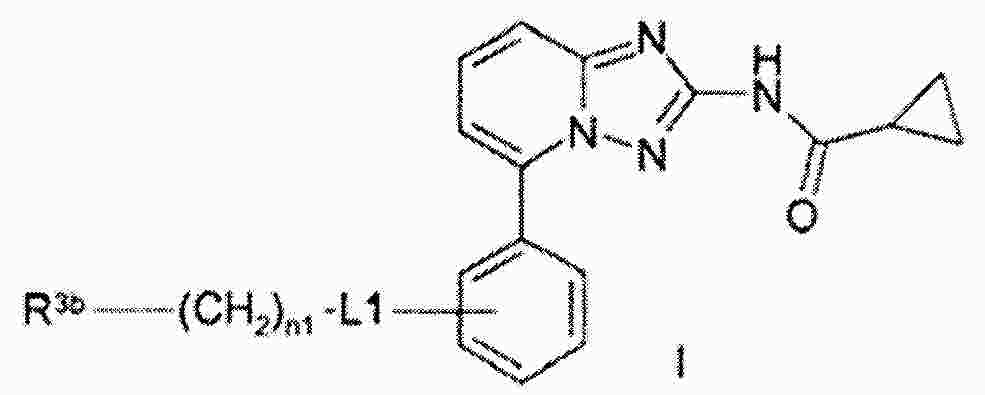
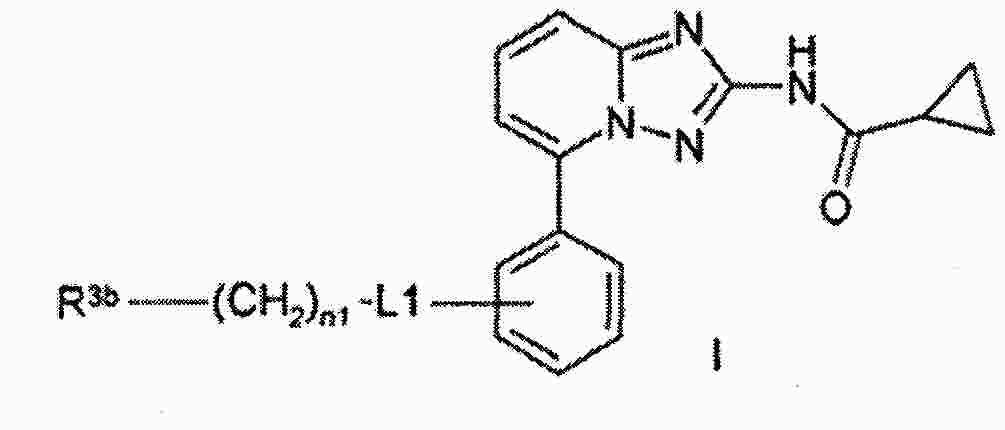
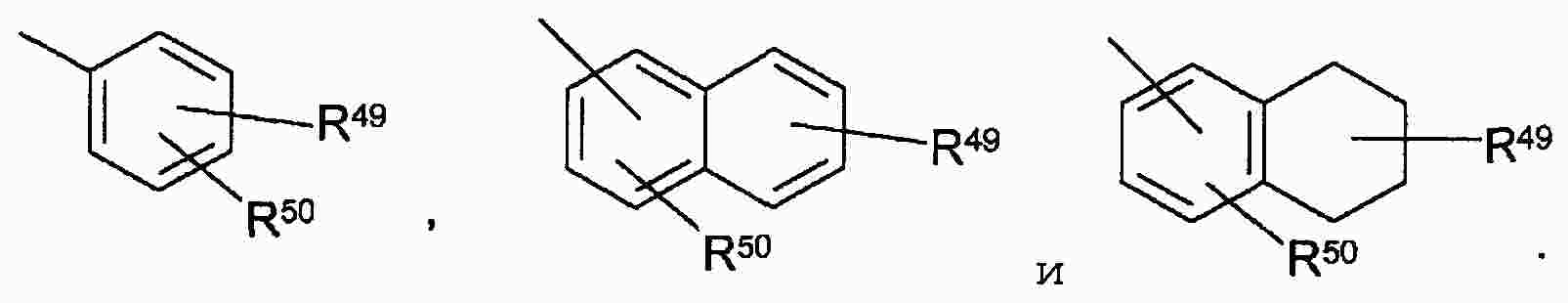
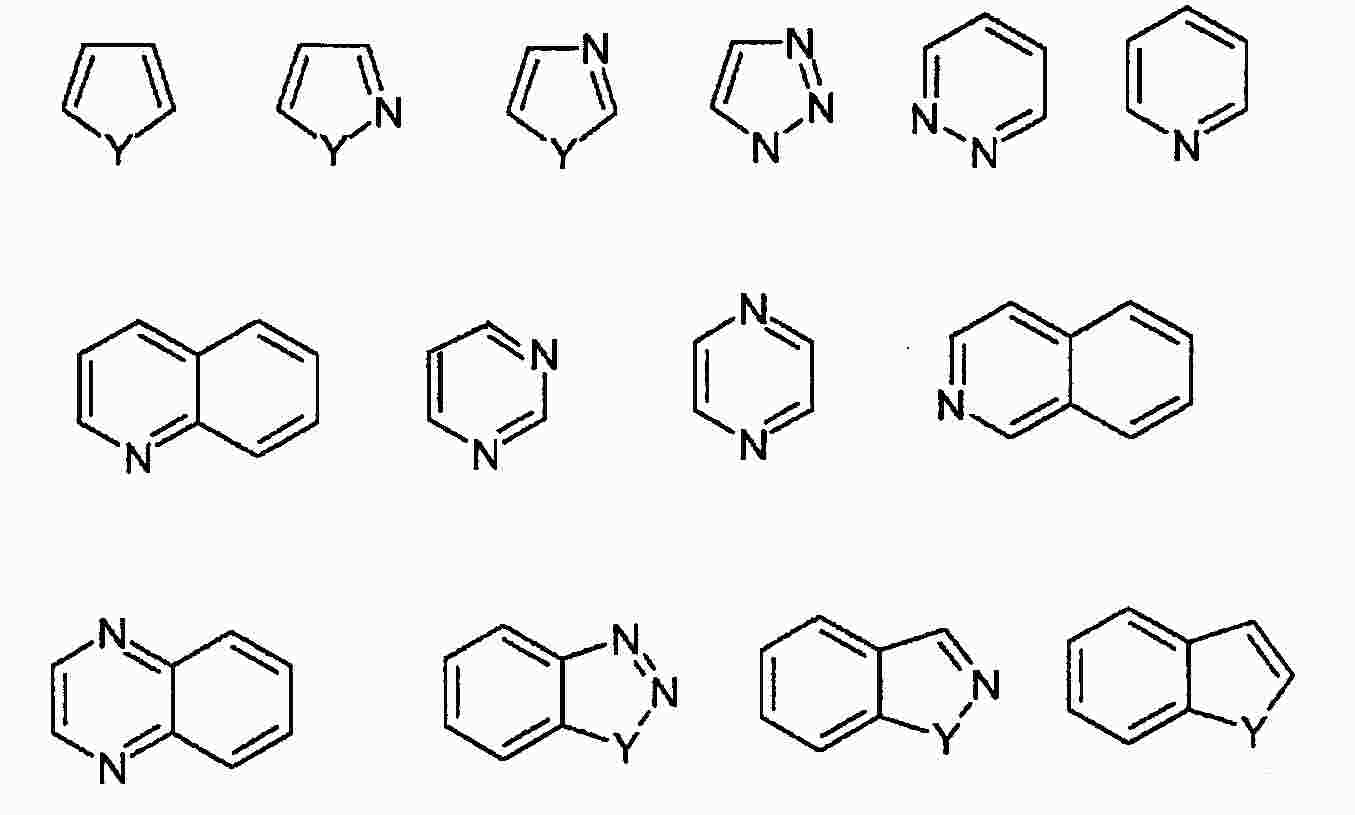
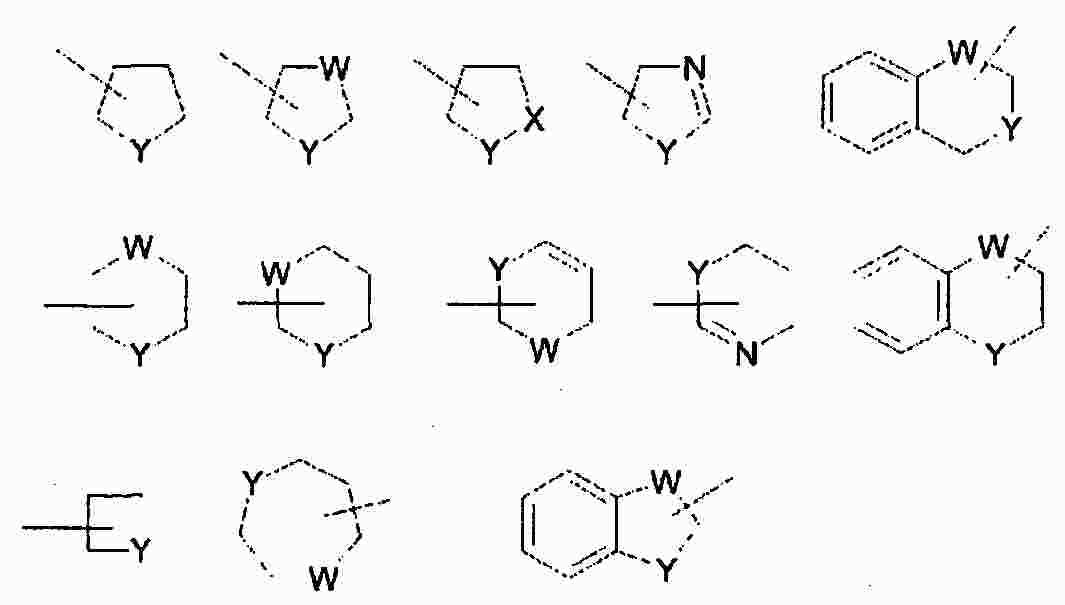
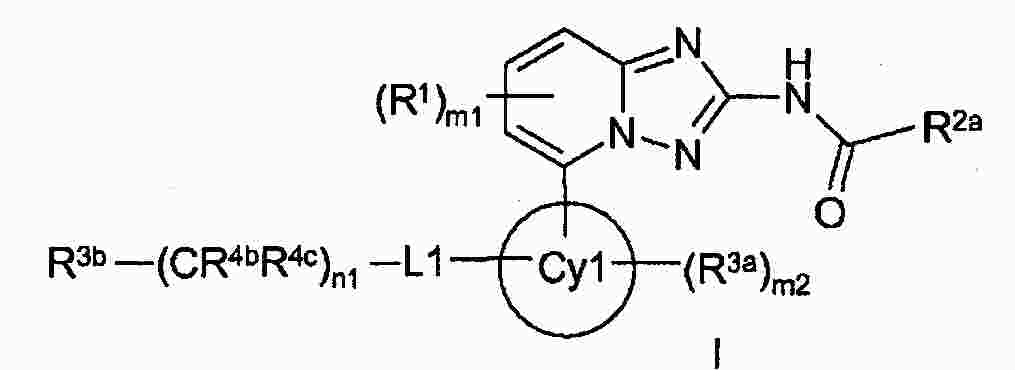
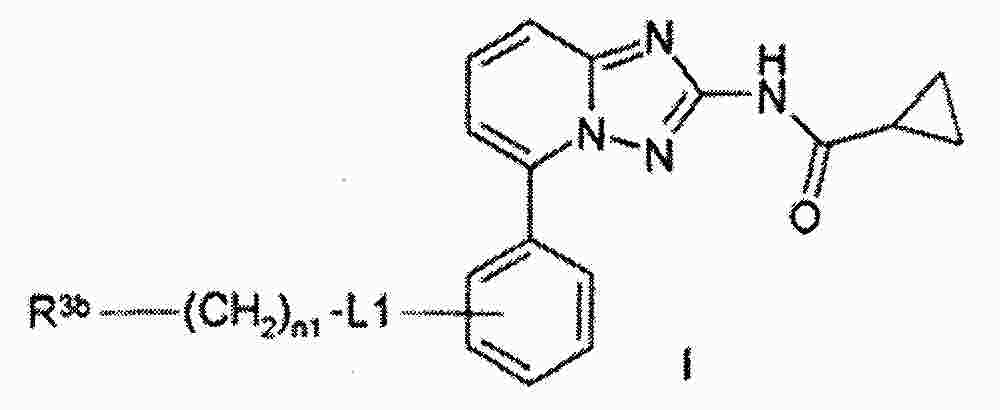
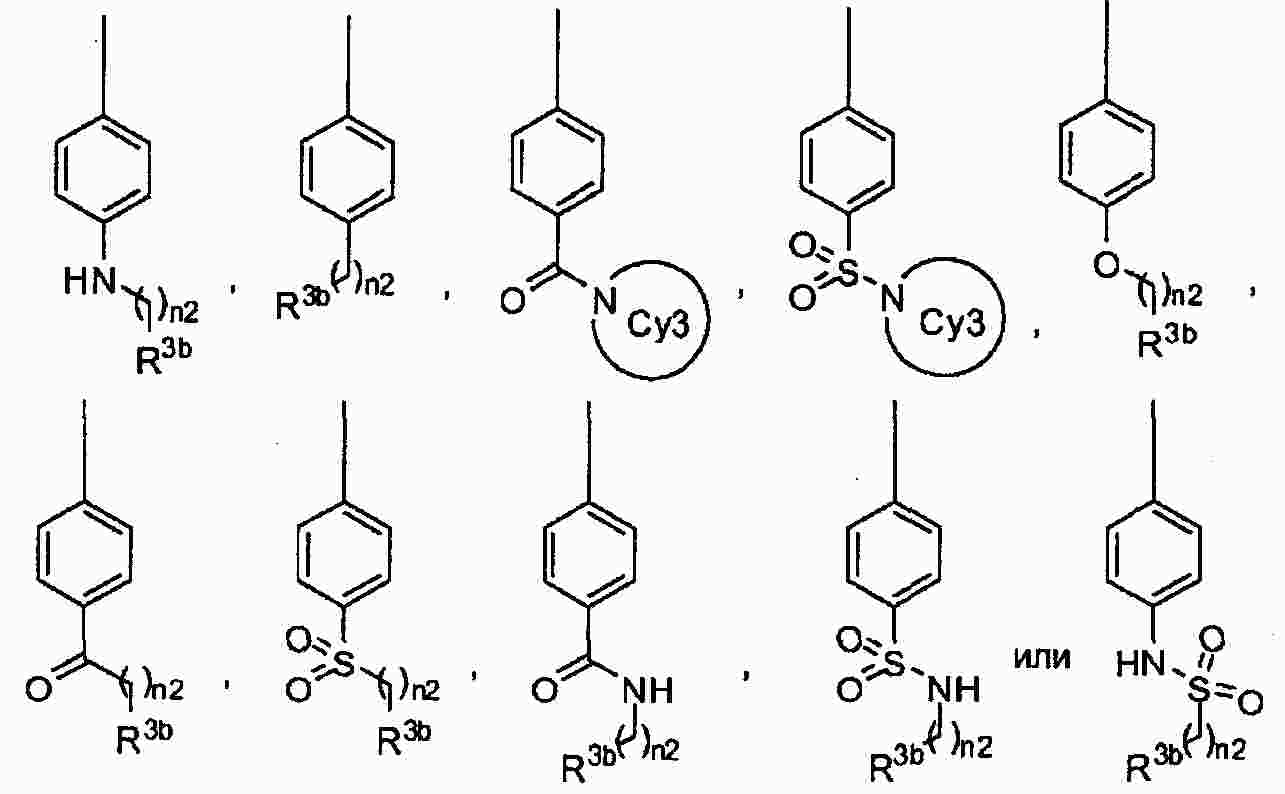
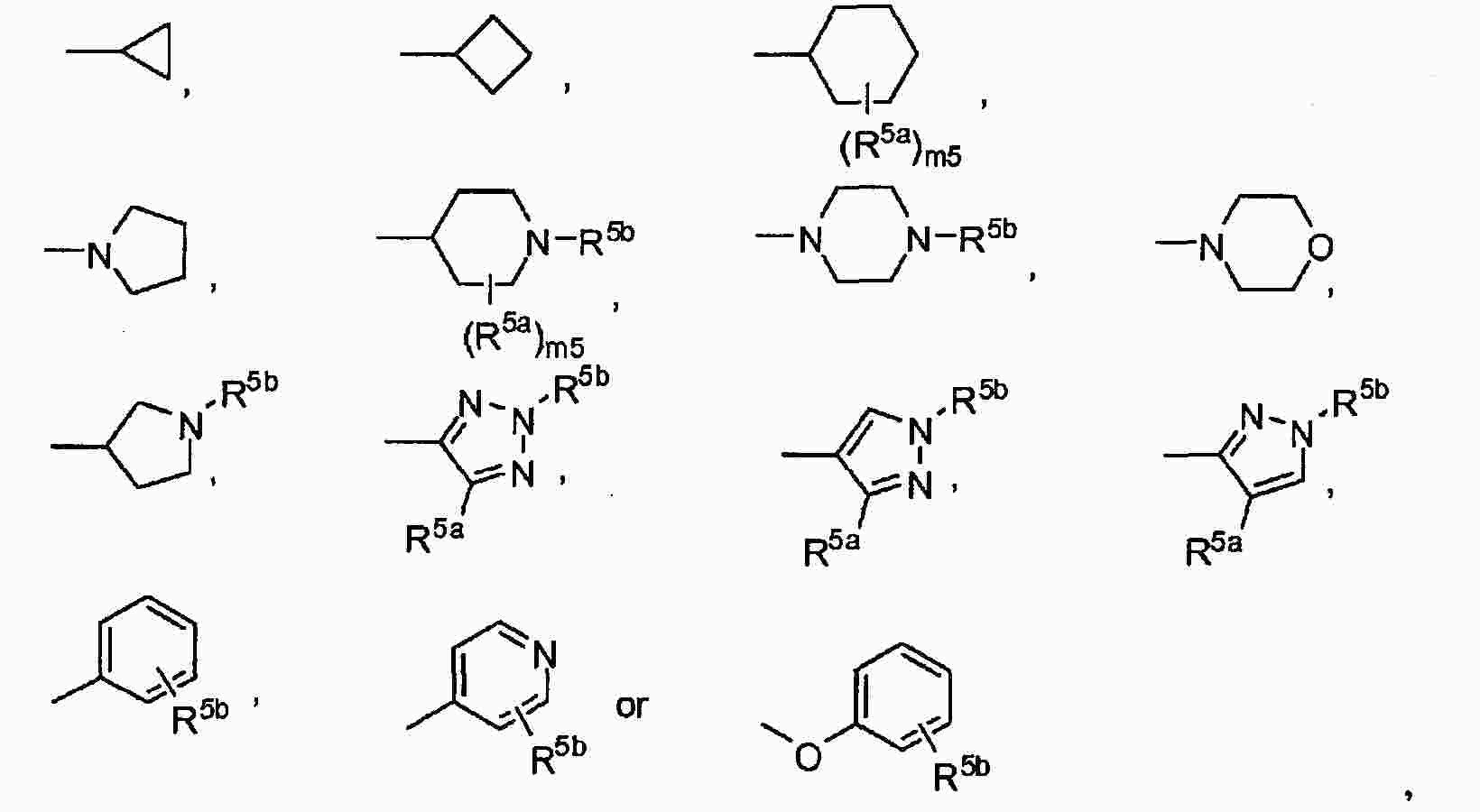
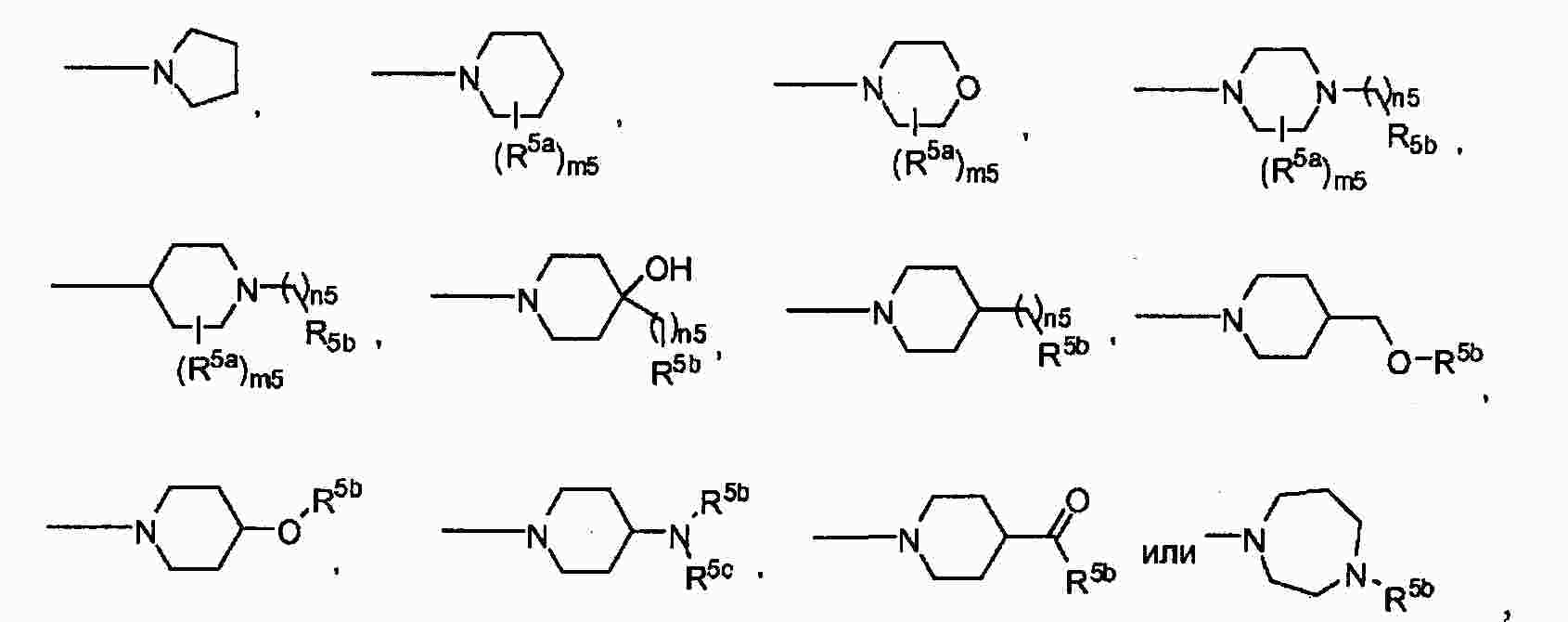
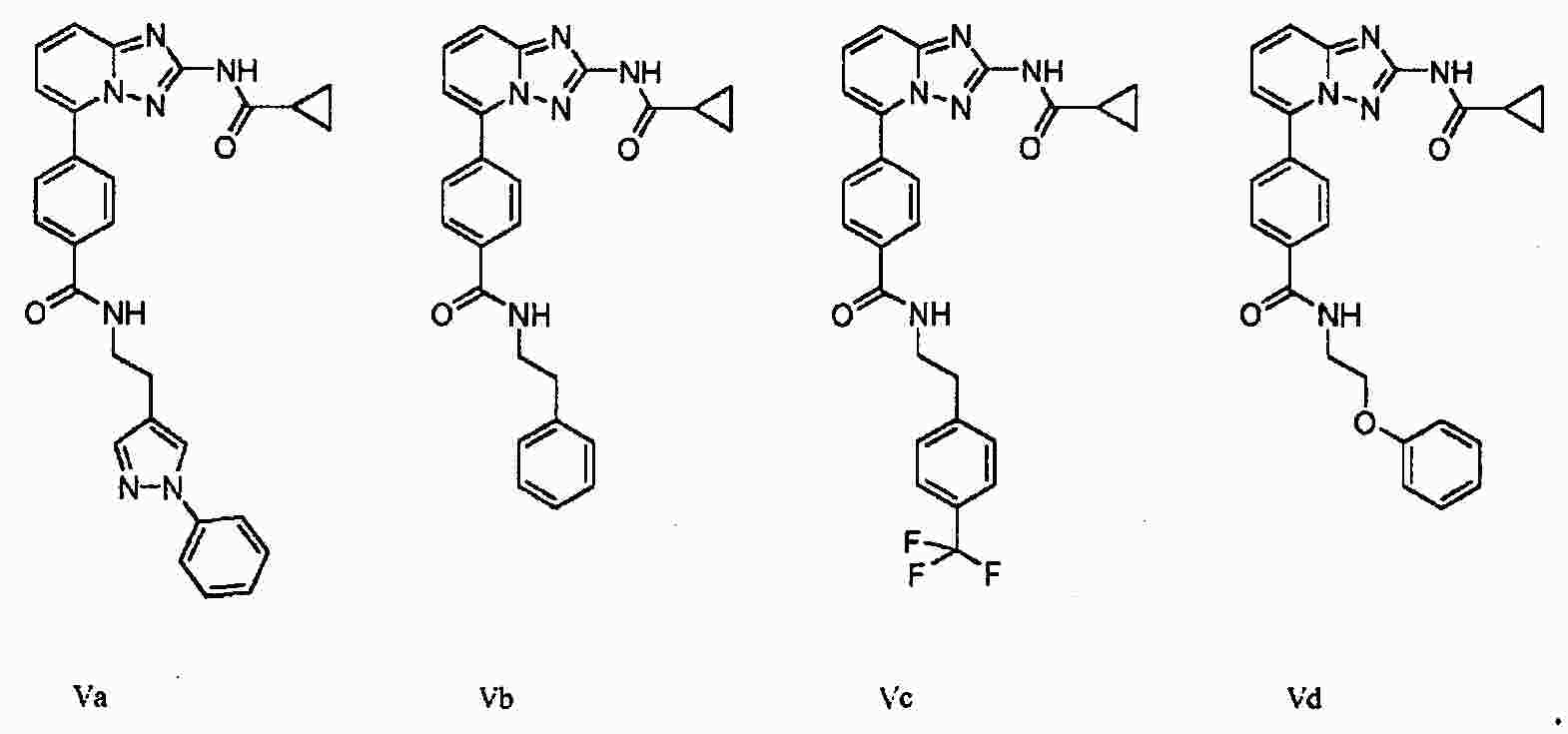
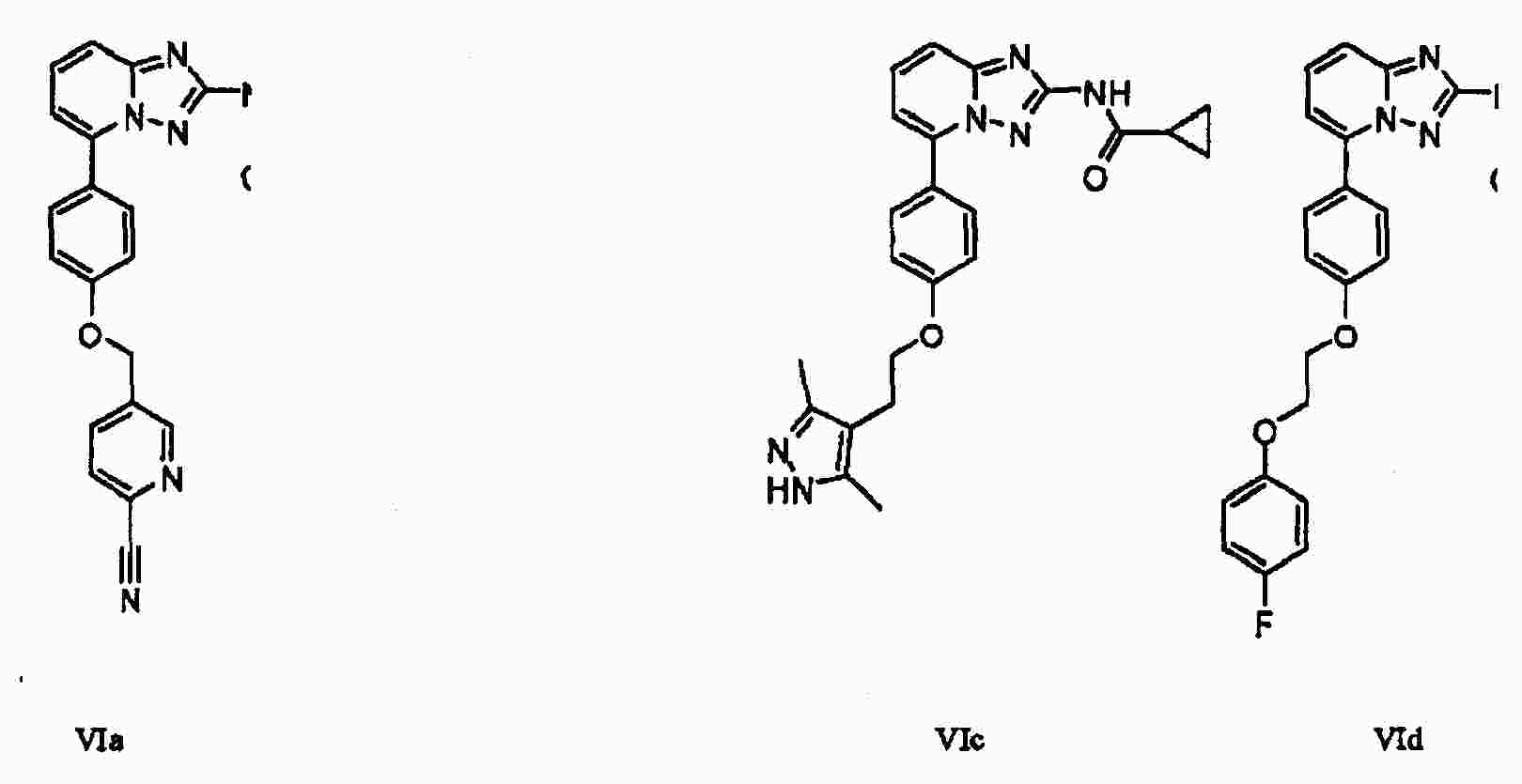
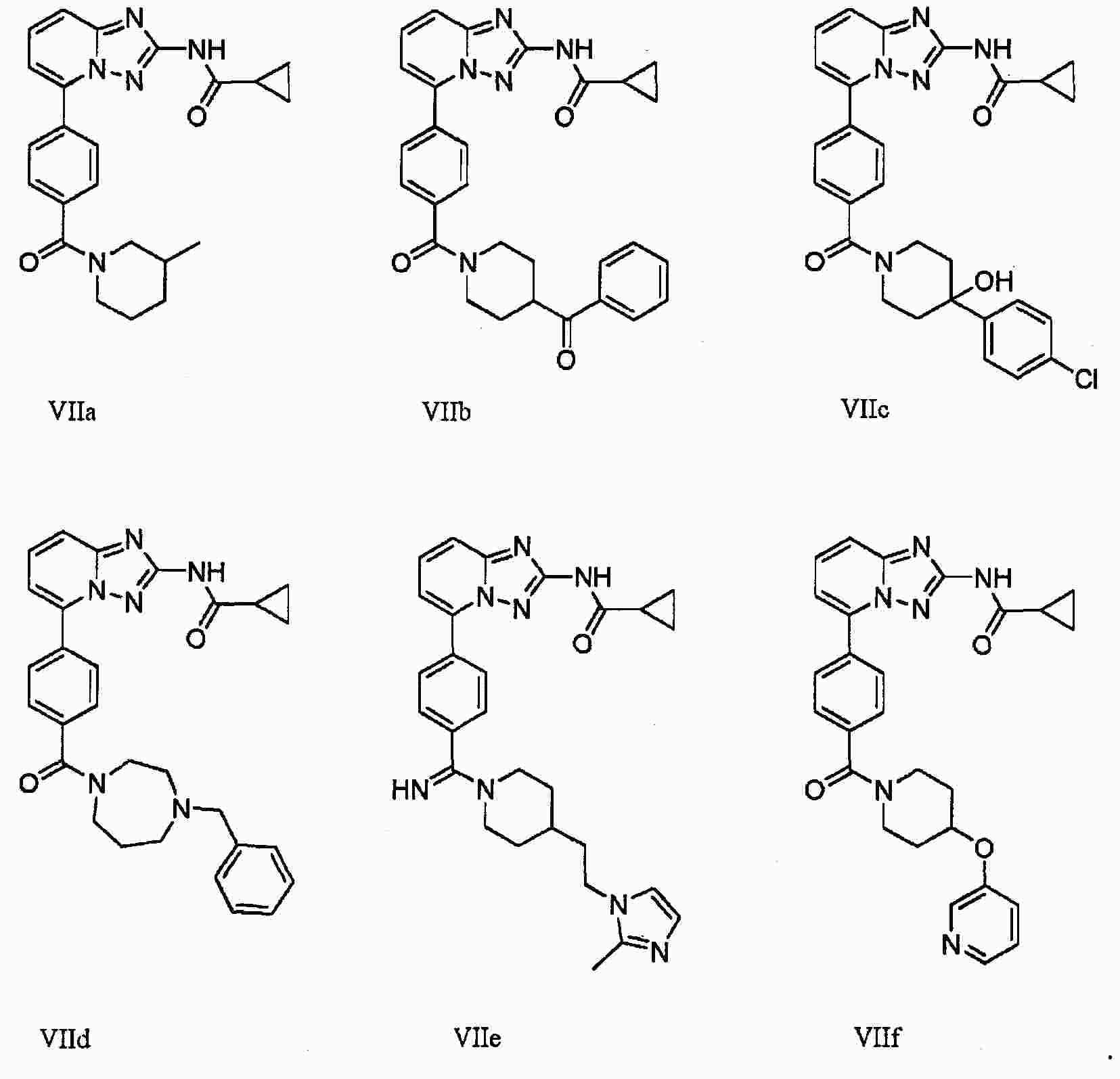
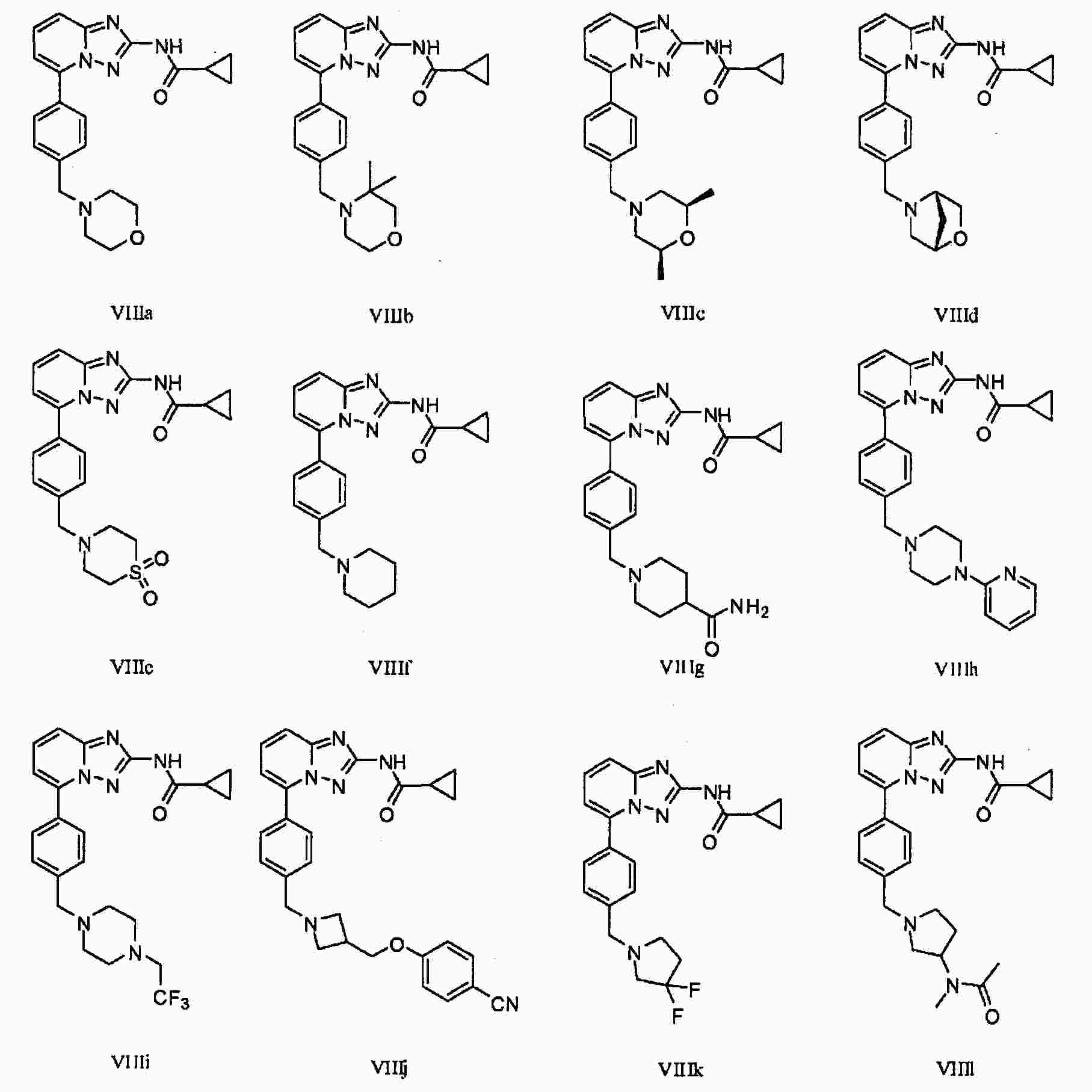
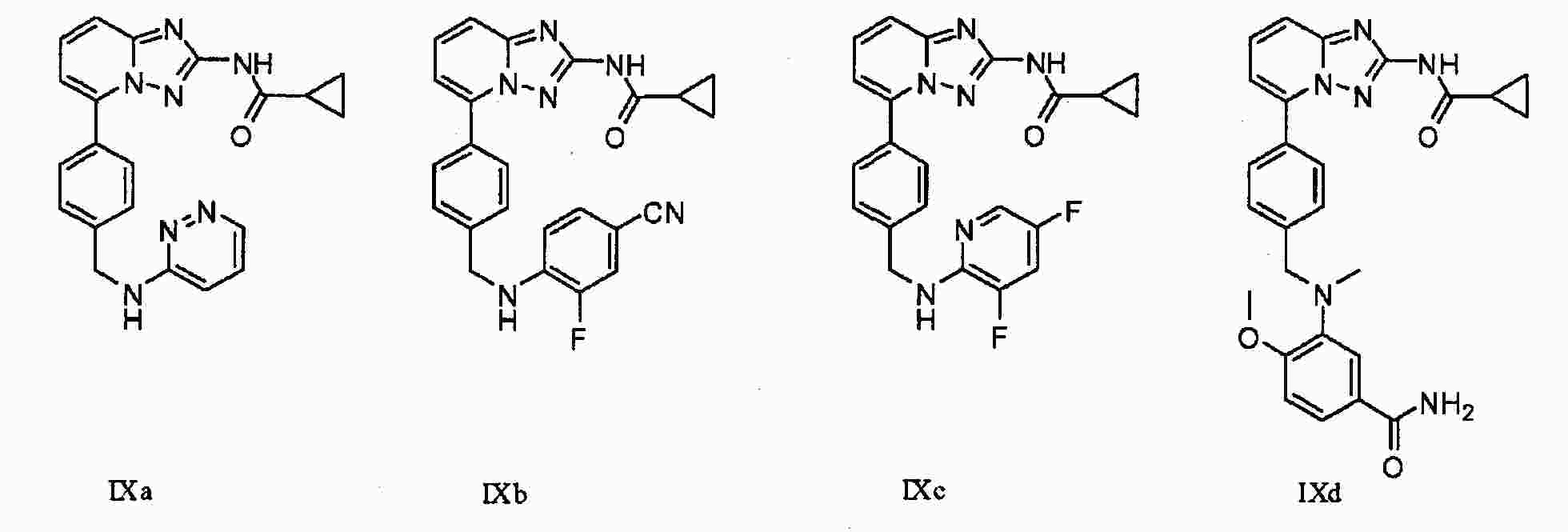
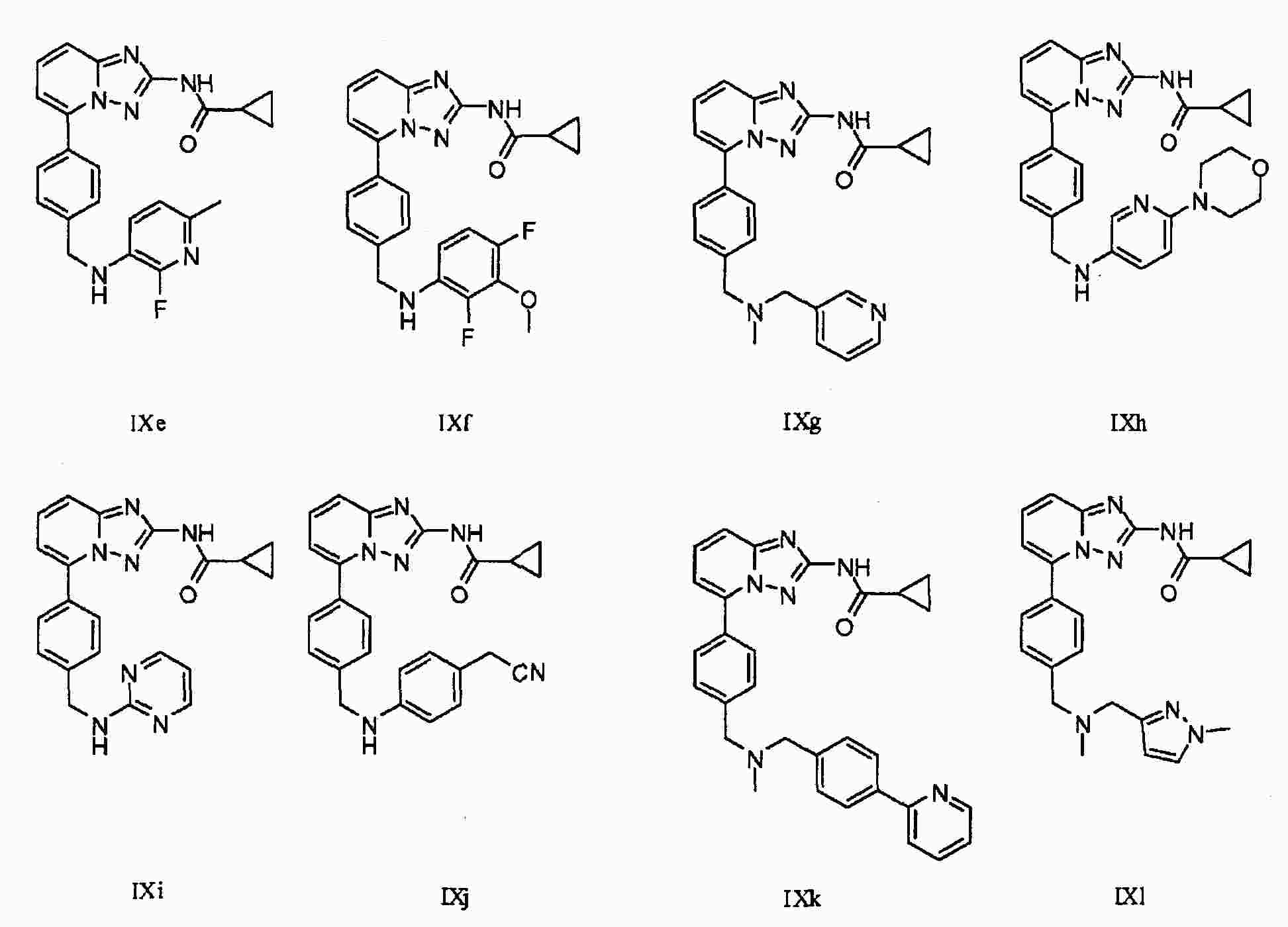
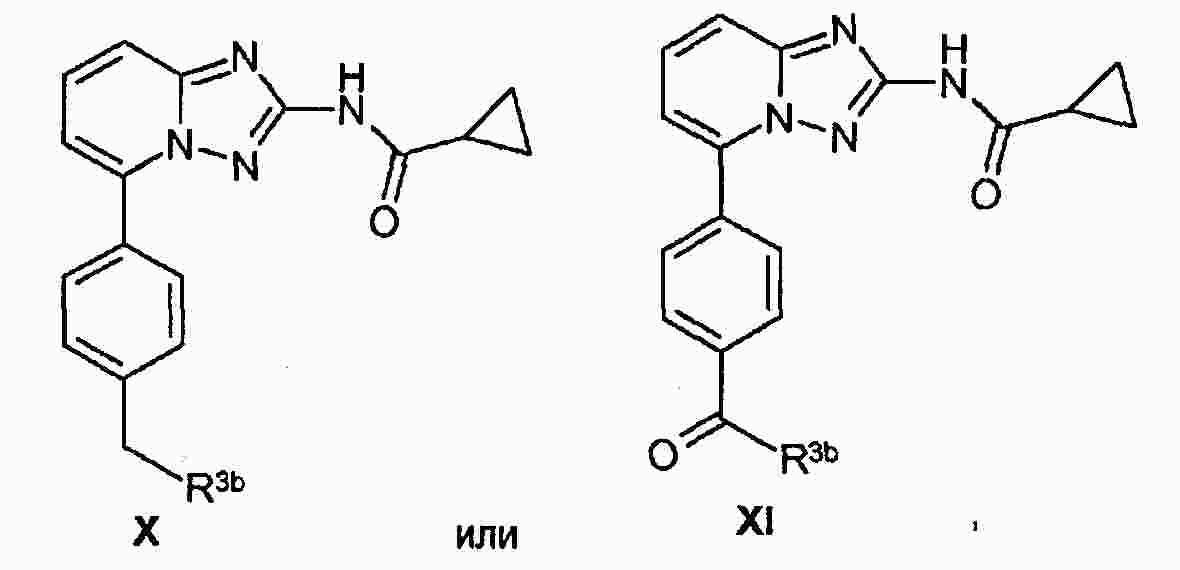
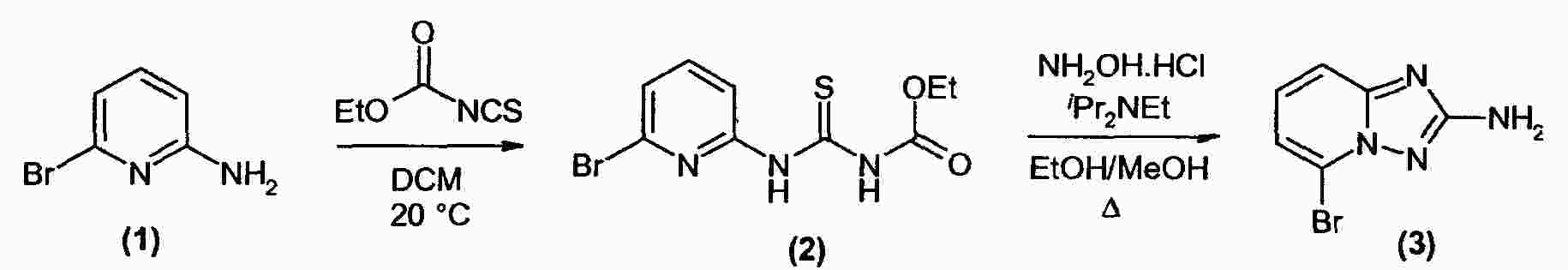
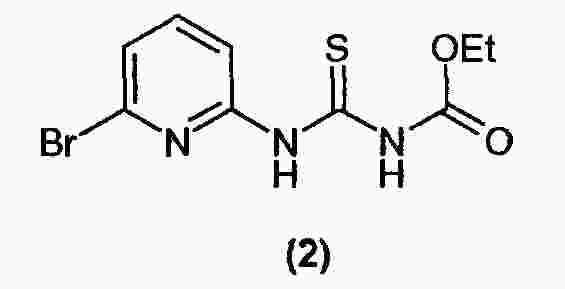
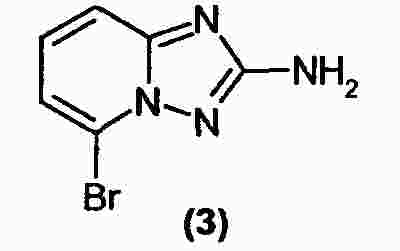
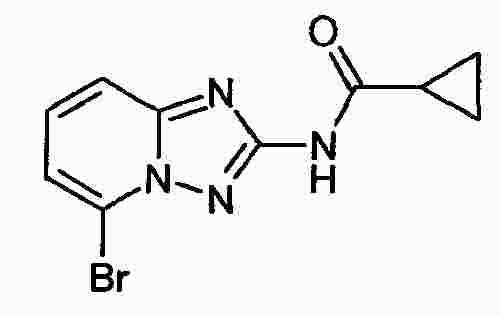
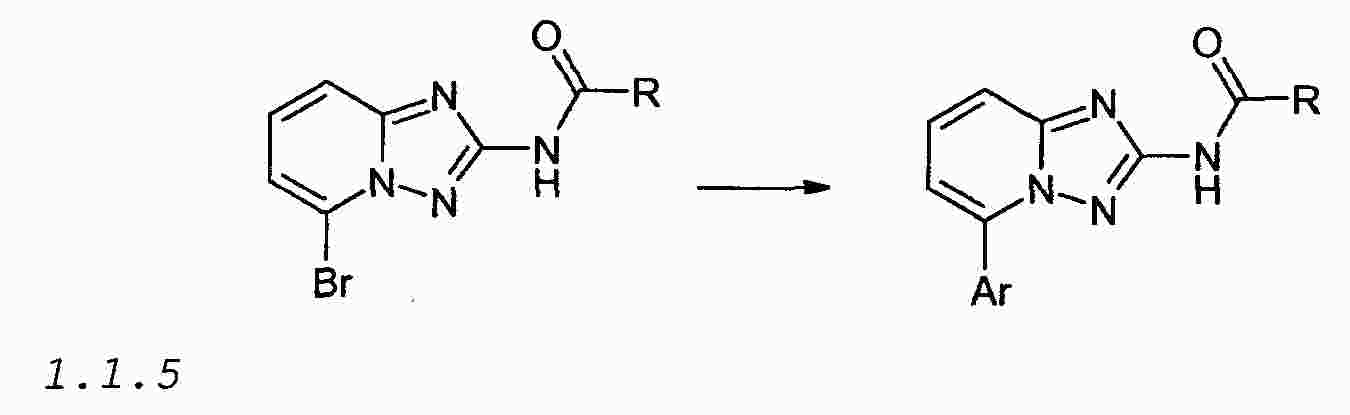
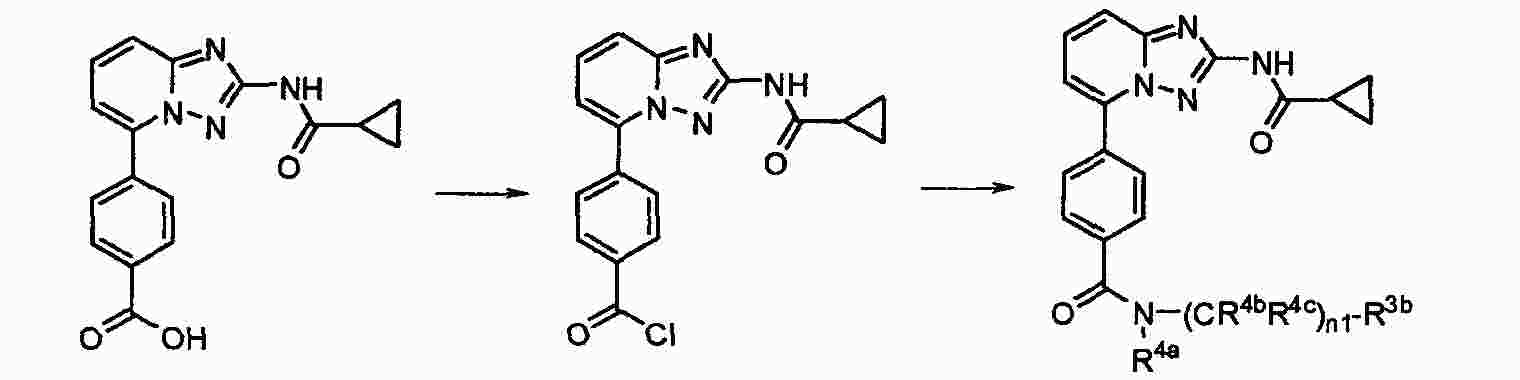
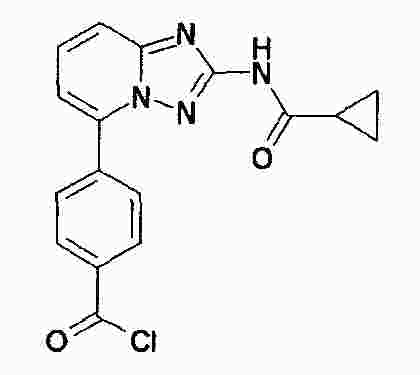
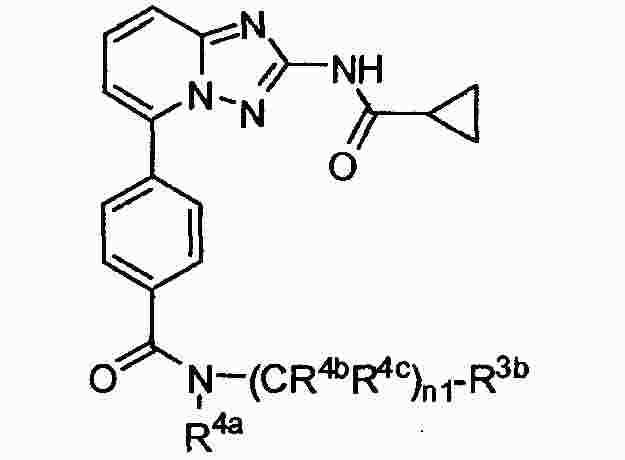
