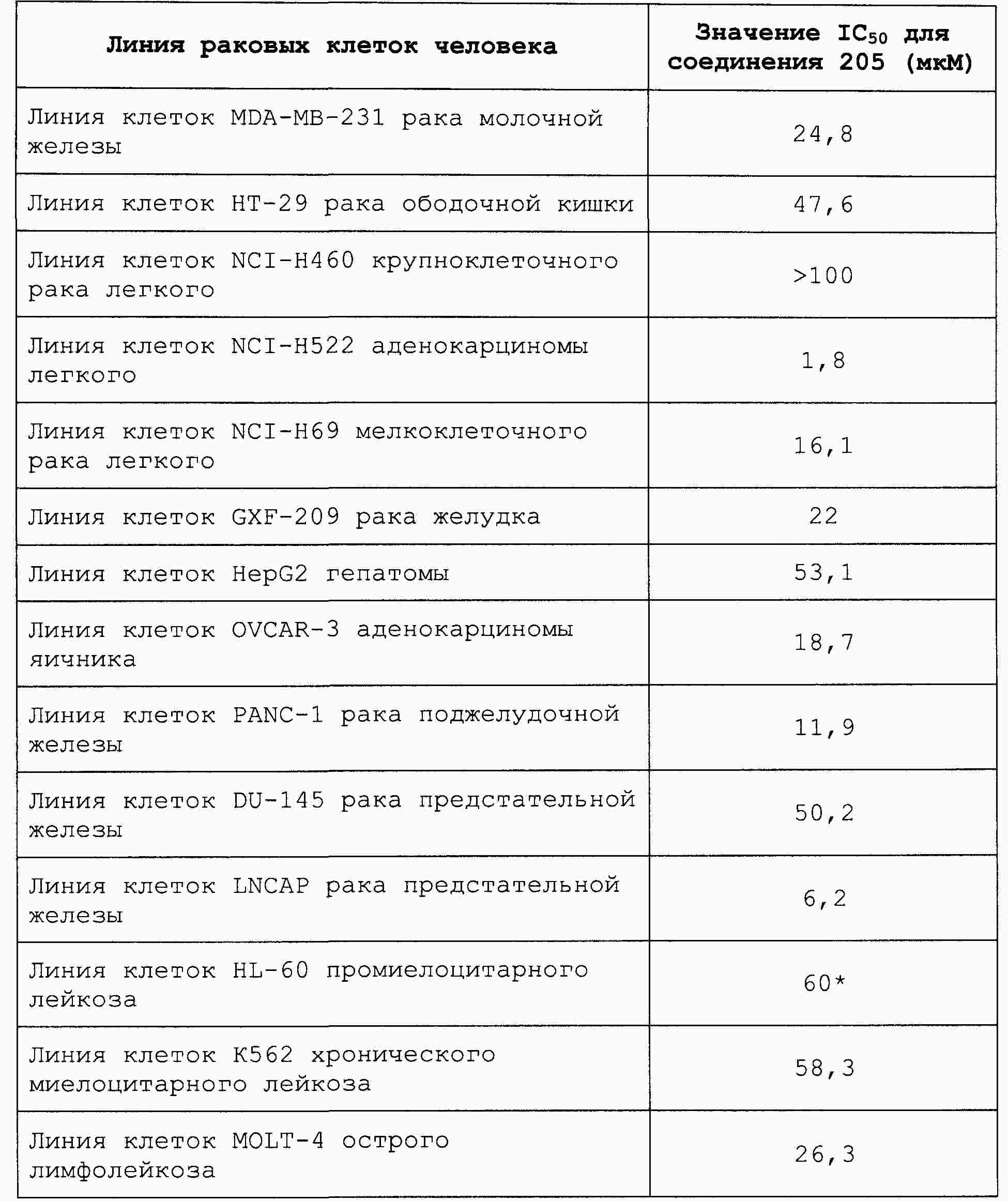
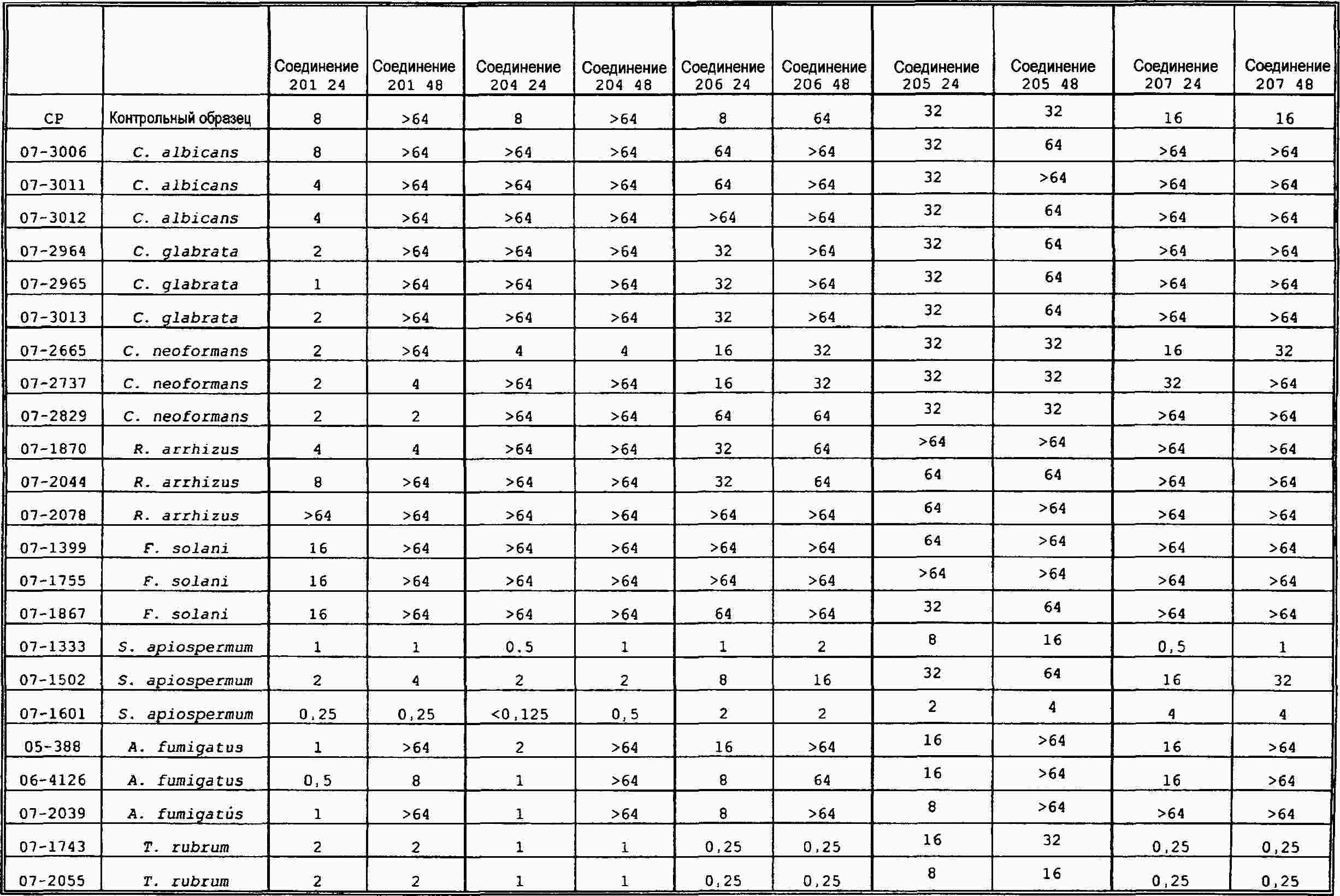
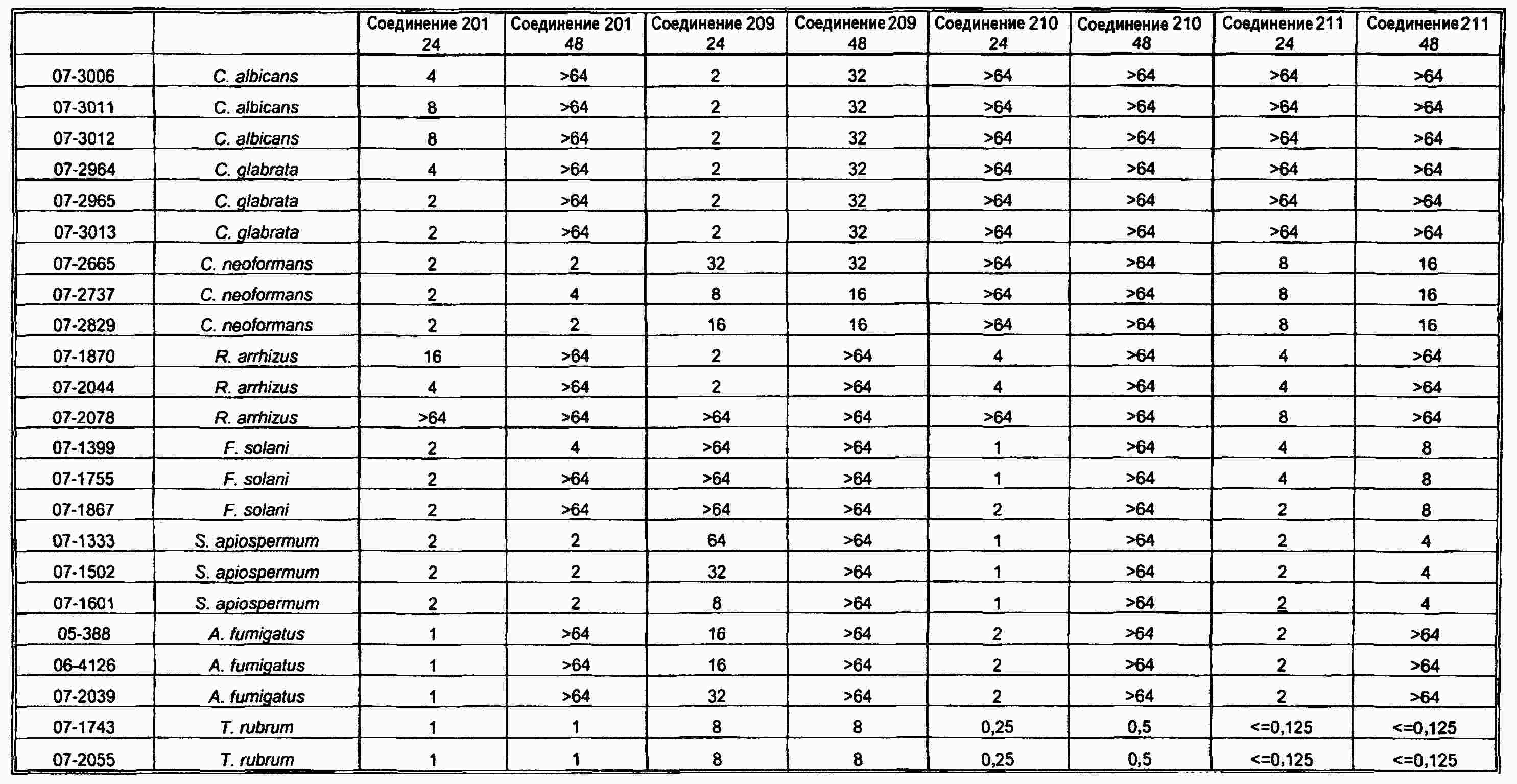
|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea000018618b*\id |
|
Термины запроса в документе Реферат Изобретение относится к соединению, имеющему структуру, в которой n равно 1-10; X означает C-R 11 или N, где R 11 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают Н, C 1 -С 6 алкил или С 3 -С 8 циклоалкил; R 5 означает ОН или SH; R 6 , R 12 , R 13 и R 14 независимо означают Н, ОН, SH, F, Cl, SO 2 R 15 , NO 2 , трифторметил, метокси или СО-R 15 , где R 15 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; или к его соли, которое предназначено для лечения опухолей. Формула [0001] Соединение, имеющее структуру [0002] Соединение, имеющее структуру [0003] Соединение по п.2, имеющее структуру [0004] Соединение по п.2, имеющее структуру [0005] Соединение по п.4, имеющее структуру [0006] Соединение по п.1, имеющее структуру [0007] Соединение по п.6, имеющее структуру [0008] Соединение по любому из пп.1-6, в котором R5 или R6 означает SH, и ароматическое кольцо, содержащее группу SH, является бензольным, аза- или полиазаароматическим пяти- или шестичленным кольцом. [0009] Соединение по любому из пп.1-7, в котором R1 и R2 означают Н, X означает СН, R5 означает SH, R6 означает Н и n равно 4. [0010] Соединение по любому из пп.1-7, в котором R1 означает SH, R2 означает Н, X означает СН, R5 означает SH, R6 означает Н и n равно 6. [0011] Соединение по любому из пп.1-7, в котором R1 и R2 означают Н, X означает N, R5 означает SH, R6 означает Н и n равно 4. [0012] Соединение по любому из пп.1-7, в котором R1 означает Н, R2 означает NR3R4, где R3 и R4 означают C1 алкил, X означает СН, R5 означает SH, R6 означает Н и n равно 4. [0013] Соединение по любому из пп.1-7, в котором R1 и R2 означают Н, X означает N, R5 означает SH, R6 означает Cl и n равно 4. [0014] Соединение по любому из пп.1-7, в котором R1 и R2 означают Н, X означает N, R5 означает SH, R6 означает Н и n равно 5. [0015] Соединение по любому из пп.1-7, в котором R1 означает Н, R2 означает NR3R4, где R3 и R4 означают Н, X означает СН, R5 означает SH, R6 означает Н, и n равно 4. [0016] Соединение по любому из пп.1-7, в котором R1 и R2 означают Н, X означает СН, R5 означает SH, R6 означает Cl и n равно 4. [0017] Соединение по любому из пп.1-7, в котором R1 и R2 означают Н, X означает СН, R5 означает SH, R6 означает метокси и n равно 4. [0018] Соединение по любому из пп.1-7, в котором R1 и R2 означают Н, X означает СН, R5 означает SH, R6 означает Н и n равно 5. [0019] Соединение по любому из пп.1-7, в котором R1 и R2 означают Н, X означает СН, R5 означает SH, R6 означает Н и n равно 6. [0020] Соединение по любому из пп.1-7, в котором R1 и R2 означают Н, X означает СН, R5 означает SH, R6 означает Н и n равно 9. [0021] Соединение по любому из пп.1-7, имеющее структуру [0022] Соединение по любому из пп.1-7, имеющее структуру [0023] Соединение, имеющее структуру [0024] Фармацевтическая композиция, включающая соединение по п.22 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. [0025] Способ уменьшения объема опухоли, сверхэкспрессирующей ядерный рецептор-корепрессор (N-CoR), включающий введение пациенту фармацевтической композиции по п.24. [0026] Способ по п.23, в котором опухоль, сверхэкспрессирующая ядерный рецептор-корепрессор (N-CoR), является опухолью головного мозга. [0027] Способ ингибирования активности гистондезацетилазы (HDAC), включающий контактирование HDAC с соединением по п.22. [0028] Способ по п.27, в котором HDAC является гистондезацетилазой 3 (HDAC3). [0029] Способ по п.27, в котором HDAC является гистондезацетилазой 4 (HDAC4). [0030] Способ ингибирования репликации ВИЧ, включающий контактирование ВИЧ-инфицированной клетки с соединением по любому из п.22. [0031] Способ ингибирования гипертрофии сердца, включающий введение пациенту количества соединения по п.22, эффективного для ингибирования гипертрофии сердца. [0032] Способ лечения пациента, страдающего раком молочной железы, раком ободочной кишки, крупноклеточным раком легкого, аденокарциномой легкого, мелкоклеточным раком легкого, раком желудка, раком печени, аденокарциномой яичника, раком поджелудочной железы, раком предстательной железы, промиелоцитарным лейкозом, хроническим миелоцитарным лейкозом или острым лимфолейкозом, включающий введение пациенту фармацевтической композиции по п.24. [0033] Способ ингибирования роста грибка, включающий контактирование грибка с соединением по п.22. [0034] Способ получения соединения, имеющего структуру [0035] Способ по п.34, предназначенный для получения соединения, имеющего структуру [0036] Способ по п.35, дополнительно включающий: [0037] Способ по п.34, предназначенный для получения соединения, имеющего структуру [0038] Способ по п.34, предназначенный для получения соединения, имеющего структуру [0039] Соединение по п.21, имеющее структуру Полный текст патента Данная заявка притязает на приоритет предварительных заявок на патент США № 61/063965, поданной 6 февраля 2008 г., № 61/008673, поданной 21 декабря 2007 г., и № 60/997338, поданной 1 октября 2007 г., которые полностью включены в настоящее описание изобретения путем ссылки. В тексте настоящей заявки приведены определенные публикации. Полный список приведенных публикаций представлен непосредственно перед формулой изобретения. Указанные публикации полностью включены в настоящую заявку путем ссылки для более полного описания уровня техники в той области, к которой относится настоящее изобретение. Гистон-дезацетилазы (далее именуются также HDAC), как известно, играют важную роль в механизме транскрипции, регулируя экспрессию генов, индуцируя гиперацетилирование гистонов и воздействуя на экспрессию генов. Поэтому мишенью для терапевтического или профилактического средства, предназначенного для лечения заболеваний, вызываемых аномальной экспрессией генов, таких как воспалительные нарушения, диабет, вызванные диабетом осложнения, гомозиготная талассемия, фиброз, цирроз, острый промиелоцитарный лейкоз (APL), отторжение трансплантатов, аутоиммунные заболевания, протозойные инфекции, опухоли и т.д., является ингибирование белков HDAC. Ацетилирование и дезацетилирование гистонов происходит под воздействием гистон-ацетилтрансфераз (HAT) и гистон-дезацетилаз (HDAC). Состояние ацетилирования гистонов является важной детерминантой транскрипции генов. Дезацетилирование обычно ассоциировано с более низкой степенью транскрипции генов, в то время как повышенное ацетилирование гистонов, индуцируемое действием ингибиторов HDAC, вызывает более высокую степень транскрипции генов. Таким образом, ингибиторы HDAC воздействуют на многие процессы, происходящие в клетке, которые, по-видимому, зависят от динамического состояния клетки в отношении ее способности к репликации и дифференцировке. Исследование ингибиторов гистон-дезацетилаз показывает, что данные ферменты играют важную роль в пролиферации и дифференцировке клеток. Ингибитор трихостатин A (TSA) (Yoshida et al., (1990) "Structural specificity for biological activity of trichostatin A, a specific inhibitor of mammalian cell cycling with potent differentiation-inducing activity in Friend leukemia cells" J. Antibiot. 43(9):1101-6) останавливает клеточный цикл как в G1, так и G2 фазах (Yoshida and Beppu, (1988) "Reversible arrest of proliferation of rat 3Y1 fibroblasts in both the G1 and G2 phases by trichostatin A" Exp. Cell Res. 177 (1) : 122-31), восстанавливает трансформированный фенотип разных линий клеток и индуцирует дифференцировку клеток лейкоза Фрейда и других. В научной литературе сообщалось, что TSA и субероиланилидгидроксамовая кислота (SAHA) ингибируют рост клеток, индуцируют терминальную дифференцировку и предотвращают образование опухолей у мышей (Finnin et al., (1999) "Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors" Nature 401, 188-193). Остановка клеточного цикла под воздействием TSA вызывает повышенную экспрессию гелсолина (Hoshikawa et al., (1994) "Trichostatin A induces morphological changes and gelsolin expression by inhibiting histone deacetylase in human carcinoma cell lines" Exp. Cell Res. 214 (1):189-97), регуляторного белка актина, содержание которого снижается в злокачественной опухоли молочной железы (Mielnicki et al., (1999) "Epigenetic Regulation of Gelsolin Expression in Human Breast Cancer Cells" Experimental Cell. Research 249(1), pp. 161-176). Было установлено, что аналогичное воздействие на клеточный цикл и дифференцировку оказывают многие ингибиторы дезацетилаз (Kim et al., (1999) "Selective Induction of Cyclin-Dependent Kinase Inhibitors and Their Roles in Cell Cycle Arrest Caused by Trichostatin A, an Inhibitor of Histone Deacetylase" Ann. N.Y. Acad. Sci. 886:200-203). В научной литературе также указывалось, что трихостатин А может быть использован при лечении фиброза, например, фиброза печени и цирроза печени. См., например, публикацию Geerts et al., (1998) "Hepatic Stellate Cells and Liver Retinoid Content in Alcoholic Liver Disease in Humans" Cliniucal and Experimental Research 22(2), 494-500. В научной литературе недавно были описаны определенные соединения, которые индуцируют клеточную дифференцировку и ингибируют гистон-дезацетилазы. Как было указано, действие нескольких экспериментальных противоопухолевых соединений, таких как трихостатин A (TSA), трапоксин, субероиланилидгидроксамовая кислота (SAHA) и фенилбутират, по меньшей мере, частично основано на ингибировании гистон-дезацетилазы (см., например, Yoshida et al. (1990) "Structural specificity for biological activity of trichostatin A, a specific inhibitor of mammalian cell cycle with potent differentiation-inducing activity in Friend leukemia cells" J. Antibiot. 43 (9):1101-6; Richon et al., (1998) "A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases" PNAS 95(6) 3003-3007; и Kijima et al., (1993) "Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase" J. Biol. Chem., 268(30) 22429-22435). Кроме того, было установлено, что диаллилсульфид и родственные молекулы (см., например, Lea et al., (1999) "Increased acetylation of histones induced by diallyl disulfide and structurally related molecules" Int J Oncol. 15 (2) : 347-52), оксамфлатин (см., например, Kim et al., (1999) "Selective Induction of Cyclin-Dependent Kinase Inhibitors and Their Roles in Cell Cycle Arrest Caused by Trichostatin A, an Inhibitor of Histone Deacetylase" Ann. N.Y. Acad. Sci. 886: 200-203), MS-27-275, синтетическое производное безамида (см., например, Saito et al., (1999) "A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors" PNAS 96(8) 4592-4597; Suzuki et al., (1999) "Synthesis and Histone Deacetylase Inhibitory Activity of New Benzamide Derivatives" J. Med. Chem., 42 (15), 3001-3003; следует отметить, что MS-27-275 позже было переименовано как MS-275); производные бутирата (см., например, Lea and Tulsyan, (1995) "Discordant effects of butyrate analogues on erythroleukemia cell proliferation, differentiation and histone deacetylase" Anticancer Res. 15 (3) :879-83), FR901228 (см., например, Nokajima et al., 1998), депудецин (см., например, Kwon et al., (1998) "Depudecin induces morphological reversion of transformed fibroblasts via the inhibition of histone deacetylase" PNAS 95(7) 3356-3361) и бисгидроксиамид м-карбоксикоричной кислоты (см., например, Richon et al., 1998) ингибируют гистон-дезацетилазы. Как показали исследования in vitro, некоторые из данных соединений ингибируют рост фибробластов, вызывая остановку клеточного цикла в фазах G1 и G2, что может привести к терминальной дифференцировке и утрате трансформирующего потенциала разных трансформированных линий клеток (см., например, Richon et al., 1996; Kim et al., 1999; Yoshida et al., 1995; Yoshida & Beppu, 1988 (указанные публикации были полностью приведены выше)). Как показали исследования in vivo, фенилбутират позволяет эффективно лечить острый промиелоцитарный лейкоз в сочетании с ретиноевой кислотой (см., например, Warrell et al., (1998) "Therapeutic Targeting of Transcription in Acute Promyelocytic Leukemia by Use of an Inhibitor of Histone Deacetylase" Journal of the National Cancer Institute, Vol. 90, № 21, 1621-1625). Было установлено, что SAHA позволяет эффективно предотвращать образование опухолей молочной железы у крыс и опухолей легкого у мышей (см., например, Desai et al., 1999). Благодаря точно установленному участию HDAC в регулировании пролиферации и дифференцировки клеток можно предположить, что аберрантная активность HDAC может играть определенную роль в развитии рака. Прямое свидетельство того, что дезацетилазы способствуют развитию рака, было получено в результате исследования разных форм острого промиелоцитарного лейкоза (APL). У большинства пациентов, страдающих APL, транслокация хромосом 15 и 17 (t(15;17) вызывает экспрессию слитого белка, содержащего N-концевую часть генного продукта PML, связанного с большинством RAR α (рецептор ретиноевой кислоты). В некоторых случаях другая транслокация (t(11;17) вызывает слияние белка цинкосодержащей пальцеобразной области PLZF с RAR α. При отсутствии лиганда рецепторы RARa дикого типа подавляют гены-мишени, связывая комплексы HDAC-peпpeccop с промоторной ДНК. В процессе нормального гемопоэза ретиноевая кислота (RA) связывается с RAR α и замещает комплекс репрессора, делая возможной экспрессию генов, участвующих в дифференцировке миелоидных клеток. RAR α-слитые белки, обнаруживаемые у пациентов, страдающих APL, больше не реагируют на физиологические уровни ретиноевой кислоты (RA) и препятствуют экспрессии RA-индуцибельных генов, которые стимулируют дифференцировку миелоидных клеток. Вследствие этого происходит размножение клона промиелоцитарных клеток и развитие лейкоза. В результате экспериментов in vitro было установлено, что TSA может восстанавливать чувствительность RA к RAR α-слитым белкам и вызывать дифференцировку миелоидных клеток. Полученные результаты позволяют установить связь между HDAC и онкогенезом и предположить, что HDAC являются потенциальными мишенями для лекарственного воздействия на пациентов, страдающих APL. (См., например, Kitamura et al., (2000) "Histone deacetylase inhibitor but not arsenic trioxide differentiates acute promyelocytic leukaemia cells with t(ll;17) in combination with all-trans retinoic acid" Brit. J. Hemat. 