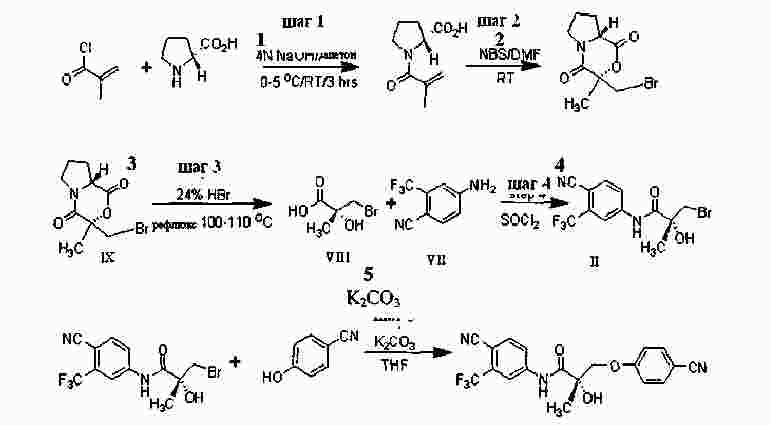
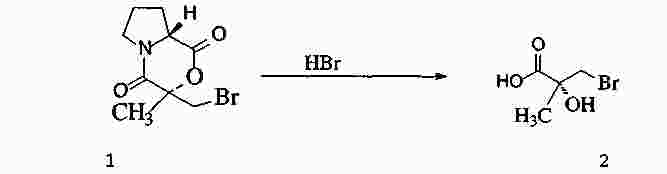
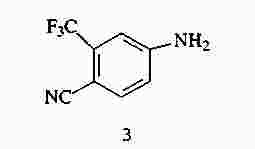
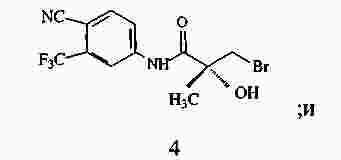
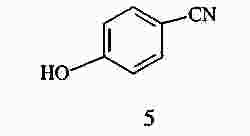
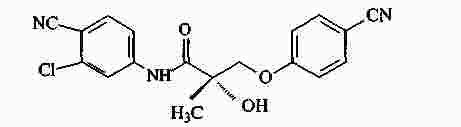
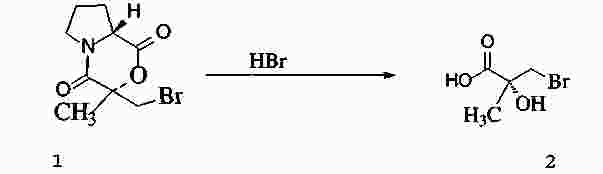
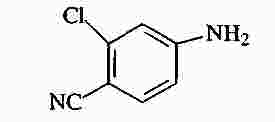
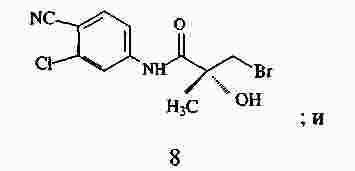
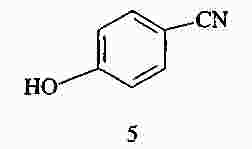
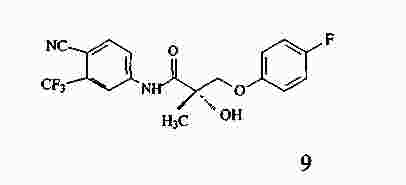
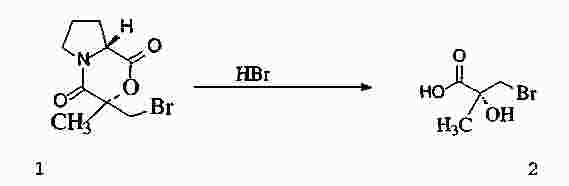
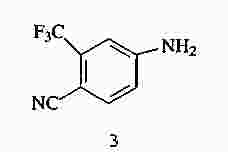
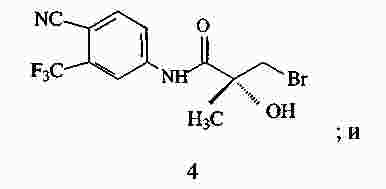
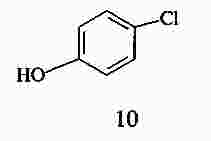
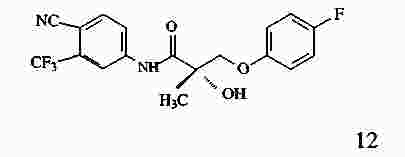
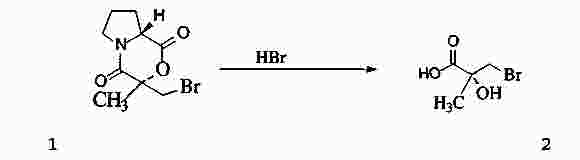
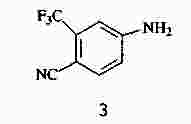
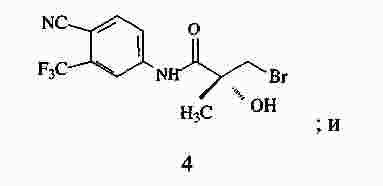
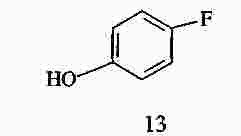
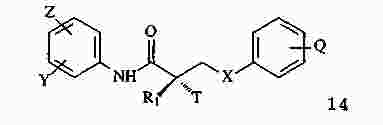
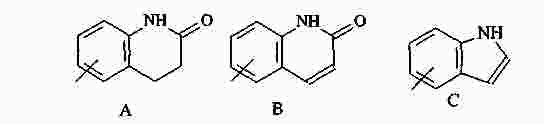
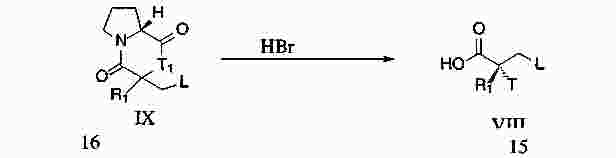
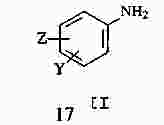
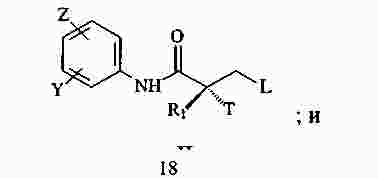
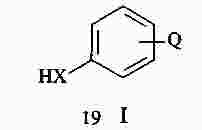
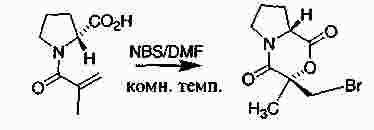
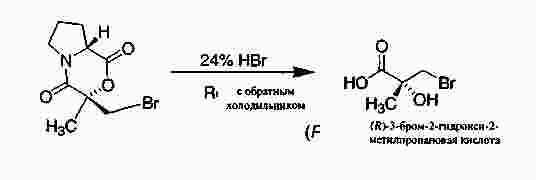
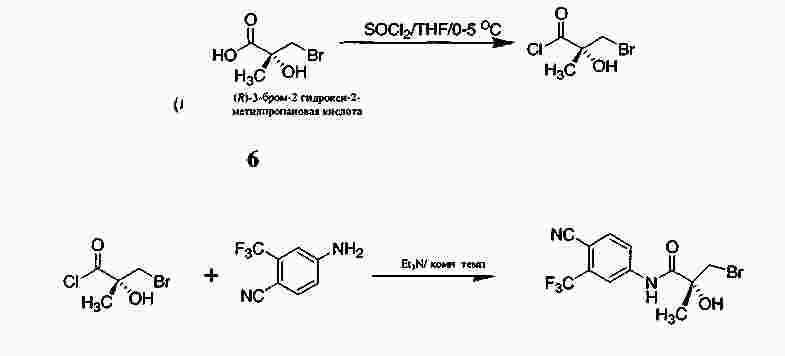
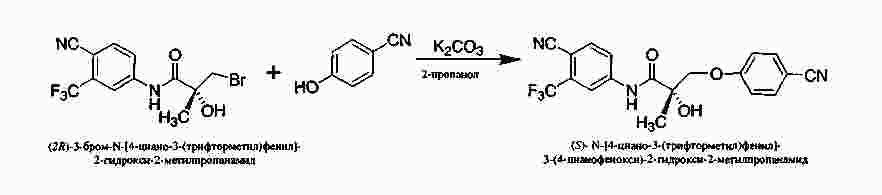
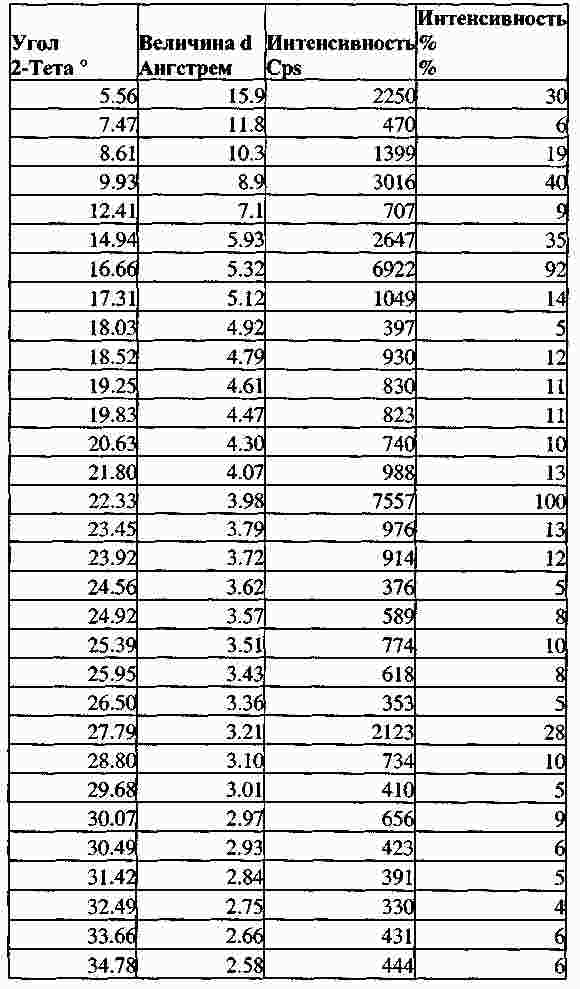
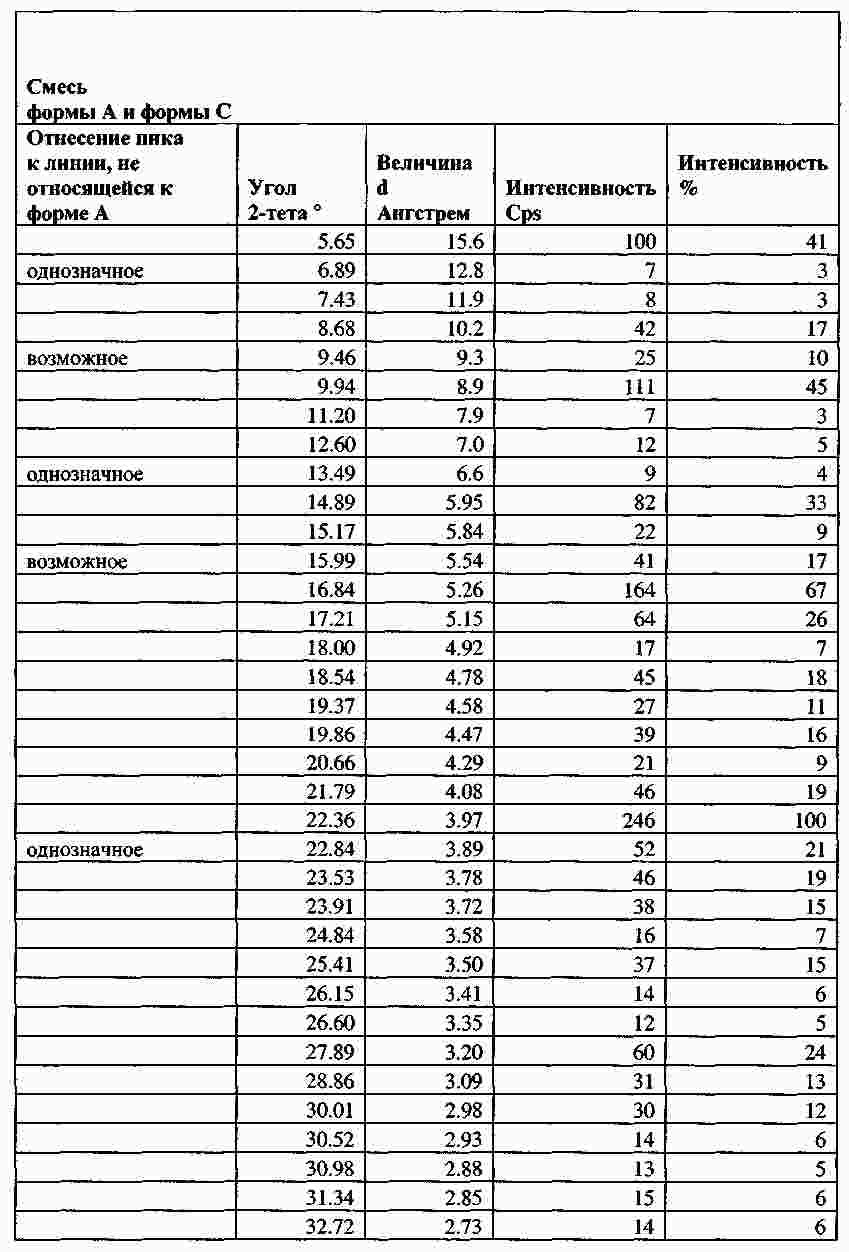
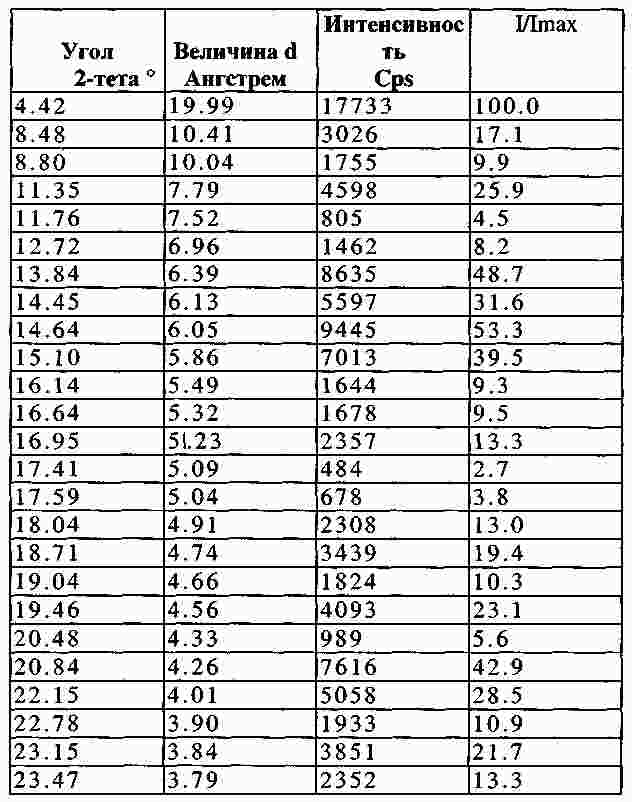
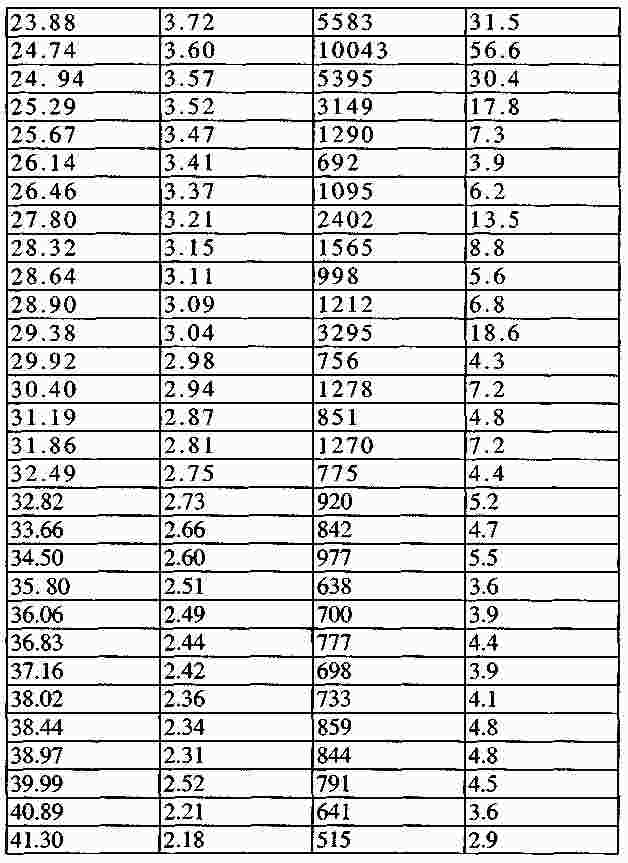
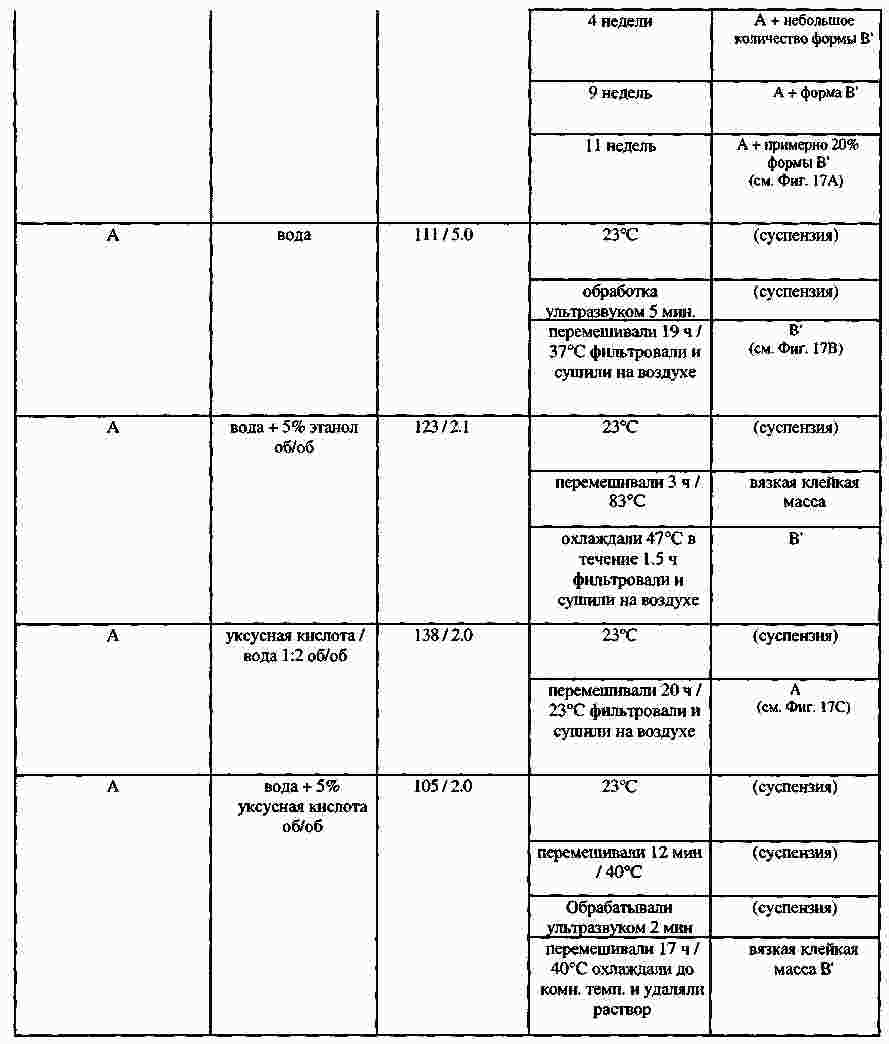
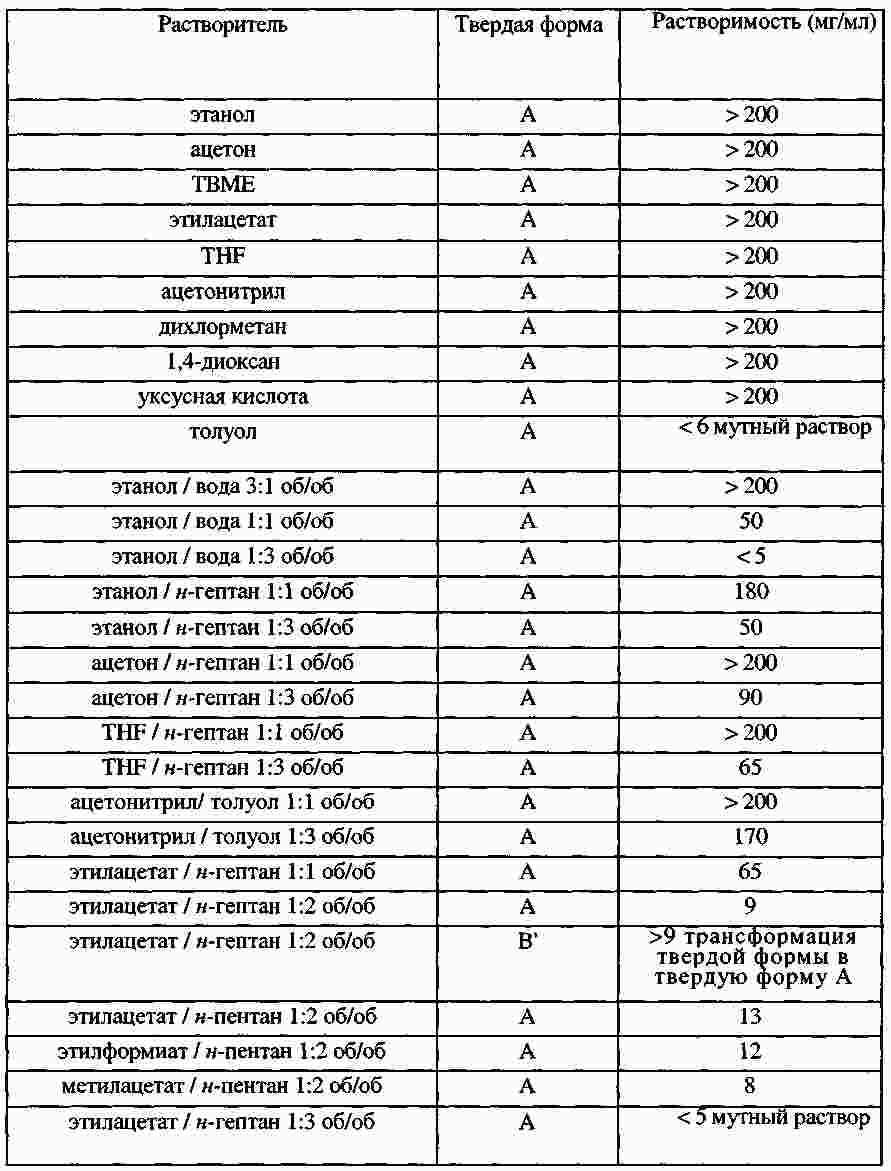
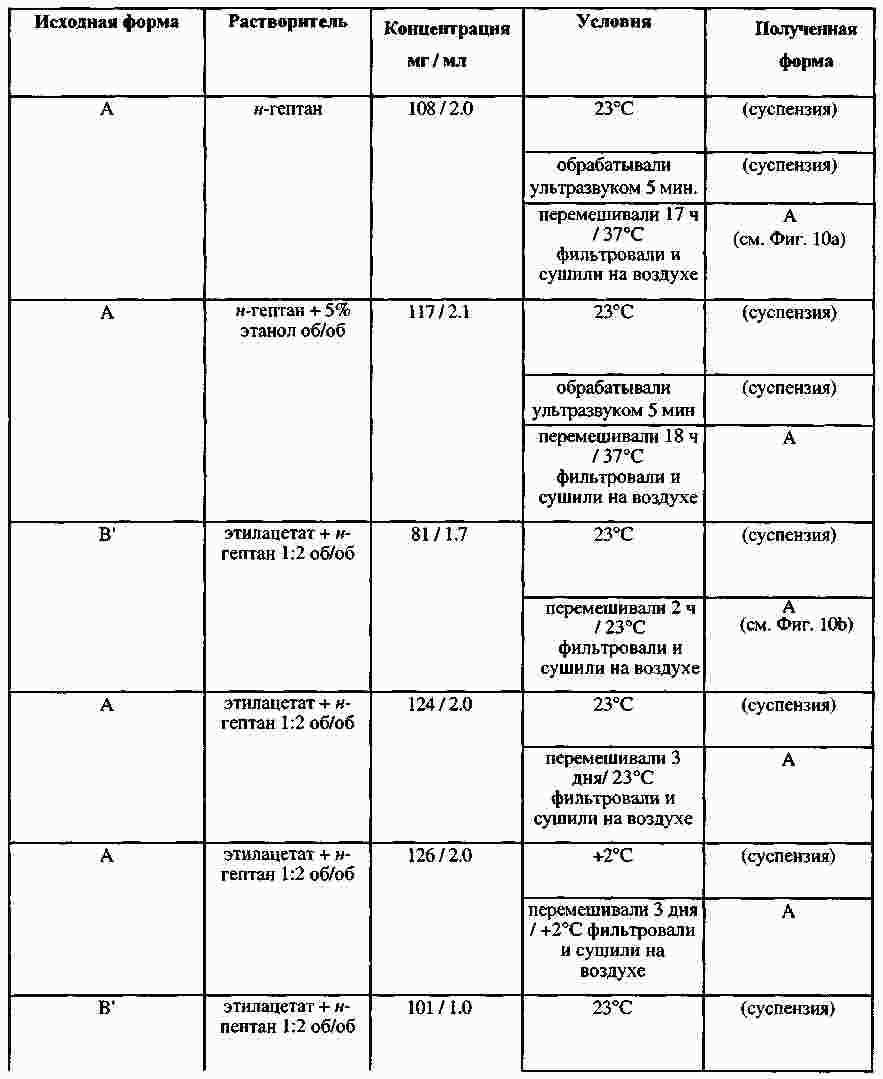
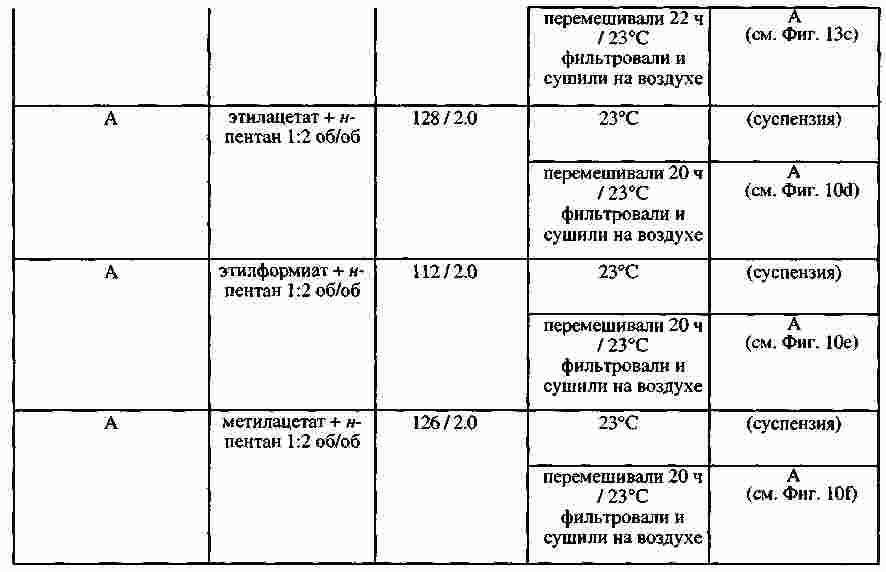
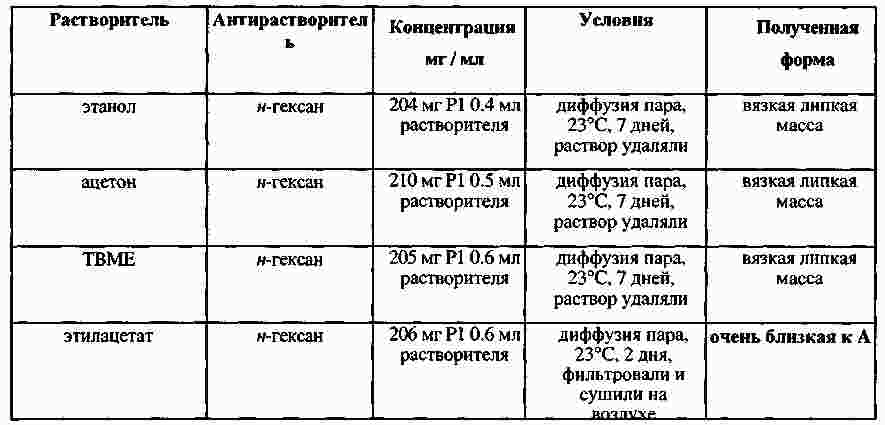
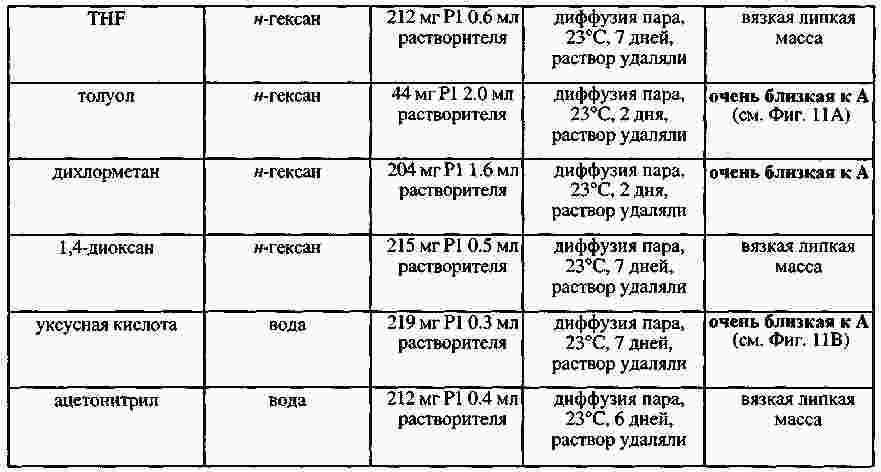
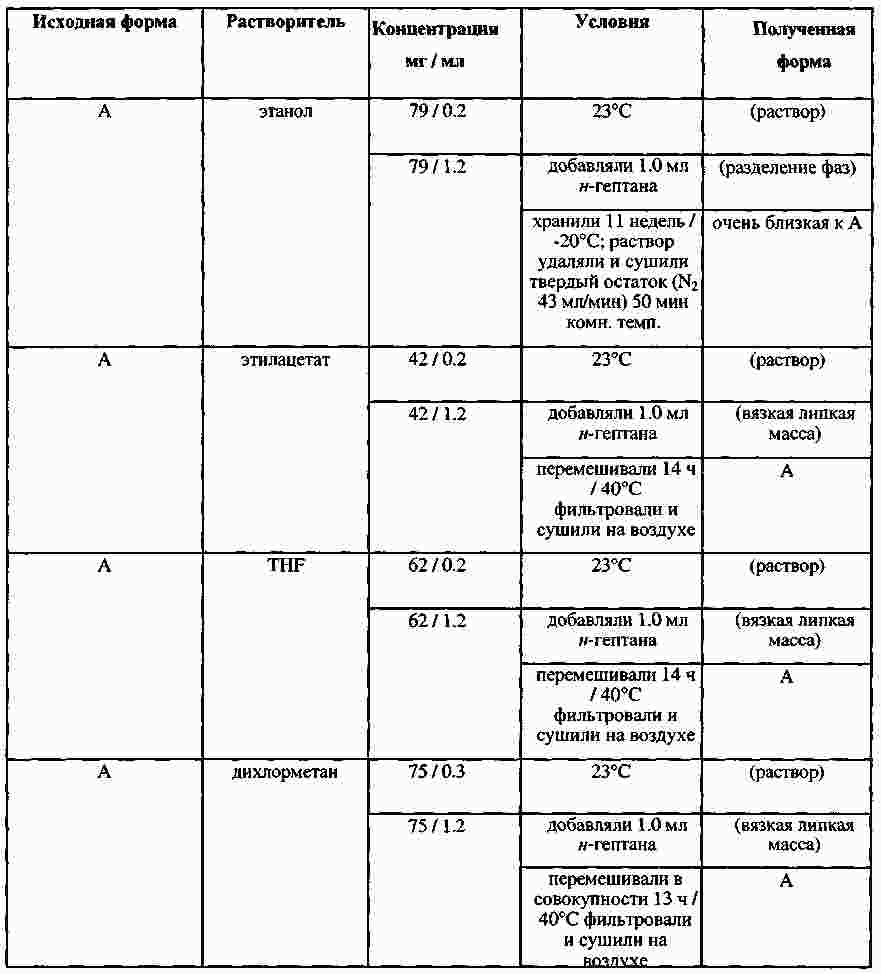
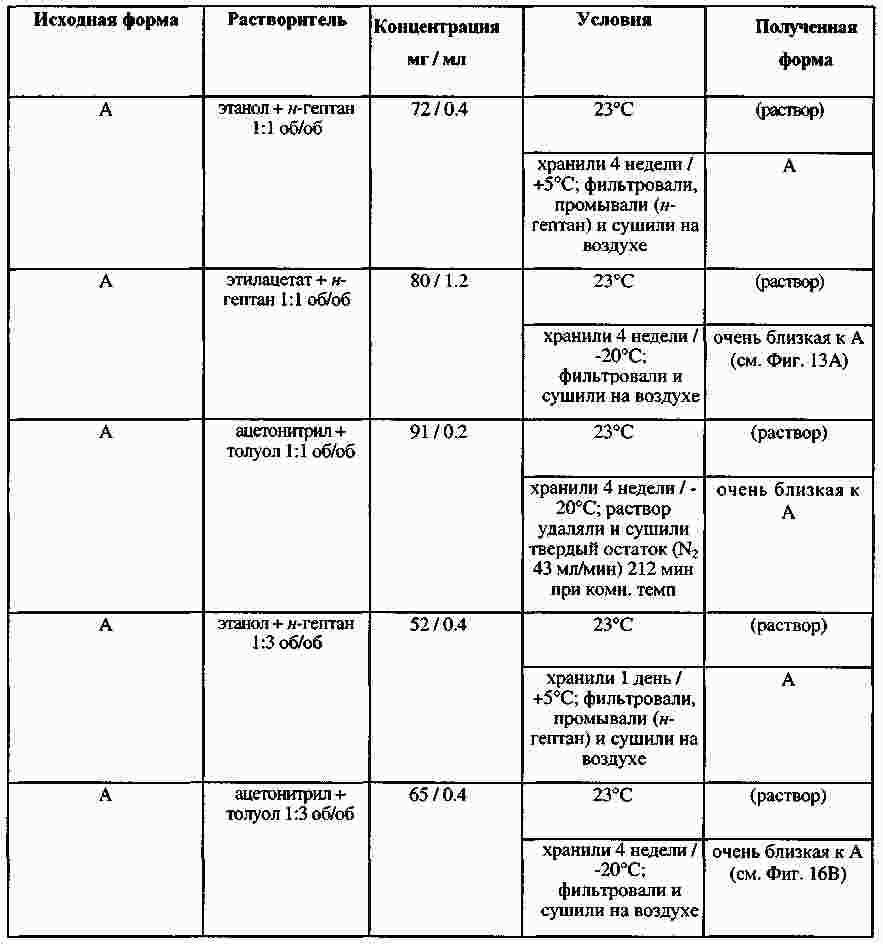
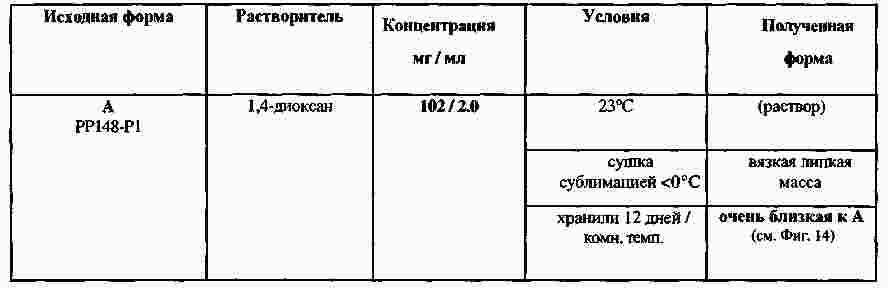
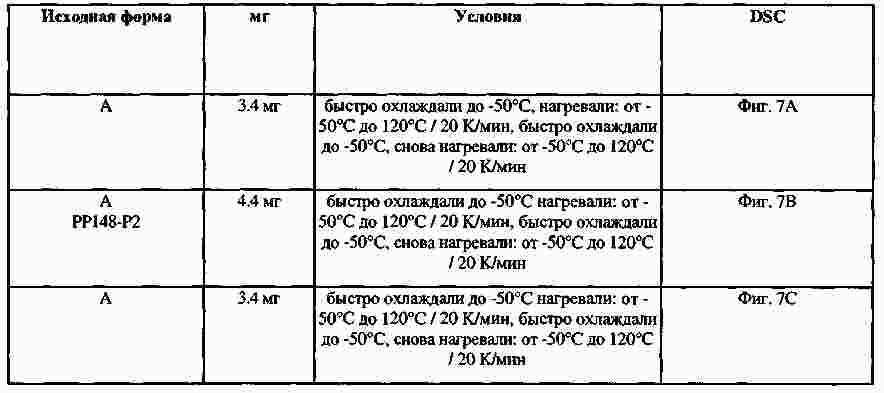
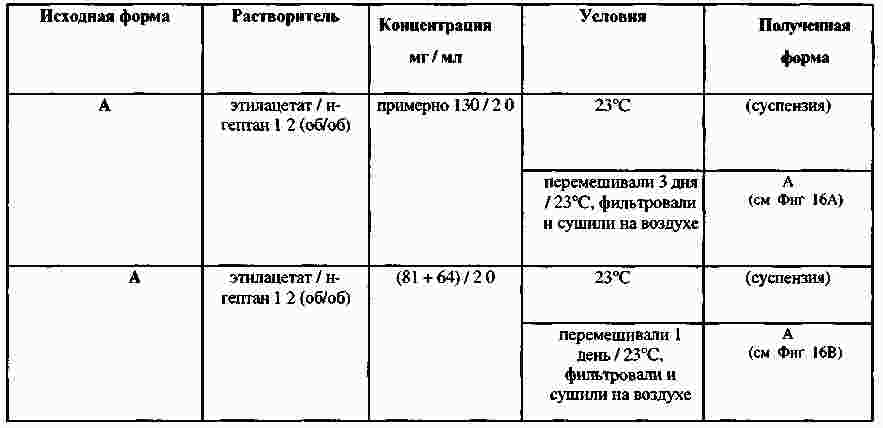
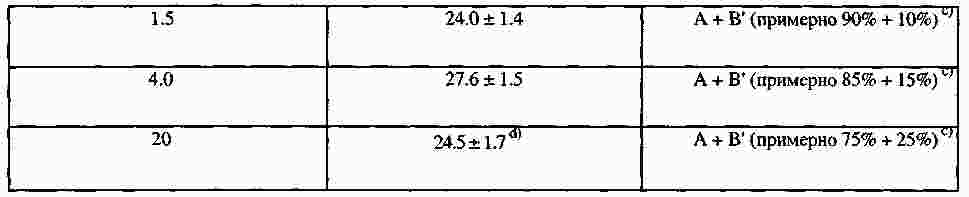
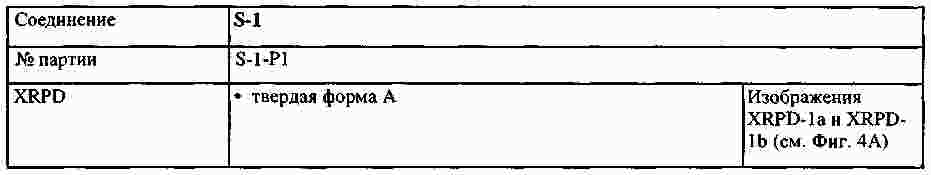
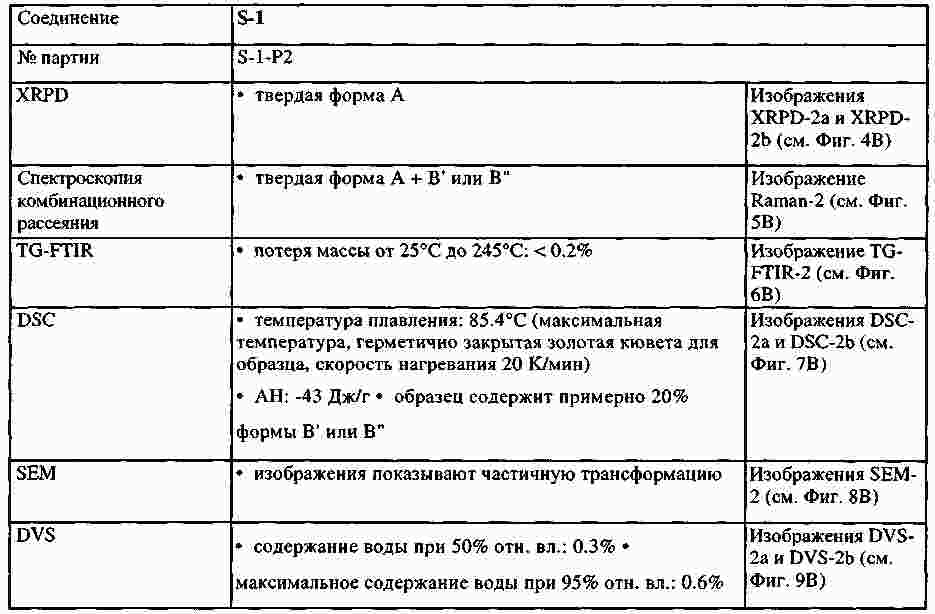
|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea000018611b*\id |
|
Термины запроса в документе Реферат Настоящее изобретение относится к твердым формам (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида и способам их производства. Формула [0001] Кристаллическая форма D (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида, которая характеризуется: [0002] Кристаллическая форма по п.1, отличающаяся тем, что указанная форма характеризуется рентгеновской порошковой дифрактограммой, изображенной на фиг. 18. [0003] Способ приготовления кристаллической формы D (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида, включающий перемешивание аморфного (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида в смеси растворитель/антирастворитель при температуре около 50 °C с получением указанной кристаллической формы. [0004] Способ по п.3, отличающийся тем, что указанным растворителем/антирастворителем является смесь этилацетат/циклогексан. [0005] Композиция, пригодная для лечения андроген-опосредованных состояний, состоящая из кристаллической формы D (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида и приемлемого носителя или растворителя. [0006] Кристаллическая форма А безводного (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида, которая характеризуется: [0007] Кристаллическая форма по п.6, отличающаяся тем, что указанная форма одновременно характеризуется свойствами, перечисленными в (а) и (б). [0008] Композиция, пригодная для лечения андроген-опосредованных состояний, включающая кристаллическую форму А безводного (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида по п.6 и приемлемый носитель или растворитель. [0009] Способ приготовления кристаллической формы А (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида, включающий растворение аморфного (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида по меньшей мере в одном органическом растворителе при температуре приблизительно от -20 до +5 °C с получением указанной кристаллической формы. [0010] Способ по п.9, отличающийся тем, что указанный органический растворитель состоит из смеси растворителя и антирастворителя, где указанная смесь включает этилформат и пентан в соотношении 1:2 (об./об.) или метилацетат и пентан в соотношении 1:2 (об./об.). [0011] Псевдокристаллическая форма (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида, отличающаяся тем, что указанная псевдокристаллическая форма характеризуется: [0012] Псевдокристаллическая форма по п.11, отличающаяся тем, что указанная форма характеризуется рентгеновской порошковой дифрактограммой, представленной на фиг. 4D. [0013] Композиция, пригодная для лечения андроген-опосредованных состояний, состоящая из псевдокристаллической формы (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида и приемлемого носителя или растворителя. [0014] Способ приготовления псевдокристаллической формы (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида, включающий перемешивание суспензии кристаллической формы (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида в воде при температуре окружающей среды приблизительно 20-30 °C в течение по меньшей мере получаса с получением псевдокристаллической формы. [0015] Композиция, пригодная для лечения андроген-опосредованных состояний, состоящая из смеси кристаллических и псевдокристаллических твердых форм (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида с приемлемым носителем или растворителем. Полный текст патента Область техники Настоящее изобретение относится к твердой форме (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида и способу его приготовления. Адрогеновый рецептор (АР) представляет собой лиганд-активируемый транскрипционный регуляторный белок, который опосредует индукцию полового развития мужчин и функционирует посредством действия на эндогенные андрогены. Андрогены широко известны в качестве мужских половых гормонов. Андрогенные гормоны представляют собой стероиды, которые вырабатываются в организме яичками и корой надпочечников, или их можно синтезировать в лаборатории. Андрогенные стероиды играют важную роль во многих физиологических процессах, включая развитие и поддержание мужских половых признаков, например мышечной массы и скелетной массы, рост простаты, сперматогенез и оволосение по мужскому типу (Matsumoto, Endocrinol. Met. Clin. N. Am. 23:857-75 (1994)). К эндогенным стероидным андрогенам относятся тестостерон и дигидротестостерон (ДГТ). Тестостерон является главным стероидом, секретируемым яичками, и представляет собой основной циркулирующий андроген, который обнаруживают в плазме мужчин. Во многих периферических тканях тестостерон превращается в ДГТ с помощью фермента 5- α-редуктазы. Поэтому считается, что ДГТ служит внутриклеточным медиатором для большинства адрогенных процессов (Zhou, et al., Molec. Endocrinol. 9:208-18 (1995)). К другим стероидным андрогенам относятся эфиры тестостерона, например ципионатный, пропионатный, фенилпропионатный, циклопентилпропионатный, изокарпоратный, энантатный и деканоатный эфиры и другие синтетические андрогены, например 7-метилнортестостерон (МЕНТ) и его ацетатный эфир (Sundaram et al., "7 Alpha-Methyl-Nortestosterone(MENT): The Optimal Androgen For Male Contraception," Ann. Med., 25:199-205 (1993) ("Sundaram"). Поскольку АР участвует в половом развитии мужчин и половой функции мужчин, АР является возможной мишенью для осуществления контрацепции мужчин или других форм заместительной гормональной терапии. В связи с этим существует необходимость в разработке новых подходов как в фундаментальной науке, так и в области клинических исследований для создания соединений, которые могли бы оказаться полезными веществами для а) контрацепции мужчин, б) лечения различных гормональных расстройств, например заболеваний, связанных с возрастным дефицитом андрогена (ВАД, ADAM), например, таких как хроническая усталость, депрессия, пониженное либидо, половая дисфункция, нарушение эрекции, гипогонадизм, остеопороз, облысение, анемия, ожирение, саркопения, остеопения, доброкачественная гиперплазия простаты, перемены настроения и нарушение познавательной способности и рак простаты; в) лечения заболеваний, связанных с дефицитом андрогенов у женщин (ДАЖ, ADIF), таких как половые расстройства, пониженное либидо, гипогонадизм, саркопения, остеопения, остеопороз, перемены настроения и нарушение познавательной способности, депрессия, анемия, облысение, ожирение, эндометриоз, рак груди, рак матки и яичников; д) лечения и/или профилактики острых и/или хронических состояний мышечного истощения; е) профилактики и/или лечения синдрома сухих глаз; ж) пероральной андрогенной заместительной терапии; и/или з) снижения частоты возникновения, остановки развития или стимулирования регрессии рака простаты. В литературе описаны различные полиморфы, сольваты и соли различных лекарств, которые придают лекарственным средствам новые свойства. У органических малых молекул лекарственных веществ наблюдается тенденция к самостоятельной сборке (самосборке) в различные полиморфные формы в зависимости от среды, которая запускает самосборку. Нагревание и действие растворителей также могут привести к изменениям, обусловливающим превращение одной полиморфной формы в другую. Для способа изготовления лекарственных средств очень важно определить, какая полиморфная форма является максимально стабильной при определенных условиях и какие процессы приводят к изменению полиморфной формы для того, чтобы быть уверенным, что конечный продукт находится в предпочтительной полиморфной форме. Различные полиморфные формы активного фармацевтического ингредиента (API) позволяют модифицировать растворимость лекарственного вещества, скорость растворения, фармакокинетику и, в итоге, его биодоступность и эффективность для пациентов. В одном варианте реализации настоящее изобретение относится к твердой форме (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида и способам его получения. В некоторых вариантах реализации такое соединения проявляет полезные свойства в качестве андрогенного и анаболического агента. (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамид представляет собой селективный модулятор андрогенного рецептора (SMARs) эффективный для а) контрацепции мужчин; б) лечения различных гормональных расстройств, например заболеваний, связанных с ВАД; в) лечения заболеваний, связанных с ДАЖ; г) лечения и/или профилактики хронического истощения мышц и/или д) снижения степени замедления регрессии или стимулирование регрессии рака простаты; е) орального замещения андрогенов и/или других клинических терапевтических и/или диагностических целей. В одном варианте реализации настоящее изобретение обеспечивает кристаллическую форму соединения (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида. В одном варианте реализации настоящее изобретение обеспечивает безводную кристаллическую форму соединения (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида. В другом варианте реализации настоящее изобретение обеспечивает композицию, включающую терапевтическое количество безводной кристаллической формы (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида и подходящий носитель или растворитель. В одном варианте реализации настоящее изобретение обеспечивает псевдокристаллическую форму соединения (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида. В другом варианте реализации настоящее изобретение обеспечивает композицию, состоящую из псевдокристаллической формы соединения (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида и подходящего носителя или растворителя. В одном варианте реализации настоящее изобретение обеспечивает процесс приготовления псевдокристаллического (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида, включающий перемешивание суспензии кристаллической формы (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида в воде при температуре окружающей среды около 20-30 °C в течение по меньшей мере 0,5 ч для получения псевдокристаллического соединения. В одном варианте реализации настоящее изобретение обеспечивает композицию, состоящую из смеси кристаллической и псевдокристаллической твердых форм соединения (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида и подходящего носителя или растворителя. Предмет изобретения подробно раскрыт и конкретно заявлен в заключительной части описания изобретения. Однако изобретение как его организация и способ осуществления, так и задачи, особенности и преимущества станут более понятны из нижеследующего подробного описания и сопутствующих чертежей, на которых на фиг. 1 представлена схема синтеза рацемическоих смесей соединения 1; на фиг. 2 - схема синтеза (S)-энантиомера соединения S-1; на фиг. 3 - график термогравиметрического анализа (ТГА) сольвата толуола (красный) и формы D (черный); на фиг. 4А-4D - паттерн рентгеновской порошковой дифракции (РПД) твердых форм соединения S-1. На фиг. 4А изображен паттерн РПД твердой формы А-партии Р1 вещества S-1; на фиг. 4В - паттерн РПД твердой формы А-партии Р2 соединения S-1; на фиг. 4С - паттерн РПД твердой формы А-партии Р3 соединения S-1; на фиг. 4D - паттерн РПД твердой формы В'-партии Р4 соединения S-1; на фиг. 5А-5D - спектры Рамана образцов партий Р1-Р4 соединения S-1 соответственно. Мощность лазера составляла 100 мВт при разрешении 2 см -1 ; на фиг. 6А-6D - спектры термогравиметрического анализа и инфракрасной спектроскопии с преобразованием Фурье (TG-FTIR) образцов партий Р1-Р4 соединения S-1 соответственно. Условия проведения анализа включают диапазон температуры в динамическом режиме 25 °C/10,0/250 °C, в атмосфере азота; на фиг. 7А-7D - спектры дифференциальной сканирующей калориметрии (ДСК) образцов партий Р1-Р4 соединения S-1 соответственно. Звездочка указывает на установочный параметр, особенность использованного оборудования; на фиг. 8А, 8В и 8С - микрофотографии SEM партий образцов Р1, Р2 и Р4 соединения S-1 соответственно; на фиг. 9А, 9В и 9С - спектры ДВС партий образцов Р1, Р2 и Р4 соединения S-1 соответственно. На фиг. 9А показан спектр ДВС формы А; на фиг. 9В - спектр ДВС формы А; на фиг. 9С - спектр ДВС формы В'; на фиг. 10 - спектр рентгеновской порошковой дифракции (РПД) соединения, полученный после изменения концентрации соединения S-1 в заданных растворителях, смены растворителя или комбинации этих действий. На фиг. А показан спектр РПД после того, как форму А соединения S-1 растворяли в н-гептане, 108 мг/2,0 мл; на фиг. В - спектр РПД после того, как форма В' соединения S-1 растворяли в этилацетате+н-гептане в соотношении 1:2 (об./об.), 81мг/1,7 мл; на фиг. С - спектр РПД после того, как форму В' соединения S-1 растворяли в этилацетате+н-пентане в соотношении 1:2 (об./об.), 101 мг/1,0 мл; на фиг. D - спектр РПД после того, как форму А соединения S-1 растворяли в этилацетате+н-пептане в соотношении 1:2 (об./об.), 128 мг/2,0 мл; на фиг. Е показан спектр РПД после того, как форму А соединения S-1 растворяли в этилацетате+н-пептане в соотношении 1:2 (об./об.), 112 мг/2,0 мл; на фиг. F - спектр РПД после того, как форму А соединение S-1 растворяли в метилацетате+н-пептане в соотношении 1:2 (об./об.), 126 мг/2,0 мл; на фиг. 11 - паттерн РПД, полученный в результате экспериментов по диффузии пара соединения S-1. На фиг. А показан спектр РПД соединения S-1 в толуоле и н-гексане при 23 °C в течение 2 суток; на фиг. В - совмещенные спектры РПД партии Р1 (форма А) и РПД, полученной на фиг. 11А; на фиг. С - спектр РПД, полученной для соединения S-1 в уксусной кислоте и воде при 23 °C в течение 7 суток; на фиг. D - совмещенные спектры РПД партии Р1 (форма А) и РПД, полученный на фиг. 11В; на фиг. 12 - паттерн РПД, представляющий результаты экспериментов по испарению, в которых растворы соединений высушивали при комнатной температуре (сухой поток N 2 ) без перемешивания. На фиг. 12А показан паттерн РПД, полученный для соединения S-1 (партия Р1) в растворе этилацетата; на фиг. 12В - совмещенные спектры РПД для партии Р1 (форма А) и РПД, полученной на фиг. 12А; на фиг. 12С - паттерн РПД, полученный для соединения S-1 (партии Р1) из тетрагидрофурана (TGF) для обеспечения формы С; на фиг. 12D - паттерны РПД смеси формы А (красный, сверху) и формы С (голубой, внизу), как показано на фиг. 12С; на фиг. 13 показан спектр РПД, представляющий результаты перекристаллизации из экспериментов по растворению, где соединение S растворяли в различных системах растворителей при комнатной температуре и охлаждали до +5 или до -20 °C; на фиг. 13А - спектры РПД, полученные для соединения S-1 (партия Р1) в этилацетате и н-гептане при 1:1 (об./об.); на фиг. 13В - совмещенные спектры РПД партии Р1 (форма А) и РПД, полученные на фиг. 13А; на фиг. 13С - спектры РПД, полученные для соединения S-1 (партия Р1) в ацетонитриле и толуоле в объемном соотношении 1:3 (об./об.); на фиг. 13D - совмещенные спектры РПД партии Р1 (форма А) и РПД, полученные на фиг. 13B; на фиг. 14 - спектры РПД, представляющие результаты эскпериментов по лиофильной сушке. На фиг. 14А показаны спектры РПД, полученные по соединению S-1 (партия Р-1) в 1,4-диоксане и охлажденные до -50 °С; на фиг. 14В - совмещенные спектры РПД партии Р1 (форма А) и РПД, полученные на фиг. 14А; на фиг. 15 - термограмма ДСК, представляющая результаты экспериментов по высушиванию, в которых соединение S-1 (партия Р4) высушивали на протяжении ночи в атмосфере сухого N 2 . Звездочка указывает на установочный параметр, особенность использованного оборудования; на фиг. 16 - спектры РПД, представляющие результаты экспериментов по относительной стабильности, где эксперименты по суспендированию проводили со смесями партий соединения S-1. На фиг. 16А показаны спектры РПД, полученные из смеси партий соединения S-1 (P1, P18, Р24, Р30, Р37 и Р38, у всех партий были свойства РПД формы А), в этилацетате+н-гептане при 1:2 (об./об.), 130 мг/2,0 мл; на фиг. 16В - совмещенные спектры РПД партии Р1 (форма А) и РПД, полученные на фиг. 16А; на фиг. 16С - спектры РПД, полученные из смесей партий соединения S-1 (P1 и Р52, при этом партия Р1 представляет собой форму А, а Р52 - форму А+С) в этилацетате+н-гептане в соотношении 1:2 (об./об.), (81+64) мг/2,0 мл; на фиг. 16D - совмещенные спектры РПД партии Р1 (форма А) и РПД, полученные на фиг. 16В; на фиг. 17 - термограмма ДСК и паттерн РПД, представляющие результаты поглощения паров воды, где соединение S-1 (партия Р1) хранили в стеклянной пробирке при 96% отн.