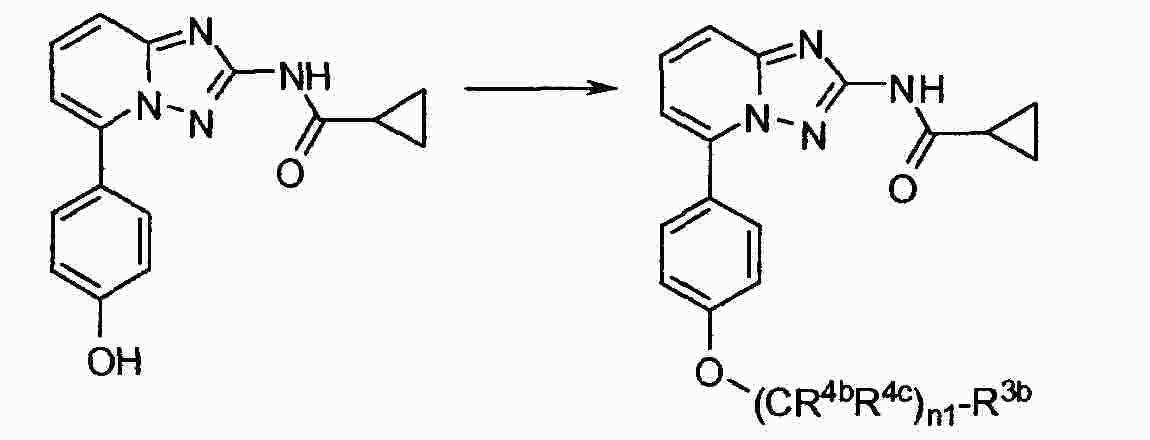
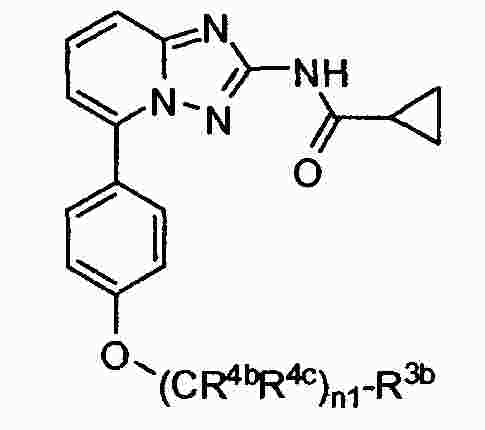
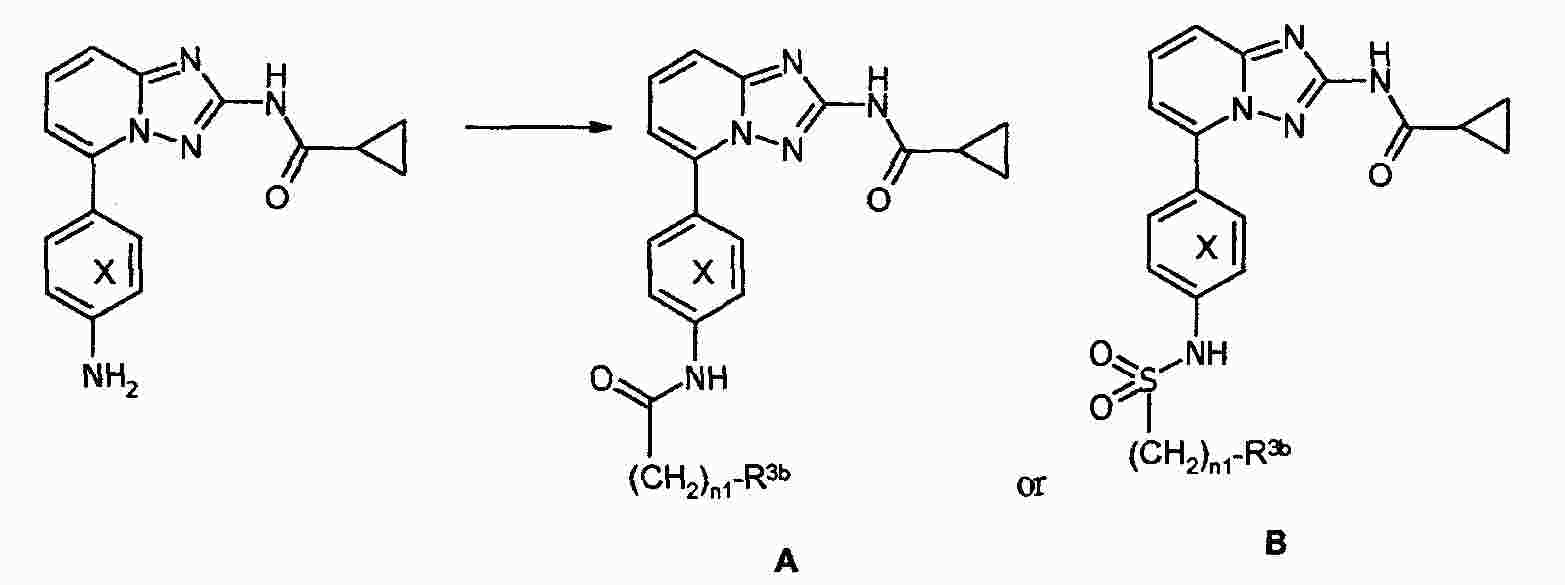
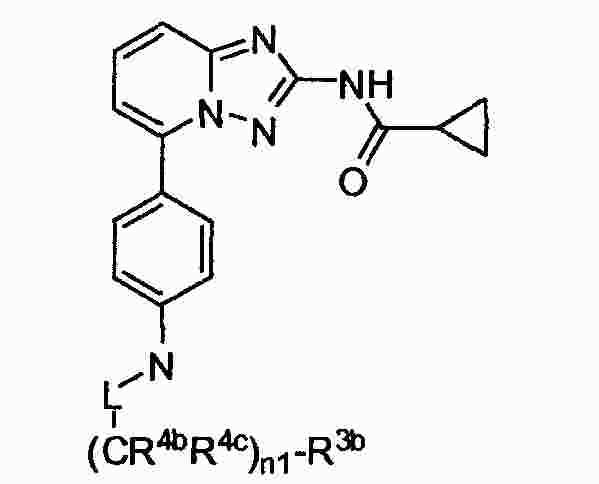
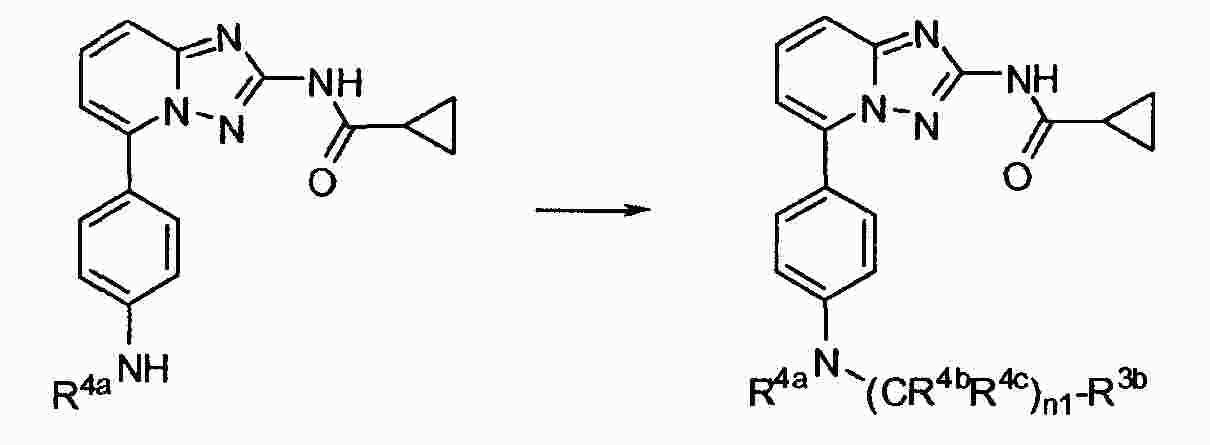
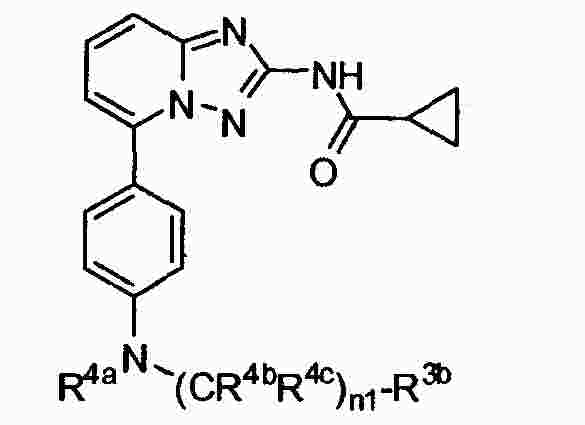
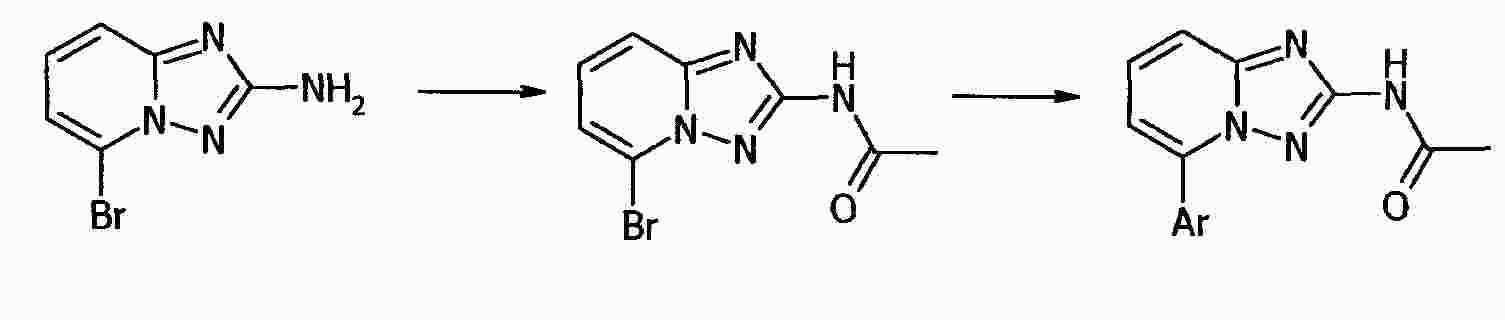
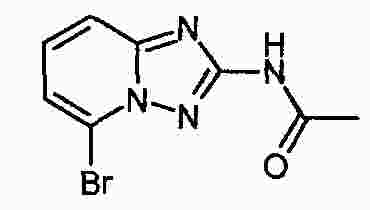
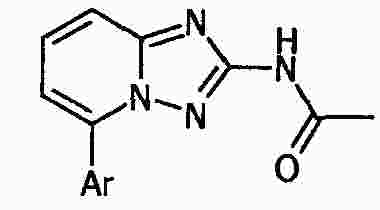
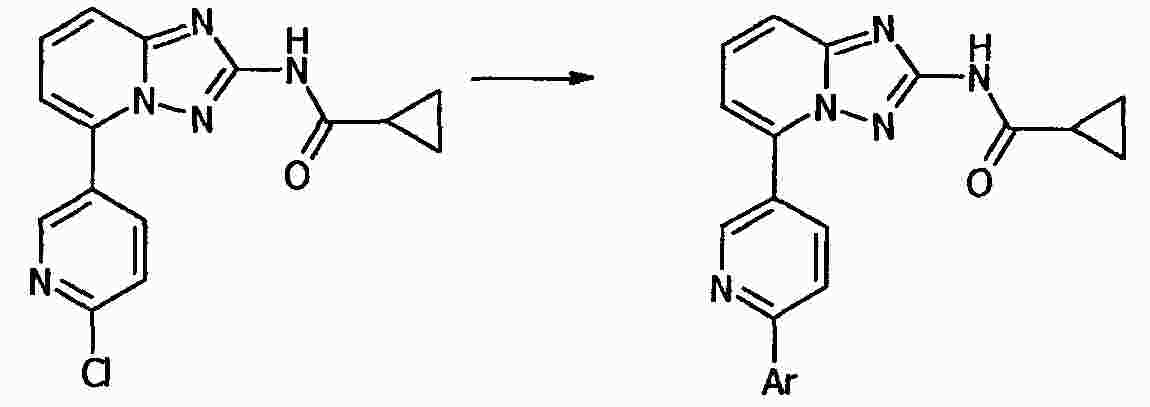
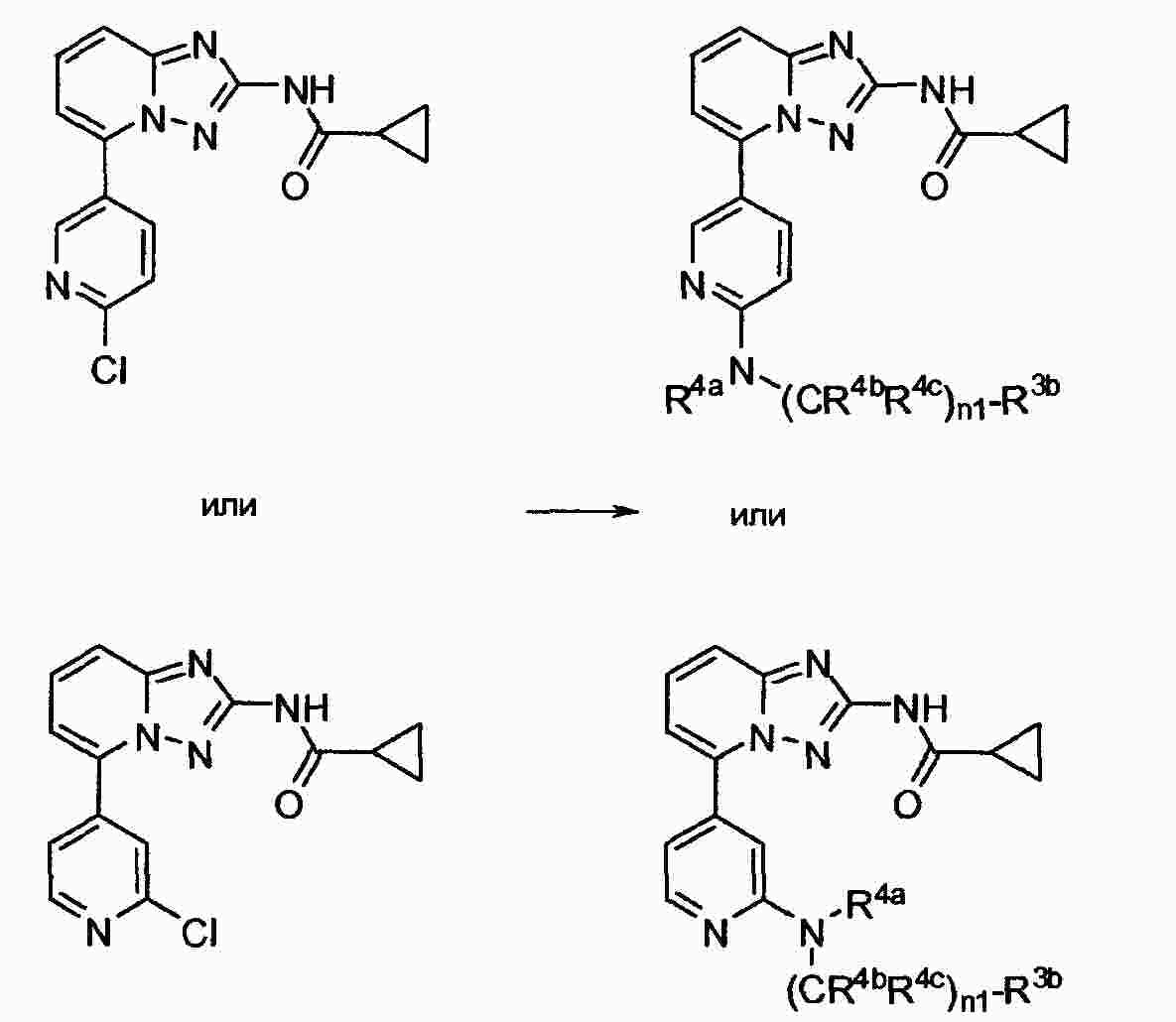
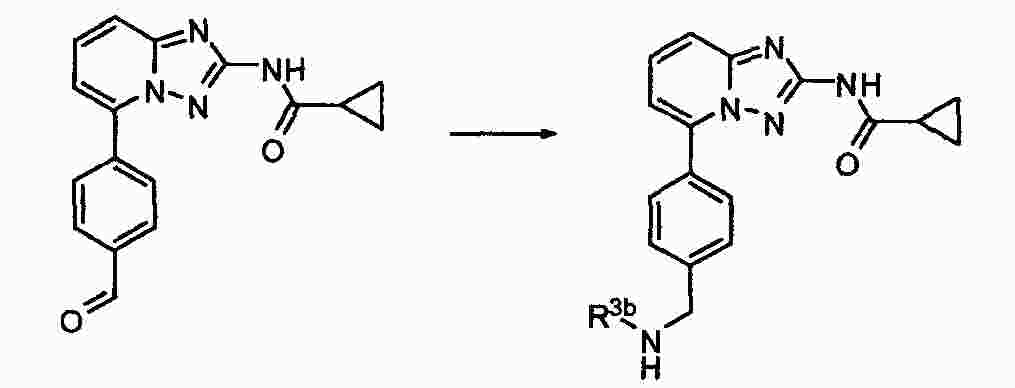
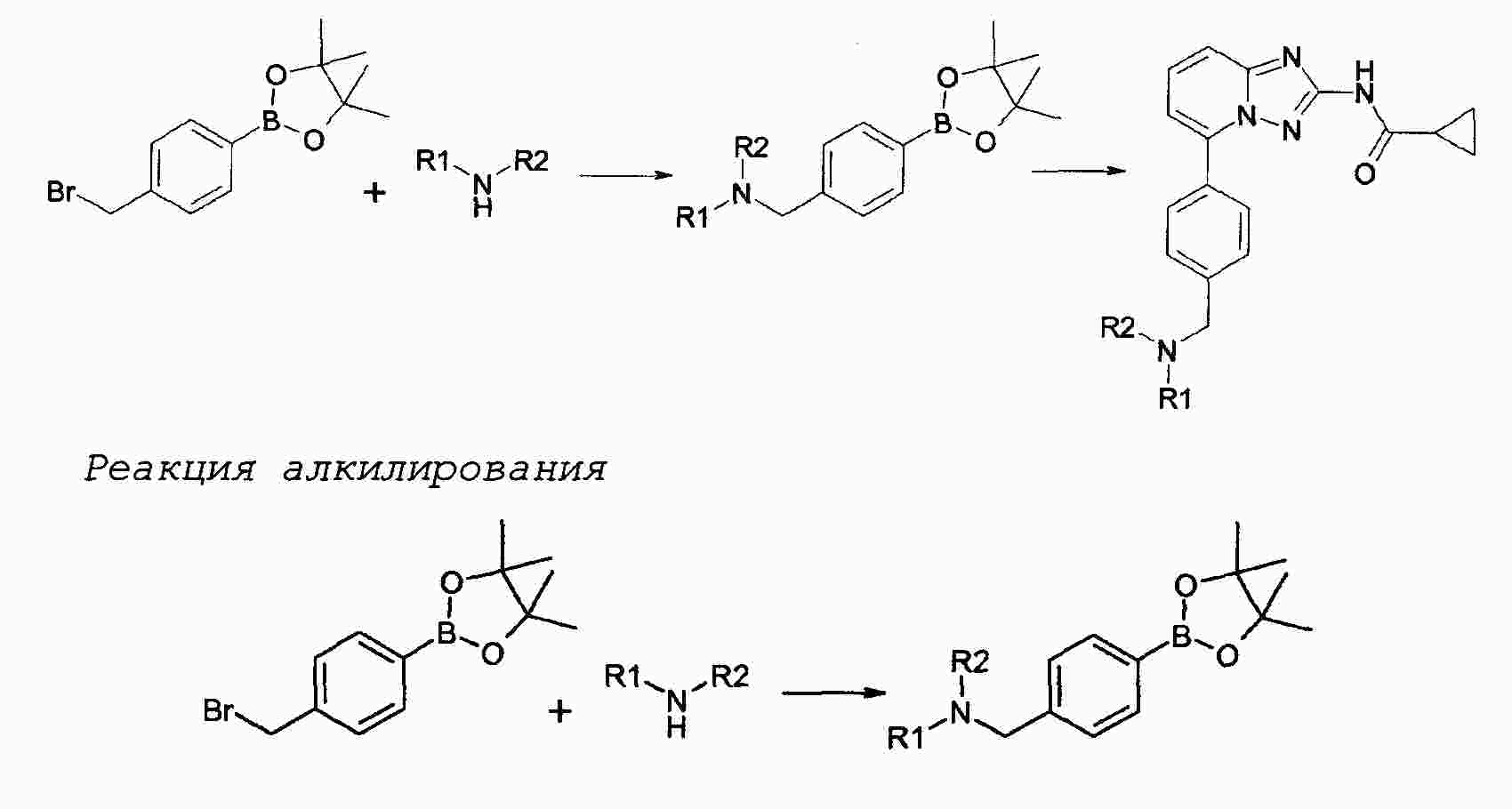
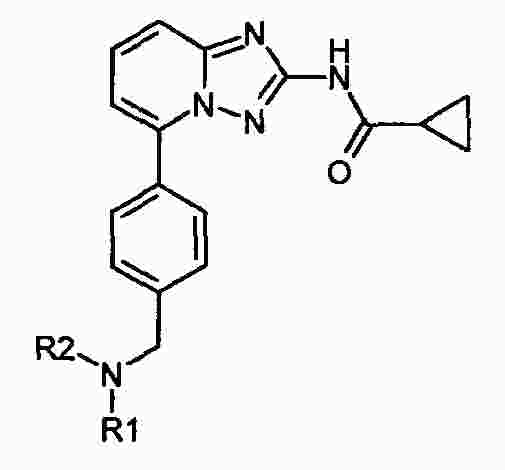
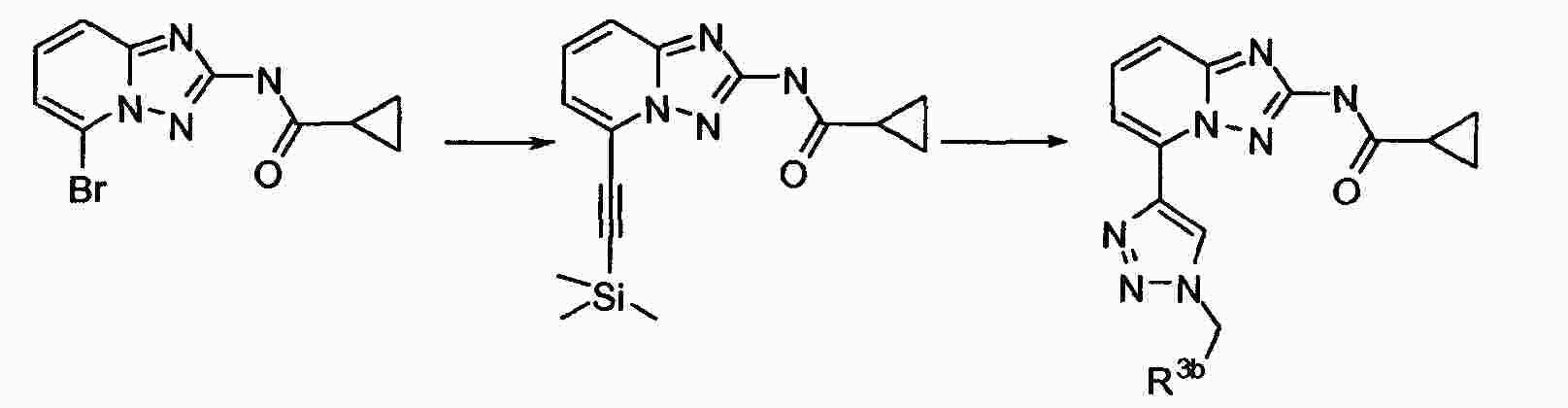
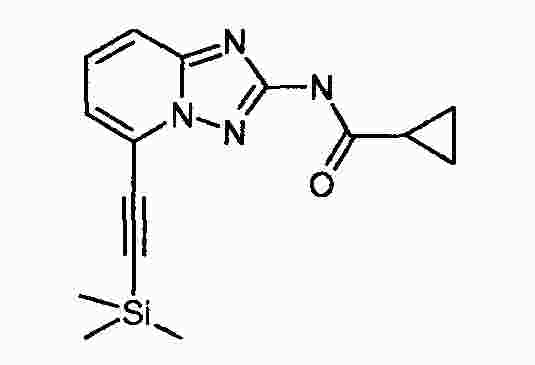
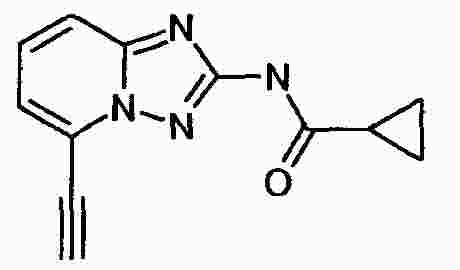
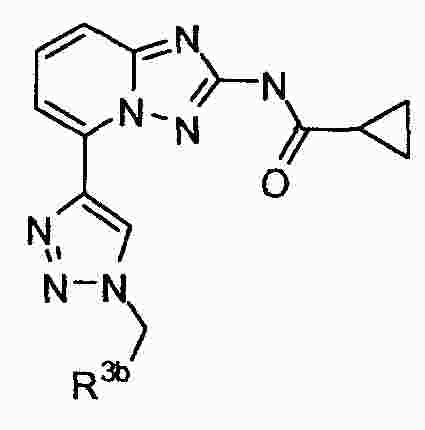
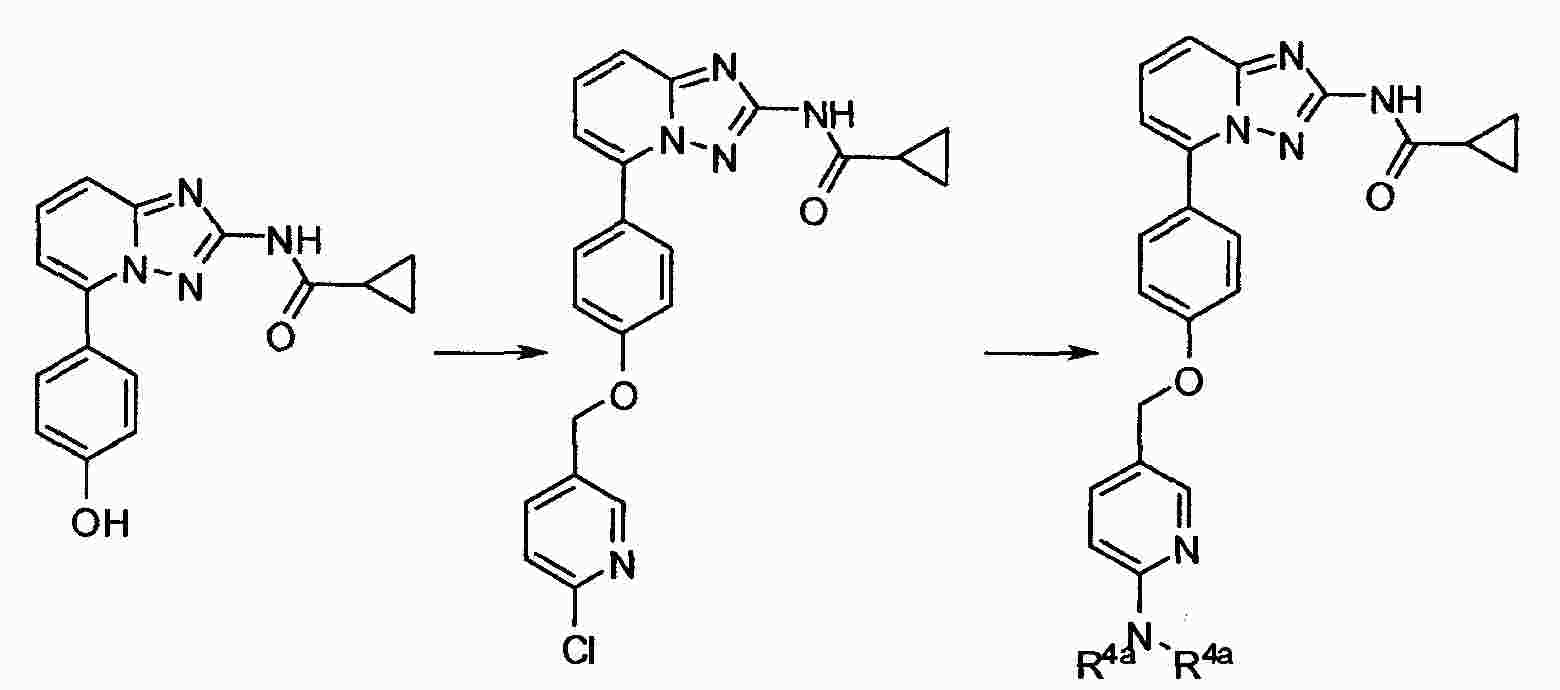
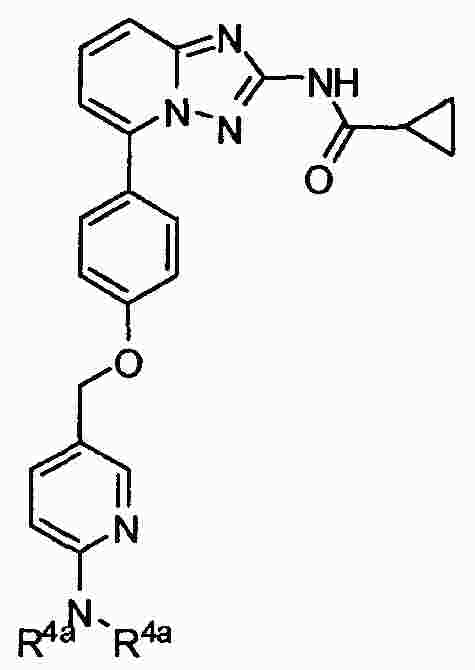
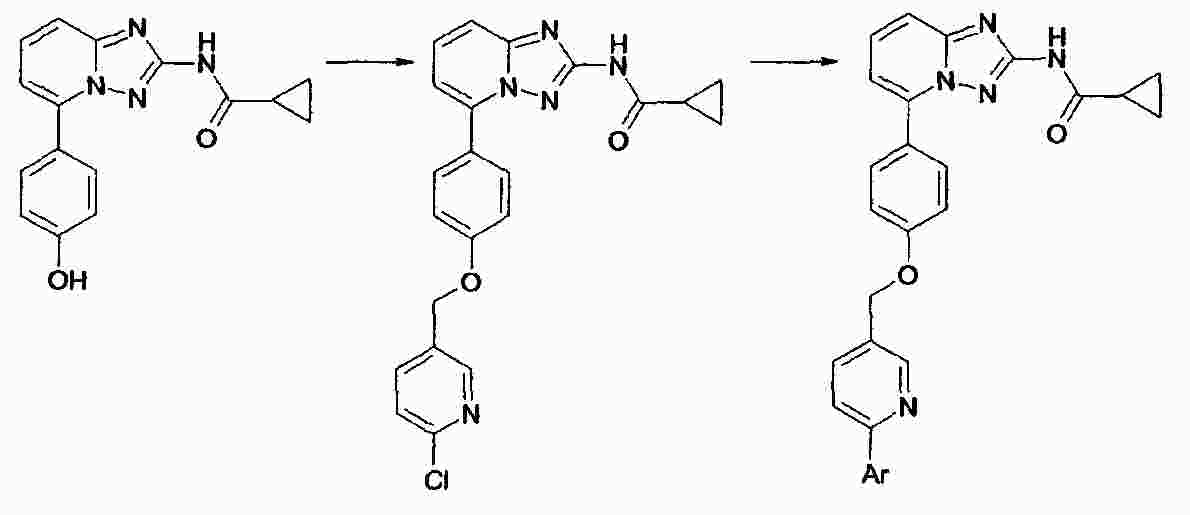
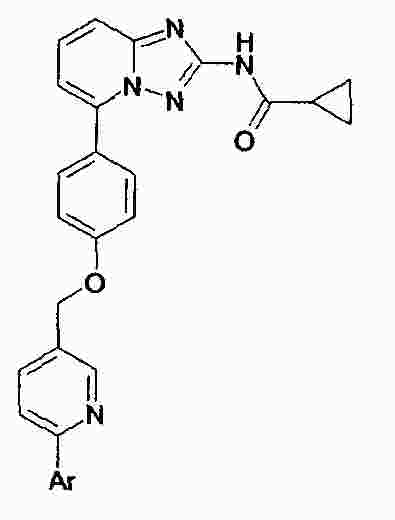
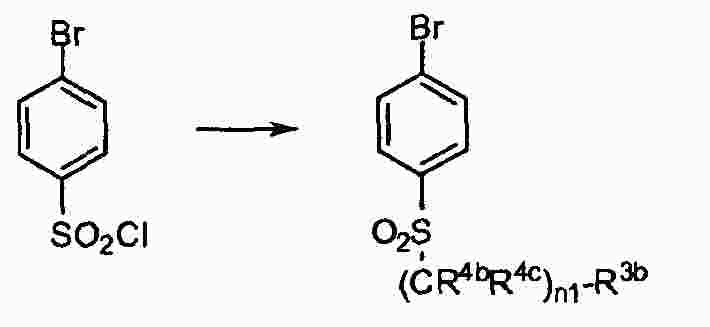
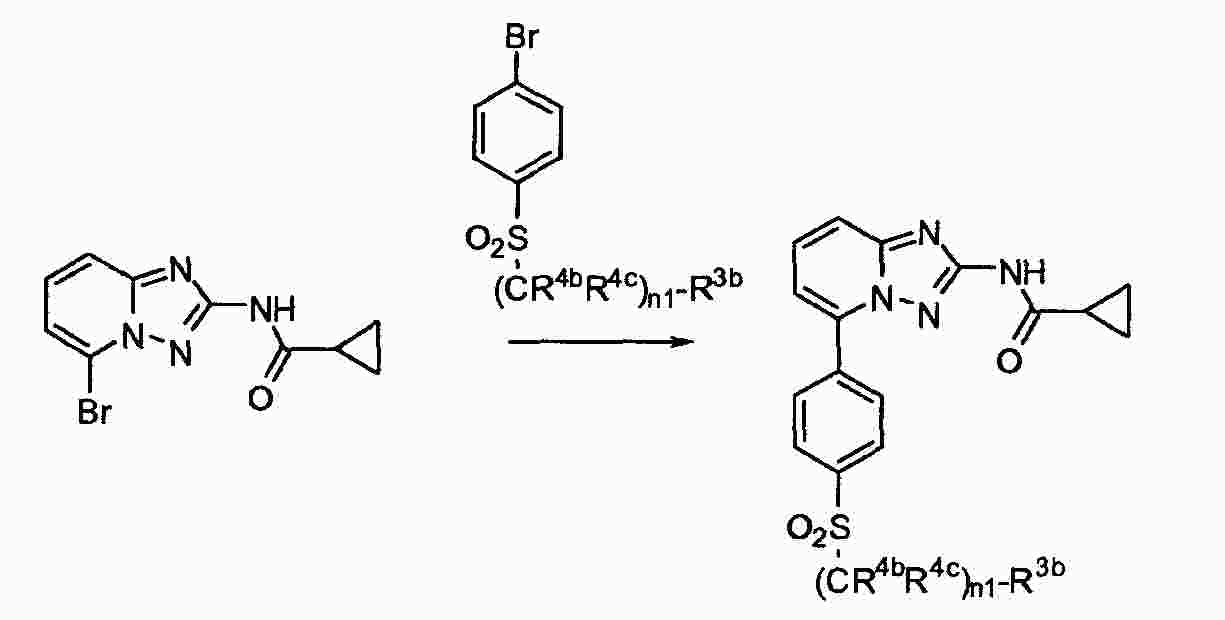