108(4) 696-702; David et al., (1998) "Histone deacetylase associated with mSin3A mediates repression by the acute promyelocytic leukemia-associated PLZF protein" Oncogene (1998) 16, 2549-2556; Lin et al., (1998) "Role of the histone deacetylase complex in acute promyelocytic leukaemia" Nature 391 ( 6669):811-4). Кроме того, полученные данные позволяют предположить, что HDAC могут быть важными терапевтическими мишенями при других типах рака. Ингибиторы HDAC вызывают дифференцировку линий клеток, выделенных из многих разных типов рака (рак предстательной железы, рак ободочной и прямой кишки, рак молочной железы, рак нервных клеток, рак печени) (Yoshida and Horinouchi, (1999) "Trichostatin and Leptomycin: Inhibition of Histone Deacetylation and Signal-Dependent Nuclear Export" Annals of the New York Academy of Sciences 886:23-35). Целый ряд ингибиторов HDAC был исследован с использованием животных моделей рака. Такие ингибиторы уменьшают рост опухоли и увеличивают продолжительность жизни мышей, несущих трансплантированные опухоли разных типов, включая меланому, лейкоз, карциномы ободочной кишки, легкого, желудка и т.д. (Ueda et al., (1994) "Serum levels of cytokines in patients with colorectal cancer: Possible involvement of interleukin-6 and interleukin-8 in hematogenous metastasis" 29(4); Kim et al., 1999). Установлено, что несколько известных ингибиторов HDAC обеспечивают защиту в разных клеточных и животных моделях острого и хронического нейродегенеративного поражения и заболевания, например, инсульт, рассеянный склероз и заболевания, вызванные повышенным уровнем полиглутамина, такие как хорея Гентингтона и мышечная атрофия, вызванная поражением спинного мозга и продолговатого мозга (SBMA) (Kozikowski et al., J. Med. Chem. (2007), 50, 3054-3061). Кроме того, недавно полученные результаты позволяют предположить, что ингибиторы HDAC могут восполнять недостаток синаптической пластичности, познавательной способности и стрессовой реакции в целом ряде неврологических и психиатрических нарушений, включая хорею Гентингтона, болезнь Паркинсона, состояние тревоги и изменение настроения, синдром Рубинстайна-Тейби и синдром Ретта (Abel, Т. and Zukin, R.S., Current Opinion in Pharmacology (2008) 8:57-64). В публикации Беглопулоса и Шена (Beglopoulos, V. and Shen. J., Trends in Pharmacological Sciences (2006) 27:33-40) указано, что ингибиторы фосфодиэстеразы 4 и гистон-дезацетилаз уменьшают потерю памяти и нейродегенеративные нарушения в животных моделях болезни Альцгеймера, воздействуя на гены сАМР-реагирующего элемента (CRE). В публикации Фишера и др. (Fischer, A. et al., Nature (2007) 447:178-182) недавно было отмечено, что познавательная способность и долговременная память могут быть восстановлены после значительной утраты нейронов и атрофии головного мозга в мышиной модели благодаря введению ингибиторов гистон-дизацетилаз (см. обзор и комментарии, приведенные в публикациях Sweat, J.D. et al., Nature (2007) 447:151-152; Mangan, К.Р. and Levenson, J.M., Cell (2007) 129:851-853; Albert, M.S., New Engl. J. Med. (2007) 357 (5):502-503; and Abel, T and Zukin, R.S., Current Opinion in Pharmacology (2008) 8:57-64). По-видимому, существует плохо изученный компонент нейродегенеративных заболеваний, относящийся к чрезмерной активности гистон-дезацетилаз, или по меньшей мере состояние пониженного ацетилирования определенных гистонов, которое устраняется повышенным ацетилированием, что позволяет улучшить познавательную способность и память. В данной связи ингибирование определенных гистон-дезацетилаз соединениями по настоящему изобретению может оказывать благоприятное воздействие при лечении нейтродегенеративных заболеваний, таких как болезнь Альцгеймера (AD). Установлено, что нейродегенеративными заболеваниями в настоящее время страдают 20 миллионов человек во всем мире. Затраты на оказание медицинской помощи пациентам, страдающим болезнью Альцгеймера, например, составили 91 миллиард долларов в 2005 г. и предположительно возрастут до 160 миллиардов долларов к 2010 г. (Burke, R.E., Pharmacology and Therapeutics (2007) 114:262-277). Несмотря на значительные исследования в области этиологии и фармакологического лечения данных болезней, до сих пор не известны методы лечения, позволяющие замедлить их развитие (Schapira, A.H.V. and Olanow, C.W., JAMA (2004) 291:358-364; Burke, R.E., Pharmacology and Therapeutics (2007) 114:262-277). Болезнь Альцгеймера (AD) и другие нейродегенеративные заболевания считаются тауопатиями, так как они характеризуются накоплением агрегатов tau-белка в нейронах. Tau-белки стимулируют сборку и стабилизацию структур микротрубочек в нейронах. Нейродегенеративные заболевания, такие как болезнь Альцгеймера, часто характеризуются нарушением познавательной способности и памяти. Механизм(ы), отвечающий за данные наиболее неприятные симптомы, ассоциирован с гибелью нейронов. Изменение организации и консолидации памяти на молекулярном уровне связано с активностью структур хроматина гистон-дезацетилаз (Korzus, E. et al., Neuron (2004) 42:961-972; Levenson, J.M. et al., The Journal of Biological Chemistry (2004) 279:40545-40559). Гистон-дезацетилазы также играют важную роль в воспалительных заболеваниях Hildemann et al. Appl Microbiol Biotechnol (2007), 75(3), 487-497; Riester et al. Appl Microbiol. Biotechnol. (2007), 75(3), 499-514; Adcock, I.M. Br J Pharmacol. (2007), 150(7), 829-831; Huang L, J Cell Physiol. (2006), 209(3), 611-616). Разнообразные клеточные функции, включающие регуляцию экспрессии генов воспалительного инфильтрата, репарацию ДНК и пролиферацию клеток, регулируются изменениями в состоянии ацетилирования гистонов и белков, не являющихся гистонами. Данные, полученные в результате недавно выполненных исследований in vitro и in vivo, показывают, что ингибиторы HDAC могут оказывать противовоспалительное действие благодаря воздействию на гибель клеток, осуществляемому путем ацетилирования белков, не являющихся гистонами. Несмотря на опасения, связанные с безопасностью данных средств при длительном применении, такие средства могут быть особенно полезными в тех случаях, когда современные противовоспалительные методы лечения оказываются недостаточно эффективными (Adcock, I.M., Br. J. Pharmacol. (2007), 150(7), 829-831). Ингибиторы гистон-дезацетилаз также считаются потенциальными анти-ВИЧ средствами, направленно воздействующими на функциональные группы Zn в цинкосодержащих пальцеобразных доменах ретровирусов, что следует из гипотезы и данных, представленных в Song et al. (2002) "Synthesis and Biological Properties of Amino Acid Amide Ligand-Based Pyridinioalkanoyl Thioesters as Anti-HIV agents" Bioorganic & Medicinal Chemistry 10, 1263-1273. Ингибиторы гистондезацетилаз также считаются потенциальными ингибиторами гипертрофии сердца, что следует из данных, приведенных в нижеследующих ссылках: WO 2007021682, WO 2006129105, WO 2007014029, WO 2006023603, публикация заявки на патент США № 2007-0004771, публикация заявки на патент США № 2007-0135433, публикация заявки на патент США № 2006-0235231, европейский патент № 1663310, публикация заявки на патент США № 2007-0135365, европейский патент № 1694688, европейский патент № 1715870, европейский патент № 1691891, патент Японии № 2007511528, европейский патент 1699436 и патент Японии № 2007514665. Основная структурная группа ингибиторов HDAC включает компонент гидроксамовой кислоты, который предположительно имеет определяющее значение для ингибирующей активности данных молекул благодаря их способности связываться с цинком. Несколько других типов групп, связывающихся с цинком, которые предназначены для использования в качестве компонентов новых ингибиторов HDAC, находятся на стадии исследования. Авторы настоящего изобретения разработали новую серию ингибиторов HDAC, используя меркаптобензаминоильную группу в качестве связующего для цинка, и считают, что данная часть может быть использована вместо гидроксамовой кислоты и других связывающихся с цинком частей во всех других ингибиторах HDAC с достижением благоприятного результата. В настоящем описании изобретения рассмотрен синтез ингибиторов HDAC. Соединения, рассмотренные в настоящем описании изобретения, являются также активными ингибиторами пролиферации раковых клеток человека. Данные соединения подавляют активность гистон-дезацетилазы 3 и гистондезацетилазы 4 (соответственно HDAC3 и HDAC4), а также воздействуют на устойчивость N-CoR в линиях клеток головного мозга человека (U-87), когда клетки подвергаются воздействию данных соединений в культуре. Изобретение относится к соединению, имеющему структуру: где n равно 1-10; X означает C-R 11 или N, где R 11 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; Z означает R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают Н, C 1 -С 6 алкил или С 3 -C 8 циклоалкил; R 5 означает ОН или SH; и R 6 , R 12 , R 13 и R 14 независимо означают Н, ОН, SH, F, Cl, SO 2 R 15 , NO 2 , трифторметил, метокси или CO-R 15 , где R 15 означает алкил, алкенил, алкинил, С 3 -C 8 циклоалкил или арил; или к его соли. Настоящее изобретение относится к способу получения соединения, имеющего структуру: где n равно 1-10; X означает C-R 11 или N, где R 11 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -C 8 циклоалкил или арил; Z означает R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают Н, C 1 -С 6 алкил или С 3 -C 8 циклоалкил; R 5 означает ОН или SH; и R 6 , R 12 , R 13 и R 14 независимо означают Н, ОН, SH, F, Cl, SO 2 R 15 , NO 2 , трифторметил, метокси или CO-R 15 , где R 15 означает алкил, алкенил, алкинил, С 3 -C 8 циклоалкил или арил; включающий: а) контактирование соединения, имеющего структуру: где R 16 означает или NH 2 , с соединением, имеющим структуру: где R 8 означает или NH 2 , R 9 означает Н или и с соединением, имеющим структуру: где R 10 означает Н или Me; m равно 1 или 2, и когда m равно 1, а отсутствует, Y означает ОН или SH; или когда m равно 2, а присутствует, и Y означает S, с образованием соединения, имеющего структуру: где Z означает Настоящее изобретение относится к способу лечения пациента, страдающего нейродегенеративным заболеванием, включающему введение пациенту соединения, имеющего структуру: где n равно 1-10; X означает C-R 11 или N, где R 11 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -C 8 циклоалкил или арил; Z означает R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают Н, С 1 -С 6 алкил или С 3 -С 8 циклоалкил; R 5 означает ОН или SH; и R 6 , R 12 , R 13 и R 14 независимо означают Н, ОН, SH, F, Cl, SO 2 R 15 , NO 2 , трифторметил, метокси или CO-R 15 , где R 15 означает алкил, алкенил, алкинил, С 3 -C 8 циклоалкил или арил; или его соли в количестве, эффективном для лечения пациента. Настоящее изобретение относится к способу уменьшения агрегации tau-белка в клетке, включающему контактирование клетки с эффективным количеством соединения, имеющего структуру: где n равно 1-10; X означает C-R 11 или N, где R 11 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; Z означает R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают Н, C 1 -С 6 алкил или С 3 -C 8 циклоалкил; R 5 означает ОН или SH; и R 6 , R 12 , R 13 и R 14 независимо означают Н, ОН, SH, F, Cl, SO 2 R 15 , NO 2 , трифторметил, метокси или CO-R 15 , где R 15 означает алкил, алкенил, алкинил, С 3 -C 8 циклоалкил или арил; или его соли, что позволяет ингибировать агрегацию tau-белка в клетке. Фиг. 1. Ингибирование роста линии клеток U373 мультиформной глиобластомы соединением 201, измеренное в 1, 3 и 7 день. Фиг. 2. Ингибирование роста линии клеток U373 мультиформной глиобластомы соединением 203, измеренное в 1, 3 и 7 день. Фиг. 3. Ингибирование роста линии клеток U373 мультиформной глиобластомы