вл. (относительная влажность) при комнатной температуре. На фиг. 17А показаны результаты ДСК, полученные для соединения S-1 партии Р1 без растворителя спустя 11 недель; на фиг. 17В - паттерн РПД соединения S-1 партии Р1 (форма А) в воде спустя 19 ч при 37 °C, что приводило к образованию формы В'; на фиг. 17С - РПД соединения S-1 партия Р1 (форма А) в уксусной кислоте+вода в соотношении 1:2 (об./об.) спустя 20 ч при 23 °C; на фиг. 17D - термограмма ДСК при нагревании образца формы А (черный), охлаждения образца после плавления (серый) и повторного нагревания образца (белый). Скорость нагревания составила 10 °С/мин, при этом скорость охлаждения составила 1 °С/мин. Нагревание формы А выше температуры плавления приводило к появлению формы В", которая не превращалась обратно в форму А, даже если образец повторно охлаждали до температуры окружающей среды; на фиг. 17Е - анализы ДСК 1 °C/мин DSC формы А (серый), В" (черный), смеси А и D (белый) и смеси В" и D (темно-серый). Формы А и В" могут подвергаться кристаллизации с получением D, но только в присутствии D, которая является затравкой кристаллизации; на фиг. 17F - графики ДСК формы А, которая хранится при температуре окружающей среды/100% отн.вл. в течение 7 суток (светло-серый), 50 °С/0% отн.вл. в течение 7 суток (темно-серый) и 50 °С/75% отн.вл. в течение 6 ч (белый), а также графики ДСК исходного образца (черный); на фиг. 17G представлены (а) графики ДСК полиморфа А, с применением в качестве затравки формы D и хранящиеся при 50 °С/75% отн.вл., (б) графики ДСК формы А, с применением в качестве затравки формы D и хранящиеся при 50 °С в воде; на фиг. 18 показаны паттерны РПД совмещенных спектров РПД формы А (наверху) и формы D (внизу) соединения S-1; на фиг. 19 показаны термограммы ДСК форм А и D. В некоторых вариантах реализации настоящее изобретение обеспечивает твердую форму (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида и способы его приготовления. Также изобретение обеспечивает фармацевтические композиции, содержащие твердую форму (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида и его применение. (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамид представляет собой направленный на андрогенный рецептор агент (ARTA), который обладает андрогенной и анаболической активностью. В некоторых вариантах реализации метилпропанамид, описанный в настоящем изобретении, является селективным модулятором андрогеновых рецепторов (CMAP, SARM), которые в некоторых вариантах реализации изобретения применяются для а) контрацепции мужчин, б) лечения различных гормональных расстройств, например заболеваний, связанных с возрастным андрогенным дефицитом (ВАД), например хронической усталости, депрессии, пониженного либидо, половой дифункции, нарушения эрекции, гипогонадизма, остеопении, остеопороза, выпадения волос, анемии, ожирения, саркопении, доброкачественной гиперплазии простаты, перемены настроения и нарушения познавательной способности и рака простаты; в) лечения заболеваний, связанных с дефицитом андрогенов у женщин, таких как половые расстройства, пониженное либидо, гипогонадизм, саркопения, остеопения, остеопороз, перемены настроения и нарушения познавательной способности, депрессия, анемия, облысение, ожирение, эндометриоз, рак груди, рак матки и яичников; д) лечения и/или профилактика острых и/или хронических состояний мышечного истощения; е) снижения степени замедления регрессии или стимулированиерегрессии рака простаты; г) пероральной андрогенной заместительной терапии; и/или для других клинических терапий и/или диагностических областей. В некоторых вариантах реализации изобретение обеспечивает полиморфную твердую форму соединения (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида согласно настоящему изобретению. В одном варианте реализации термин "полиморф" относится к особой форме CMAP (SARM) соединений согласно настоящему изобретению, например, полиморфы могут представлять собой кристаллические формы, которые различаются между собой по фармацевтически значимым физическим свойствам, например при различных условиях кристаллизации, условиях окружающей среды, гигроскопической активности веществ и т.д. В одном варианте реализации изобретение обеспечивает кристаллическую форму соединения (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида. В одном варианте реализации изобретение обеспечивает кристаллическую форму безводного (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида. В другом варианте реализации кристаллическая форма соединения (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида (соединение S-1), характеризуется: а) рентгеновской порошковой дифрактограммой, имеющей пики при углах °2 θ (d выражается в Å), составляющих около 5.6 (15.9), 7.5 (11.8), 8.6 (10.3), 9.9 (8.9), 12.4 (7.1), 15.0 (5.9), 16.7 (5.3), 17.3 (5.1), 18.0 (4.9), 18.5 (4.8), 19.3 (4.6), 19.8 (4.5), 20.6 (4.3), 21.8 (4.1), 22.3 (4.0), 23.4 (3.8), 23.9 (3.7), 24.6 (3.6), 24.9 (3.6), 25.4 (3.5), 26.0 (3.4), 26.5 (3.4), 27.8 (3.2); и б) точкой плавления приблизительно 80 °C. Согласно этому аспекту и в другом варианте реализации такую кристаллическую форму соединения S-1, обладающую всеми или частью свойств, перечисленных в (а) и (б), обозначают в настоящем изобретении, как кристаллическая форма А. В другом варианте реализации растворимость формы А в воде составляет 20-30 мг/л при 22 °C. В другом варианте реализации растворимость формы А в воде составляет 23-27 мг/л при 22 °C. В одном варианте реализации настоящее изобретение обеспечивает псевдокристаллическую форму соединения (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида. В одном варианте реализации псевдокристаллическая форма (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида (соединение S-1) характеризуется: а) рентгеновской порошковой дифрактограммой, демонстрирующей широкое гало с двумя гармоничными пиками при углах °2 θ между 15-25, и б) температурой стеклования приблизительно 55 °C. Согласно этому аспекту и в другом варианте реализации такую псевдокристаллическую форму соединения S-1, обладающую всеми или частью свойств, перечисленных в (а) и (б), в настоящем изобретении обозначают, как псевдокристаллическая форма В'. В одном варианте реализации термин "псевдокристаллический" обозначает состояние материала, обладающего ближним порядком без дальнего порядка, например жидкокристаллический или другой тип ламеллярных структур. В другом варианте реализации "форма В'" соединения S-1 является псевдокристаллической формой. В другом варианте форму А соединения S-1 можно трансформировать целиком или частично в псевдокристаллическую форму В' соединения S-1. В другом варианте реализации изобретения растворимость формы В' в воде соствляет 20-30 мг/л при 22 °C. В другом варианте реализации растворимость формы В' в воде 23-27 мг/л при 22 °C. В другом варианте реализации изобретение обеспечивает псевдокристаллическую форму В" (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида, для которой характерны: а) рентгеновская порошковая дифрактограмма, демонстрирующая широкое гало с двумя гармоничными пиками при углах °2 θ между 15-25, и б) температура стеклования приблизительно 55 °C. В одном варианте реализации настоящее изобретение обеспечивает кристаллическую форму С (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида, которая характеризуется: а) рентгеновской порошковой дифрактограммой, состоящей из одиночных пиков при углах °2 θ (d выражается в Å), составляющих около 6.9 (12.8), 9.5 (9.3), 13.5 (6.6), 16.0 (5.6), 22.8 (3.9). В другом варианте реализации кристаллическую форму С соединения S-1 получают в виде смеси формы А и С посредством выпаривания А из тетрагидрофурана (THF). В другом варианте реализации настоящее изобретение обеспечивает кристаллическую форму D (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида, которая характеризуется: а) рентгеновской порошковой дифрактограммой, состоящей из одиночных пиков при углах °2 θ (d выражается в Å), составляющих около 4.4 (19.9), 8.5 (10.4), 8.8 (10.0), 11.3 (7.8), 12.7 (6.9), 13.8 (6.4), 14.4 (6.1), 14.6 (6.0), 15.1 (5.8), 16.1 (5.5), 16.6 (5.3), 16.9 (5.2), 18.0 (4.9), 18.7 (4.7), 19.0 (4.6), 19.4 (4.55), 20.8 (4.25), 22.1 (4.0), 22.7 (3.9), 23.1 (3.8), 23.4 (3.8), 24.7 (3.6), 24.9 (3.56), 25.3 (3.51), 27.8 (3.2), 29.3 (3.0); и б) температурой плавления приблизительно 130 °C. В другом варианте изобретения кристаллическая форма D соединения S-1 стабильна при 50 °С/75% отн.вл. (относительная влажность), а также при других условиях: температуры окружающей среды/75% отн.вл., температуры окружающей среды/100% отн.вл., 30 °С/75% отн.вл. и 50 °С/0% отн.вл. В другом варианте реализации характеристики различных твердых форм S-1 представлены в примере 2 и на фиг. 3-19. Твердые формы согласно настоящему изобретению можно анализировать с помощью любого метода, известного из уровня техники, например, как в одном варианте реализации, рентгеновской порошковой дифрактометрией. В другом варианте реализации анализ твердых форм согласно изобретению может включать спектроскопию Рамана (спектроскопия комбинационного рассеяния). В другом варианте реализации анализ твердых форм согласно настоящему изобретению может включать TG-FTIR (термогравиметрический инфракрасный анализ с Фурье-преобразованием). В другом варианте реализации анализ твердых форм согласно изобретению может включать преобразование FT-Raman (Фурье-Рамана). В другом варианте реализации анализ твердых форм согласно изобретению может включать ДСК, DSC (дифференциальную сканирующую калориметрию). В другом варианте реализации анализ твердых форм согласно настоящему изобретению может включать ДСП, DVS (динамическая сорбция паров). В другом варианте реализации анализ твердых форм согласно изобретению может включать растровую сканирующую электронную микроскопию (РЭМ, SEM). В одном варианте реализации изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм А и В' в соотношении от 95:5 до 85:15 соответственно. В другом варианте реализации соотношение составляет приблизительно от 85:15 до 75:25 соответственно. В другом варианте реализации соотношение составляет приблизительно от 75:25 до 65:35 соответственно. В другом варианте реализации соотношение составляет от 95:5 до 90:10 соответственно. В другом варианте реализации соотношение составляет от 90:10 до 85:15 соответственно. В другом варианте реализации соотношение составляет от 97:3 до 93:7 соответственно. В другом варианте реализации соотношение составляет приблизительно от 85:15 до 80:20. В другом варианте реализации соотношение составляет приблизительно от 70:20 до 60:20. В другом варианте реализации соотношение составляет 50:50 соответственно. В другом варианте реализации изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм А, В' и С в соотношении между примерно 90:5:5 и 80:10:10 соответственно. В другом варианте реализации соотношение составляет приблизительно от 80:10:10 до 75:15:10 соответственно. В другом варианте реализации соотношение составляет приблизительно от 95:3:2 до 90:7:3 соответственно. В другом варианте реализации соотношение составляет приблизительно от 75:15:10 до 65:20:15. В другом варианте реализации соотношение составляет приблизительно от 70:20:10 до 60:20:20. В другом варианте реализации настоящее изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм А, В' и D в соотношении от 5:5:90 до 10:10:80 соответственно. В другом варианте реализации соотношение составляет приблизительно от 10:10:80 до 10:15:75 соответственно. В другом варианте реализации соотношение составляет приблизительно от 2:3:95 до 3:7:90 соответственно. В другом варианте реализации соотношение составляет приблизительно от 10:15:75 до 15:20:65. В другом варианте реализации соотношение составляет приблизительно от 10:20:70 до 20:20:60. В другом варианте реализации настоящее изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм А и С в соотношении примерно от 98:2 до 95:5 соответственно. В одном варианте реализации изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм А и С в соотношении от 95:5 до 90:10 соответственно. В одном варианте реализации настоящее изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм А и С в соотношении примерно от 90:10 до 85:15 соответственно. В одном варианте реализации настоящее изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм А и С в соотношении примерно от 85:15 до 80:20 соответственно. В одном варианте реализации настоящее изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм А и С в соотношении примерно 50:50 соответственно. В одном варианте реализации настоящее изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм А и D в соотношении примерно от 2:98 до 5:95 соответственно. В одном варианте реализации настоящее изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм А и D в соотношении примерно от 5:95 до 10:90 соответственно. В одном варианте реализации настоящее изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм А и D в соотношении примерно от 10:90 до 15:85 соответственно. В одном варианте реализации настоящее изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм А и D в соотношении примерно от 15:85 до 20:80 соответственно. В одном варианте реализации настоящее изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм А и D в соотношении примерно 50:50 соответственно. В одном варианте реализации настоящее изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм В' и D в соотношении приблизительно от 2:98 до 5:95 соответственно. В одном варианте реализации настоящее изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм В' и D в соотношении приблизительно от 5:95 до 10:90 соответственно. В одном варианте реализации настоящее изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм В' и D в соотношении приблизительно от 10:90 до 15:85 соответственно. В одном варианте реализации настоящее изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм В' и D в соотношении приблизительно от 15:85 до 20:80 соответственно. В одном варианте реализации настоящее изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм В' и D в соотношении приблизительно 50:50 соответственно. В одном варианте реализации настоящее изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм В' и С в соотношении приблизительно от 98:2 до 95:5 соответственно. В одном варианте реализации настоящее изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм В' и С в соотношении приблизительно от 95:5 до 90:10 соответственно. В одном варианте реализации настоящее изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм В' и С в соотношении приблизительно от 90:10 до 85:15 соответственно. В одном варианте реализации настоящее изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм В' и С в соотношении приблизительно от 85:15 до 80:20 соответственно. В одном варианте реализации настоящее изобретение обеспечивает полиморфную смесь, состоящую из кристаллических форм В' и С в соотношении приблизительно 50:50 соответственно. В другом варианте реализации соотношение между кристаллической формой А и кристаллической формой В' составлет приблизительно от 95:5 до 85:15. В другом варианте реализации соотношение между кристаллической формой А и кристаллической формой В' составлет приблизительно от 98:2 до 95:5. В другом варианте реализации соотношение между кристаллической формой А и кристаллической формой В' составлет приблизительно от 85:15 до 75:25. В другом варианте реализации соотношение между кристаллической формой А и кристаллической формой В' составлет приблизительно от 75:25 до 65:35 соответственно. В одном варианте реализации образец (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида может содержать смесь твердых форм А, В', В", С и D. В другом варианте реализации содержание нескольких твердых форм в образце (например, содержание твердых форм А и В в образце) можно определить с помощью Modulated DSC (модулированной дифференциальной сканирующей калориметрии) при скорости нагрева 3 °C/мин с 10 до 130 °C, с последующей линейной интеграцией твердой формы А и/или В для получения энтальпии каждой. В одном варианте реализации твердая форма соединения CMAP (SARM) может влиять на его биодоступность, стабильность, технологичность и его простоту производства и применения, которые следует рассматривать как часть настоящего изобретения. В одном варианте реализации настоящее изобретение обеспечивает процесс приготовления кристаллической формы (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида, включающий растворение аморфного (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида по меньшей мере в одном органическом растворителе при температуре от -20 до +30 °C при условиях, благоприятных для кристаллизации, посредством чего получают кристаллическую форму. В одном варианте релизации настоящее изобретение обеспечивает процесс приготовления кристаллической формы A (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида, включающий растворение аморфного (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида по меньшей мере в одном органическом растворителе при температуре от -20 до +30 °C при условиях, благоприятных для кристаллизации, посредством чего получают кристаллическую форму. В другом варианте реализации температура кристаллизации (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида составляет около 5 °C. В другом варианте реализации температура равна приблизительно -20 °C. В другом варианте реализации температура равна приблизительно 20 °C. В другом варианте реализации температура составляет приблизительно от 20 до 50 °C. В другом варианте реализации температура составляет приблизительно от -10 до 0 °C. В другом варианте реализации температура составляет приблизительно от 0 до 5 °C. В другом варианте реализации температура составляет приблизительно от -10 до -20 °C. В другом варианте реализации форму A (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида (соединение S-1) получают посредством кристаллизации из органического растворителя, состоящего из смеси растворителей. В другом варианте реализации смесь состоит из двух растворителей в соотношении 1:2 об./об. соответственно. В другом варианте реализации смесь состоит из этилформата и пентана в соотношении 1:2 об./об. соответственно. В другом варианте смесь состоит из метилацетата и пентана в соотношении 1:2 об./об. соответственно. В другом варианте реализации форму А получают посредством кристаллизации из смеси растворителя/антирастворителя при температуре окружающей среды. В другом варианте реализации изобретения растворителями являются этилацетат, этанол, дихлорметан или ацетонитрил, а антирастворителями являются н-гексан, н-пентан, н-гептан и циклогескан и т.д. В другом варианте реализации изобретения кристаллическую форму А соединения S-1 получают посредством образования суспензии псевдокристаллической формы соединения формулы S-1 в смеси растворителя/антирастворителя. В другом варианте реализации твердую форму А посредством образования суспензии псевдокристаллической формы соединения формулы S-1 в смеси этилацетата и гептана в соотношении 1:2 об./об. соответственно. В другом варианте реализации твердую форму А получают посредством образования суспензии псевдокристаллической формы соединения формулы S-1 в смеси этилацетата и пентана в соотношении 1:2 об./об. соответственно. В другом варианте реализации изобретения кристаллическую форму А соединения S-1 получают посредством приготовления суспензии формы В' в смеси растворителя/антирастворителя в концентрациях выше уровня насыщения при температуре 23 °С в течение нескольких часов с последующим высушиванием для получения формы А. В другом варианте реализации форму D (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида (соединение S-1) получают посредством кристаллизации из смеси растворителя/антирастворителя при 50 °С с использованием этилацетат и циклогексана в качестве растворителя и антирастворителя соответственно. В другом варианте форму D получают из других полиморфных форм посредством затравки, используя образец с небольшим количеством D, и хранением при 110 °С/0% отн.вл. в течение 7 дней или при 50 °С в воде в течение 24 ч с последующим высушиванием. В другом варианте нагревание форм А и/или В" до 110 °С в присутствии D приводит к преобразованию форм А и В" в форму D. В другом варианте реализации форма D в присутствии влаги действует как затравка кристаллизации и запускает превращение форм А и В' в D. В другом варианте реализации изобретения на фиг. 17G показано время полиморфной модификации А, содержащей затравку небольшим количеством D при 50 °С/75% отн.вл. Количество полиморфа D, изначально добавленное в образец, очень мало и не определяется с помощью ДСК при скорости нагрева 10 °С/мин. После 24 ч большинство полиморфной формы А превратилось в В', но небольшое количество образца также превратилось в D, и количество образца в D увеличивается с течением времени. Процесс превращения ускоряется на фиг. 17G посредством хранения образца в воде при 50 °С. Форма А превратилась как в форму В', так и в форму D через 6 ч, но образец находится преимущественно в виде формы D после 24 ч. В одном варианте реализации настоящее изобретение обеспечивает процесс приготовления псевдокристаллической формы (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида, включающий перемешивание суспензии кристаллической формы (R) или (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида в воде при темпратуре окружающей среды 20-30 °C в течение по меньшей мере получаса для получения псевдокристаллического соединения. В одном варианте реализации настоящее изобретение обеспечивает процесс приготовления псевдокристаллической формы В' (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида, включающий перемешивание суспензии кристаллической формы (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида в воде при температуре окружающей среды 20-30 °C в течение по меньшей мере получаса для получения псевдокристаллического соединения. В одном варианте реализации настоящее изобретение обеспечивает процесс приготовления псевдокристаллической формы В' (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида, включающий перемешивание суспензии кристаллической формы A (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида в воде при температуре окружающей среды 20-30 °C в течение по меньшей мере получаса для получения псевдокристаллического соединения. В другом варианте псевдокристаллическую форму В' получают посредством перемешивания суспензии кристаллической формы А при 50 °C в воде в течение 24 ч. В другом варианте реализации псевдокристаллическую форму В' получают посредством перемешивания суспензии кристаллической формы А при 37 °С в течение ночи для получения псевдокристаллической формы В'. В одном варианте реализации изобретения твердую форму В' (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида получают посредством хранения твердой формы А при 40 °C и 75% относительной влажности в течение 1-2 ч. В другом варианте реализации твердая форма А хранится при 40 °C и 75% отн.вл. в течение 2-4 ч. В другом варианте реализации твердая форма А хранится при 40 °C и 75% отн.вл. в течение 4-10 ч. В другом варианте реализации твердая форма А хранится при 40 °C и 75% отн.вл. в течение 10-15 ч. В другом варианте реализации твердая форма А хранится при 40 °C и 75% отн.вл. в течение 15-24 ч. В другом варианте реализации твердая форма А хранится при 40 °C и 75% отн.вл. в течение 24 ч. В другом варианте реализации твердая форма А хранится при 40 °C и 75% отн.вл. в течение 30 дней. В одном варианте реализации твердую форму В' (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида получают посредством хранения твердой формы А при 40 °C и 75% отн.вл. В другом варианте реализации твердая форма А хранится при температуре в интервале 30-40 °C и отн.вл. 50-75%. В другом варианте реализации твердая форма А хранится при температуре в интервале 40-50 °C и отн.вл. 60-80%. В другом варианте реализации твердая форма А хранится при температуре в интервале 40-50 °C и относительной влажности 60-80%. В одном варианте реализации форма В' рассматривается в качестве лиотропной жидкокристаллической формы ввиду опосредованного растворителем образования. В одном варианте реализации изобретения жидкокристаллическую форму В" (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида получают посредством плавления или нагревания твердой формы A (S)-N-(4-циано-3-(трифторметил)фенила)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида до 80 °C с последующим охлаждением. В одном варианте реализации изобретения форму В" (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида получают посредством плавления или нагревания до 130 °C твердой формы D (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида с последующим охлаждением. В одном варианте реализации изобретения при выпаривании (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида из растворителей, например этанола, без антирастворителя получают форму В". В одном варианте реализации изобретения форма В" рассматривается в качестве термотропной жидкокристаллической формы ввиду термального способа ее получения. В одном варианте реализации изобретение обеспечивает процесс приготовления твердой формы С (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида, включающий растворение кристаллической формы A (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида в тетрагидрофуране (THF) с последующим выпариванием для получения твердой формы С. В другом варианте реализации изобретения форму С получают в виде смеси с формой А. В одном варианте реализации настоящее изобретение обеспечивает процесс приготовления твердой формы сольвата толуола (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида, включающий использование способа кристаллизации с применением растворителя/антирастворителя, при котором толуол используется в качестве антирастворителя. В другом варианте реализации изобретения, твердая форма сольвата толуола имеет температуру плавления около 100 °С с энтальпией плавления 70 ±5 Дж/г. В другом варианте реализации изобретения график термогравиметрического анализа (ТГА) сольвата толуола на фиг. 3 показывает, что содержание толуола в сольвате составляет около 7%, что соответствует одной молекуле толуола на каждые три молекулы S-1. В другом варианте реализации остатки молекул толуола находятся скорее внутри структуры элементарной ячейки, а не в каналах или за пределами пространственной решетки. В другом варианте реализации твердая форма сольвата толуола является наиболее стабильной формой толуола. В некоторых вариантах реализации изобретения кристаллические формы CMAP согласно настоящему изобретению включают изменение данной кристаллической формы на структурно похожую, однако, не идентичную исходной форме. В одном варианте реализации изобретения такие изменения в кристаллической форме могут привести к появлению структурно более стабильной формы, чем исходная форма. В некоторых вариантах реализации кристаллические формы согласно изобретению включают измененные кристаллические формы, а также исходные, в одном препарате. В некоторых вариантах реализации такие измененные кристаллические формы могут включать небольшое количество целого препарата соединения CMAP, например до 1 % или 5, 10, 15, 25% препарата. В другом варианте реализации такие измененные формы могут включать большое количество препарата соединения CMAP и могут включать 55%, или в другом варианте реализации 65%, или в другом варианте реализации 75%, или 80, или 85, или 90, или 95, или до 100% препарата соединения CMAP. В одном варианте реализации предпочтительная кристаллическая форма является термодинамически подходящей. В другом варианте реализации подходящая кристаллическая форма является результатом изменения влажности. В другом варианте реализации подходящая кристаллическая форма является результатом изменения температуры. В другом варианте реализации подходящая кристаллическая форма является результатом смены растворителей. В некоторых вариантах реализации в результате процесса приготовления полиморфа соединения (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида получают различные кристаллические формы. В одном варианте реализации в результате процесса получают смесь кристаллических/псевдокристаллических форм А, В', С и D. В одном варианте реализации в результате процесса получают смесь кристаллических/псевдокристаллических форм А, В', В", С и D. В другом варианте реализации в результате процесса получают смесь кристаллических форм А и С. В другом варианте реализации в результате процесса получают смесь кристаллических/псевдокристаллических форм А и В'. В одном варианте реализации в результате процесса получают смесь кристаллических форм А и D. В другом варианте реализации в результате процесса получают смесь кристаллических/псевдокристаллических форм В' и D. В другом варианте реализации в результате процесса получают смесь кристаллических/псевдокристаллических форм В" и D. В другом варианте реализации в результате процесса получают смесь кристаллических форм С и D. В другом варианте реализации в результате процесса получают смесь кристаллических/псевдокристаллических форм В' и С. В другом варианте реализации в результате процесса получают смесь кристаллических/псевдокристаллических форм А и В". В другом варианте реализации в результате процесса получают смесь псевдокристаллических форм В' и В". В другом варианте реализации в результате процесса получают смесь кристаллических/псевдокристаллических форм С и В". В другом варианте реализации в результате процесса получают смесь кристаллических/псевдокристаллических форм А, С и В". В другом варианте реализации в результате процесса получают смесь кристаллических/псевдокристаллических форм A, D и В". В другом варианте реализации в результате процесса получают смесь кристаллических/псевдокристаллических форм В', В" и С. В другом варианте реализации в результате процесса получают смесь кристаллических/псевдокристаллических форм А, В' и В". В другом варианте реализации в результате процесса получают смесь кристаллических/псевдокристаллических форм D, В' и В". В одном варианте реализации твердые формы соединений согласно настоящему изобретению высушивают из раствора под вакуумом при комнатной температуре с последующим постепенным повышением температуры. В другом варианте реализации твердые формы соединений согласно настоящему изобретению фильтруют из раствора. В одном варианте реализации под термином "температура окружающей среды" понимают комнатную температуру. В другом варианте реализации под термином "температура окружающей среды" подразумевается 20-25 °C. В другом варианте реализации под термином "температура окружающей среды" подразумевается 25-30 °C. В другом варианте реализации изобретения форма D является наиболее термодинамически устойчивой полиморфой как при сухих условиях, так и в присутствии воды при температуре окружающей среды, достигающей температуры плавления при 130 °С. В другом варианте реализации на фиг. 19 изображена термограмма дифференциальной сканирующей калориметрии (ДСК) формы А и формы D, на которой форма А плавится при температуре около 80 °C, и форма D плавится при температуре около 130 °C. В другом варианте реализации энтальпия плавления для формы А составляет 40 ±5 Дж/г, а энтальпия плавления для формы D составляет 75 ±5 Дж/г. В другом варианте реализации форма А является стабильной, сохраняя форму А по меньшей мере 7 дней при условиях хранения при температуре окружающей среды и 75% отн.