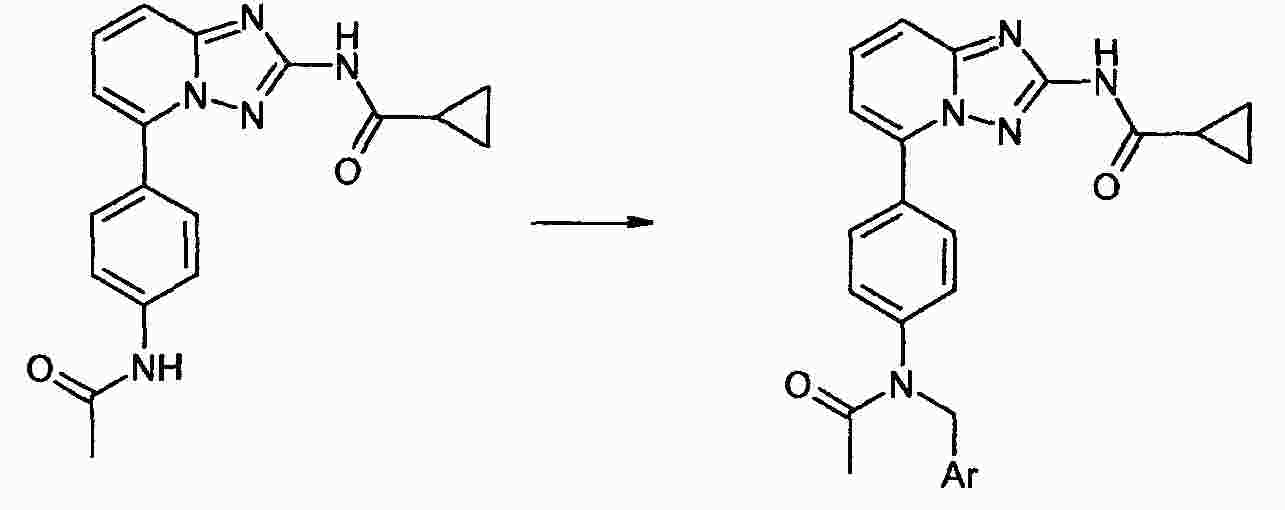
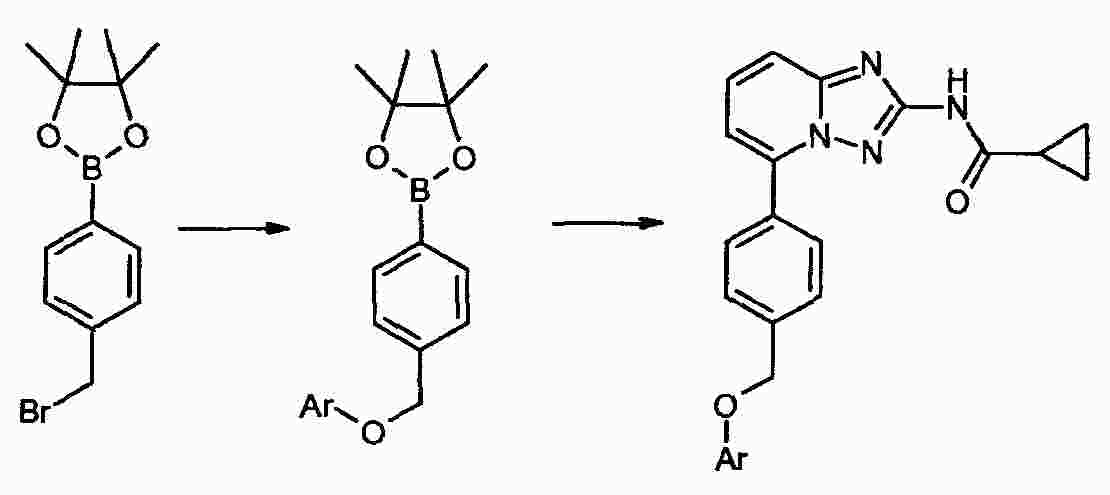
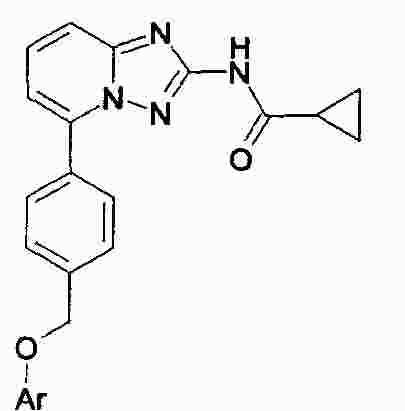
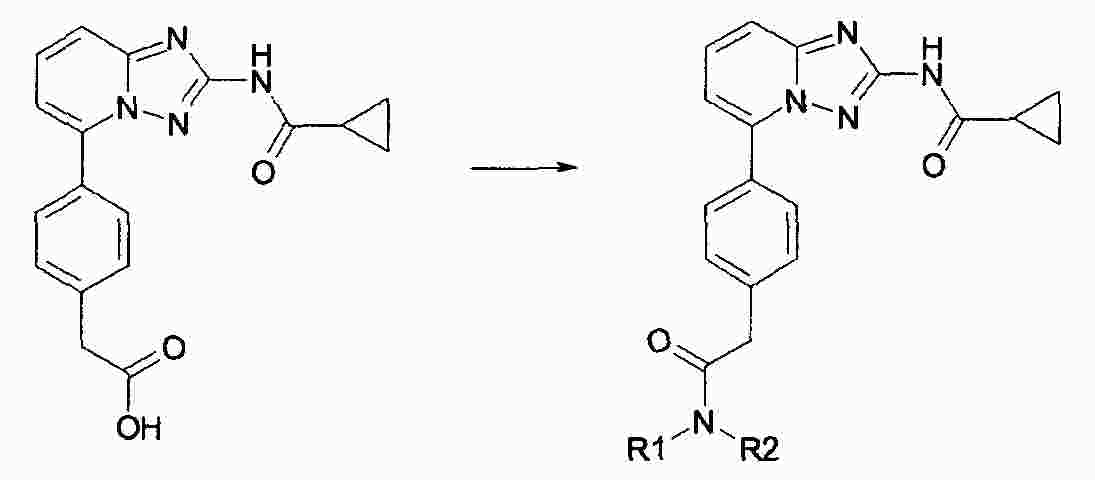
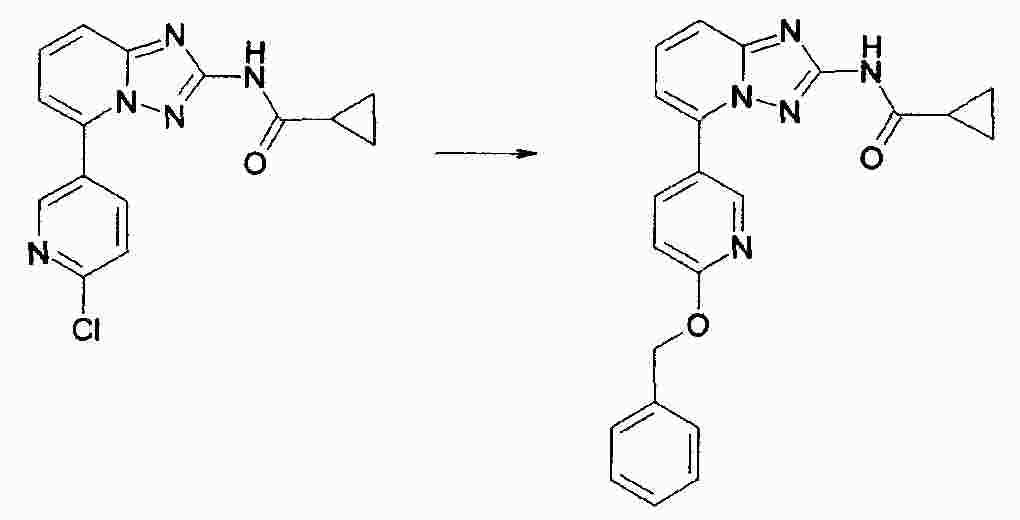
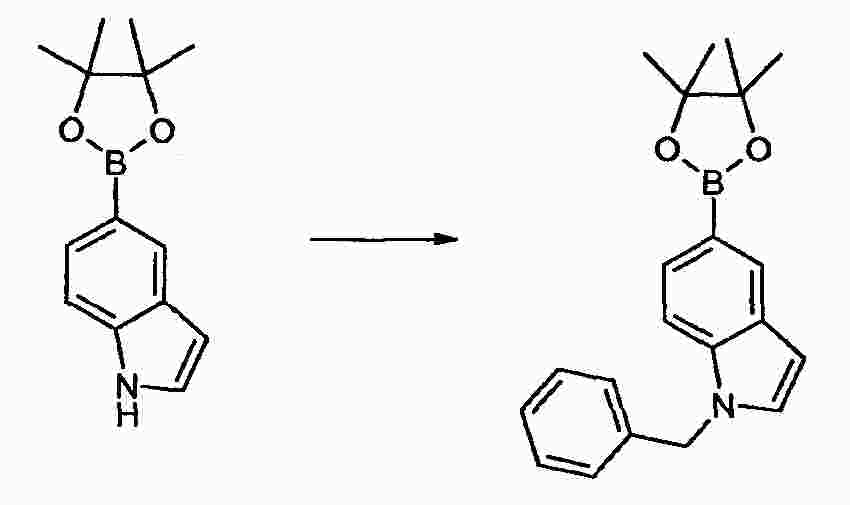
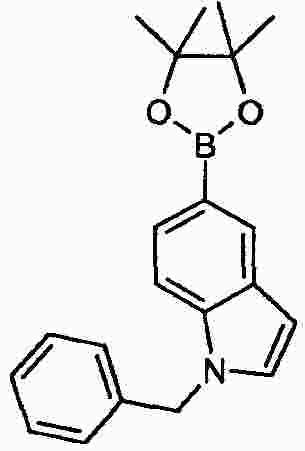
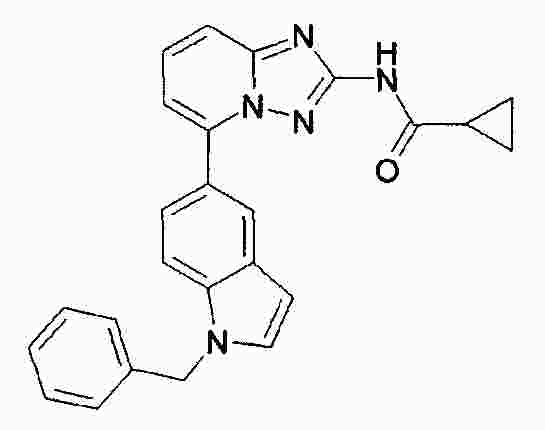
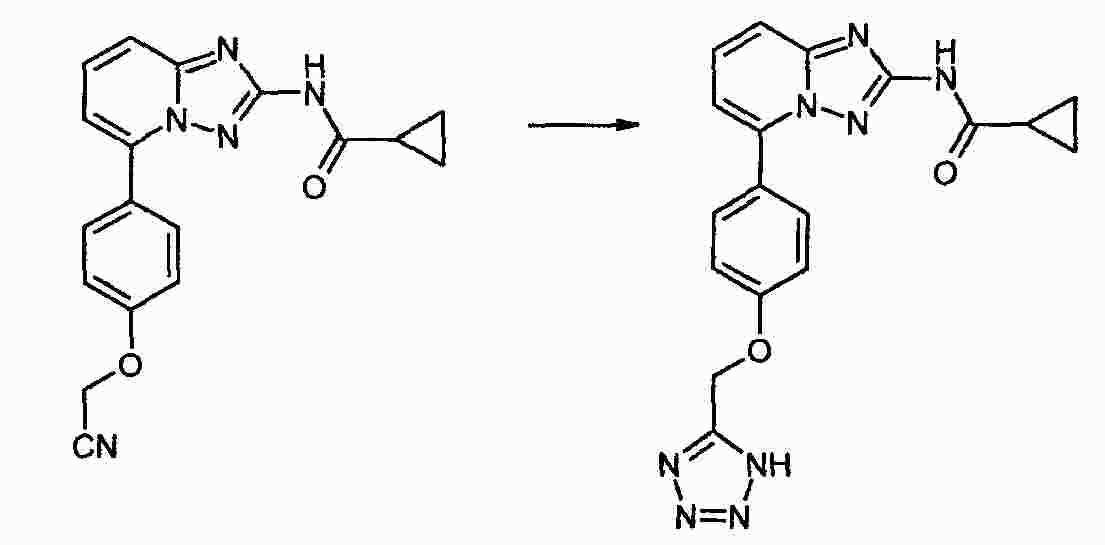
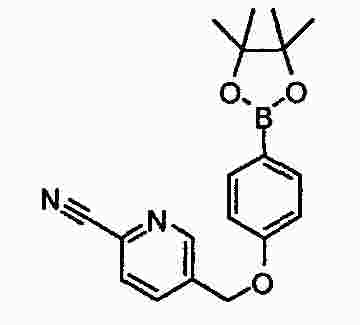
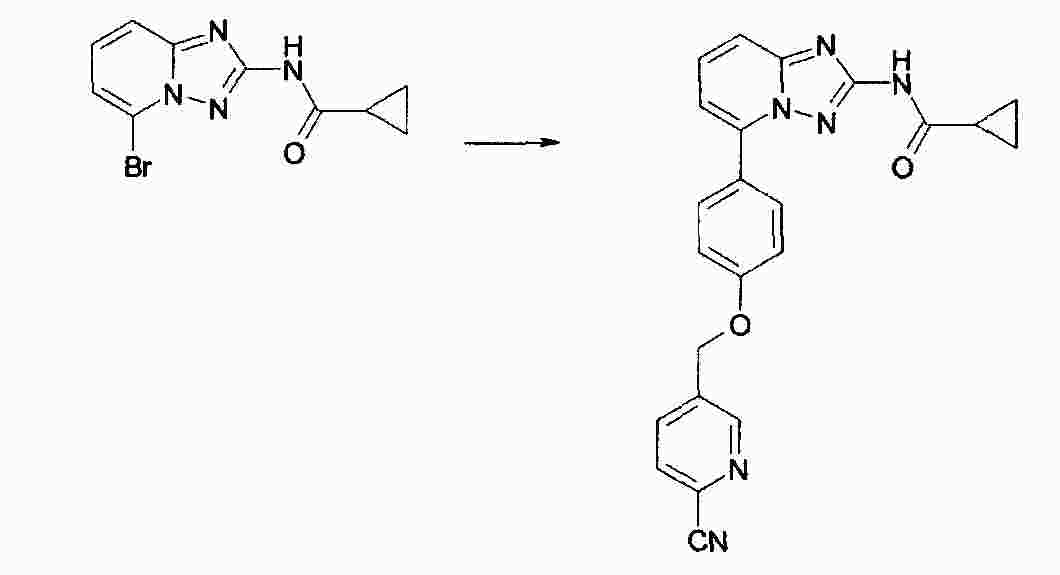
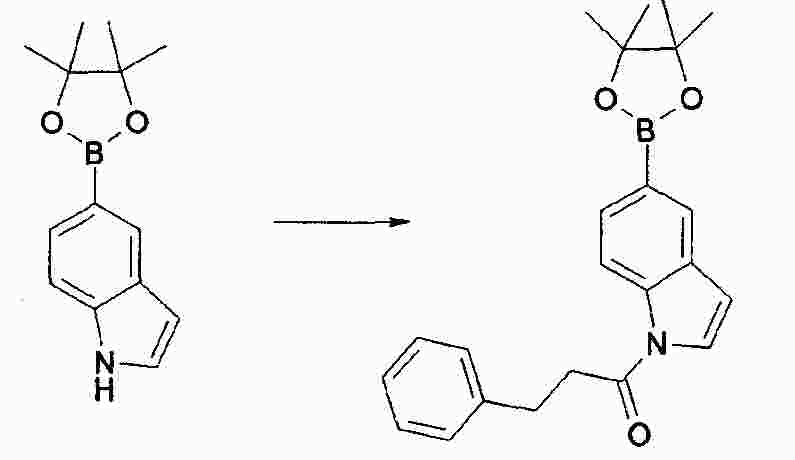
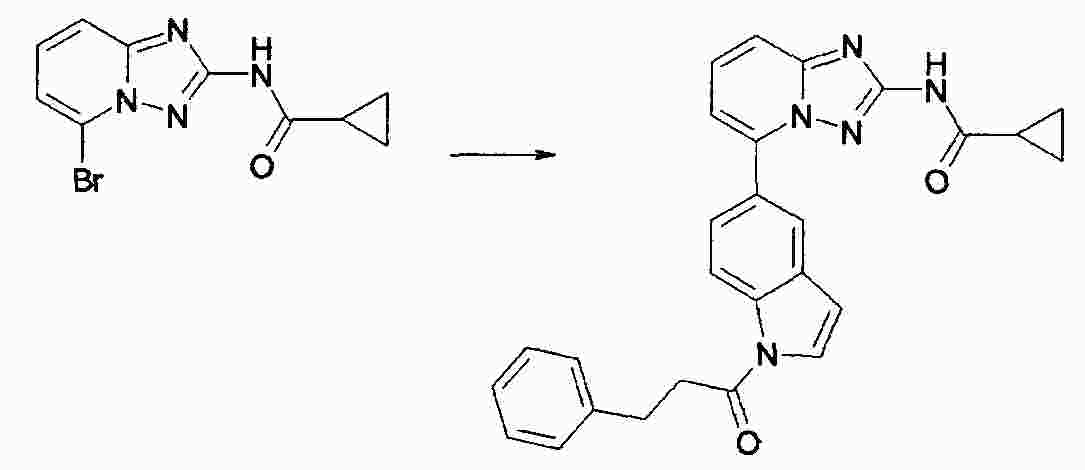
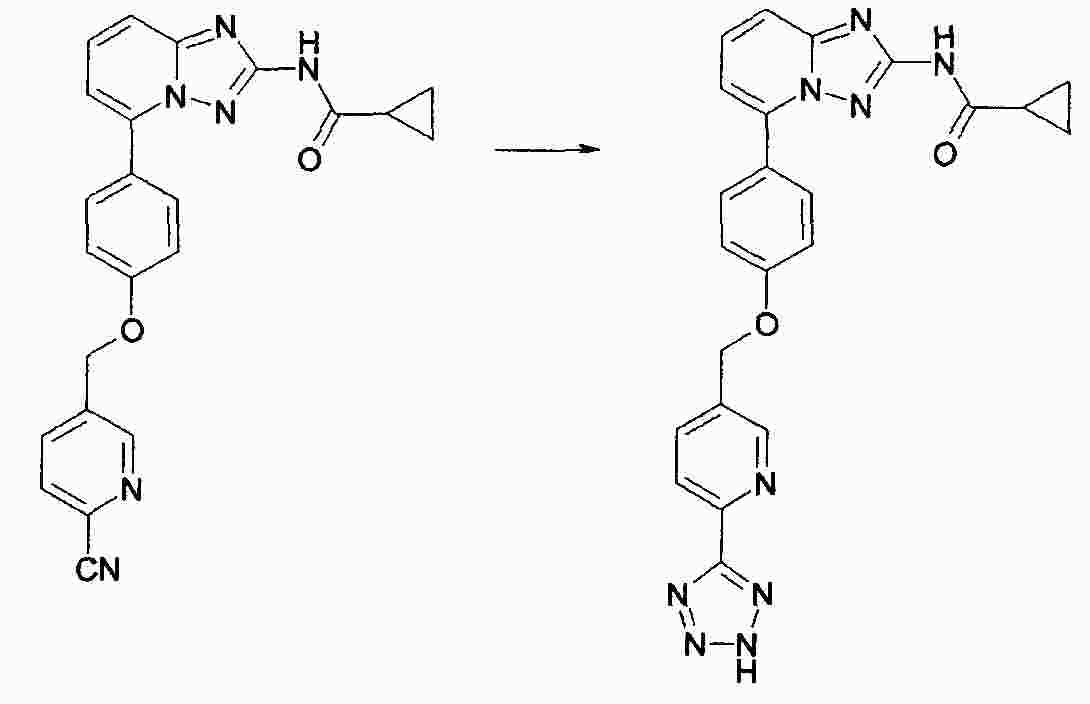
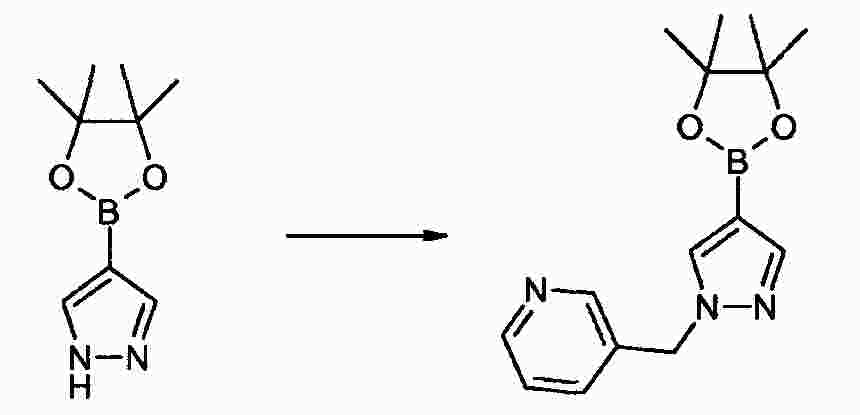
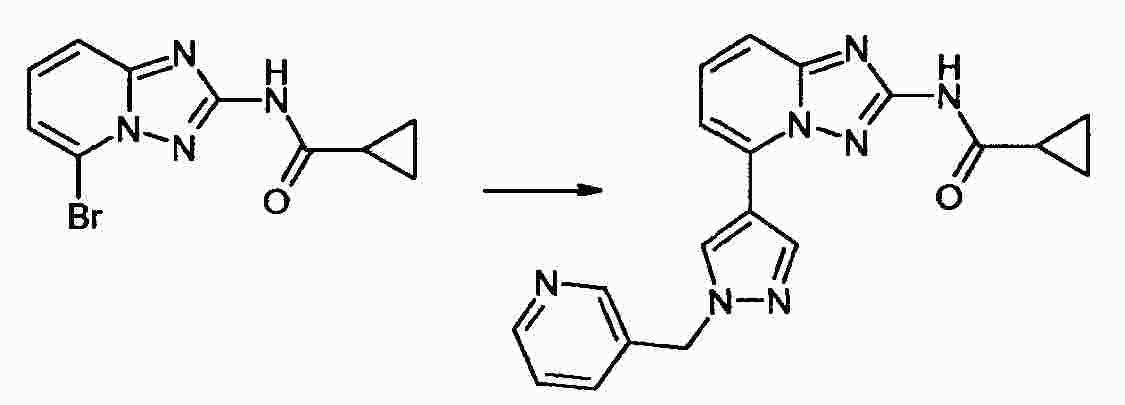
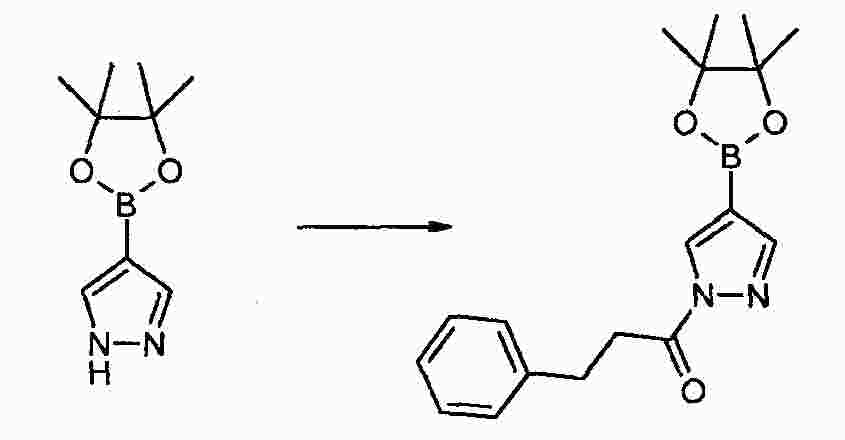
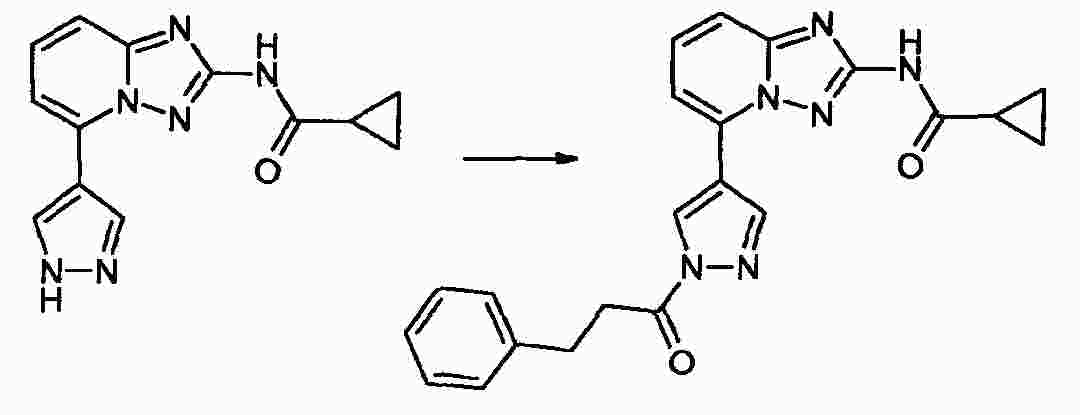
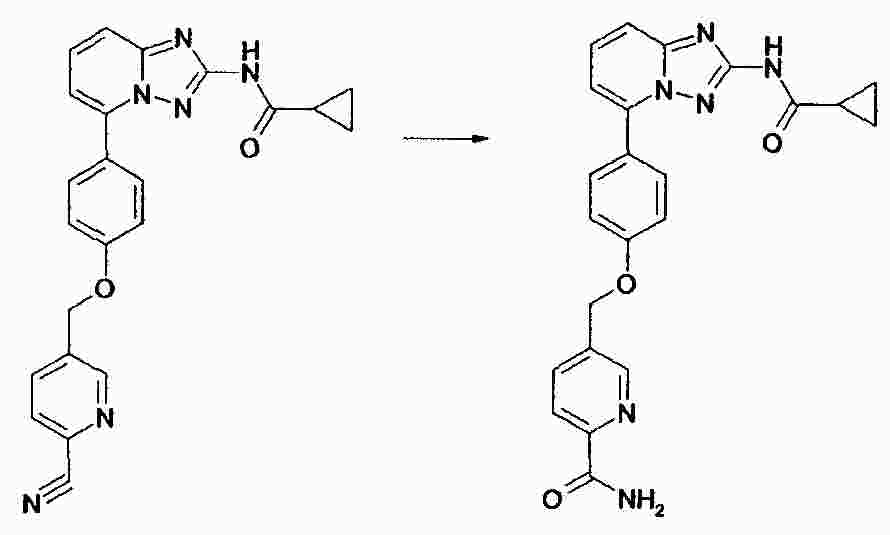
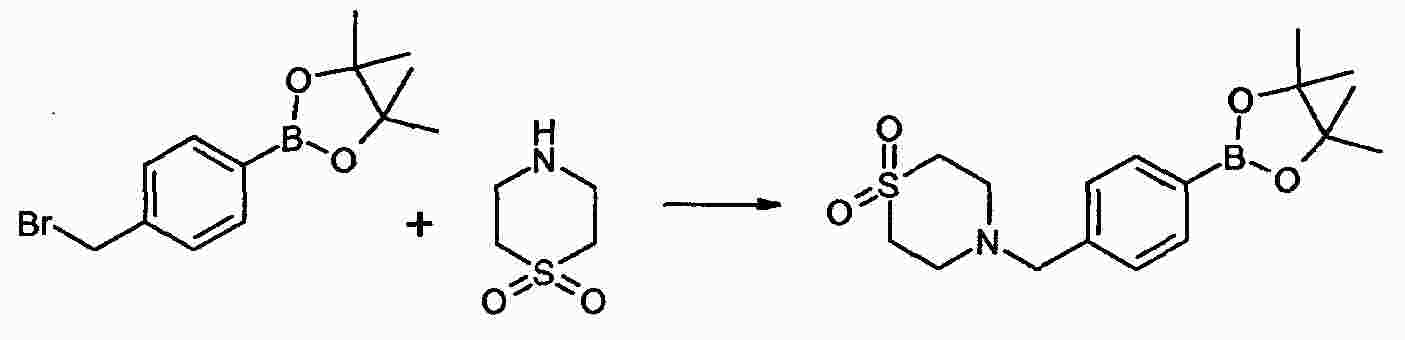
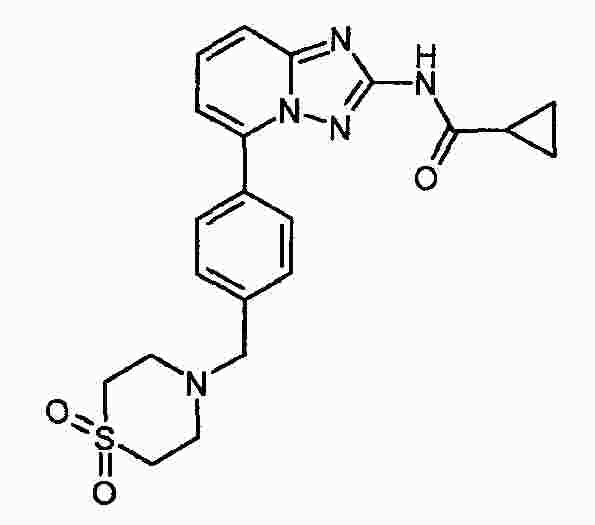
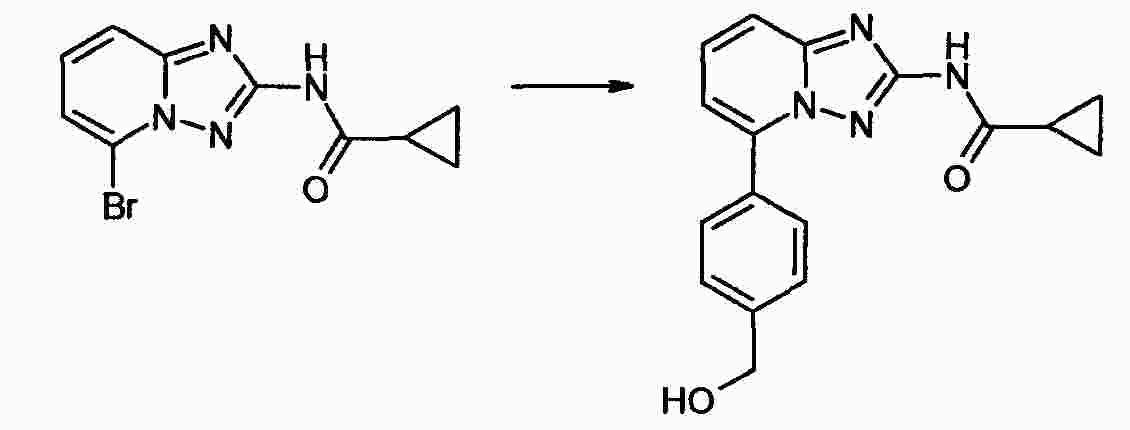
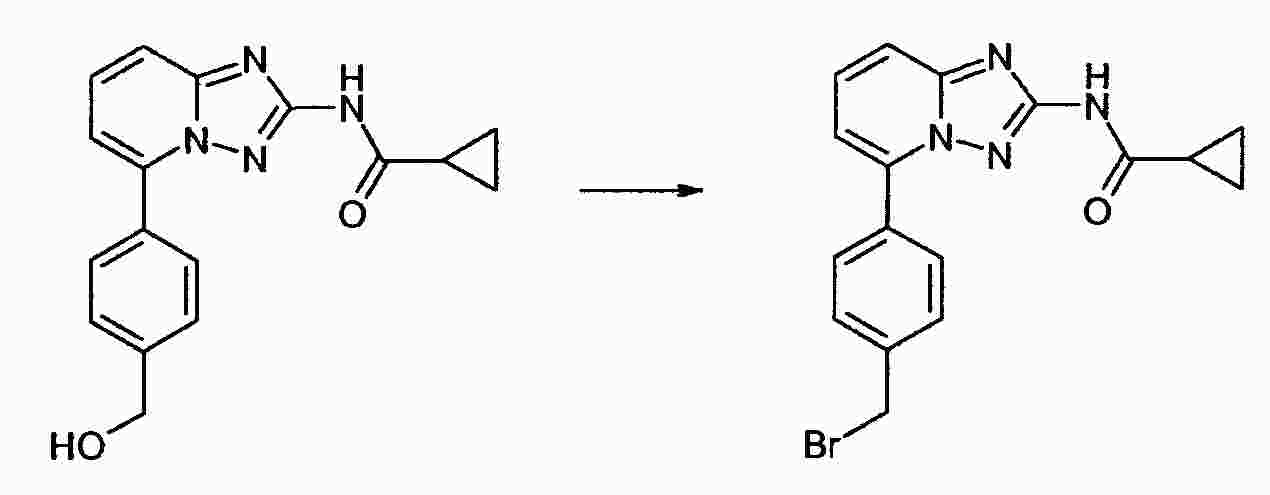
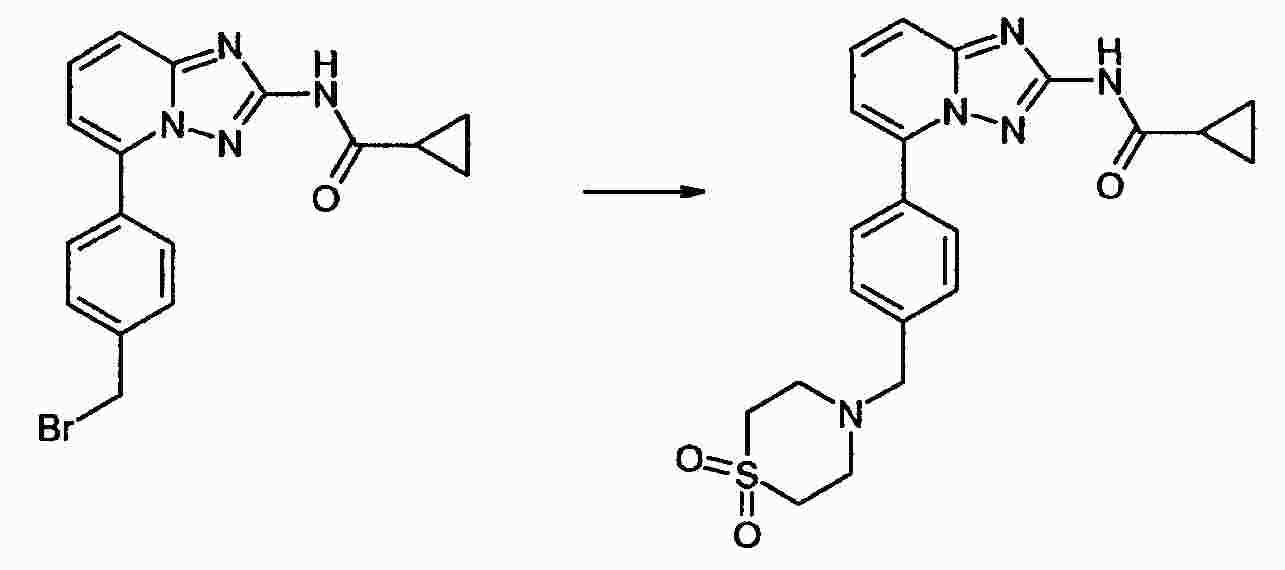
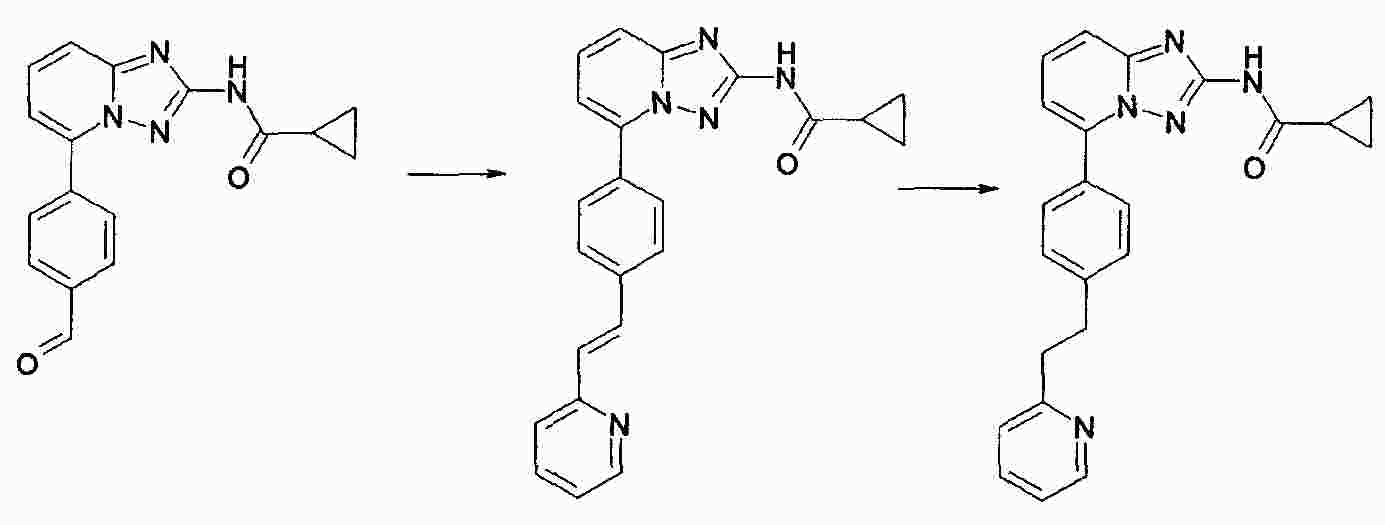