соединением 204, измеренное в 1, 3 и 7 день. Фиг. 4. Ингибирование роста линии клеток U373 мультиформной глиобластомы соединением 205, измеренное в 1, 3 и 7 день. Фиг. 5. Ингибирование роста линии клеток U373 мультиформной глиобластомы соединением 206, измеренное в 1, 3 и 7 день. Фиг. 6. Ингибирование соединением 205, соединением 206 или SAHA объема ксенотрансплантата опухоли GBM, измеренное в 0, 7, 14, 21 и 28 день. Фиг. 7. Ингибирование роста линии клеток DAOY медуллобластомы соединением 205 или соединением 205 в комбинации с ATRA, измеренное в 1, 3 и 7 день. Фиг. 8А. Ингибирование роста линии клеток MDA-MB-231 рака молочной железы: графическое представление (А) и кривая соответствуют значению IC 50 (В) данных, полученных после воздействия на клетки MDA-MB-231 соединения 205 при помощи анализа CellTiter-Glo. На А также показано воздействие 10 мкМ доксорубицина, использованного в качестве положительного контрольного образца. Каждая точка представляет собой среднее значение ± стандартное отклонение, полученное по меньшей мере для трех одинаковых образцов. Фиг. 8В. Ингибирование роста линии клеток НТ-29 рака ободочной кишки: графическое представление (А) и кривая соответствуют значению IC 50 (В) данных, полученных после воздействия на клетки НТ-29 соединения 205 при помощи анализа CellTiter-Glo. На А также показано воздействие 10 мкМ доксорубицина, использованного в качестве положительного контрольного образца. Каждая точка представляет собой среднее значение ± стандартное отклонение, полученное по меньшей мере для трех одинаковых образцов. Фиг. 8С. Ингибирование роста линии клеток NCI-H460 крупноклеточного рака легкого: графическое представление (А) и кривая соответствуют значению IC 50 (В) данных, полученных после воздействия на клетки NCI-H460 соединения 205 при помощи анализа CellTiter-Glo. На А также показано воздействие 10 мкМ доксорубицина, использованного в качестве положительного контрольного образца. Каждая точка представляет собой среднее значение ± стандартное отклонение, полученное по меньшей мере для трех одинаковых образцов. Фиг. 8D. Ингибирование роста линии клеток NCI-H522 аденокарциномы легкого: графическое представление (А) и кривая соответствуют значению IC 50 (В) данных, полученных после воздействия на клетки NCI-H522 соединения 205 при помощи анализа CellTiter-Glo. На А также показано воздействие 10 мкМ доксорубицина, использованного в качестве положительного контрольного образца. Каждая точка представляет собой среднее значение ± стандартное отклонение, полученное по меньшей мере для трех одинаковых образцов. Фиг. 8Е. Ингибирование роста линии клеток NCI-H69 мелкоклеточного рака легкого: графическое представление (А) и кривая соответствуют значению IC 50 (В) данных, полученных после воздействия на клетки NCI-H69 соединения 205 при помощи анализа CellTiter-Glo. На А также показано воздействие 10 мкМ доксорубицина, использованного в качестве положительного контрольного образца. Каждая точка представляет собой среднее значение ± стандартное отклонение, полученное по меньшей мере для трех одинаковых образцов. Фиг. 8F. Ингибирование роста линии клеток GXF-209 рака желудка: графическое представление (А) и кривая соответствуют значению IC 50 (В) данных, полученных после воздействия на клетки GXF-209 соединения 205 при помощи анализа CellTiter-Glo. На А также показано воздействие 10 мкМ доксорубицина, использованного в качестве положительного контрольного образца. Каждая точка представляет собой среднее значение ± стандартное отклонение, полученное по меньшей мере для трех одинаковых образцов. Фиг. 8G. Ингибирование роста линии клеток HepG2 рака печени (гепатома): графическое представление (А) и кривая соответствуют значению IC 50 (В) данных, полученных после воздействия на клетки HepG2 соединения 205 при помощи анализа CellTiter-Glo. На А также показано воздействие 10 мкМ доксорубицина, использованного в качестве положительного контрольного образца. Каждая точка представляет собой среднее значение ± стандартное отклонение, полученное по меньшей мере для трех одинаковых образцов. Фиг. 8Н. Ингибирование роста линии клеток OVCAR-3 аденокарциномы яичника: графическое представление (А) и кривая соответствуют значению IC 50 (В) данных, полученных после воздействия на клетки OVCAR-3 соединения 205 при помощи анализа CellTiter-Glo. На А также показано воздействие 10 мкМ доксорубицина, использованного в качестве положительного контрольного образца. Каждая точка представляет собой среднее значение ± стандартное отклонение, полученное по меньшей мере для трех одинаковых образцов. Фиг. 81. Ингибирование роста линии клеток PANC-1 рака поджелудочной железы: графическое представление (А) и кривая соответствуют значению IC 50 (В) данных, полученных после воздействия на клетки PANC-1 соединения 205 при помощи анализа CellTiter-Glo. На А также показано воздействие 10 мкМ доксорубицина, использованного в качестве положительного контрольного образца. Каждая точка представляет собой среднее значение ± стандартное отклонение, полученное по меньшей мере для трех одинаковых образцов. Фиг. 8J. Ингибирование роста линии клеток DU-145 рака предстательной железы: графическое представление (А) и кривая соответствуют значению IC 50 (В) данных, полученных после воздействия на клетки DU-145 соединения 205 при помощи анализа CellTiter-Glo. На А также показано воздействие 10 мкМ доксорубицина, использованного в качестве положительного контрольного образца. Каждая точка представляет собой среднее значение ± стандартное отклонение, полученное по меньшей мере для трех одинаковых образцов. Фиг. 8К. Ингибирование роста линии клеток LNCAP рака предстательной железы: графическое представление (А) и кривая соответствуют значению IC 50 (В) данных, полученных после воздействия на клетки LNCAP соединения 205 при помощи анализа CellTiter-Glo. На А также показано воздействие 10 мкМ доксорубицина, использованного в качестве положительного контрольного образца. Каждая точка представляет собой среднее значение ± стандартное отклонение, полученное по меньшей мере для трех одинаковых образцов. Фиг. 8L-N. Ингибирование роста линий клеток HL-60 (фиг. 8b), К562 (фиг. 8М) и MOLT4 (фиг. 8N) лейкоза: графическое представление (А) и кривая соответствуют значению IC 50 (В) данных, полученных после воздействия на клетки HL-60 (фиг. 8b), К562 (фиг. 8М) и MOLT4 (фиг. 8N) соединения 205 при помощи анализа CellTiter-Glo. На А также показано воздействие 10 мкМ доксорубицина, использованного в качестве положительного контрольного образца. Каждая точка представляет собой среднее значение ± стандартное отклонение, полученное по меньшей мере для трех одинаковых образцов. Фиг. 9. Ингибирование соединениями 205 и 206 ксенотрансплантата U87 GBM. Фиг. 10. Ингибирование соединением 205 ксенотрансплантата SHSY-5Y. Фиг. 11. Ингибирование соединением 205 ксенотрансплантата DAOY. Фиг. 12: Ингибирование соединением 205 и трихостатином активности HDAC в клетках DAOY. Фиг. 13. Ингибирование соединением 205 и SAHA активности HDAC в ксенотрансплантатах DAOY. Фиг. 14. Ингибирование соединениями 201 и 2 05 активности HDAC в ксенотрансплантатах SHSY-5Y. Фиг. 15. Воздействие соединений 201 и 205 на нормальную ткань головного мозга мышей. Фиг. 16. Воздействие соединений 205 и 212 на мультиформную глиобластому (U373). Фиг. 17. Воздействие соединений 205 и 213 на мультиформную глиобластому (U373). Фиг. 18. Воздействие соединения 214 на линию клеток U373 мультиформной глиобластомы. Фиг. 19. Воздействие соединений 205 и 214 на мультиформную глиобластому (U373). Фиг. 20. Воздействие соединения 208 на линию клеток U373 мультиформной глиобластомы. Фиг. 21. Воздействие соединения 213 на линию клеток U373 мультиформной глиоболастомы. Фиг. 22. Воздействие соединения 212 на линию клеток U373 мультиформной глиобластомы. Настоящее изобретение относится к соединению, имеющему структуру: где n равно 1-10; X означает C-R 11 или N, где R 11 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; Z означает R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают Н, C 1 -С 6 алкил или С 3 -C 8 циклоалкил; R 5 означает ОН или SH; и R 6 , R 12 , R 13 и R 14 независимо означают Н, ОН, SH, F, Cl, SO 2 R 15 , NO 2 , трифторметил, метокси или CO-R 15 , где R 15 означает алкил, алкенил, алкинил, С 3 -C 8 циклоалкил или арил; или к его соли. В одном варианте осуществления изобретения соединение имеет структуру: где n равно 1-9; X означает C-R 11 или N, где R 11 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -C 8 циклоалкил или арил; R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают Н, C 1 -С 6 алкил или С 3 -С 8 циклоалкил; R 5 означает ОН или SH; и R 6 , R 12 , R 13 и R 14 независимо означают Н, ОН, SH, F, Cl, SO 2 R 15 , NO 2 , трифторметил, метокси или CO-R 15 , где R 15 означает алкил, алкенил, алкинил, С 3 -C 8 циклоалкил или арил. В одном варианте осуществления изобретения соединение имеет структуру: где n равно 1-8; X означает СН или N; R 1 означает Н или ОН; R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают Н, C 1 -С 6 алкил или С 3 -C 8 циклоалкил; R 5 означает ОН или SH; и R 6 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил. В одном варианте осуществления изобретения соединение имеет структуру: где n равно 1-9; X означает C-R 11 или N, где R 11 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -C 8 циклоалкил или арил; R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают Н, C 1 -С 6 алкил или С 3 -C 8 циклоалкил; R 5 означает ОН или SH; и R 6 , R 12 , R 13 и R 14 независимо означают Н, ОН, SH, F, Cl, трифторметил, метокси или CO-R 15 , где R 15 означает алкил, алкенил, алкинил, С 3 -C 8 циклоалкил или арил. В одном варианте осуществления изобретения соединение имеет структуру: где n равно 1-8; X означает СН или N; R 1 означает Н или ОН; R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают C 1 -С 6 алкил или С 3 -С 8 циклоалкил; R 5 означает ОН или SH; и R 6 означает Н, ОН, SH, F, Cl, трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил или С 3 -С 8 циклоалкил, или арил. В одном варианте осуществления изобретения соединение имеет структуру: где n равно 1-8; X означает C-R 11 или N, где R 11 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают C 1 -С 6 алкил или С 3 -С 8 циклоалкил; и R 5 означает ОН или SH; и R 6 , R 12 , R 13 и R 14 независимо означают Н, ОН, SH, F, Cl, SO 2 R 15 , NO 2 , трифторметил, метокси или СО-R 15 , где R 15 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; или является его солью. В одном варианте осуществления изобретения соединение имеет структуру: где n равно 1-8; X означает СН или N; R 1 означает Н, ОН или SH; R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают C 1 -С 6 алкил или С 3 -С 8 циклоалкил; и R 5 означает ОН или SH; и R 6 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил. В некоторых вариантах осуществления изобретения соединение по настоящему изобретению имеет структуру, в которой R 5 или R 6 означает SH, и ароматическое кольцо, содержащее группу SH, является бензольным, аза- или полиаза-ароматическим пяти- или шестичленным кольцом. В одном варианте осуществления изобретения R 1 и R 2 означают Н, X означает СН, R 5 означает SH, R 6 означает Н и n равно 4. В одном варианте осуществления изобретения R 1 означает ОН, R 2 означает Н, X означает СН, R 5 означает ОН, R 6 означает Н и n равно 6. В одном варианте осуществления изобретения R 1 означает SH, R 2 означает Н, X означает СН, R 5 означает SH, R 6 означает Н и n равно 6. В одном варианте осуществления изобретения R 1 и R 2 означают Н, X означает N, R 5 означает SH, R 6 означает Н, и n равно 4. В одном варианте осуществления изобретения R 1 означает Н, R 2 означает NR 3 R 4 , где R 3 и R 4 означают C 1 алкил, X означает СН, R 5 означает SH, R 6 означает Н и n равно 4. В одном варианте осуществления изобретения R 1 и R 2 означают Н, X означает N, R 5 означает SH, R 6 означает Cl и n равно 4. В одном варианте осуществления изобретения R 1 и R 2 означают Н, X означает N, R 5 означает SH, R 6 означает Н и n равно 5. В одном варианте осуществления изобретения R 1 означает Н, R 2 означает NR 3 R 4 , где R 3 и R 4 означают Н, X означает СН, R 5 означает SH, R 6 означает Н и n равно 4. В одном варианте осуществления изобретения R 1 и R 2 означают Н, X означает СН, R 5 означает SH, R 6 означает Cl и n равно 4. В одном варианте осуществления изобретения R 1 и R 2 означают Н, X означает СН, R 5 означает SH, R 6 означает метокси, и п равно 4. В одном варианте осуществления изобретения R 1 и R 2 означают Н, X означает СН, R 5 означает SH, R 6 означает Н и n равно 5. В одном варианте осуществления изобретения R 1 и R 2 означают Н, X означает СН, R 5 означает SH, R 6 означает Н и n равно 6. В одном варианте осуществления изобретения R 1 и R 2 означают Н, X означает СН, R 5 означает SH, R 6 означает Н и n равно 9. В некоторых вариантах осуществления изобретения соединение имеет структуру: В некоторых вариантах осуществления изобретения соединение имеет структуру: где R 8 означает