вл., при температуре окружающей среды/100% отн.вл., 30 °С/75% отн.вл. и 50 °С/0% отн.вл. В другом варианте реализации форма А превращается в форму В при хрании при 50 °С/75% отн.вл. В другом варианте реализации форма А превращается в форму В' при хранении при 40 °С/75% отн.вл. в течение месяца. В другом варианте реализации форма А, которая хранится при 25 °С/60% отн.вл. и 30 °С/65% отн.вл., стабильна в течение 36 и 9 месяцев соответственно. В одном варианте реализации (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамид получают с помощью хирального синтеза. В одном варианте реализации (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамид можно получить с помощью процесса согласно приведенной ниже схеме синтеза В одном варианте реализации изобретения процесс, изображенный на приведенной выше схеме, включает взаимодействие ациланилида на 5 этапе с цианофенолом, такую реакцию можно проводить в присутствии карбоната калия, карбоната натрия или карбоната цезия. В одном варианте реализации изобретения реакция в присутствии карбоната калия может неожиданно приводить к образованию продукта с меньшим количеством примесей, чем реакция в присутствии карбоната цезия. Этот процесс представляет собой улучшенный и более эффективный процесс синтеза для производства конечного продукта, который позволяет минимизировать необходимость дополнительных этапов очистки. Этот обнаруженный факт также можно использовать для получения других соединений, например приведенных ниже 6, 9, 12 и 14. В одном варианте реализации изобретение обеспечивает процесс получения (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида, при этом процесс включает стадии: а) приготовления карбоновой кислоты формулы 1 посредством раскрытия кольца циклического соединения формулы 2 в присутствии HBr б) взаимодействия амина формулы 3 с карбоновой кислотой формулы 2 в присутствии агента реакции присоединения с получением амида формулы 4 в) взаимодействия амида формулы 4 с соединением формулы 5 при этом стадию (в) проводят в присутствии карбоната калия и тетрагидрофурана. В одном варианте реализации изобретение обеспечивает процесс получения соединения формулы 6 При этом указанный процесс включает стадии: а) получения карбоновой кислоты формулы 1 посредством раскрытия кольца циклического соединения формулы 2 в присутствии HBr б) взаимодействия амина формулы 7 с карбоновой кислотой формулы 2 в присутствии агента реакции присоединения с получением амида формулы 8 в) взаимодействия амида формулы 8 с соединением формулы 5 при этом стадию (в) проводят в присутствии карбоната калия и тетрагидрофурана. В одном варианте реализации изобретение обеспечивает процесс получения соединения формулы 9 При этом указанный процесс включает стадии: а) получения карбоновой кислоты формулы 1 посредством раскрытия кольца циклического соединения формулы 2 в присутствии HBr б) взаимодействия амина формулы 3 с карбоновой кислотой формулы 2 в присутствии агента реакции присоединения с получением амида формулы 4 в) взаимодействия амида формулы 4 с соединением формулы 10 при этом стадию (в) проводят в присутствии карбоната калия и тетрагидрофурана. В одном варианте реализации изобретение обеспечивает процесс получения соединения формулы 12 при этом указанный процесс включает стадии: а) получения карбоновой кислоты формулы 1 посредством раскрытия кольца циклического соединения формулы 2 в присутствии HBr б) взаимодействия амина формулы 3 с карбоновой кислотой формулы 2 в присутствии агента реакции присоединения с получением амида формулы 4 в) взаимодействия амида формулы 4 с соединением формулы 13 при этом стадию (в) проводят в присутствии карбоната калия и тетрагидрофурана. В одном варианте реализации изобретение обеспечивает процесс получения соединения формулы 14 где X представляет собой О, NH, Se, PR или NR; T представляет собой ОН, OR, NHCOCH 3 или NHCOR; Z представляет собой NO 2 , CN, СООН, COR, NHCOR или CONHR; Y представляет собой CF 3 , F, I, Br, Cl, CN, CR 3 или SnR 3 ; Q представляет собой алкил, галоген, CF 3 , CN, CR 3 , SnR 3 , NR 2 , NHCOCH 3 , NHCOCF 3 , NHCOR, NHCONHR, NHCOOR, OCONHR, CONHR, NHCSCH 3 , NHCSCF 3 , NHCSR NHSO 2 CH 3 , NHSO 2 R, OR, COR, OCOR, OSO 2 R, SO 2 R, SR; или Q вместе с бензольным кольцом, с которым оно связано, является конденсированной циклической системой, представленной структурой А, В или С R представляет собой алкил, галоалкил, дигалоалкил, тригалоалкил, CH 2 F, CHF 2 , CF 3 , CF 2 CF 3 , арил, фенил, галоген, алкенил или ОН; и R 1 является СН 3 , CH 2 F, CHF 2 , CF 3 , CH 2 CH 3 или CF 2 CF 3 . При этом указанный процесс включает стадии: а) получения карбоновой кислоты формулы 15 посредством раскрытия кольца циклического соединения формулы 16 в присутствии HBr при этом L, R 1 и Т такие, как описано выше, и T 1 представляет собой О или NH; б) взаимодействия амина формулы 17 при этом Z и Y такие, как описано выше, с карбоновой кислотой формулы 17 в присутствии связующего реагента для получения амида формулы 18 в) связывания амида формулы II с соединением формулы 19 при этом Q и X такие, как описано выше, и стадия (в) проводится в присутствии карбоната калия и тетрагидрофурана. В одном варианте реализации кристаллические и псевдокристаллические формы согласно настоящему изобретению получают с помощью любого процесса, который находится в компетенции специалиста в данной области техники, в том числе, как представлено в настоящем изобретении, но не ограничиваясь указанными примерами. В одном варианте реализации для такого процесса необходим исходный материал для приготовления кристаллических и псевдокристаллических форм согласно настоящему изобретению, который, в свою очередь, в некоторых вариантах реализации получают по способу, схематически изложенному выше. В некоторых вариантах реализации приготовление исходного материала включает специфическую реакцию амида формулы 4 с соединением формулы 5 в присутствии карбоната калия и полярного растворителя, например, как в некоторых вариантах реализации, тетрагидрофурана, что приводит к получению очень чистых препаратов, что, в свою очередь, может повысить скорость кристаллизации. В некоторых вариантах реализации применение чистых препаратов, как описано в настоящем изобретении, зависящее от применяемых условий кристаллизации, может приводить к получению различных соотношений кристаллических форм. В некоторых вариантах реализации применение чистых препаратов, как описано в настоящем изобретении, зависящее от применяемых условий кристаллизации, может приводить к получению различных соотношений кристаллических форм, и скорости, с которой эти формы получают. В одном варианте реализации процесс дополнительно включает стадию превращения соединения селективного модулятора андрогенового рецептора (CMAP) (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида в его аналог, изомер, полиморф, полиморфную форму А, псевдокристаллическую форму В', сольват, метаболит, производное, фармацевтически приемлемую соль, фармацевтический продукт, N-оксид, гидрат, полугидрат или любую их комбинацию. В одном варианте реализации изобретение обеспечивает процесс приготовления аналога соединения селективного андрогенового модулятора согласно настоящему изобретению. В другом варианте реализации изобретение обеспечивает процесс приготовления изомера соединения селективного андрогенового модулятора согласно настоящему изобретению. В другом варианте реализации изобретение обеспечивает процесс приготовления метаболита соединения селективного андрогенового модулятора согласно настоящему изобретению. В другом варианте реализации настоящее изобретение обеспечивает процесс приготовления производного соединения селективного андрогенового модулятора согласно настоящему изобретению. В другом варианте реализации изобретение обеспечивает процесс приготовления фармацевтически приемлемой соли соединения селективного андрогенового модулятора согласно настоящему изобретению. В другом варианте реализации изобретение обеспечивает процесс приготовления фармацевтического продукта соединения селективного андрогенового модулятора согласно настоящему изобретению. В другом варианте реализации настоящее изобретение обеспечивает процесс приготовления N-оксида соединения селективного андрогенового модулятора согласно настоящему изобретению. В другом варианте реализации изобретение обеспечивает процесс приготовления гидрата соединения селективного андрогенового модулятора согласно настоящему изобретению. В другом варианте реализации изобретение обеспечивает процесс приготовления полиморфа соединения селективного андрогенового модулятора согласно настоящему изобретению. В другом варианте реализации изобретение обеспечивает процесс приготовления полиморфной формы А, описанной в настоящем изобретении, соединения селективного андрогенового модулятора согласно настоящему изобретению. В другом варианте реализации изобретение обеспечивает процесс приготовления псевдокристаллической формы В', описанной в настоящем изобретении, соединения селективного андрогенового модулятора согласно настоящему изобретению. В другом варианте реализации изобретение обеспечивает процесс приготовления полиморфной формы С, описанной в настоящем изобретении, соединения селективного андрогенового модулятора согласно настоящему изобретению. В другом варианте реализации изобретение обеспечивает процесс приготовления полиморфной формы D, описанной в настоящем изобретении, соединения селективного андрогенового модулятора согласно настоящему изобретению. В другом варианте реализации изобретение обеспечивает процесс приготовления псевдокристаллической формы соединения селективного андрогенового модулятора согласно настоящему изобретению. В другом варианте реализации изобретение обеспечивает процесс приготовления сольвата соединения селективного андрогенового модулятора согласно настоящему изобретению. В другом варианте реализации изобретение обеспечивает процесс приготовления комбинации любого аналога, изомера, метаболита, производного, полиморфа, полиморфной формы А, псевдокристаллической формы В', псевдокристаллической формы, сольвата, фармацевтически приемлемой соли, N-оксида и/или гидратов соединения селективного андрогенового модулятора согласно настоящему изобретению. В одном варианте реализации настоящее изобретение включает любое соединение, полученное таким образом. В одном варианте реализации термин "изомер" включает без ограничения оптические изомеры и аналоги, структурные изомеры и аналоги, комформационные изомеры и аналоги и т.д. В одном варианте реализации CMAP представляет собой чистый (S)-энантиомер. В одном варианте реализации выделение оптически активного (R)- или (S)-энантиомера из рацемической смеси соединений CMAP согласно изобретению включает способы кристаллизации. В другом варианте реализации способы кристаллизации включают фракционную кристаллизацию энантиомеров. В другом варианте реализации способы кристаллизации включают фракционную кристаллизацию диастереоизомерных солей (винные соли или соли хинина). В другом варианте реализации способы кристаллизации включают фракционную кристаллизацию хиральных дополнительных производных (эфиров ментола и т.п.). В другом варианте реализации выделение оптически активного (R)- или (S)-энантиомера из рацемической смеси соединений CMAP согласно изобретению включает взаимодействие рацемической смеси с другой хиральной группой, образование диастереоизомерной смеси с последующим разделением диастереоизомеров и удалением дополнительной хиральной группы для получения чистых энантиомеров. В другом варианте реализации выделение оптически активного (R)- или (S)-энантиомера из рацемической смеси соединений CMAP согласно изобретению включает хиральный синтез. В другом варианте реализации выделение оптически активного (R)- или (S)-энантиомера из рацемической смеси соединений CMAP согласно изобретению включает биологическое разделение. В другом варианте реализации выделение оптически активного (R)- или (S)-энантиомера из рацемической смеси соединений CMAP согласно изобретению включает ферментативное разделение. В другом варианте реализации выделение оптически активного (R)- или (S)-энантиомера из рацемической смеси соединений CMAP согласно изобретению включает хроматографическое разделение с использованием хиральной стационарной фазы. В другом варианте реализации выделение оптически активного (R)- или (S)-энантиомера из рацемической смеси соединений CMAP согласно изобретению включает аффинную хроматографию. В другом варианте реализации выделение оптически активного (R)- или (S)-энантиомера из рацемической смеси соединений CMAP согласно изобретению включает капиллярный электрофорез. В другом варианте реализации выеление оптически активного (R)- или (S)-энантиомера из рацемической смеси соединений CMAP согласно изобретению включает образование эфирной группы из гидроксильной группы хирального углерода с оптически активной кислотой, например (-)-камфановой кислотой, разделение полученных диастереоизомерных эфиров с помощью фракционной кристаллизации или предпочтительно флеш-хроматографии и последующий гидролиз каждого отдельного эфира до спирта. В другом варианте реализации S-энантиомер соединения CMAP согласно изобретению можно превратить в R-энантиомер или его рацемат. В другом варианте реализации R-энантиомер соединения CMAP можно превратить в S-энантиомер или его рацемат. В одном варианте реализации один энантиомер можно превратить в другой энантиомер или его рацемат посредством использования хирального реагента, растворителя, биокатализатора, хирального катализатора, ассимметричной гидрогенизации, фермента или их комбинации. В некоторых вариантах реализации твердые соединения согласно настоящему изобретению включают сольваты. В одном варианте реализации термином "сольват" обозначаются соединения веществ CMAP с растворителями, например сольват этилацетата, который является частью полиморфной структуры соединения CMAP. К таким растворителям относят этанол, ацетон, этилацетат, тетрагидрофуран, ацетонитрил, дихлорметан, 1,4-диоксан, уксусная кислота, толуол, вода, н-гептан, толуол, н-пентан ТВМЕ или их сочетания. В другом варианте реализации процесс согласно настоящему изобретению дополнительно включает стадию превращения соединения CMAP в его фармацевтически приемлемую соль. В одном варианте реализации фармацевтически приемлимые соли включают соли аминозамещенных соединений с органическими и неорганическими солями, например лимонной кислотой и соляной кислотой. Также изобретение включает N-оксиды аминозаместителей соединений, описанных в настоящем изобретении. Фармацевтически приемлемые соли также можно приготовить из фенольных соединений посредством обработки неорганическими основаниями, например гидроксидом натрия. Также эфиры фенольных соединений можно приготовить с помощью алифатических и ароматических карбоновых кислот, например эфиры уксусной и бензойной кислоты. Изобретение включает "фармацевтически приемлемые соли" соединений согласно настоящему изобретению, которые можно получить по реакции соединений согласно настоящему изобретению с кислотой или основанием. Подходящие фармацевтически приемлемые соли аминов формулы I можно приготовить из неорганических кислот или органических кислот. В одном варианте реализации изобретения примерами неорганических солей аминов являются бисульфаты, бораты, бромиды, хлориды, гемисульфаты, гидроброматы, гидрохлораты, 2-гидроксиэтилсульфонаты (гидроксиэтансульфонаты), иодаты, йодиты, изотионаты, нитраты, персульфаты, фосфаты, сульфаты, сульфаматы, сульфанилаты, сульфоновые кислоты (алкилсульфонаты, арилсульфонаты, галогензамещенные алкилсульфонаты, галогензамещенные арилсульфонаты), сульфонаты и тиоцианаты. В одном варианте реализации изобретения к примерам органических солей аминов относят соли алифатических, циклоалифатических, ароматических, аралифатических, гетероциклических, карбоновых и сульфоновых классов органических кислот, примерами которых являются ацетаты, аргинины, аспартаты, аскорбаты, адипаты, антранилаты, альгинаты, алкановые карбоксилаты, замещенные карбоксилаты алканов, альгинаты, бензолсульфонаты, бензоаты, бисульфаты, бутираты, бикарбонаты, битартраты, карбоксилаты, цитраты, камфораты, камфорсульфонаты, циклогексилсульфаматы, циклопентанпропионаты, эдетаты кальция, камзилаты, карбонаты, клавулонаты, циннаматы, дикарбоксилаты, диглюконаты, додецилсульфонаты, дигидрохлориды, деканоаты, энантуаты, этансульфонаты, эдетаты, эдизилаты, эстолаты, эзилаты, фумараты, форматы, флюориды, галактуронаты, глюконаты, глутаматы, гликолаты, глюкораты, глюкогептаноаты, глицерофосфаты, глюцептаты, гликолиларсанилаты, глютараты, глутамат, гептаноаты, гексаноаты, гидроксималеаты, гидроксикарбоновые кислоты, гексиресорцинаты, гидроксибензоаты, гидроксинафтоат, гидрофлюорат, лактаты, лактобионаты, лаураты, малаты, малеаты, метиленебис(бета-оксинафтоат), малонаты, манделаты, мезилаты, сульфонаты метана, метилбромиды, метилнитраты, метилсульфонаты, монокалийные малеаты, мукаты, монокарбоксилаты, митраты, нафталенсульфонаты, 2-нафталенсульфонаты, никотинаты, напсилаты, N-метилглюкамины, оксалаты, октаноаты, олеаты, памоаты, фенилацетаты, пикраты, фенилбензоаты, пивалаты, пропионаты, фталаты, фенилацетаты, пектинаты, фенилпропионаты, пальмитаты, пантотенаты, полигалактураты, пируваты, хинаты, салицилаты, сукцинаты, стеараты, сульфанилаты, субацетаты, тартараты, теофиллинацетаты, п-толуолсульфонаты (тозилаты), трифторацетаты, терефталаты, таннаты, теоклаты, тригалоацетаты, триэтйодиды, трикарбоксилаты, ундеканоаты или валераты. В одном варианте реализации к примерам неорганических солей карбоновых кислот или фенолов относят аммоний, щелочные металлы, включая литий, натрий, калий, цезий, щелочно-земельные металлы, включая кальций, магний, алюминий, цинк, барий, холины или четвертичные аммонии. В другом варианте реализации к примерам органических солей карбоновых кислот или фенолов относят аргинин, органические амины, включая алифатические органические амины, алициклические органические амины, ароматические органические амины, бензатины, т-бутиламины, бенэтамины, (N-бензилфенэтиламин), дициклогексиламины, диметиламины, диэтаноламины, этаноламины, этилендиамины, гидрабамины, имидазолы, лизины, метиламины, мегламины, N-метил-D-глюкамины, N,N'-дибензилэтилендиамины, никотинамиды, органические амины, орнитины, пиридины, пиколинаты, пиперазины, прокаин, трис(гидроксиметил)метиламины, триэтиламины, триэтаноламины, триметиламины, трометамины или мочевину. В одном варианте реализации соли можно образовать с помощью традиционных методов, например, посредством взаимодействия свободного основания или свободной кислоты, получая продукт с одни или более эквивалентами соответствующей кислоты или основания в растворителе или среде, в которой соль нерастворима, или в растворителе, например воде, который удаляют под вакуумом (in vacuo) лиофильной сущкой или посредством обмена ионов существующей соли на другой ион или используя соответствующую ионообменную смолу. В одном варианте реализации изобретение также включает N-оксиды аминозаместителей соединений, описанных в настоящем изобретении. Также эфиры фенольных соединений можно получить с помощью алифатических и ароматических карбоновых кислот, например эфиры уксусной и бензойной кислоты. Настоящее изобретение также включает способ приготовления производных соединений CMAP. В некоторых вариантах реализации термин "производное" обозначает без ограничения производные простых эфиров, кислот, амидов, сложных эфиров и т.д. Способы приготовления производных известны специалистам в данной области. Например, производные простых эфиров получают посредством связывания соответствующих спиртов. Производные амидов и сложных эфиров получают из соответствующей карбоновой кислоты по реакции с аминами и спиртами соответственно. В некоторых вариантах реализации изобретение включает процесс приготовления гидратов соединений CMAP. В одном варианте реализации термин "гидрат" включает без ограничения полугидрат, моногидрат, дигидрат, тригидрат и т.п. Гидраты соединений CMAP можно приготовить посредством взаимодействия соединения CMAP с водой при соответствующих условиях для получения предпочтительного гидрата. Термин "полугидрат" означает гидрат, в котором молекулярное соотношение между молекулами воды и безводным соединением составляет 1:2. Настоящее изобретение также включает процесс приготовления фармацевтического продукта соединений CMAP. Под термином "фармацевтический продукт" понимают композицию, подходящую для фармацевтического использования (фармацевтическая композиция), как определено в настоящем описании. В некоторых вариантах реализации настоящее изобретение включает процесс приготовления аналогов соединений CMAP. В одном варианте реализации термин "аналог" относится к соединениям со структурой похожей, но не идентичной структуре указанного соединения. В другом варианте реализации термин "аналог" означает изомер или производное соединения CMAP. В другом варианте реализации под термином "аналог соединения CMAP" согласно настоящему изобретению понимается соединение с различными заместителями в каждом или в обоих фенильных кольцах соединения. В другом варианте реализации под термином "аналог" понимают встраивание различных ароматических колец, например пиридильных колец, вместо одного или обоих бензольных колец. В другом варианте реализации под термином "аналог" понимают встраивание атома серы вместо каждой или обоих простой эфирной и кето-групп. В некоторых вариантах реализации настоящее изобретение включает метаболит соединений CMAP. В некоторых вариантах реализации изобретения под термином "метаболит" понимают любое вещество, полученное из другого вещества посредством имитации или непосредственно с помощью метаболического процесса. В некоторых вариантах реализации такие метаболиты можно приготовить синтетически, и они являются активными in situ, поскольку они аналогичны метаболитам, полученными естественным путем. В одном варианте реализации настоящее изобретение обеспечивает композицию, включающую кристаллическую форму безводного (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида и подходящий носитель или растворитель. В другом варианте реализации настоящее изобретение обеспечивает композицию, включающую кристаллическую форму А безводного (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида и подходящий носитель или растворитель. В одном варианте реализации настоящее изобретение обеспечивает композицию, включающую псевдокристаллическую форму (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида и подходящий носитель или растворитель. В одном варианте реализации настоящее изобретение обеспечивает композицию, включающую псевдокристаллическую форму В' (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида и подходящий носитель или растворитель. В одном варианте реализации настоящее изобретение обеспечивает композицию, включающую смесь любых твердых форм соединеия (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида согласно настоящему изобретению и подходящий носитель или растворитель. В другом варианте реализации настоящее изобретение обеспечивает композицию, включающую смесь кристаллических и псевдокристаллических твердых форм соединения (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида и подходящий носитель или растворитель. В другом варианте реализации настоящее изобретение обеспечивает композицию, включающую смесь кристаллической формы А и псевдокристаллической твердой формы В' (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида и подходящий носитель или растворитель. В одном варианте реализации настоящее изобретение обеспечивает композиции, включающие различные формы (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида, которые могут присутствовать в различных соотношениях или по отдельности в композиции, которая обладает свойствами, полезными для лечения андроген-обусловленных заболеваний, описанных в настоящем изобретении. В другом варианте настоящее изобретение включает композиции, содержащие различные изомеры N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида, которые могут присутствовать в различных соотношениях или по отдельности (один изомер) в композиции, которая обладает свойствами полезными для лечения андроген-обусловленных заболеваний, описанных в настоящем изобретении. В некоторых вариантах реализации под термином "фармацевтическая композиция" понимают "терапевтически эффективное количество" активного ингредиента, т.е. соединения CMAP, вместе с фармацевтически приемлемым носителем или растворителем. В некоторых вариантах реализации под фразой "терапевтически эффективное количество" понимается количество, которое обеспечивает терапевтический эффект для данных условий и режима введения. Фармацевтические композиции, содержащие агент CMAP можно вводить субъекту с помощью любого способа, известного специалисту в данной области техники, например парентерально, параканцерально, трансмукозально, трансдермально, внутримышечно, внутривенно, внутрикожно, подкожно, внутрибрюшинно, интравентрикулярно, интракраниально или внутриопухолево. В другом варианте реализации настоящее изобретение обеспечивает композицию твердых форм согласно настоящему изобретению и подходящий носитель или растворитель. В одном варианте реализации фармацевтические композиции вводят перорально и поэтому выпускают в форме, пригодной для перорального применения, например в форме твердого препарата. К подходящим твердым пероральным составам относятся таблетки, капсулы, пилюли, гранулы, пеллеты и т.д. В одном варианте реализации изобретения соединения CMAP выпускаются в виде капсул. Согласно этому варианту реализации композиции согласно настоящему изобретению включают в дополнение к активному соединению CMAP и инертному носителю или растворителю твердую желатиновую капсулу. Пероральные составы, содержащие настоящий полиморф, могут включать любые традиционно используемые пероральные формы, включая таблетки, капсулы, буккальные формы, таблетки для рассасывания или пастилки. Капсулы могут содержать смесь кристаллической формы А в необходимом процентном содержании вместе с любым полиморфом (полиморфами) CMAP или аморфным CMAP. Капсулы или таблетки необходимой кристаллической формы заданного процентного содержания можно также комбинировать со смесями других активных соединений или инертными наполнителями и/или растворителями, например фармацевтически приемлемыми крахмалами (например, кукурузным, картофельным или крахмалом тапиоки), сахарами, искусственными подсластителями, порошковой целлюлозой, например кристаллической или микрокристаллической целлюлозой, различной мукой, желатинами, камедями и т.д. Таблетированные составы можно приготовить с помощью традиционных способов компрессии, сырой грануляции или сухой грануляции, с применением фармацевтически примемлемых растворителей (наполнителей), связующих агентов, лубрикантов, дезинтеграторов, суспендирующих или стабилизирующих агентов, включая без ограничения стеарат магния, стериновую кислоту, тальк, лаурилсульфат натрия, микрокристаллическую целлюлозу, кальций карбоксиметилцеллюлоза, поливинилпирролидон, желатин, альгиновую кислоту, аравийскую камедь, ксантановую камедь, цитрат натрия, комплексные силикаты, карбонат кальция, глицин, декстрин, сахарозу, сорбит, дикальцийфосфат, сульфат кальция, лактозу, каолин, маннит, хлорид натрия, тальк, сухие крахмалы и порошковый сахар. В пероральных составах в некоторых вариантах реализации используются стандартные образцы или составы с замедленным высвобождением или спансулы. К примерам систем наполнителей, подходящих для приготовления составов представленного полиморфа, относятся один или более наполнителей, дезинтеграторов, лубрикантов. Наполнитель может представлять собой любой наполнитель, известный в данной области техники, включая без ограничения лактозу, микрокристаллическую целлюлозу, сахарозу, маннит, фосфат кальция, карбонат кальция, порошковую целлюлозу, мальтодекстрин, сорбит, крахмал или ксилит. Дезинтегрант, подходящий для использования в настоящих составах, можно выбрать из известных в области техники, включая прежелатинизированный крахмал и гликолят крахмала натрия. К другим применяемым дезинтеграторам относятся кроскармеллоза натрия, кросповидон, крахмал, альгиновая кислота, альгинат натрия, глины (например, вигум или ксантановая камедь), целлюлозный флок, ионообменные смолы или шипучие системы, например, с использованием пищевых кислот (лимонной, винной, яблочной, фумаровой, молочной, адипиновой, аскорибновой, аспартатовой, эриторбовой, глютаминовой и янтарной) и щелочного карбонатного компонента (например, бикарбоната натрия, кальция, магния, калия, аммония и т.д.). Дезинтегратор(ы), использованные в настоящем описании, могут составлять от приблизительно 4 до приблизительно 40 мас.% композиции, предпочтительно от приблизительно 15 до приблизительно 35%, более предпочтительно от приблизительно 20 до приблизительно 35%. Фармацевтические составы могут также содержать антиоксиданты или их смесь, например аскорбиновую кислоту. Другие антиоксиданты, которые можно использовать, включают аскорбат натрия и аскорбил пальмитат предпочтительно в сочетании с аскорбиновой кислотой. Например, содержание антиоксиданта(ов) варьирует от примерно 0,5 до примерно 15 мас.%, наиболее предпочтительно от приблизительно 0,5 до приблизительно 5 мас.%. В некоторых вариантах реализации изобретния активный фармакологический агент (агенты) содержатся в количестве от приблизительно 0,5 до примерно 20 мас.% в расчете на общую композицию или в некоторых вариантах реализации от примерно 1 до примерно 5%, и покрывающая оболочка или капсула составляет до приблизительно 8 мас.% в расчете на общую композицию. Составы, описанные в настоящем изобретении, можно применять в виде безоболочечных или некапсулированных твердых форм. В некоторых вариантах реализации фармакологические композиции возможно покрыты пленочным покрытием, например, составляющими от примерно 0,3 до примерно 8 мас.% в расчете на общую композицию. Пленочные покрытия, которые используются в представленных составах, известны в данной области техники и обычно включают полимер (обычно целлюлозного типа), краситель и пластификатор. В составы с пленочным покрытием можно включать дополнительные ингридиенты, например увлажняющие средства, сахара, ароматизаторы, масла и лубриканты, для придания определенных свойств пленочному покрытию. Композиции и составы, описанные в настоящем изобретении, также можно объединять и применять целиком, если поместить их в капсулу, например в желатиновую капсулу. В другом варианте реализации активный компонент можно доставлять в везикуле, например липосоме (см. Langer, Science 249:1527-1533 (1990); Treat et al. in Liposomes in the Therapy of Infectious Disease and Cancer, Lopez-Berestein and Fidler (eds.), Liss, New York, pp. 353-365 (1989); Lopez-Berestein, тот же источник, pp. 317-327). В настоящем описании "фармацевтически приемлемые носители или растворители" хорошо знакомы специалисту в данной области. Носитель или растворитель могут представлять собой твердый носитель или растворитель для твердых составов. Твердые носители/растворители включают без ограничения камедь, крахмал (например, крахмал кукурузы, прежелатинизированный крахмал), сахар (например, сахароза, лактоза, маннит, декстроза), целлюлозный материал (например, микрокристаллическая целлюлоза), акрилат (полиметилакрилат), карбонат кальция, оксид магния, тальк или их смеси. Более того, композиции дополнительно могут содержать связующий агент (например, гуммиарабик, кукурузный крахмал, желатин, карбомер, этилцеллюлозу, гуаровую камедь, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, повидон), дезинтегрирующие агенты (например, кукурузный крахмал, картофельный крахмал, альгиновую кислоту, диоксид кремния, кроскармелозу натрия, кросповидон, гуаровую камедь, натрия гликолят крахмал), буферы (например, Трис-HCl, ацетатный, фосфатный) различных pH и ионной силы, добавки, например альбумин или желатин для предотвращения абсорбции к поверхностям, детергенты (например, Tween 20, Tween 80, Pluronic F68, соли желчной кислоты), ингибиторы протеаз, поверхностно-активные вещества (например, лаурил сульфат натрия), усилители проницаемости, солюбилизирующие агенты (например, глицерин, полиэтилен глицерин), антиоксиданты (например, аскорбиновая кислота, метабисульфит натрия, бутилированный гидроксианизол), стаблизаторы (например, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза), усилители вязкозти (например, карбомер, коллоидный диоксид крмния, этилцеллюлоза, гуаровая камедь), подсластители (аспартам, лимонная кислота), консерванты (например, Тимерозал, бензиловый спирт, парабены), лубриканты (например, стеариновая кислота, стеарат магния, полиэтиленгликоль, лаурил сульфат натрия), вещества, способствующие текучести (наример, коллоидный диоксид кремния), пластификаторы (например, диэтилфталат, триэтилцитрат), эмульгаторы (например, карбомер, гидроксипропилцеллюлоза, лаурил сульфат натрия), полимерные покрытия (например, полоксамеры или полоксамины), агенты, образующие оболочки и пленки (например, этилцелюллоза, акрилаты, полиметакрилаты) и/или адъюванты. В одном варианте реализации изобретения фармацевтические композиции, описанные в настоящем изобретении, представляют собой композиции с контролируемым высвобождением, т.