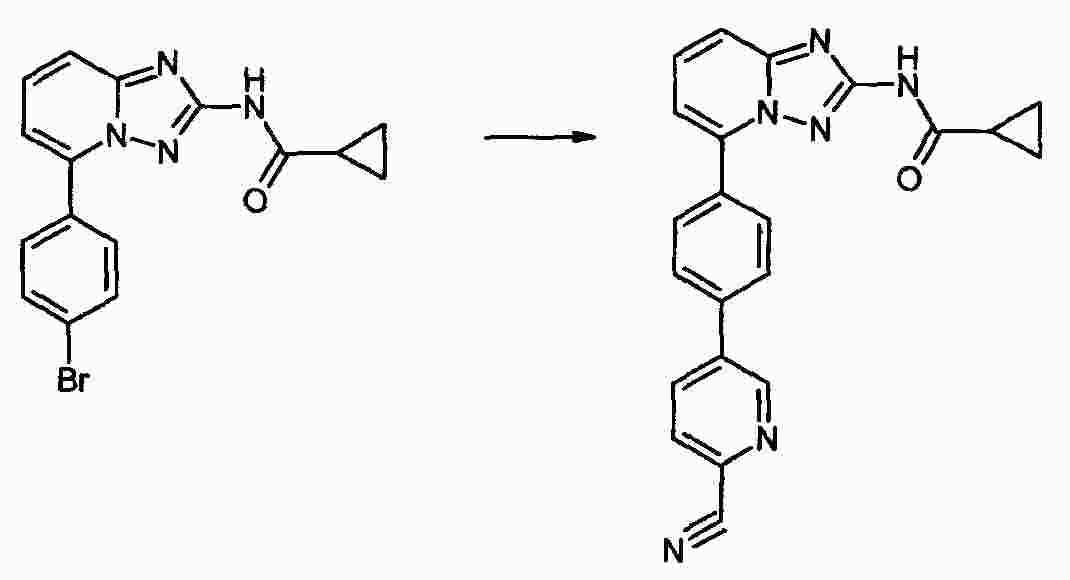
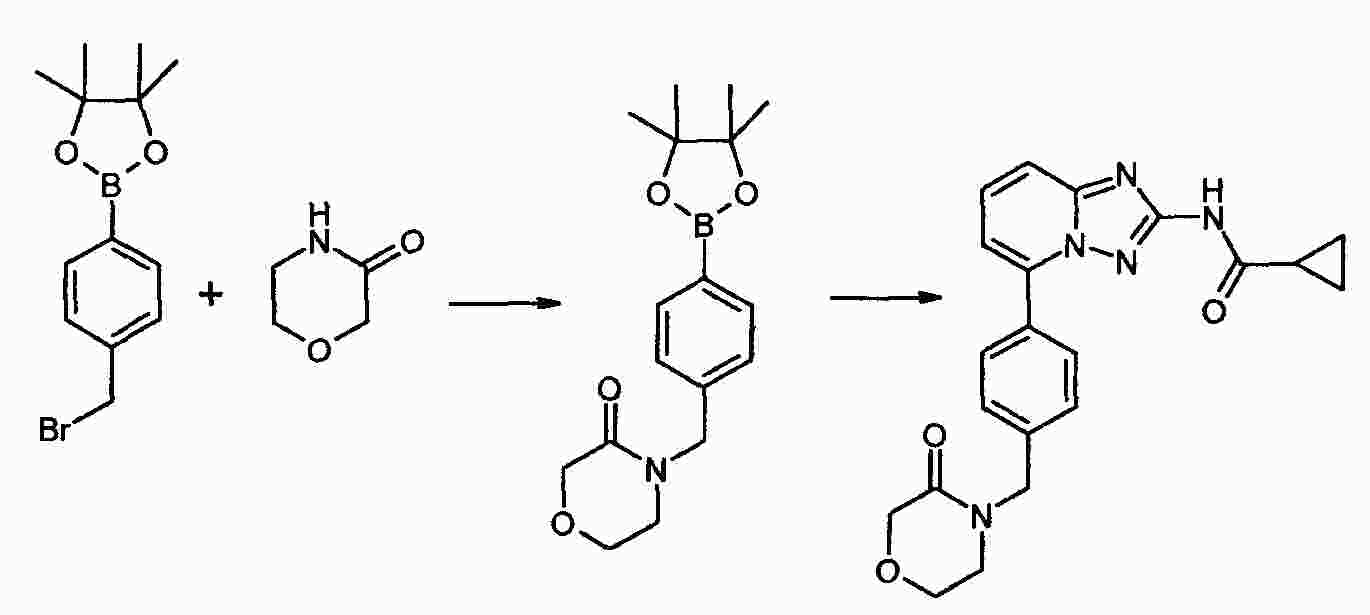
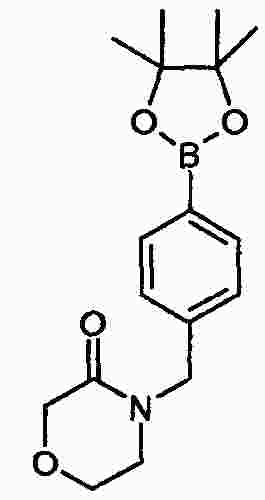
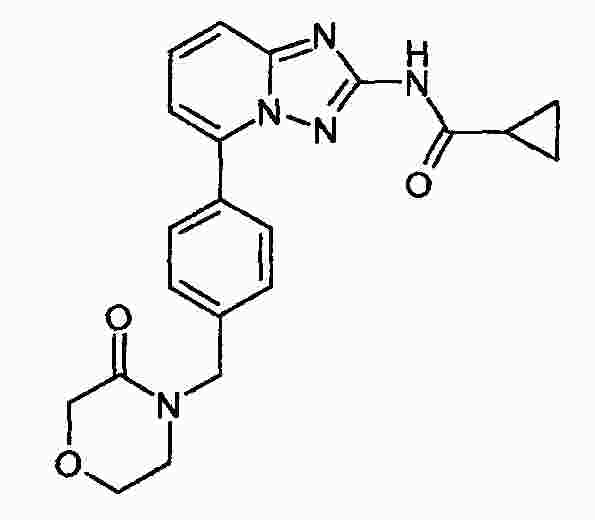
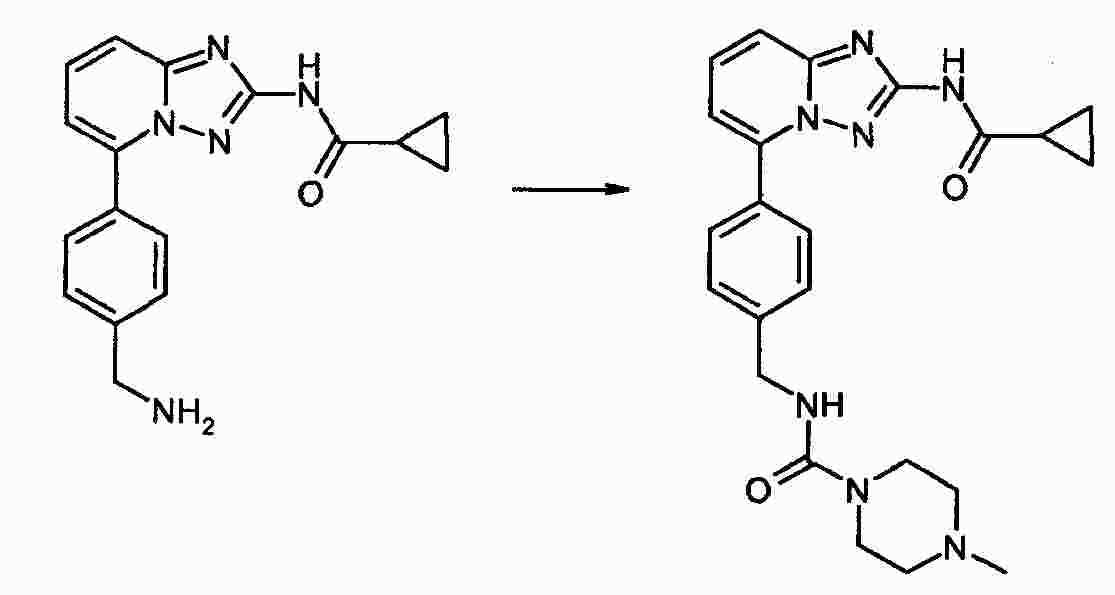
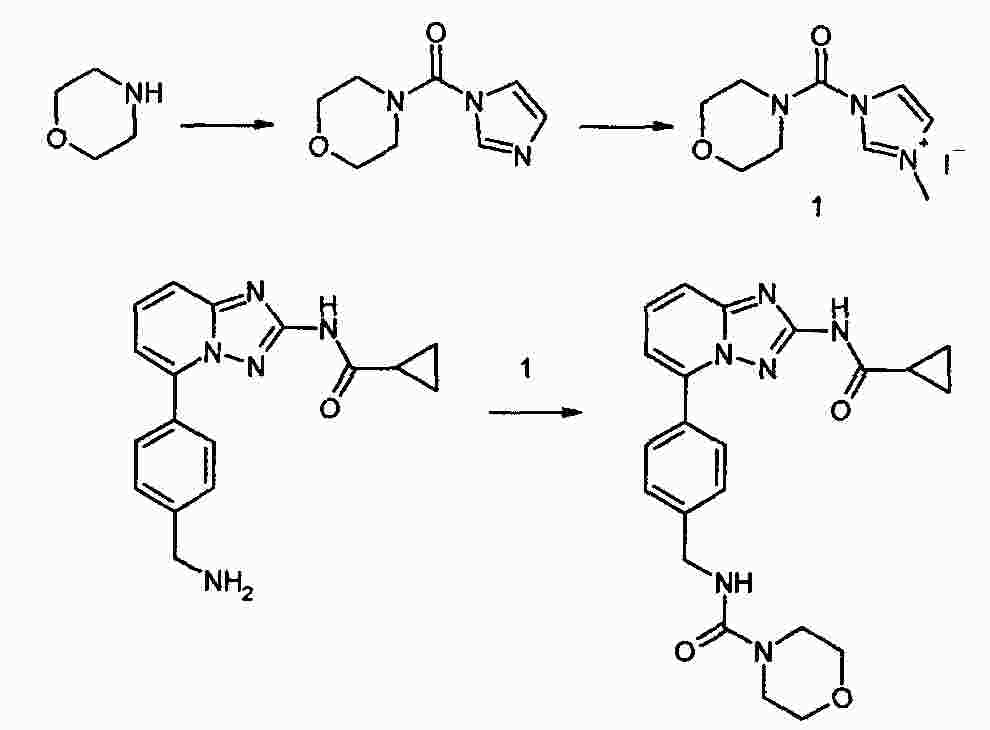
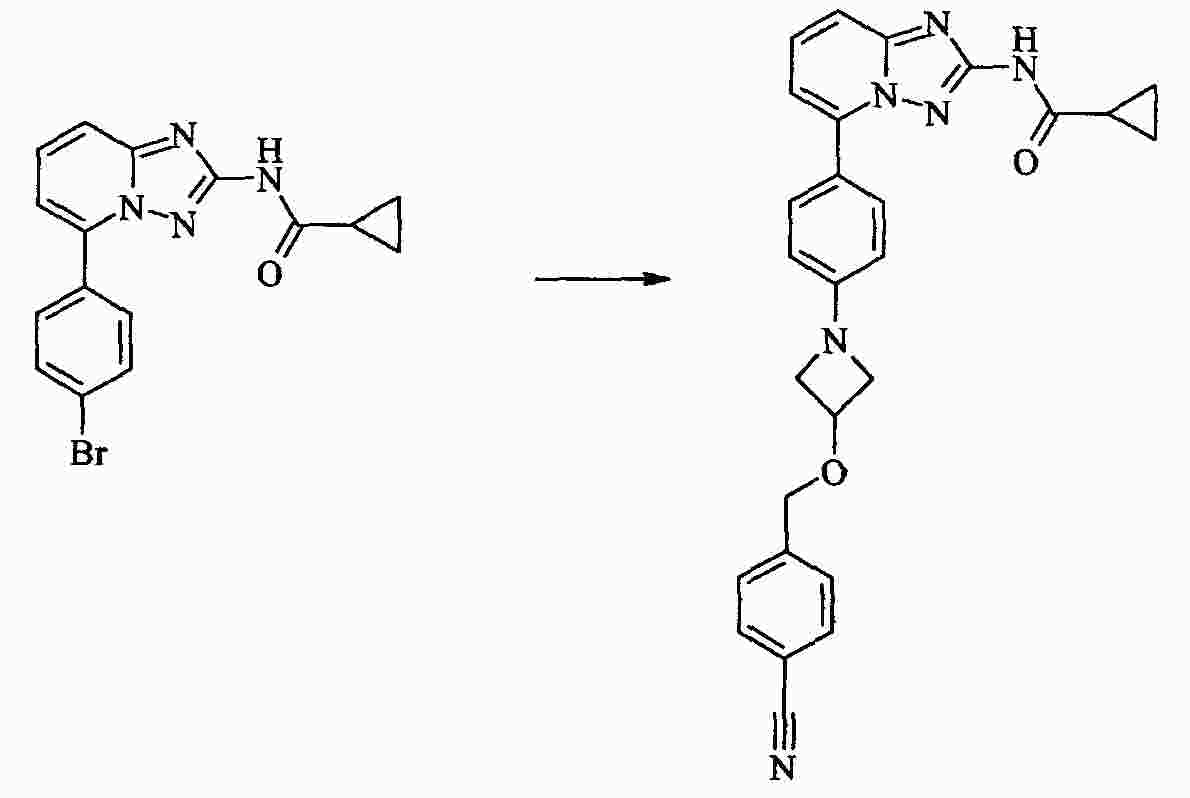
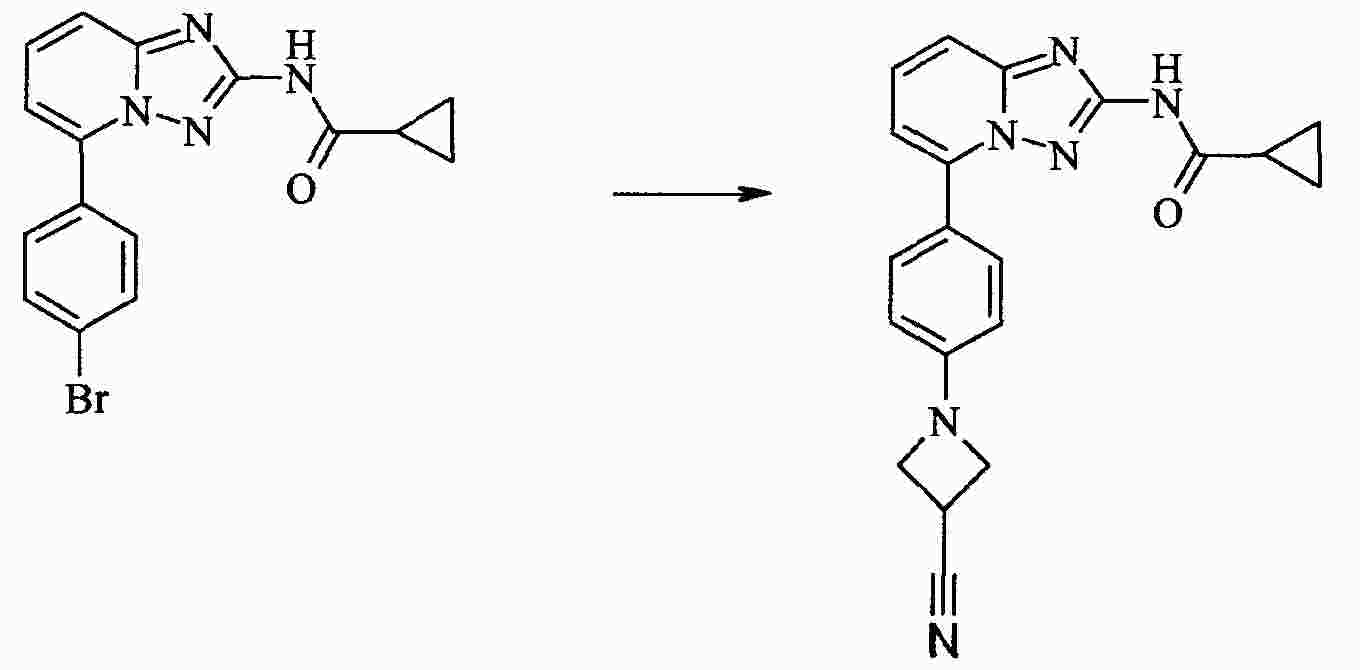
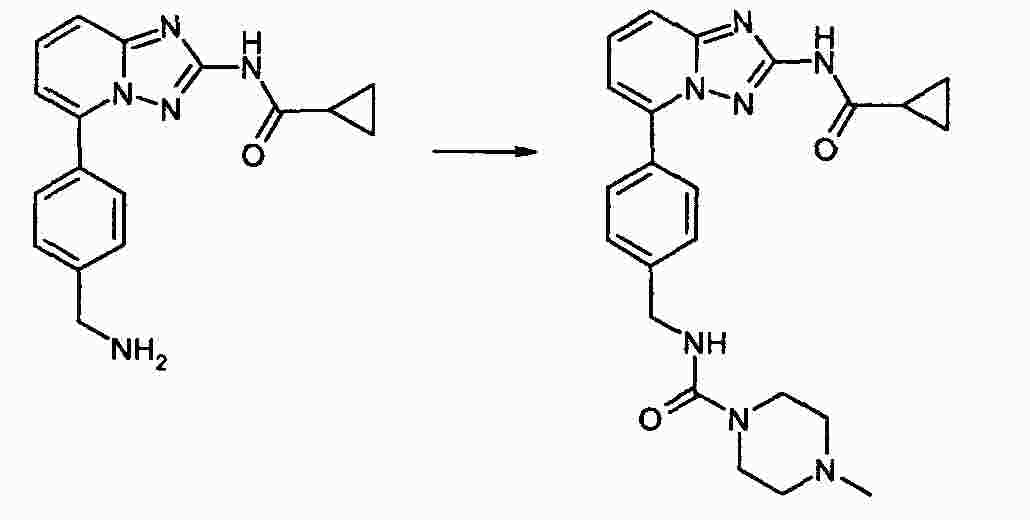
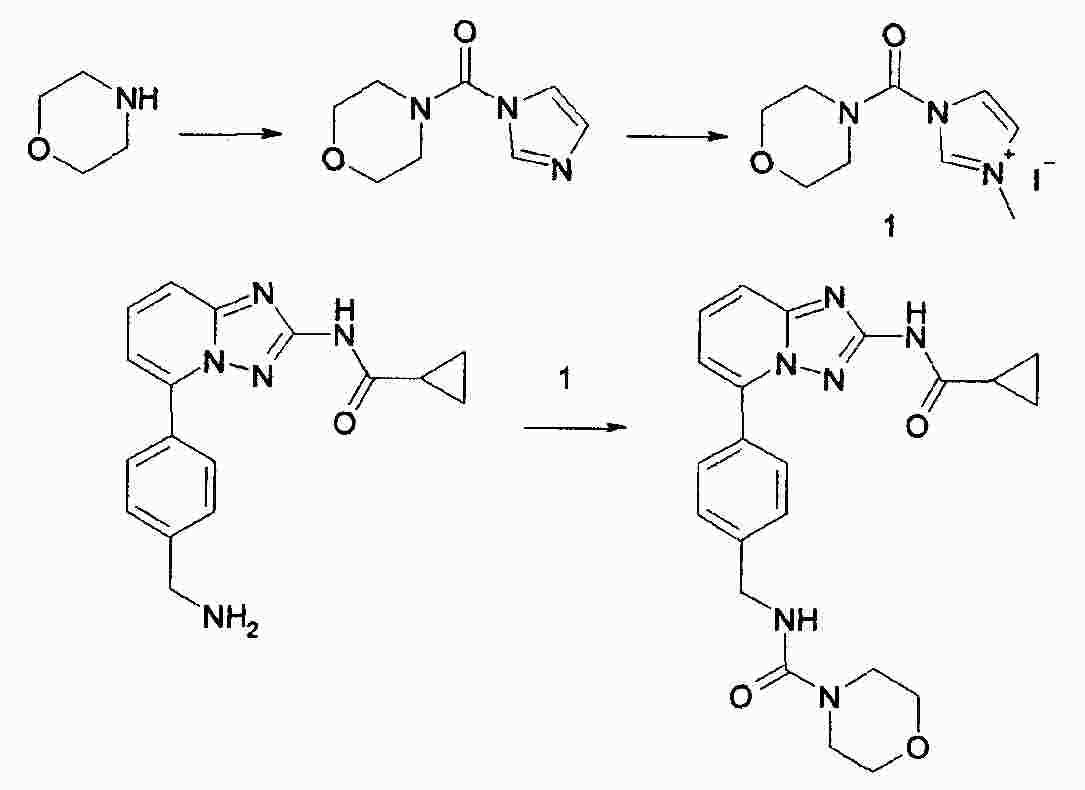
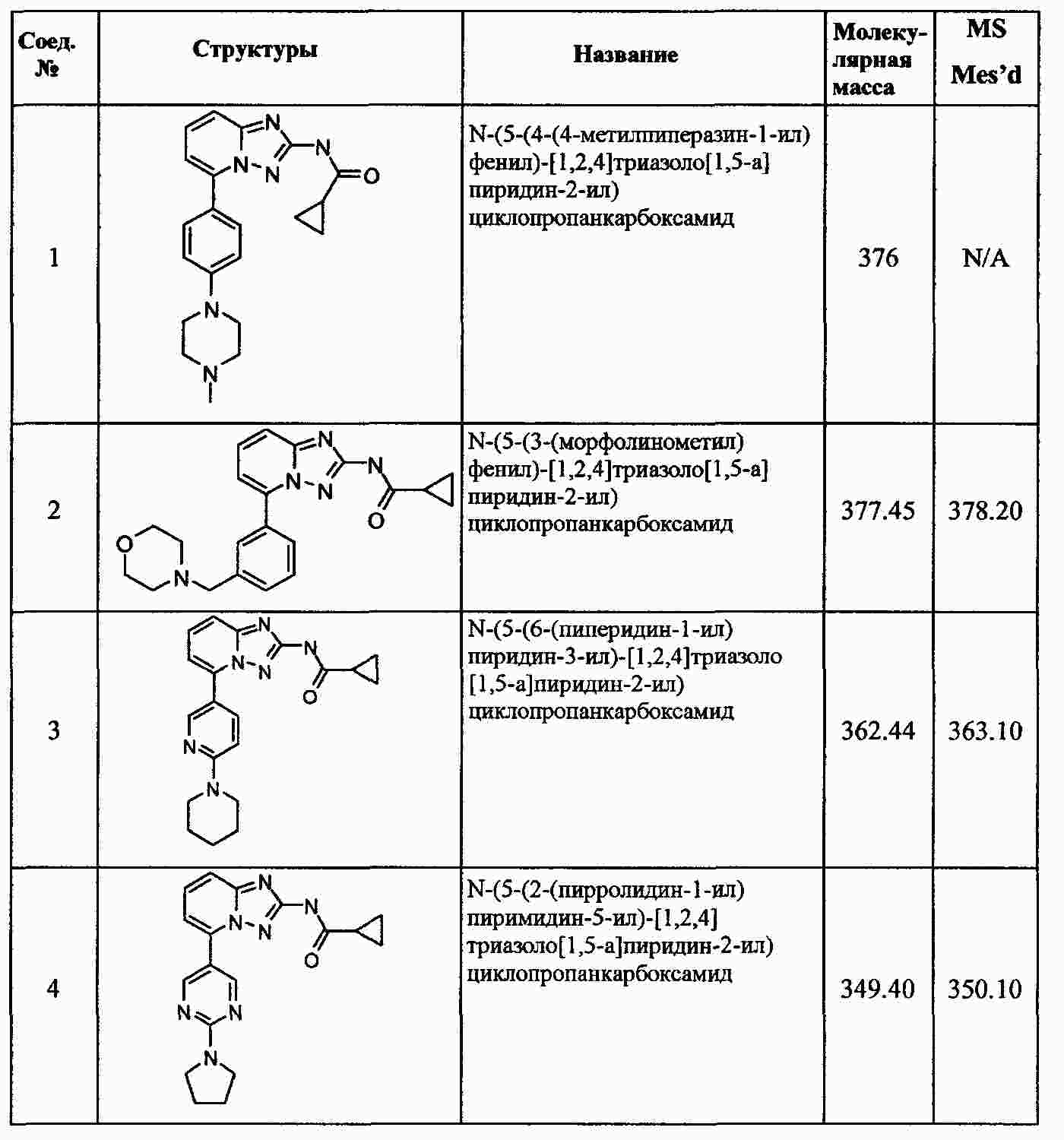
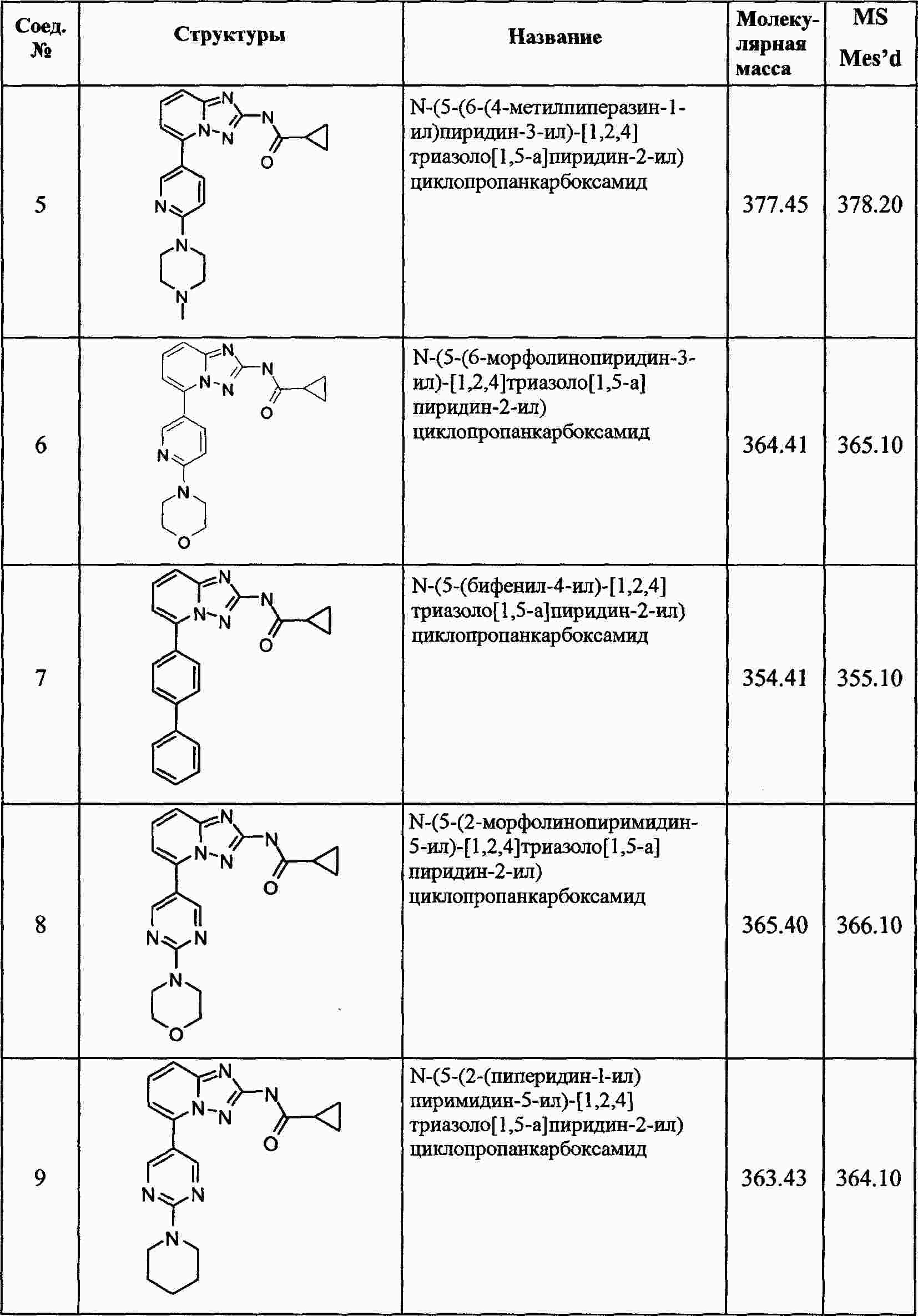
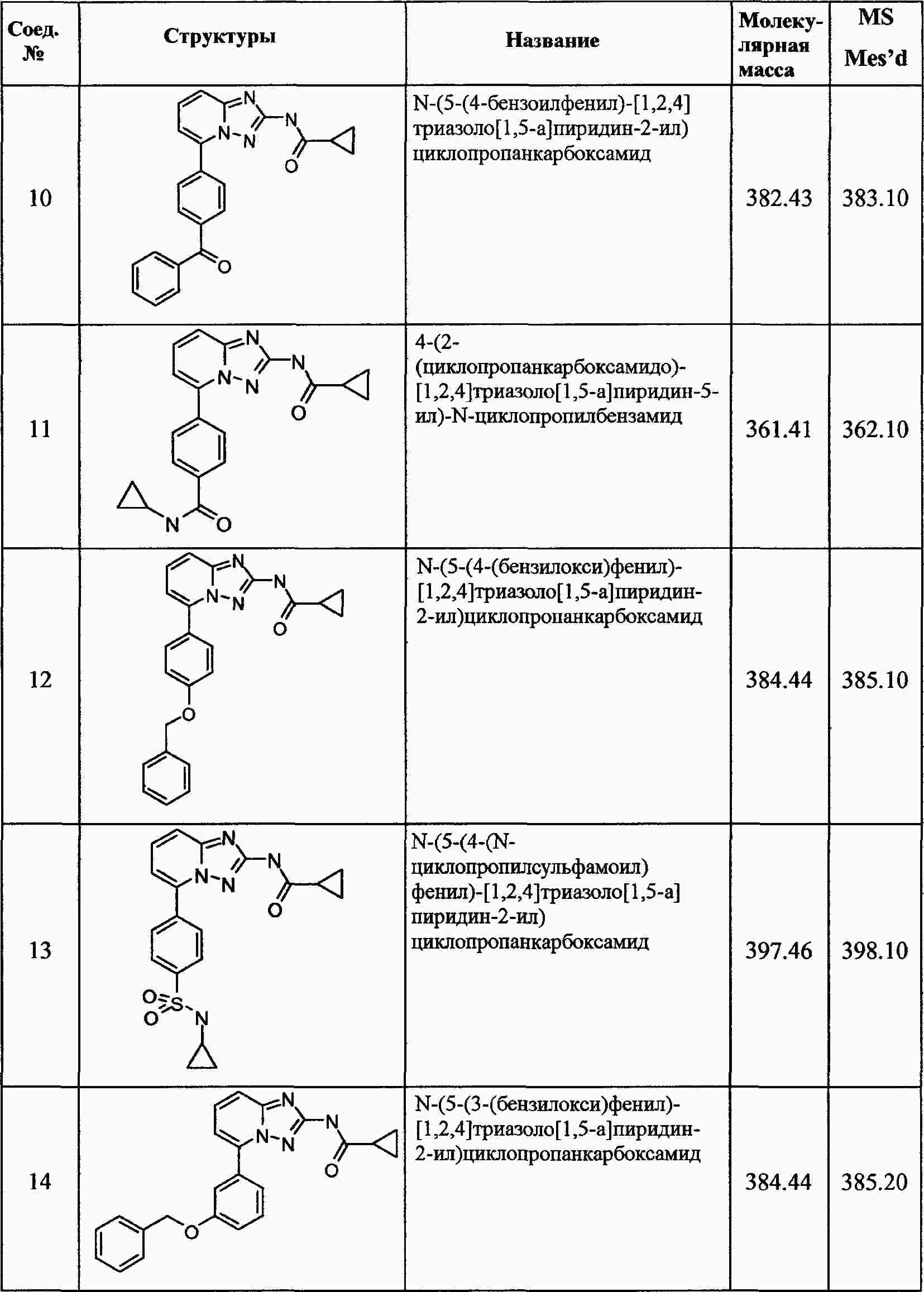
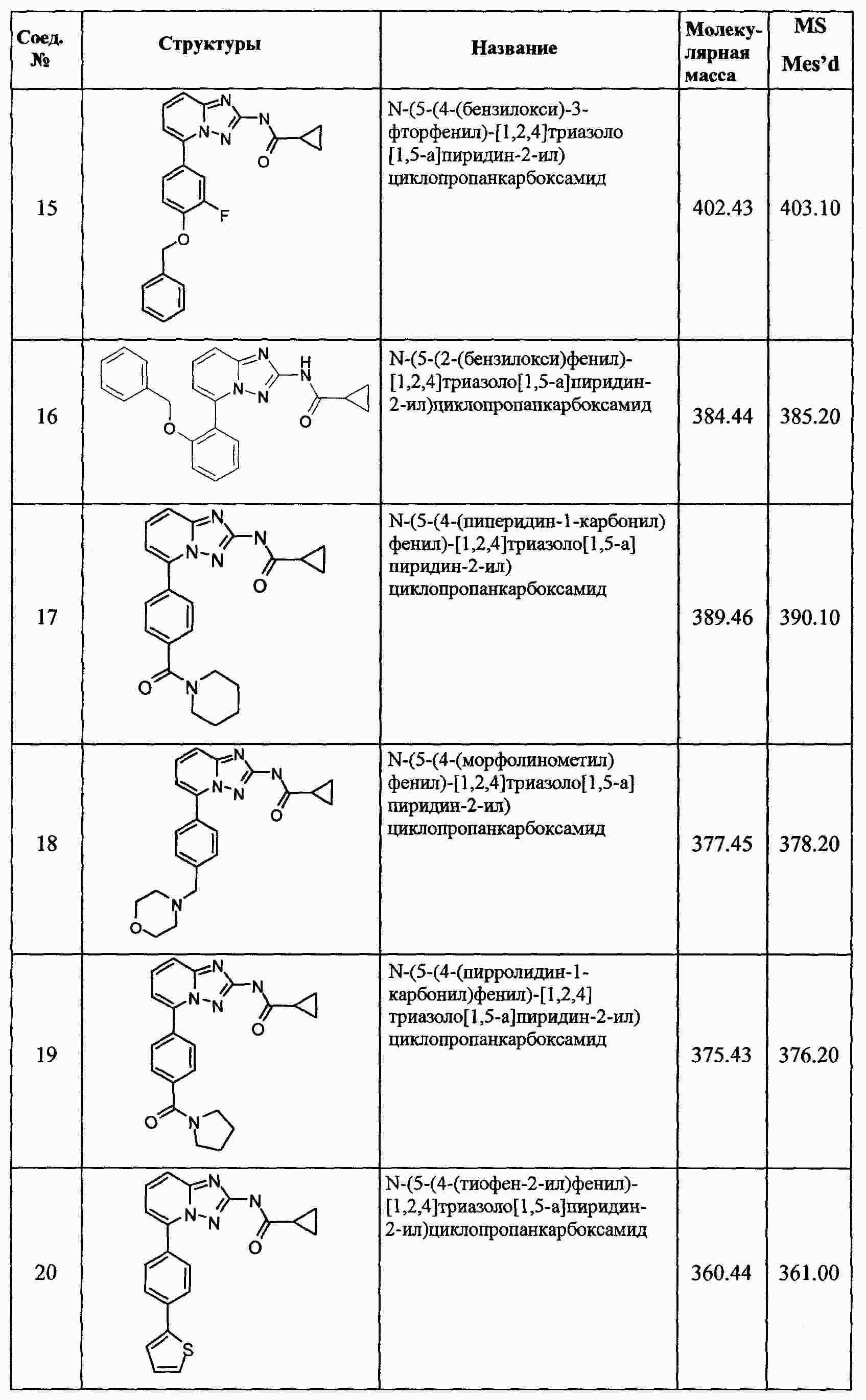
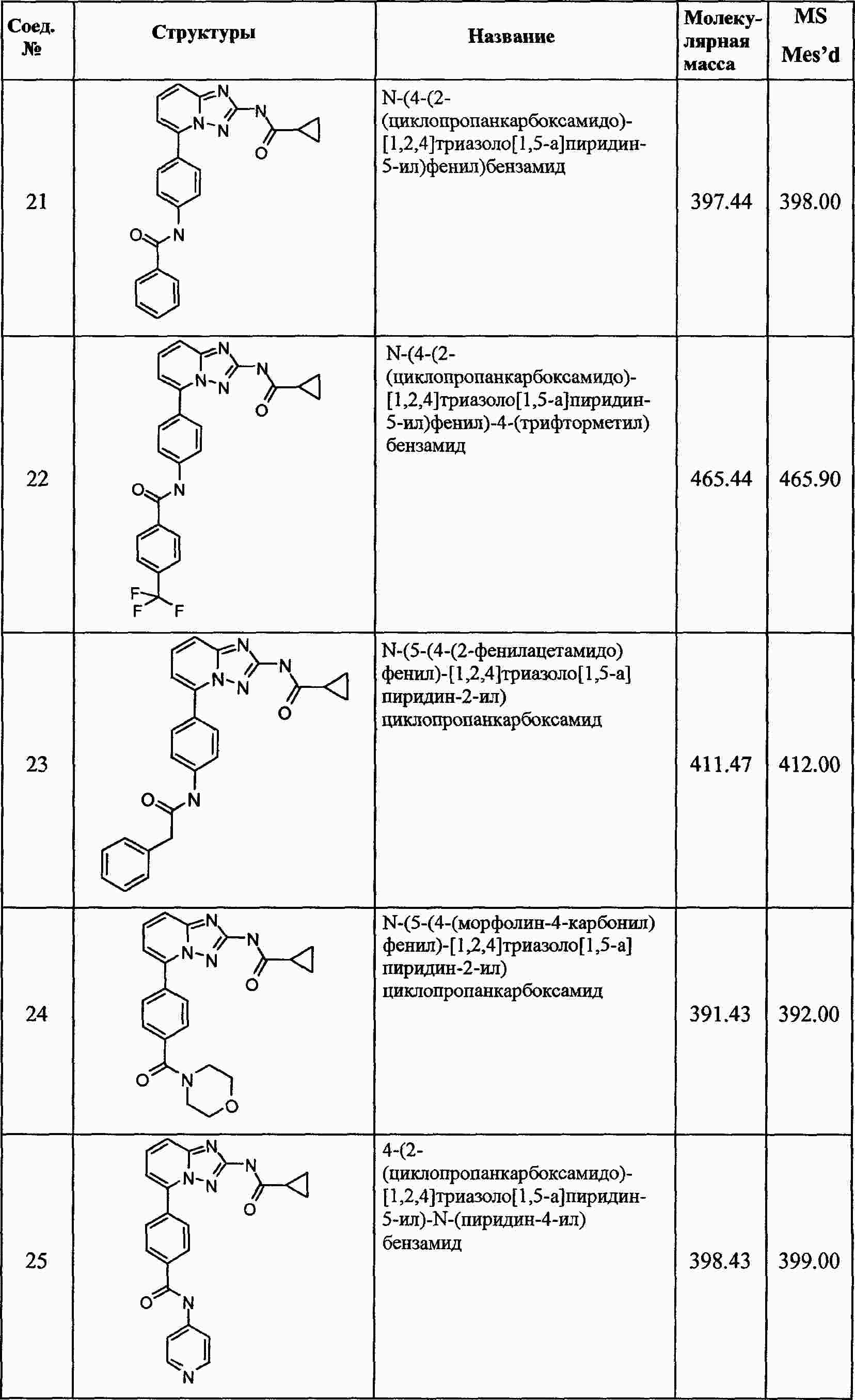
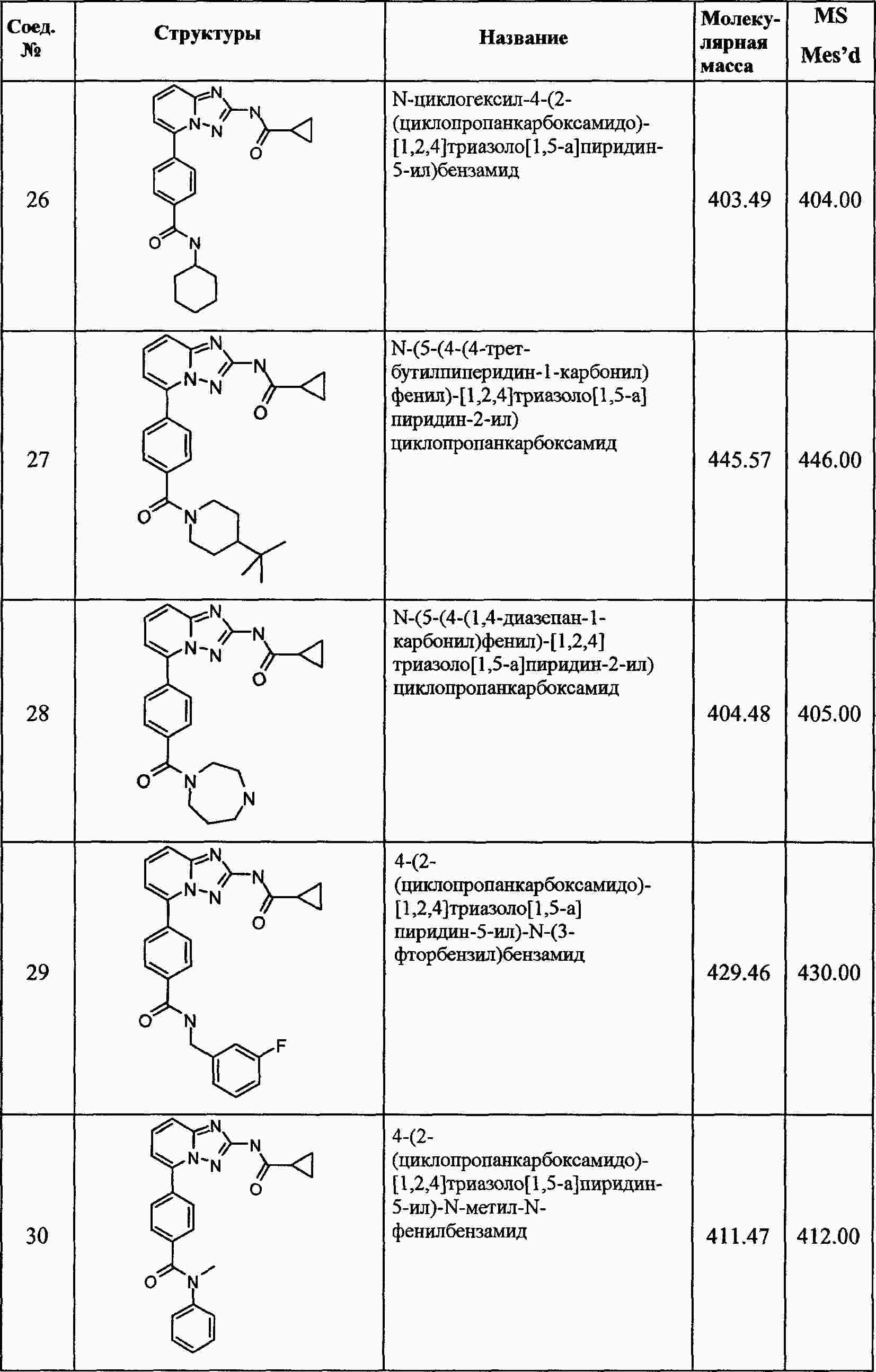
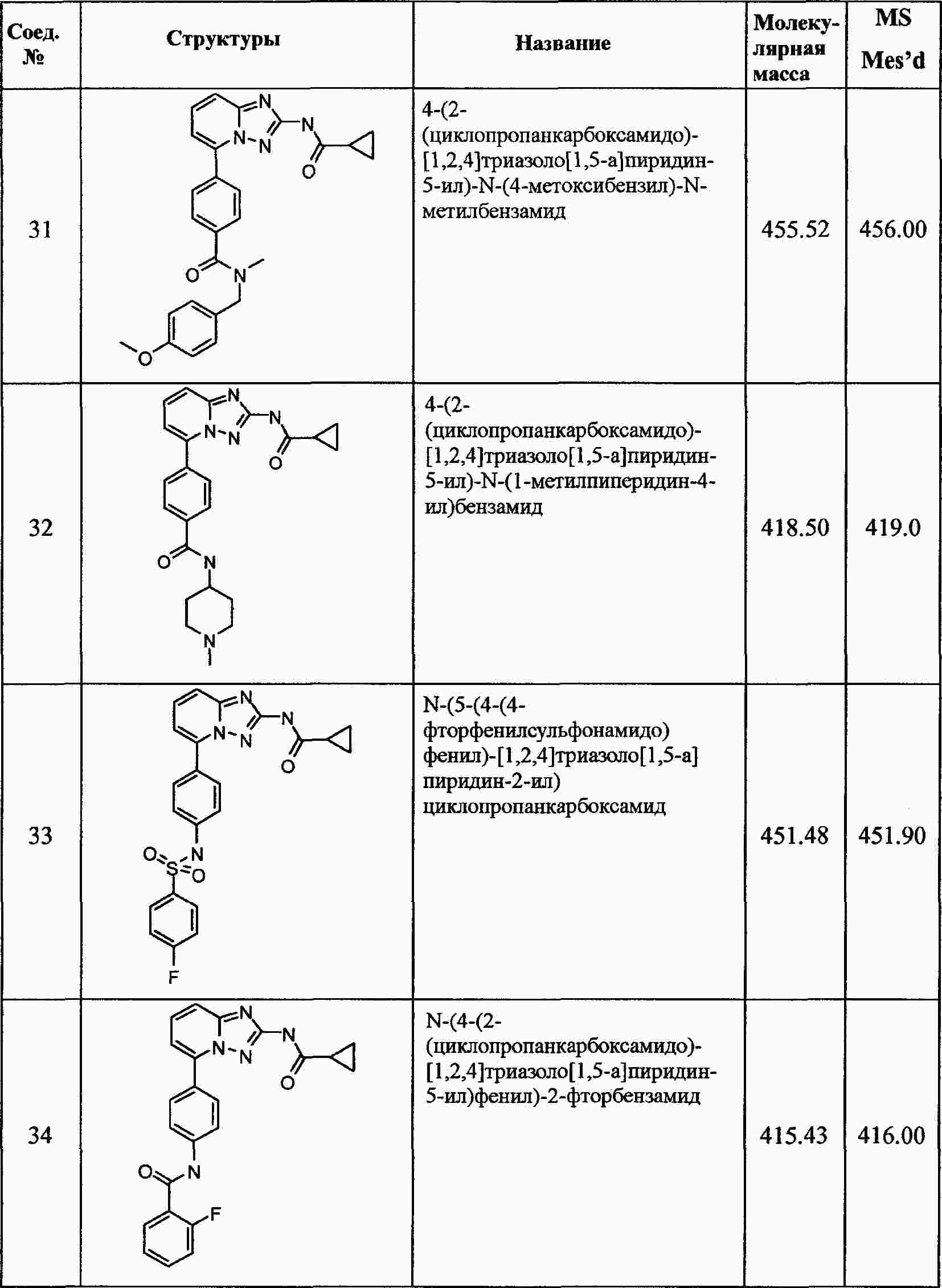
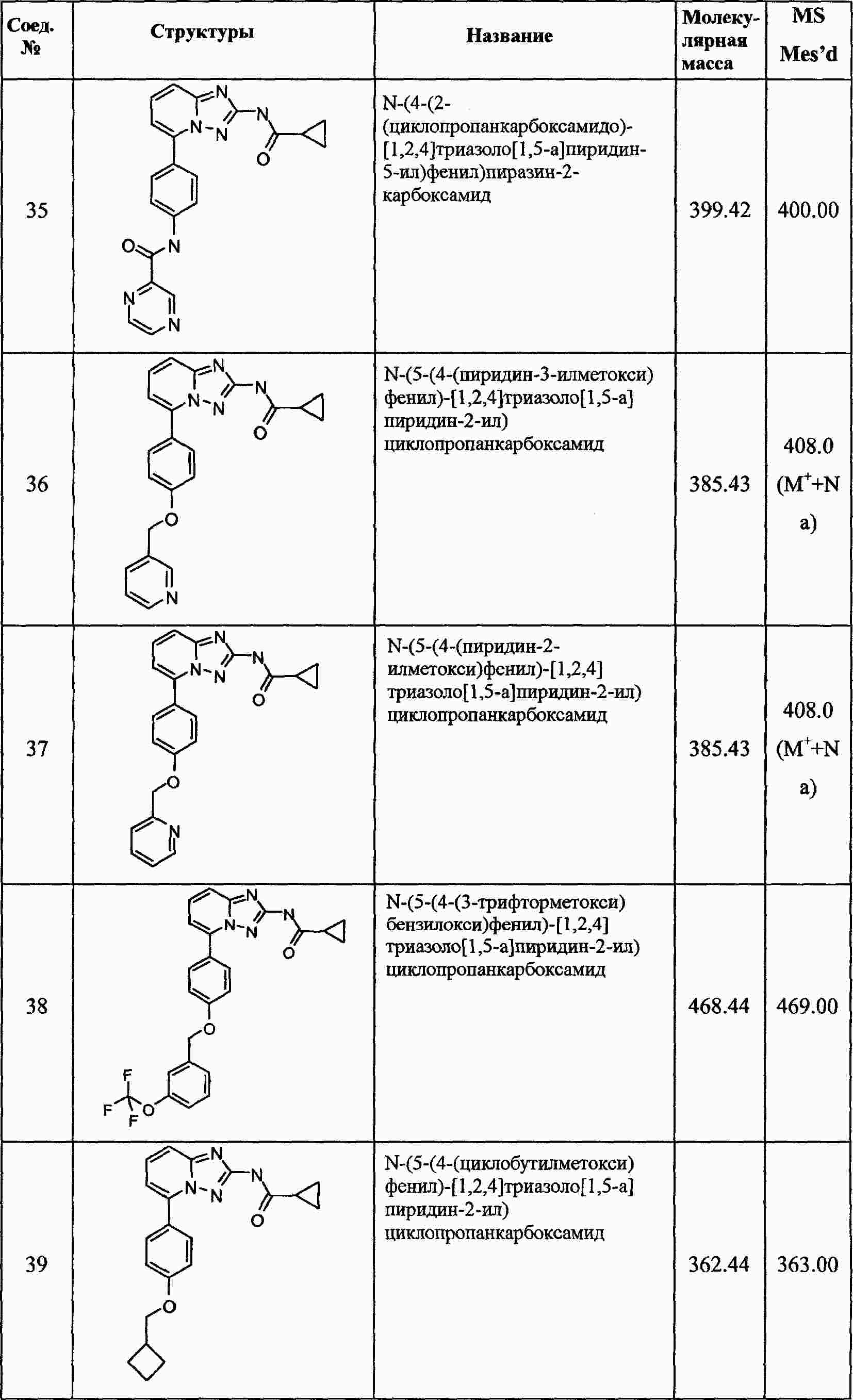
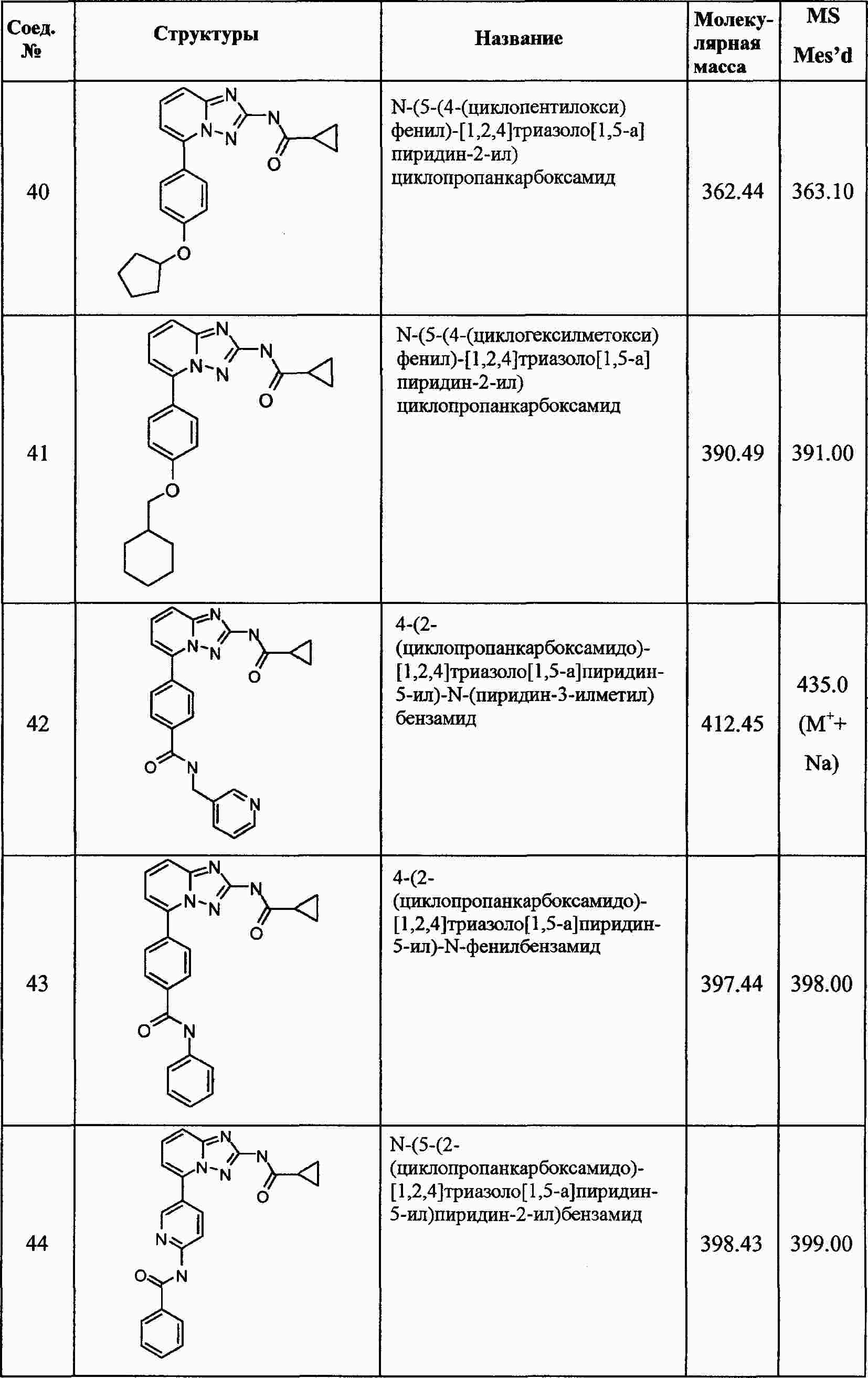
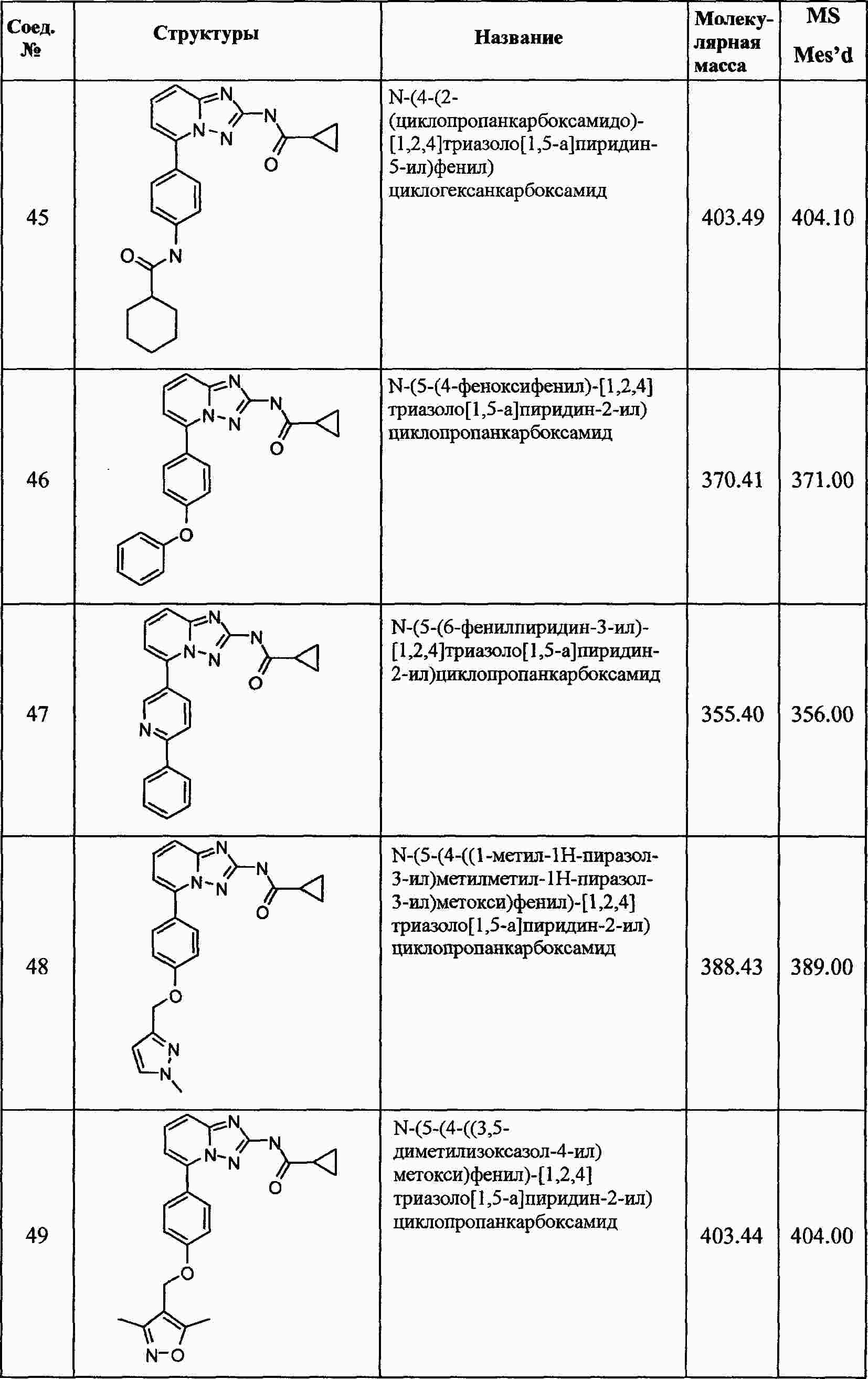
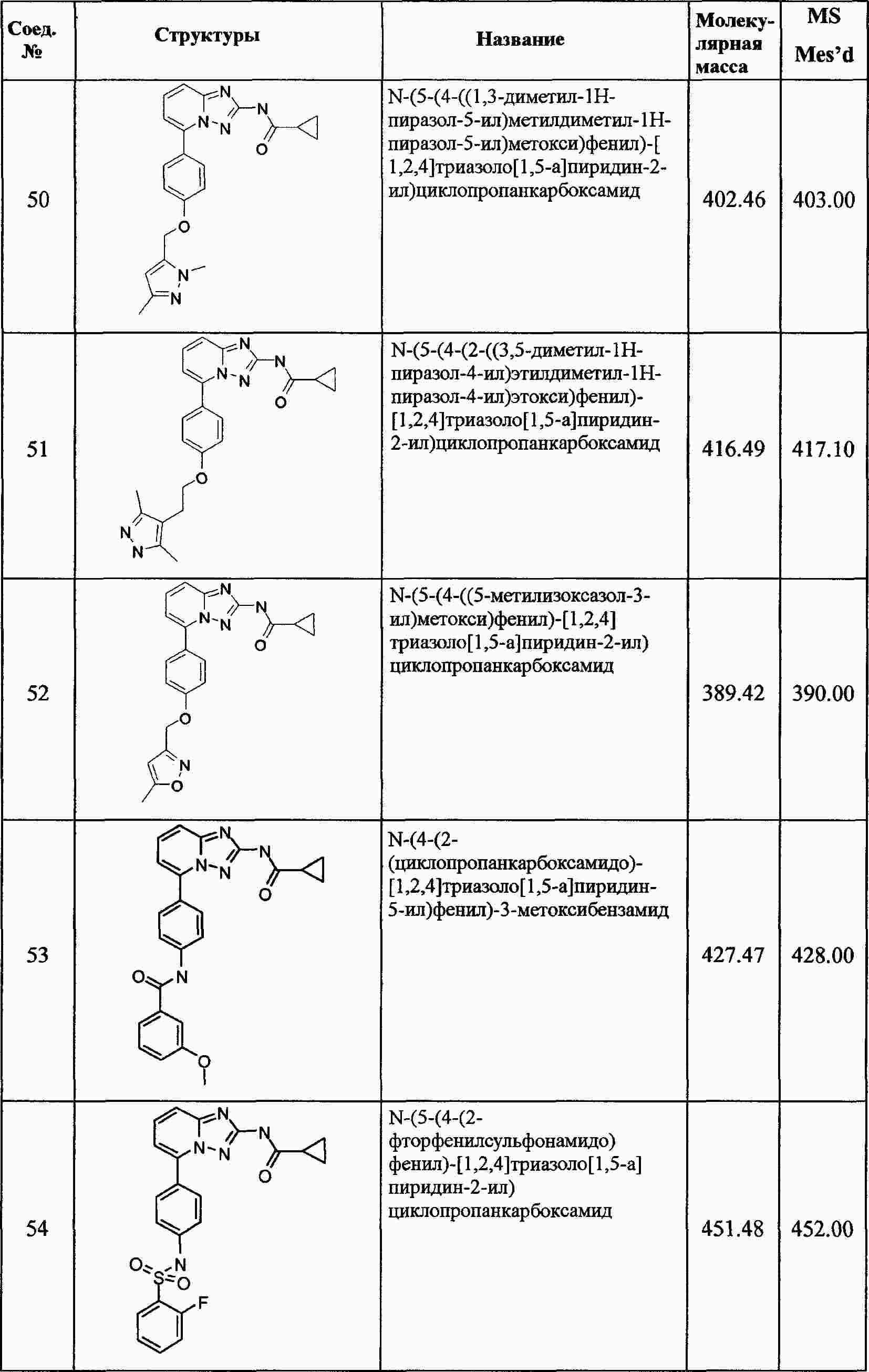
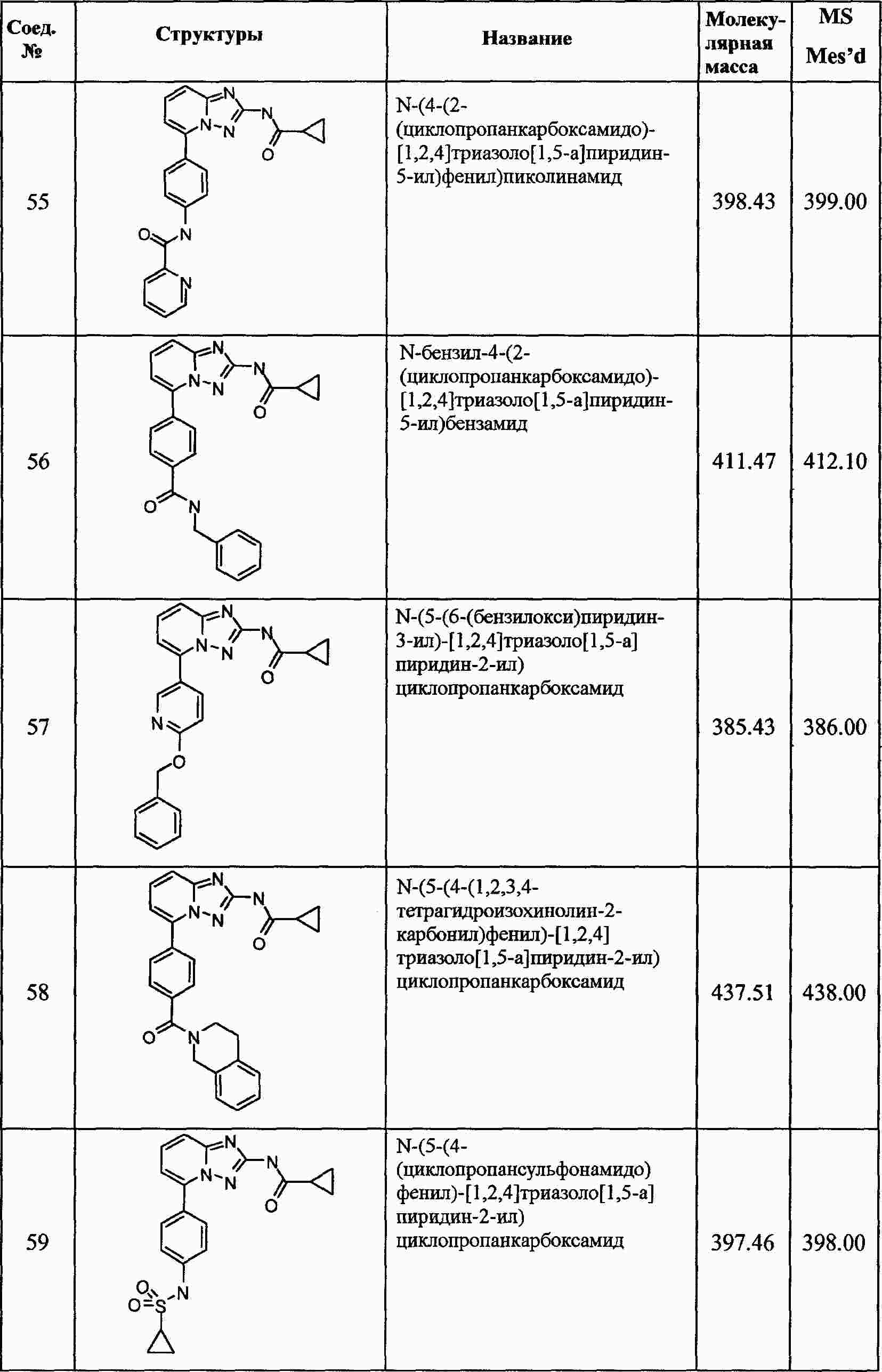
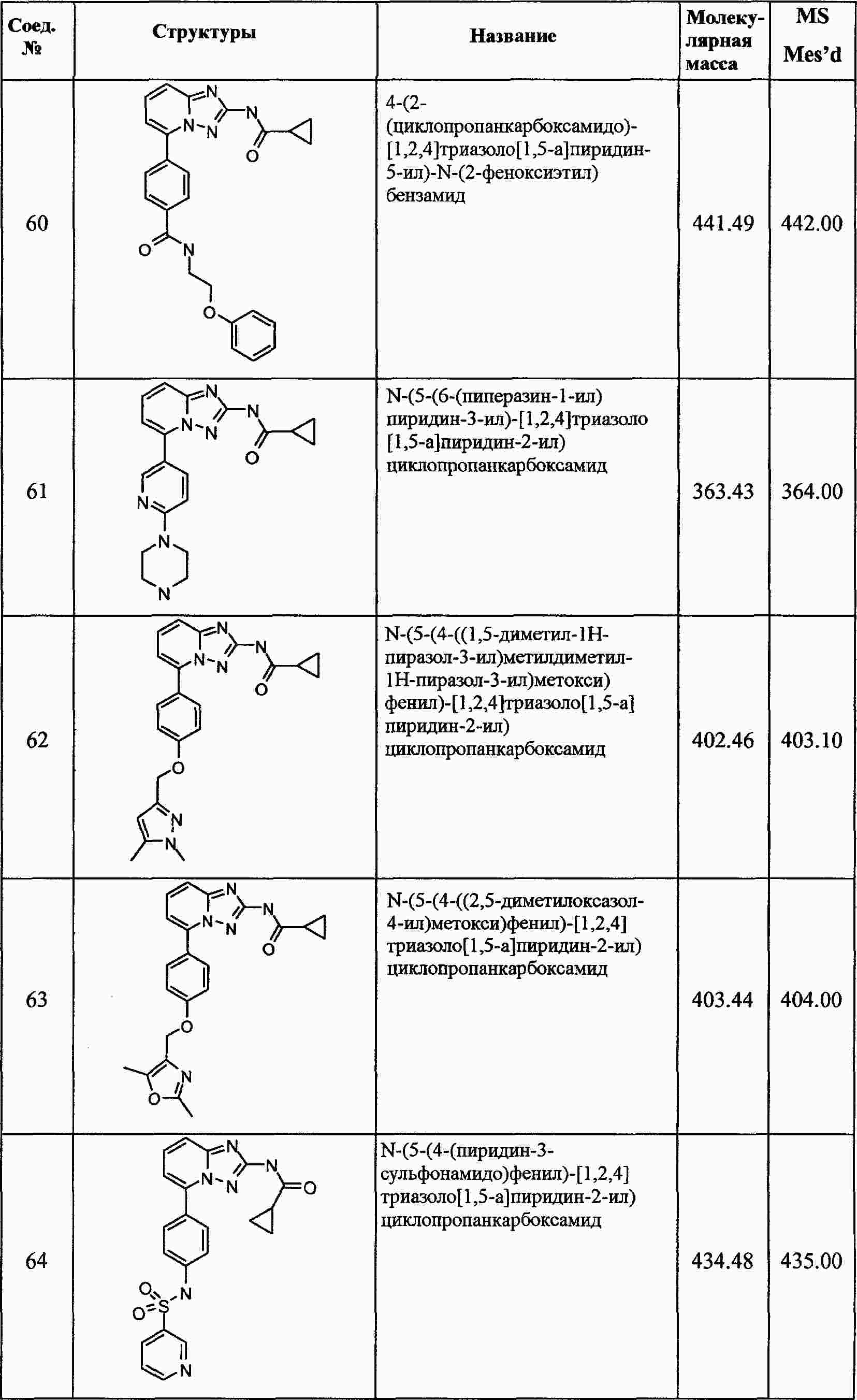
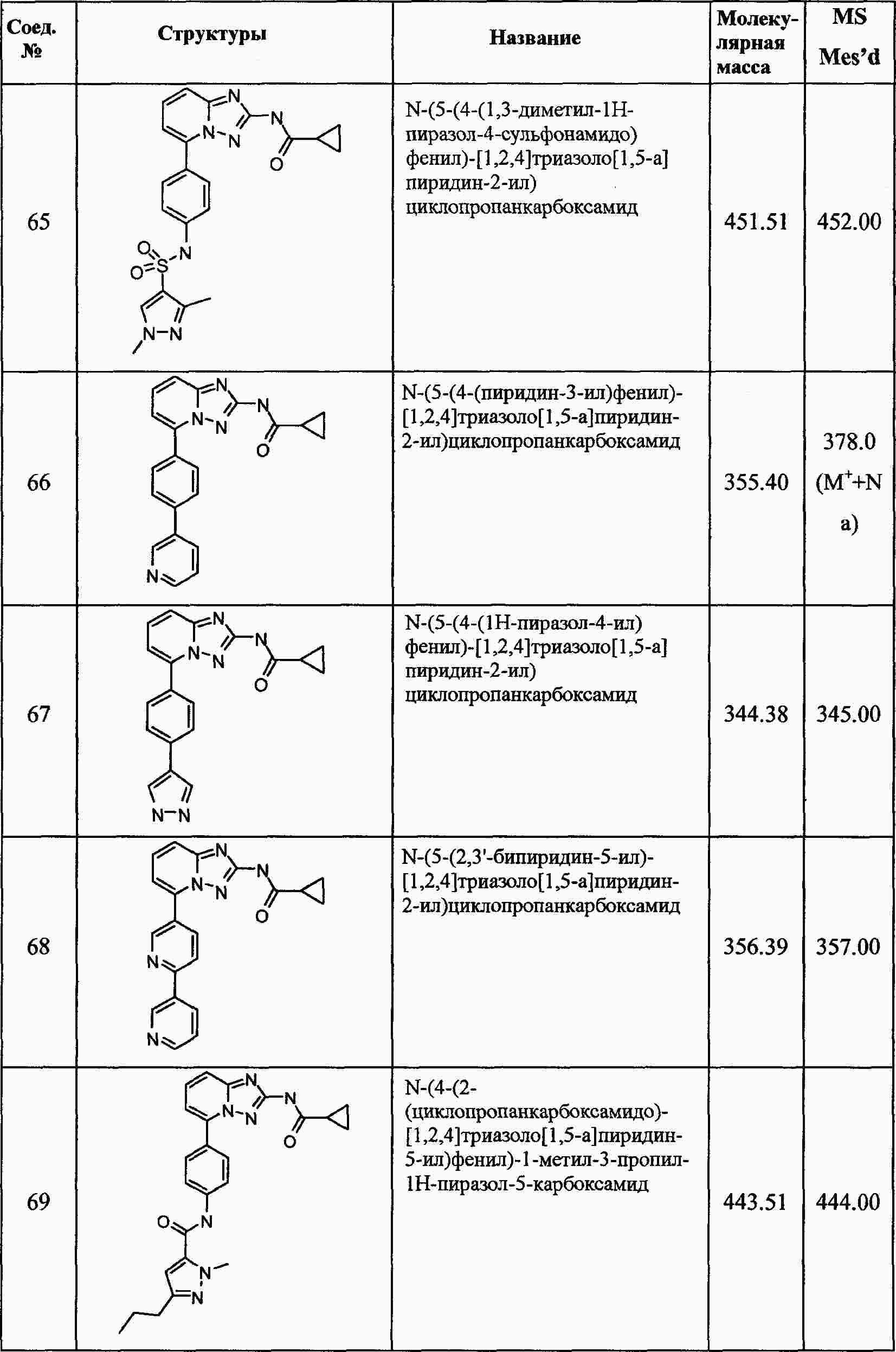