Н, алкил или арил, или Настоящее изобретение относится к фармацевтической композиции, включающей одно или несколько вышеуказанных соединений или их фармацевтически приемлемых солей и фармацевтически приемлемый носитель. Настоящее изобретение относится к способу уменьшения объема опухоли, сверхэкспрессирующей ядерный рецептор-корепрессор (N-CoR), включающему введение пациенту вышеуказанной фармацевтической композиции с целью уменьшения объема опухоли. В одном варианте осуществления способа по настоящему изобретению опухоль, сверхэкспрессирующая ядерный рецептор-корепрессор (N-CoR), является опухолью головного мозга. Настоящее изобретение относится к способу ингибирования активности гистондезацетилазы (HDAC), включающему контактирование HDAC с одним или несколькими вышеуказанными соединениями с целью подавления активности гистондезацетилазы. В одном варианте осуществления изобретения HDAC является гистон-дезацетилазой 3 (HDAC3). В одном варианте осуществления изобретения HDAC является гистон-дезацетилазой 4 (HDAC4). Настоящее изобретение относится к способу ингибирования репликации ВИЧ, включающему контактирование ВИЧ-инфицированной клетки с одним или несколькими вышеуказанными соединениями с целью ингибирования репликации ВИЧ. Настоящее изобретение относится к способу ингибирования гипертрофии сердца, включающему введение пациенту количества одного или нескольких вышеуказанных соединений, эффективного для ингибирования гипертрофии сердца. Настоящее изобретение относится к способу лечения пациента, страдающего раком молочной железы, раком ободочной кишки, крупноклеточным раком легкого, аденокарциномой легкого, мелкоклеточным раком легкого, раком желудка, раком печени, аденокарциномой яичника, раком поджелудочной железы, раком предстательной железы, промиелоцитарным лейкозом, хроническим миелоцитарным лейкозом или острым лимфолейкозом, включающему введение пациенту вышеуказанной фармацевтической композиции с целью лечения данного пациента. Настоящее изобретение относится к способу ингибирования роста грибка, включающему контактирование грибка с одним или несколькими вышеуказанными соединениями с целью ингибирования роста грибка. Настоящее изобретение относится к способу получения соединения, имеющего структуру: где n равно 1-10; X означает C-R 11 или N, где R 11 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; Z означает R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают Н, C 1 -С 6 алкил или С 3 -С 8 циклоалкил; R 5 означает ОН или SH; и R 6 , R 12 , R 13 и R 14 независимо означают Н, ОН, SH, F, Cl, SO 2 R 15 , NO 2 , трифторметил, метокси или CO-R 15 , где R 15 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; включающему: а) контактирование соединения, имеющего структуру: где R 16 означает или NH 2 , с соединением, имеющим структуру: где R 8 означает или NH 2 , R 9 означает Н или и с соединением, имеющим структуру: где R 10 означает Н или Me; m равно 1 или 2, и когда га равно 1, а отсутствует, Y означает ОН или SH; или когда m равно 2, а присутствует, и Y означает S, с образованием соединения, имеющего структуру: где Z означает Один вариант осуществления изобретения относится к способу получения соединения, имеющего структуру: где n равно 1-9; X означает СН или N; R 1 означает Н или ОН; R.2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают Н, C 1 -С 6 алкил или С 3 -С 8 циклоалкил; R 5 означает ОН или SH; и R 6 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -C 8 циклоалкил или арил; включающему: а) контактирование соединения, имеющего структуру с соединением, имеющим структуру в присутствии одного или нескольких первых приемлемых реагентов, образующих амидную связь, первого приемлемого основания и первого приемлемого растворителя с образованием соединения, имеющего структуру b) обработку продукта, полученного на стадии а), в приемлемых условиях снятия защиты с образованием соединения, имеющего структуру которое получают в виде свободного основания или соли; с) контактирование продукта, полученного на стадии b), с соединением, имеющим структуру в присутствии одного или нескольких вторых приемлемых реагентов, образующих амидную связь, второго приемлемого основания и второго приемлемого растворителя с образованием соединения, имеющего структуру где m равно 1 или 2, и когда m равно 1, α отсутствует, Y означает ОН или SH; или когда m равно 2, α присутствует и Y означает S. В одном варианте осуществления изобретения способ по настоящему изобретению дополнительно включает: i) осуществление взаимодействия продукта, полученного на стадии с), с цинком в присутствии хлористо-водородной кислоты с образованием соединения, имеющего структуру когда m равно 2, α присутствует и Y означает S. Один вариант осуществления изобретения относится к способу получения соединения, имеющего структуру где n равно 1-8; R 1 и R 2 оба означают ОН или SH; включающему: а) контактирование соединения, имеющего структуру по меньшей мере с двумя эквивалентами соединения, имеющего структуру с образованием соединения, имеющего структуру В одном варианте осуществления изобретения способ получения соединения, имеющего структуру включает a) нагревание метил-2-гидроксибензоата и 1,6-диаминогексана в колбе с насадкой Дина-Старка для отгонки метанола; b) охлаждение колбы до комнатной температуры и растирание содержимого колбы в воде; c) сбор твердого вещества, находящегося в колбе, фильтрованием; и d) перекристаллизацию твердого вещества из спирта. В одном варианте осуществления изобретения способ получения соединения, имеющего структуру: включает: a) объединение 3-аминопиридина, 6-трет-бутоксикарбониламиногексановой кислоты в метиленхлориде; b) добавление HOBt, EDC*HCl и DIPEA к смеси, полученной на стадии а); и c) перемешивание смеси, полученной на стадии b), в течение 3 ч при комнатной температуре с образованием соединения d) осуществление взаимодействия соединения, полученного на стадии с), в условиях снятия защиты с образованием соединения e) объединение 2,2'-дитиодибензойной кислоты, HOBt, EDC*HCl и ДМФА; f) добавление соединения, полученного на стадии d), к смеси, полученной на стадии е), и DIPEA и перемешивание реакционной смеси при комнатной температуре в течение ночи; g) выливание продукта, полученного на стадии f), в воду и экстракцию этилацетатом; h) промывание органического слоя насыщенным раствором соли, сушку над сульфатом натрия и концентрирование; i) очистку неочищенного остатка колоночной хроматографией с образованием соединения j) растворение соединения, полученного на стадии i), в охлаждаемой льдом смеси метанола и метиленхлорида и добавление концентрированной HCl и цинковой пыли; k) перемешивание смеси, полученной на стадии j), в течение 4 ч и разбавление смеси водой и метиленхлоридом; l) отделение водного слоя и добавление насыщенного водного раствора бикарбоната натрия, и затем охлаждение; m) сбор твердого вещества фильтрованием и последующую сушку в течение ночи; n) экстракцию сухого твердого вещества смесью горячего метанола и метиленхлорида; о) фильтрование горячего раствора через фильтровальную бумагу из стекловолокна; и р) упаривание фильтрата досуха и растирание в этилацетате с образованием соединения: Настоящее изобретение относится к способу лечения пациента, страдающего нейродегенеративным заболеванием, который включает введение пациенту соединения, имеющего структуру: где n равно 1-10; X означает C-R 11 или N, где R 11 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; Z означает R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают Н, C 1 -С 6 алкил или С 3 -С 8 циклоалкил; R 5 означает ОН или SH; и R 6 , R 12 , R 13 и R 14 независимо означают Н, ОН, SH, F, Cl, SO 2 R 15 , NO 2 , трифторметил, метокси или CO-R 15 , где R 15 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; или его соли в количестве, эффективном для лечения пациента. В одном варианте осуществления изобретения способ включает введение пациенту соединения, имеющего структуру: где n равно 1-9; X означает C-R 11 или N, где R 11 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают Н, C 1 -С 6 алкил или С 3 -С 8 циклоалкил; R 5 означает ОН или SH; и R 6 , R 12 , R 13 и R 14 независимо означают Н, ОН, SH, F, Cl, SO 2 R 15 , NO 2 , трифторметил, метокси или CO-R 15 , где R 15 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; или его соли. В одном варианте осуществления изобретения способ включает введение пациенту соединения, имеющего структуру: где n равно 1-9; X означает СН или N; R 1 означает Н или ОН; R2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают Н, C 1 -С 6 алкил или С 3 -С 8 циклоалкил; R 5 означает ОН или SH; и R 6 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -C 8 циклоалкил или арил; или его соли. В одном варианте осуществления изобретения способ включает введение пациенту соединения, имеющего структуру: где n равно 1-9; X означает C-R 11 или N, где R 11 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают Н, C 1 -С 6 алкил или С 3 -С 8 циклоалкил; R 5 означает ОН или SH; и R 6 , R 12 , R 13 и R 14 независимо означают Н, ОН, SH, F, Cl, трифторметил, метокси или CO-R 15 , где R 15 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; или его соли. В одном варианте осуществления изобретения способ включает введение пациенту соединения, имеющего структуру где n равно 1-9; X означает СН или N; R 1 означает Н или ОН; R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают Н, C 1 -С 6 алкил или С 3 -С 8 циклоалкил; R 5 означает ОН или SH; и R 6 означает Н, ОН, SH, F, Cl, трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; или его соли. В одном варианте осуществления изобретения способ включает введение пациенту соединения, имеющего структуру где n равно 1-8; X означает C-R 11 или N, где R 11 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают C 1 -С 6 алкил или С 3 -С 8 циклоалкил; R 5 означает ОН или SH; и R 6 , R 12 , R 13 и R 14 независимо означают Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; или его соли. В одном варианте осуществления изобретения способ включает введение пациенту соединения, имеющего структуру где n равно 1-8; X означает СН или N; R 1 означает Н, ОН или SH; R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают C 1 -С 6 алкил или С 3 -С 8 циклоалкил; и R 5 означает ОН или SH; и R 6 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -C 8 циклоалкил или арил; или его соли. В некоторых вариантах осуществления способа по настоящему изобретению пациент является человеком. В некоторых вариантах осуществления способа по настоящему изобретению нейродегенеративное заболевание является болезнью Альцгеймера, легкой степенью нарушения познавательной способности, болезнью Паркинсона, лобно-височным слабоумием, слабоумием или слабоумием с тельцами Леви. В одном варианте осуществления изобретения нейродегенеративное заболевание является болезнью Альцгеймера. В одном варианте осуществления изобретения способы дополнительно включают введение пациенту антагониста рецептора NMDA, ингибитора ацетилхолинэстеразы, антиамилоидного антитела, антигониста 5-НТ6, ингибитора гамма-секретазы, ингибитора бета-секретазы или ингибитора агрегации амилоид- β-пептида. В некоторых вариантах осуществления изобретения способ дополнительно включает введение пациенту ингибитора агрегации tau-белка. Примеры ингибиторов агрегации tau-белка включают хлорид метилтиониния, производные нафтохинона и антрахиноны. В одном варианте осуществления изобретения нейродегенеративное заболевание является болезнью Паркинсона. В одном варианте осуществления изобретения способы дополнительно включают введение пациенту агониста рецептора допамина. Настоящее изобретение относится к способу уменьшения агрегации tau-белка в клетках, включающему контактирование клетки с эффективным количеством соединения, имеющего структуру где n равно 1-10; X означает C-R 11 или N, где R 11 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; Z означает R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают Н, C 1 -С 6 алкил или С 3 -С 8 циклоалкил; R 5 означает ОН или SH; и R 6 , R 12 , R 13 и R 14 независимо означают Н, ОН, SH, F, Cl, SO 2 R 15 , NO 2 , трифторметил, метокси или CO-R 15 , где R 15 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; или его соли с целью ингибирования агрегации tau-белка в клетке. В одном варианте осуществления изобретения способ включает введение пациенту соединения, имеющего структуру где n равно 1-9; X означает C-R 11 или N, где R 11 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают Н, C 1 -С 6 алкил или С 3 -С 8 циклоалкил; R 5 означает ОН или SH; и R 6 , R 12 , R 13 и R 14 независимо означают Н, ОН, SH, F, Cl, SO 2 R 15 , NO 2 , трифторметил, метокси или CO-R 15 , где R 15 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; или его соли. В одном варианте осуществления изобретения способ включает введение пациенту соединения, имеющего структуру где n равно 1-9; X означает СН или N; R 1 означает Н или ОН; R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают Н, C 1 -С 6 алкил или