е. композиции, в которых соединение CMAP высвобождается с течением времени после введения. В другом варианте реализации композиции представляют собой композиции с незамедлительным высвобождением, т.е. композиции, в которых соединение CMAP полностью высвобождается непосредственно после введения. Также в другом варианте реализации изобретения фармацевтические композиции могут доставляться в виде системы контролируемого высвобождения. Например, агент можно вводить с помощью липосом или с помощью других способов перорального введения. Композиции могут также включать встраивание активного материала в или на определенные препараты полимерных соединений, например как полимолочная кислота, полигликолевая кислота, гидрогели и т.д., или на липосомы, микроэмульсии, мицеллы, моноламеллярные или полиламеллярные везикулы, "тени" эритроцитов или сферопласты. Такие композиции будут оказывать влияние на физическое состояние, растворимость, стабильность, скорость высвобождения in vivo и скорость очистки in vivo. Приготовление фармацевтических комопзиций, содержащих активный компонент, известно в данной области техники, например перемешивание, гранулирование или таблетирование. Активный терапевтический ингридиент часто смешивают с фармацевтически приемлемыми наполнителями, совместимыми с активным ингридиентом. Для перорального введения агенты CMAP или их физиологически толерантные производные, например соли, N-оксиды и эфиры и т.д., смешиваются с добавками, предназначенными для этих целей, например средами, стабилизаторами, или инертными растворителями, и с помощью стандартных способов превращаются в подходящие для введения формы, например таблетки, покрытые таблетки, твердые или мягкие желатиновые капсулы, водные, спиртовые или масляные растворы. Активный компонент можно включать в состав композиции в качестве нейтрализованных фармацевтически приемлемых солей. Фармацевтически приемлемые соли включают соли присоединения кислоты (образованными свободными аминогруппами полипептида или молекулы антитела), образованными с неорганической кислотой, например соляной или фосфорной, или с органической кислотой, например уксусной, щавелевой, винной, миндальной и т.д. Соли, образованные свободными карбоксильными группами, также можно получить из неорганических оснований, например гидроксидов натрия, калия, аммония, кальция или железа, а также органических оснований, например изопропиламин, триметиламин, 2-этиламиноэтанол, гистидин, прокаин и т.д. Соли CMAP для использования в медицине представляют собой фармацевтически приемлемые соли. Другие соли, однако, можно использовать для приготовления соединений согласно настоящему изобретению или их фармацевтически приемлемых солей. Подходящие фармацевтически приемлемые соли соединений согласно изобретению включают соли присоединения кислоты, которые можно получить посредством, например, смешивания раствора соединения согласно настоящему изобретению с раствором фармацевтически приемлемой кислоты, например соляной, серной, метансерной, фумаровой, малеиновой, янтарной, уксусной, банзойной, щавелевой, лимонной, винной, угольной или фосфорной кислот. Твердые формы и процессы их получения, приведенные в настоящем описании, в некоторых вариантах реализации изобретения относятся к селективным модуляторам андрогеновых рецепторов (CMAP), которые применимы для пероральной тестостеронзаместительной гормональной терапии, имеющей неожиданную in vivo андрогенную и анаболическую активность. В некоторых вариантах реализации соединения с соответствующими заместителями эффективны для лечения рака простаты и визуализации рака простаты. Как рассмотрено в настоящем описании, соответственно замещенные соединения CMAP согласно настоящему изобретению эффективны для а) контрацепции мужчин; б) лечения различных гормональных заболеваний, например заболеваний, связанных с возрастным андрогенным дефицитом (ВАД), например хроническая усталость, депрессия, пониженное либидо, половая дисфункция, нарушение эрекции, гипогонадизм, остеопороз, облысение, анемия, ожирение, саркопения, остеопения, доброкачественная гиперплазия простаты, перемены настроения и нарушение познавательной способности и рак простаты; в) лечения заболеваний, связанных с дефицитом андрогенов у женщин (ДАЖ), таких как половые расстройства, пониженное либидо, гипогонадизм, саркопения, остеопения, остеопороз, перемены настроения и нарушение познавательной способности, депрессия, анемия, облысение, ожирение, эндометриоз, рак груди, рак матки и яичников; г) лечения и/или профилактика хронических состояний мышечного истощения; д) снижения степени замедления регрессии или стимулированиерегрессии рака простаты; е) пероральной андрогенной заместительной терапии; и/или других видов терапевтического лечения или диагностики. В представленном описании изобретения рецепторы внеклеточных сигнальных молекул собирательно обозначают "рецепторами клеточного сигналлинга (сигнальной системы клетки)". Многие рецепторы клеточного сигналлинга являются трансмембранными белками на поверхности клеток, когда они связывают внеклеточные сигнальные молекулы (например, лиганд), они активируются и вызывают каскад внутриклеточных сигналов, которые изменяют поведение клетки. И, наоборот, в некоторых случаях, рецепторы находятся внутри клетки, и сигнальный лиганд должен проникнуть в клетку, чтобы их активировать; поэтому эти сигнальные молекулы должны быть достаточно малы и гидрофобны, чтобы диффундировать через цитоплазматическую мембрану клетки. В настоящем описании такие рецепторы собирательно называют "внутриклеточными рецепторами клеточного сигналлинга". Стероидные гормоны являются примером малых гидрофобных молекул, которые непосредственно диффундируют через цитоплазматическую мембрану целевой клетки и связываются с внутриклеточными рецепторами клеточного сигналлинга. Эти рецепторы структурно связаны и составляют суперсемейство внутриклеточных рецепторов (или суперсемейство рецепторов стероидных горомонов). Рецепторы стероидных гормонов включают рецепторы прогестерона, эстрогена, андрогенов, глюкокортикоидов и минералокортикоидов. Настоящее изобретение направлено, в частности, на рецепторы андрогенов. Помимо связывания лиганда с рецептором, рецепторы можно заблокировать для предотвращения связывания лиганда. Когда вещество связывается с рецептором, пространственная структура вещества соответствует пространству, которое создается пространственной структурой рецептора в шарнирной конфигурации. Согласно одному варианту реализации настоящее изобретение направлено на процесс приготовления твердых форм соединений селективных модуляторов андрогеновых рецепторов, которые являются агонистами. Таким образом, в одном варианте реализации соединения CMAP согласно настоящему изобретению используются для связывания и активации рецепторов стероидных гормонов. В одном варианте реализации агонист согласно настоящему изобретению является агонистом, который связывает рецептор андрогена. В другом варианте реализации соединение обладает высоким сродством к андрогеновому рецептору. В другом варианте реализации агонист также обладает анаболическим действием. В другом варианте реализации настоящее изобретение обеспечивает соединения селективных андрогеновых модуляторов, которые обладают агонистическим и анаболическим действием нестероидного соединения на андрогеновый рецептор. Согласно одному варианту реализации настоящее изобретение направлено на процессы приготовления твердых форм соединений селективных модуляторов андрогеновых рецепторов, которые являются антагонистами. Так, в одном варианте реализации твердые формы соединений CMAP согласно настоящему изобретению используются для связывания и инактивации рецепторов стероидных гормонов. В другом варианте реализации твердые формы согласно изобретению обладают высоким сродством к андрогеновому рецептору. В другом варианте реализации твердые формы согласно изобретению также обладают анаболическим действием. В другом варианте реализации твердые формы соединений CMAP необратимо связываются с андрогеновым рецептором. В другом варианте реализации изобретения твердые формы соединеий CMAP представляют собой алкилирующие агенты. В другом варианте реализации твердые формы соединений CMAP согласно настоящему изобретению можно классифицировать как частичные агонисты/антагонисты андрогеновых рецепторов (AR). Твердые формы CMAP в некторорых тканях являются агонистами андрогеновых рецепторов и вызывают усиление транскрипции генов, отвечающих за андрогеновые рецепторы (например, анаболическое действие на мышцы). В других тканях эти соединения служат ингибиторами андрогеновых рецепторов для предотвращения агонистического действия природных андрогенов. Специалисту в данной области хорошо известны исследования по определению, являются ли соединения согласно настоящему изобретению агонистами или антагонистами андрогеновых рецепторов. Например, агонистическое действие AR можно определить, контролируя способность твердых форм соединений CMAP поддерживать и/или стимулировать рост тканей, содержащих андрогеновые рецепторы, например везикулы простаты или семенные пузырьки, по измерению массы. Антагонистическое действие AR можно определить, контролируя способность соединений CMAP подавлять рост тканей, содержащих андрогеновые рецепторы. В одном варианте реализации твердые формы соединений CMAP необратимо связываются с андрогеновыми рецепторами млекопитающих, например людей. Таким образом, согласно одному варианту реализации соединения согласно настоящему изобретению могут содержать функциональную группу (например, аффинную метку), которая допускает алкилирование андрогенового рецептора (т.е. образование ковалентной связи). Таким образом, в данном случае, эти соединения представляют собой алкилирующие агенты, которые необратимо связываются с рецептором и, соответственно, не могут быть заменены на стероид, например эндогенные лиганды ДГТ и тестостерон. В настоящем описании "алкилирующий агент" определяется как агент, который алкилирует (образует ковалентную связь) с компонентом клетки, например ДНК, РНК или белком. Он является высокоактивным химическим соединением, который вводит алкильные радикалы в биологически активные молекулы и таким образом блокирует их нормальное функционирование. Алкилирующая группа является электрофильной группой, которая взаимодействует с нуклеофильными группами в клеточных компонентах. Согласно одному варианту реализации настоящего изобретения обеспечивается способ связывания твердых форм соединений CMAP согласно настоящему изобретению с андрогеновым рецептором посредством контакта рецептора с твердыми формами соединений CMAP, например полиморфной формой А, полиморфной формой С, полиморфной формой D, псевдокристаллической формой В', псевдокристаллической формой В", их сольватом, полиморфом, метаболитом и т.д. или любыми их комбинациями при условиях, позволяющих соединению селективных модуляторов андрогенового рецептора связываться с андрогеновым рецептором. Связывание твердых форм соединений селективных модуляторов андрогеновых рецепторов с андрогеновым рецептором позволяет использовать соединения согласно настоящему изобретению в качестве мужского контрацептива и в ряде гормональных терапий. Агонисты связываются и активируют андрогеновый рецептор. Антагонисты связываются и инактивируют андрогеновый рецептор. Связывание агониста или антагониста может быть как обратимым, так и необратимым. В одном варианте реализации твердые формы соединений CMAP согласно настоящему изобретению вводят как единственный активный ингридиент. Однако в рамки настоящего изобретения также включены способы гормональной терапии, лечения рака простаты, замедления прогрессии рака простаты и предотвращения и/или лечения рецидивов рака простаты, которые включают введение твердых форм соединений CMAP в сочетании с одним или более терапевтическими агентами. Такие агенты включают без ограничения аналоги LHRH, обратимые антиандрогены, антиэстрогены, противораковые лекарственные средства, ингибиторы 5- α-редуктазы, ингибиторы ароматазы, прогестины, агенты, действующие через другие ядерные гормональные рецепторы, селективные модуляторы эстрогеновых рецепторов (SERM), прогестерон, эстроген, ингибиторы PDE5, апоморфин, бисфосфат и одна или более твердых форм CMAP, например, обладающая агонистическим действием AR. Таким образом, согласно одному варианту реализации настоящее изобретение обеспечивает композиции и фармацевтические композиции, содержащие твердые формы соединений селективных модуляторов андрогеновых рецепторов в сочетании с аналогом LHRH. Согласно другому варианту реализации настоящее изобретение обеспечивает композиции и фармацевтические композиции, содержащие твердые формы соединений селективных модуляторов андрогеновых рецепторов в сочетании с обратимым антиандрогеном. В другом варианте реализации настоящее изобретение обеспечивает композиции и фармацевтические композиции, содержащие твердые формы соединений селективных модуляторов андрогеновых рецепторов в сочетании с антиэстрогеном. В другом варианте реализации настоящее изобретение обеспечивает композиции и фармацевтические композиции, содержащие твердые формы соединений селективных модуляторов андрогеновых рецепторов в сочетании с противораковым лекарственным средством. В другом варианте реализации настоящее изобретение обеспечивает композиции и фармацевтические композиции, содержащие твердые формы соединений селективных модуляторов андрогеновых рецепторов в сочетании с ингибитором 5-редуктазы. В другом варианте реализации настоящее изобретение обеспечивает композиции и фармацевтические композиции, содержащие твердые формы соединений селективных модуляторов андрогеновых рецепторов в сочетании с ингибитором ароматазы. В другом варианте реализации настоящее изобретение обеспечивает композиции и фармацевтические композиции, содержащие твердые формы соединений селективных модуляторов андрогеновых рецепторов в сочетании с прогестином. В другом варианте реализации настоящее изобретение обеспечивает композиции и фармацевтические композиции, содержащие твердые формы соединений селективных модуляторов андрогеновых рецепторов в сочетании с агентом, действующим через другие ядерные гормональные рецепторы. В другом варианте реализации настоящее изобретение обеспечивает композиции и фармацевтические композиции, содержащие твердые формы соединений селективных модуляторов андрогеновых рецепторов в сочетании с селективными модуляторами эстрогеновых рецепторов (СМЭР). В другом варианте реализации настоящее изобретение обеспечивает композиции и фармацевтические композиции, содержащие твердые формы соединений селективных модуляторов андрогеновых рецепторов в сочетании с прогестероном. В другом варианте реализации настоящее изобретение обеспечивает композиции и фармацевтические композиции, содержащие твердые формы соединений селективных модуляторов андрогеновых рецепторов в сочетании с эстрогеном. В другом варианте реализации настоящее изобретение обеспечивает композиции и фармацевтические композиции, содержащие твердые формы соединений селективных модуляторов андрогеновых рецепторов в сочетании с ингибиторами PDE5. В другом варианте реализации настоящее изобретение обеспечивает композиции и фармацевтические композиции, содержащие твердые формы соединений селективных модуляторов андрогеновых рецепторов в сочетании с апоморфином. В другом варианте реализации настоящее изобретение обеспечивает композиции и фармацевтические композиции, содержащие твердые формы соединений селективных модуляторов андрогеновых рецепторов в сочетании с бисфосфонатом. В другом варианте реализации настоящее изобретение обеспечивает композиции и фармацевтические композиции, содержащие твердые формы соединений селективных модуляторов андрогеновых рецепторов в сочетании с одним или более дополнительных CMAP. Приведенные ниже примеры представлены исключительно с целью более полной иллюстрации предпочтительных вариантов реализации изобретения, но не ограничивают притязаний изобретения. Пример 1. Синтез соединения S-1. (2R)-1-метакрилоилпирролидин-2-карбоновая кислота. Растворяли D-пролин (14,93 г, 0,13 моль) в 71 мл 2N NaOH и охлаждали на ледяной бане; полученный щелочной раствор разбавляли ацетоном (71 мл). Ацетоновый раствор (71 мл) метакрилоилхлорида (13,56 г, 0,13 моль) и 2N раствор NaOH (71 мл) добавляли одновременно в течение 40 мин к водному раствору D-пролина на ледяной бане. В ходе добавления метакрилоилхлорида pH смеси выдерживали в пределах 10-11 °С. После перемешивания (3 ч, комнатная температура), смесь выпаривали in vacuo при температуре 35-45 °C для удаления ацетона. Поученный раствор промывали с помощью этилового эфира и подкисляли до pH 2 с помощью концентрированной HCl. Кислую смесь насыщали NaCl и экстрагировали с помощью EtOAc (100 мл × 3). Объединенный экстракты высушивали над Na 2 SO 4 , фильтровали через целит и выпаривали in vacuo с получением неочищенного продукта в виде бесцветного масла. В результате перекристаллизации масла из этилового эфира и гексана получали 16,2 (68%) желаемого соединения в виде бесцветных кристаллов: mp 102-103 °C (лит. источник [214]: mp 102,5-103,5 °C); спектр ЯМР этого вещества доказывает существование двух ротамеров соединения, указанного в заголовке. 