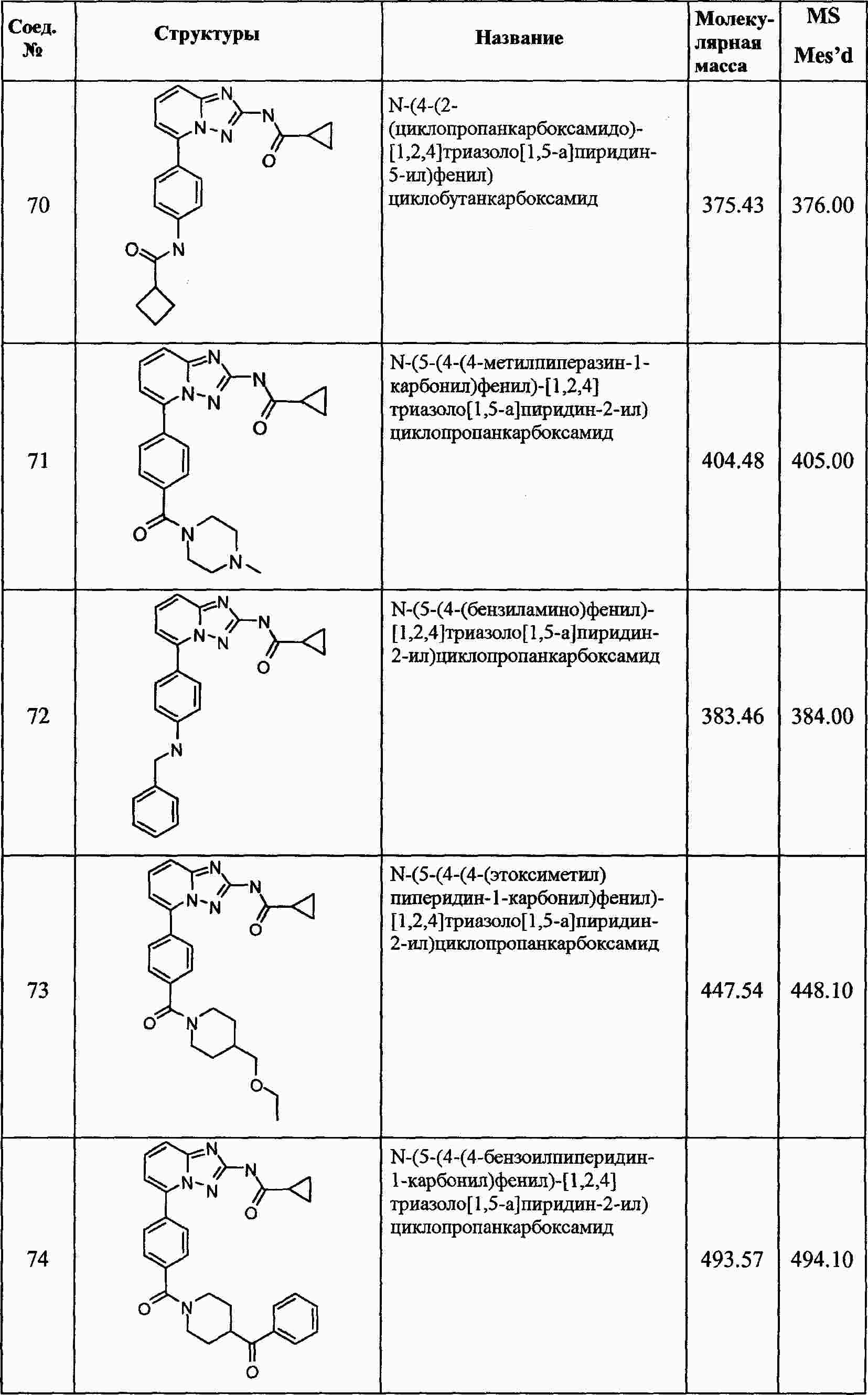
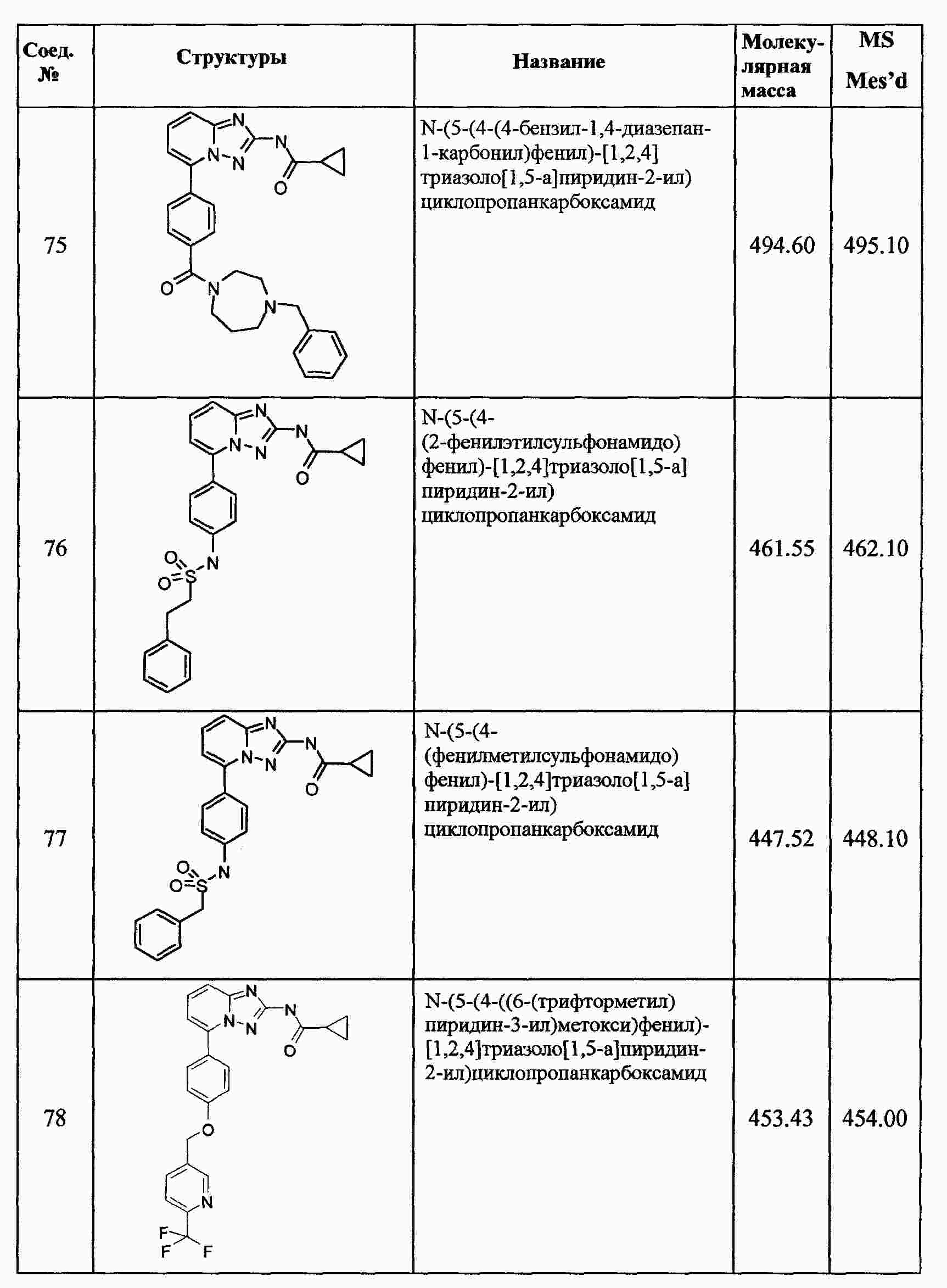
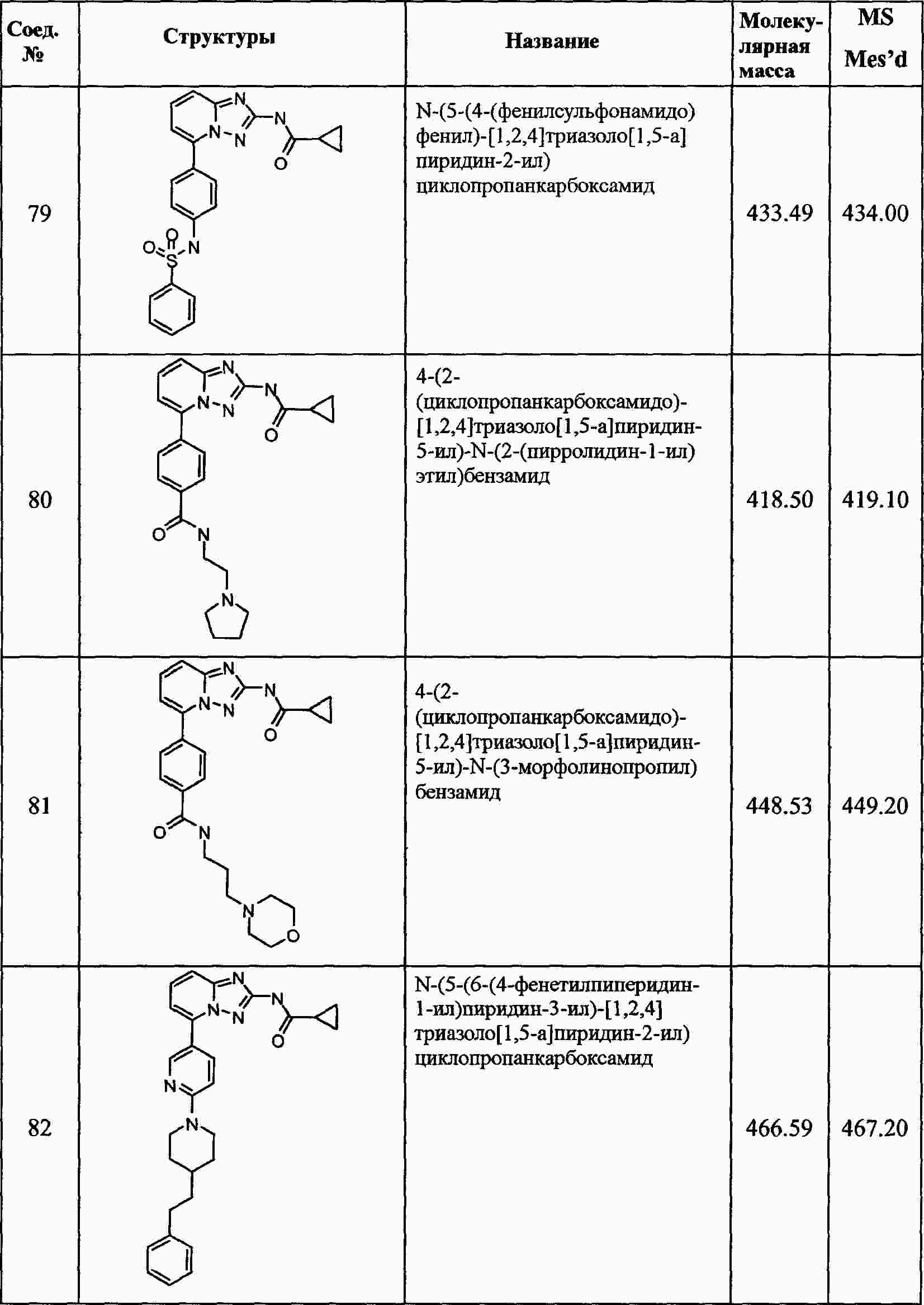
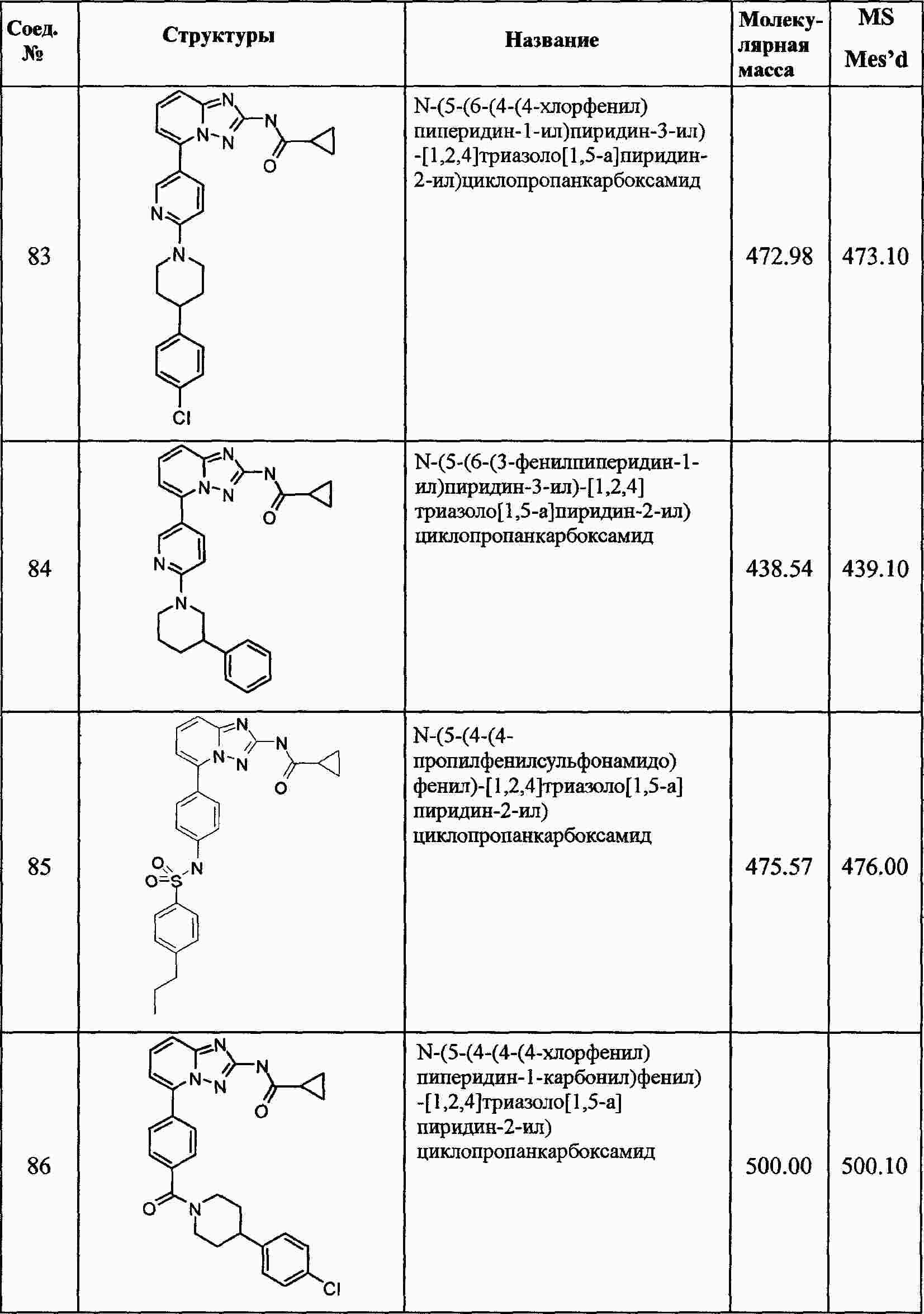
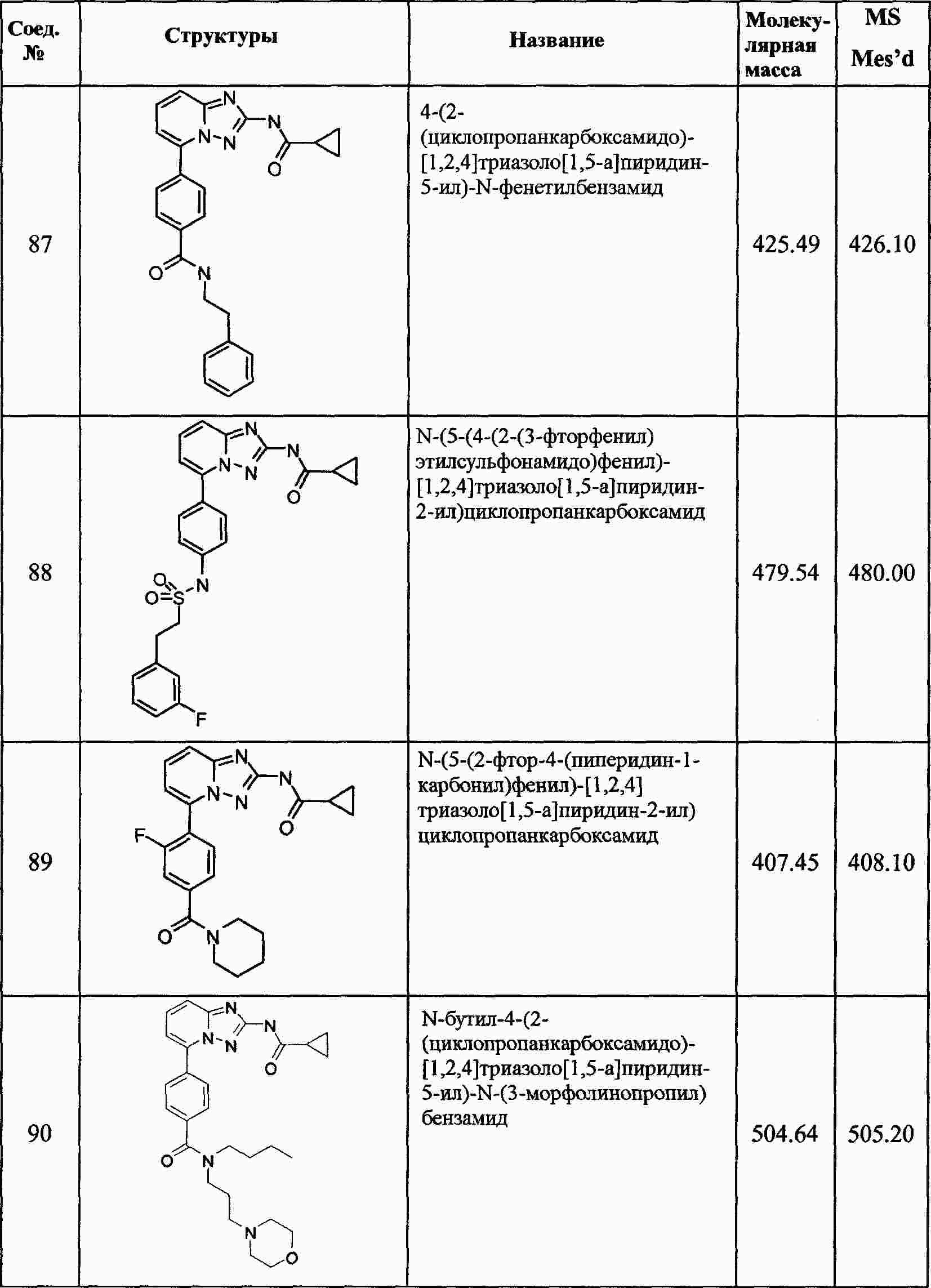
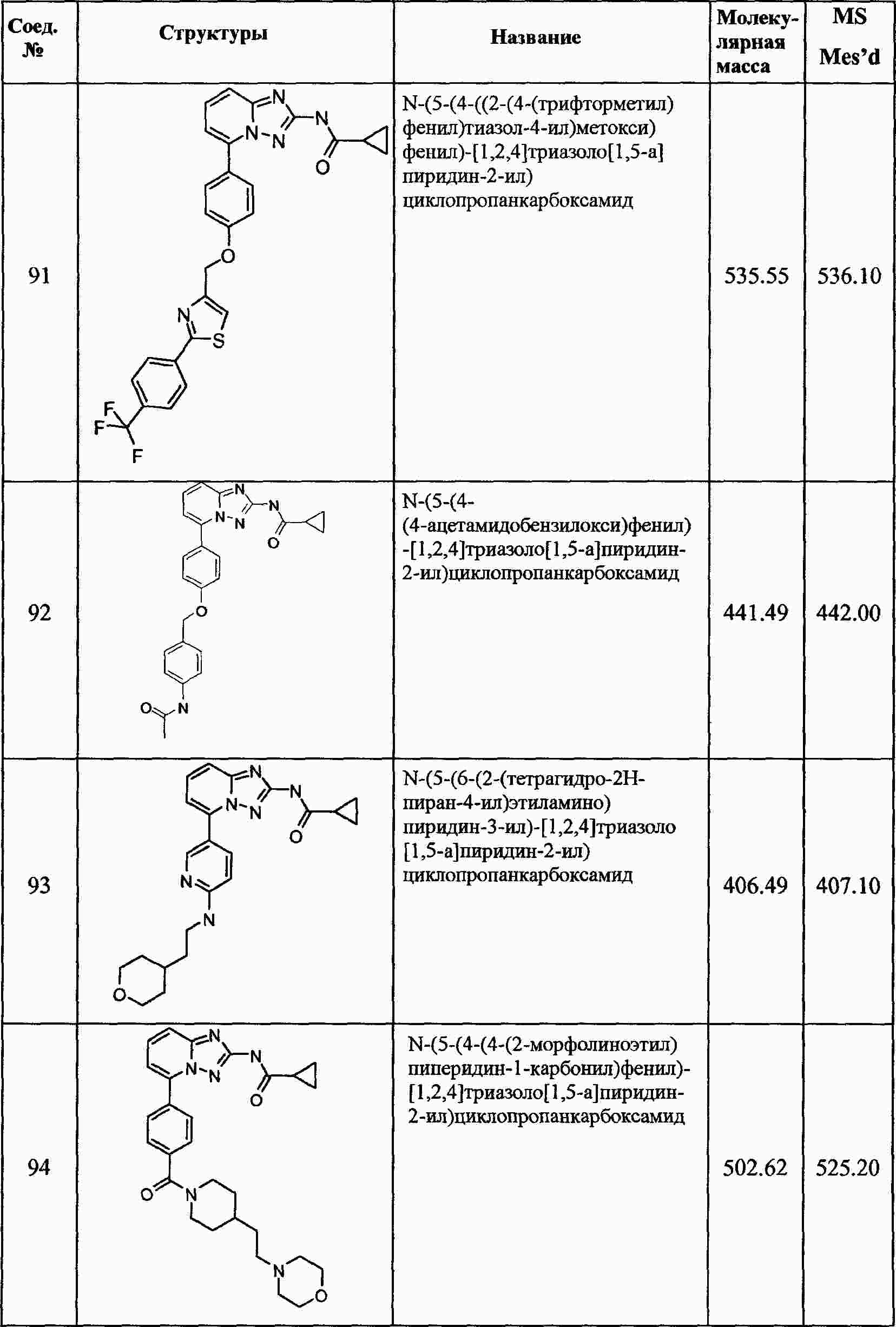
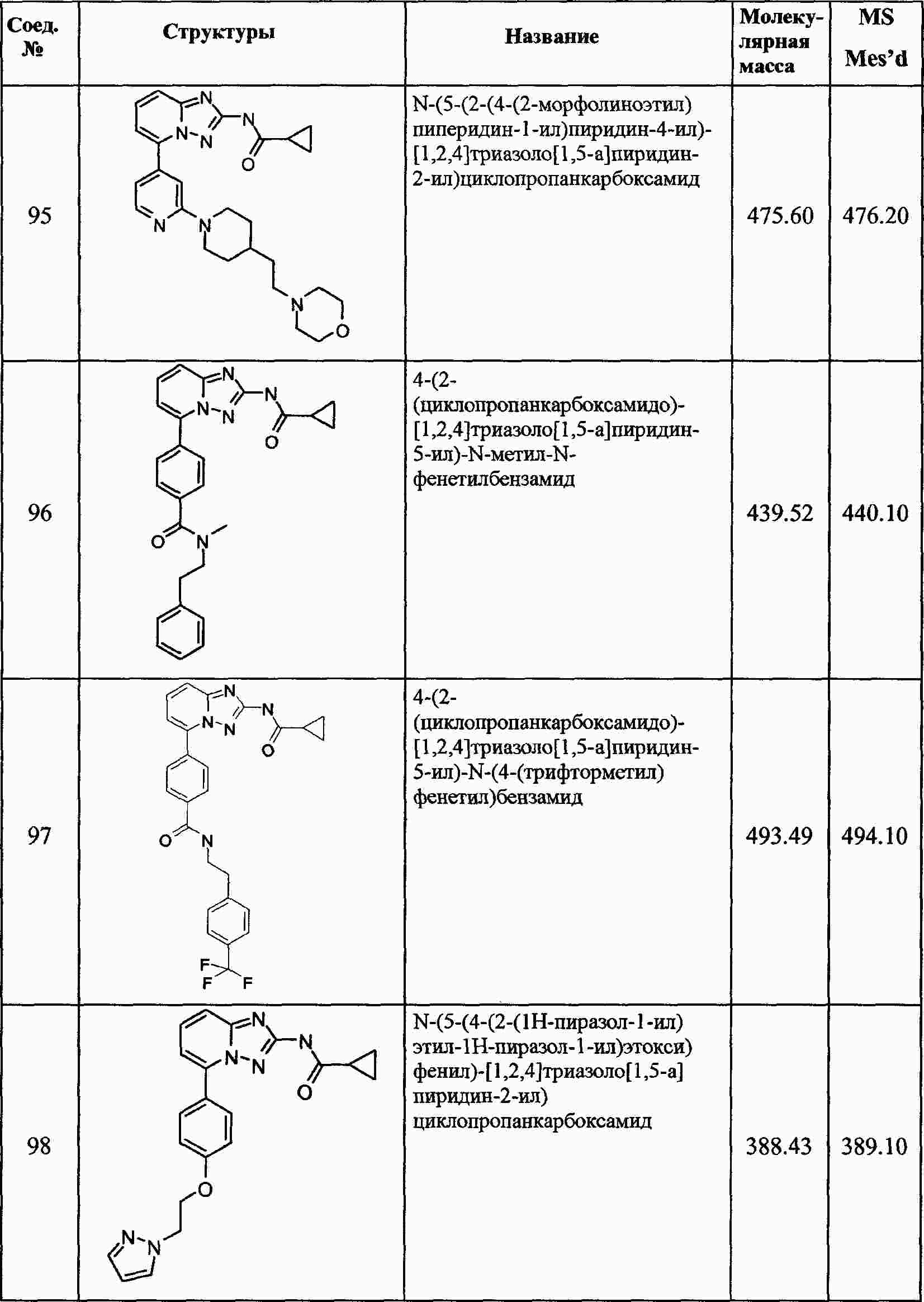
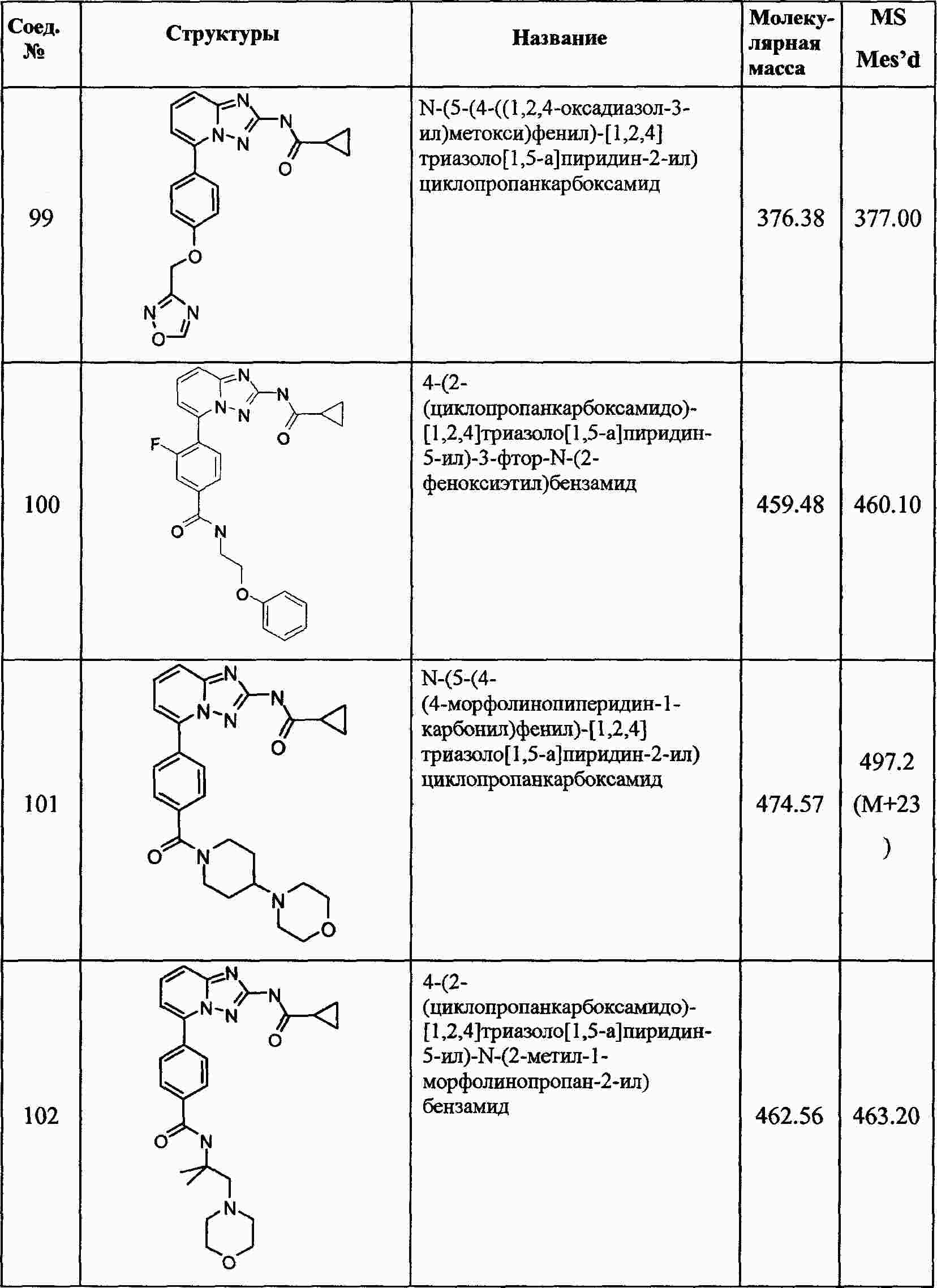
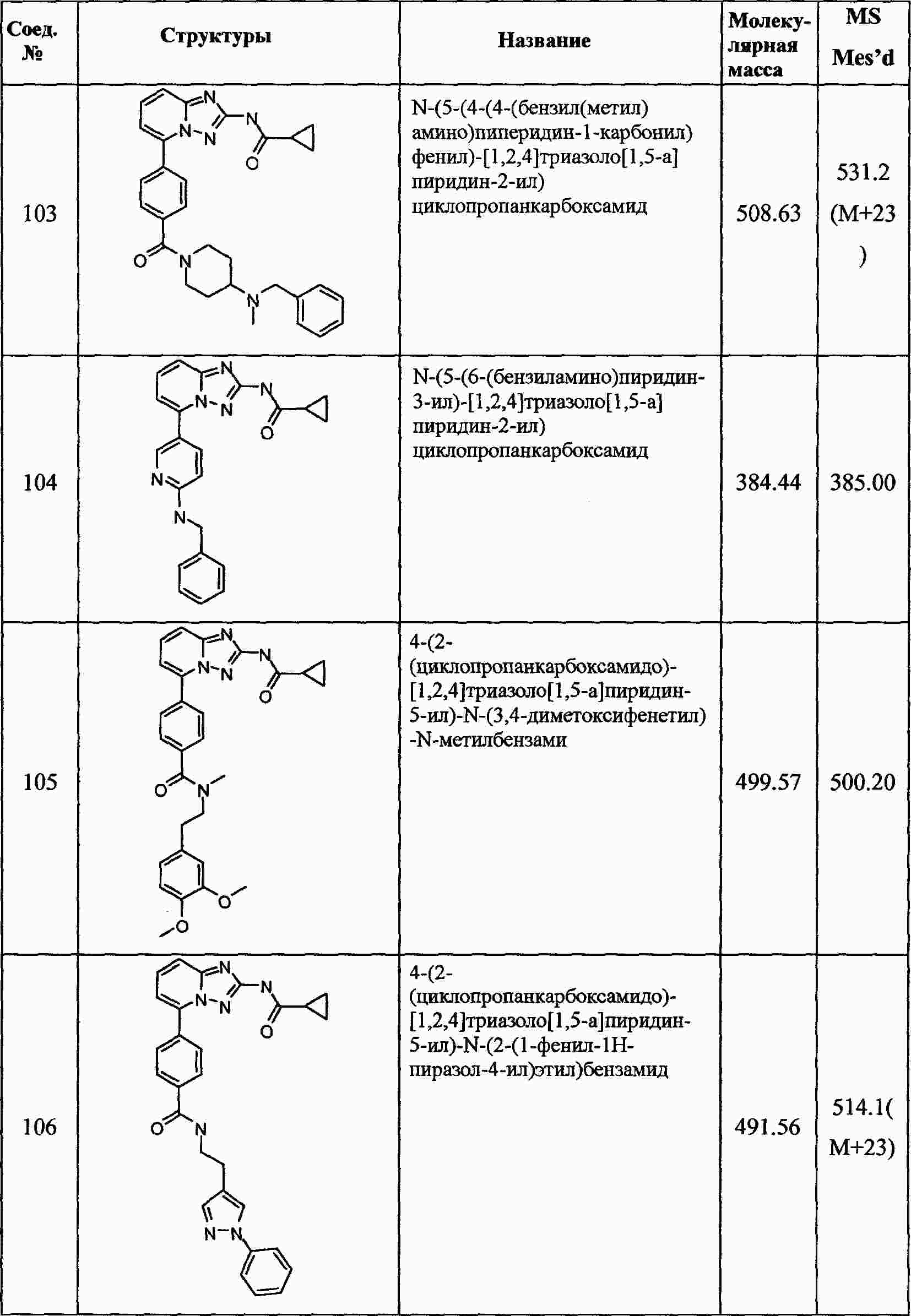
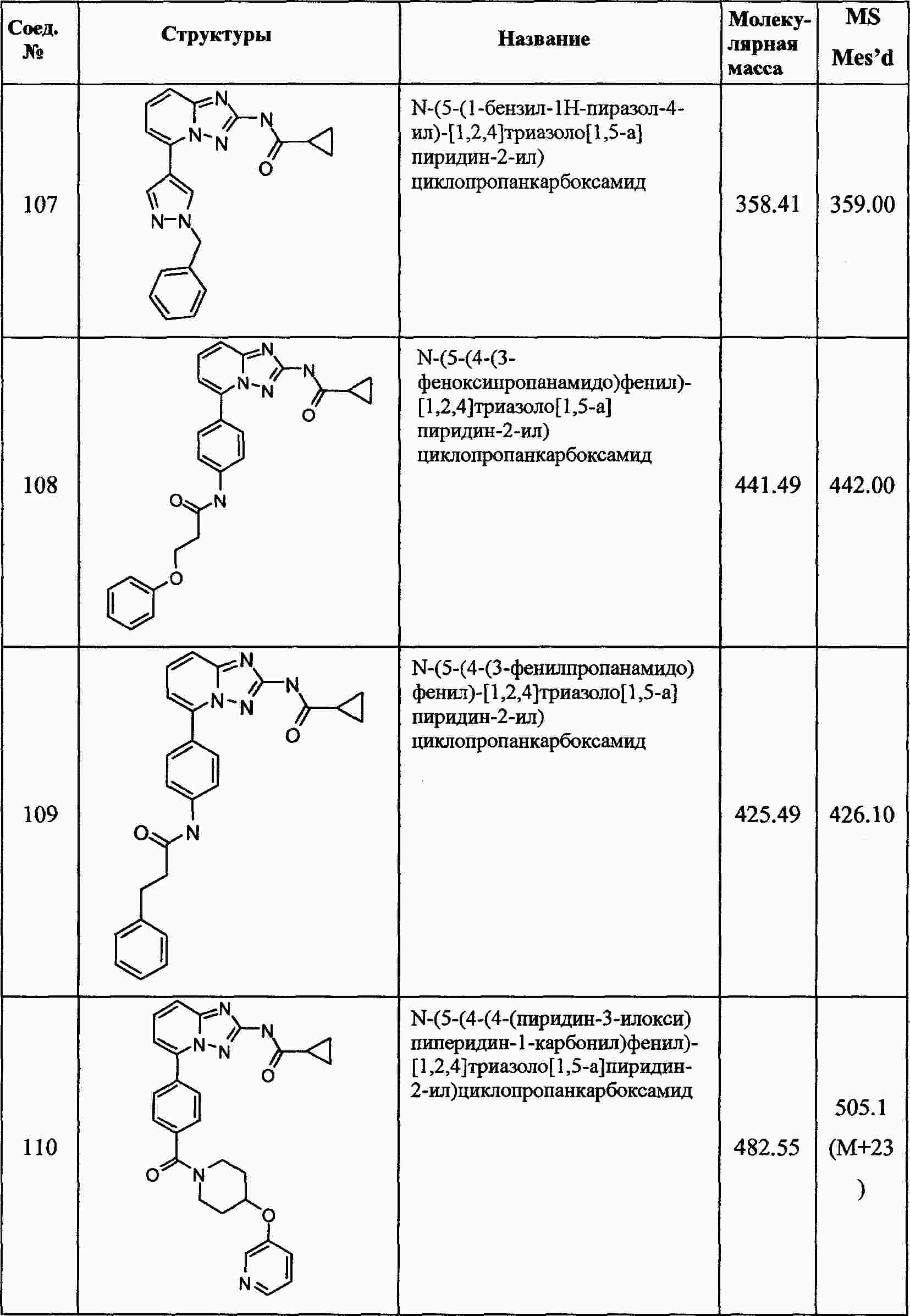
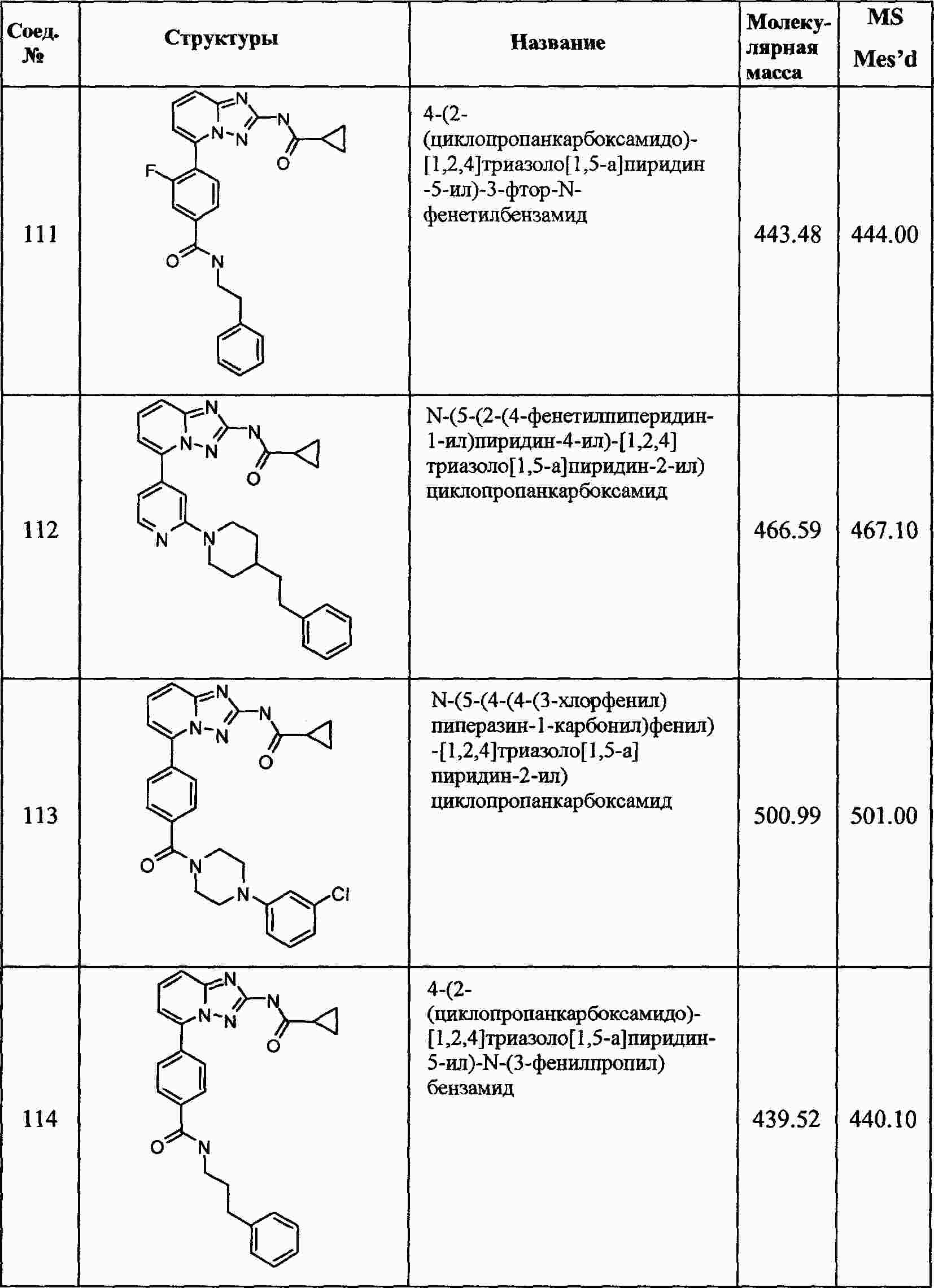
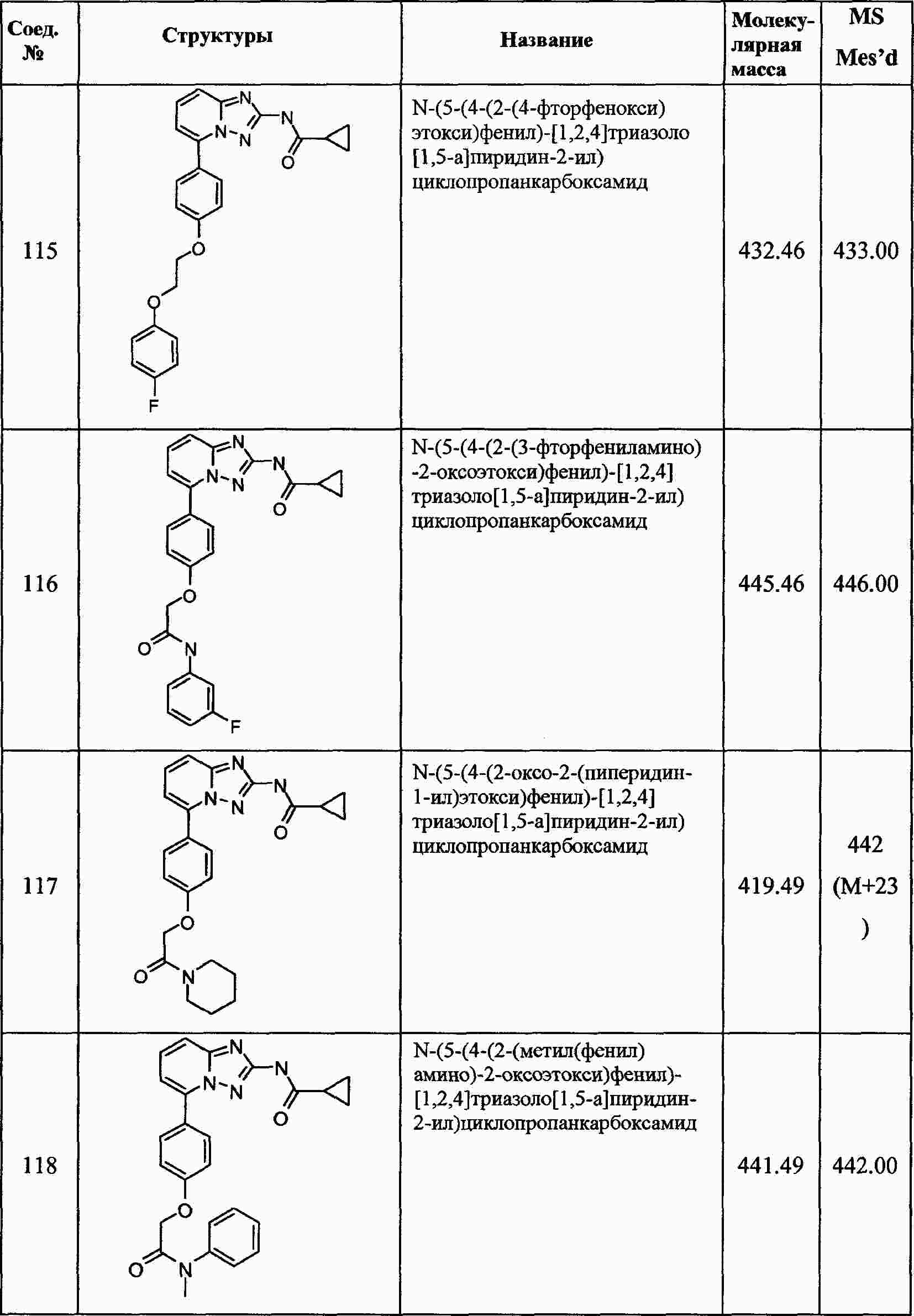
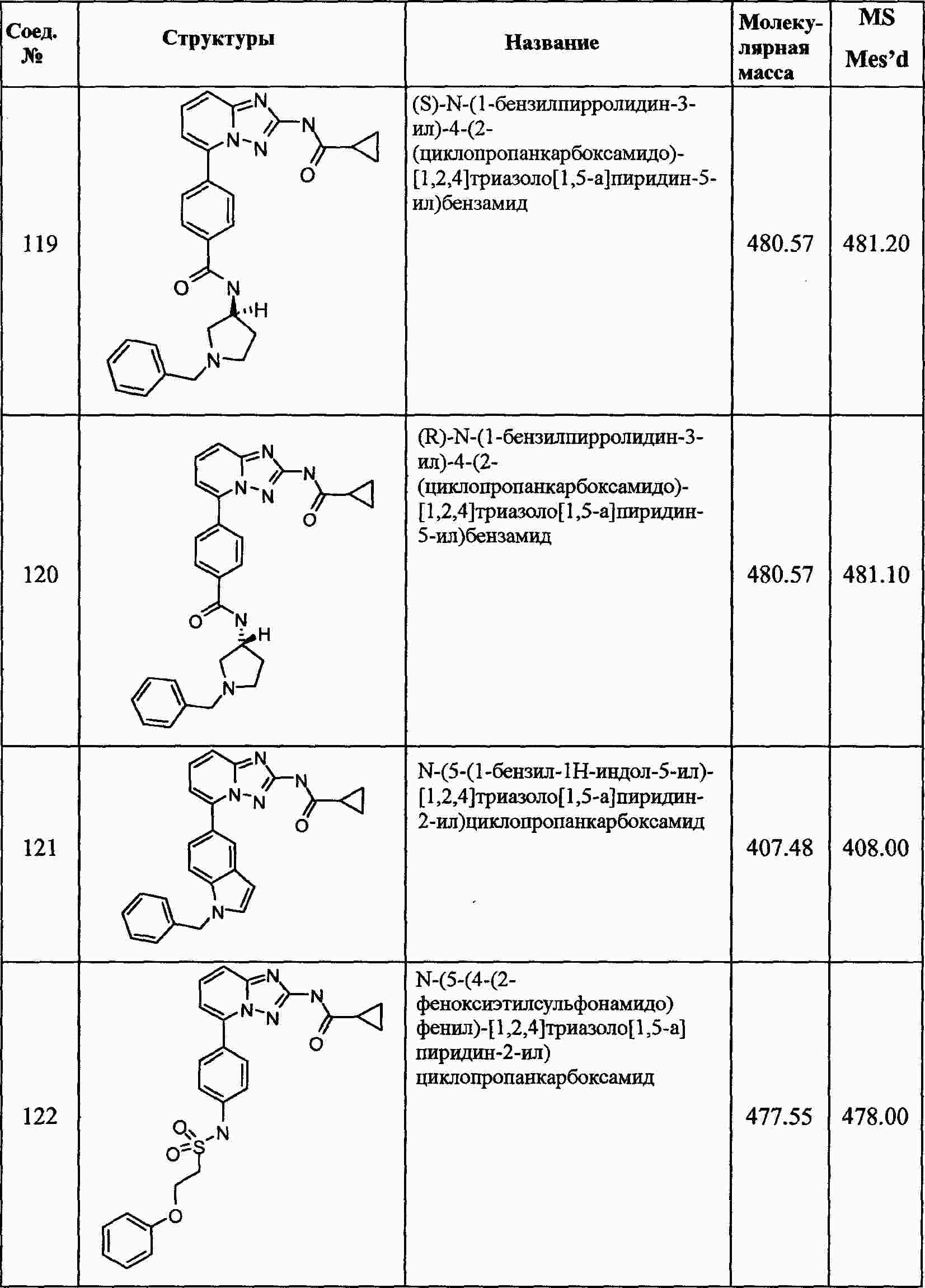
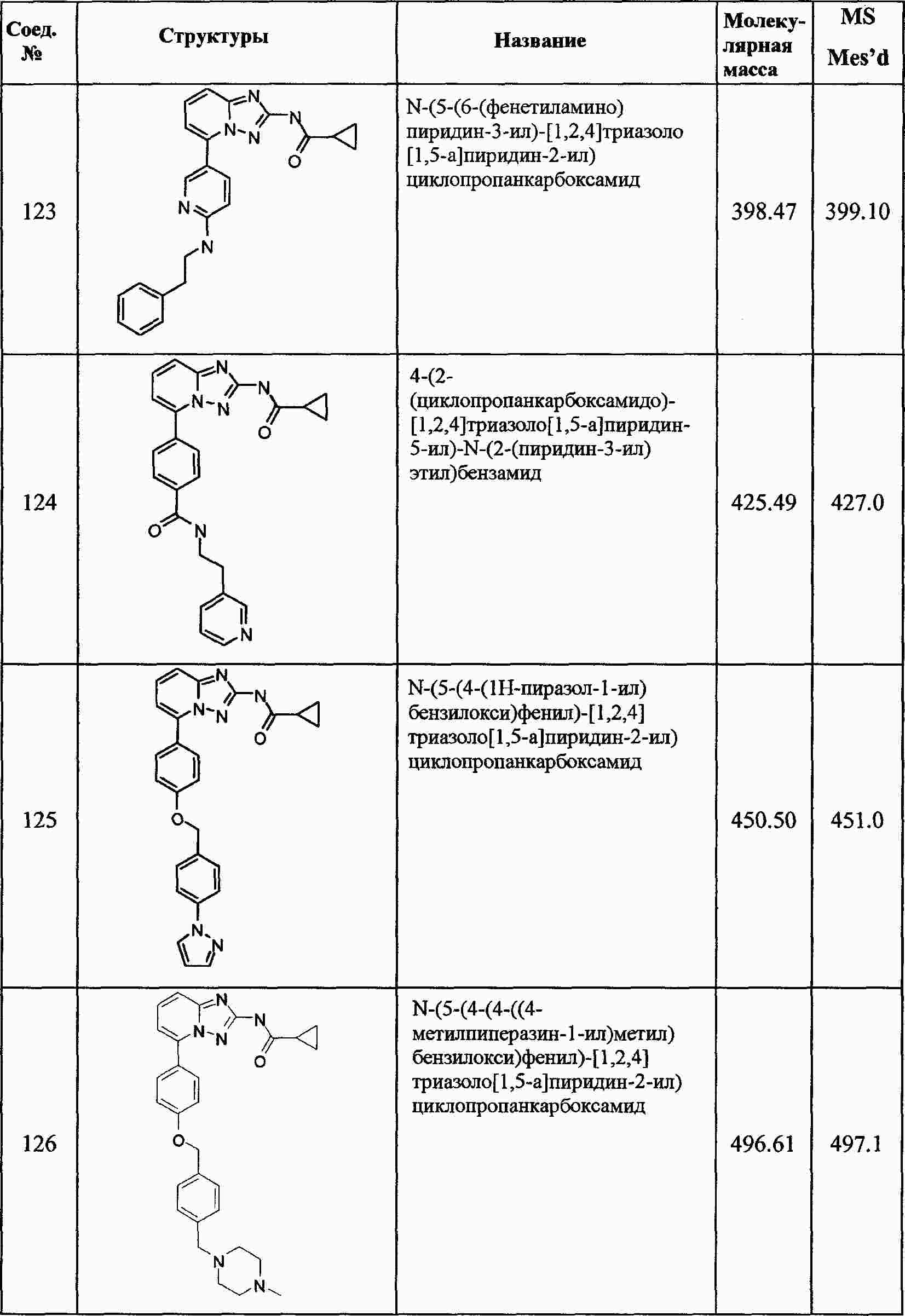
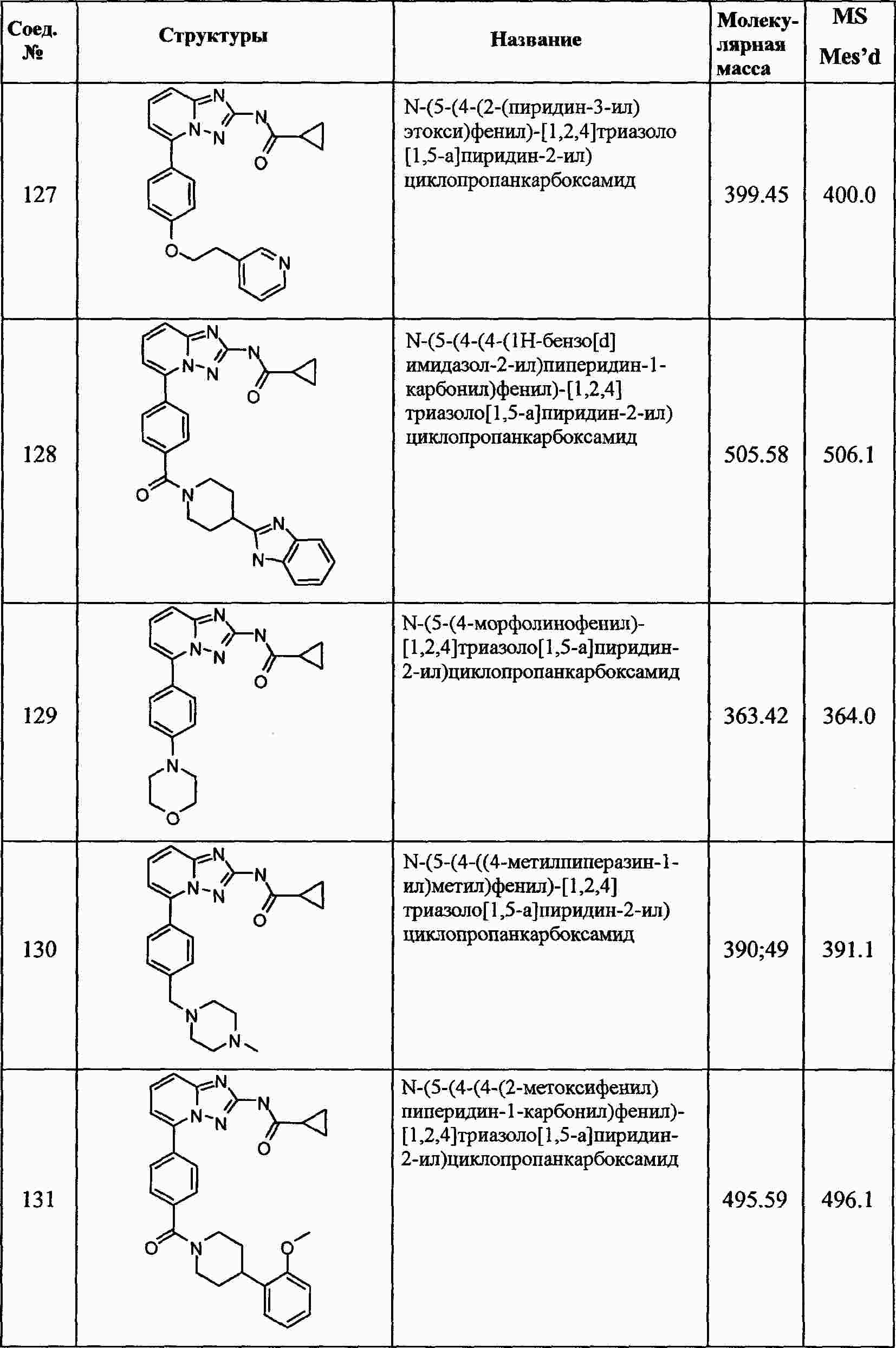
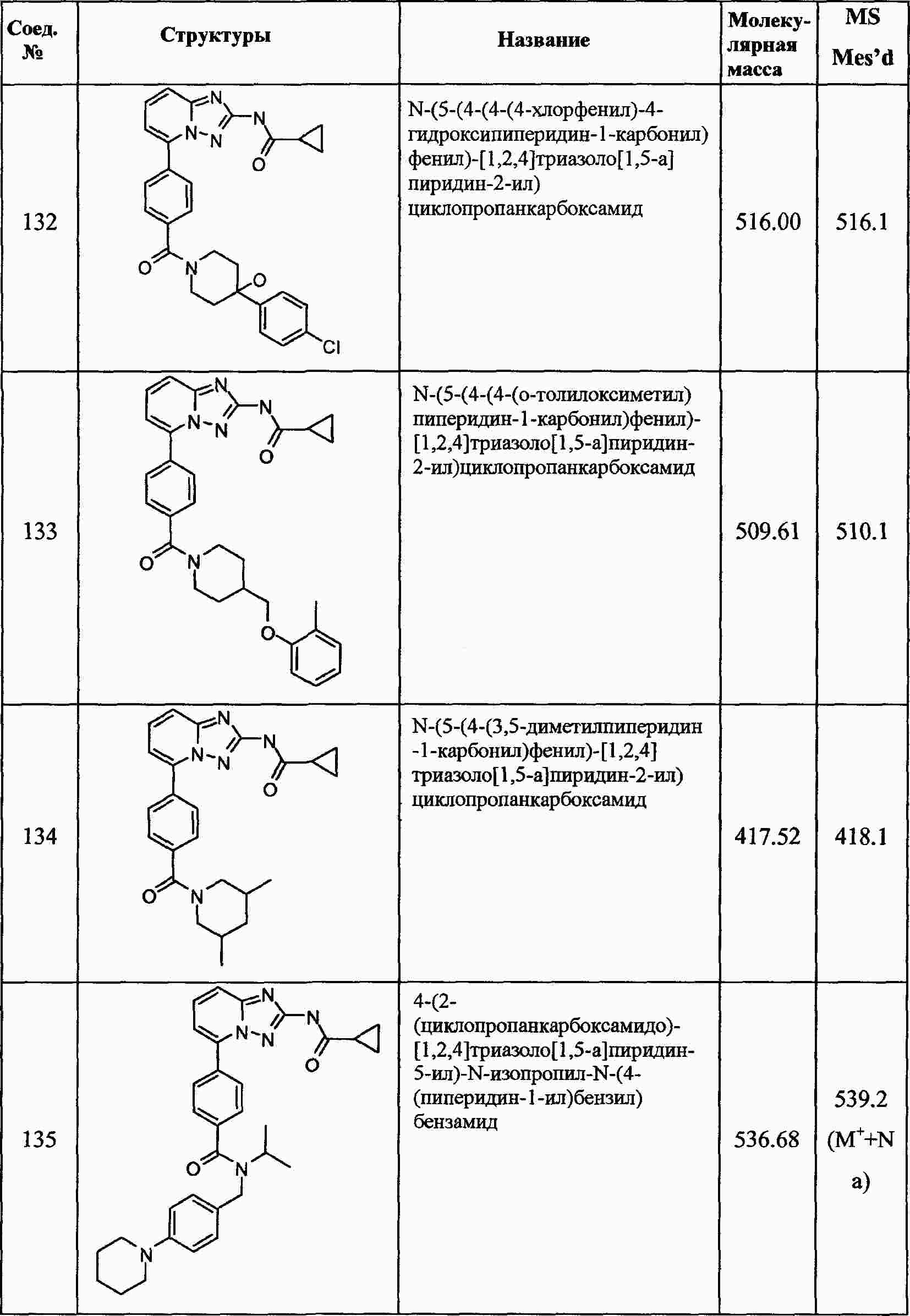
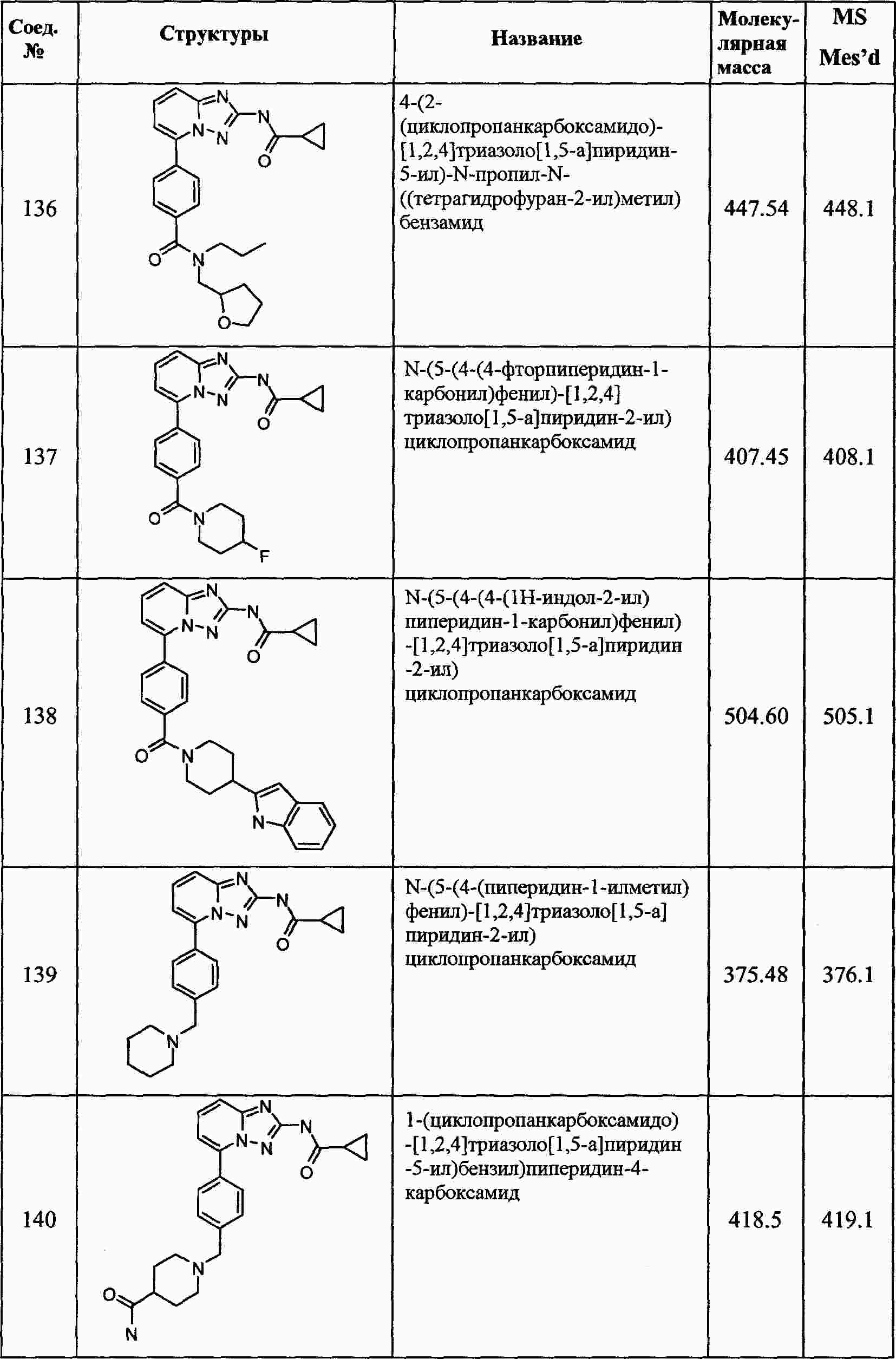
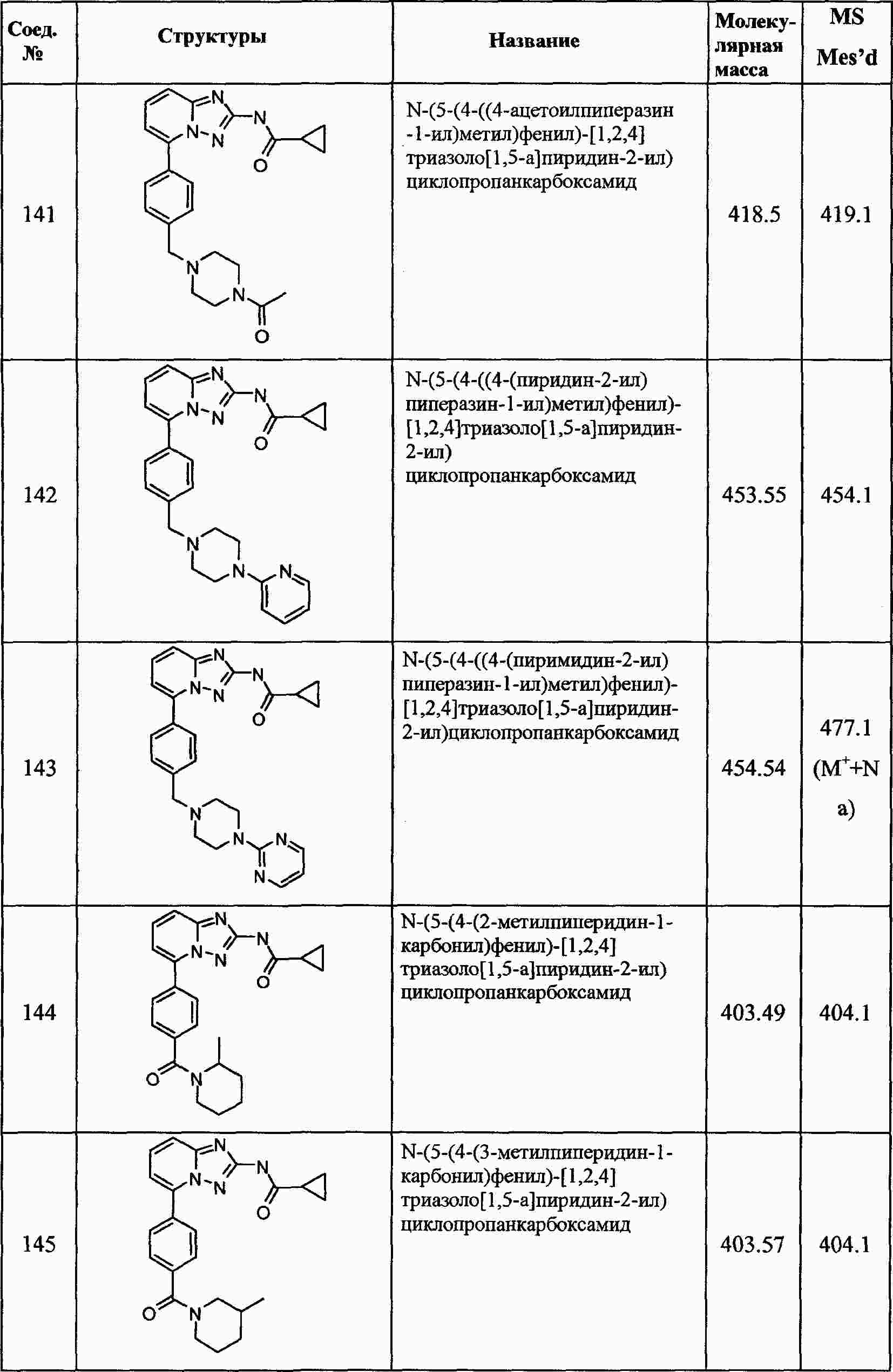
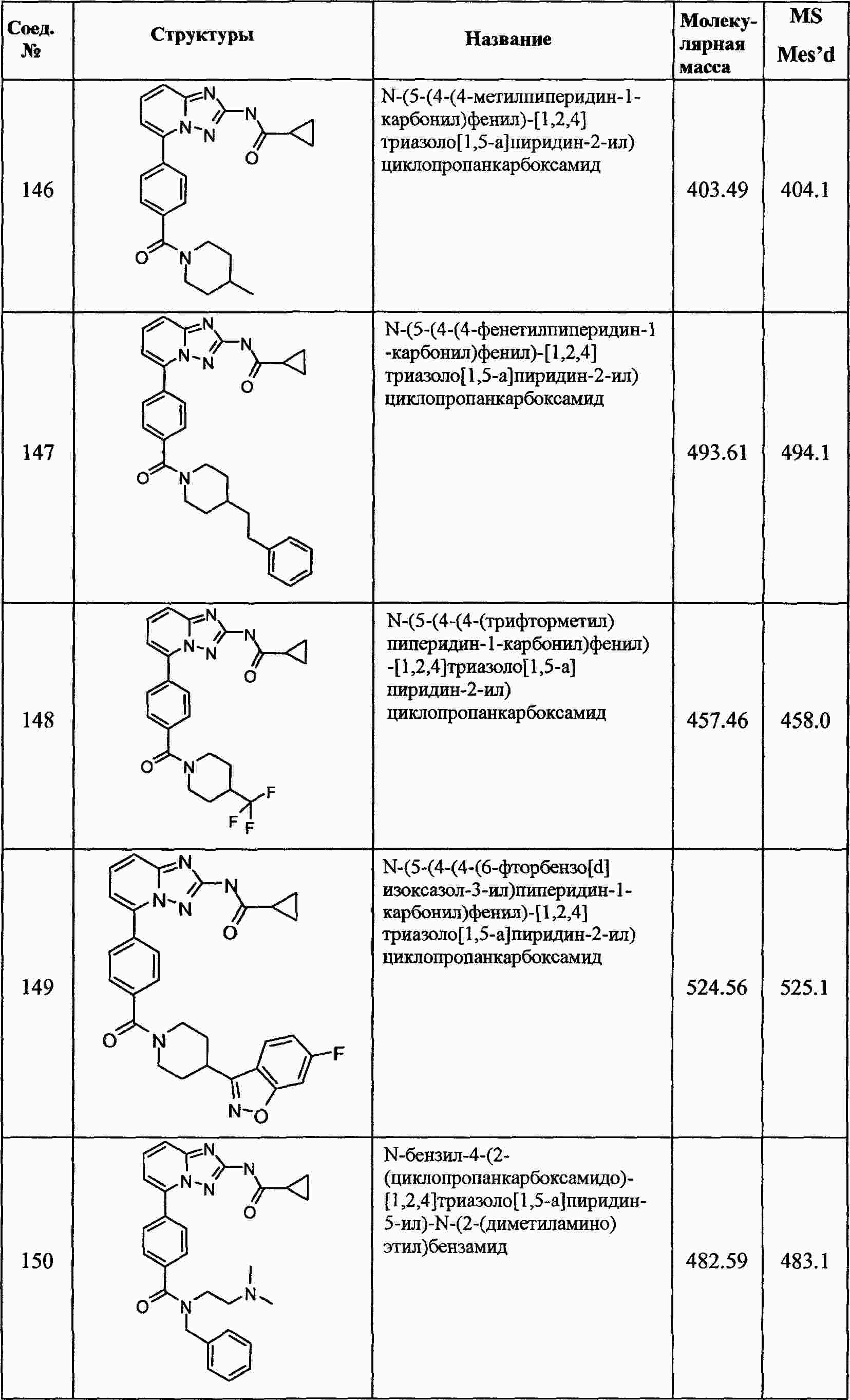
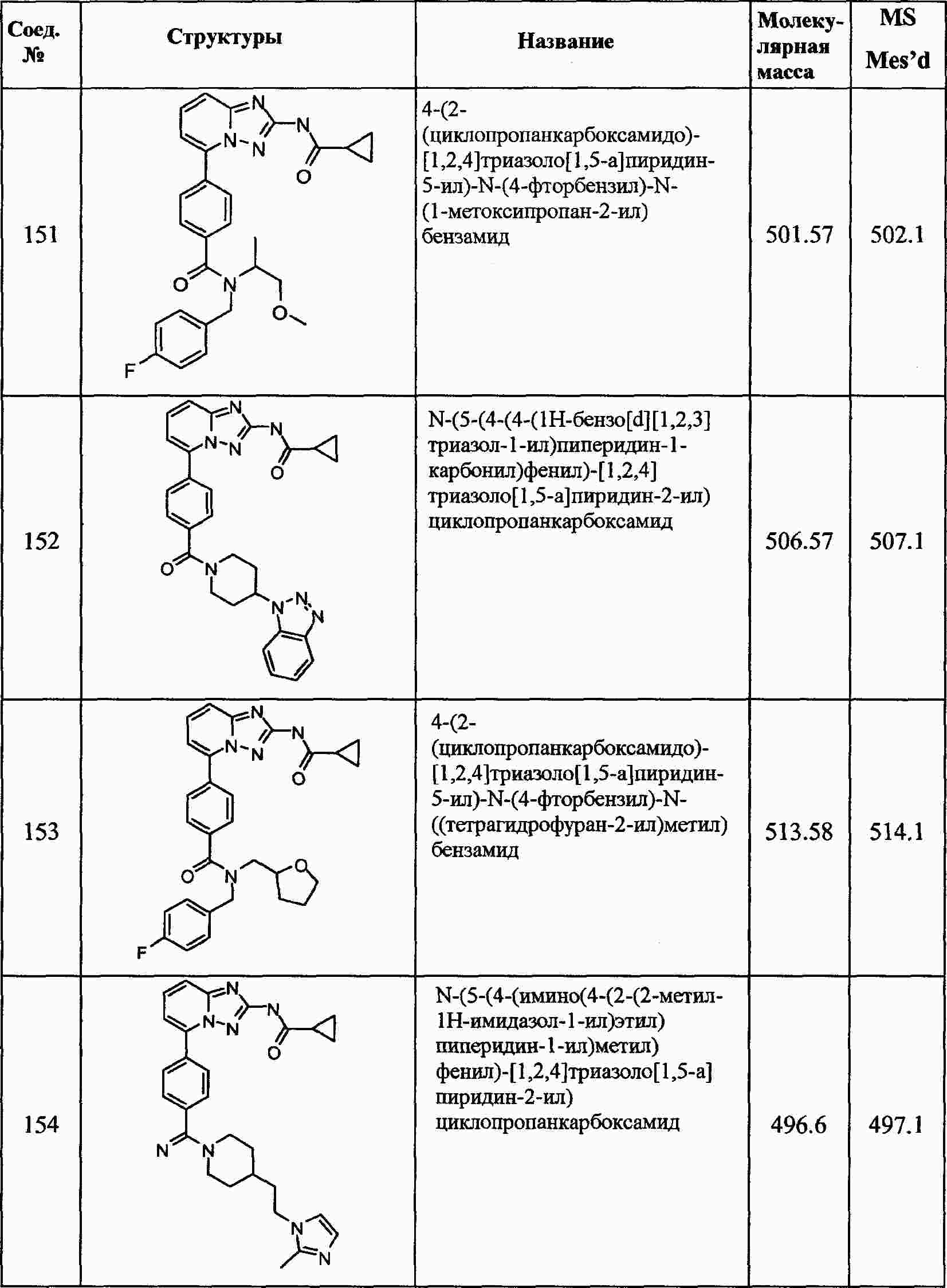
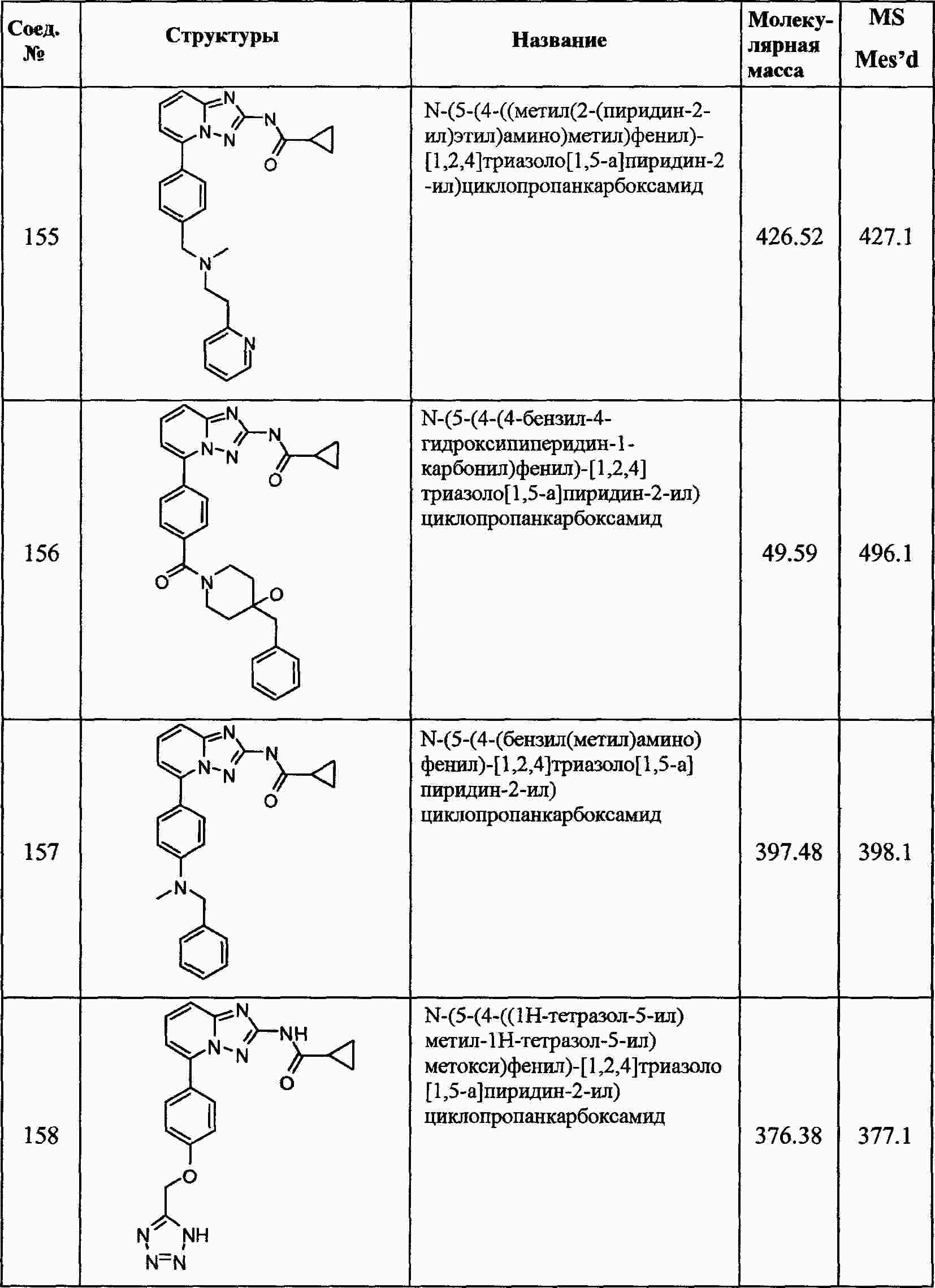
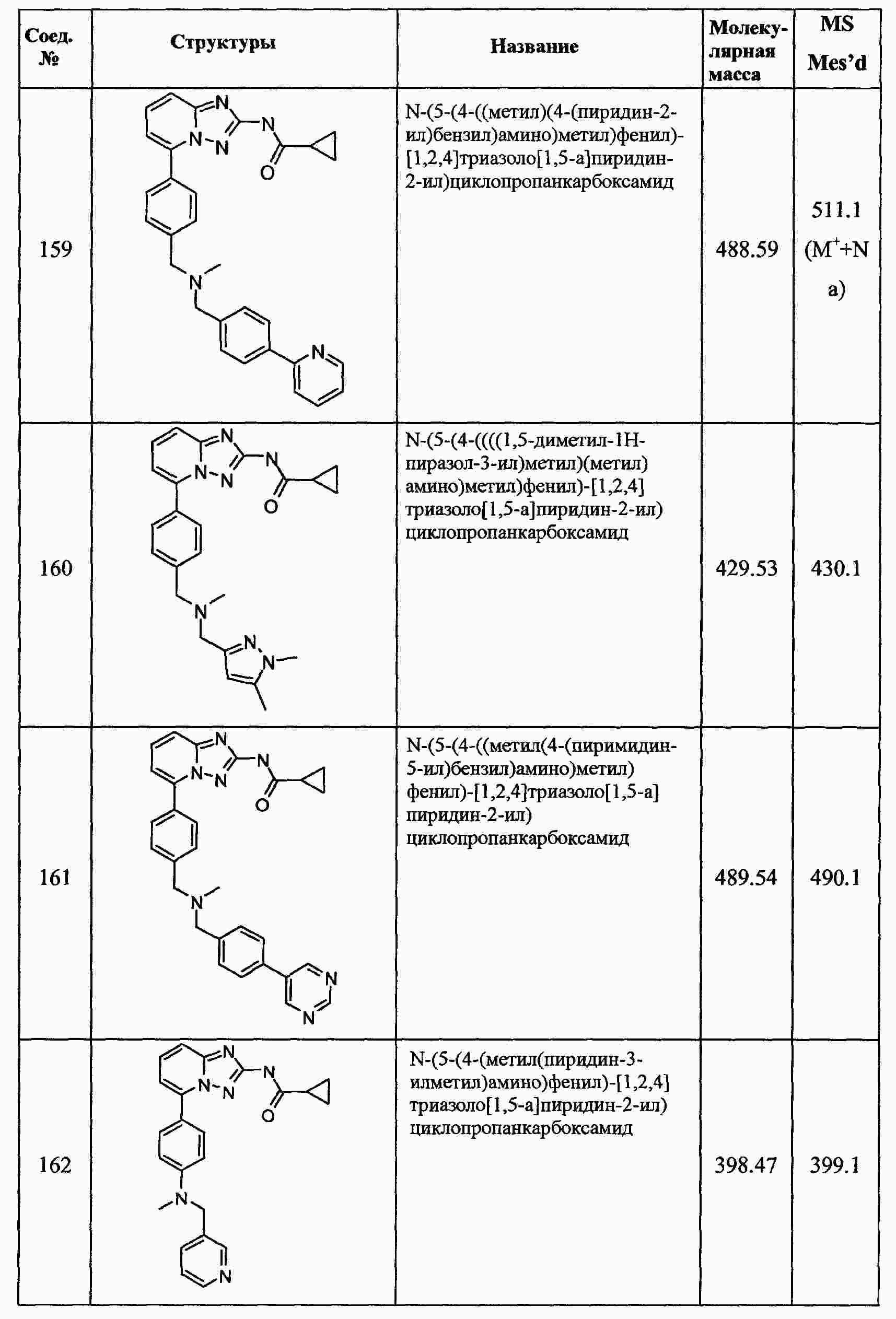
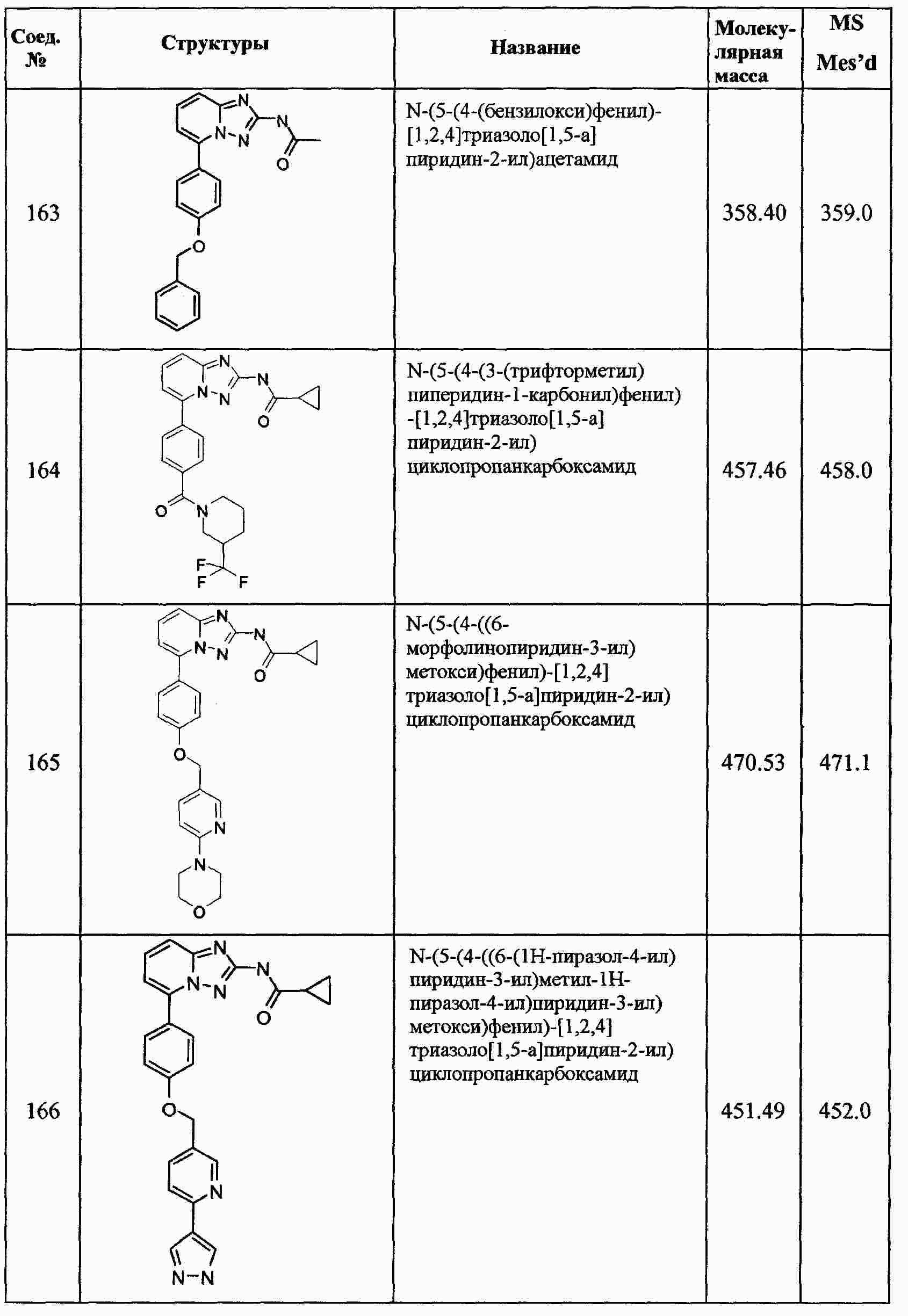
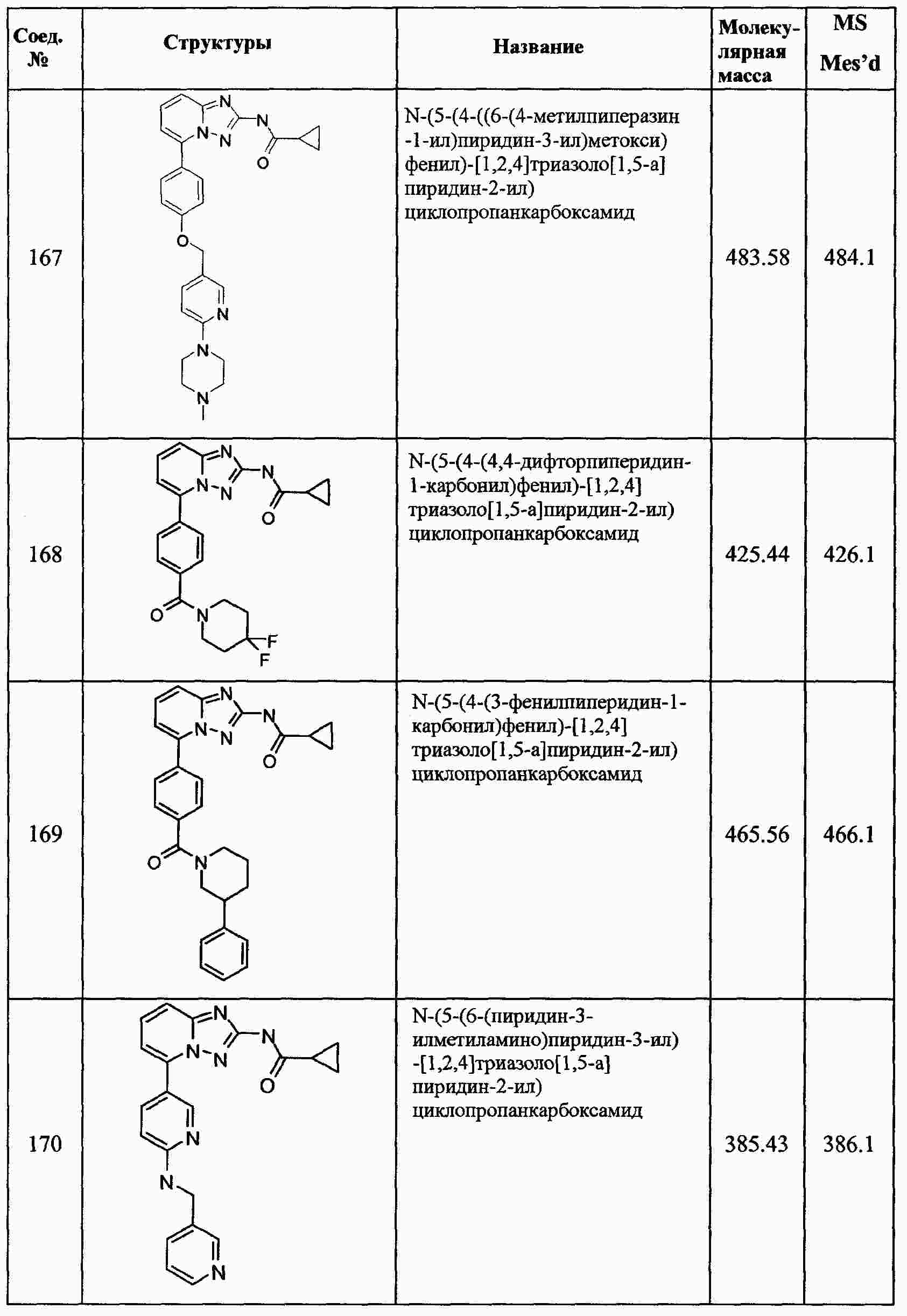
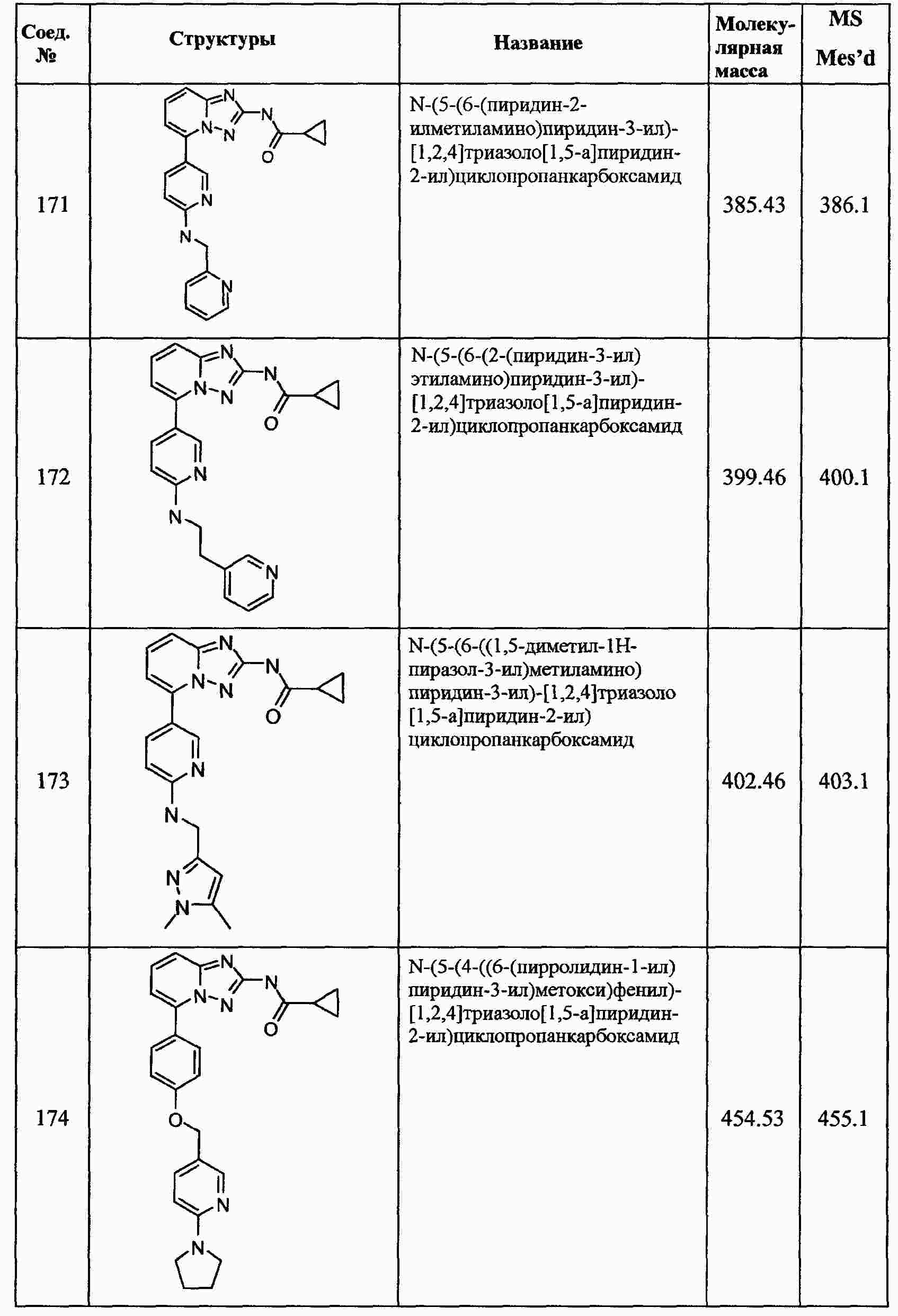