С 3 -С 8 циклоалкил; R 5 означает ОН или SH; и R 6 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -C 8 циклоалкил или арил; или его соли. В одном варианте осуществления изобретения способ включает введение пациенту соединения, имеющего структуру где n равно 1-9; X означает C-R 11 или N, где R 11 означает Н, ОН, SH, F, Cl, SO 2 R 7 ; NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают Н, C 1 -С 6 алкил или С 3 -С 8 циклоалкил; R 5 означает ОН или SH; и R 6 , R 12 , R 13 и R 14 независимо означают Н, ОН, SH, F, Cl, трифторметил, метокси или CO-R 15 , где R 15 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; или его соли. В одном варианте осуществления изобретения способ включает введение пациенту соединения, имеющего структуру: где n равно 1-9; X означает СН или N; R 1 означает Н или ОН; R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают Н, C 1 -С 6 алкил или С 3 -С 8 циклоалкил; и R 5 означает ОН или SH; и R 6 означает Н, ОН, SH, F, Cl, трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; или его соли. В одном варианте осуществления изобретения способ включает введение пациенту соединения, имеющего структуру: где n равно 1-8; X означает C-R 11 или N, где R 11 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают C 1 -С 6 алкил или С 3 -С 8 циклоалкил; R 5 означает ОН или SH; и R 6 , R 12 , R 13 и R 14 независимо означают Н, ОН, SH, F, Cl, SO 2 R 15 , NO 2 , трифторметил, метокси или CO-R 15 , где R 15 означает алкил, алкенил, алкинил, С 3 -C 8 циклоалкил или арил; или его соли. В одном варианте осуществления изобретения способ включает введение пациенту соединения, имеющего структуру: где n равно 1-8; X означает СН или N; R 1 означает Н, ОН или SH; R 2 означает Н или NR 3 R 4 , где R 3 и R 4 независимо означают C 1 -С 6 алкил или С 3 -С 8 циклоалкил; R 5 означает ОН или SH; и R 6 означает Н, ОН, SH, F, Cl, SO 2 R 7 , NO 2 , трифторметил, метокси или CO-R 7 , где R 7 означает алкил, алкенил, алкинил, С 3 -С 8 циклоалкил или арил; или его соли. В некоторых вариантах осуществления способа по настоящему изобретению клетка является нервной клеткой. В некоторых вариантах осуществления изобретения клетка находится в организме пациента. Настоящее изобретение относится к способу лечения пациента, имеющего опухоль, сверхэкспрессирующую N-CoR, включающему введение пациенту одного или нескольких соединений по настоящему изобретению отдельно или в комбинации с одним или несколькими лигандами рецептора ретиноида, с одним или несколькими лигандами гистон-дезацетилазы либо с теми и другими вместе, в количестве, эффективном для лечения данного пациента. Соединения по настоящему изобретению могут быть использованы в комбинации с другими соединениями, ингибирующими фермент гистондезацетилазу (HDAC). Ферменты HDAC вызывают посттрансляционную модификацию гистонов (публикация патента США № 2004/0197888, Armour et al.). Гистоны представляют собой группы белков, которые связываются с ДНК в эукариотических клетках с образованием уплотненных структур, именуемых хроматином. Такое уплотнение позволяет локализовать в ядре эукариотической клетки большое количество ДНК, но плотная структура хроматина ограничивает доступ факторов транскрипции к ДНК. Ацетилирование гистонов уменьшает плотность хроматина, позволяя факторам транскрипции связываться с ДНК. Дезацетилирование, катализируемое гистон-дезацетилазами (HDAC), увеличивает плотность хроматина, уменьшая доступ факторов транскрипции к ДНК. Поэтому ингибиторы гистон-дезацетилаз предотвращают уплотнение хроматина, позволяя факторам транскрипции связываться с ДНК, и увеличивают экспрессию генов. В способах по настоящему изобретению оценка процентного значения клеток с N-CoR в цитоплазме по отношению к процентному значению клеток с N-CoR в ядре является показателем отношения числа более дифференцированных клеток к числу менее дифференцированных клеток в данной ткани. В способе по настоящему изобретению опухоли, сверхэкспрессирующие N-CoR, могут включать мультиформную глиобластому, рак молочной железы, рак ободочной и прямой кишки, мелкоклеточный рак легкого или рак яичника. Настоящее изобретение относится также к способу ингибирования у пациента роста опухоли, сверхэкспрессирующей N-CoR, ввключающему введение пациенту одного или нескольких соединений по настоящему изобретению отдельно или в комбинации с одним или несколькими лигандами рецептора ретиноида, одним или несколькими лигандами гистон-дезацетилазы либо с теми и другими вместе, в количестве, эффективном для воздействия на N-CoR с целью индукции дифференцировки клеток опухоли, сверхэкспрессирующей N-CoR, и подавления роста опухоли у данного пациента. В значении, использованном в настоящем описании изобретения, термин "терапевтически эффективное количество" означает количество, достаточное для лечения пациента, страдающего каким-либо заболеванием (например, имеющего опухоль, сверхэкспрессирующую N-CoR), или уменьшения интенсивности симптома или осложнения, ассоциированного с данным заболеванием, без возникновения вредных побочных эффектов (таких как токсичность, раздражение или аллергическая реакция) и с обоснованным соотношением пользы/риска при использовании в соответствии с настоящим изобретением. Конкретное эффективное количество может изменяться в зависимости от таких факторов, как определенное заболевание, подлежащее лечению, физическое состояние пациента, вид млекопитающего, нуждающегося в лечении, продолжительность лечения, характер одновременно проводимого лечения (если такое лечение проводится), применяемые препараты и структура соединений или их производных. В используемом здесь значении термин "лечение" означает замедление, прекращение или обратное развитие заболевания, в частности, опухолей, сверхэкспрессирующих N-CoR. В используемом здесь значении термин "алкил" означает насыщенные алифатические углеводородные группы с разветвленной и прямой цепью, содержащие конкретное число атомов углерода. Таким образом, C 1 -C n в термине "C 1 -C n алкил" означает группы, содержащие 1, 2, ..., n-1 или п атомов углерода, расположенных в виде линейной или разветвленной цепи. Например, C 1 -C 6 в термине "C 1 -С 6 алкил" означает группы, содержащие 1, 2, 3, 4, 5 или 6 атомов углерода, расположенных в виде линейной или разветвленной цепи, и, в частности, включает метил, этил, пропил, бутил, пентил, гексил и тому подобные. В одном варианте осуществления изобретения алкил представляет собой C 1 (метил). Термин "циклоалкил" означает циклические кольца алканов, содержащих в общей сложности три-восемь атомов углерода или любое число в данном интервале (то есть циклопропил, циклобутил, циклопентил, циклогексил, циклогептил или циклооктил). В используемом здесь значении термин "алкенил" означает неароматический углеводородный радикал с прямой или разветвленной цепью, содержащий по меньшей мере одну углерод-углеродную двойную связь, при этом в радикале может присутствовать максимально возможное число неароматических углерод-углеродных двойных связей. Например, "С 2 -С 6 алкенил" означает алкенильный радикал, содержащий 2, 3, 4, 5 или 6 атомов углерода и имеющий соответственно 1, 2, 3, 4 или 5 углерод-углеродных двойных связей. Алкенильные группы включают этенил, пропенил, бутенил и циклогексенил. Термин "алкинил" означает углеводородный радикал с прямой или разветвленной цепью, имеющий по меньшей мере одну углерод-углеродную тройную связь, при этом в радикале может присутствовать максимально возможное число неароматических углерод-углеродных тройных связей. Таким образом, "С 2 -С 6 алкинил" означает алкинильный радикал, содержащий 2 или 3 атома углерода и одну углерод-углеродную тройную связь, или содержащий 4 или 5 атомов углерода и до двух углерод-углеродных тройных связей, или содержащий 6 атомов углерода и до трех углерод-углеродных тройных связей. Алкинильные группы включают этинил, пропинил и бутинил. В используемом здесь значении термин "арил" означает любое устойчивое моноциклическое, бициклическое или полициклическое углеродное кольцо, содержащее до 10 атомов в каждом кольце, при этом по меньшей мере одно кольцо является ароматическим. Примеры таких арильных групп включают фенил, нафтил, тетрагидронафтил, инданил, бифенил, фенантрил, антрил или аценафтил. В тех случаях, когда арильный заместитель является бициклическим и одно кольцо является неароматическим, присоединение происходит при помощи ароматического кольца. Специалисту в данной области должно быть понятно, что при выборе соединений по настоящему изобретению используемые в них разные заместители, то есть R 1 , R 2 и т.д., следует выбирать в соответствии с хорошо известными принципами образования химических структур. Разные группы R, присоединяемые к ароматическим кольцам соединений по настоящему изобретению, могут быть добавлены к кольцам стандартными методами, например, описанными в Advanced Organic Chemistry: Part B: Reaction and Synthesis, Francis Carey and Richard Sundberg, (Springer) 5 th ed., (2007), которая включена в настоящее описание изобретения путем ссылки. Соединения по настоящему изобретению могут быть в форме соли. В используемом здесь значении термин "соль" означает соль соединений по настоящему изобретению, которые были модифицированы с образованием кислых или основных солей. В случае соединений, используемых для лечения рака, соль является фармацевтически приемлемой. Примеры фармацевтически приемлемых солей включают, не ограничиваясь ими, соли минеральных или органических кислот основных остатков, таких как амины; соли щелочных металлов или органические соли кислотных остатков, таких как фенолы. Соли могут быть получены с использованием органической или неорганической кислоты. Такие кислые соли являются хлоридами, бромидами, сульфатами, нитратами, фосфатами, сульфонатами, формиатами, тартратами, малеатами, малатами, цитратами, бензоатами, салицилатами, аскорбатами и тому подобными. Феноляты являются солями щелочно-земельных металлов, натрия, калия или лития. Термин "фармацевтически приемлемая соль" означает относительно нетоксичные неорганические и органические кислотно-аддитивные или основно-аддитивные соли соединений по настоящему изобретению. Соли могут быть получены in situ в процессе конечного выделения и очистки соединений по настоящему изобретению или в результате отдельного взаимодействия очищенного соединения по настоящему изобретению в форме свободного основания или свободной кислоты с приемлемой органической или неорганической кислотой или основанием, и выделения полученной таким образом соли. Типичные соли включают гидробромид, гидрохлорид, сульфат, бисульфат, фосфат, нитрат, ацетат, валерат, олеат, пальмитат, стеарат, лаурат, бензоат, лактат, фосфат, тозилат, цитрат, малеат, фумарат, сукцинат, тартрат, нафтилат, мезилат, глюкогептонат, лактобионат, лаурилсульфонат и тому подобные. (См., например, публикацию Berge et al. (1977) "Pharmaceutical Salts", J. Pharm. Sci. 66:1-19). Композиции по настоящему изобретению можно вводить в разных формах, включая формы, подробно рассмотренные в настоящем описании изобретения. Лечение соединением по настоящему изобретению может быть одним из компонентов комбинированной или дополнительной терапии, то есть пациенту, нуждающемуся в лечении определенным лекарственным средством, вводят также другое лекарственное средство в сочетании с одним или несколькими соединениями по настоящему изобретению. Такая комбинированная терапия может быть последовательной, когда пациенту сначала вводят одно лекарственное средство и затем другое лекарственное средство, либо оба лекарственных средства пациенту вводят одновременно. Лекарственные средства могут быть введены независимо одинаковым способом введения либо двумя или большим числом разных способов введения в зависимости от используемых лекарственных форм. В используемом здесь значении термин "фармацевтически приемлемый носитель" означает фармацевтически приемлемый растворитель, суспендирующий агент или наполнитель, предназначенный для доставки соединений по настоящему изобретению в организм животного или человека. Носитель может быть жидким или твердым и может быть выбран в зависимости от предполагаемого способа введения. Липосомы также являются фармацевтически приемлемым носителем. Доза соединений, вводимых в процессе лечения, может изменяться в зависимости от таких факторов как фармакодинамические характеристики конкретного химиотерапевтического средства и способа его введения, возраста, пола, интенсивности обмена вещества, абсорбирующей способности, состояния здоровья и массы тела реципиента; характера и интенсивности проявления симптомов; вида одновременно проводимого лечения; частоты введения и желаемого терапевтического эффекта. Лекарственная форма соединений по настоящему изобретению может включать одно соединение или смесь такого соединения с противораковыми соединениями, ингибиторами роста опухоли или другими соединениями, также используемыми при лечении поражения нейритов. Соединения по настоящему изобретению могут быть введены в виде лекарственных форм для перорального