1 H ЯМР (300 МГц, DMSO-d 6 ) δ 5.28 (с.) и 5.15 (с.) для первого ротамера, 5.15 (с.) и 5.03 (с.) для второго ротамера (итого 2Н для обоих ротамеров, винильная СН 2 ), 4.48-4.44 для первого ротамера, 4.24-4.20 (м.) для второго ротамера (итого 1Н для обоих ротамеров, CH в хиральном центре), 3.57-3.38 (м., 2Н, СН 2 ), 2.27-2.12 (1H, СН), 1.97-1.72 (м., 6Н, СН 2 , СН, Me); 13 C ЯМР (75 МГц, DMSO-d 6 ) δ для основного ротамера 173.3, 169.1, 140.9, 116.4, 58.3, 48.7, 28.9, 24.7, 19.5: для минорного ротамера 174.0, 170.0, 141.6, 115.2, 60.3, 45.9, 31.0, 22.3, 19.7; ИК (KBr) 3437 (OH), 1737 (C=O), 1647 (CO, COOH), 1584, 1508, 1459, 1369, 1348, 1178 см -1 ; [ α] D 26 +80.8 ° (с = 1, MeOH); по аналитическому расчету для C 9 H 13 NO 3 : С 59.00, Н 7.15, N 7.65. Найдено: С 59.13, Н 7.19, N 7.61. (3R,8aR)-3-бромметил-3-метилтетрагидропирроло[2,1-с][1,4]оксазин-1,4-дион. Раствор NBS (23,5 г, 0,132 моль) в 100 мл DMF добавляли по каплям к перемешиваемому раствору (метилакрилоил)пирролидина (16,1 г, 88 ммоль) в 70 мл DMF в аргоне при комнатной температуре и полученную смесь перемешивали 3 дня. Растворитель удаляли in vacuo, и образовывался желтый осадок. Осадок суспендировали в воде, перемешивали в течение ночи при комнатной температуре, фильтровали и сушили с получением 18,6 (81%) (меньший вес в высушенном состоянии ~34%) соединения, указанного в заголовке, в виде желтого осадка: mp 152-154 °C (лит. источник [214]: mp 107-109 °C для S-изомера); 1 H ЯМР (300 МГц, DMSO-d 6 ) δ 4.69 (д. д., J = 9.6 Гц, J = 6.7 Гц, 1H, СН в хиральном центре), 4.02 (д., J=11.4 Гц, 1H, CHH a ), 3.86 (д., J = 11.4 Гц, 1H, CHH b ), 3.53-3.24 (м., 4Н, СН 2 ), 2.30-2.20 (м., 1H, СН), 2.04-1.72 (м., 3H, СН 2 и СН), 1.56 (с, 2Н, Me); 13 C ЯМР (75 МГц, DMSO-d 6 ) δ 167.3, 163.1, 83.9, 57.2, 45.4, 37.8, 29.0, 22.9, 21.6; ИК (KBr) 3474, 1745 (C=O), 1687 (C=O), 1448, 1377, 1360, 1308, 1227, 1159, 1062см -1 ; [ α] D 26 +124.5 ° (с = 1.3, хлороформ); по аналитическому расчету для C 9 H 12 BrNO 3 : С 41.24, Н 4.61, N 5.34. Найдено: С 41.46, Н 4.64, N 5.32. (2R)-3-бром-2-гидрокси-2-метилпропановая кислота. Смесь бромлактона (18,5 г, 71 ммоль) в 300 мл 24% HBr нагревали в колбе с обратным холодильником в течение 1 ч. Полученный раствор разбавляли солевым раствором (200 мл) и экстрагировали с помощью этилацетата (100 мл × 4). Объединенные экстракты промывали насыщенным раствором NaHCO 3 (100 мл × 4). Водный раствор подкисляли концентрированной HCl до pH 1, который в свою очередь экстрагировали с помощью этилацетата (100 мл × 4). Объединенный органический раствор сушили над Na 2 SO 4 , фильтровали через целит и выпаривали in vacuo досуха. Перекристаллизация из толуола дала 10,2 г (86%) желаемого соединения в виде бесцветных кристаллов: mp 107-109 °C (лит. источник [214]: mp 109-113 °C для S-изомера); 1 Н ЯМР (300 МГц, DMSO-d 6 ) δ 3.63 (д., J = 10.1 Гц, 1H, СНН а ), 3.52 (д., J = 10.1 Гц, 1H, CHH b ), 1.35 (с, 3H, Me); IR (KBr) 3434 (ОН), 3300-2500 (COOH), 1730 (C=O), 1449, 1421, 1380, 1292, 1193, 1085 см -1 ; [ α] D 26 +10.5 ° (с = 2.6, MeOH); по аналитическому расчету для C 4 H 7 BrO 3 : С 26.25, Н 3.86. Найдено: С 26.28, Н 3.75. Синтез (2R)-3-бром-N-[4-циано-3-(трифторметил)фенил]-2-гидрокси-2-метилпропанамида. Хлористый тионил (46,02 г, 0,39 моль) добавляли по каплям к охлажденному раствору (менее 4 °С) соединения 6 (51,13 г, 0,28 моль) в 300 мл THF в атмосфере аргона. Полученную смесь перемешивали 3 ч при тех же условиях. Добавляли к ней Et 3 N (39,14 г, 0,39 моль) и перемешивали 20 мин при тех же условиях. Через 20 мин добавляли 5-амино-2-цианобензотрифторид (40,0 г, 0,21 моль), 400 мл THF и затем смесь оставляли перемешиваться на ночь при комнатной температуре. Растворитель удаляли при пониженном давлении с получением твердого остатка, который разводили в 300 мл Н 2 О и экстрагировали с помощью EtOAc (2 × 400 мл). Объединенные органические экстракты промывали насыщенным раствором NaHCO 3 (2 ×300 мл) и солевым раствором (300 мл). Органический слой высушивали над MgSO 4 и концентрировали при пониженном давлении с получением твердого остатка, который очищали на хроматографической колонке с использованием CH 2 Cl 2 /EtOAc (80:20) с получением твердого вещества. Это вещество перекристаллизовывали из CH 2 Cl 2 /гексана с получением 55,8 г 73,9%) (2R)-3-бром-N-[4-циано-3-(трифторметил)фенил]-2-гидрокси-2-метилпропанамида в виде светло-желтых кристаллов. 1 H ЯМР (CDCl 3 /TMS) δ 1.66 (с, 3H, CH 3 ), 3.11 (с, 1H, ОН), 3.63 (д., J = 10.8 Гц, 1H, СН 2 ), 4.05 (д., J = 10.8 Гц, 1H, СН 2 ), 7.85 (д., J = 8.4 Гц, 1Н, ArH), 7.99 (д. д., J = 2.1, 8.4 Гц, 1H, ArH), 8.12 (д., J = 2.1 Гц, 1Н, ArH), 9.04 (уш. с, 1Н, NH). Рассчитанная масса: 349.99, [M-H] - 349.0. М.р.: 124-126 °C. Синтез (S)-H-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида. Смесь бромамида ((2R)-3-бром-H-[4-циано-3-(трифторметил)фенил]-2-гидрокси-2-метилпропанамида, 50 г, 0,14 моль), безводного K 2 CO 3 (59,04 г, 0,43 моль) и 4-цианофенола (25,44 г, 0,21 моль) в 500 мл 2-пропанола нагревали в колбе с обратным холодильником 3 ч, а затем концентрировали при пониженном давлении с образованием твердого вещества. Полученный осадок разводили в 500 мл Н 2 О и затем экстрагировали с помощью EtOAc (2 × 300 мл). Объединенные экстракты EtOAc промывали 10% NaOH (4 × 200 мл) и солевым раствором. Органический слой сушили над MgSO 4 и затем концентрировали при пониженном давлении с получением масла, которое обрабатывали с помощью 300 мл этанола и активированного угля. Реакционную смесь нагревали в колбе с обратным холодильником в течение 1 ч и затем горячую смесь фильтровали через целит. Фильтрат концентрировали при пониженном давлении с получением масла. Это масло очищали с помощью колоночной хроматографии с использованием CH 2 Cl 2 /EtOAc (80:20) с получением масла, которое кристаллизовали из CH 2 Cl 2 /гексана с получением 33,2 г (59,9%) (S)-N-(4-циано-3-(трифторметил)фенил)-3-(4-цианофенокси)-2-гидрокси-2-метилпропанамида в виде бесцветного твердого вещества (волокнистого типа). 1 Н ЯМР (CDCl 3 /TMS) δ 1.63 (с, 3H, СН 3 ), 3.35 (с, 1H,OH), 4.07 (д., J = 9.04 Гц, 1H, CH), 4.51 (д., J = 9.04 Гц, 1H, CH), 6.97 - 6.99 (м., 2Н, ArH), 7.57-7.60 (м., 2Н, ArH), 7.81 (д., J = 8.55 Гц, 1H, ArH), 7.97 (д. д., J = 1.95, 8.55 Гц, 1H, ArH), 8.12 (д., J = 1.95 Гц, 1H, ArH), 9.13 (уш. с, 1Н, NH). Рассчитанная масса: 389.10, [M-H] - 388.1. Мр: 92-94 °C. Пример 2. Кристаллизация соединения S-1 SARM. Материалы и методы. Методы. Рентгеновская порошковая дифрактометрия (XRPD). Применяли XRPD для определения кристаллической структуры или распознавания жидкокристаллических материалов в частично кристаллических смесях. XRPD проводили с помощью рентгеновского дифрактометра PW 1710 (PANalytical), где использовался трубчатый медный анод с Ka-излучением. Рентгенограмму накапливали в режиме пошагового сканирования (размер шага 0,02 °2 θ, время счета 2,4 с/шаг). Измерения в образцах проводили без какой-либо специальной обработки, помимо применения к ним легкого давления для получения гладкой поверхности. Измерения проводили в атмосфере воздуха окружающей среды. Спектроскопия комбинационного рассеяния (СКР). Записывали спектры комбинационного рассеяния с преобразованием Фурье с помощью системы для СКР с Фурье-преобразованием Bruker RFS 100, оснащенной лазером ближнего инфракрасного диапазона Nd:YAG, работающим при 1064 нм, и охлаждаемым жидким азотом германиевым детектором. Для каждого образца собирали 64 сканирования с разрешением 2 см -1 . Используемая мощность лазера составляла 100 мВт. Измерения комбинационного рассеяния проводили с использованием алюминиевых держателей образцов или герметически закрытых стеклянных трубок при комнатной температуре. Термогравиметрический инфракрасный анализ с Фурье-преобразованием (TG-FTIR) и термогравиметрический анализ. Аппаратура TG-FTIR состоит из гравиметрического анализатора (TG) с Фурье-инфракрасным спектрометром (FTIR) для анализа выделяющихся газов, например паров H 2 O, по потере их массы в совокупности с характеристикой выделяющихся компонентов. Термогравиметрические измерения проводили с помощью Netzsch Thermo-Microbalance TG 209, соединенного со спектрометром Bruker FTIR Vector 22. Были использованы кюветы для образцов с отверстием в атмосфере N 2 при скорости нагревания 10 K/мин, с температурным диапазоном от 25 до 250 °С. Дополнительный термогравиметрический анализ проводили с помощью ТА Instruments Q500 TGA в различных условиях. Дифференциальная сканирующая калориметрия (DSC). Термический анализ проводили с помощью аппаратуры Perkin Elmer DSC7 при следующих экспериментальных условиях: масса образца от 3 до 6 мг, закрытая золотая кювета для образца, температурный диапазон от -50 до 120 °С, скорость нагревания 20 K/мин. Образцы взвешивали в атмосфере воздуха или сухого N 2 . Проводили дополнительный термический анализ с помощью аппаратуры ТА Instruments Q1000 DSC, с применением герметических алюминиевых кювет при различных условиях. Динамическая сорбция пара (DVS). Количественное определение динамической сорбции пара касается определения массы воды, абсорбированной и в последующем десорбированной в ходе процесса кристаллизации. Для того чтобы определить, являются ли партии Р1, Р2 и Р4 гидратированными полиморфами, проводили измерения DVS (фиг. 9). Образец (от 13 до 14 мг) помещали на платиновую кювету и оставляли образец для уравновешивания при 25 °С/50% относительной влажности перед началом предварительно заданной программы изменения влажности (1,0 ч 50%, от 50 до 95% отн. вл.: 5% отн. вл./ч, 10 ч при 95% отн. вл., от 95 до 0% отн. вл.: 5% отн. вл./ч, 10 ч при 0% отн. вл., от 0 до 50% отн. вл.: 5% отн. вл./ч, 1 ч при 50% отн. вл. Растровая электронная микроскопия (SEM). Были сделаны снимки партий S-1 Р1, Р2 и Р4 (фиг. 8) с помощью системы SEM CamScan CS24. Фильтрация. Стадию фильтрации проводили в ходе следующих экспериментов: уравновешивание суспензии, эксперимент по осаждению, перекристаллизация, эксперименты по относительной стабильности, эксперимент по растворимости в воде. Центрифужные фильтрующие устройства: Ultrafree-CL (0.22 |a,m), Millipore; центрифуга типа Eppendorf 5804R были использованы при температуре 22 °С, с программой центрифугирования 2 мин 3000 об/мин. Высокоэффективная жидкостная хроматография (HPLC). HPLC использовали для анализа чистоты соединения S-1. Использовали хроматограф HP 1090M при следующих условиях: Колонка: Symmetry Shield RP18, 3,9 × 150 мм, 5 мкм Температура колонки: 35 °С Объем пробы: 10 мкл Растворитель: ацетонитрил + вода 1:1 об./об. Подвижная фаза А: 0,1% TFA - вода Подвижная фаза В: 0,1% TFA - ацетонитрил Скорость потока: 1 мл/мин Детектирование: УФ при 271 нм Время прогона: 21 мин Время удерживания (S-1): 10,7 мин. Материалы. Растворители. Во всех экспериментах использовали растворители марки Fluka или Merck. Вода: деионизированная (Fluka № 95305). Реактивы. Соединение S-1 было синтезировано, как описано в примере 1. Результаты. Четыре партии соединения S-1, обозначенные, соответственно, (S-1-P1), (S-1-Р2), (S-1-P3) и (S-1-P4), были выбраны для определения характеристик. S-1-P1, S-1-P2 и S-1-P3 представляли собой индивидуальные партии, приготовленные в ходе синтетического процесса, описанного в примере 1. Партия S-1-P4 представляла собой образец партии S-1-P1, которую хранили при 40 °C/75%. Были проведены следующие эксперименты для определения стабильности, растворимости и характеристик различных твердых форм соединения S-1. Следующая таблица представляет результаты дифракции рентгеновских лучей формы А соединения S-1, изображенные на фиг. 4А. На следующей таблице представлены результаты дифракции рентгеновских лучей формы А+С соединения S-1, изображенные на фиг. 12D, где были идентифицированы углы дифракции формы С В одном варианте реализации форма С имеет дополнительные линии, которые перекрываются сигналами от формы А. Проводили поиск пиков и вычисление величины d с помощью программы EVA, версии 10, 0, 0, 0; пик Cu K α2 удалялся программой, и в списке присутствовали только линии вплоть до 35 ° 2 θ. Измерения в образце РР148-Р1 производили на держателе образцов толщиной 0,1 мм на дифракторметре PANalytical PW1710. Измерения в образце РР148-Р52 производили на держателе образцов толщиной 0,1 мм на дифракторметре Bruker D8 Advance. Следующая таблица представляет результаты дифракции рентгеновских лучей формы D соединения S-1, изображенные на фиг. 18 (внизу): Сорбция паров воды (камера влажности). Вещество помещали в стеклянную пробирку при 96% отн. вл. (относительная влажность) в камеру влажности при комнатной температуре. Через различные промежутки времени хранения проводили измерения комбинационного рассеяния с использованием герметически закрытых стеклянных пробирок. Результаты обобщены в табл. 1. Измерения приблизительной растворимости. Для определения приблизительной растворимости при комнатной температуре добавляли пошагово растворитель к твердому веществу. После каждого добавления образец тщательно размешивали. Добавление растворителя продолжали до тех пор, пока не происходило полное растворение или пока не было добавлено 15 мл растворителя. Растворимость твердой формы А и В' при 23 °C представлена в табл. 2. Эксперименты по уравновешиванию суспензии. Эксперименты по уравновешиванию суспензии проводили с 81-128 мг вещества. Суспензию перемешивали на магнитной мешалке. Образцы, полученные после фильтрации, высушивали на воздухе при температуре окружающей среды в течение лишь небольшого периода времени для того, чтобы предотвратить возможную десольвацию лабильных гидратов или сольватов. Результаты экспериментов по уравновешиванию суспензии твердой формы А и В' представлены в табл. 3. Эксперименты по диффузии пара. Эксперименты по диффузии пара проводили с раствором вещества в различных растворителях. Растворы помещали в небольшие открытые контейнеры, которые хранили в больших сосудах, содержащих удобосмешиваемые, летучие антирастворители. Большие сосуды затем плотно закрывали. Антирастворители (осадители) диффундировали через паровую фазу в растворы, что приводило к