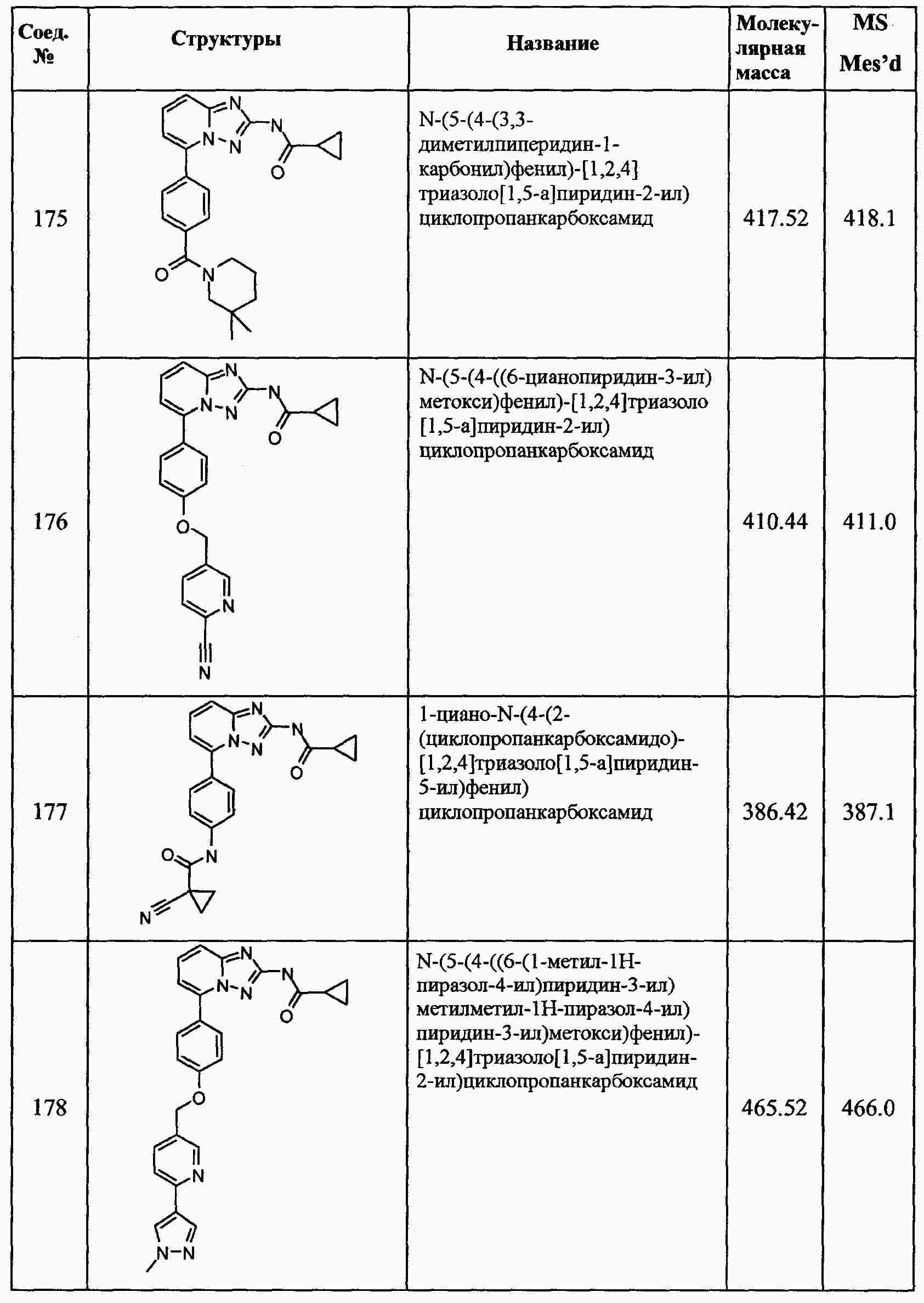
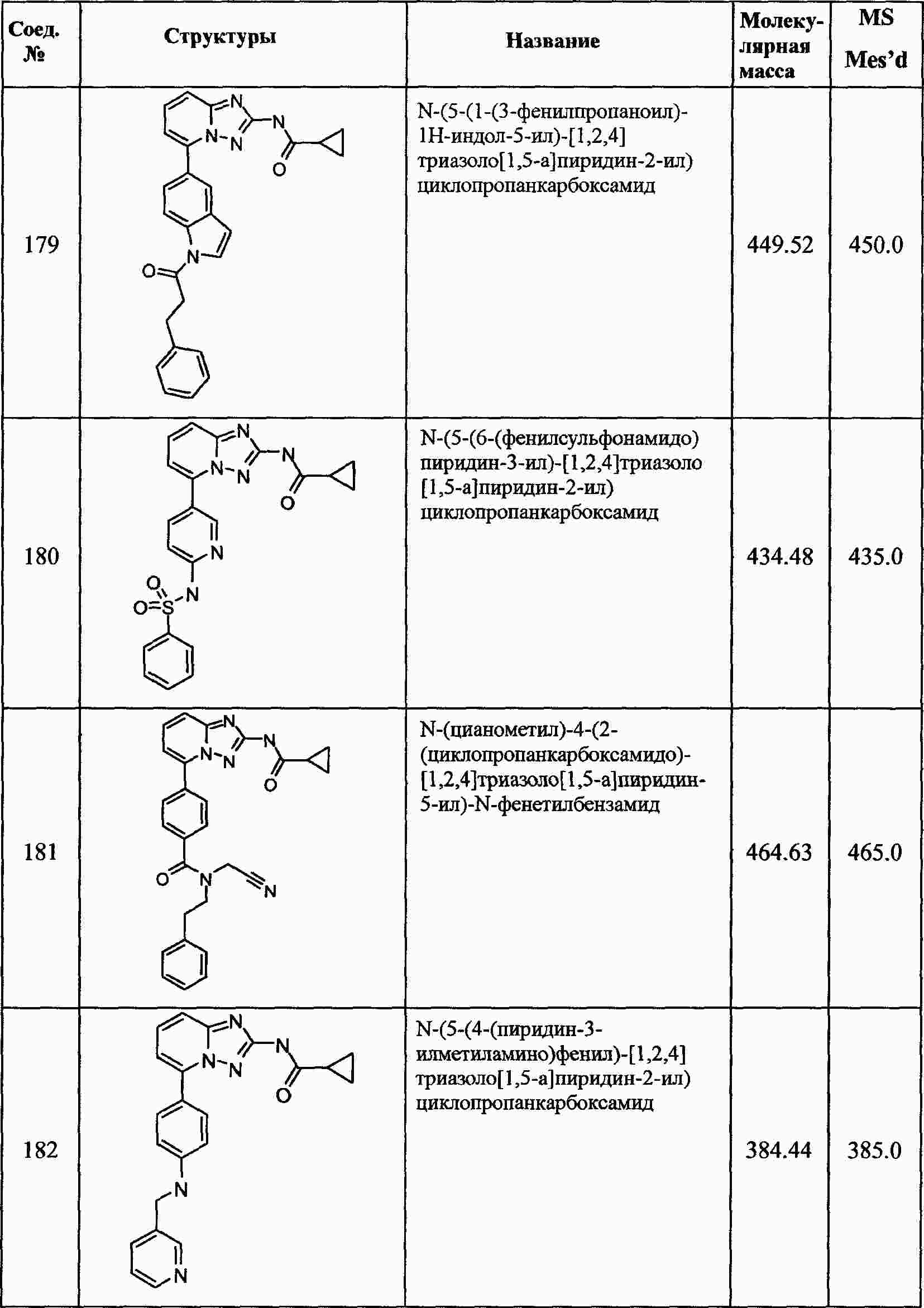
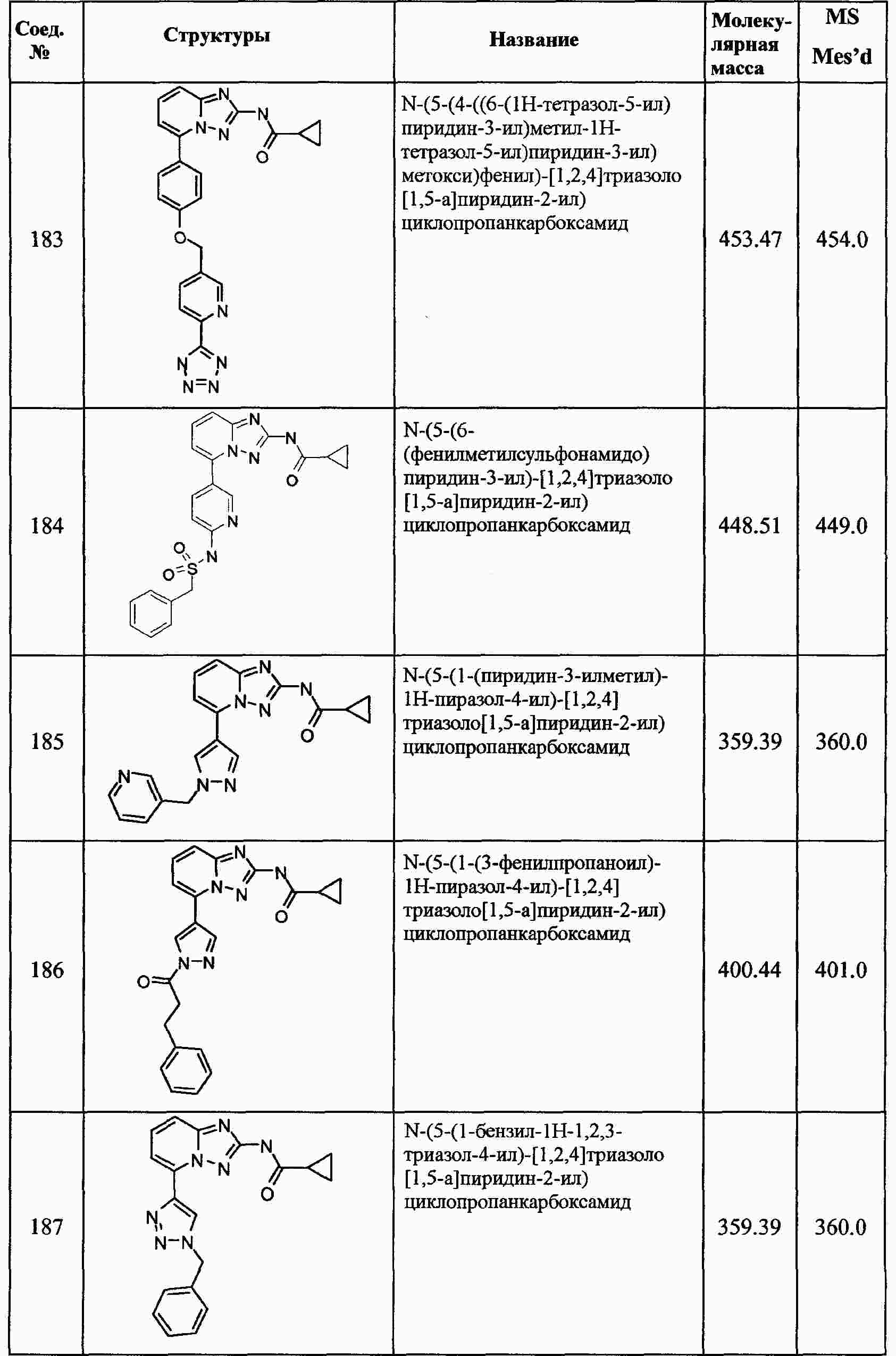
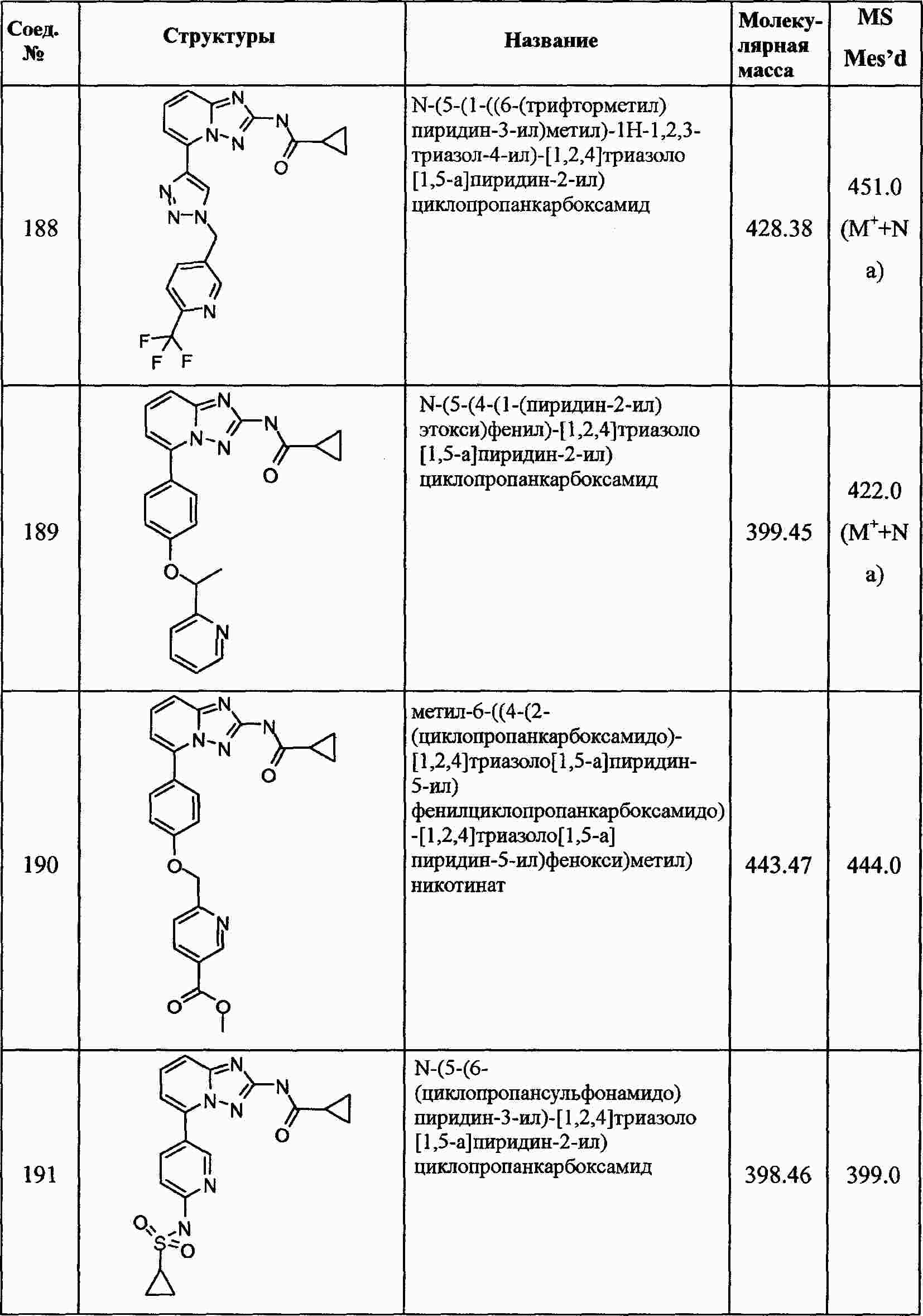
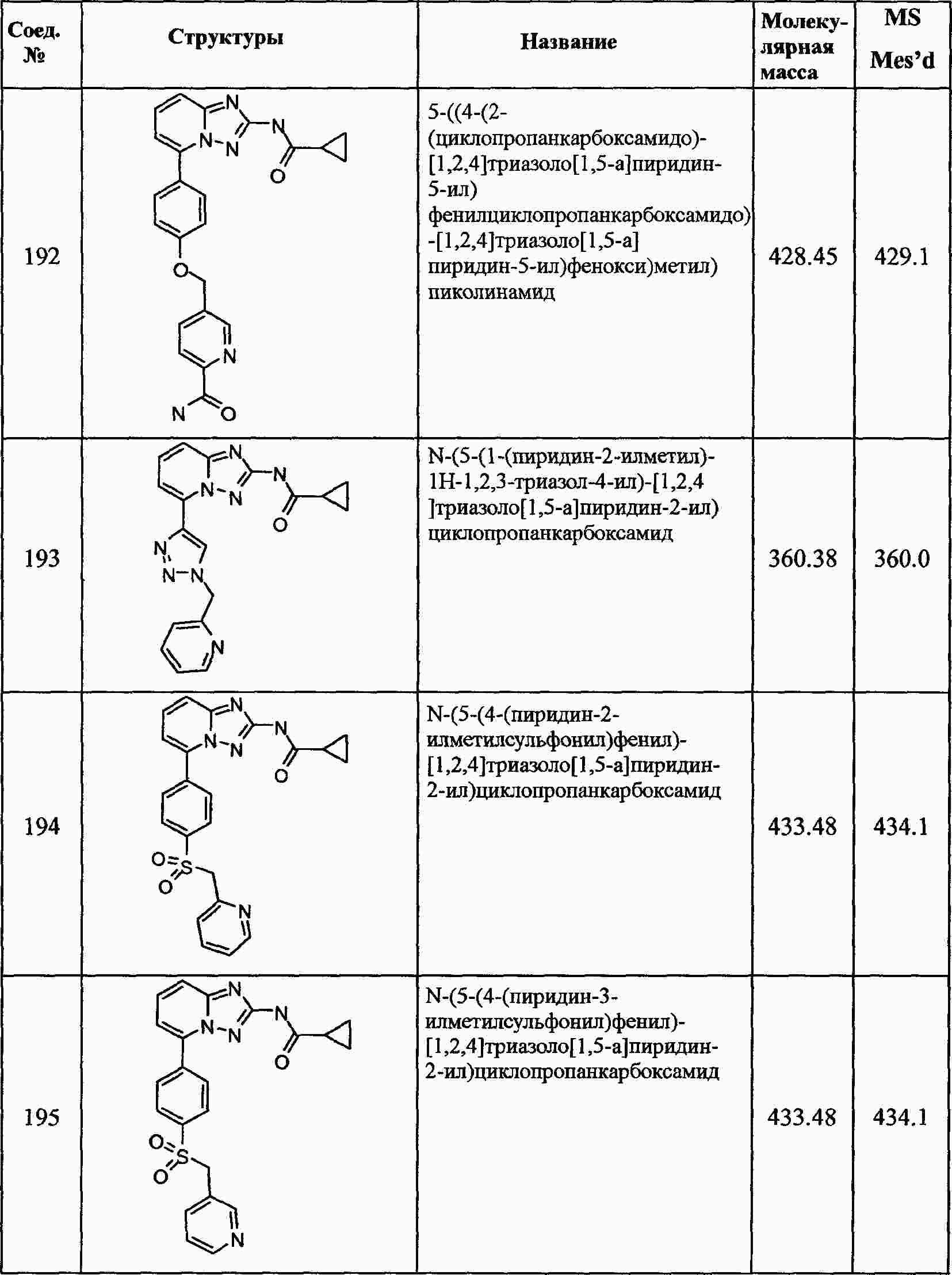
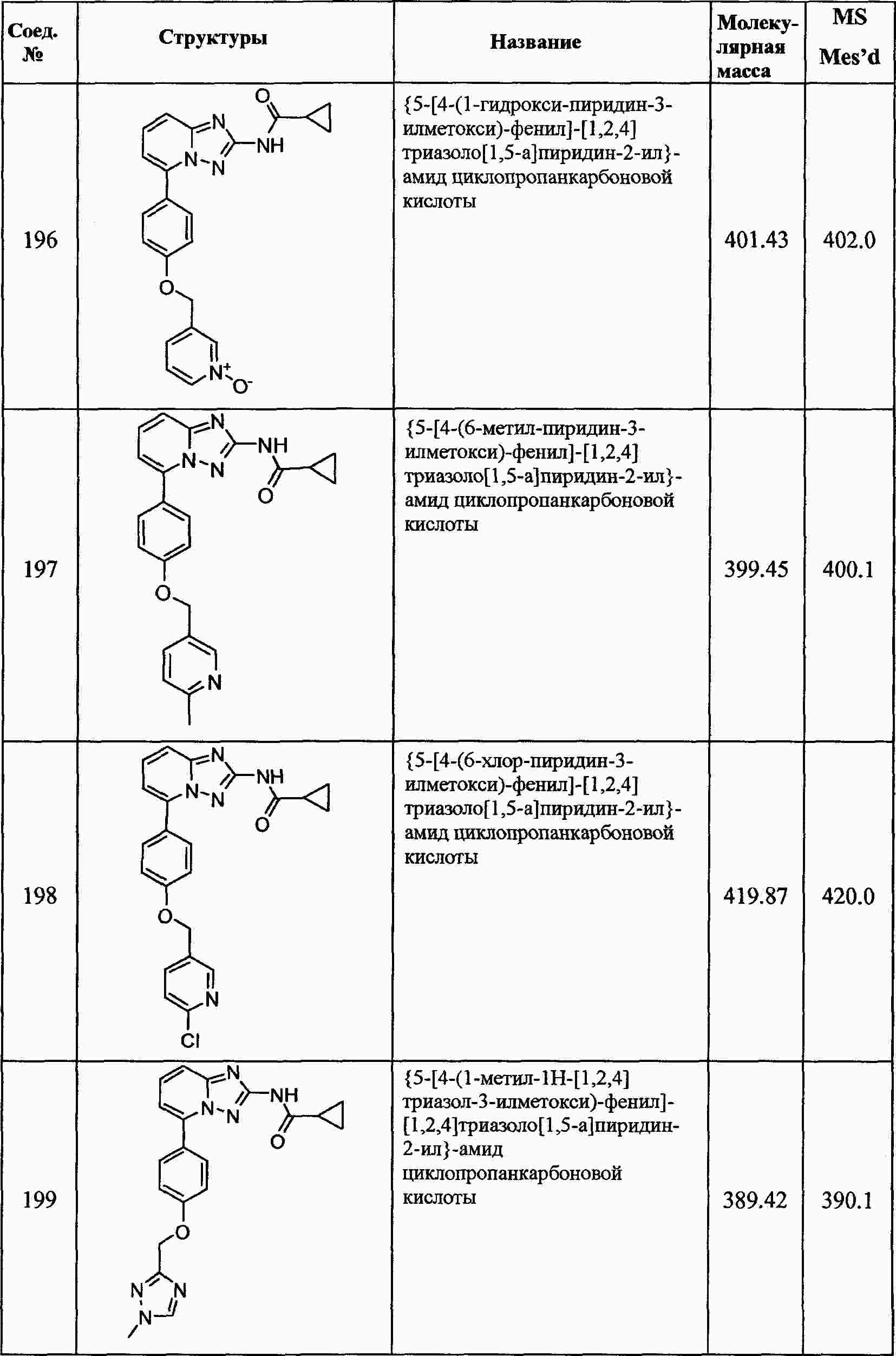
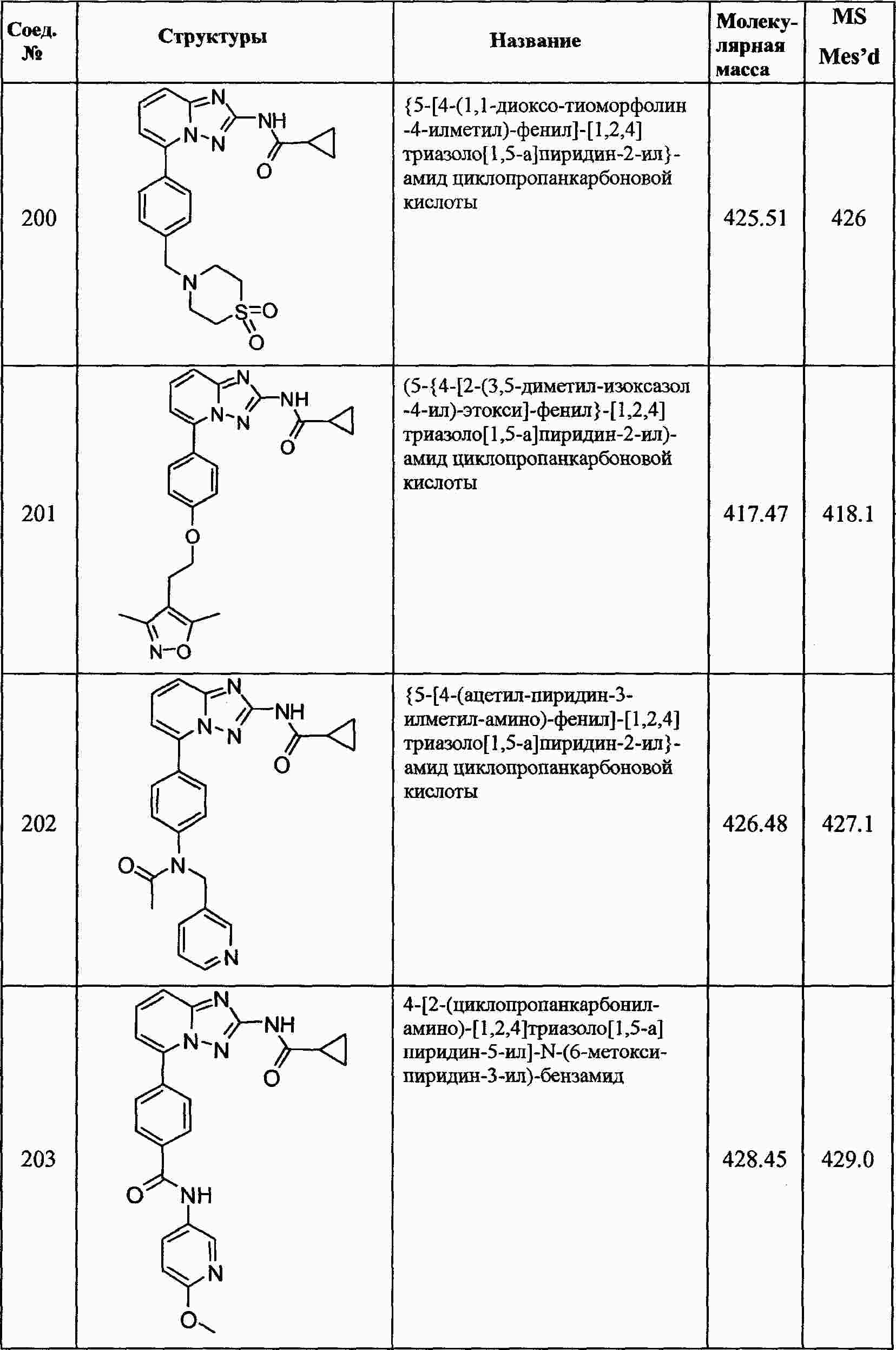
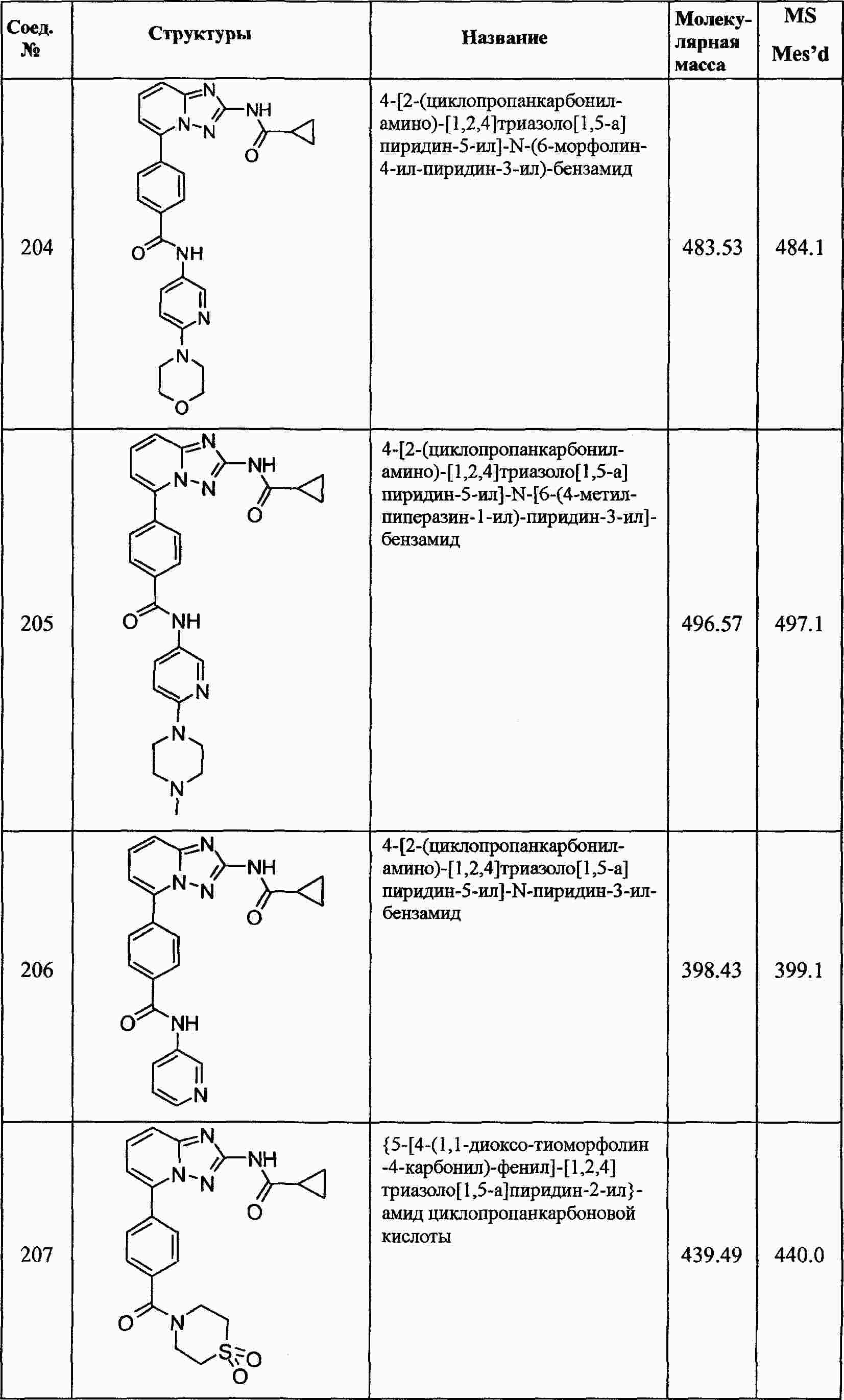
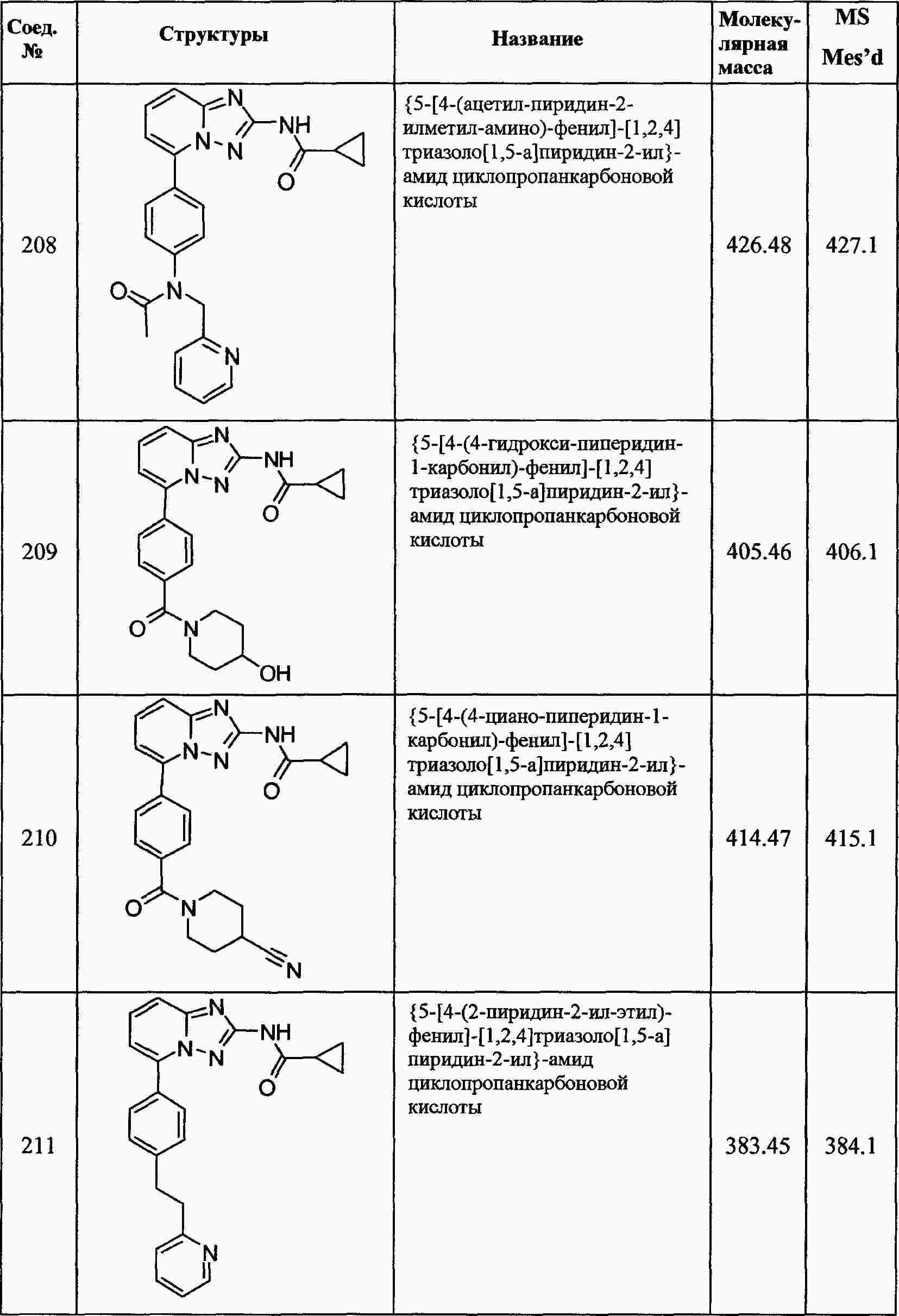
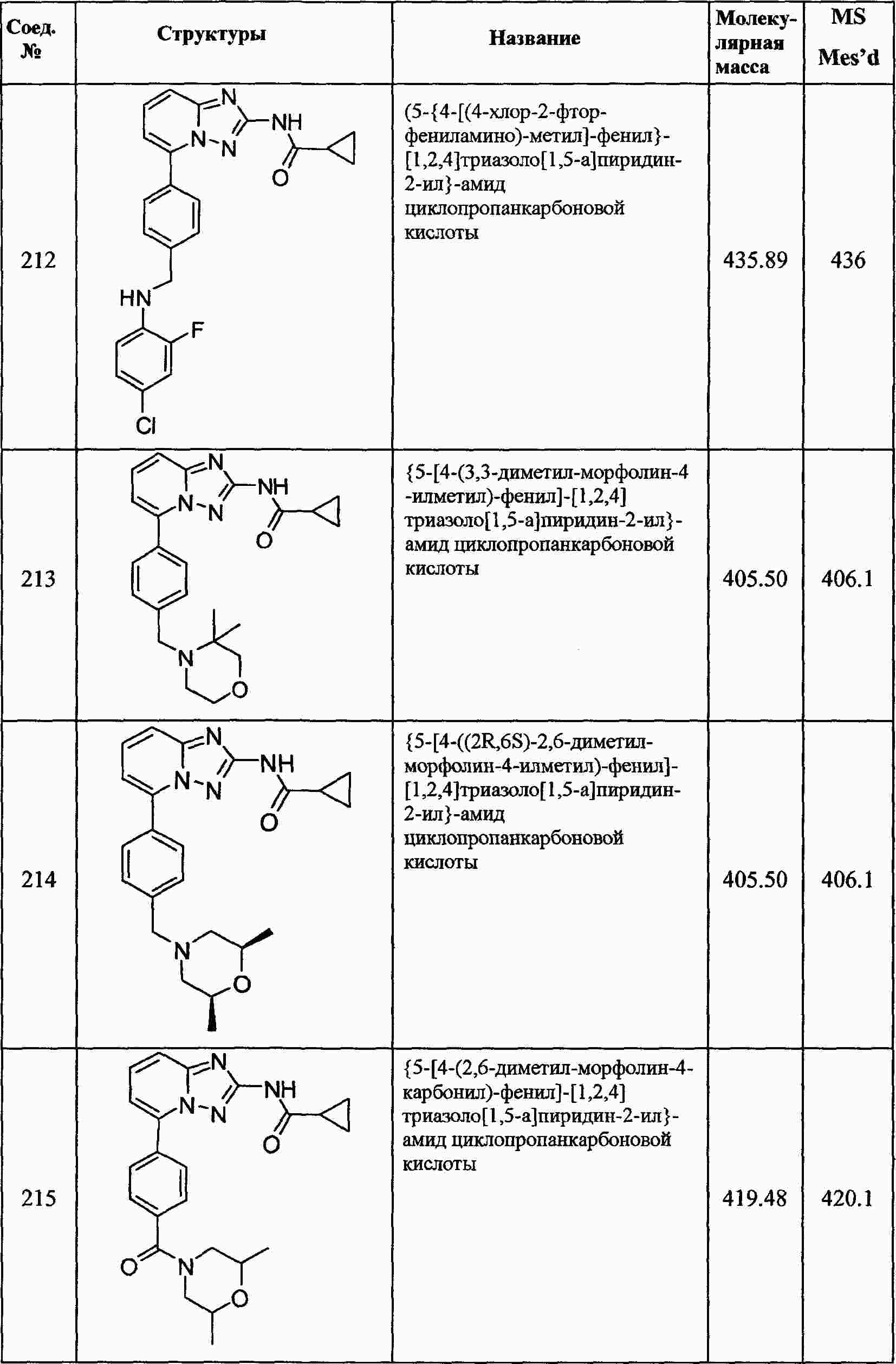
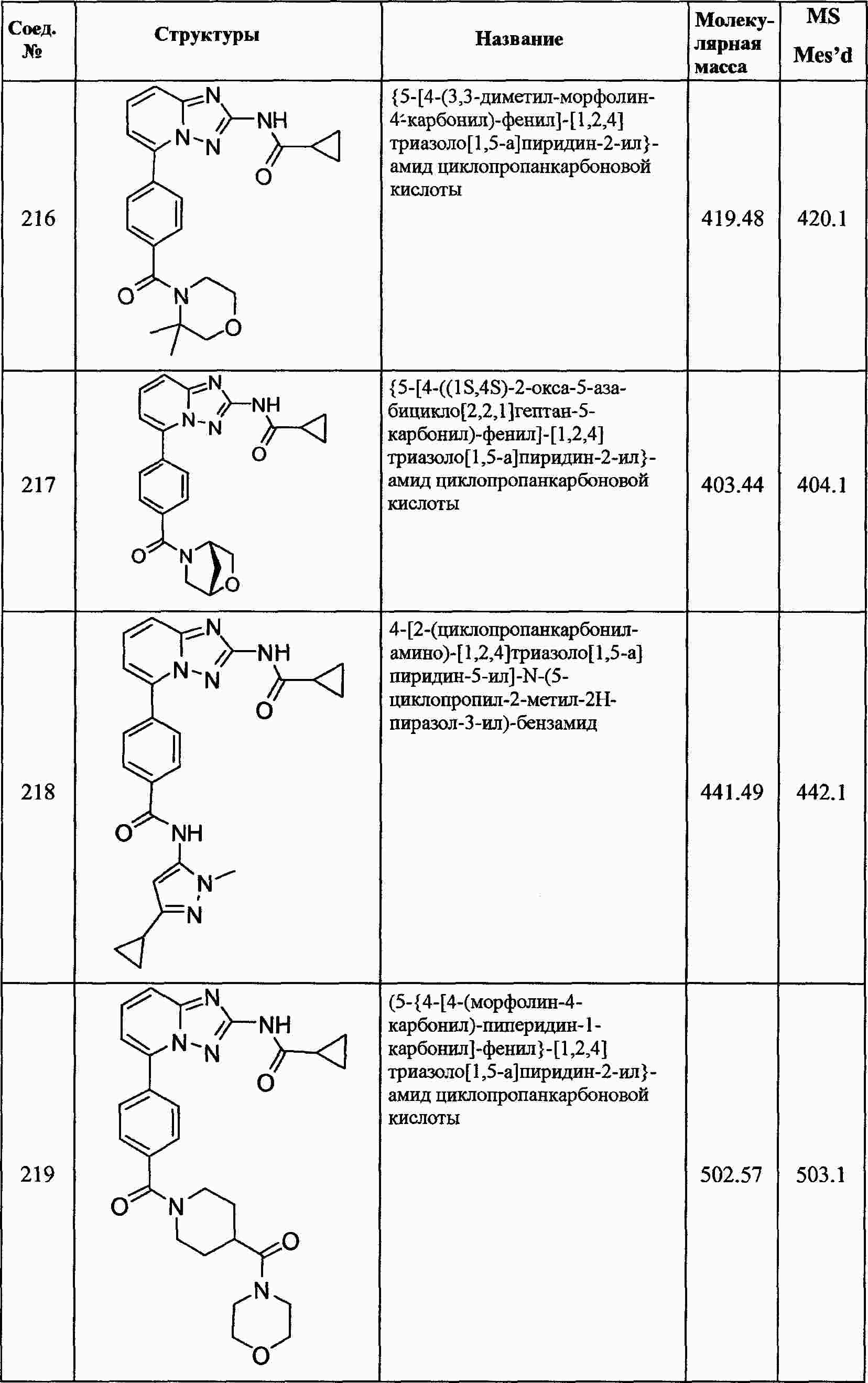
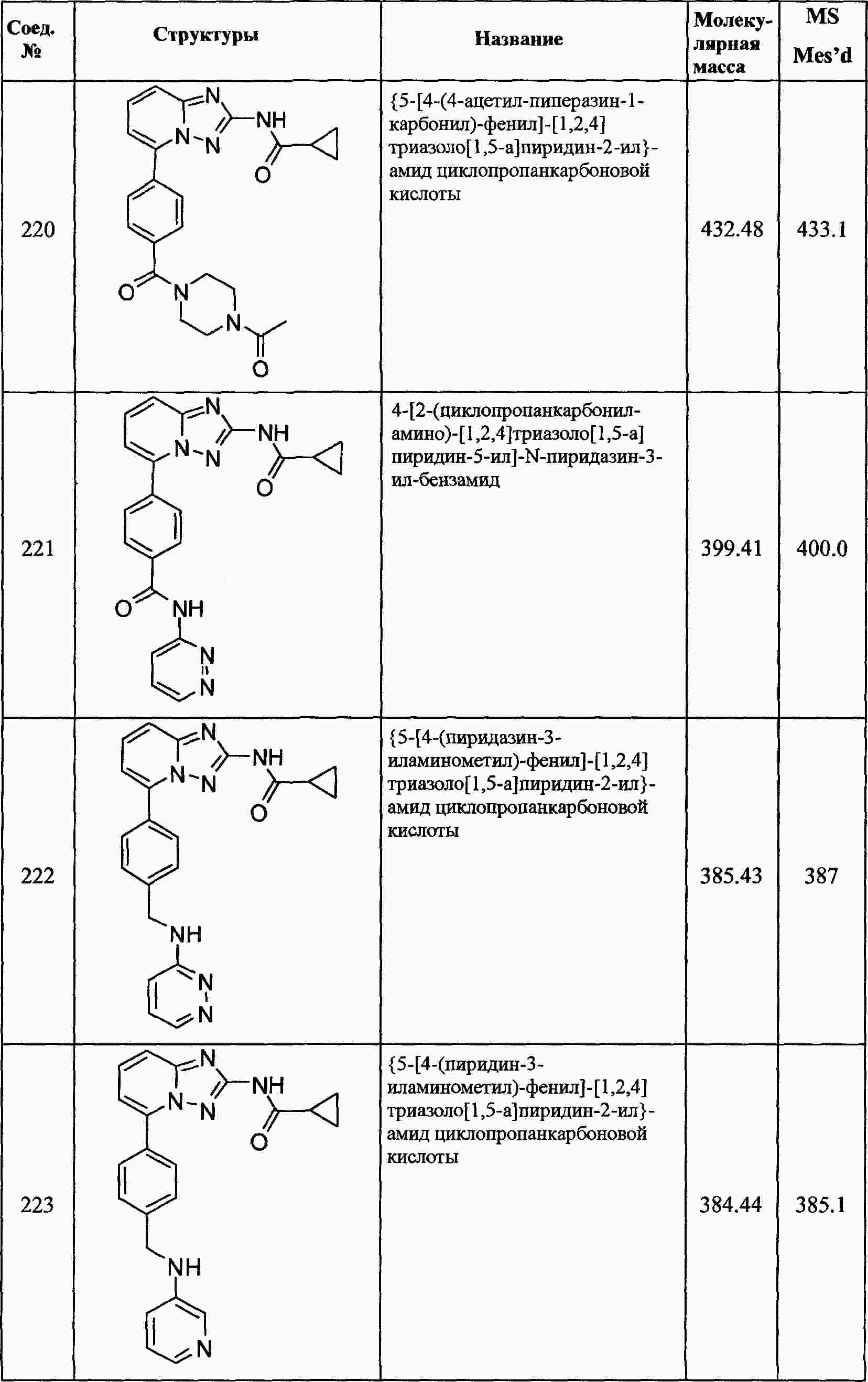
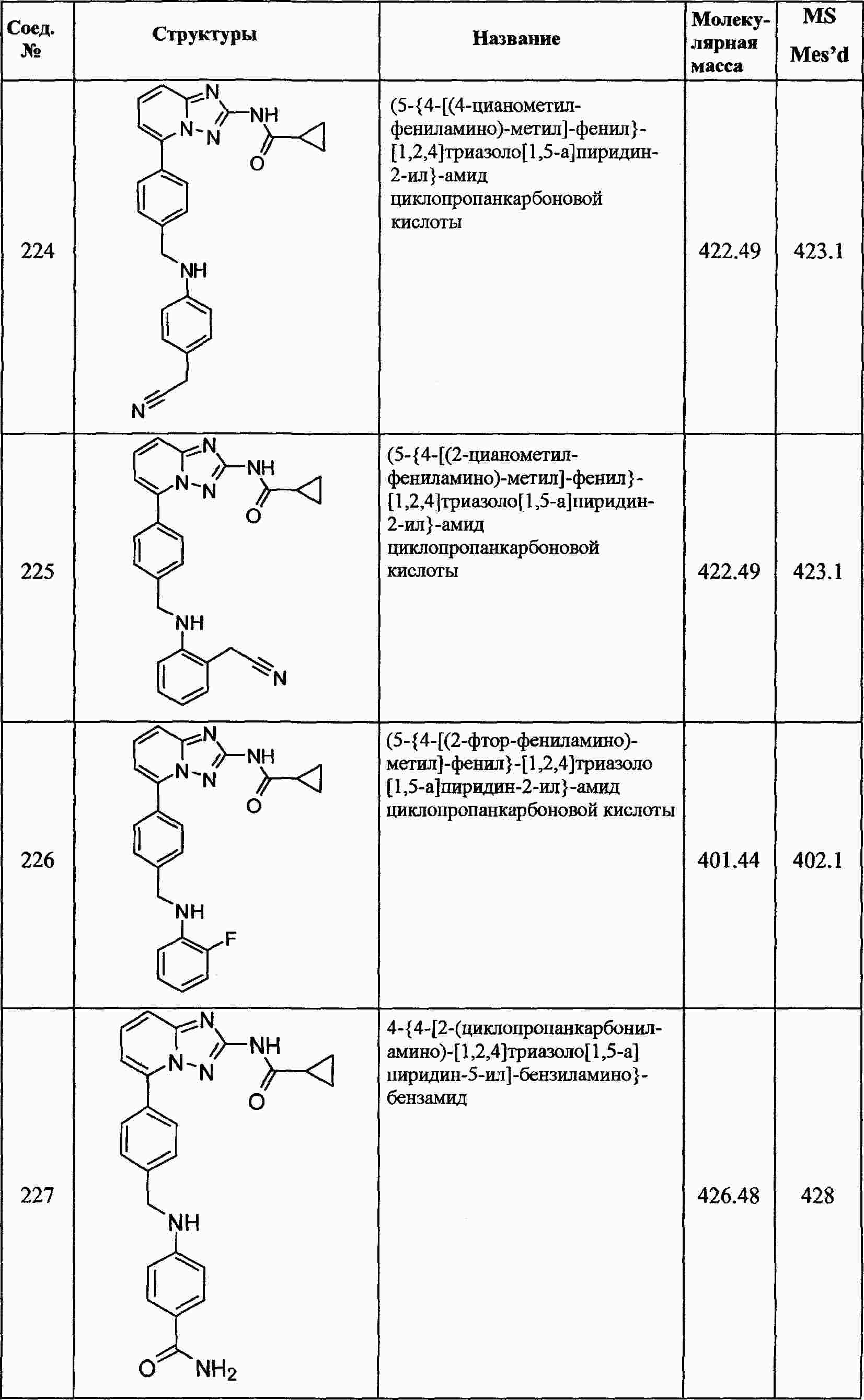
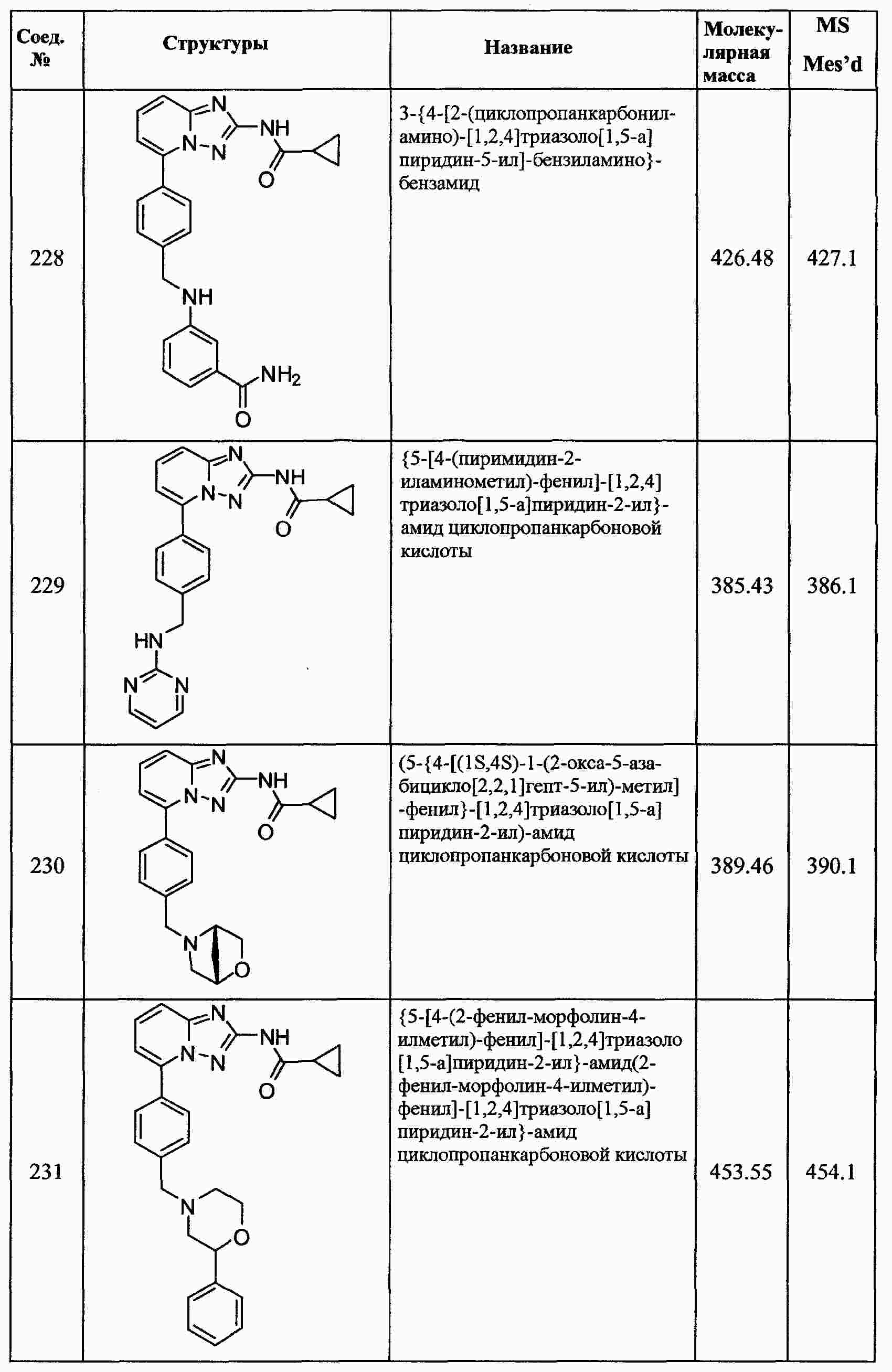
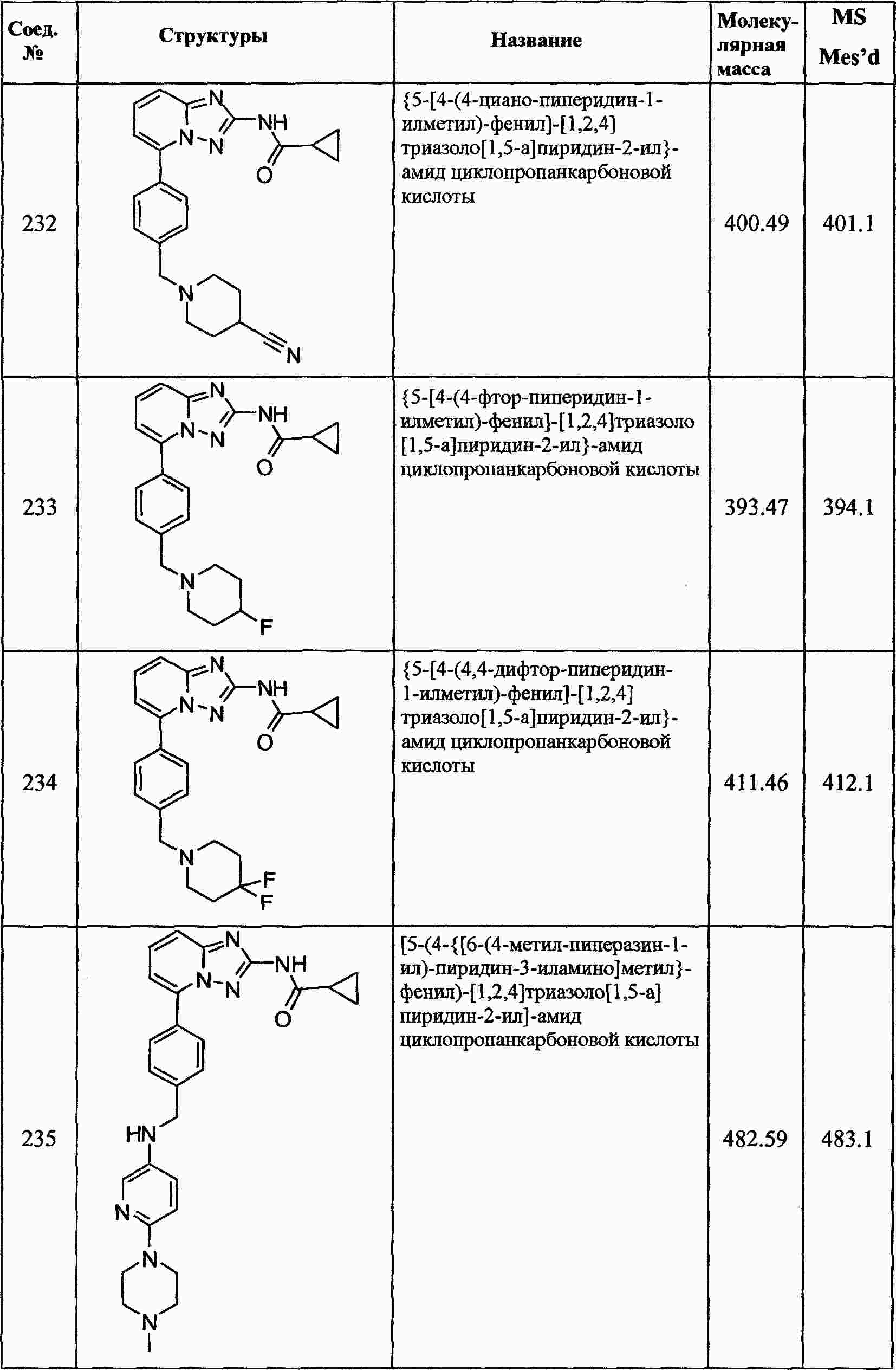
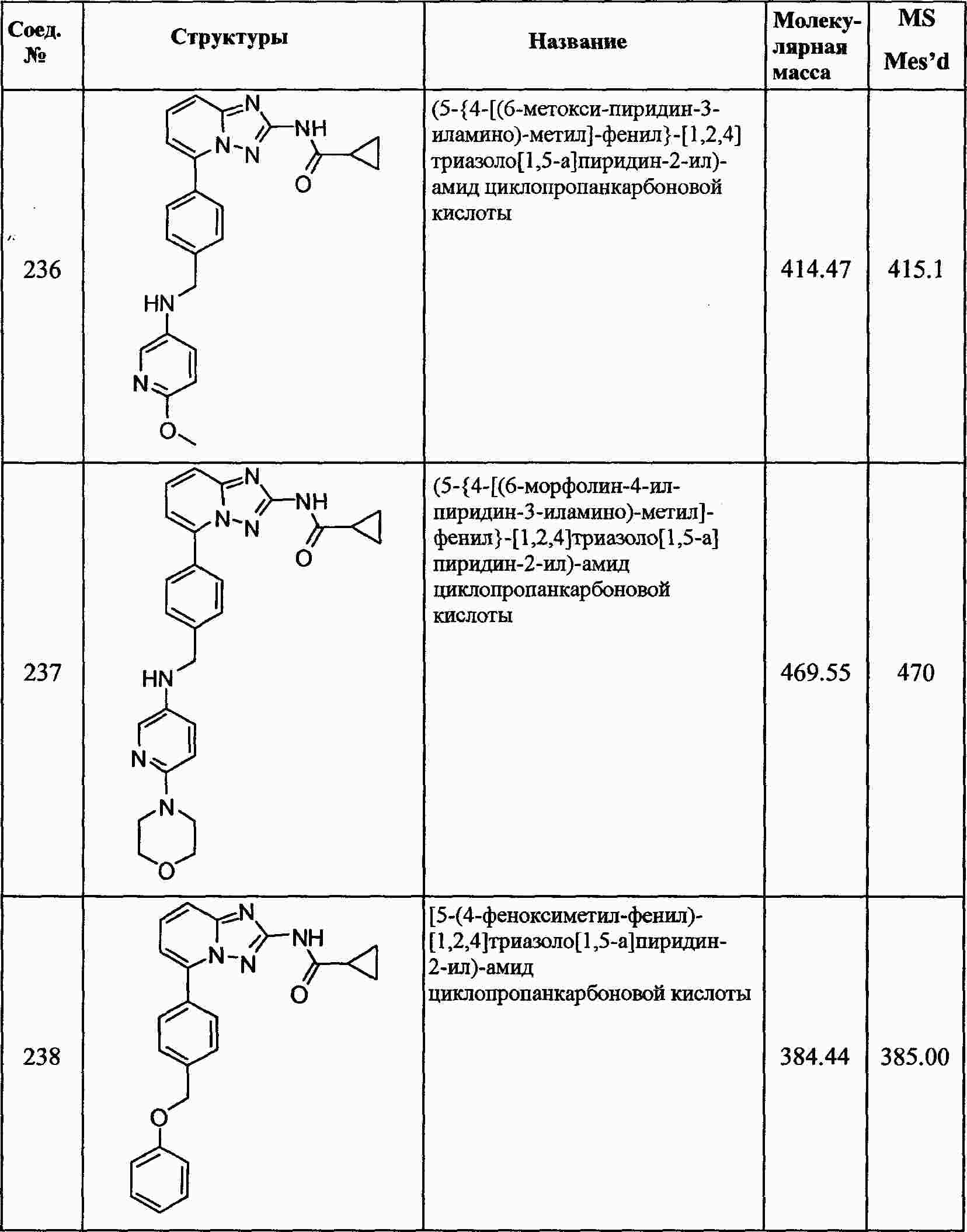
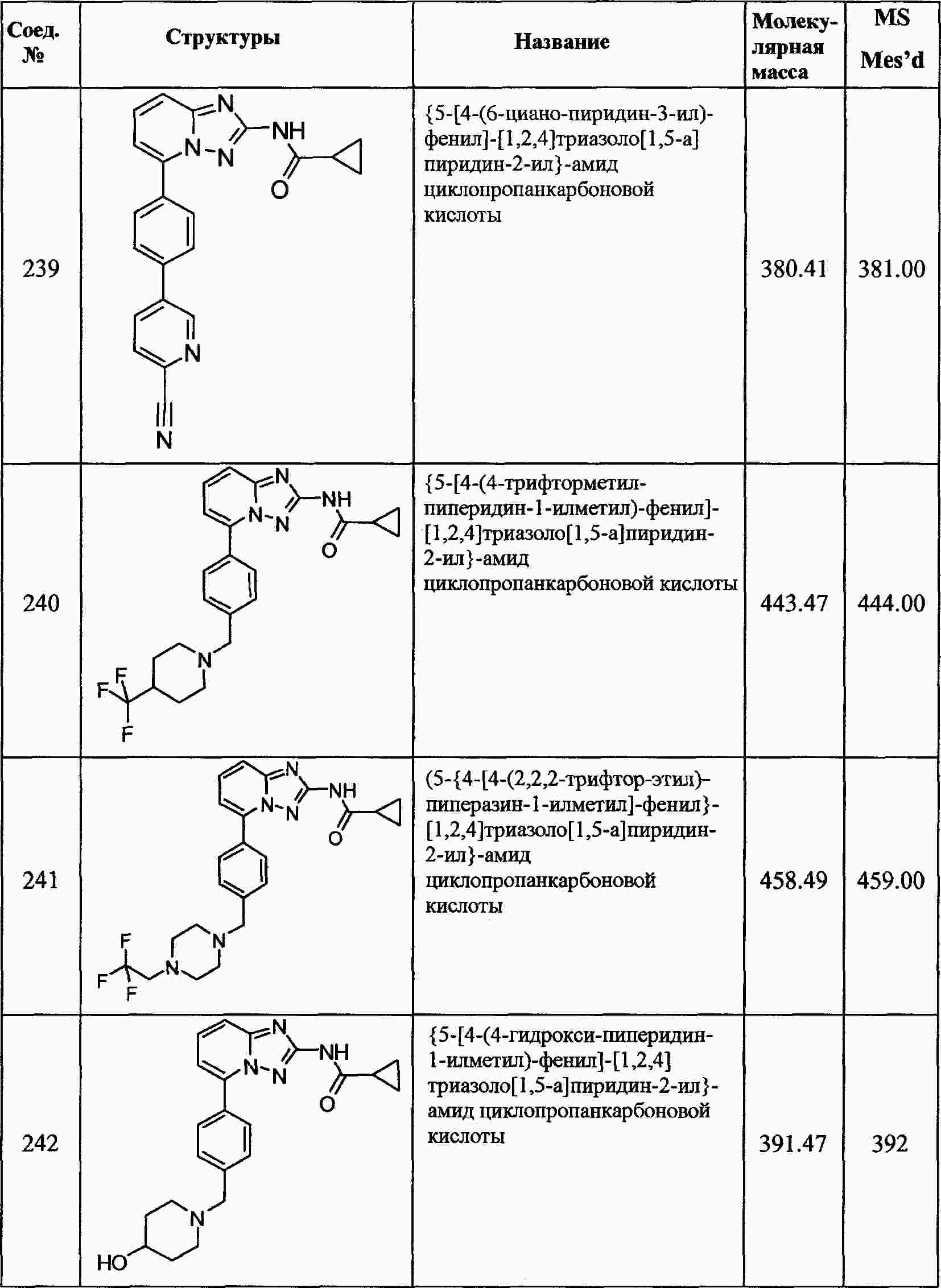
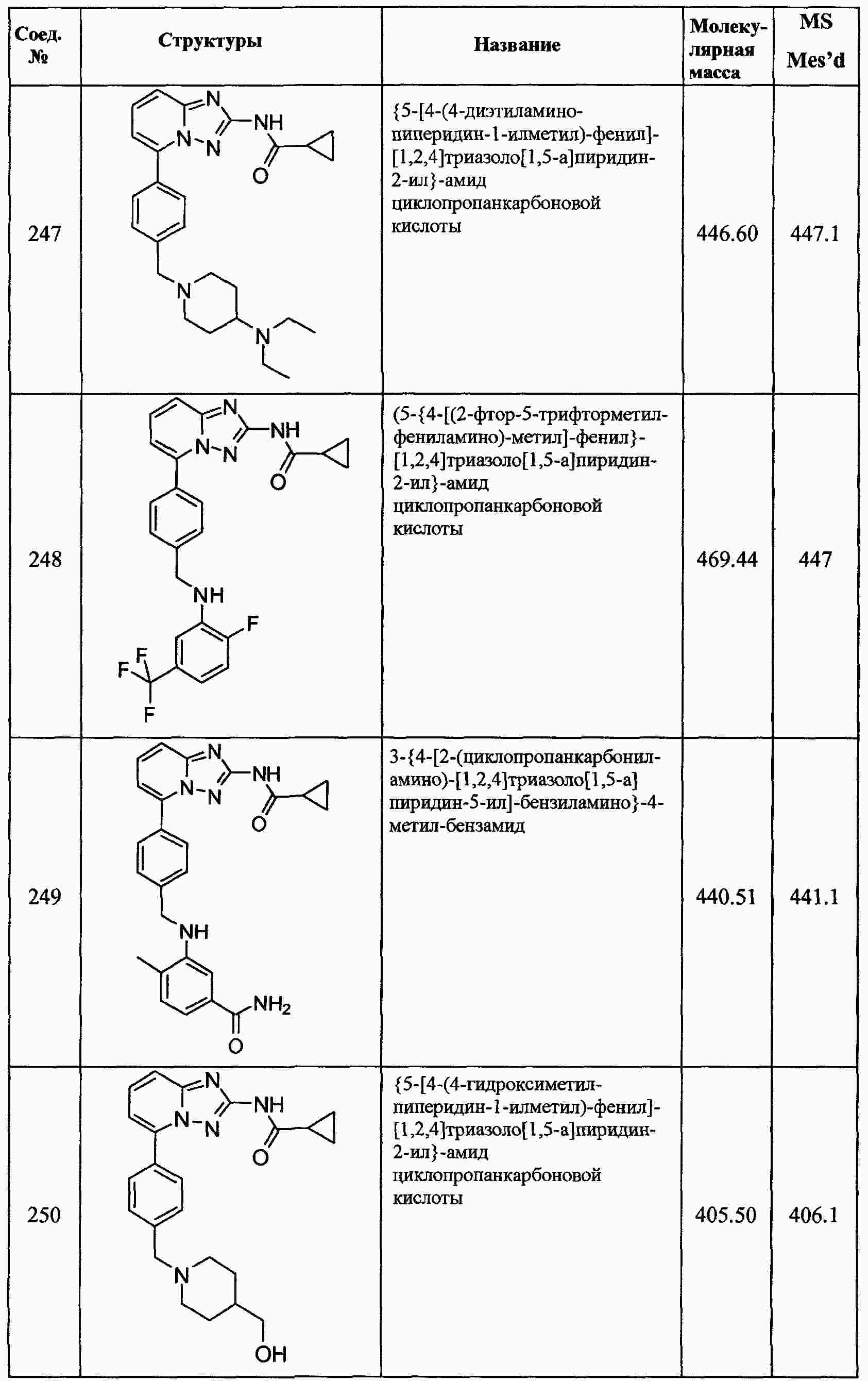
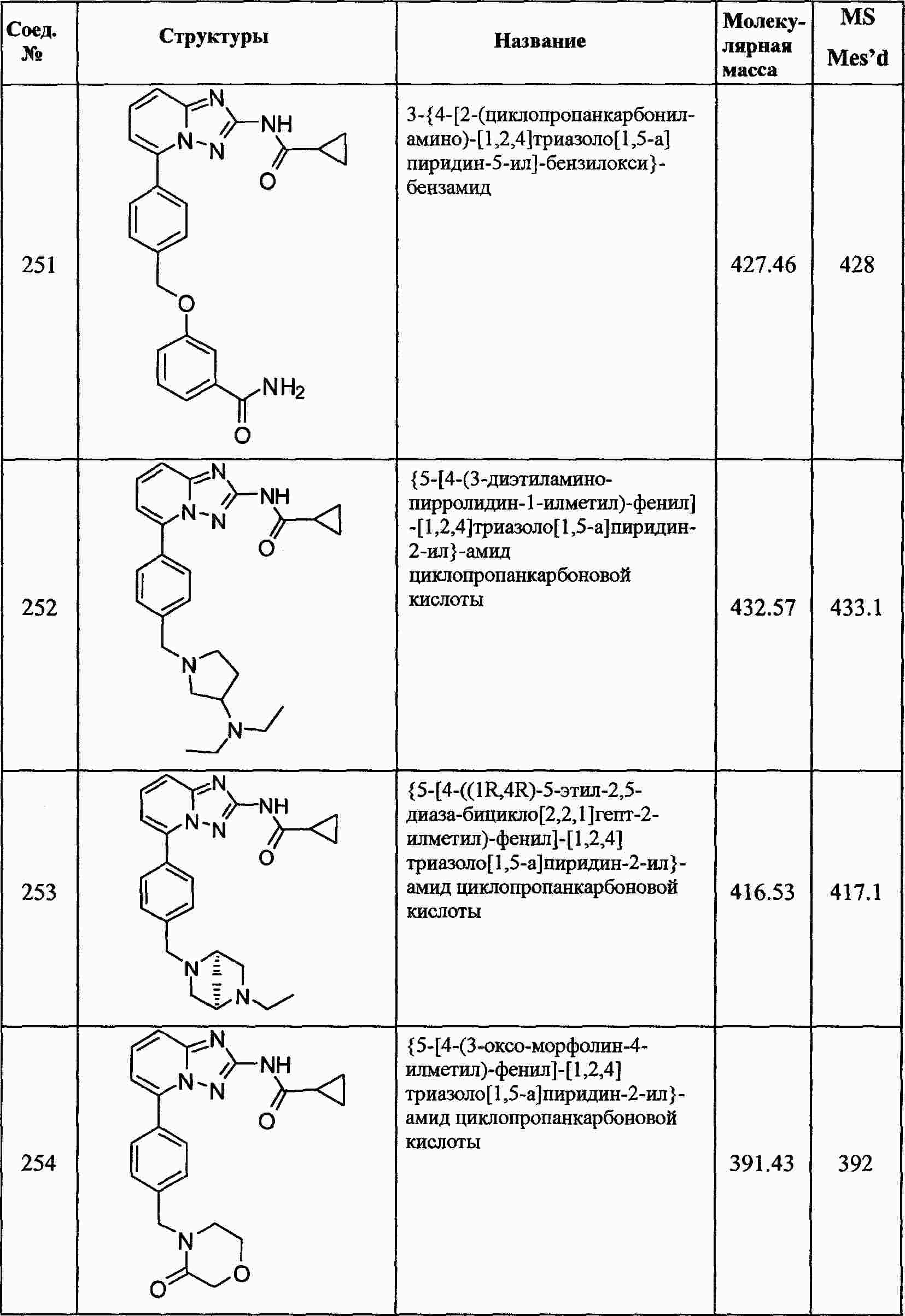
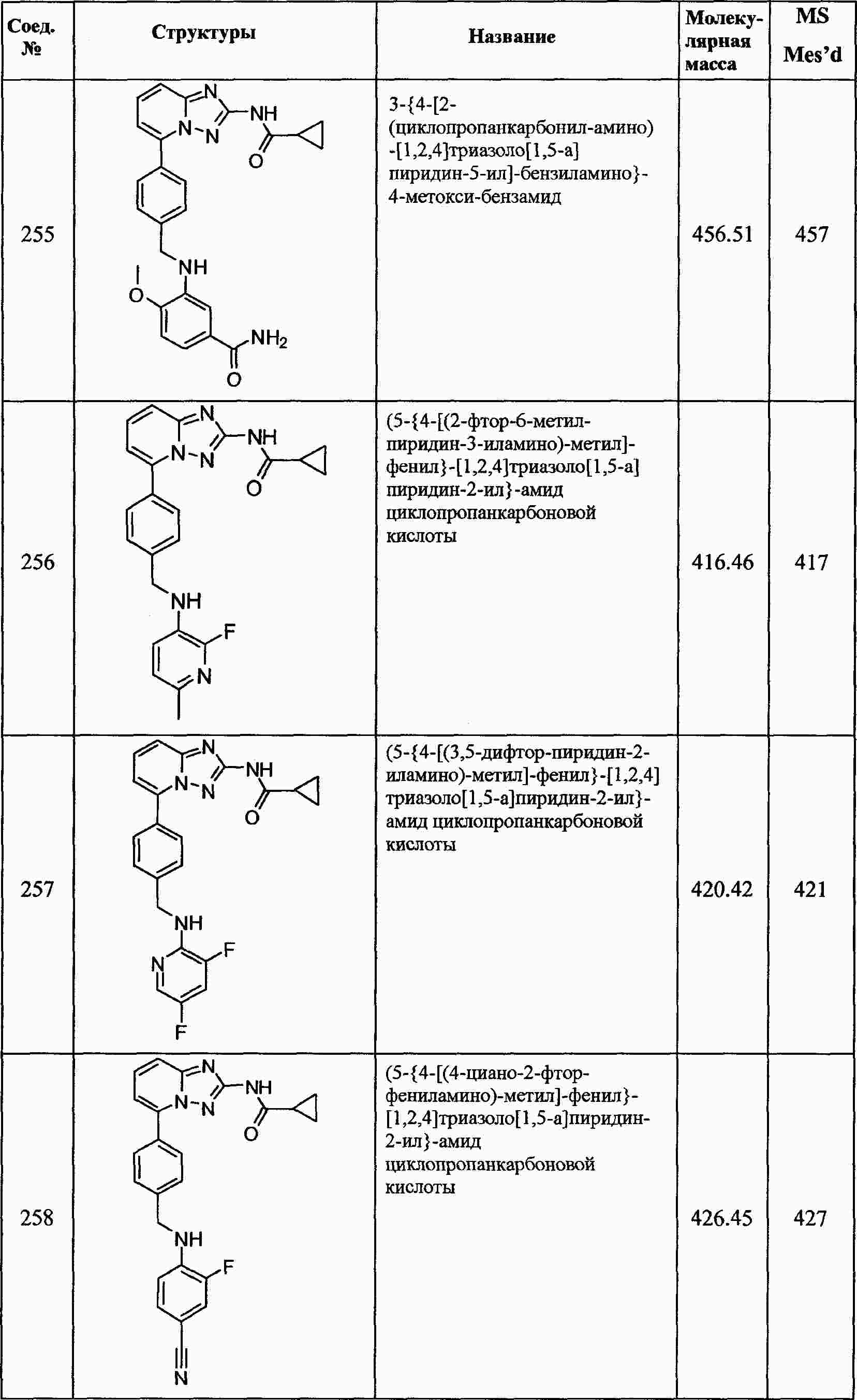
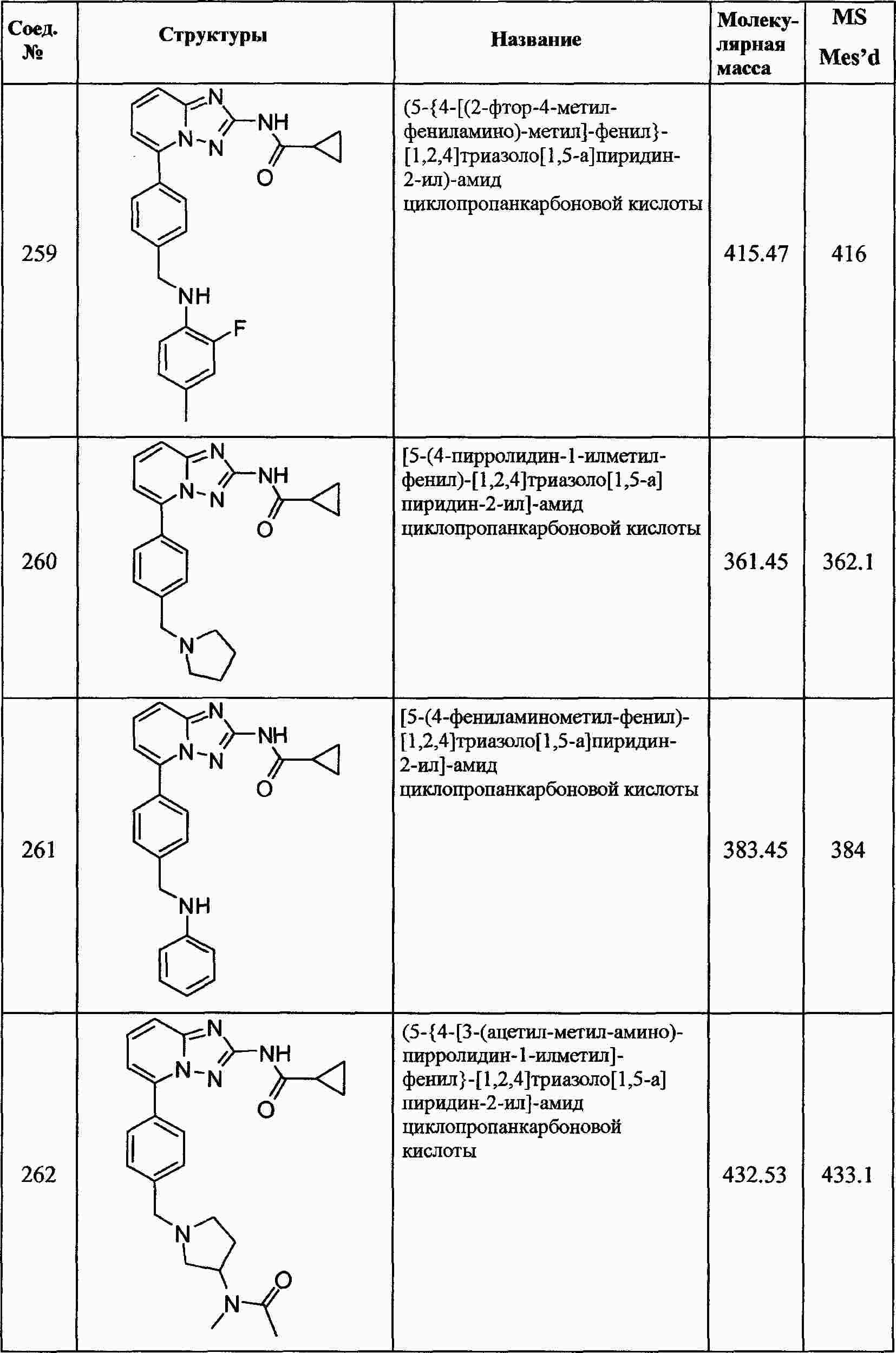
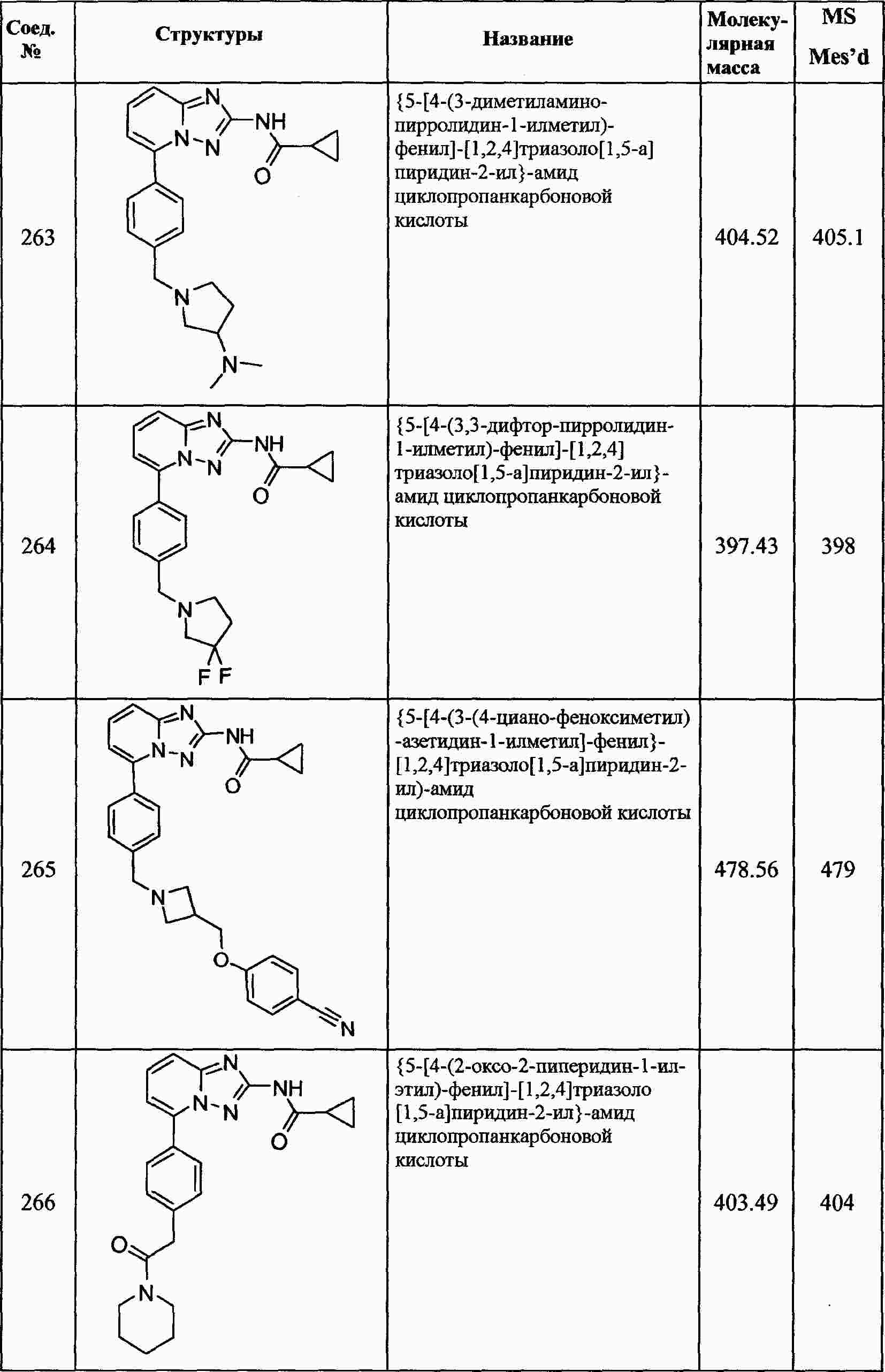
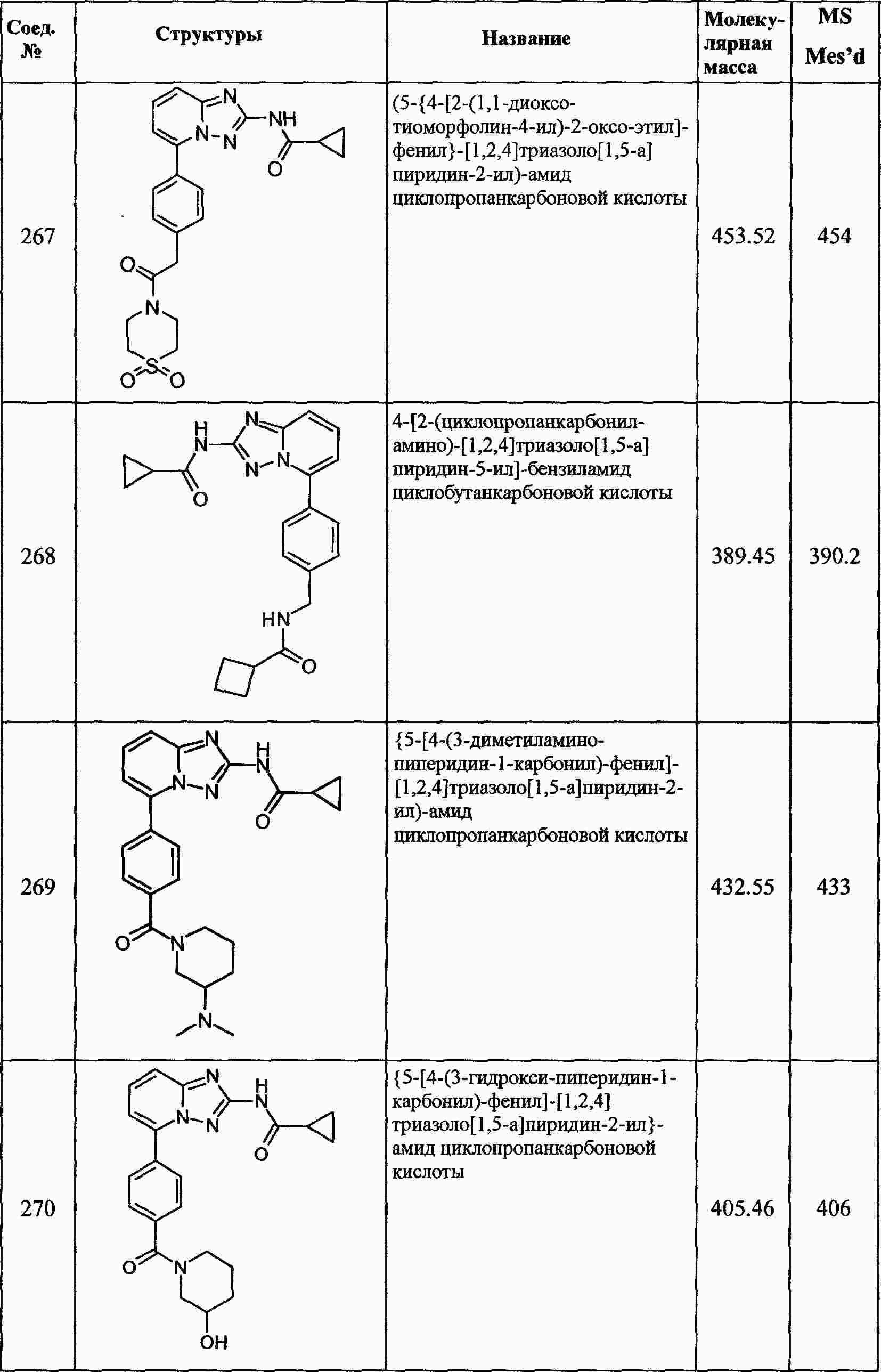
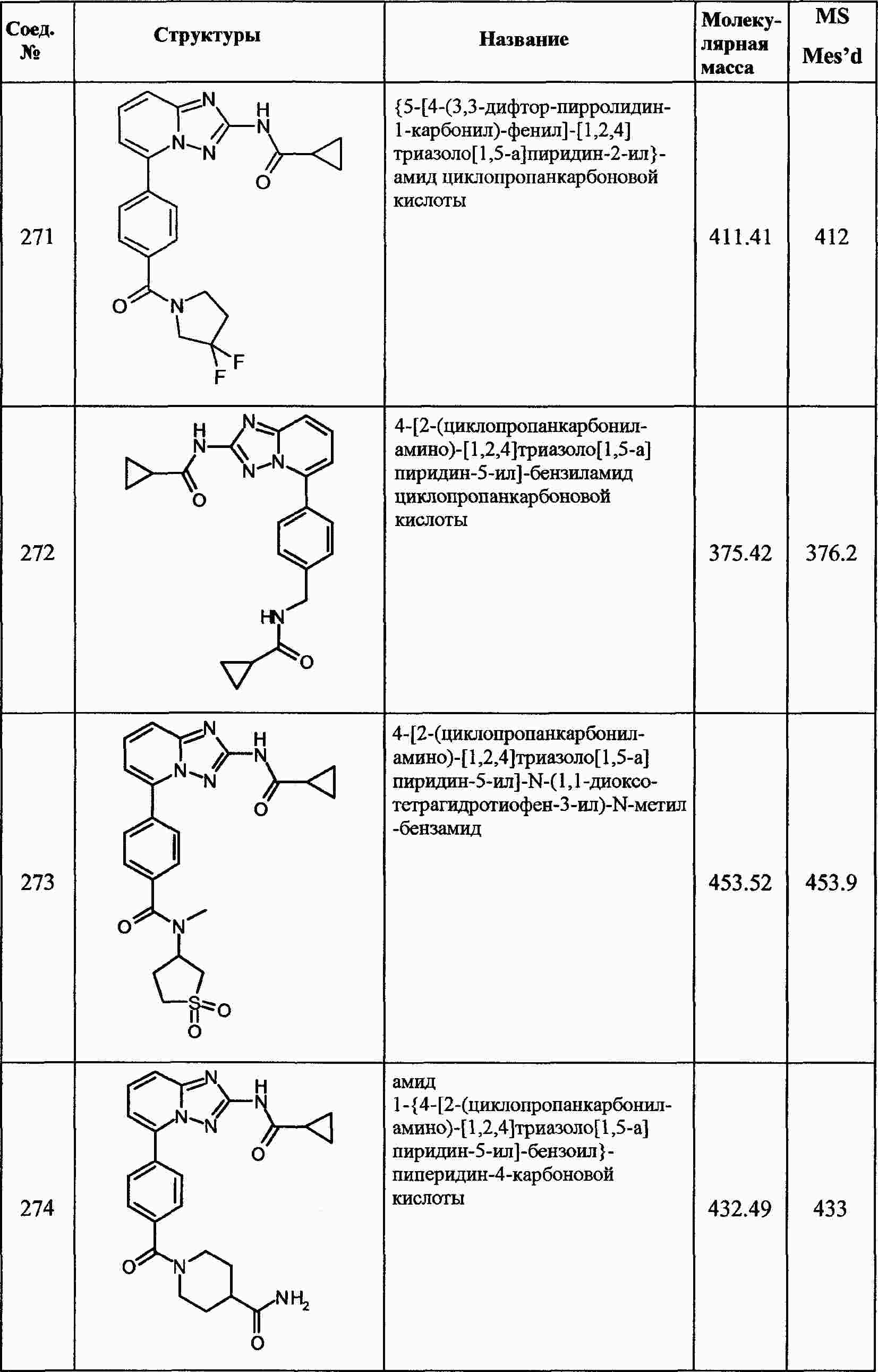
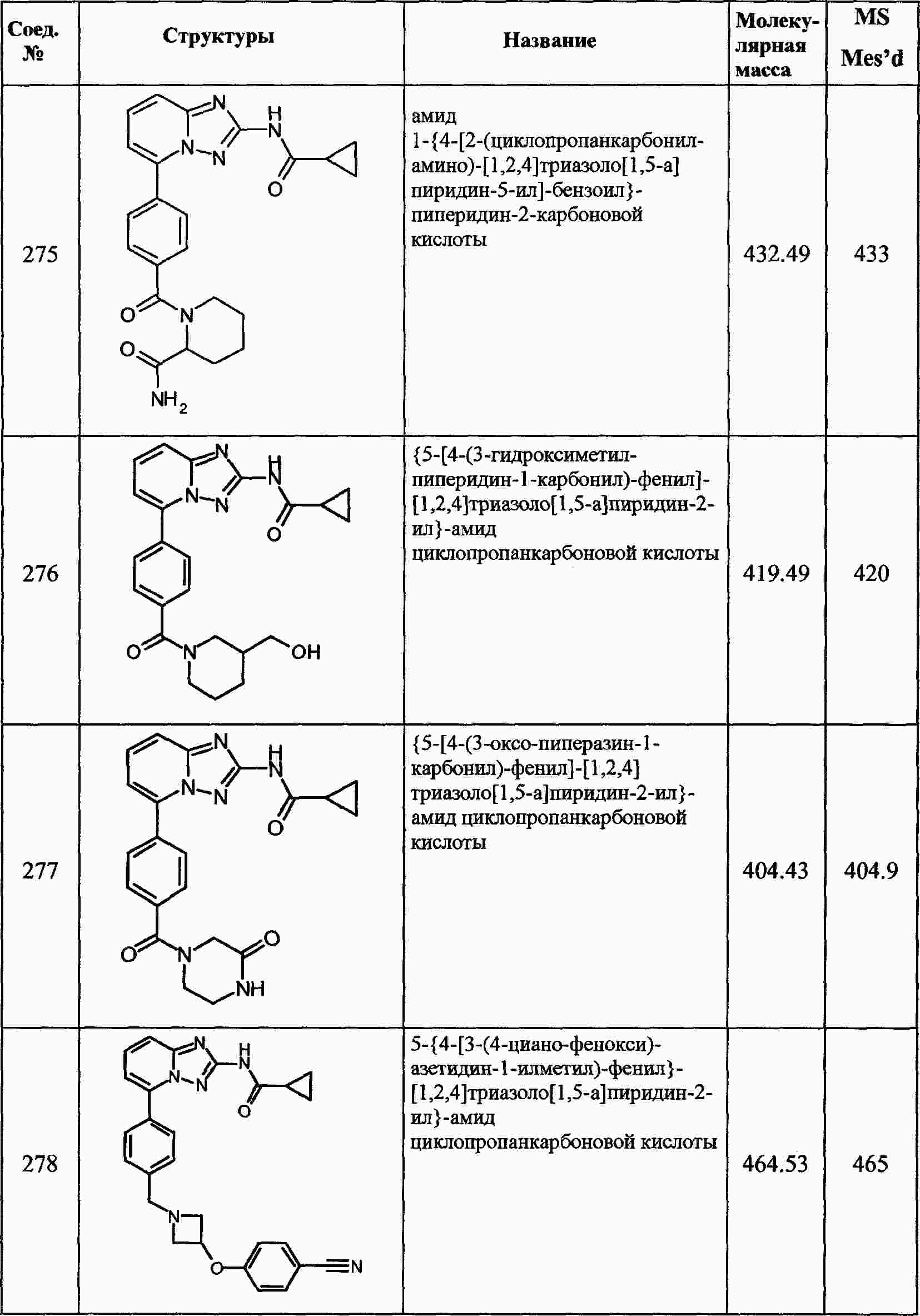