введения, таких как таблетки, капсулы, пилюли, порошки, гранулы, эликсиры, настойки, суспензии, сиропы и эмульсии. Соединения могут быть также введены внутривенно (в виде болюса или инфузии), внутрибрюшинно, подкожно или внутримышечно либо прямо в опухоль, например, при помощи инъекции или других методов, с использованием лекарственных форм, хорошо известных специалистам в фармации. Соединения по настоящему изобретению могут быть введены в смеси с приемлемыми фармацевтическими разбавителями, наполнителями, эксципиентами или носителями (которые в настоящем описании изобретения обобщенно именуются фармацевтически приемлемым носителем), выбираемыми в соответствии с предполагаемой формой введения и совместимости со стандартной фармацевтической практикой. Лекарственная форма может быть предназначена для перорального, ректального, местного, внутривенного или парентерального введения, а также для прямой инъекции. Соединения могут быть введены отдельно, но обычно их смешивают с фармацевтически приемлемым носителем. Носитель может быть твердым или жидким, и тип носителя обычно выбирают в зависимости от используемого способа введения. В одном варианте осуществления изобретения носитель может быть моноклональным антителом. Активный агент может быть введен в форме таблетки или капсулы, липосомы, агломерированного порошка или в жидкой форме. Примеры приемлемых твердых носителей включают лактозу, сахарозу, желатин и агар. Капсулу или таблетку можно легко получить, ее можно легко проглотить или прожевать; другие твердые формы включают гранулы и порошки. Таблетки могут содержать приемлемые связывающие вещества, смазывающие вещества, разбавители, дезинтеграторы, красители, отдушки, вещества, сообщающие текучесть, и расплавляющие вещества. Примеры приемлемых жидких лекарственных форм включают растворы или суспензии в воде, фармацевтически приемлемых жирах и маслах, спиртах или других органических растворителях, включающих сложные эфиры, эмульсии, сиропы или эликсиры, суспензии, растворы и/или суспензии, восстанавливаемые из нешипучих гранул, и шипучие препараты, восстанавливаемые из шипучих гранул. Такие жидкие лекарственные формы могут содержать, например, приемлемые растворители, консерванты, эмульгаторы, суспендирующие агенты, разбавители, подсластители, загустители и расплавляющие вещества. Лекарственные формы для перорального введения необязательно содержат ароматизаторы и красители. Лекарственные формы для парентерального и внутривенного введения могут также включать минералы и другие вещества, делающие их совместимыми с выбранным типом инъекции или системы доставки. Конкретные примеры фармацевтически приемлемых носителей и наполнителей, которые могут быть использованы для получения лекарственных форм для перорального введения по настоящему изобретению, описаны в патенте США № 3903297, выданном Роберту 2 сентября 1975 г. Методы получения и композиции, используемые для получения лекарственных форм, пригодных для применения в настоящем изобретении, описаны в следующих публикациях: 7 Modern Pharmaceutics, Chapters 9 and 10 (Banker & Rhodes, Editors, 1979); Pharmaceutical Dosage Forms: Tablets (Lieberman et al., 1981); Ansel, Introduction to Pharmaceutical Dosage Forms 2nd Edition (1976); Remington's Pharmaceutical Sciences, 17th ed. (Mack Publishing Company, Easton, Pa., 1985); Advances in Pharmaceutical Sciences (David Ganderton, Trevor Jones, Eds., 1992); Advances in Pharmaceutical Sciences Vol.7. (David Ganderton, Trevor Jones, James McGinity, Eds., 1995); Aqueous Polymeric Coatings for Pharmaceutical Dosage Forms (Drugs and the Pharmaceutical Sciences, Series 36 (James McGinity, Ed., 1989); Pharmaceutical Particulate Carriers: Therapeutic Applications: Drugs and the Pharmaceutical Sciences, Vol.61 (Alain Rolland, Ed., 1993); Drug Delivery to the Gastrointestinal Tract (Ellis Horwood Books in the Biological Sciences. Series in Pharmaceutical Technology; J. G. Hardy, S. S. Davis, Clive G. Wilson, Eds.); Modem Pharmaceutics Drugs and the Pharmaceutical Sciences, Vol.40 (Gilbert S. Banker, Christopher T. Rhodes, Eds.). Все вышеуказанные публикации включены в настоящее описание изобретения путем ссылки. Таблетки могут содержать приемлемые связывающие вещества, смазывающие вещества, дезинтеграторы, красители, отдушки, вещества, сообщающие текучесть, и расплавляющие вещества. Например, лекарственная форма для перорального введения в виде таблетки или капсулы может содержать активный лекарственный компонент вместе с нетоксичным, фармацевтически приемлемым, инертным носителем, предназначенным для перорального введения, таким как лактоза, желатин, агар, крахмал, сахароза, глюкоза, метилцеллюлоза, стеарат магния, дикальцийфосфат, сульфат кальция, маннит, сорбит и тому подобные. Приемлемые связывающие вещества включают крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза, кукурузные подсластители, природные и синтетические камеди, такие как аравийская камедь, трагант или альгинат натрия, карбоксиметилцеллюлозу, полиэтиленгликоль, воски и тому подобные. Смазывающие вещества, используемые в лекарственных формах, включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и тому подобные. Дезинтеграторы включают, без каких-либо ограничений, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и тому подобные. Соединения по настоящему изобретению могут быть введены в виде систем для доставки липосом, таких как мелкие однослойные пузырьки, крупные однослойные пузырьки и многослойные пузырьки. Липосомы могут быть получены из разных фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины. Соединения по настоящему изобретению могут быть введены в виде компонентов эмульсий, направленно воздействующих на ткани. Соединения по настоящему изобретению могут быть также связаны с растворимыми полимерами, используемыми в качестве носителей для направленной доставки лекарственного средства или пролекарства. Такие полимеры включают поливинилпирролидон, сополимер пирана, полигидроксилпропилметакриламидфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенный пальмитоильными остатками. Кроме того, соединения могут быть связаны с поддающимися биологическому разложению полимерами, используемыми для достижения регулируемого высвобождения лекарственного средства, такими как, например, полимер молочной кислоты, полигликолевая кислота, сополимеры полимера молочной кислоты и полигликолевой кислоты, полиэпсилонкапролактон, полигидроксимасляная кислота, сложные полиортоэфиры, полиацетали, полидигидропираны, полицианоацилаты и перекрестно сшитые или амфипатические блоксополимеры гидрогелей. Активный ингредиент может быть введен перорально в твердых лекарственных формах, таких как капсулы, таблетки и порошки, или в жидких лекарственных формах, таких как эликсиры, сиропы и суспензии. Активный ингредиент может быть также введен парентерально в стерильных жидких лекарственных формах. Желатиновые капсулы могут содержать соединения, являющиеся активным ингредиентом, и порошкообразные носители, такие как лактоза, крахмал, производные целлюлозы, стеарат магния, стеариновая кислота и тому подобные. Аналогичные разбавители могут быть использованы для получения прессованных таблеток. Как таблетки, так и капсулы могут быть получены в виде продуктов с немедленным высвобождением или продуктов с замедленным высвобождением, обеспечивающих непрерывное высвобождение лекарственного средства в течение нескольких часов. На прессованные таблетки может быть нанесено сахарное покрытие или пленочное покрытие, маскирующее неприятный вкус и защищающее таблетку от воздействия окружающей среды, либо энтеросолюбильное покрытие, обеспечивающее избирательную дезинтеграцию в желудочно-кишечном тракте. Компоненты лекарственного средства для перорального введения в жидкой лекарственной формы объединяют с любым нетоксичным, фармацевтически приемлемым, инертным носителем, предназначенным для перорального введения, таким как этанол, глицерин, вода и тому подобные. Примеры приемлемых жидких лекарственных форм включают растворы или суспензии в воде, фармацевтически приемлемых жирах и маслах, спиртах или других органических растворителях, включающих сложные эфиры, эмульсии, сиропы или эликсиры, суспензии, растворы и/или суспензии, восстанавливаемые из нешипучих гранул, и шипучие препараты, восстанавливаемые из шипучих гранул. Такие жидкие лекарственные формы могут содержать, например, приемлемые растворители, консерванты, эмульгаторы, суспендирующие агенты, разбавители, подсластители, загустители и расплавляющие вещества. Жидкие лекарственные формы для перорального введения могут содержать красители и отдушки, увеличивающие привлекательность для пациента. Как правило, приемлемыми носителями для растворов, предназначенных для парентерального введения, являются вода, приемлемое масло, физиологический раствор, водный раствор декстрозы (глюкозы) и растворы родственных cахаров, а также гликоли, такие как пропиленгликоль или полиэтиленгликоли. Растворы для парентерального введения предпочтительно содержат водорастворимую соль активного ингредиента, приемлемые стабилизирующие вещества и, в случае необходимости, буферы. Приемлемыми стабилизирующими веществами являются антиоксиданты, такие как бисульфит натрия, сульфит натрия или аскорбиновая кислота, используемые отдельно или в комбинации. В качестве стабилизирующих веществ используют также лимонную кислоту и ее соли и натриевую соль EDTA. Кроме того, растворы для парентерального ведения могут содержать консерванты, такие как хлорид бензалкония, метил- или пропилпарабен и хлорбутанол. Приемлемые фармацевтические носители описаны в публикации Remington's Pharmaceutical Sciences, Mack Publishing Company, которая является справочником в данной области. Соединения по настоящему изобретению могут быть также введены в виде лекарственной формы для назального введения благодаря использованию приемлемых наполнителей для назального введения или при помощи чрезкожных способов введения с использованием чрезкожных пластырей, хорошо известных специалистам в данной области. Введение лекарственного средства в виде системы для чрезкожной доставки является непрерывным в отличие от периодического введения в соответствии с определенной схемой введения. Лекарственные формы для парентерального и внутривенного введения могут также включать минералы и другие вещества, делающие их совместимыми с выбранным типом инъекций или системы доставки. Соединения и композиции по настоящему изобретению могут быть нанесены на стенты, предназначенные для временной или постоянной имплантации в сердечно-сосудистую систему пациента. Настоящее изобретение будет лучше понято со ссылкой на приведенную ниже экспериментальную часть, но специалистам в данной области должно быть понятно, что экспериментальная часть лишь иллюстрирует изобретение, которое более полно описано в нижеследующей формуле изобретения. Метил-2-гидроксибензоат (17,6 г, 115,65 ммоль) и 1,6-диаминогексан (6,4 г, 55, 32,1 ммоль) нагревали в 100 мл круглодонной колбе, помещенной в нагревательный кожух, с насадкой Дина-Старка. Через 20 мин происходила отгонка метанола из реакционной смеси в насадку Дина-Старка. Реакционную смесь продолжали нагревать в течение 40 мин, затем смесь охлаждали до комнатной температуры и растирали в воде (100 мл). Отделившееся твердое вещество удаляли фильтрованием и подвергали воздушной сушке в течение ночи. Твердое вещество перекристаллизовывали из спирта, в результате чего была получена первая порция указанного в заголовке соединения (4 г) и вторая порция, равная 1,8 г, температура плавления которых была равна 141-142 °С (критическая температура плавления 141-142 °С). Общий выход 5,8 г (30%). 1 Н ЯМР (CDCl 3 ) δ 12,30 (ушир.с, 2Н), 7,40 (м, 4Н), 6,98 (д, 2Н), 6,82 (т, 2Н), 6,40 (ушир.с, 2Н), 3,42 (кв, 4Н), 1,70 (м, 4Н), 1,41 (м, 4Н). Эксперимент был выполнен в соответствии с методом, описанным в научной литературе (J. Med. Chem. (1981); 24, 1245-1249). Синтез бис-1,6- (2-меркаптобензоиламино)гексана (204) Указанное в заголовке соединение было синтезировано в результате выполнения двух стадий с использованием в качестве исходных веществ 1,6-диаминогексана и 2,2'-дитиодибензоилхлорида, как показано на схеме 1. К раствору 1,6-диаминогексана (465 мг, 4 ммоль) в метиленхлориде (20 мл) добавляли пиридин (3 мл) и затем раствор 2,2'-дитиодибензоилдихлорида (1,03 г, 3 ммоль) в метиленхлориде (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь упаривали досуха, и продукт очищали колоночной хроматографией, используя 2% метанола в метиленхлориде для элюирования чистого дисульфидного соединения. Выход 0,365 г (31%, т.пл. 220 °С). К охлаждаемому льдом раствору дисульфидного производного (0,365 г, 0,95 ммоль) в смеси метанола (2 мл) и метиленхлорида (5 мл) порциями добавляли концентрированную HCl (0,57 мл, 6 ммоль) и цинковую пыль (285 мг, атомная масса 4,4 мг) в течение 10 мин. Реакционную смесь перемешивали при 0-10 °С в течение 2 часов, остаток цинка удаляли фильтрованием и промывали горячей смесью метанола и метиленхлорида (50 мл). Фильтрат концентрировали досуха, вновь растворяли в горячем метаноле (5 мл) и добавляли воду (25 мл). Отделенное твердое вещество фильтровали, промывали водой и сушили, получая при этом бис-1,6-(2-меркаптобензоиламино)гексан. Выход 100 мг (27%, т.