насыщению или перенасыщению. Результаты экспериментов по диффузии пара твердой формы А и В' представлены в табл. 4. Эксперименты по испарению. Растворы вещества сушили при комнатной температуре (в сухом потоке азота) без перемешивания. Результаты экспериментов по испарению твердой формы А представлены в табл. 5. Эксперименты по осаждению. Эксперименты по осаждению проводили с 42-79 мг вещества. В раствор добавляли осадитель. Образцы, полученные после фильтрации (стеклянный фильтр, пористость Р4), сушили на воздухе при температуре окружающей среды, в течение небольшого периода времени, с целью предотвращения возможной десольвации лабильных гидратов или сольватов. Результаты экспериментов по осаждению твердой формы А представлены в табл. 6. Перекристаллизация из раствора. Вещество растворяли в различных системах растворителей при комнатной температуре и охлаждали до +5 или до -20 °С. Образцы, полученные после фильтрации (стеклянный фильтр, пористость Р4), сушили на воздухе при температуре окружающей среды в течение лишь небольшого периода времени с целью предотвращения возможной десольвации лабильных гидратов или сольватов. Результаты экспериментов по перекристаллизации твердой формы А представлены в табл. 7. Эксперимент по сублимационной сушке. Вещество растворяли в 1,4-диоксане и раствор охлаждали до -50 °С. В ходе сублимации растворителя температура твердого вещества составляла <0 °С, что отображено в табл. 8. Эксперимент по высушиванию. Образец сушили в течение ночи в атмосфере сухого N 2 при комнатной температуре, после чего закрывали DSC-кювету с образцом. Результаты обобщены в табл. 9. Эксперименты по охлаждению и повторному нагреванию расплава. После нагревания в DSC до 120 °С образцы охлаждали до -50 °С и снова нагревали до 120 °С. Результаты обобщены в табл. 10. Эксперименты по относительной стабильности. Эксперименты с суспензией проводили с 130-145 мг вещества. Суспензии перемешивали на магнитной мешалке и фильтровали через заранее заданный промежуток времени. Образцы, полученные после фильтрации (стеклянный фильтр, пористость Р4), сушили на воздухе при температуре окружающей среды. Результаты обобщены в табл. 11. Растворимость в воде твердых форм А и В'. Суспензии твердых форм (25 или 50 мг в 3,5 или 7,0 мл бидистиллированной воды) встряхивали (800 об/мин) и фильтровали через 0,5, 1,5, 4 и 20 ч. После фильтрации твердый остаток исследовали с помощью спектроскопии комбинационного рассеяния, а концентрации в прозрачных растворах определяли с помощью HPLC. Растворимость в воде твердой формы А соединения S-1 при 22 °C приведена в табл. 12. a) Среднее значение двух измерений ( ± стандартное отклонение) b) По результатам спектроскопии комбинационного рассеяния c) Грубая оценка d) pH раствора 8,7 Растворимость в воде формы В' соединения S-1 при 22 °C приведена в табл. 13. a) Среднее значение двух измерений ( ± стандартное отклонение) b) По результатам спектроскопии комбинационного рассеяния c) Грубая оценка d) pH раствора 8,7 Характеристика формы А соединения S-1-P1. Исходное вещество для исследования полиморфизма, партия № S-1-P1, является кристаллическим и представляет собой кристаллическую форму А. По данным TG-FTIR, потеря массы вплоть до 200 °С очень невелика ( <0,2%), и таким образом партия № S-1-P1 не является гидратом или сольватом. Вещество из партии № S-1-P1 плавится при 82 °С (максимальная температура DSC, скорость нагревания 20 K/мин). После плавления и быстрого охлаждения до -50 °С в DSC была получена безводная жидкокристаллическая форма. Температура фазового перехода образца была определена как примерно 52 °С и не наблюдалась перекристаллизация образца при нагревании в DSC. Вещество S-1-P1 могло содержать небольшое количество (по грубым оценкам 5%) формы В' или В". Измерения DVS формы А при 25 °С не дают каких-либо доказательств образования классического гидрата в условиях эксперимента. Максимальное содержание воды при относительной влажности 93% составляет 1,5%. Очень слабо выраженный гистерезис, по всей вероятности, вызван вязким слоем (вероятно, состоящим из твердой формы В') на поверхности частиц, который влияет на скорость обмена воды. Действительно, в результате хранения формы А при относительной влажности 96% при комнатной температуре в течение 11 недель спектроскопия комбинационного рассеяния и DSC показали образование примерно 20% формы В'. Характеристика твердой формы В'. Исследования с помощью DSC и XRPD указывают на то, что твердая форма, которая образуется при хранении твердой формы А при 40 °С и 75% относительной влажности (партия S-1-P4; 40 °С/75% отн. вл.), является псевдокристаллической формой с ограниченным ближним порядком. По всей вероятности, именно этот ограниченный порядок обуславливает эндотермальный пик в DSC в районе 55 °С и широкое плечо в районе 17 ° в дифракционной картине. Твердая форма партии S-1-P4 при 40 °С/75% относительной влажности представляет собой форму В'. Характеристика твердой формы В' при DVS при 25 °С представляет собой нетипичную сорбционную характеристику гидрата. Максимальное содержание воды при 94% относительной влажности составляет примерно 2,4%. Даже несмотря на то, что определенно наблюдается гистерезис, на сорбционной кривой нет четкой стадии, которая бы однозначно указывала на существование классического гидрата. Образование твердой формы В'. В дополнение к наблюдаемой трансформации при высокой относительной влажности твердую форму В' можно получить путем перемешивания суспензии твердой формы А в воде при 37 °С в течение ночи. Образование твердой формы В". Способами получения твердой формы В" являются плавление и охлаждение расплава, а также медленное выпаривание растворов в таких растворителях, как этанол. Полиморфную модификацию В" можно получить из полиморфов А и D путем их нагревания до температуры выше их точек плавления, соответственно, 80 и 130 °С. Формы В' и В" неразличимы каким-либо аналитическим методом, использованным до сих пор, но их можно различить на основании пути их образования. В' определяется как лиотропная жидкокристаллическая форма из-за ее образования с участием растворителя, в то время как В" определяется как термотропная жидкокристаллическая форма, получаемая термальным способом. Выпаривание из растворителя лекарственной формы, например этанола, без осадителя также приводит к образованию В". Образование твердой формы С. Полиморфная модификация С может быть получена только в виде смеси с формой А путем растворения и последующего выпаривания лекарства из THF при температуре окружающей среды. Образование твердой формы D. Полиморфная модификация D была изначально получена путем кристаллизации из смеси растворитель/осадитель при 50 °С с помощью этилацетата и циклогексана в качестве растворителя и осадителя соответственно. Форму D можно также получить из других полиморфных форм путем "засевания" образца небольшим количеством D и его хранения при 110 °С/0% отн. вл. в течение 7 дней или при 50 °С в воде в течение 24 ч и высушивания. Образование твердой формы сольвата толуола. Сольват толуола был получен любым способом кристаллизации с помощью растворителя/осадителя, где в качестве осадителя применяли толуол. Растворимость в воде твердых форм А и В'. Растворимость форм А и В' соединения S-1 в воде при 22 °С составляет 24,0 ±1,4 мг/1000 мл и 27,3 ± 0,8 мг/1000 мл; данные значения получены на момент времени уравновешивания суспензии, составляющий 1,5 ч. Эти значения растворимости очень близки из-за быстрой трансформации формы А в форму В' на поверхности частиц в ходе экспериментов по растворимости. Характеристика различных партий твердой формы А. Для образцов партий S-1-P1, S-1-P2 и S-1-P3 характерна одинаковая дифракционная картина. Измерения DSC показывают, что они с наибольшей вероятностью содержат некоторый процент твердой формы В' или В", на что указывают изменения теплоемкости в районе 50 °С. Для образца S-1-P2 характерно наибольшее содержание твердой формы В' или В" (примерно 20%). Для лучшего понимания результатов DSC были получены микрофотографии на растровом электронном микроскопе (SEM) образцов S-1-P1 и S-1-P2. В то время как изображения образца S-1-P1 указывают на довольно хорошо сформированные частицы, изображения образца S-1-P2 показывают частичную трансформацию, вероятно вызванную слишком высокой температурой высушивания или частичным контактом с водой. Частичное образование твердой формы В' или В" могло быть также вызвано быстрым осаждением и относительно высоким соотношением осадитель/растворитель после осаждения. Другими объяснениями могут быть высушивание при высоких температурах или хранение в условиях высокой влажности. Системы растворителей для кристаллизации твердой формы А. Кристаллическая форма А обладает высокой растворимостью в ряде растворителей, обычно используемых для кристаллизации. Ввиду ее высокой растворимости, для кристаллизации необходимы смеси растворитель/осадитель. В ходе экспериментов по уравновешиванию суспензии при комнатной температуре было обнаружено, что твердая форма В' (партия S-1-P4; 40 °С/75% отн. вл.) может быть трансформирована в твердую форму А при перемешивании суспензий в этилацетате/гептане 1:2 об./об. или этилацетате/пентане 1:2. Кроме того, эксперименты по уравновешиванию суспензий с использованием твердой формы А в этилформиате/пентане 1:2 об./об. и метилацетате/пентане 1:2 об./об. показали отсутствие трансформации твердой формы А. Таким образом, подобные смеси растворитель/осадитель класса 3 можно применять для кристаллизации формы А. Преимуществом таких систем растворителей является значительно более низкая температура кипения и, следовательно, более низкие, по всей вероятности, температуры высушивания. Подробные характеристики соединения S-1-P1, твердой формы А, даны в табл. 14. Подробные характеристики соединения S-1-P2, твердой формы А, приведены в табл. 15. Подробные характеристики соединения S-1-P3, твердой формы А, приведены в табл. 16. Подробные характеристики соединения S-1-P4, твердой формы В', приведены в табл.17. В различных партиях P1, P2 и P3 соединения S-1 выявлена кристаллическая форма А с одинаковыми характеристиками по результатам XRPD, спектроскопии комбинационного рассеяния, TG FTIR, DVS и DSC. В партии Р4 выявлена псевдокристаллическая твердая форма, охарактеризованная по результатам XRPD, спектроскопии комбинационного рассеяния, TG FTIR, DVS и DSC, приведенным выше в настоящем описании. Относительная стабильность полиморфных форм в сухих условиях. Термограмма DSC для А и D на фиг. 19 показывает, что А плавится около 80 °С, тогда как точка плавления D находится около 130 °С. Энтальпия плавления для А составляет 40 ±5 Дж/г, в то время как энтальпия плавления для D составляет 75 ±5 Дж/г. Температура плавления и энтальпия плавления свидетельствуют о том, что D обладает большей стабильностью, по сравнению с формой А. Фиг. 17D показывает, что плавление полиморфных модификаций А или D приводит к образованию жидкокристаллического полиморфа В" вместо истинно изотропной жидкой фазы. Образование истинной жидкой фазы не наблюдалось даже после нагревания образца до 200 °С. Охлаждение полиморфа В" до температуры окружающей среды не приводило к перекристаллизации обратно в форму А или D. Это подтверждается отсутствием эндотермы плавления на кривой DSC (фиг. 17D) образца, повторно нагретого после того, как он был последовательно расплавлен и охлажден до температуры окружающей среды. Кривая DSC также показывает, что форма В" претерпевает фазовый переход в районе 55 °С. Подобное стеклование наблюдается для В', что в совокупности с широким плечом в районе 17 ° на фиг. 4D является основанием для их отнесения к жидкокристаллическим фазам. По результатам XRPD видны гармонические пики для В' в совокупности с широким плечом в районе 17 ° на фиг. 4D, что является основанием для их отнесения к жидкокристаллическим фазам. Фиг. 17Е показывает, что нагревание полиморфов А и В" до 110 °С в присутствии D вызывает превращение форм А и В" в форму D. Это является подтверждением того, что А и В" являются метастабильными фазами при температуре ниже 130 °С, которые могут переходить в форму D. Тем не менее, вероятно из-за высокого энергетического барьера перехода, скорости превращения А или В" в D очень малы при изначальном отсутствии какого-либо количества D в качестве затравки для кристаллизации. Таким образом, формы А и В" можно считать практически стабильными при температуре окружающей среды. При температуре выше 130 °С форма D плавится и переходит в В", которая теперь становится наиболее устойчивой формой. Микронизация частиц полиморфной модификации А в сухих условиях также приводит к ~25%-ному превращению в В". Относительная стабильность полиморфных форм во влажных условиях. Полиморфная модификация А остается стабильной в своей форме А по меньшей мере в течение 7 дней в условиях хранения при температуре окружающей среды /75% отн. вл. (относительной влажности), температуре окружающей среды/100% отн. вл., 30 °С/75% отн. вл. и 50 °С/0% отн. вл. Но она превращается в В', если ее хранить при 50 °С/75% отн. вл. Некоторые результаты отображены на фиг. 17F. В действительности полиморфные модификации А, которые хранили при 25 °С/60% отн. вл. и 30 °С/65% отн. вл., были стабильны в течение 36 месяцев и 9 месяцев, соответственно, в то время как образец, который хранили при 40 °С/75% отн. вл., превращался в В' в течение одного месяца. Эти результаты указывают на то, что полиморфная модификация А переходит в В' в присутствии влаги. Полиморфная модификация D, с одной стороны, остается стабильной при 50 °С/75% отн. вл., равно как и в других условиях: темп. окр. среды/75% отн. вл., темп, окр. среды/100% отн. вл., 30 °С/75% отн. вл. и 50 °С/0% отн. вл. Действительно, полиморфная модификация D в присутствии влаги действует как затравка для процесса кристаллизации и вызывает трансформацию полиморфных модификаций А и В' в D, по аналогии с ее ролью в качестве затравки для А при ее кристаллизации в D в сухих условиях. Фиг. 17G(a) показывает изменение во времени полиморфной модификации А, содержащей затравку небольшим количеством D при 50 °С/75% отн. вл. Количество полиморфа D, изначально добавленного к образцу, настолько мало, что оно не детектируется в DSC при скорости нагревания 10 °С/мин. Через 24 ч большая часть полиморфной формы А превращается в В', но небольшое количество образца также превращается в D, и количество образца в модификации D возрастает со временем. На фиг. 17G(b) представлен ускоренный процесс трансформации при хранении образца в воде при 50 °С. Форма А превращается в обе формы В' и D через 6 ч, но образец преимущественно находится в форме D через 24 ч. В этом состоит отличие от превращения в В' чистой формы А, которая далее не превращается в D. До сих пор неясно, может ли А непосредственно переходить в D с помощью затравки в воде, или же она превращается только в В' (которая затем превращается в D в воде). Дальнейшие исследования показали, что А и В' переходят в D в присутствии влаги при более низких температурах, хотя и при более низких скоростях. Относительная стабильность солъвата толуола в толуоле. Перекристаллизация вещества S-1 из системы растворитель/осадитель, где применяется толуол в качестве осадителя, приводит к получению сольвата толуола. Точка плавления сольвата толуола лежит около 100 °С, а энтальпия плавления составляет 70 ±5 Дж/г. График TGA сольвата толуола на фиг. 3 показывает, что содержание толуола в сольвате составляет ~7%, что соответствует одной молекуле толуола на каждые три молекулы S-1. Отношение масс растворителя/лекарства оставалось неизменным для каждой полученной партии, и это свидетельствует о том, что остатки молекул толуола находятся внутри структуры элементарной ячейки, а не в каналах или слоях за пределами пространственной решетки. Из-за низкой растворимости S-1 в толуоле ( <2 мг/мл), значимая трансформация из формы D в сольват толуола не наблюдалась после суспендирования (50 мг/мл) в толуоле в течение 4 дней, как при температуре окружающей среды, так и при 50 °С. Обработка суспензии ультразвуком в течение 10 мин все же приводило к частичной трансформации в сольват толуола. Квалифицированному в данной области техники специалисту будет понятно, что настоящее изобретение не ограничивается тем материалом, который был частично представлен и описан выше. Напротив, объем изобретения определяется следующей формулой изобретения. |