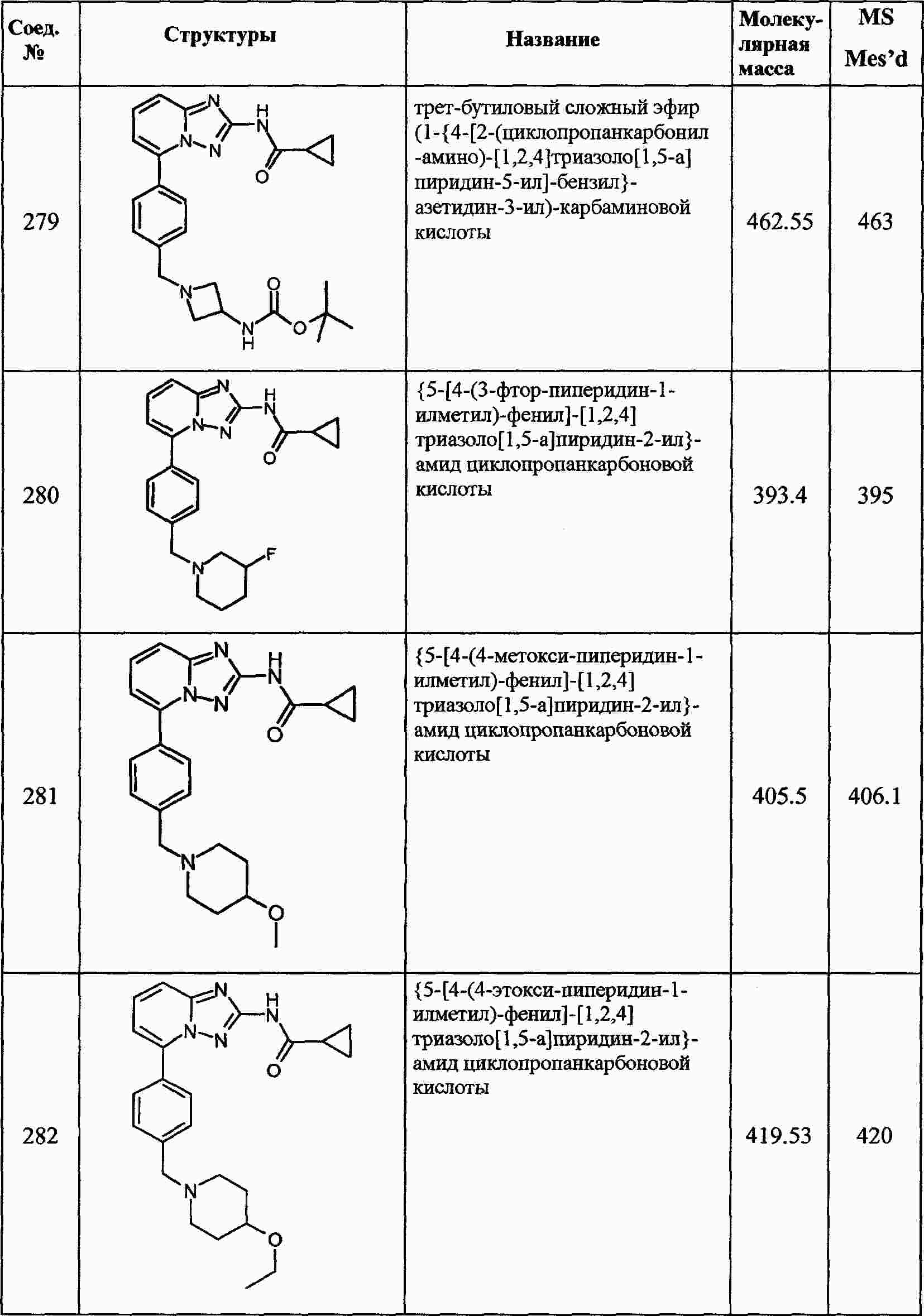
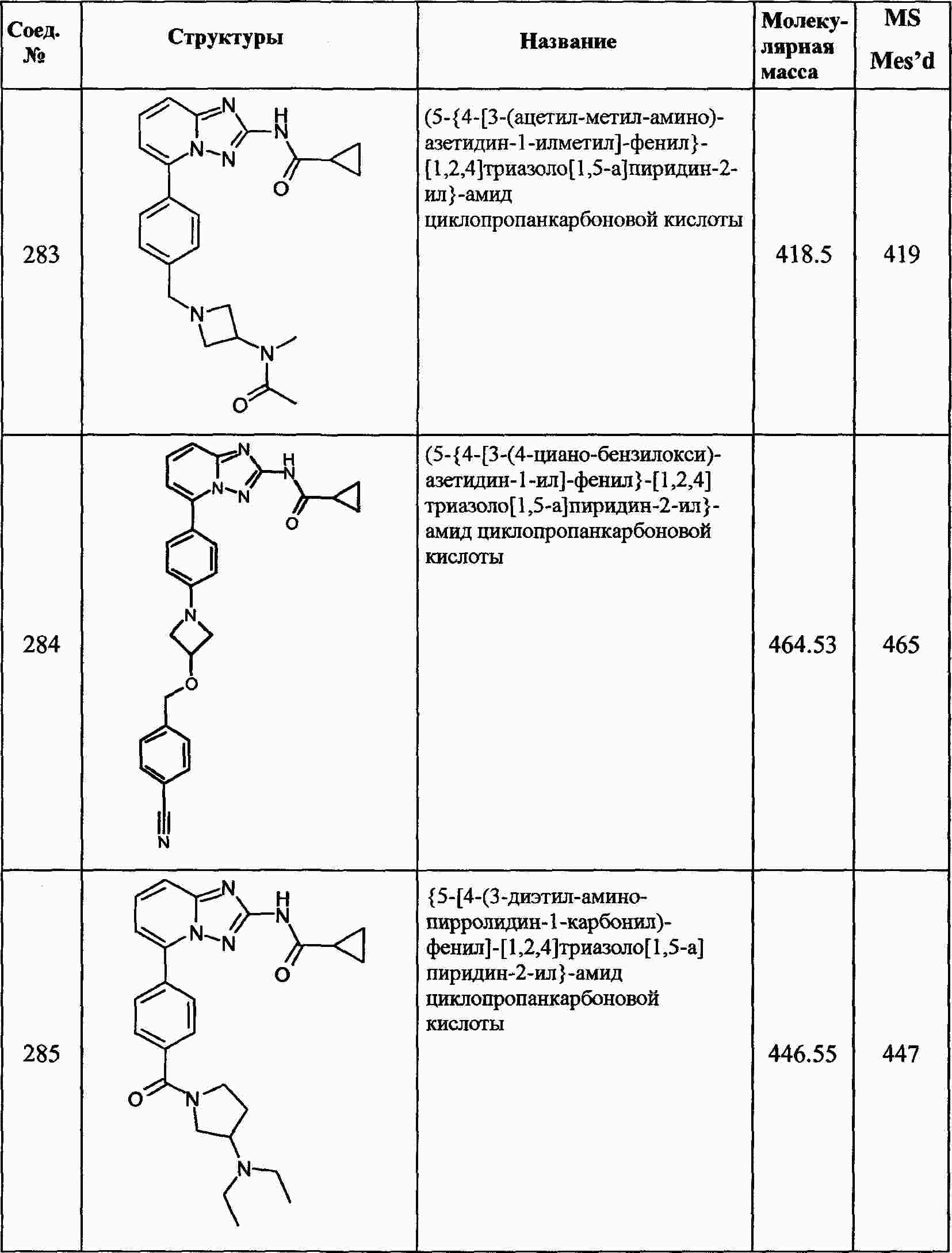
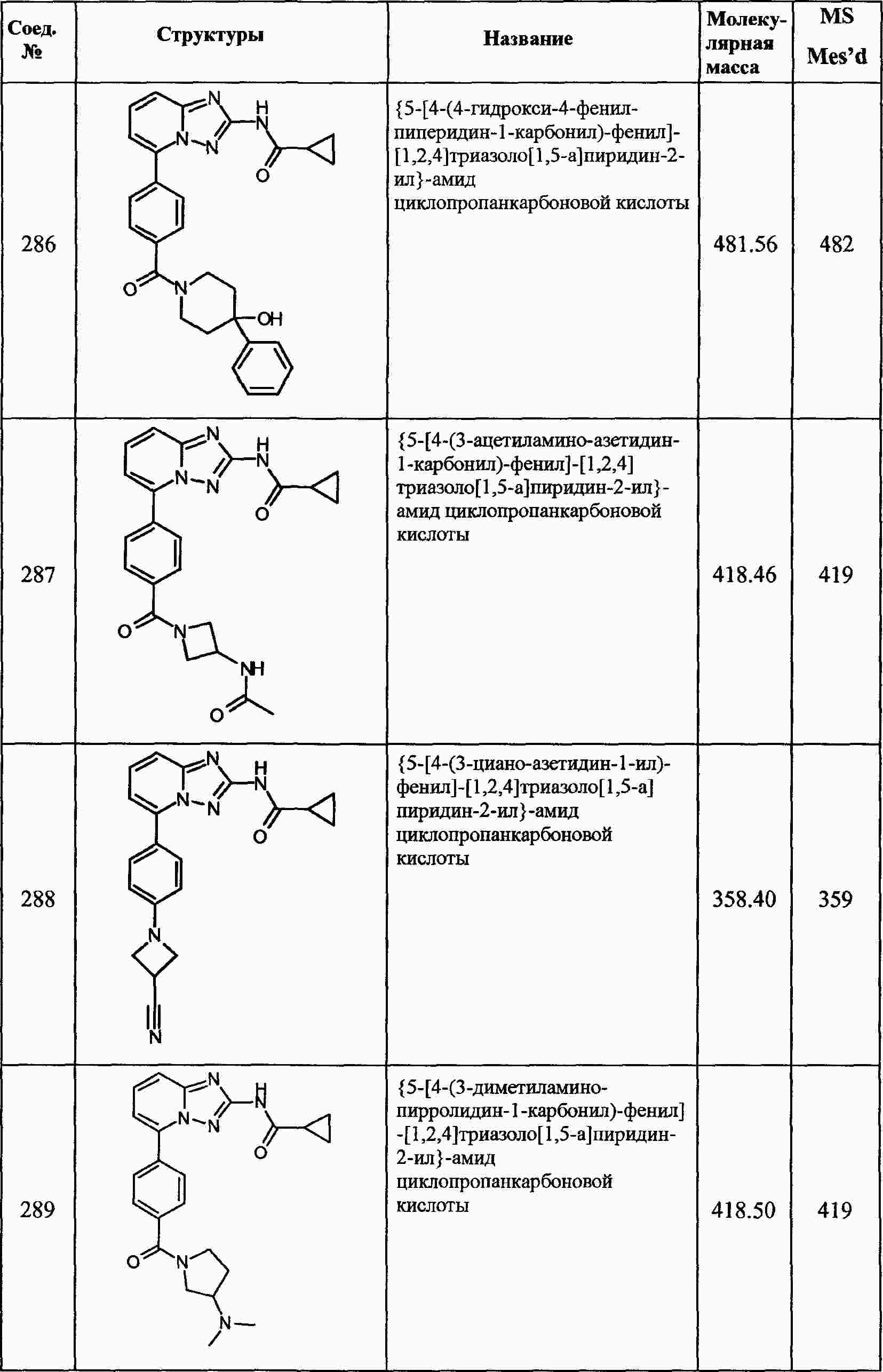
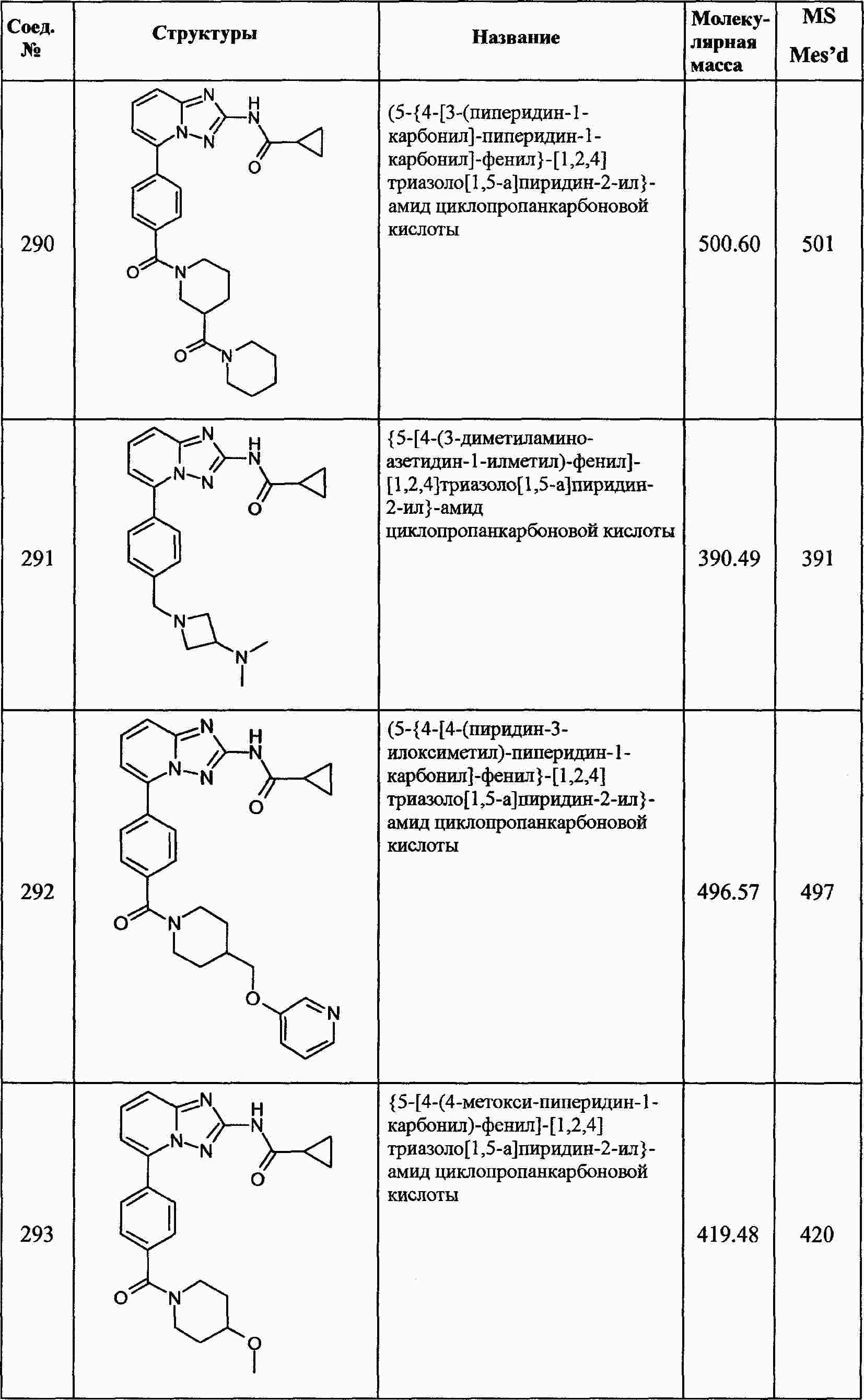
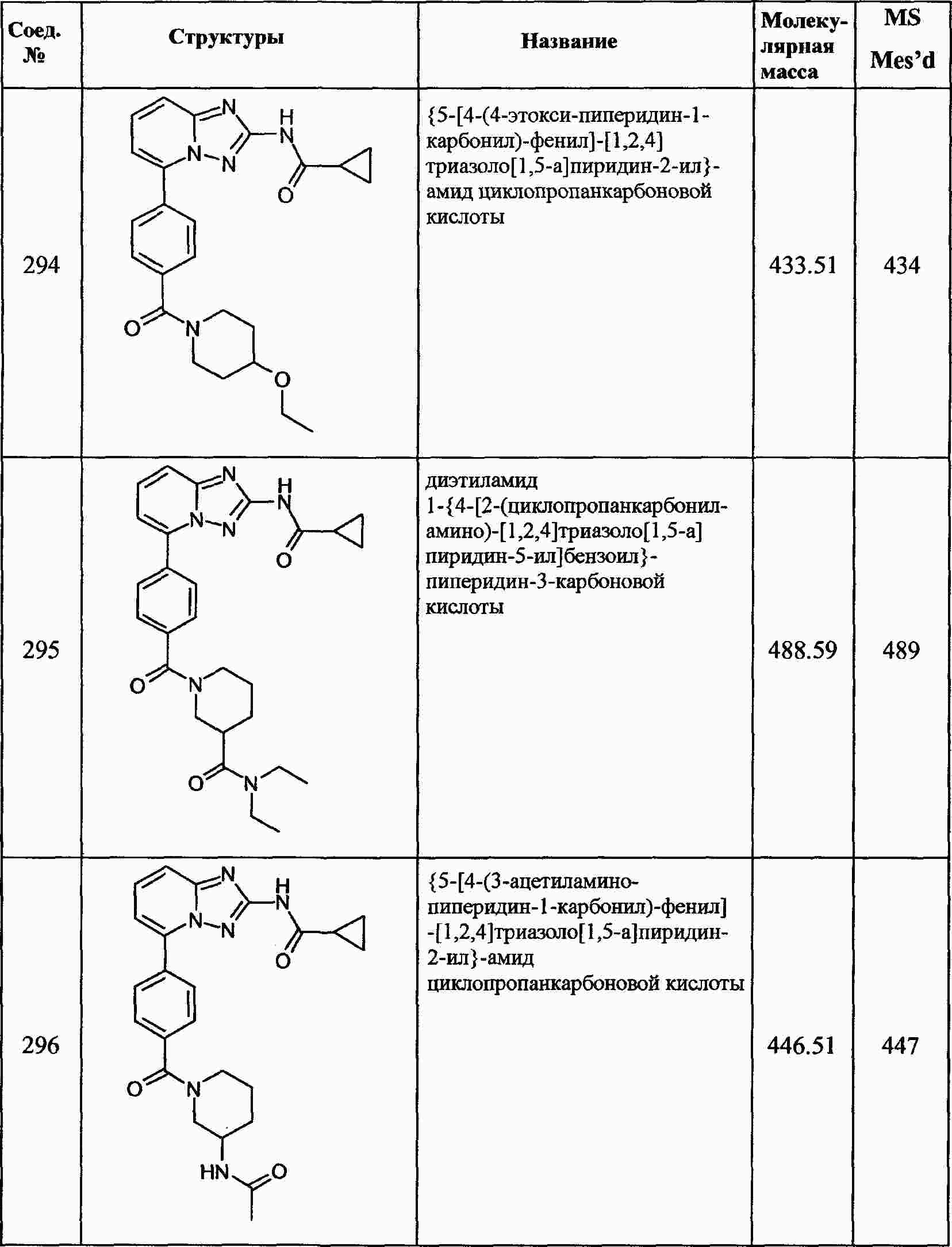
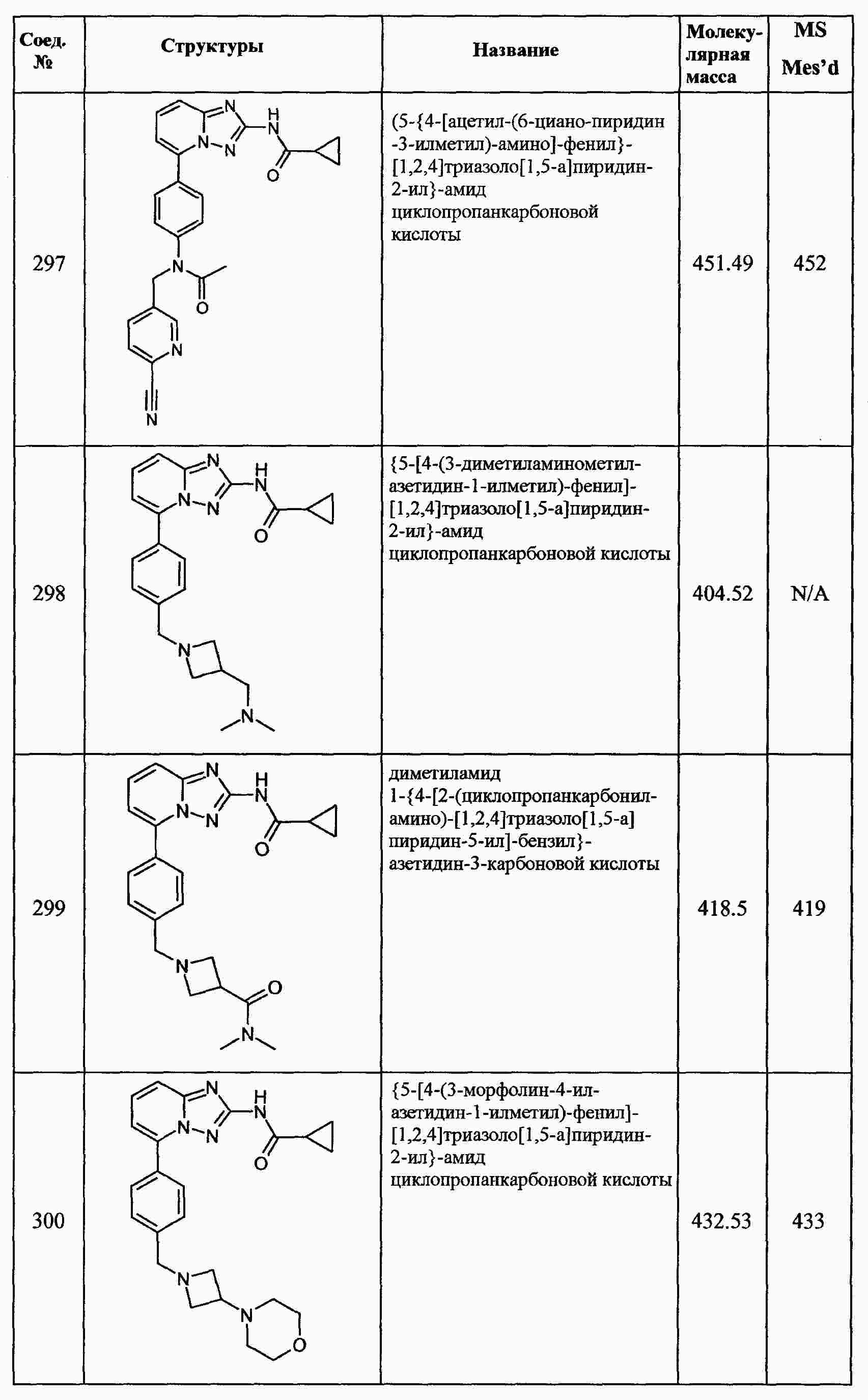
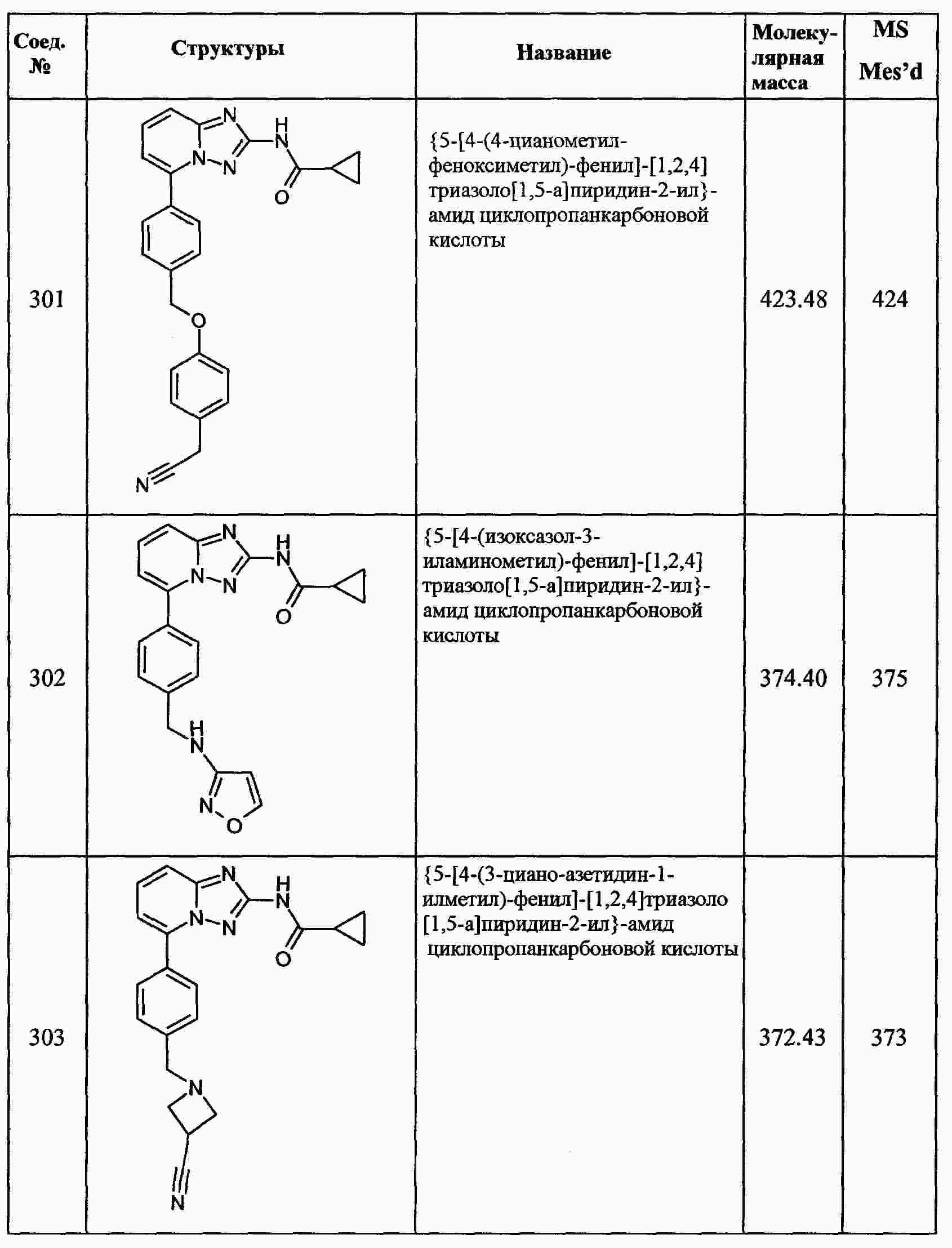
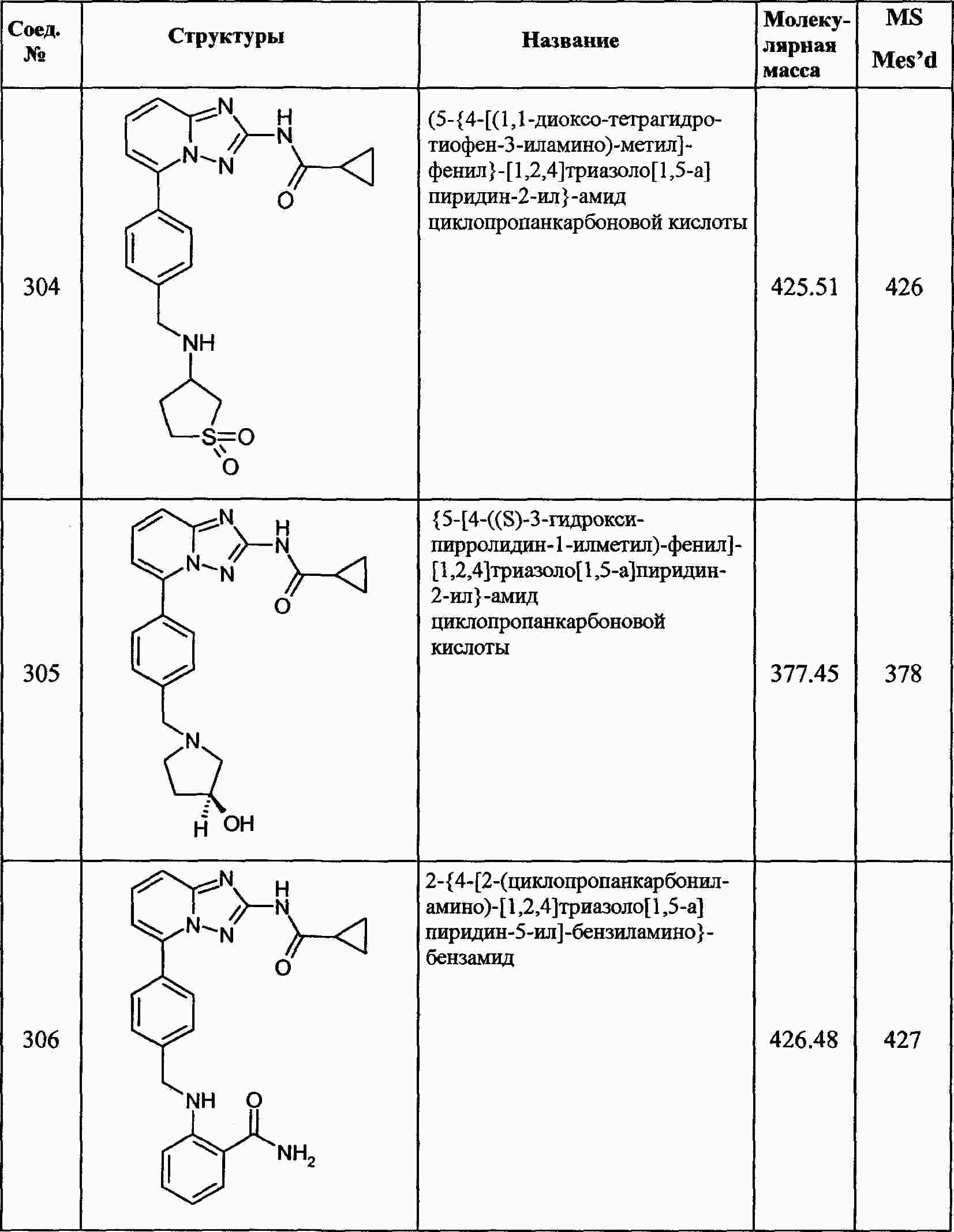
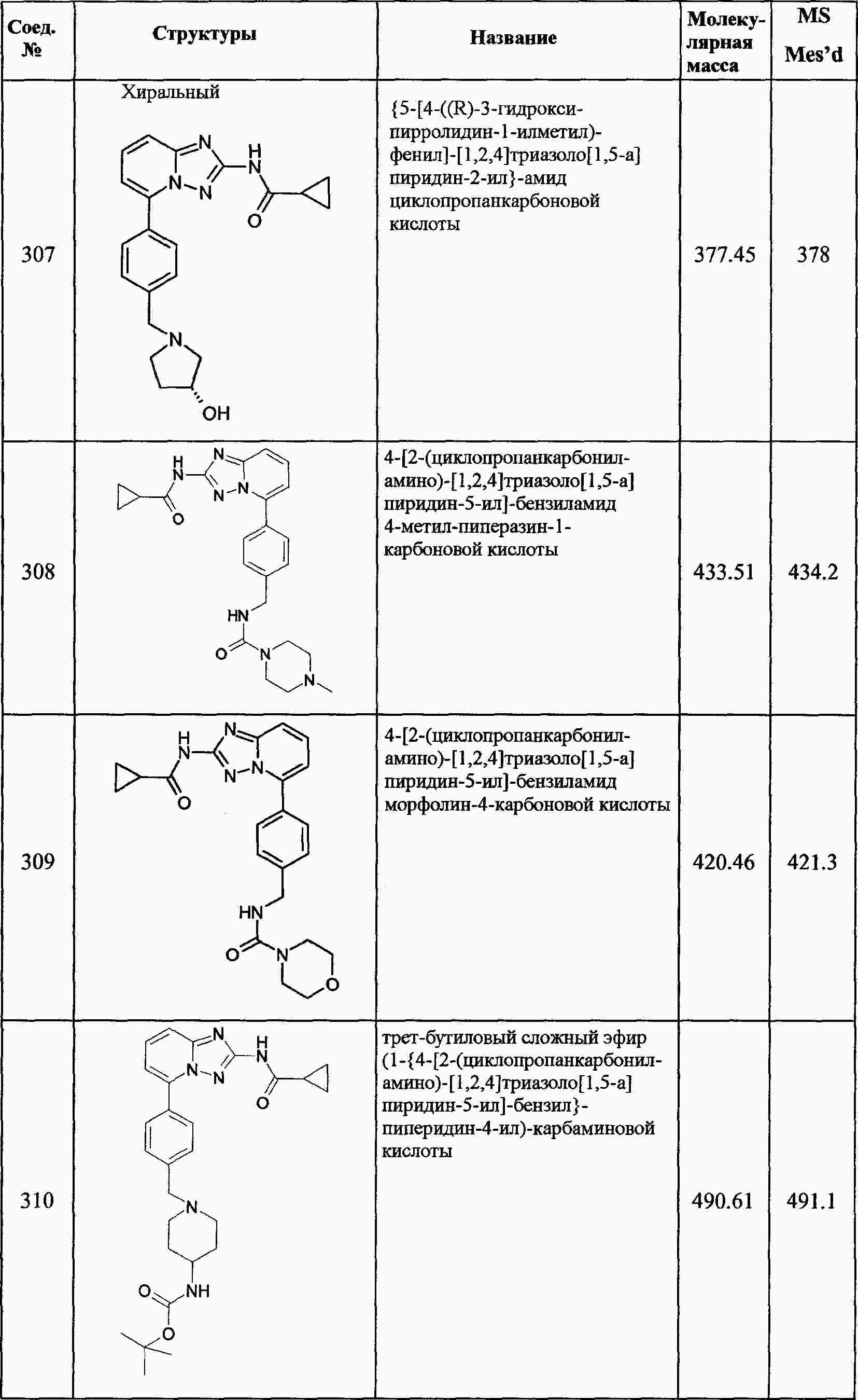
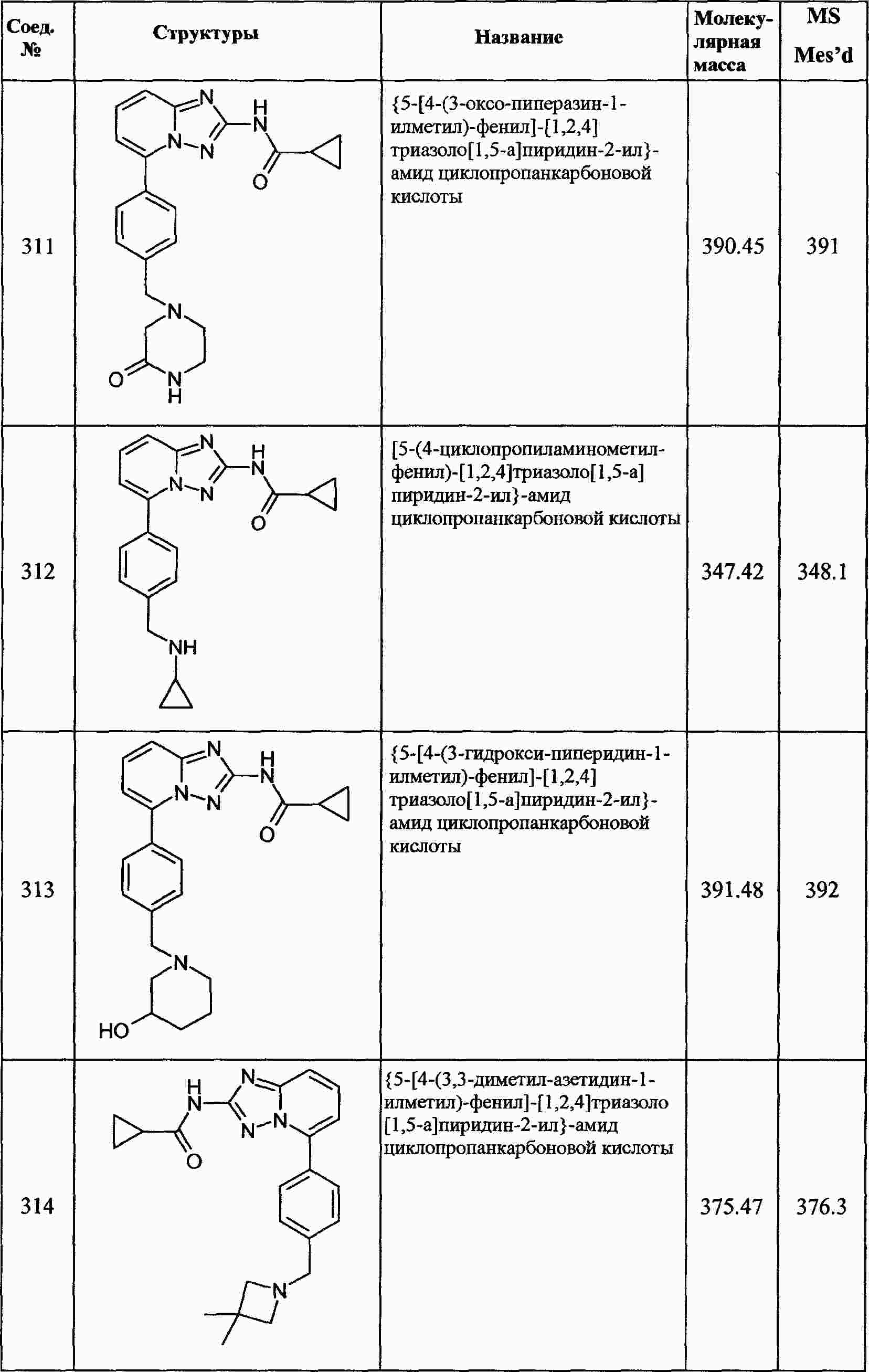
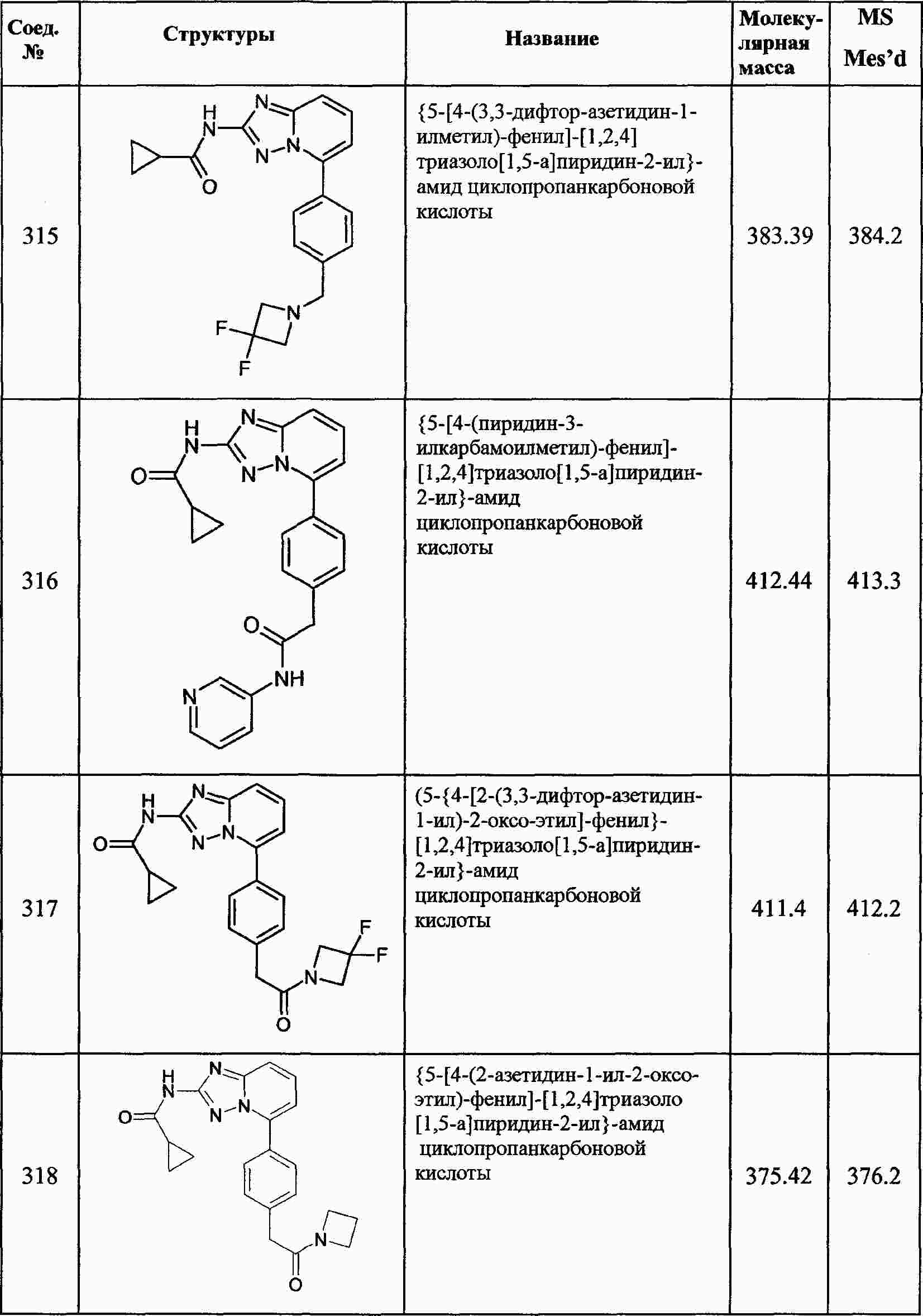
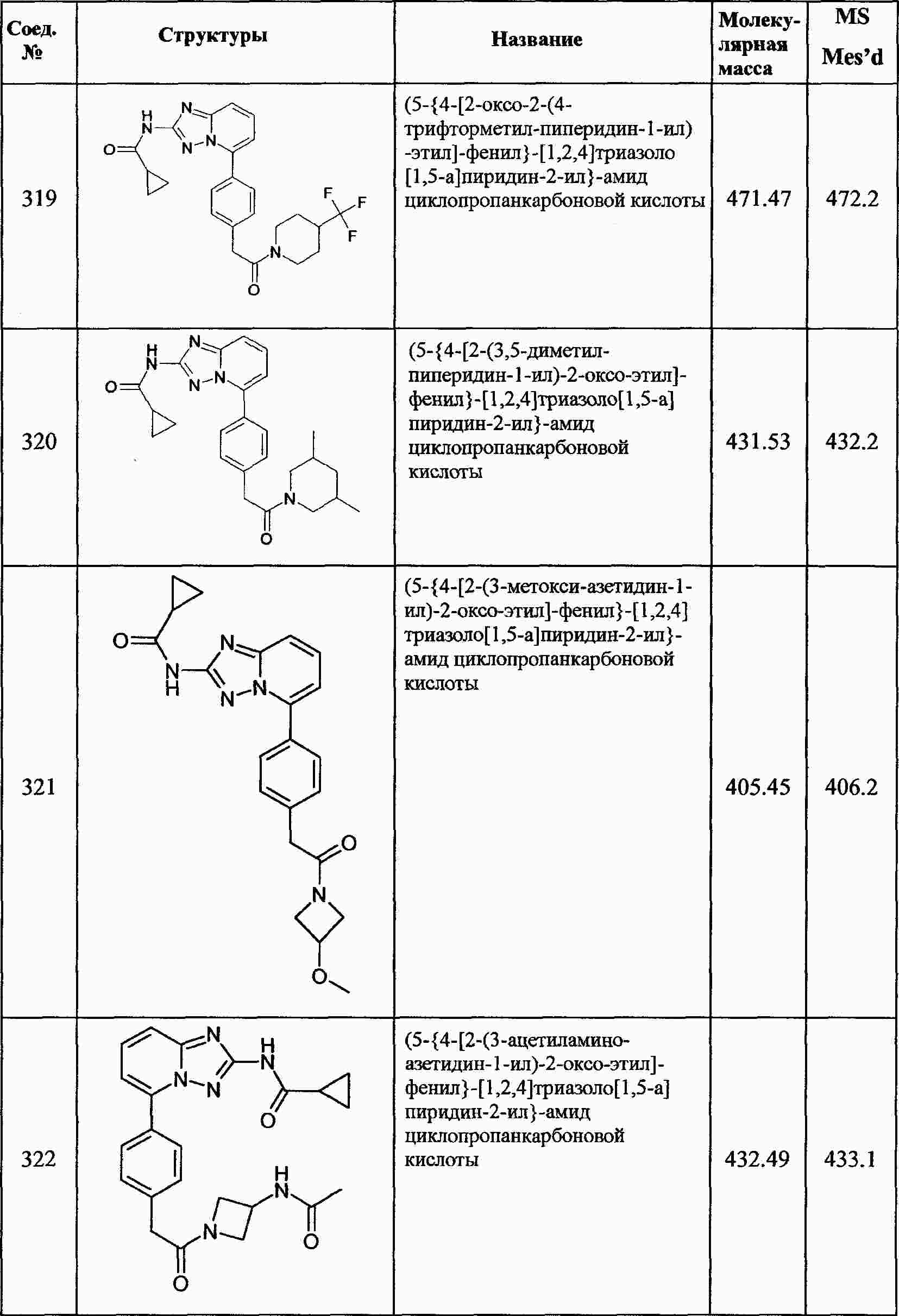
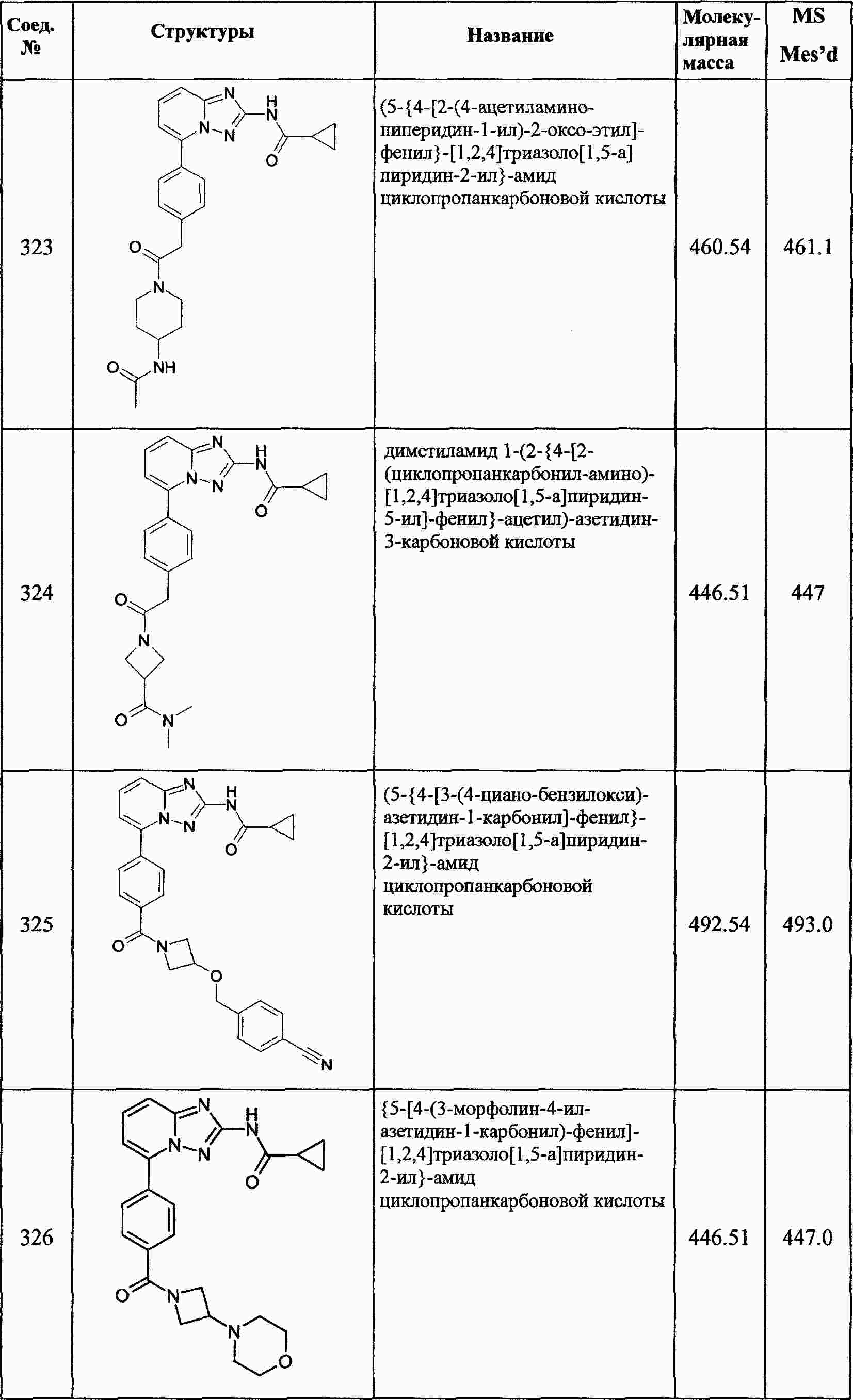
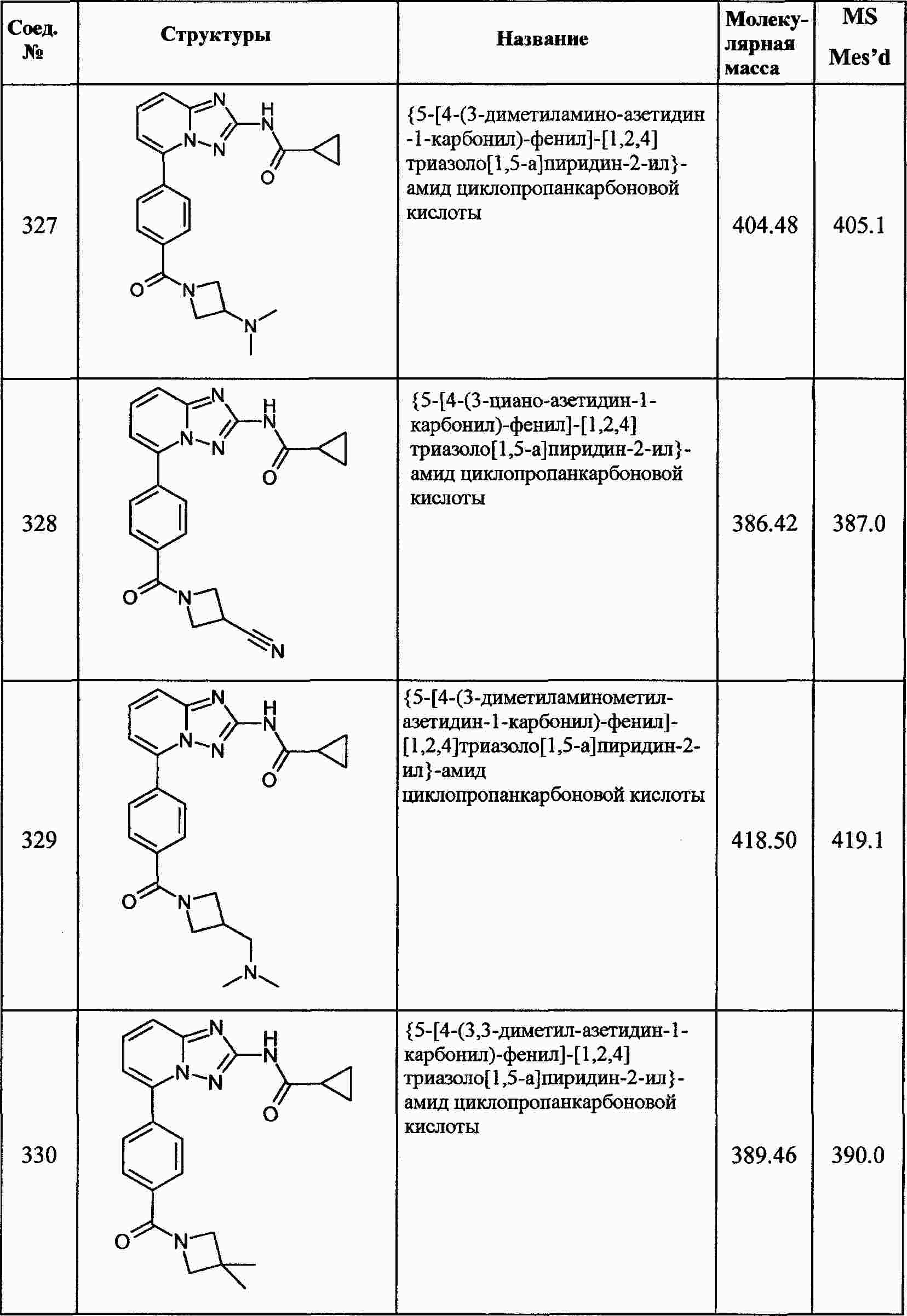
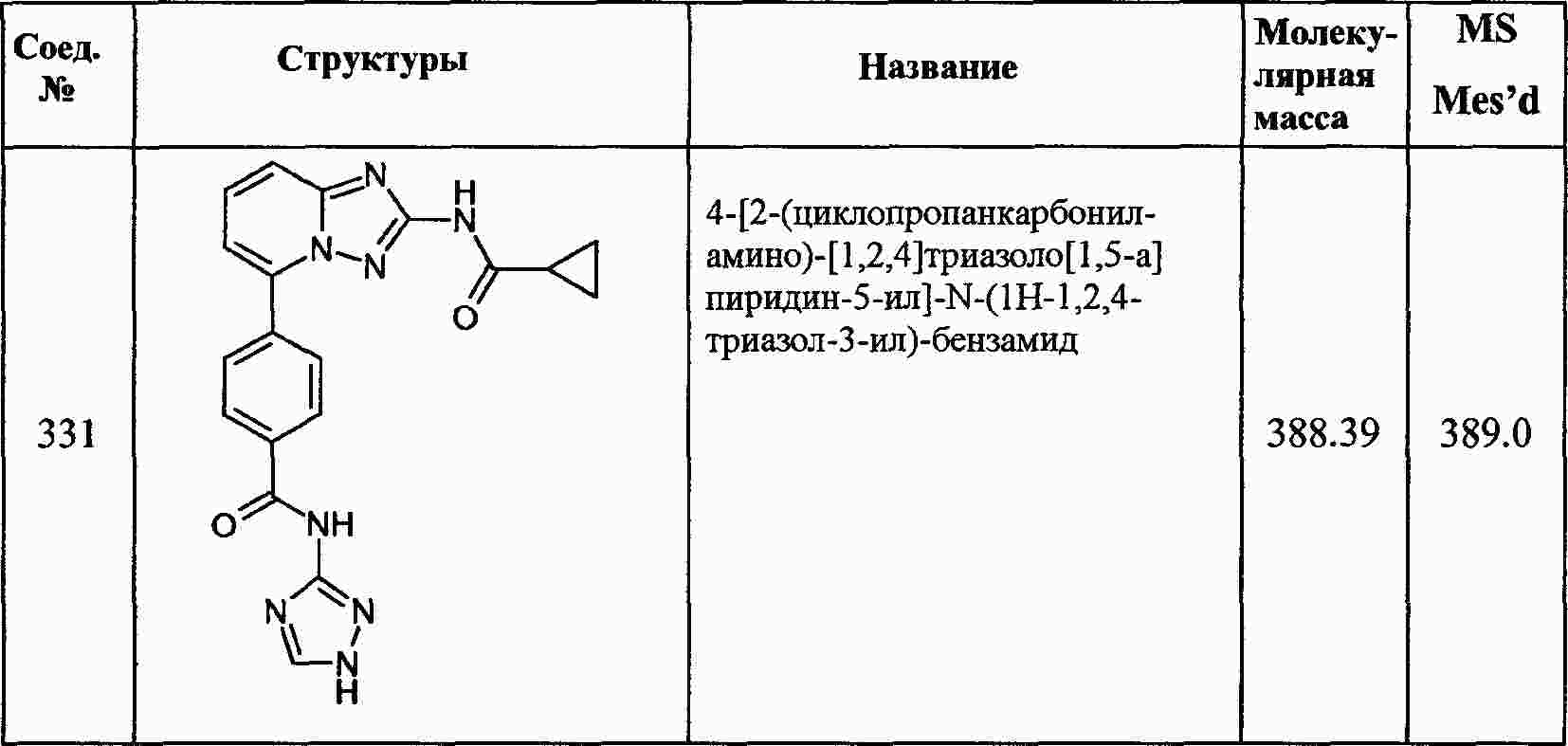
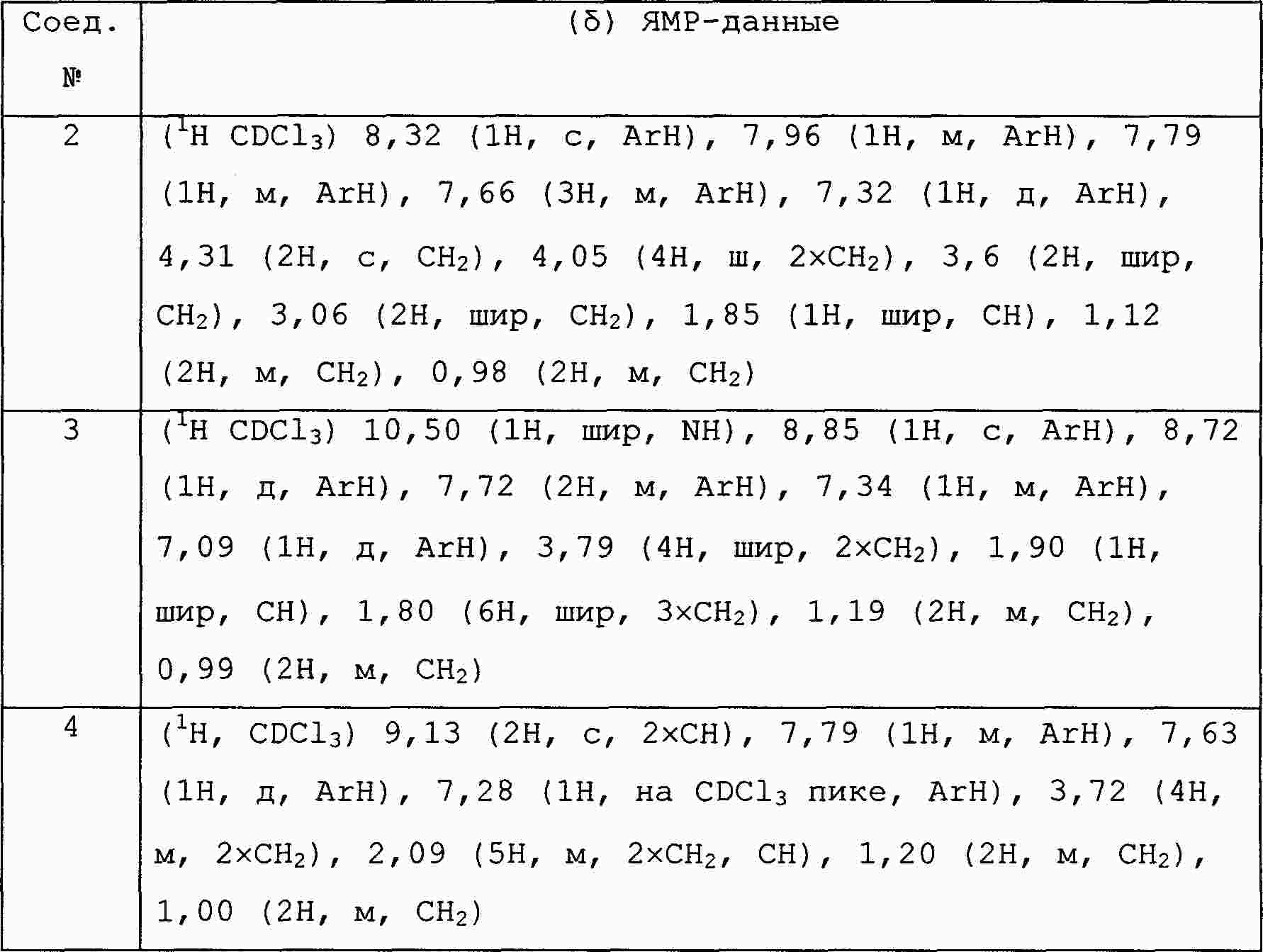
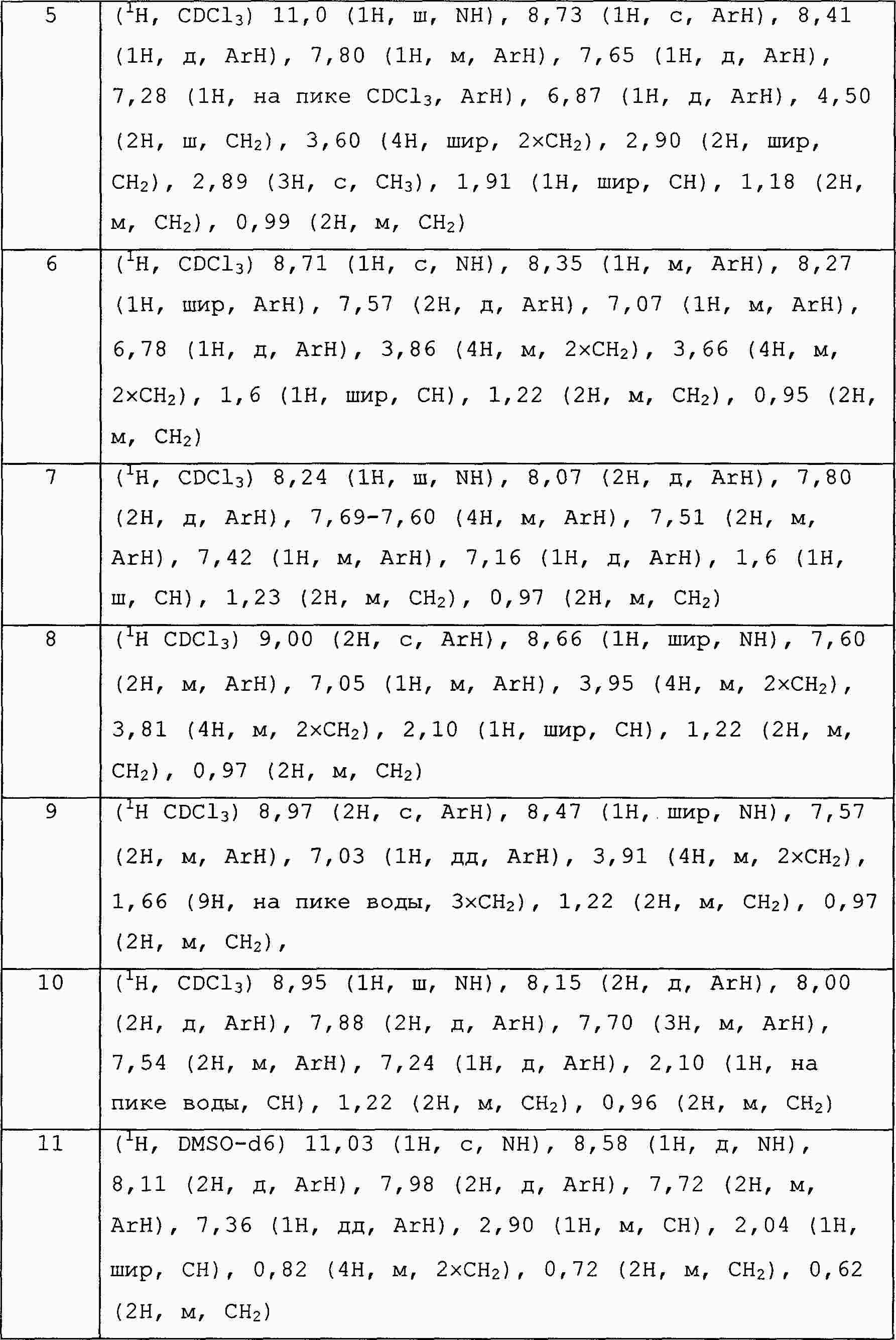
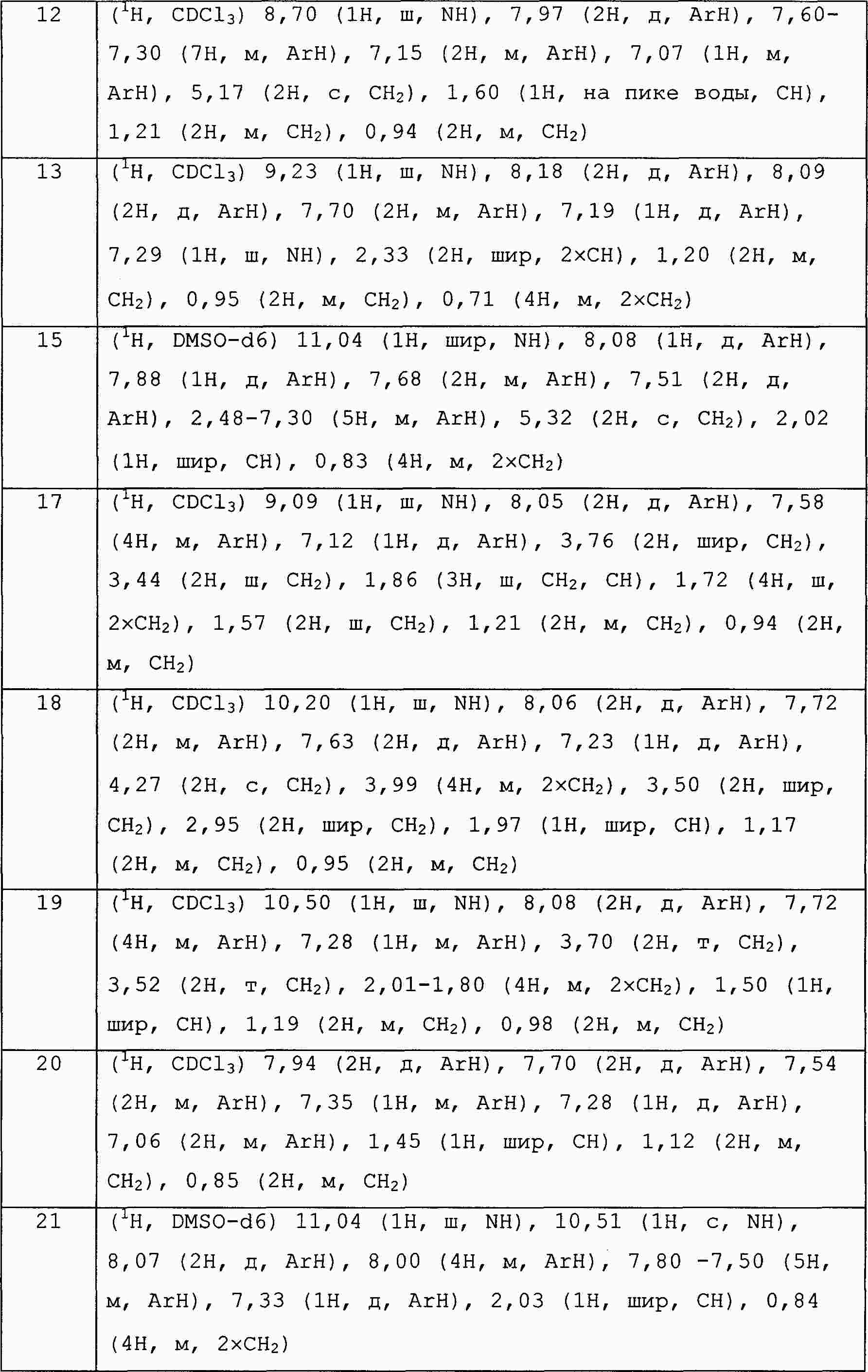
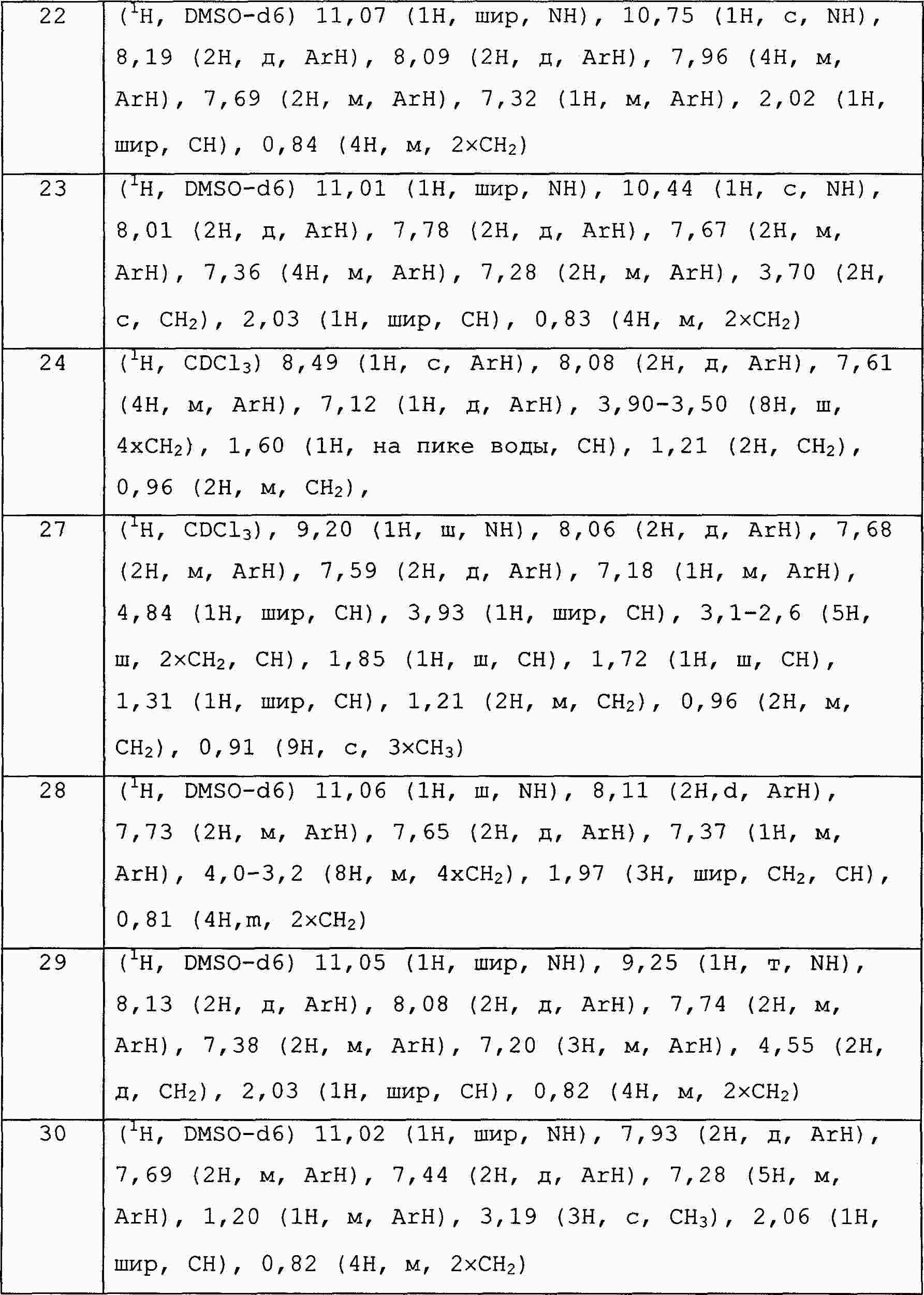
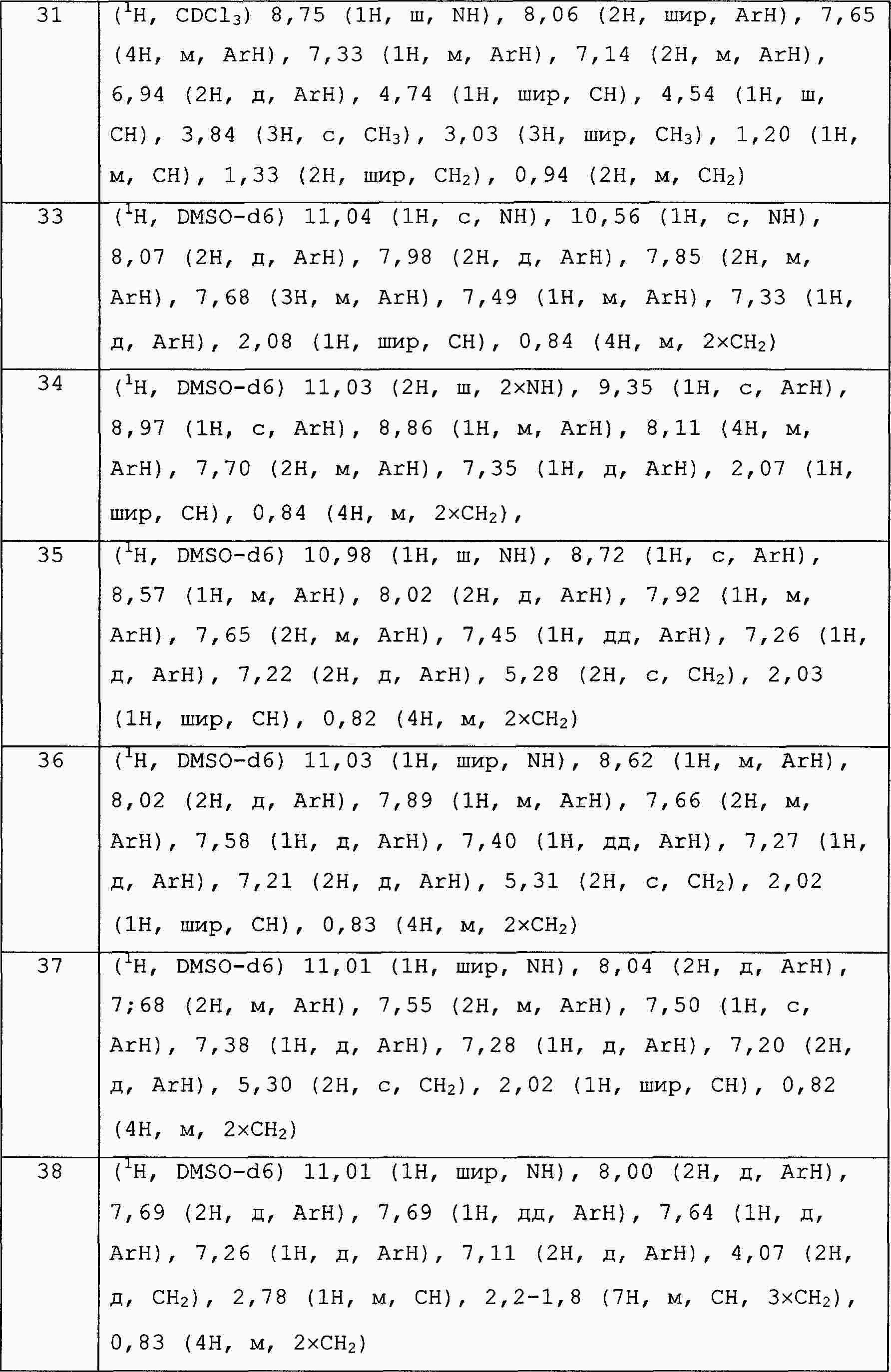
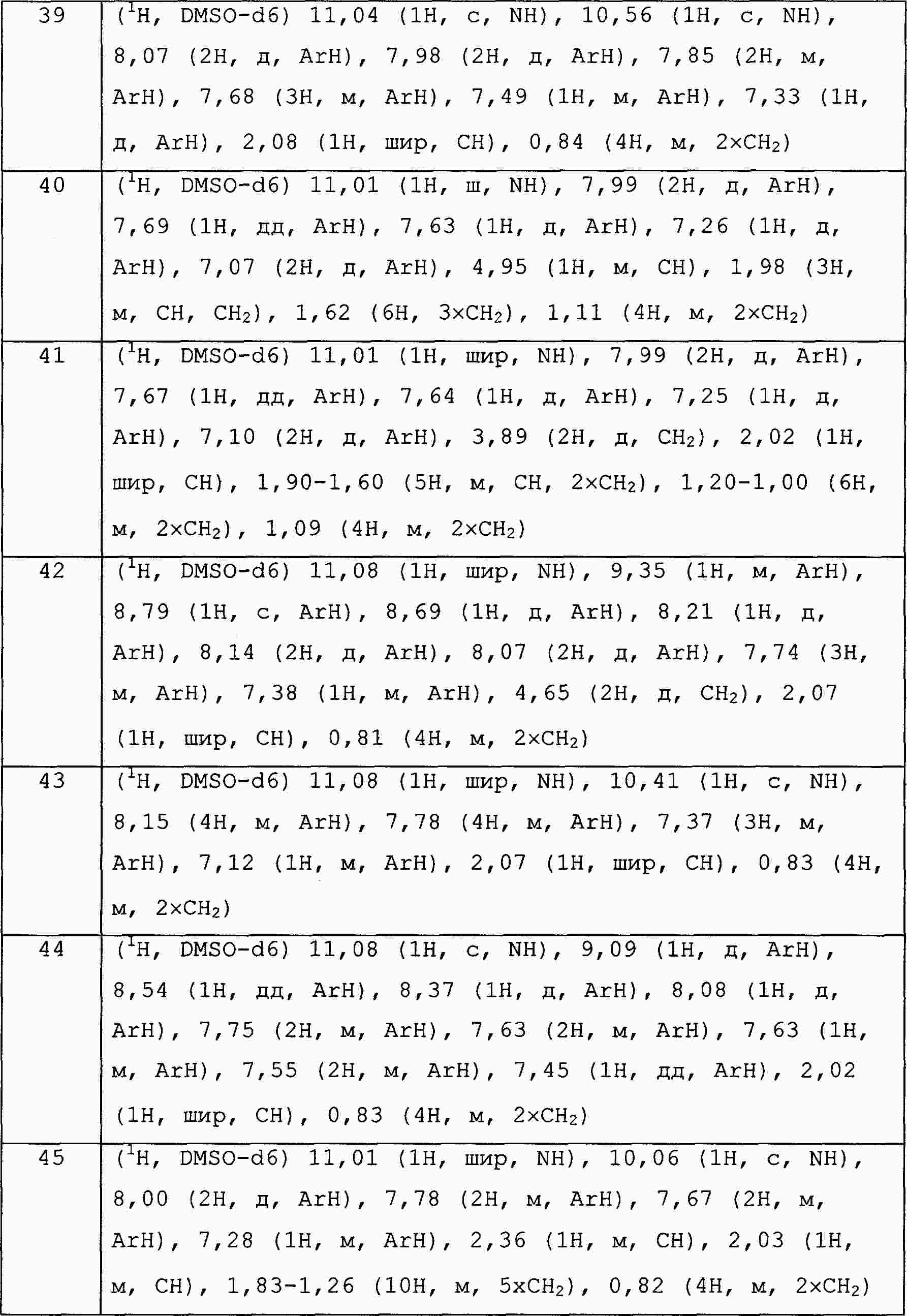
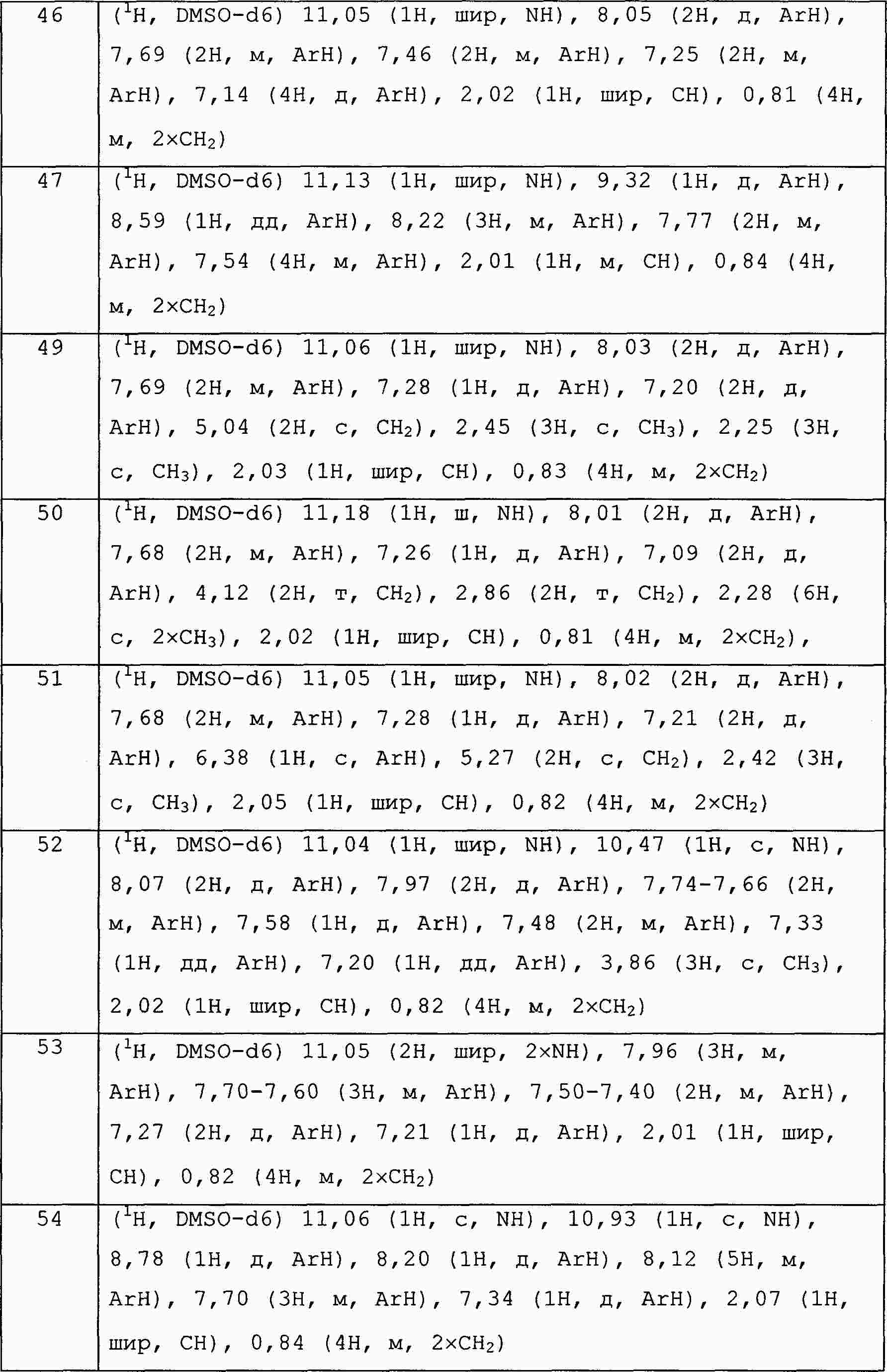
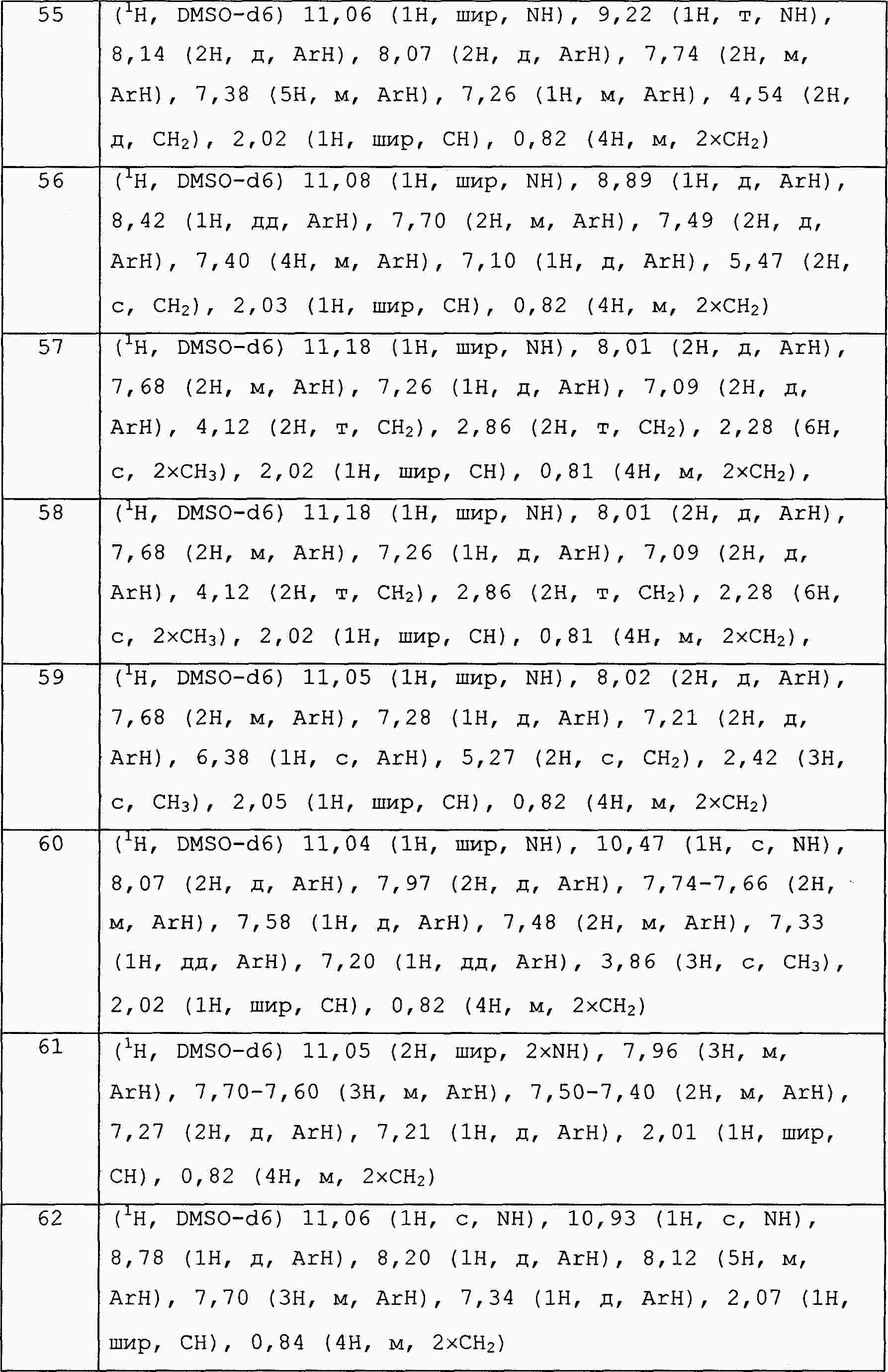
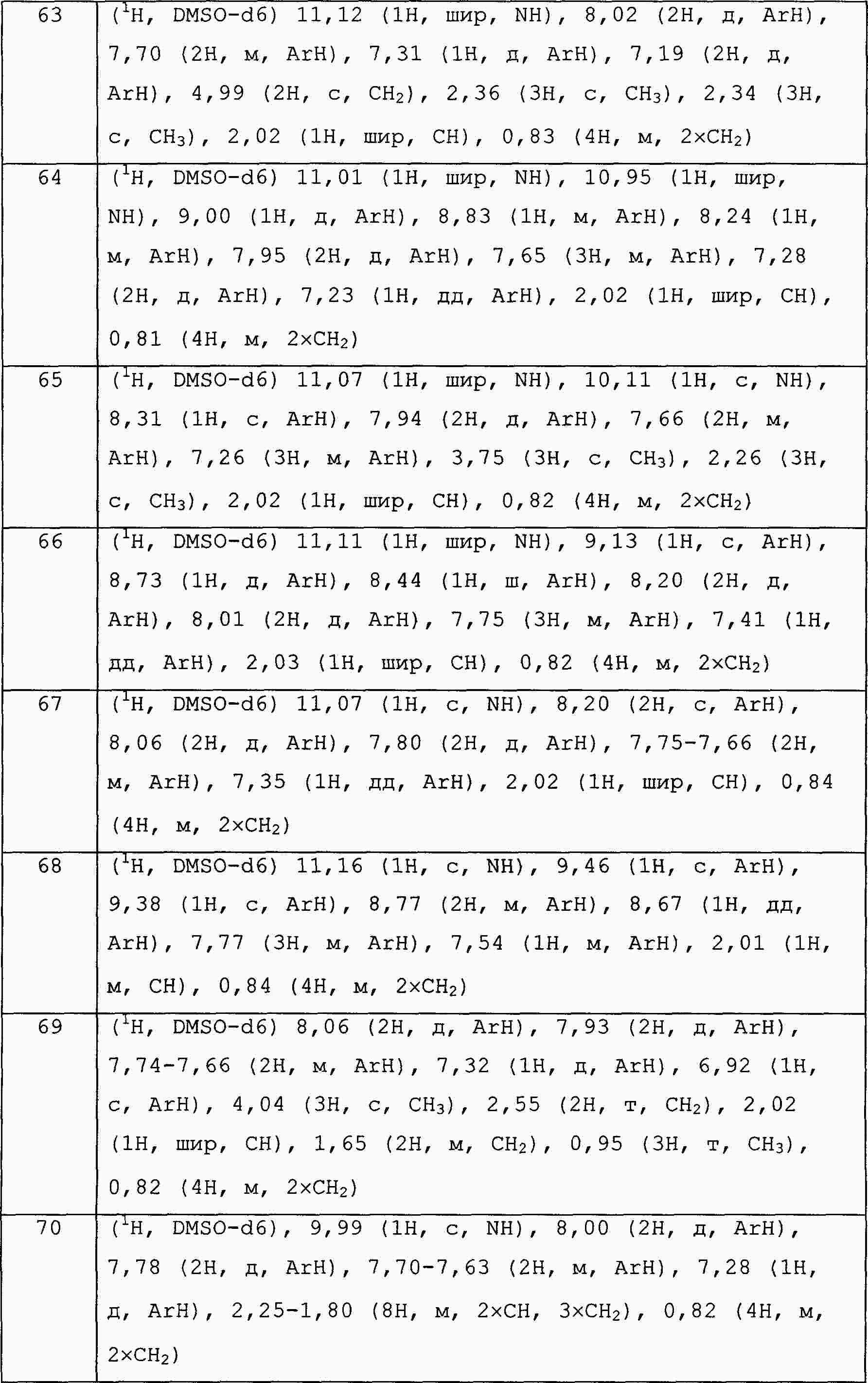