п.133-135 °С (разложение)). 1 Н ЯМР (ДМСО-d 6 -D 2 O) 8 7,01-7,40 (м, 8Н), 3,22 (м, 4Н), 1,26-1,62 (м, 8Н). FAB (МН + ) 389. Синтез 2-меркапто-N-[5-(пиридин-3-илкарбамоил)пентил]бензамида (205). Стадия 1. Трет-бутиловый эфир [5-(пиридин-3-илкарбамоил) пентил] карбаминовой кислоты (3). К смеси 3-аминопиридина (1, 2,82 г, 30 ммоль) и 6-трет-бутоксикарбониламиногексановой кислоты (2, 9,2 г, 40 ммоль) в метиленхлориде (50 мл) добавляли HOBt (135 мг, 1 ммоль), EDC ∙HCl (7,6 г, 40 ммоль) и DIPEA (10,45 мл, 60 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. В данный момент времени результаты ТСХ свидетельствовали об исчезновении исходного вещества. Реакционный раствор промывали водой (3x25 мл), водным раствором бикарбоната натрия (25 мл), насыщенным раствором соли, затем сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный остаток очищали колоночной хроматографией, используя в качестве элюента 1% метанола в метиленхлориде, что позволило получить чистый продукт в виде маслянистого остатка. Остаток растирали в гексане, получая при этом соединение 3 в виде бесцветного твердого вещества (6,3 г, 68%, т.п.96-98 °С). 1 Н ЯМР (CDCl 3 ) δ 8,68 (ушир.с, 2Н), 8,48 (м, 1Н), 8,30 (м, 2Н), 7,32 (м, 1Н), 4,62 (ушир.с, 1Н), 3,16 (м, 2Н), 2,40 (м, 2Н), 2,78 (м, 2Н), 1,50 (м, 4Н), 1,40 (с, 9Н). Стадия 2. Дигидрохлорид 6-амино-N-(пиридин-3-ил)гексаноамида (4). К охлаждаемой льдом смеси трет-бутилового эфира [5-(пиридин-3-илкарбамоил)пентил]карбаминовой кислоты (3, 3,07 г, 10 ммоль) в метиленхлориде (30 мл) добавляли раствор HCl в диоксане (4 М, 10 мл). Смесь перемешивали при комнатной температуре в течение ночи. Отделенное твердое вещество фильтровали, промывали метиленхлоридом и сушили в вакуумном сушильном шкафу, получая при этом продукт 4 в виде хлористо-водородной соли (2,7 г, 96%). Спектр 1 H ЯМР чистого твердого вещества соответствовал структуре соединения 4. 1 H ЯМР (D 2 O) δ 9,20 (с, 2Н), 7,91 (м, 1Н), 2,90 (т, 2Н), 2,42 (т, 2Н), 2,62 (м, 4Н), 1,36 (м, 2Н). ВОС-защитная группа может быть удалена в альтернативных условиях реакции. Соединение 4 может быть получено в стандартных условиях удаления аминозащитной группы (например, при использовании 3,0 эквивалентов 0,75 М раствора HCl (в эфире) при перемешивании в течение 12 часов. (См. P. Cali, M. Begtrup, Synthesis, 2002, 63-64). Вышеописанным способом были также синтезированы два следующих соединения, а именно дигидрохлорид 6-амино-N-(фенил)гексаноамида и дигидрохлорид 6-амино-N-(4-диметиламинофенил)гексаноамида. [ 1 Н ЯМР (CDCl 3 ) δ 7,32 (д, 2Н), 6,67 (д, 2Н), 2,91 (с, 6Н), 2,68 (м, 2Н), 2,31 (м, 2Н), 1,72 (м, 2Н), 1,43 (ушир.м, 4Н)]. Стадия 3. 2,2'-Дитиобис{N-[5-(пиридин-3-илкарбамоил)пентил]бензамид (6). К смеси дисульфида 2-тиобензойной кислоты (5, 0,765 г, 2,5 ммоль), HOBt (0,665 г, 4,9 ммоль), EDC ∙HCl (2 г, 10 ммоль) в ДМФА (40 мл) добавляли производное 4 амина (1,5 г, 5 ммоль) и DIPEA (3,5 мл, 20 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь выливали в воду и экстрагировали этилацетатом (5 ×30 мл). Объединенные органические слои промывали насыщенным раствором соли, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный остаток очищали колоночной хроматографией, используя 2-5% метанола в метиленхлориде для элюирования требуемого продукта 6 (1 г, 27%), который был получен в виде бесцветного твердого вещества. 1 Н-ЯМР (ДМСО-d 6 ) δ: 10,01 (ушир.с, 2Н), 8,67, (с, 2Н), 8,21 (д, 2Н), 7,98 (м, 4Н), 7,83 (д, 2Н), 7,65 (т, 2Н), 7,42 (т, 2Н), 7,30 (м, 2Н), 3,81 (т, 4Н), 2,30 (т, 4Н), 1,46 (м, 4Н), 1,30 (м, 4Н). Вышеуказанным способом были также синтезированы два следующих соединения, а именно 2,2'-дитиобис{N-[5-(фенилкарбамоил)пентил]бензамид и 2,2'-дитиобис{N-[5-(4-диметиламинофенилкарбамоил)пентил]бензамид}. [ 1 Н ЯМР (CDCl 3 ) δ 8,00 (д, 2Н), 7,56 (м, 4Н), 7,35 (м, 6Н), 6,66 (д, 4Н), 3,90 (т, 4Н), 2,90 (с, 12Н), 2,51 (т, 4Н), 1,76 (м, 12Н), 1,45 (м, 4Н)]. Спектры 1 Н ЯМР двух указанных соединений соответствуют их структурам. Стадия 4. 2-Меркапто-N-[5-(пиридин-3-илкарбамоил)пентил]бензамид (205). К охлаждаемому льдом раствору дисульфидного производного 6 (0,85 г, 1,2 ммоль) в смеси метанола (10 мл) и метиленхлорида (25 мл) порциями добавляли концентрированную HCl (3,4 мл) и цинковую пыль (1,2 г) в течение 10 мин. После перемешивания при комнатной температуре в течение 4 часов смесь разбавляли водой (30 мл) и метиленхлоридом (25 мл). Водный слой отделяли и подщелачивали насыщенным водным раствором бикарбоната натрия при одновременном охлаждении смеси. Отделенное твердое вещество фильтровали и подвергали воздушной сушке в течение ночи. Сухое твердое вещество экстрагировали в смесь горячего метанола и метиленхлорида (200 мл, соотношение 2:3). Горячий раствор фильтровали через фильтровальную бумагу из стекловолокна. Фильтрат упаривали досуха, и остаток растирали в этилацетате, получая при этом чистый требуемый продукт 205 (555 мг, 65%, т.п. 233-237 °С) в виде бесцветного твердого вещества. 1 Н-ЯМР (ДМСО-d 6 ) δ 10,06 (ушир.с, 1Н), 9,41 (ушир.с, 1Н), 8,76 (д, 1Н), 8,21 (д, 1Н), 8,02 (д, 1Н), 7,40 (м, 2Н), 7,32 (м, 1Н), 7,02 (т, 1Н), 6,91 (т, 1Н), 3,24 (кв, 2Н), 2,30 (т, 2Н), 1,60 (м, 4Н), 1,38 (м, 2Н). FAB (МН + ) 344. Вышеуказанным способом были синтезированы два следующих соединения, а именно, 2-меркапто-N-[5-(фенилкарбамоил)пентил]бензамид (201), т.п.110-112 °С и 2-меркапто-N-[5-(4-диметиламинокарбамоил)пентил]бензамид (206), т.пл. 108-110 °С. Спектры 1 Н ЯМР указанных соединений соответствуют нижеследующим структурам: 5-Хлор-2-меркапто-N-[5-(пиридин-3-илкарбамоил)пентил]бензамид (207а) 5-Хлор-2-меркапто-N-[5-(пиридин-3-илкарбамоил)пентил]бензамид (207а), т.п. 238-240 °С, был получен вышеуказанным способом путем обработки амина 4 на стадии 3 4,4'-дихлор-2,2'-дитиодибензойной кислотой (9а) вместо 2,2'-дитиодибензойной кислоты (5). 1 H-ЯМР (ДМСО-d 6 ) δ 10,06 (ушир.с, 1Н), 9,47 (ушир.с, 1Н), 8,71 (с, 1Н), 8,20 (д, 1Н, J=3,2 Гц), 8,02 (д, 1Н, J=8,4 Гц), 7,30 (м, ЗН), 7,04 (д, 1Н, J=7,2 Гц), 3,14 (м, 2Н), 2,30 (т, 2Н), 1,60 (м, 4Н), 1,34 (м, 2Н). FAB (МН + ) 378. 2-Меркапто-N-[5-(фенил-3-илкарбамоил)пентил]бензамид (201) 2-Меркапто-N-[5-(фенил-3-илкарбамоил)пентил]бензамид (201), т.п.110-112 °С, был получен вышеуказанным способом при использовании в качестве исходных веществ анилина и 6-трет-бутоксикарбониламиногексановой кислоты (2). 1 Н ЯМР (CDCl 3 ) δ 7,69 (ушир.с, 1Н), 7,58 (д, 2Н, J=8 Гц), 7,49 (дд, 1Н, J=6,3, 1,5 Гц), 7,35 (м, 4Н), 7,16 (м, 2Н), 6,41 (ушир.с, 1Н), 4,71 (с, 1Н), 3,51 (кв, 2Н, J=6,6), 2,43 (т, 2Н, J=7,2 Гц), 1,88-1,66 (м, 4Н), 1,52 (м, 2Н). EIMS (МН + ) 343. 2-Меркапто-N-[5-(4-диметиламино-3-илкарбамоил)пентил]бензамид (206) 2-Меркапто-N-[5-(4-диметиламино-3-илкарбамоил)пентил]бензамид (206), т.п. 108-110 °С, был получен аналогичным способом при использовании в качестве исходных веществ 4-диметиламиноанилина и 6-трет-бутоксикарбониламиногексановой кислоты [2, 1 Н ЯМР (CDCl 3 ) δ 7,36 (д, 2Н), 6,69 (д, 2Н), 3,10 (м, 2Н), 2,91 (с, 6Н), 2,31 (т, 2Н), 1,75 (м, 2Н), 1,44 (м, 13Н) ] в результате выполнения четырех стадий. Соединение 206: 1 Н ЯМР (ДМСО-d 6 ) δ 9,52 (ушир.с, 1Н), 9,38 (ушир.с, 1Н), 7,37 (д, 4Н, J=9 Гц), 7,13 (м, 1Н), 6,99 (м, 1Н), 6,65 (д, 2Н, J=9,00 Гц), 3,24 (м, 2Н), 2,23 (т, 2Н, J=7,4, Гц), 1,55 (м, 4Н), 1,33 (м, 2Н). Бомбардировка ускоренными атомами (FAB) в режиме отрицательных ионов (М-Н + ) 384. 2-Меркапто-N-[5-(4-аминофенилкарбамоил)пентил]бензамид (209) 2-Меркапто-N-[5-(4-аминофенилкарбамоил)пентил]бензамид (209) был синтезирован аналогичным способом при использовании в качестве исходных веществ 4-нитроанилина и 6-трет-бутоксикарбониламиногексановой кислоты (2). Нитрогруппа была восстановлена в аминогруппу на стадии 4 в процессе восстановления Zn/HCl. 1 Н ЯМР (ДМСО-d 6 ) δ 9,39 (ушир.с, 2Н), 7,41 (д, 1Н, J=8,00 Гц), 7,32 (д, 1Н, J=8,0 Гц), 7,14 (м, ЗН), 6,99 (м, 1Н), 6,47 (д, 2Н, J=8,7 Гц), 4,79 (с, 2Н), 3,28 (м, 2Н), 2,21 (т, 2Н, J=7,5 Гц), 1,58 (м, 4Н), 1,36 (м, 2Н). ESMS (МН + ) 358. 5-Хлор-2-меркапто-N- (5-фенилкарбамоил)пентил) бензамид (210) 5-Хлор-2-меркапто-N-(5-фенилкарбамоил)пентил)бензамид (210) был получен аналогичным способом при использовании анилина и 6-трет-бутоксикарбониламиногексановой кислоты (2). Соответствующий амин на стадии 3 подвергали взаимодействию с 4,4'-дихлор-2,2'-дитиодибензойной кислотой (9а) вместо 2,2'-дитиодибензойной кислоты (5), в результате чего на конечной стадии был получен требуемый продукт 210. 1 Н-ЯМР (ДМСО-d 6 ) δ 9,82 (ушир.с, 1Н), 8,56 (ушир.с, 1Н), 8,71 (с, 1Н), 7,51 (м, 4Н), 7,40 (м, 1Н), 7,34 (м, 1Н), 7,28 (м, 1Н), 5,41 (ушир.с, 1Н), 3,20 (м, 2Н), 2,30 (т, 2Н, J=7,2 Гц), 1,61-1,50 (м, 4Н), 1,35 (м, 2Н). ESMS (МН + ) 377. 2-Меркапто-5-метокси-N-(5-фенилкарбамоил)пентил)бензамид (211) 2-Меркапто-5-метокси-N-(5-фенилкарбамоилпентил)бензамид (211) был также получен путем обработки производного амина на стадии 3 4,4' -диметокси-2,2'-дитиодибензойной кислотой (9b). 1 Н ЯМР (CDCl 3 ) δ 8,30 (ушир.с, 1Н), 7,53 (д, 2Н, J=7,8 Гц), 7,44 (д, 1Н, J=8,4 Гц), 7,24 (м, 2Н), 7,08 (м, 2Н), 6,87 (дд, 1Н, J=3,0, 5,7 Гц), 6,49 (ушир.с, 1Н), 3,37 (кв, 2Н, J=6,5 Гц), 2,39 (т, 2Н, J=7,5 Гц), 1,75 (м, 2Н), 1,56 (м, 2Н), 1,46 (м, 2Н). ESMS (М Н + ) 373. 2-Меркапто-N-(6-фенилкарбамоилгексил)бензамид (212) 2-Меркапто-N-(6-фенилкарбамоилгексил)бензамид (212) был синтезирован аналогичным способом при использовании в качестве исходных веществ анилина и 7-трет-бутоксикарбониламиногептановой кислоты (16) вместо 6-трет-бутоксикарбониламиногексановой кислоты (2) на стадии 1. 1 Н-ЯМР (ДМСО-d 6 ) δ 9,82 (ушир.с, 1Н), 8,38 (ушир.с, 1Н), 7,56 (д, 2Н, J=7,8 Гц), 7,41 (м, 2Н), 7,26 (м, 3Н), 7,25 (м, 1Н), 6,99 (т, 1Н, J=7,5 Гц), 5,34 (ушир.с, 1Н), 3,20 (м, 2Н), 2,29 (т, 2Н, J=7,2 Гц), 1,61-1,48 (м, 4Н), 1,34 (м, 2Н). m/е 356. 2-Меркапто-N-(7-фенилкарбамоилгептил)бензамид (213) 2-Меркапто-N-(7-фенилкарбамоилгептил)бензамид (213) был получен при использовании в качестве исходных веществ анилина и 8-трет-бутоксикарбониламинооктановой кислоты (18) вместо 6-трет-бутоксикарбониламиногексановой кислоты (2) на стадии 1. 1 H-ЯМР (ДМСО-d 6 ) δ 9,82 (ушир.с, 1Н), 8,38 (ушир.с, 1Н), 7,57 (д, 2Н, J=7,5 Гц), 7,40 (м, 2Н), 7,25 (м, 3Н), 7,15 (м, 1Н), 6,99 (т, 1Н, J=7,2 Гц), 5,34 (ушир.с, 1Н), 3,20 (м, 2Н), 2,28 (т, 2Н, J=7,2 Гц), 1,60-1,49 (м, 4Н), 1,31 (м, 2Н). m/е 370. 2-Меркапто-N-(10-фенилкарбамоилдецил)бензамид (214) 2-Меркапто-N-(10-фенилкарбамоилдецил)бензамид (214) был получен при использовании в качестве исходных веществ анилина и 11-трет-бутоксикарбониламиноундекановой кислоты (20) на стадии 1 вместо 6-трет-бутоксикарбониламиногексановой кислоты (2), как было указано выше. 1 H-ЯМР (ДМСО-d 6 ) δ 9,82 (ушир.с, 1Н), 8,37 (ушир.с, 1Н), 7,57 (д, 2Н, J=8,00 Гц), 7,41 (м, 2Н), 7,24 (м, 3Н), 7,17 (м, 1Н), 6,99 (т, 1Н, J=7,5 Гц), 5,34 (ушир.с, 1Н), 3,20 (м, 2Н), 2,27 (т, 2Н, J=7,5 Гц), 1,56 (м, 2Н), 1,48 (м, 2Н), 1,34 (м, 2Н), m/е 412. Линии клеток MDA-MB-231, HT-29, NCI-H460, NCI-H522, NCI-Н69, GXF-209, HepG2, OVAR-3, PANC-1, DU-145, LNCAP, HL-60, K-562 и MOLT-4 были получены из Американской коллекции типовых культур (АТСС; Manassas, VA) или из Национального института рака (NCI; Frederick, MD). Среда RPMI, L-глутаминдипептид (HyQ SG-200) и HEPES были получены из компании Hyclone (Logan, UT). Фетальная телячья сыворотка (FBS) была получена из компании Sigma-Aldrich (St. Louis, МО). ДМСО был приобретен в компании Fisher Chemicals (Fair Lawn, NJ). Реагент для люминесцентного анализа жизнеспособности клеток CellTiter-Glo был приобретен в корпорации Promega (Madison, WI). Вся пластиковая посуда для культур тканей была приобретена в компании Corning Incorporated (New York, NY). Соединение 100 и соединение 102 были предоставлены компанией Lixte Biotechnology Holdings, Inc. (East Setauket, NY). Все линии клеток культивировали стандартными методами дважды за неделю в среде RPMI-1640, содержащей 2 мМ L-глутаминдипептида, 10 мМ HEPES и 10% FBS. Адгезивные линии клеток MDA-MB-231, HT-29, NCI-H460, NCI-Н522, GXF-209, HepG2, OVAR-3, PANC-1, DU-145 и LNCAP высевали на два 96-луночных планшета в количестве 2500 клеток/лунку в общем объеме, равном 50 мкл, и инкубировали во влажной камере для культур клеток при 37 °С и 5% CO 2 в течение ночи. Взвесь линий клеток NCI-H69, HL-60, К-562 и MOLT-4 высевали на два 96-луночных планшета в количестве 10000 клеток/лунку в общем объеме, равном 50 мкл, и инкубировали во влажной камере при 37 °С и 5% СО 2 в течение ночи. Получали 20 мМ исходного раствора соединения 205 в ДМСО. Из раствора получали двукратные исходные растворы в требуемых конечных концентрациях в среде RPMI-1640. 50 мкл двукратных исходных растворов добавляли в соответствующие лунки, содержащие 50 мкл клеток и среду, с достижением конечных концентраций, указанных в приложении. В лунки, содержащие среду и контрольные клетки, добавляли 50 мкл среды, и в лунки, содержащие контрольный наполнитель, добавляли 50 мкл ложного двукратного исходного раствора в ДМСО. Одновременно с добавлением лекарственных средств к клеткам каждую линию клеток на одном из планшетов исследовали при помощи описанного ниже анализа CellTiter-Glo для получения значений для каждой линии клеток в 0-й день. После инкубации в течение 72 ч при помощи анализа CellTiter-Glo исследовали клетки на другом планшете. Данный анализ выполняли в соответствии с инструкциями производителя. Планшеты удаляли из камеры, помещали на стол и выдерживали при комнатной температуре в течение 30 мин. Планшеты не ставили друг на друга. После инкубации при комнатной температуре в течение 30 мин в каждую лунку планшета добавляли 100 мкл реагента для анализа CellTiter-Glo и смешивали в течение 2 мин, затем снова инкубировали в течение 10 мин при комнатной температуре. Люминесценцию регистрировали при помощи сцинтилляционного и люминесцентного счетчика Microbeta компании PerkinElmer (Trilux). Исследования выполняли в соответствии с описанием, приведенным в разделе "Вещества и методы", используя первичные данные и установки для планшетов, указанные в приложении. Значения IC 50 для соединения 205 в каждой линии клеток приведены в таблице 2. Графическое представление воздействия указанных соединений на каждую линию клеток наряду с построением сопоставимых кривых проиллюстрировано на фиг. 8A-N. Большинство испытанных линий клеток было восприимчиво к воздействию соединения 205 в низком диапазоне мкМ (таблица 2). Такими линиями клеток являются рак молочной железы; рак ободочной кишки; два основных типа рака легкого, аденокарцинома и мелкоклеточный рак легкого (крупноклеточный рак легкого был восприимчив в наименьшей степени); рак желудка; рак печени (гепатома); аденокарцинома яичника; рак поджелудочной железы, два типа рака предстательной железы и три типа лейкоза, промиелоцитарный лейкоз, хронический миелоцитарный лейкоз и острый лимфолейкоз (таблица 2). Соединение 205 в наибольшей степени воздействовало на аденокарциному легкого, рак желудка, рак поджелудочной железы и рак молочной железы. * Значение IC 50 было определено на основании данных, так как построение кривой по точкам было невозможно. Пример 1. Для идентификации новых терапевтических мишеней при лечении мультиформной глиобластомы (GBM) оценивали способность соединений по настоящему изобретению ингибировать клетки мультиформной глиобластомы in vitro и in vivo. Все соединения ингибировали рост GBM in vivo в зависимости от дозы, как показано на фиг. 1-5 и 16-22. Соединения 205 и 206 ингибировали увеличение объема ксенотрансплантата опухоли GBM у мышей SCID, как показано на фиг. 6. На основании графиков роста линии клеток U373 мультиформной глиобластомы (GBM) в зависимости от воздействия разных доз лекарственного средства в течение 7 дней было измерено соотношение с контрольным образцом, представленное в графической форме. Все соединения активно воздействуют на линию клеток U-87, однако, соединения 201, 205, 206, 212 и 213 оказывают наиболее сильное воздействие при определении на эквимолярной основе. Соединения 205 и 206 характеризуются значительным ингибированием линии клеток U-87 ксенотрансплантатов GBM по сравнению с контрольным образцом (фиг. 9). Было установлено, что соединение 205 оказывает активное воздействие на линию клеток DAOY медуллобластомы (фиг. 11-13). Кроме того, было установлено, что соединение 205 в комбинации с ATRA также активно воздействует на линию клеток DAOY медуллобластомы (фиг. 7). Соединение 205 активно воздействует на рост ксенотрансплантатов SHSY-5Y (фиг. 10 и 14), не подавляя при этом рост нормальных клеток, о чем свидетельствует высокая жизнеспособность нормальных клеток головного мозга мышей по сравнению с контрольным образцом (фиг. 15). Кроме того, было установлено, что соединение 205 активно воздействует на линии клеток разных типов рака человека в низких микромолярных концентрациях. Типы рака включают рак молочной железы, рак ободочной кишки, рак легкого (3 типа), рак желудка, рак печени (гепатома), рак яичника, рак поджелудочной железы, рак предстательной железы и три типа лейкоза, а именно промиелоцитарный лейкоз, хронический миелоцитарный лейкоз и острый лимфолейкоз (табл. 2, фиг. 8А-N). Было также установлено, что соединение 205 активно воздействует на мультиформную глиобластому (GBM) человека в мышиной модели in vivo при подкожной имплантации опухолевых клеток мышам SCID. Кроме того, было установлено, что в данном случае соединение 205 является более активным по сравнению с эквимолярной дозой SAHA. Поэтому с учетом противораковой активности соединений по настоящему изобретению можно также предположить, что соединения благодаря своей структуре должны обладать активностью в качестве анти-ВИЧ средств, обеспечивая направленную доставку функциональных групп Zn в цинкосодержащие пальцеобразные домены ретровируса. Пример 2. На рынок лекарственных средств продолжают поступать новые противогрибковые средства, многие из которых обладают более высокой активностью по сравнению с имеющимися в настоящее время лекарственными средствами. Было исследовано в общей сложности 23 штамма, включающих 3 штамма Candida albicans, 3 штамма Candida glabrata, 3 штамма Cryptococcus neoformans, 3 штамма Aspergillus fumigatus, 3 штамма Rhizopus oryzae, 3 штамма Fusarium solani, 3 штамма Pseudallescheria boydii и 2 штамма Trichosporon rubrum. Bce штаммы были клиническими штаммами, направленными в лабораторию по исследованию грибов для оценки. Противогрибковое действие исследовали методами, описанными в публикациях Национального комитета по стандартам для клинических лабораторий, М-27А2, "Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts; Approved Standard" и М38-А "Reference Method for Broth Dilution Antifungal Susceptibility Testing of Conidium-Forming Filamentous Fungi; Approved Standard". Методы включают испытание в среде RPMI-1640 с глутамином и без бикарбоната, при величине инокулята 0,5-2,5 ×10 3 для дрожжей или 1-5 ×10 4 для плесневых грибов и инкубации при 35 °С в течение 24 и 48 ч. Минимальную ингибирующую концентрацию (MIC) определяли в виде наименьшей концентрации, вызывающей 50% уменьшение помутнения по сравнению с содержимым контрольной пробирки без добавления лекарственного средства для дрожжей и 80% ингибирование для плесневых грибов. Концентрации лекарственных средств были равны 0,125-64 мкг/мл. При испытании соединений по настоящему изобретению конечные результаты были определены для большинства видов. Все большее число пациентов с ослабленным иммунитетом вследствие трансплантации, ВИЧ/ СПИДа и рака, главным образом лейкоза, подвержено возникновению тяжелых микозов. Грибки, которые наиболее часто выделяют из мест поражения микозом у пациентов, включают Aspergillus spp. и Candida spp. Существуют эффективные методы лечения Candida spp., но озабоченность вызывает лечение микозов, обусловленных Aspergillus spp., для которых характерна высокая смертность пациентов с ослабленным иммунитетом. Такие инфекции плохо поддаются лечению в данной группе пациентов, что увеличивает потребность в лекарственных средствах, обладающих хорошей активностью в отношении указанных грибков. С учетом изменения характера микозов и отсутствия методов лечения данных инфекций соединения по настоящему изобретению предположительно могут быть использованы в качестве потенциальных противогрибковых средств. Для испытания использовали стандартную партию каждого средства. При испытании соединения 205 отвешивали 10 мг порцию каждого порошка и добавляли к 1 мл 10 0% ДМСО. Полученную концентрацию, равную 10 мкг/мл, разводили до рабочей концентрации, равной 6400 мкг/мл соединения 205 в 100% ДМСО. Все последующие разведения также производили с использованием соответствующих разбавителей. Конечные испытуемые концентрации находились в пределах 0,125-64 мкг/мл. В качестве контрольной идентификационной среды была использована среда RPMI-1640, рекомендованная NCCLS (Hardy Diagnostics, Santa Monica, CA). Все штаммы, использованные в данном исследовании, представляли собой клинические штаммы, полученные из лаборатории по исследованию грибов, и были предназначены для стандартного исследования и/или идентификации противогрибковой активности. Было подтверждено, что штаммы, не отправленные на идентификацию, гарантированно обладали необходимой для исследования чистотой. Было исследовано в общей сложности 23 штамма, которые включали 3 штамма Candida albicans, 3 штамма Candida glabrata, 3 штамма Cryptococcus neoformans, 3 штамма Aspergillus fumigatus, 3 штамма Rhizopus oryzae, 3 штамма Fusarium solani, 3 штамма Pseudallescheria boydii и 2 штамма Trichosporon rubrum. Минимальные ингибирующие концентрации (MIC) определяли в течение первых 24 ч, когда можно было определить рост культуры в контрольной пробирке, не содержащей лекарственного средства. Минимальную ингибирующую концентрацию (MIC) для дрожжей определяли в виде наименьшей концентрации, вызывающей 50% уменьшение помутнения, по сравнению с контролем роста, тогда как MIC для плесневых грибов определяли в виде наименьшей концентрации, вызывающей 80% уменьшение помутнения. Был создан новый класс ингибиторов гистон-дезацетилаз. Новизна ингибиторов состоит в использовании 2-меркапто-N-бензоиламиногруппы, связанной с замещенной или незамещенной 5-фенил-3-илкарбамоильной концевой группой или 5-пиридин-3-илкарбамоильной группой, отделенной 5-10 метиленовыми группами. Новизна соединений по настоящему изобретению состоит в использовании меркаптобензоиламиногруппы для образования хелатного соединения с цинком. Было продемонстрировано, что ингибиторы HDAC данного класса активно воздействуют на широкий спектр раковых клеток человека, выращиваемых в культуре и в виде ксенотрансплантатов у иммуносупрессорных мышей. Далее было продемонстрировано, что группа ингибиторов HDAC подавляет гистон-дезацетилазы как класса I (гистон 3), так и класса II (гистон 4). Соединения данного типа ингибируют HDAC в ксенотрансплантатах, выращиваемых у голых мышей, и при систематическом введении подавляют активность HDAC в нормальных тканях головного мозга мышей. Таким образом, соединения данного типа ингибируют предполагаемую мишень in vivo и достигают нормальной ткани головного мозга в дозах лекарственного средства, не вызывающих видимой токсичности у мышей при значительном ингибировании роста опухоли. Важным преимуществом данной группы лекарственных средств является то, что на их основе могут быть легко получены производные как в положении меркаптобензамоильной части, так и у противоположного конца молекулы, благодаря чему могут быть образованы многочисленные структуры, включая приведенные примеры, которые состоят из замещенных и незамещенных пиридинильных и фенильных частей и новой симметричной молекулы с двумя меркаптобензоиламиногруппами бис-1,6-(меркаптобензоиламино)гексан (соединение 204). Другим потенциальным преимуществом данной группы лекарственных средств является то, что механизм воздействия ингибиторов HDAC отличается от всех стандартных противораковых средств. Весьма вероятно, что ингибиторы HDAC по настоящему изобретению могут быть объединены с другими лекарственными средствами для достижения более высокой противораковой активности без увеличения токсичности. Приведенный пример показывает, что комбинация транс-ретиноевой кислоты с соединением 205 обладает более чем аддитивной активностью в отношении ингибирования роста клеток нейробластомы (фиг. 7). Результаты исследования соединений по настоящему изобретению представлены в табл. 2. Соединения по настоящему изобретению действительно обеспечивают достижение конечного результата. С учетом уровней соединений, достигаемых в организме человека, профилей безопасности и других факторов, принимаемых во внимание при создании лекарственных средств, соединения по настоящему изобретению могут претендовать на применение в качестве противогрибковых средств при лечении инфекций у человека и растений, так как было продемонстрировано, что члены класса ингибиторов HDAC обладают значительной антипролиферативной активностью в отношении важных в медицинском отношении патогенных грибков (суммированных в табл. 3 и 4). Кроме того, поскольку члены данной группы соединений проникают через гематоэнцефалический барьер и подавляют активность HDAC в нормальных тканях головного мозга, и поскольку во многих научных публикациях указано, что такая невральная активность оказывает благоприятное воздействие в нескольких моделях нейродегенеративных заболеваний, можно предположить, что соединения по настоящему изобретению могут быть полезны в качестве нейрозащитных средств и потенциально пригодны для лечения хронических нейродегенеративных заболеваний неизвестного происхождения, таких как тауопатии, и травм головного мозга. Кроме того, вполне вероятно, что соединения данного класса могут быть пригодны для лечения хронических и, возможно, острых воспалительных заболеваний, ассоциированных с фиброзом, таких как гипертрофия сердца и ревматоидный артрит. |