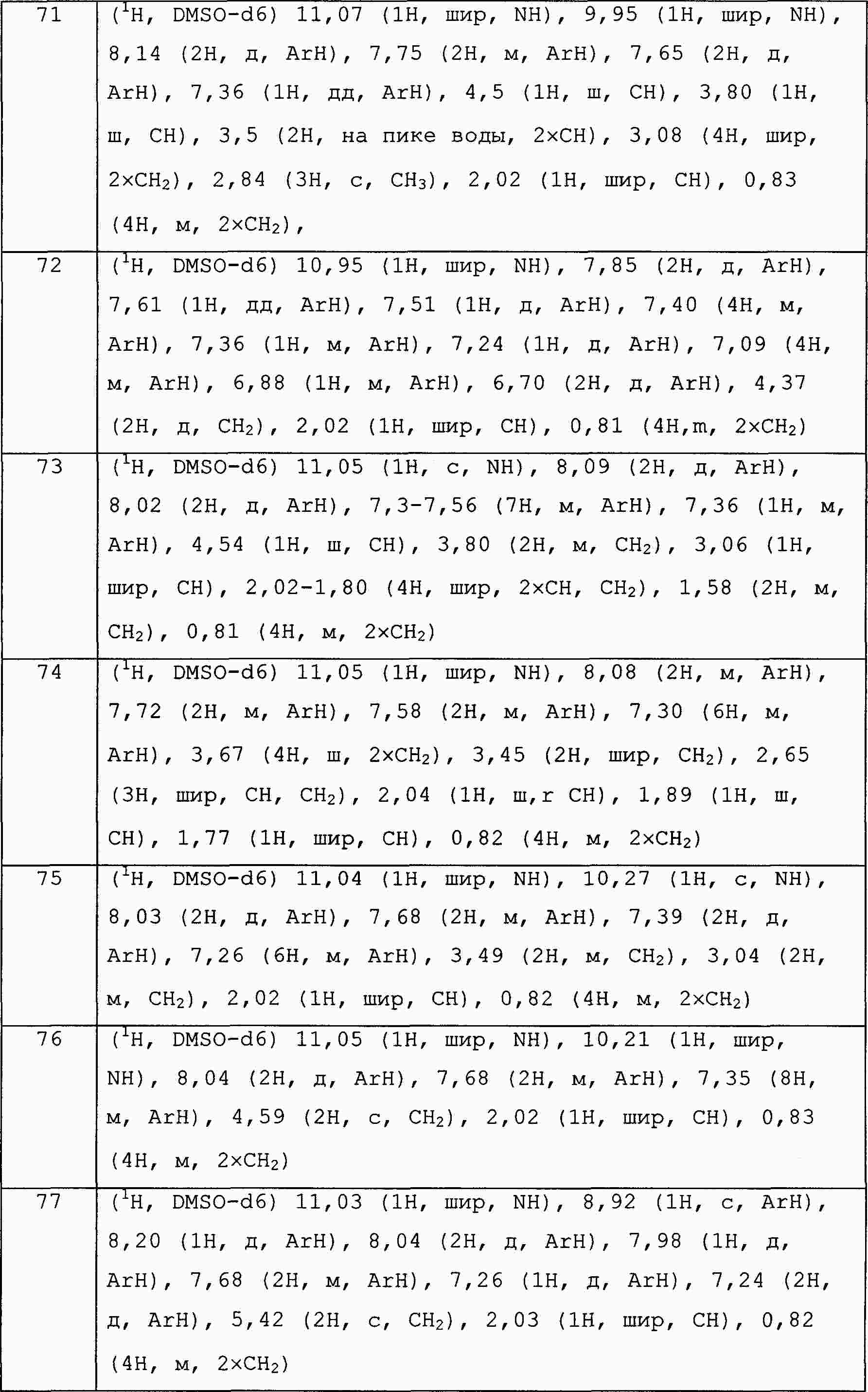
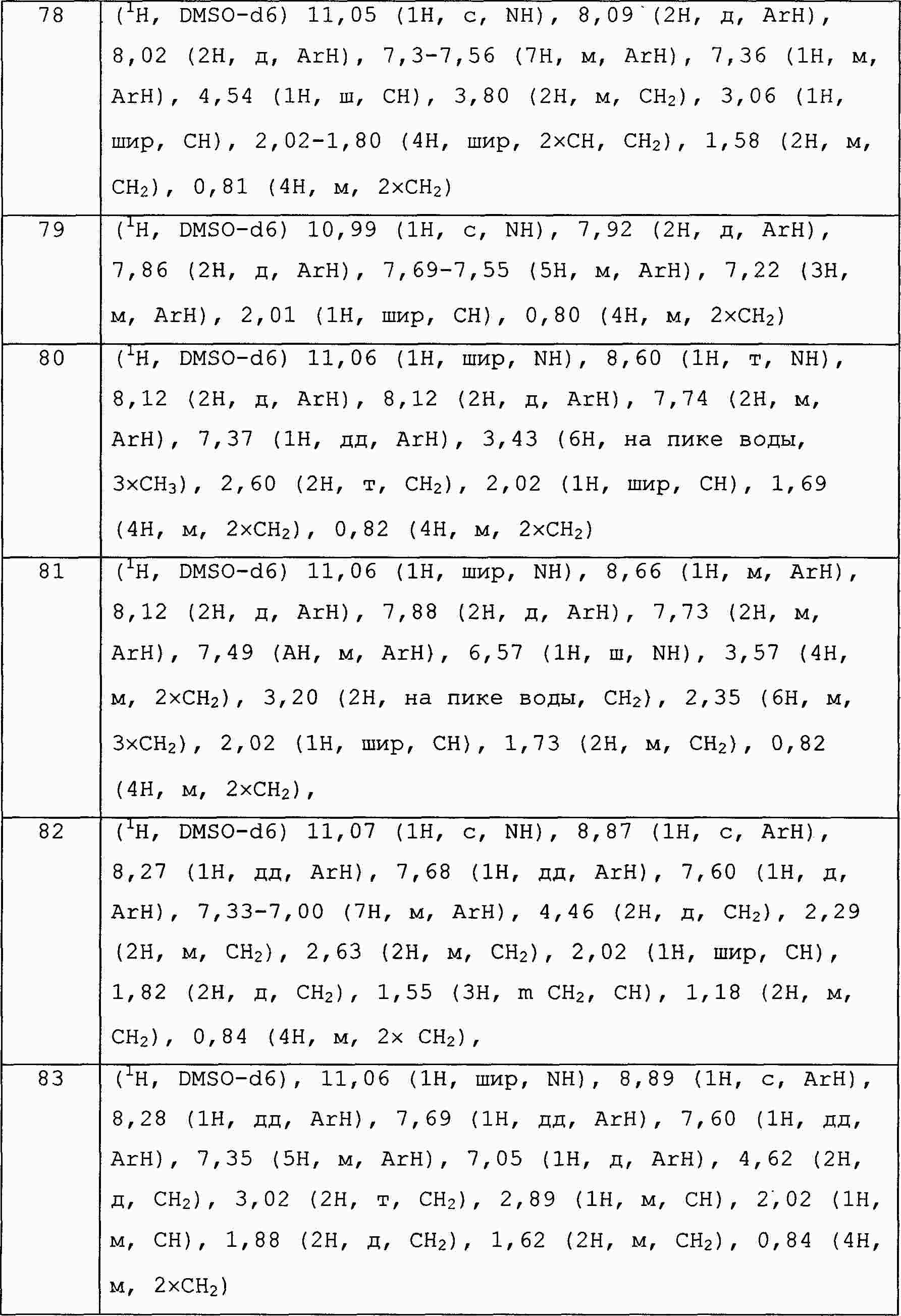
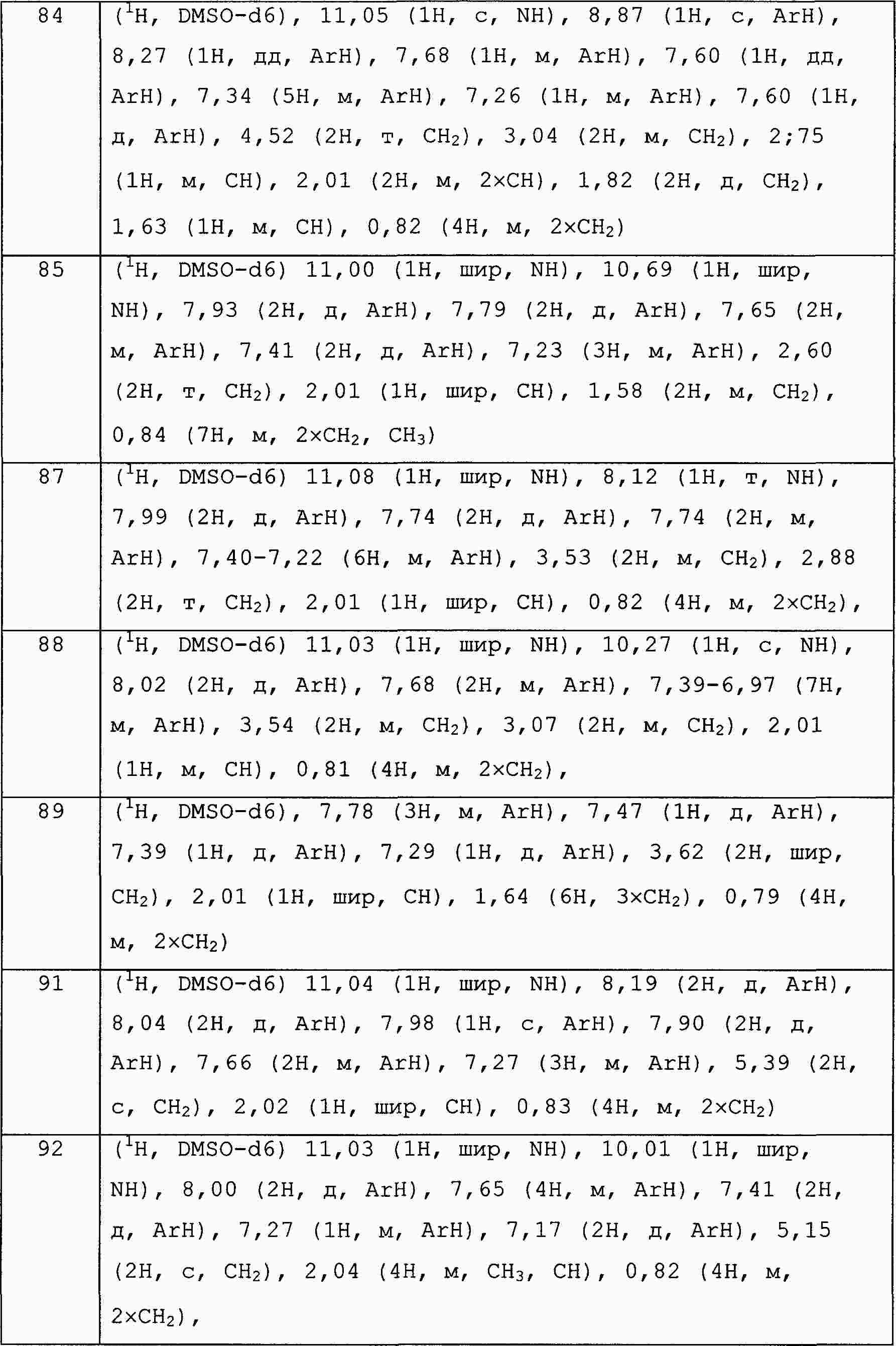
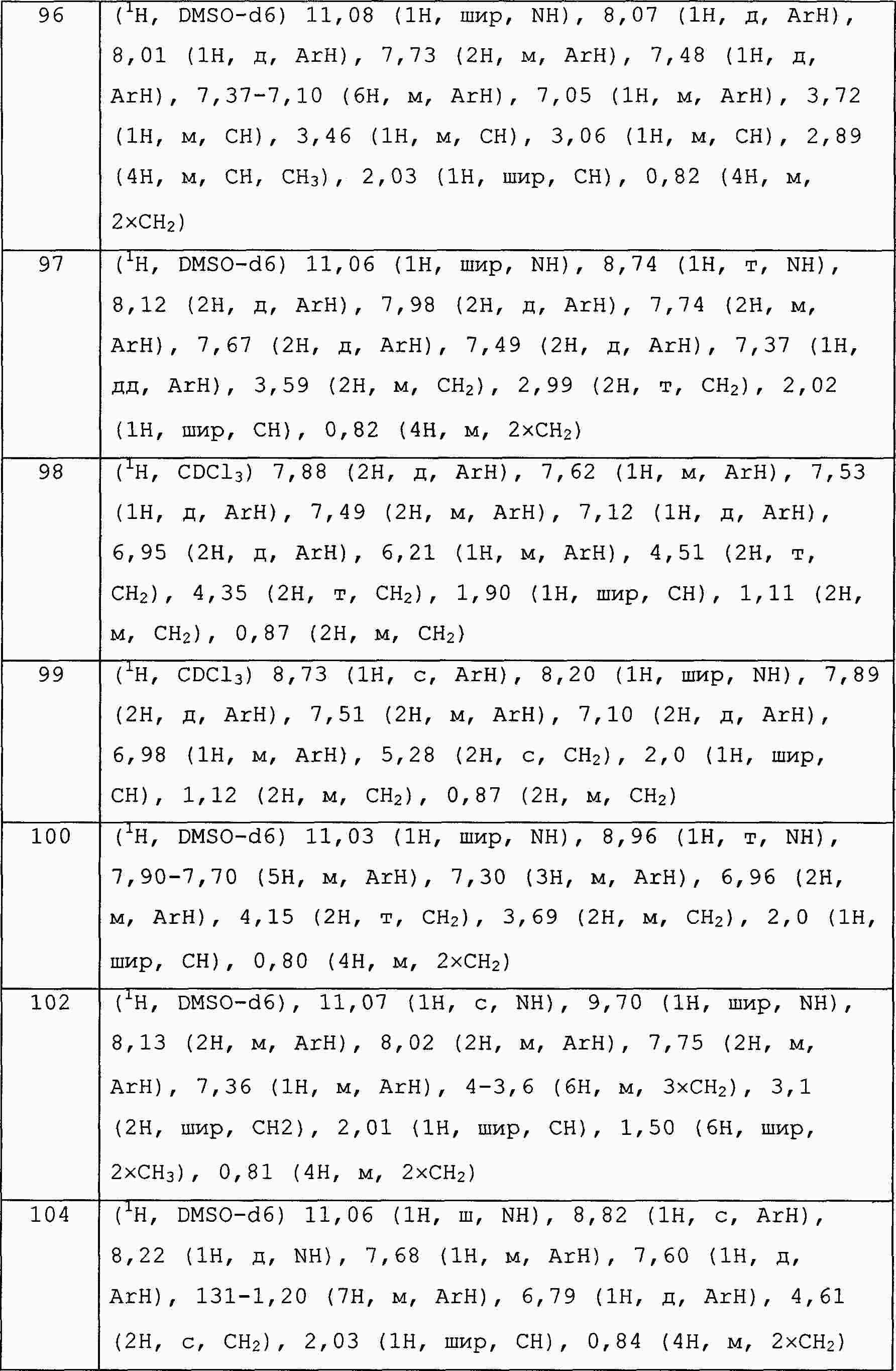
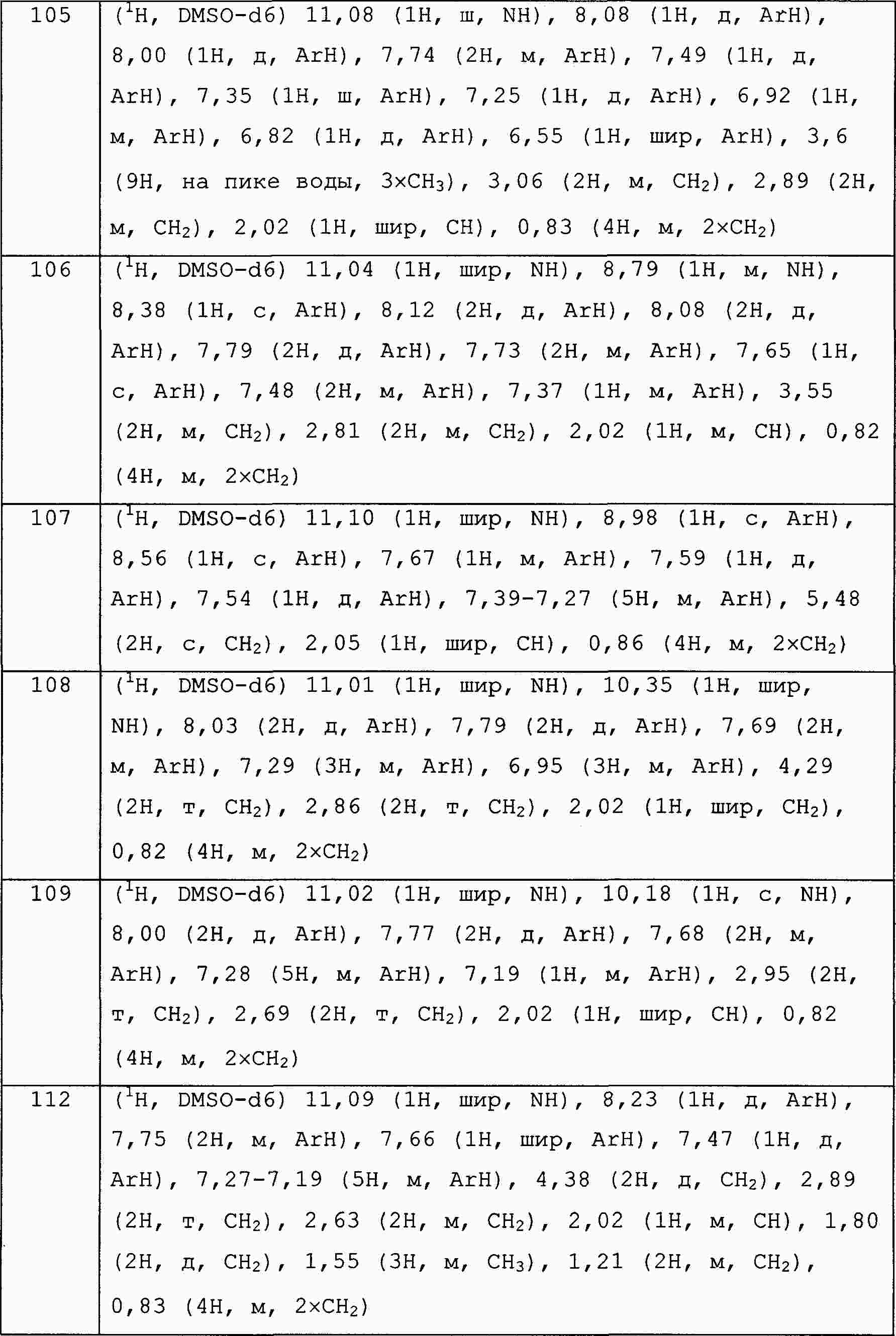
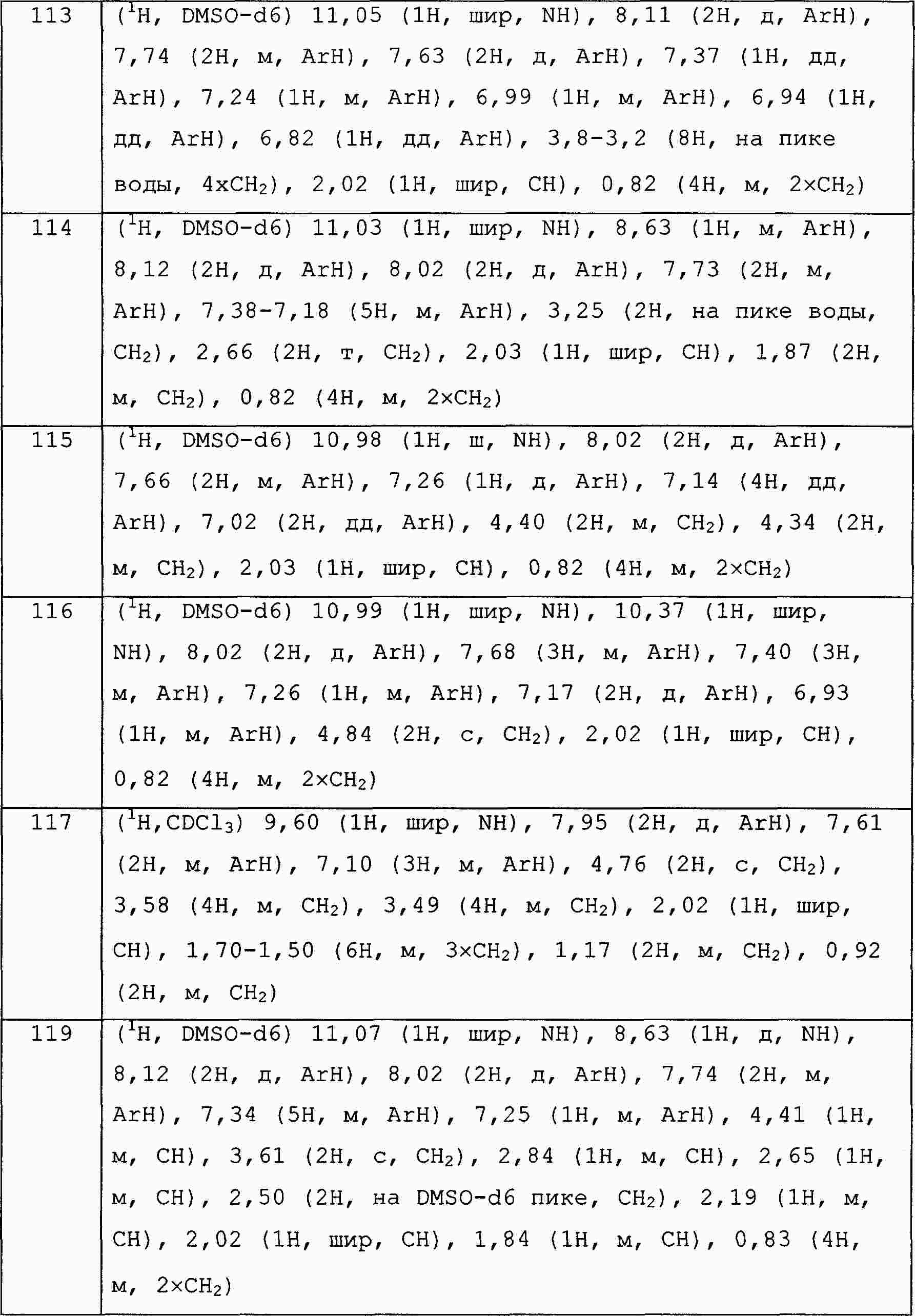
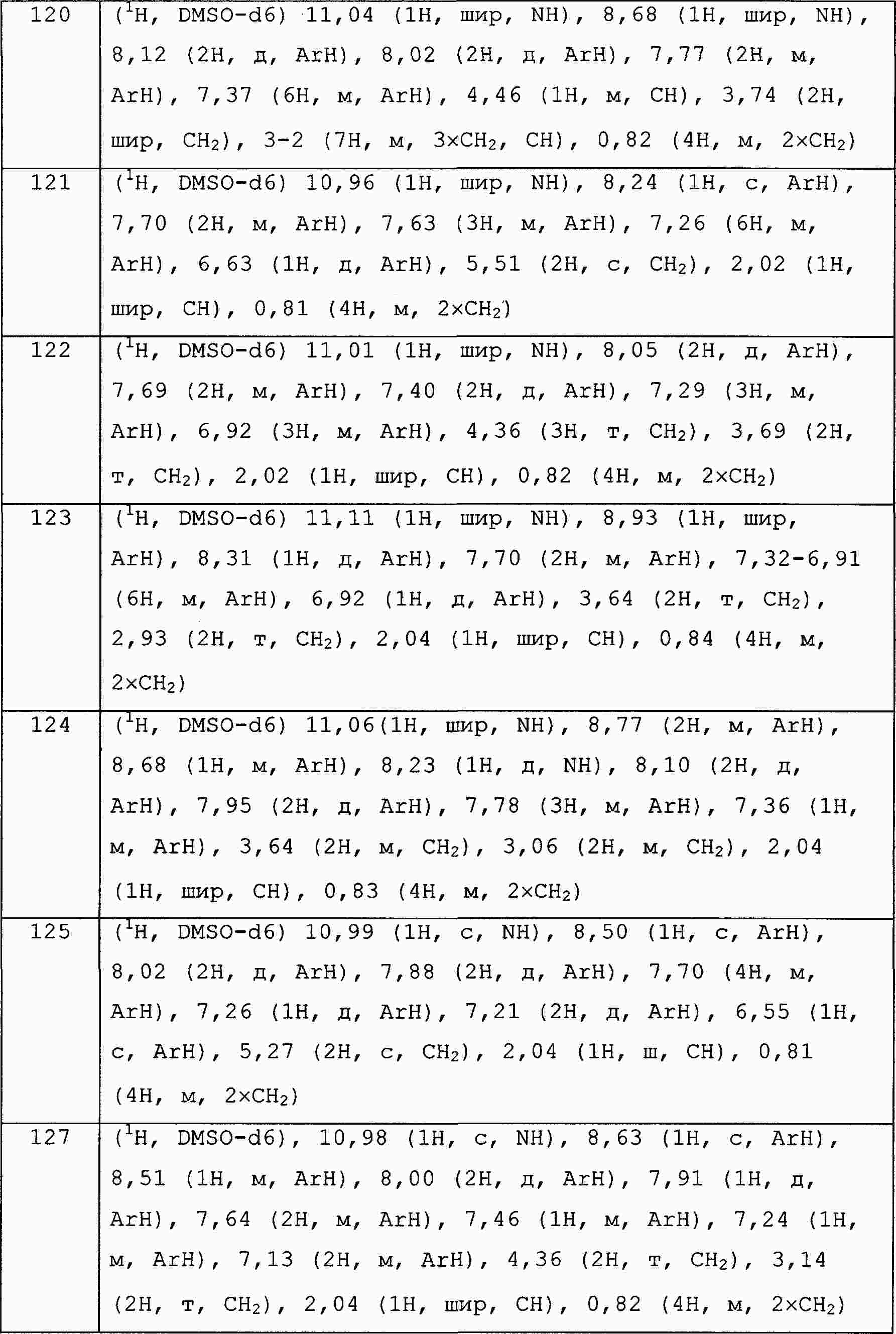
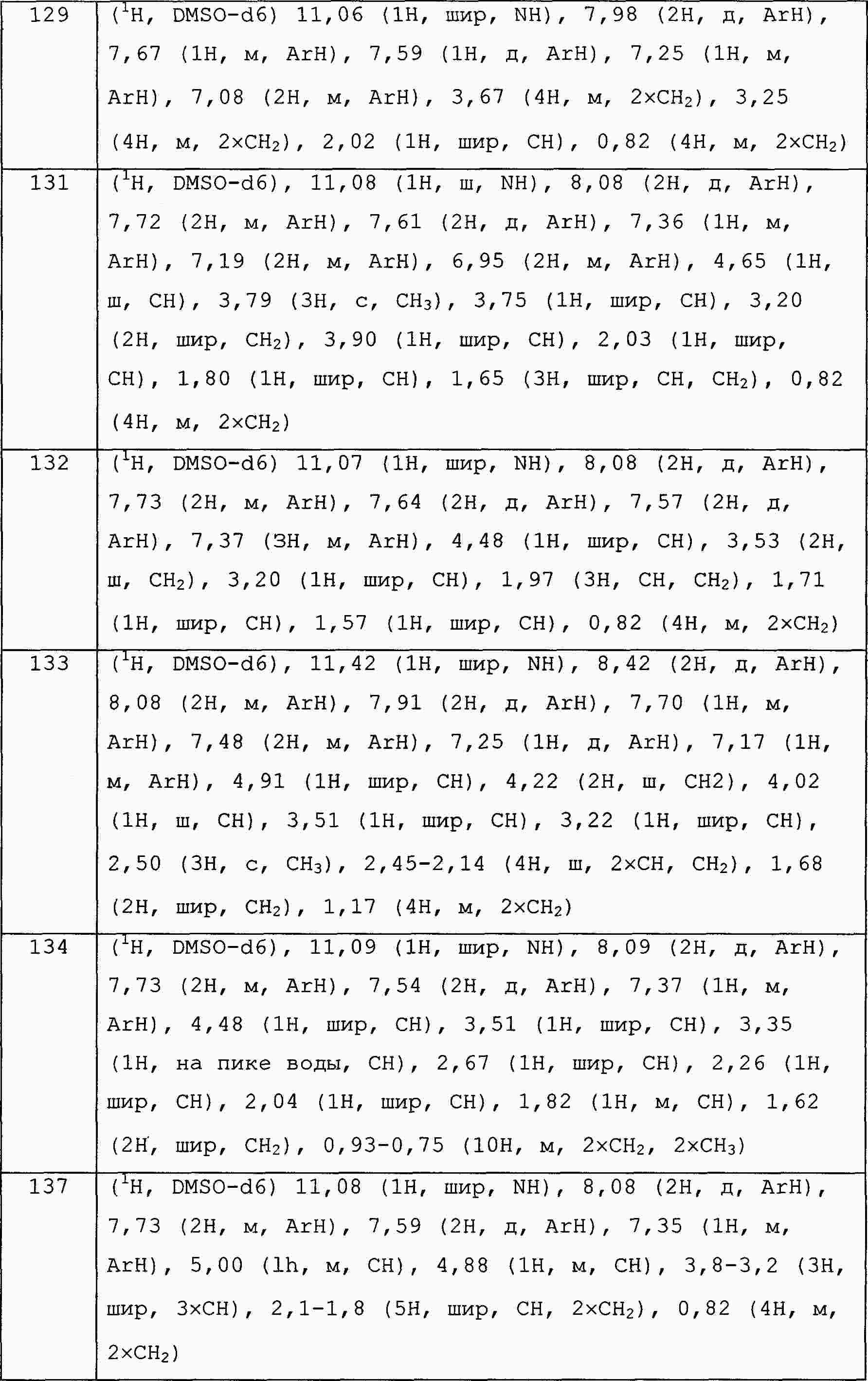
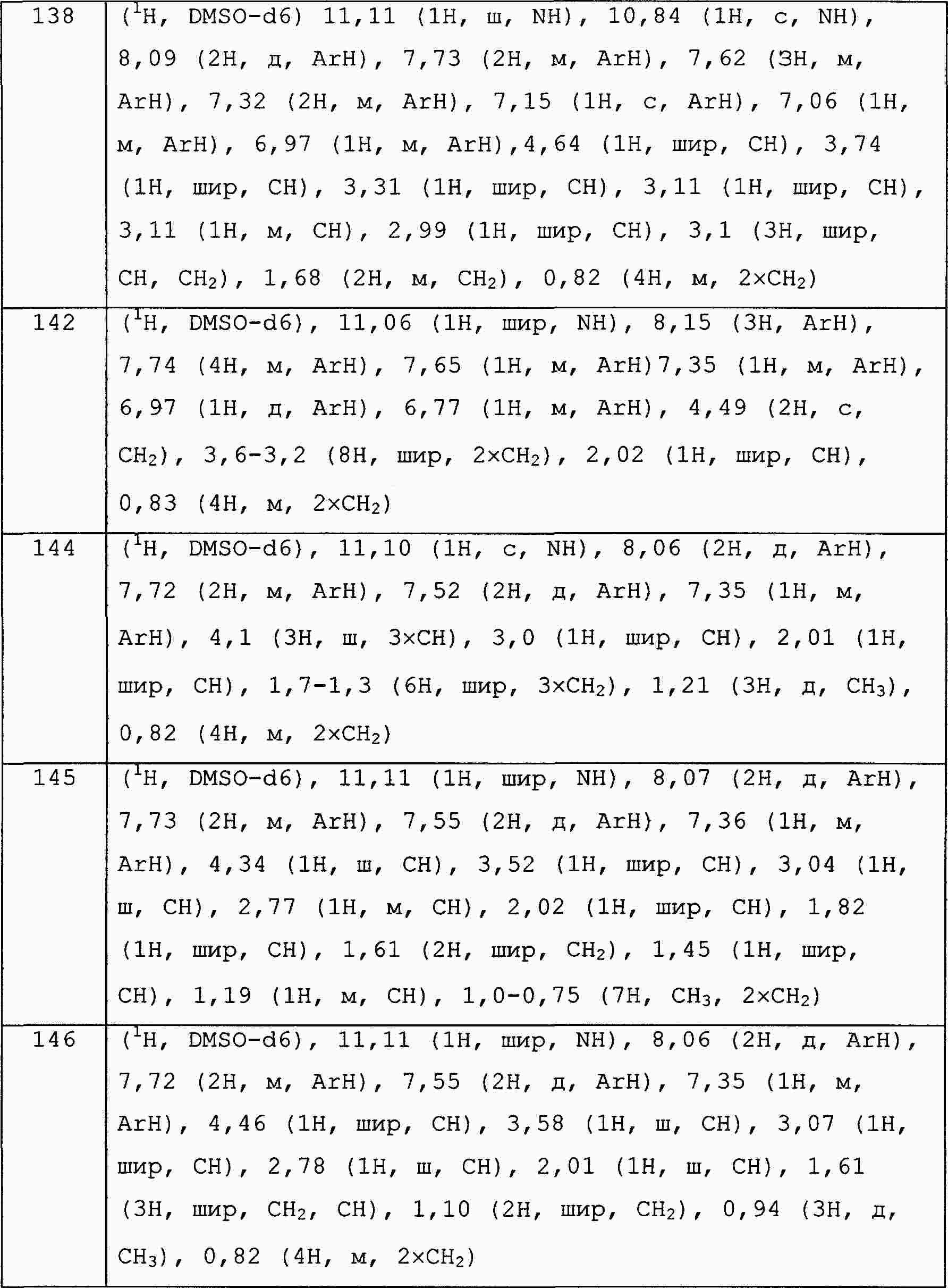
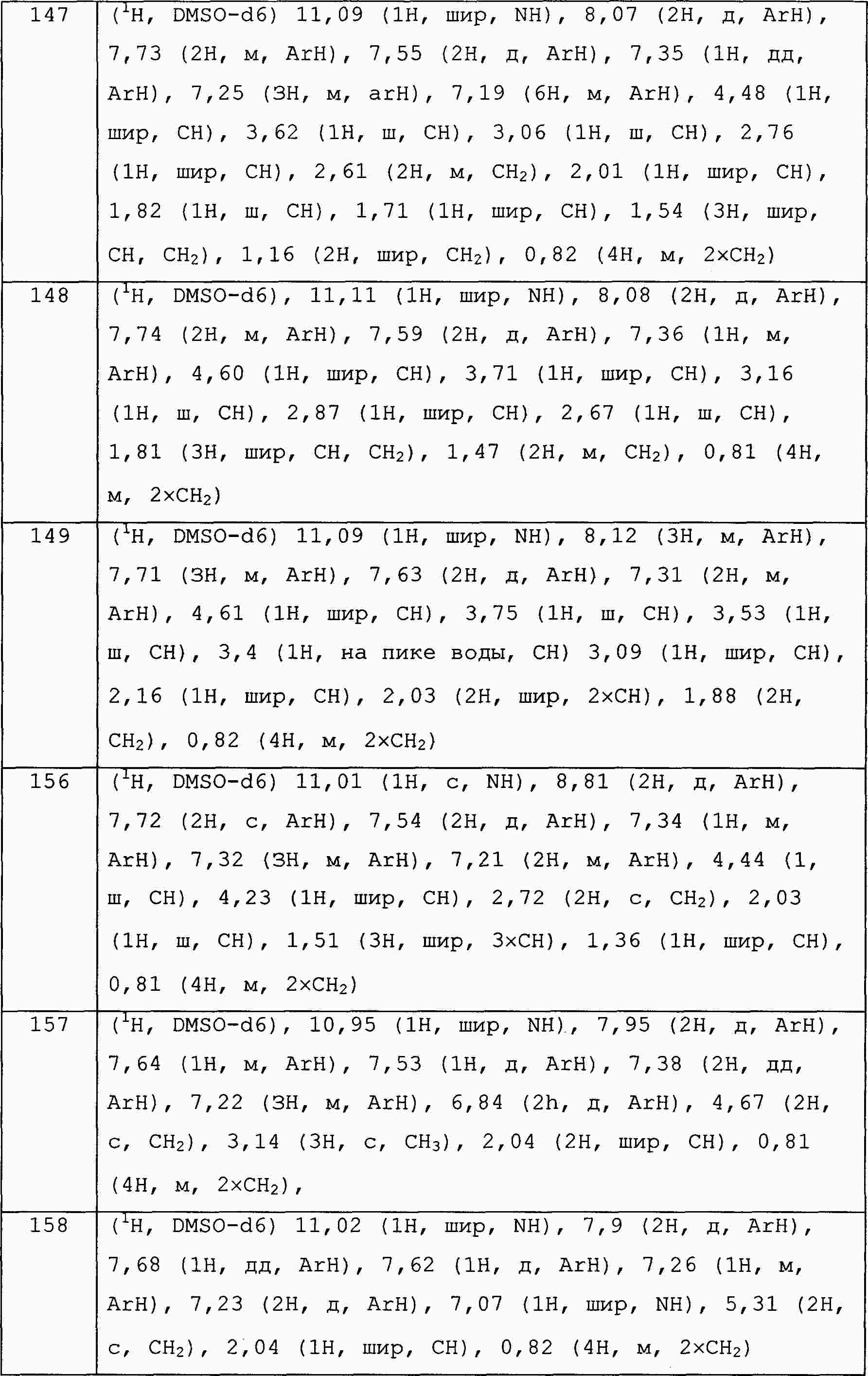
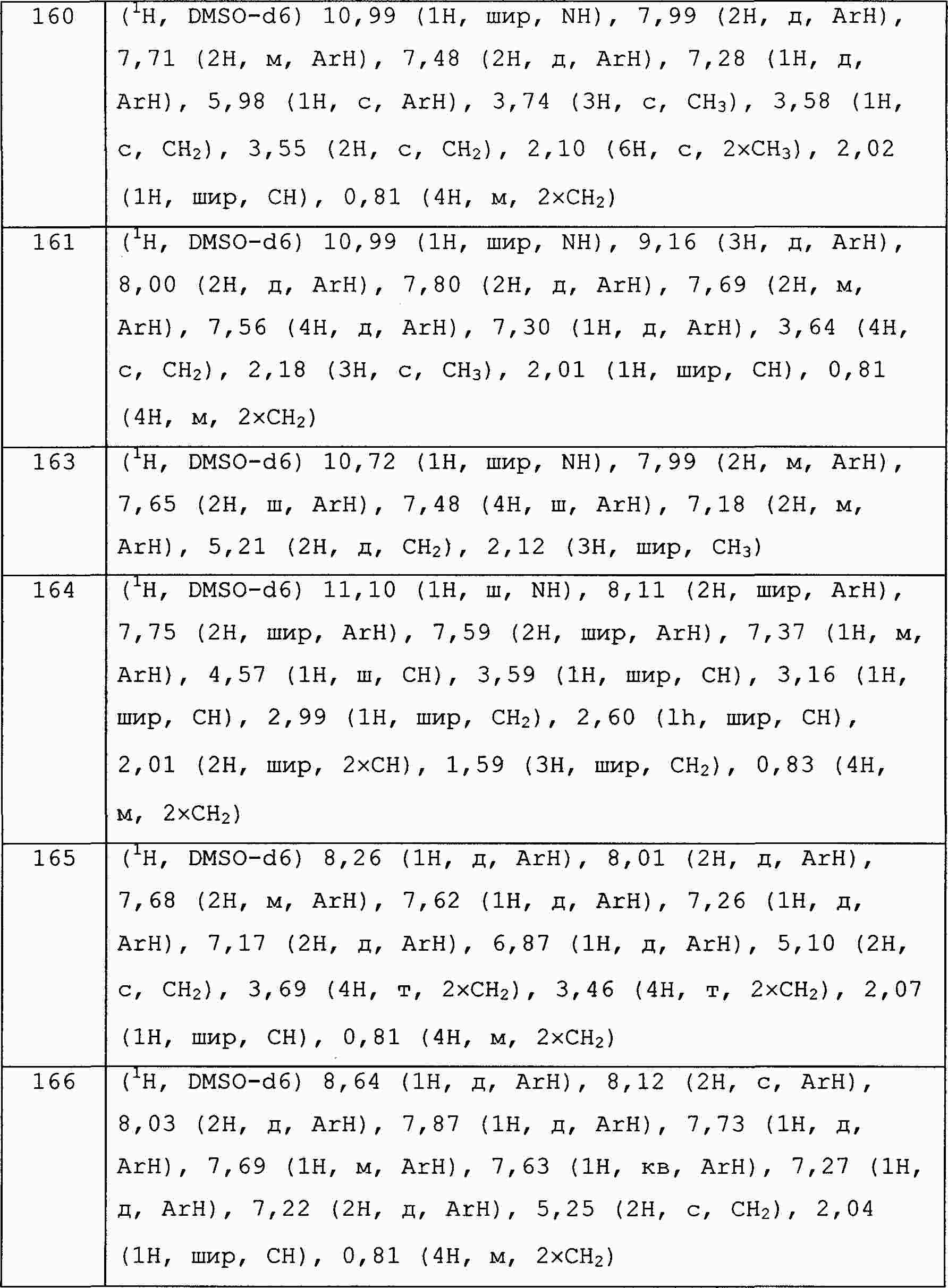
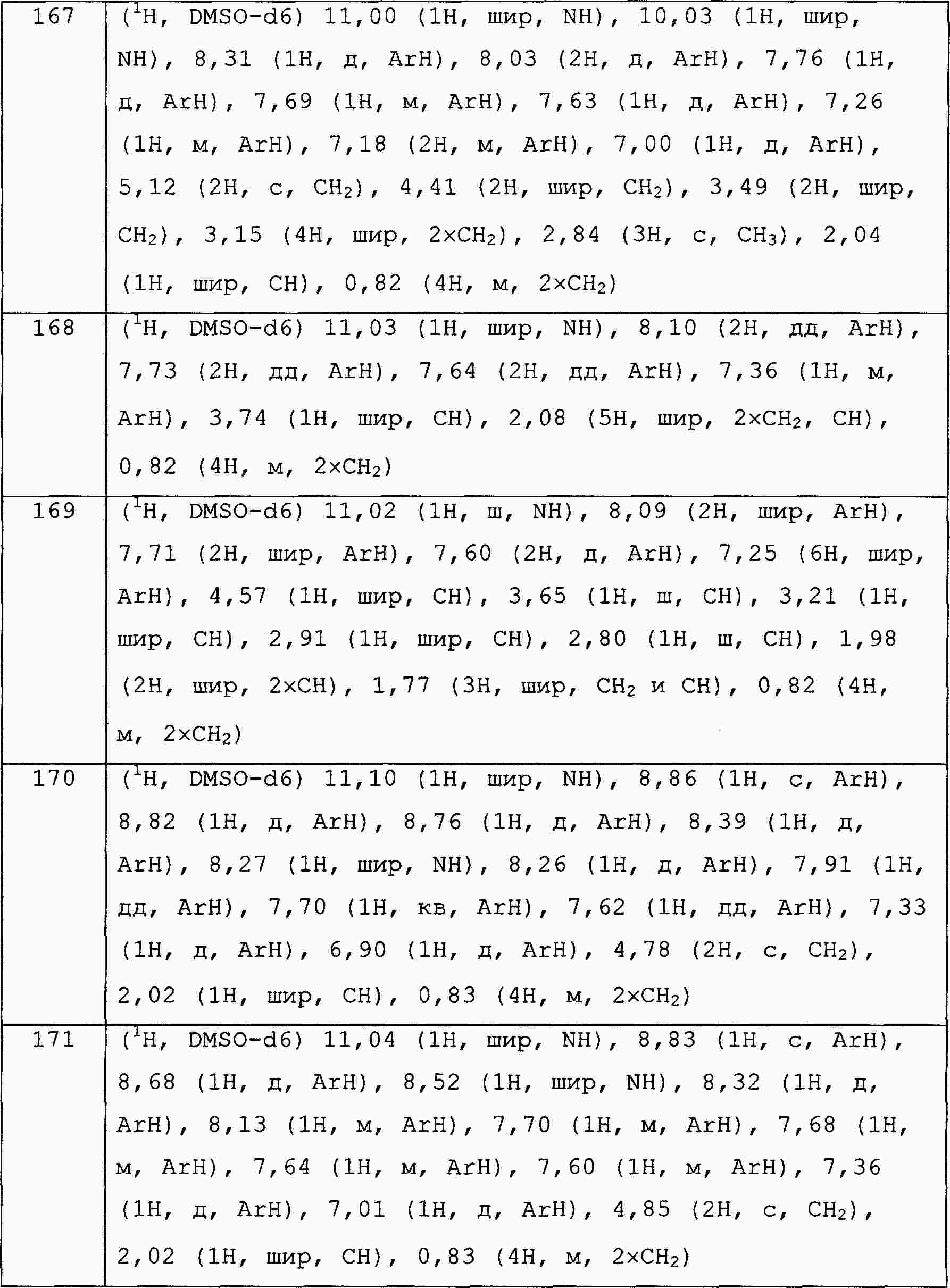
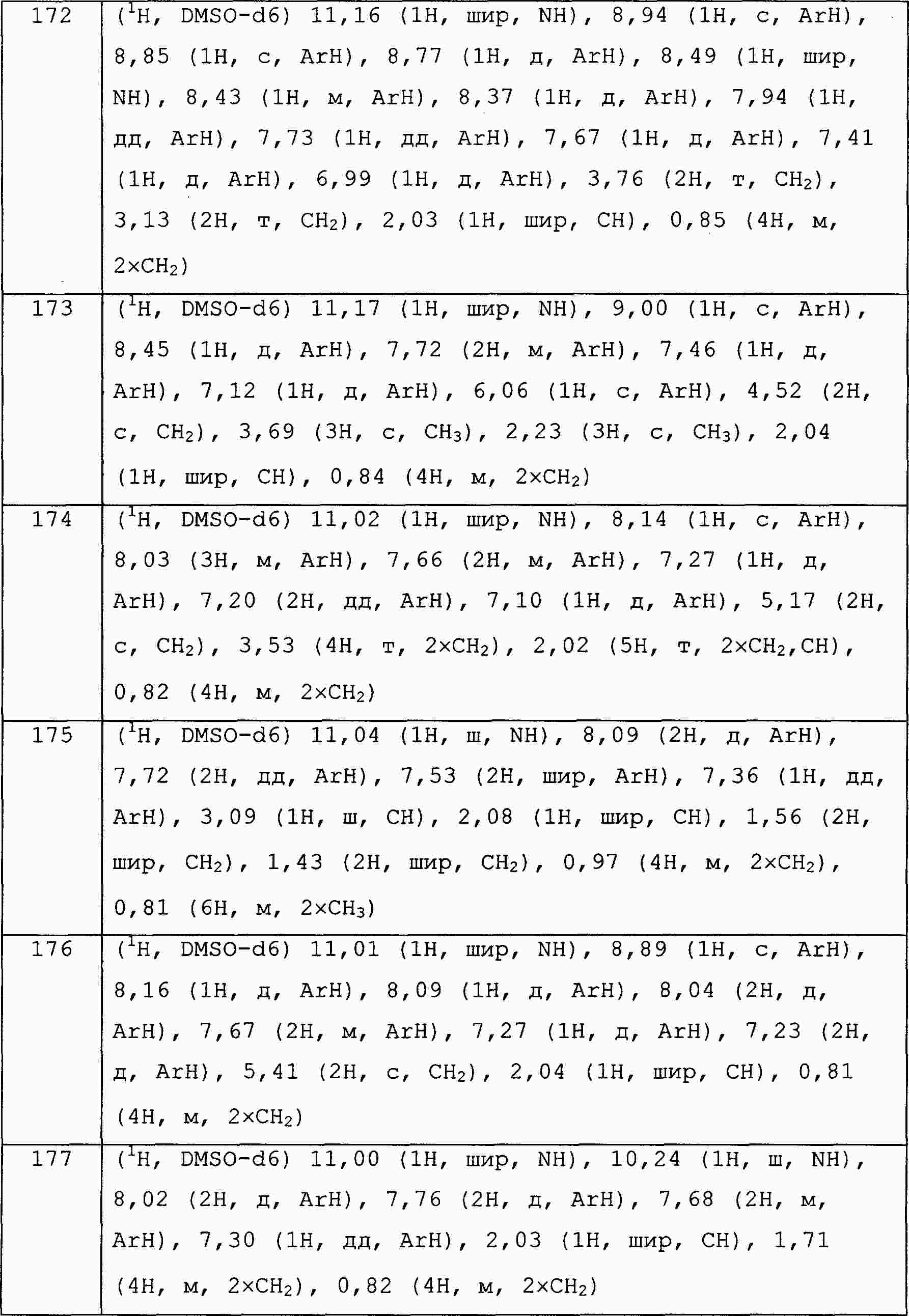
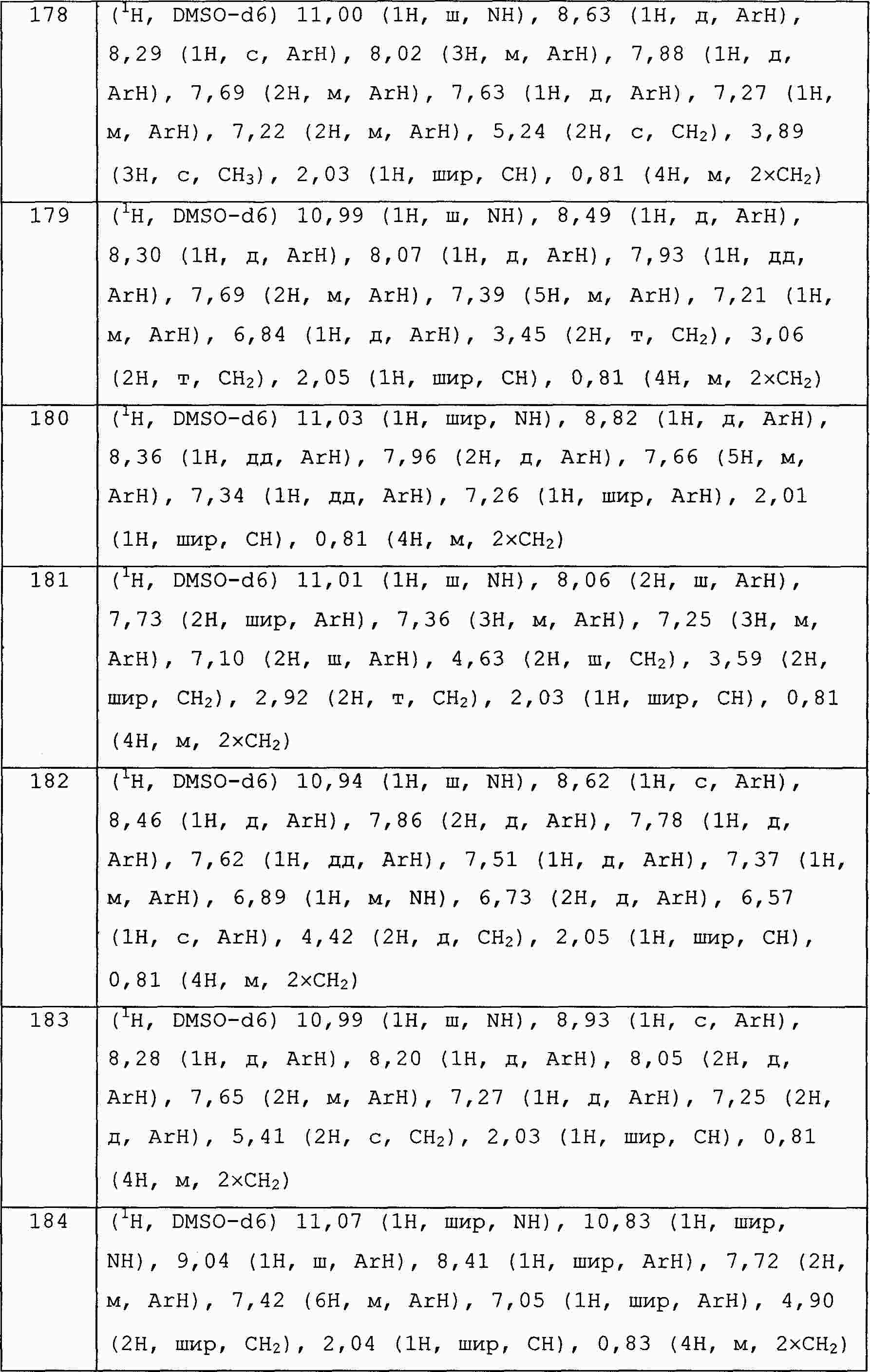
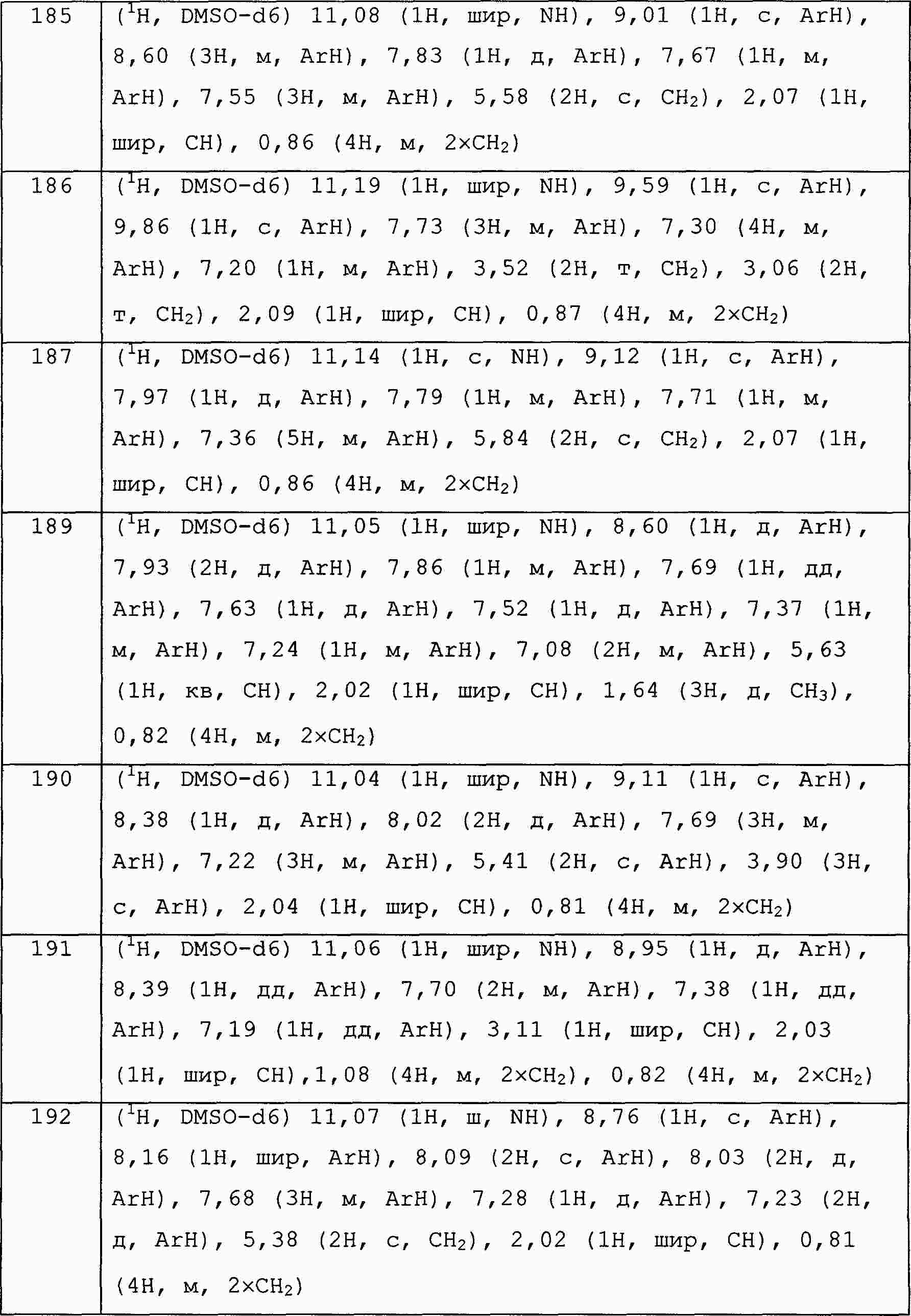
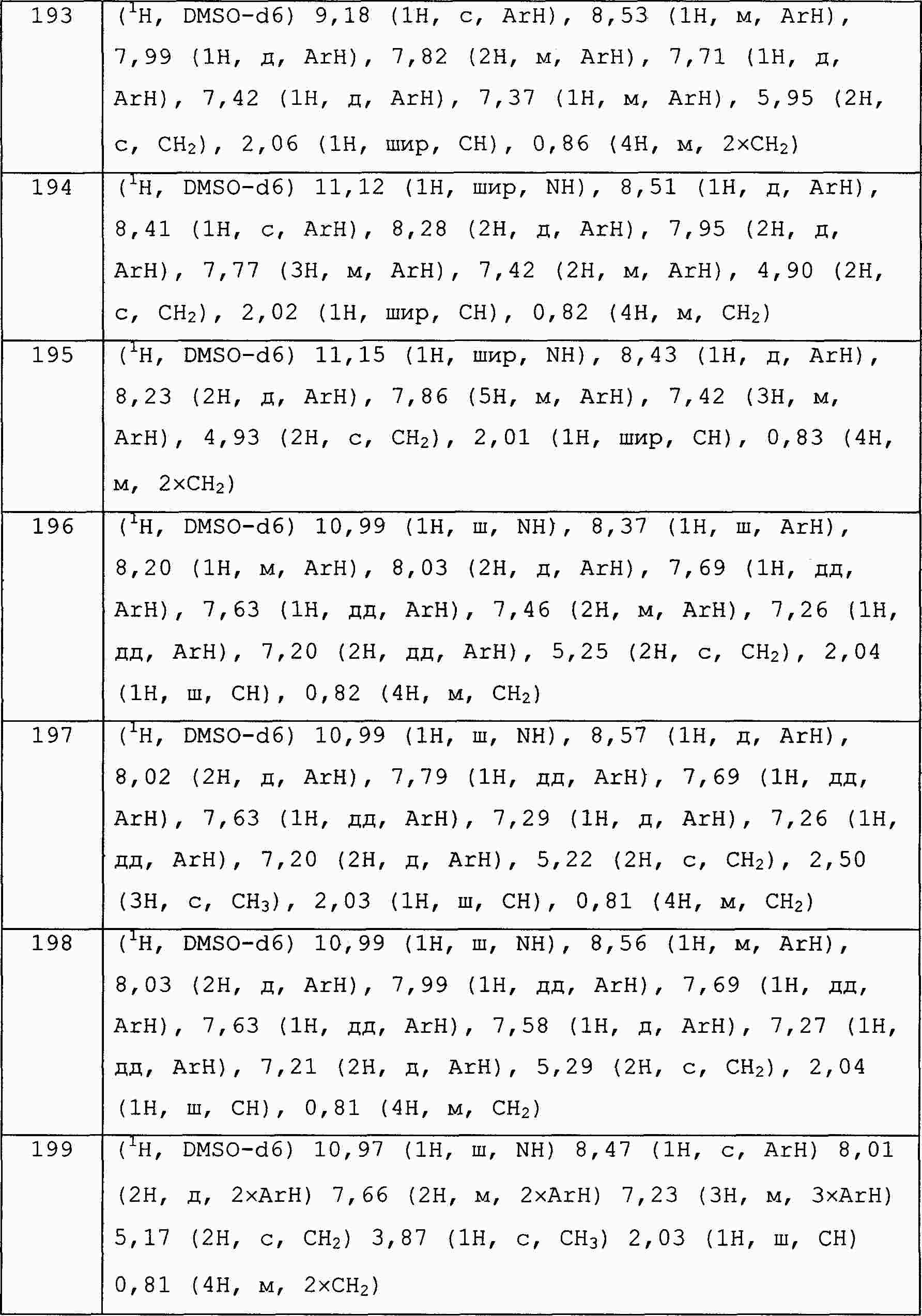
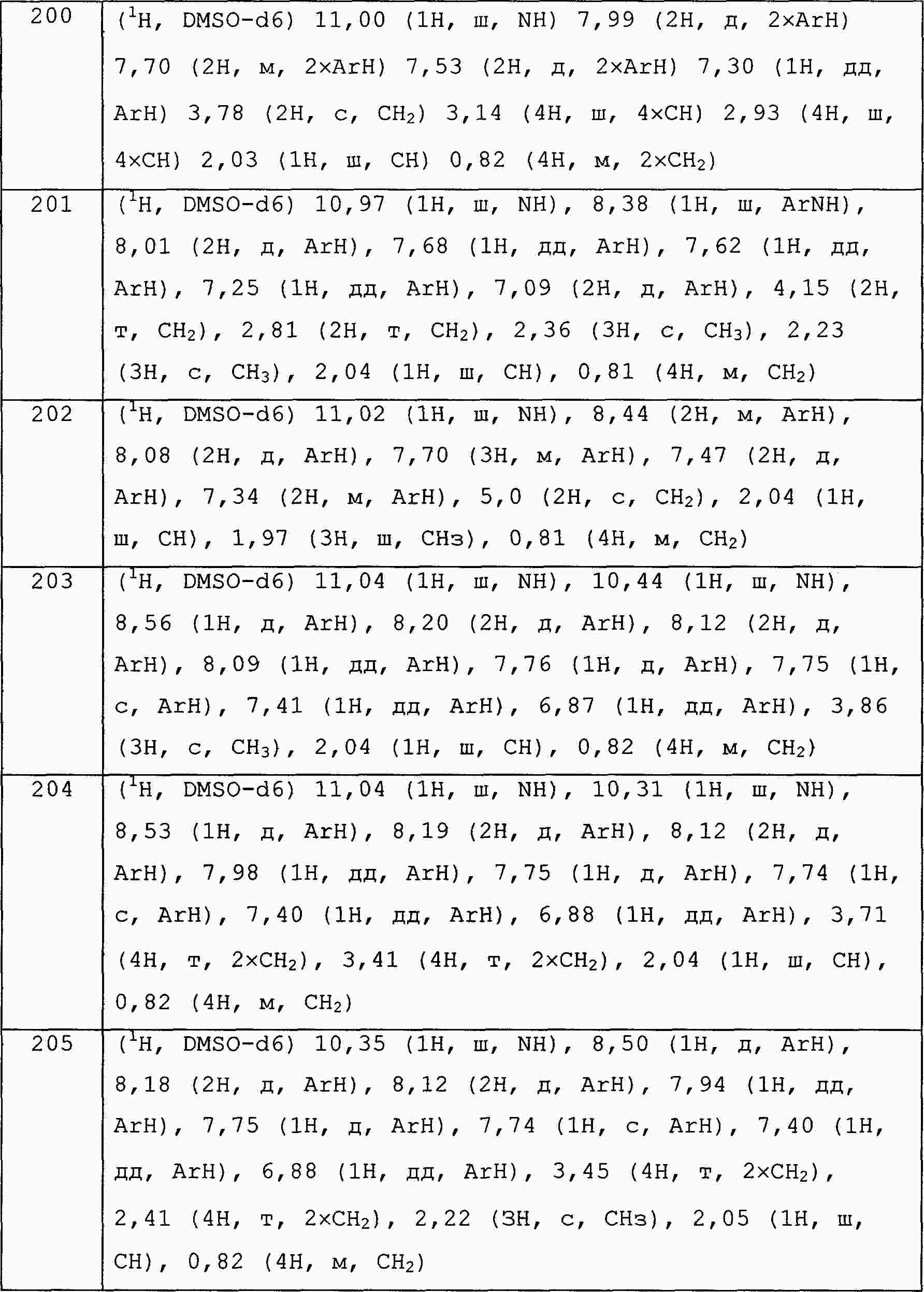
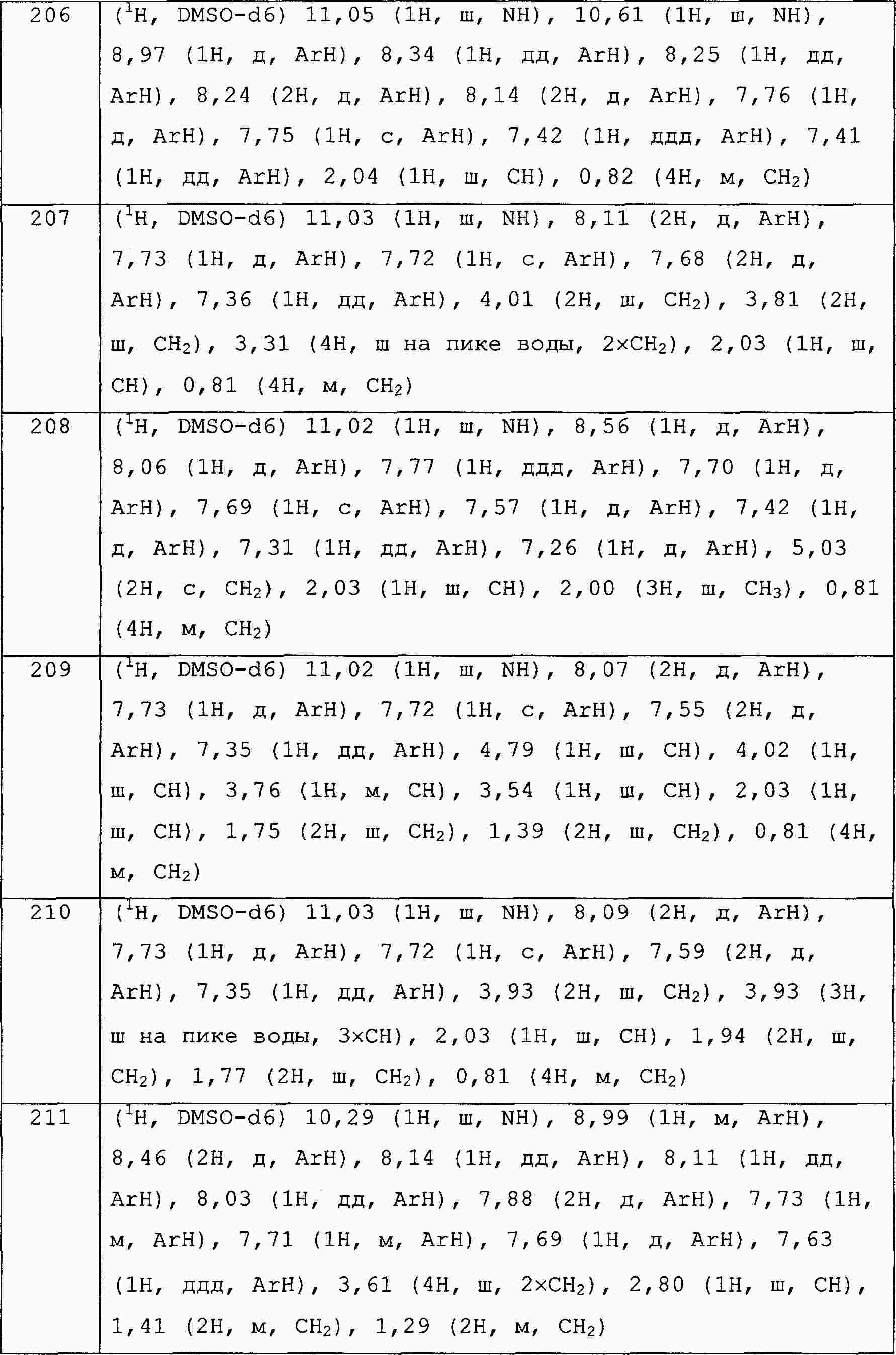
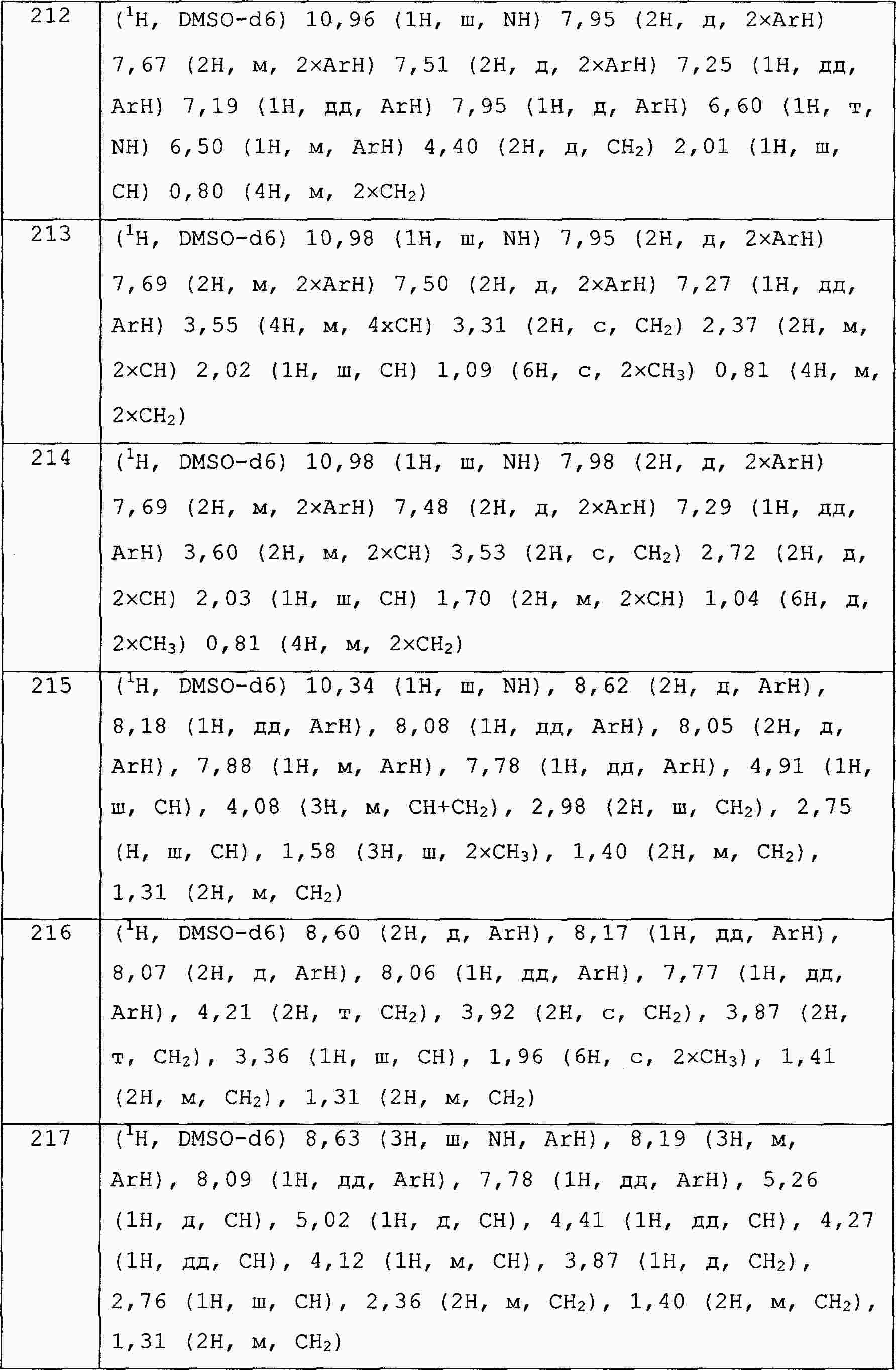
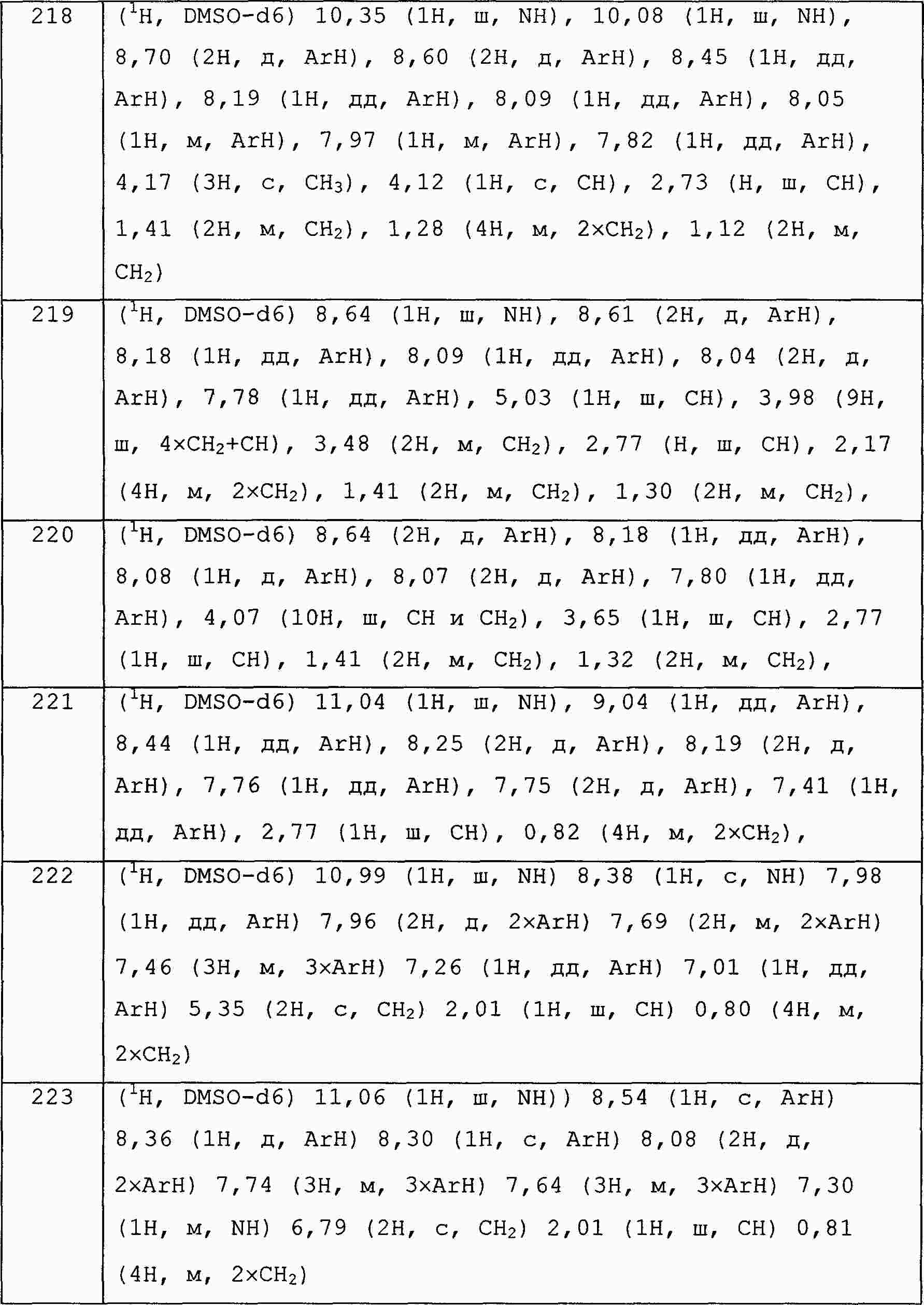
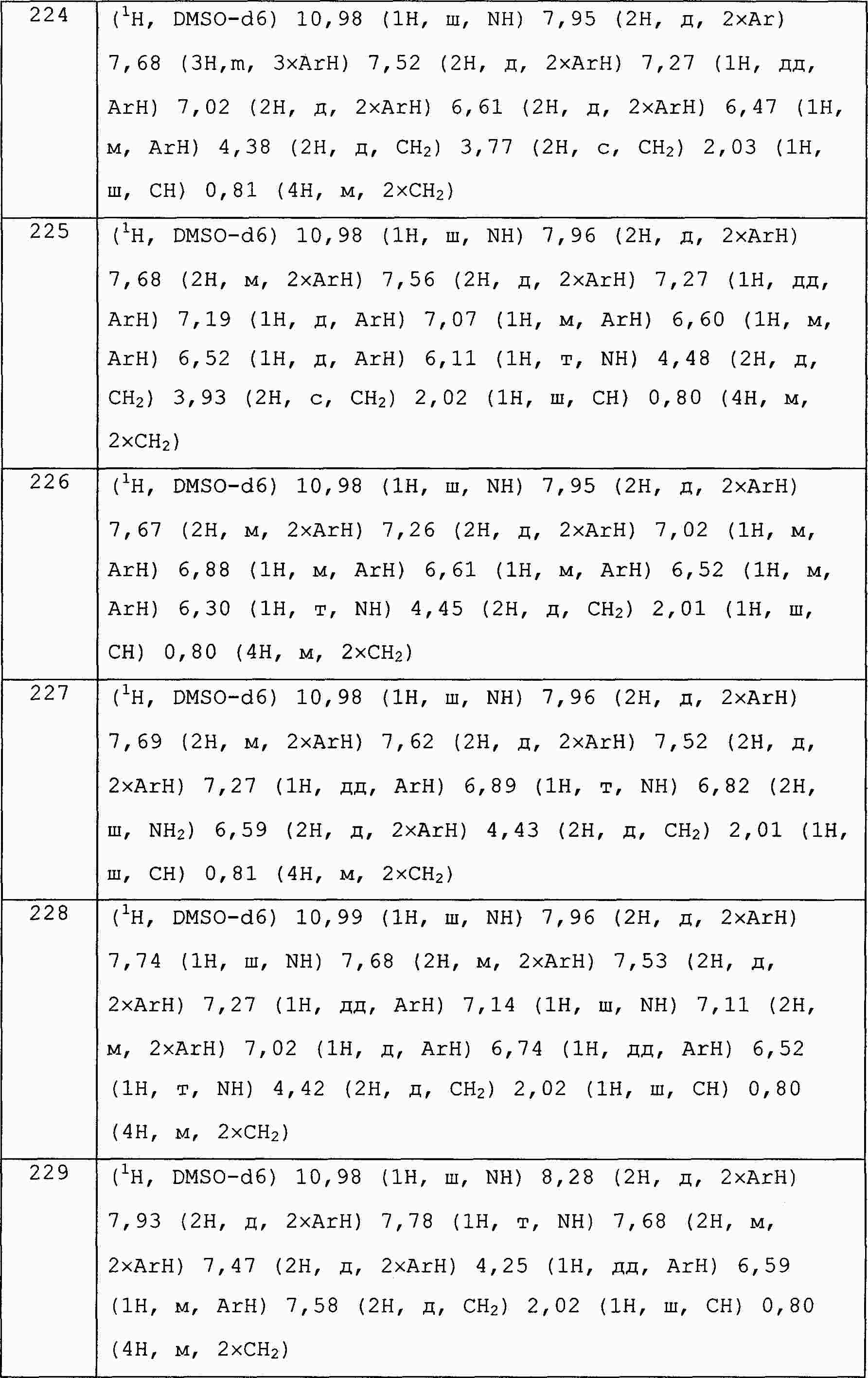
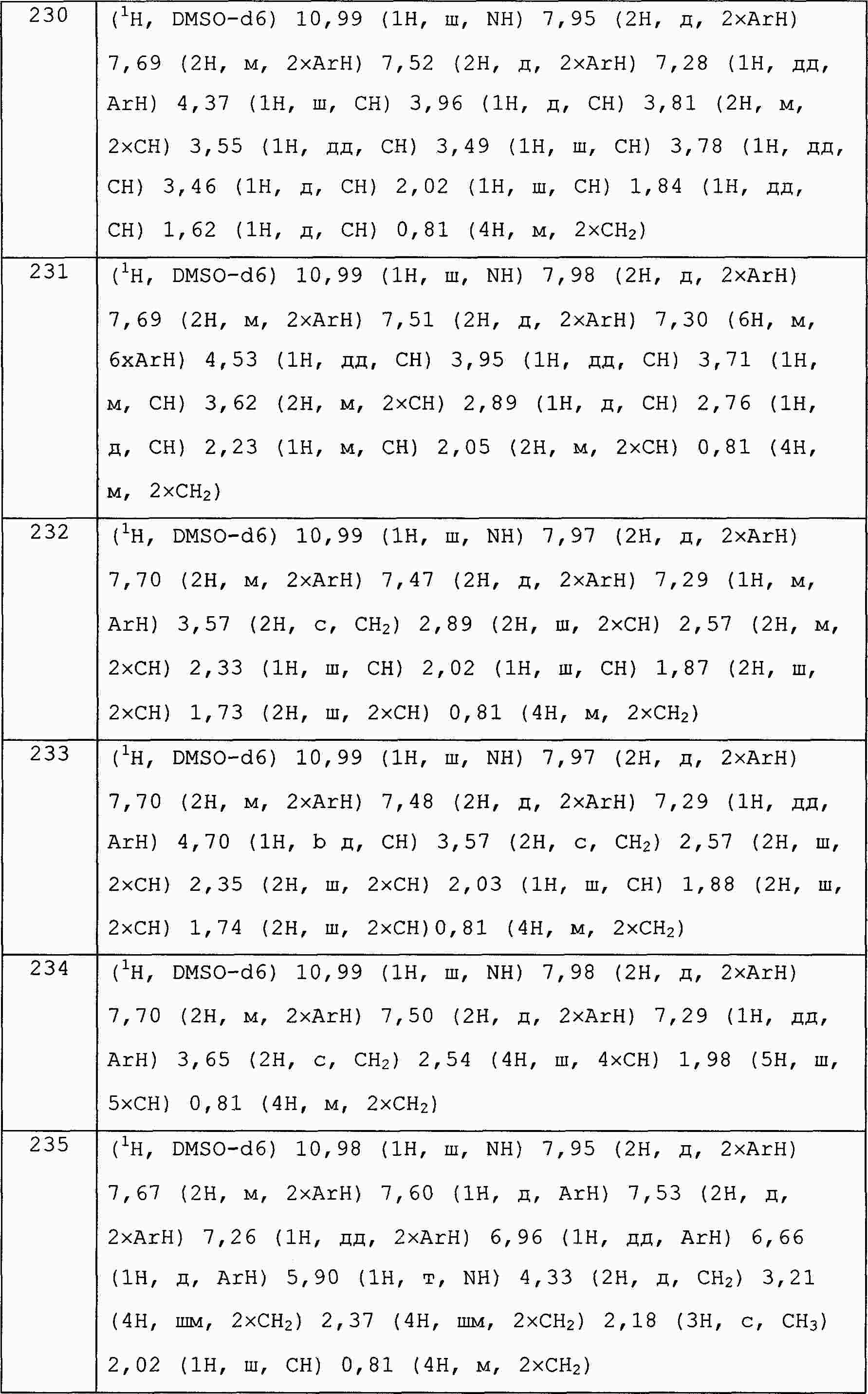
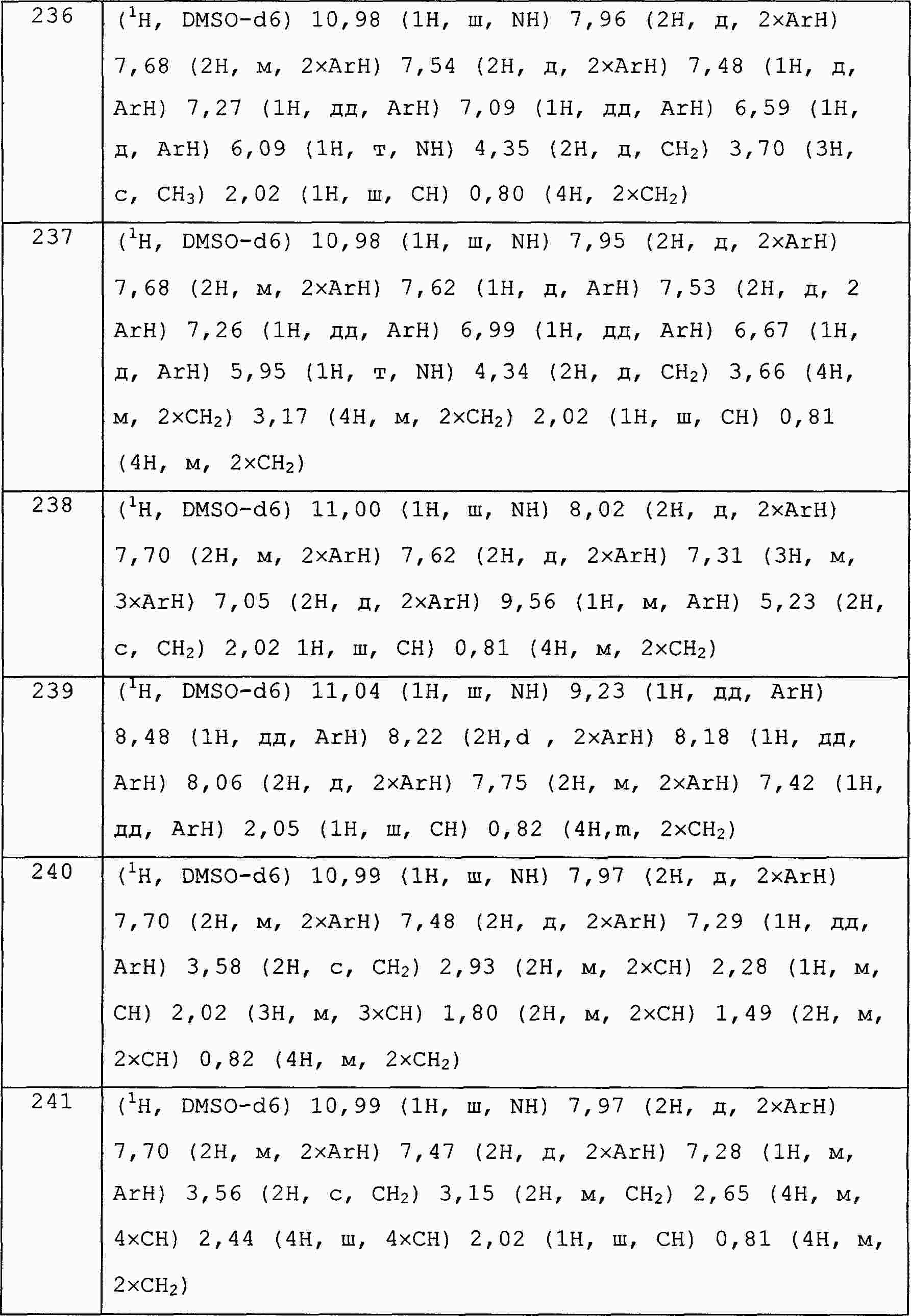
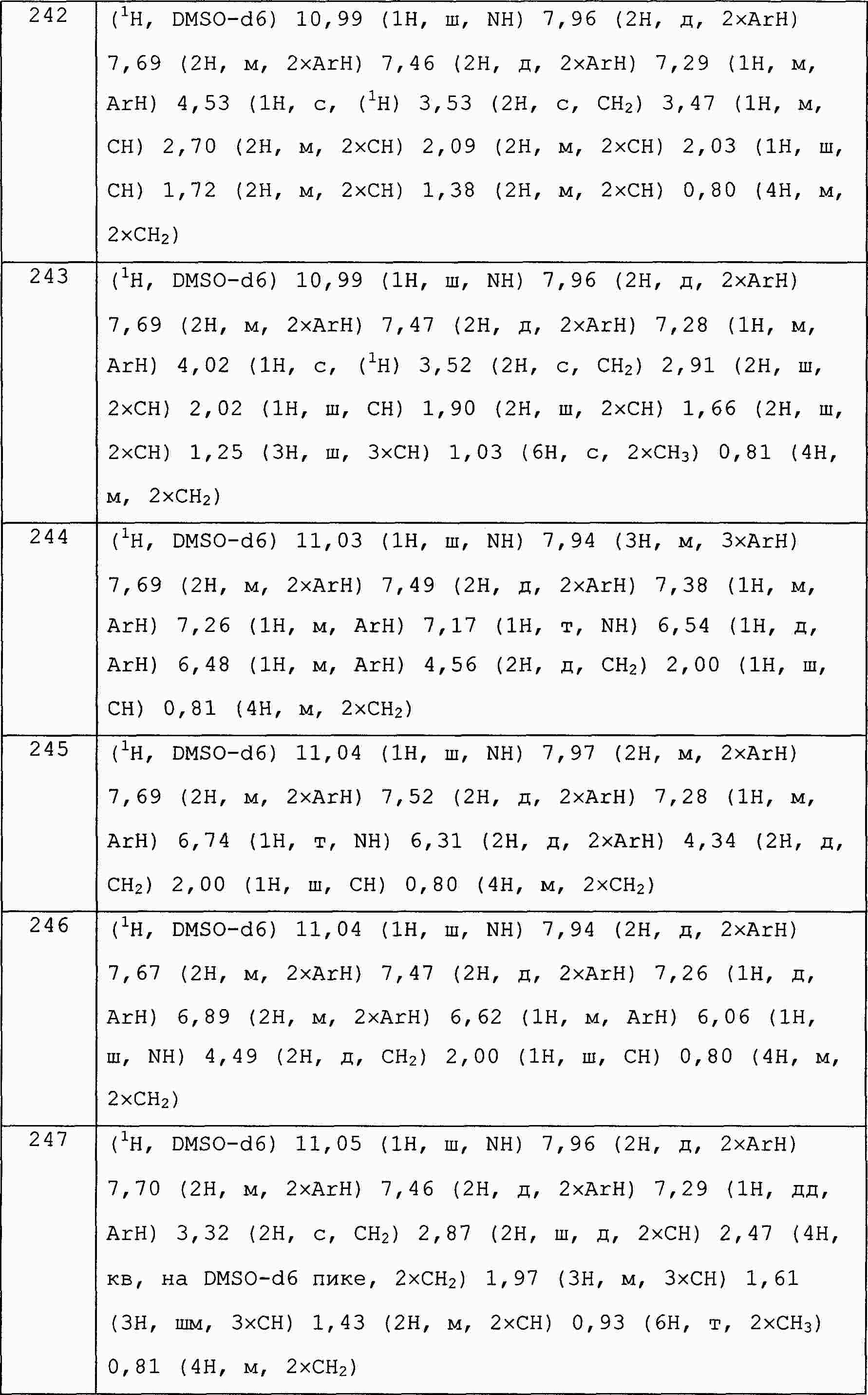
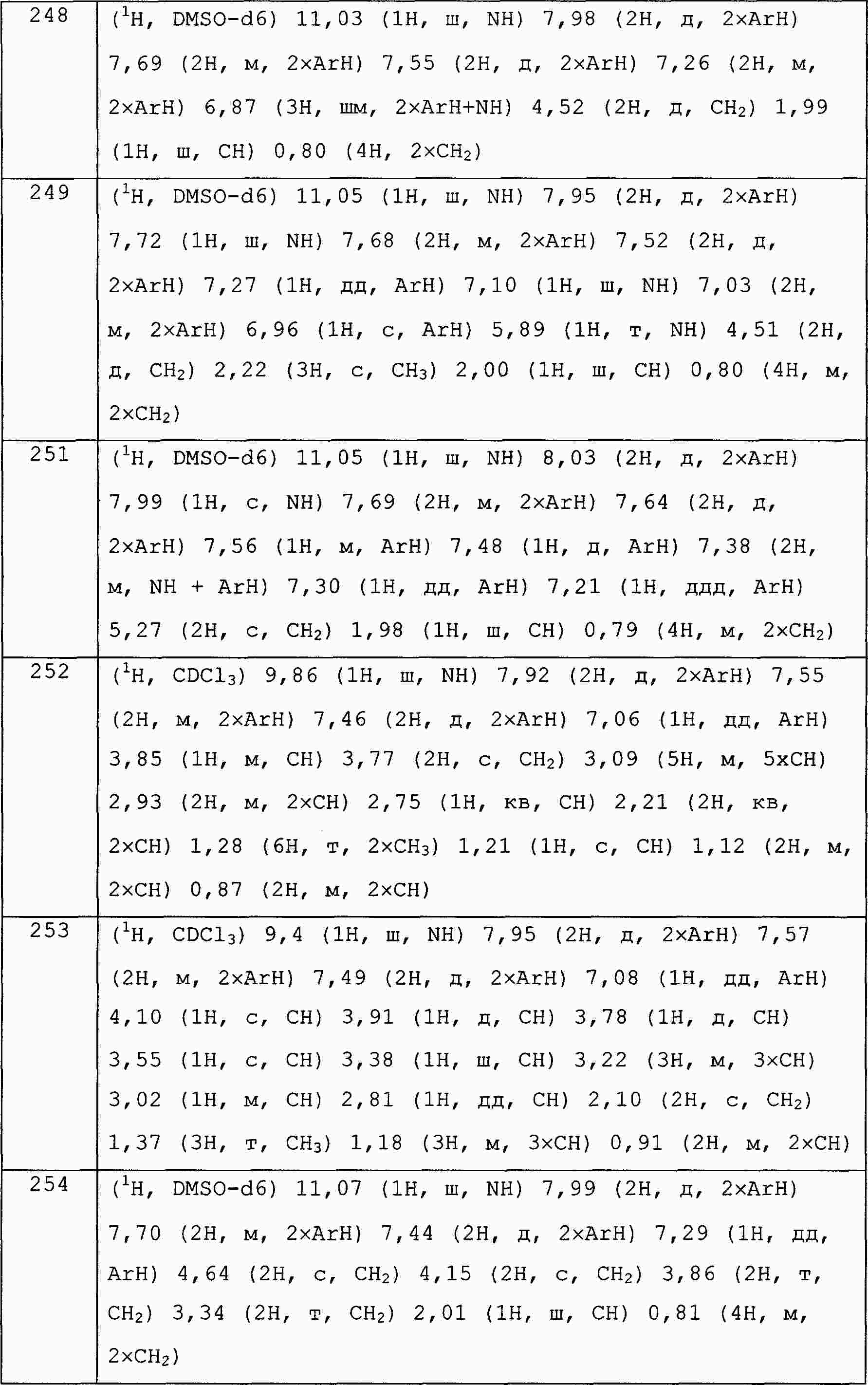
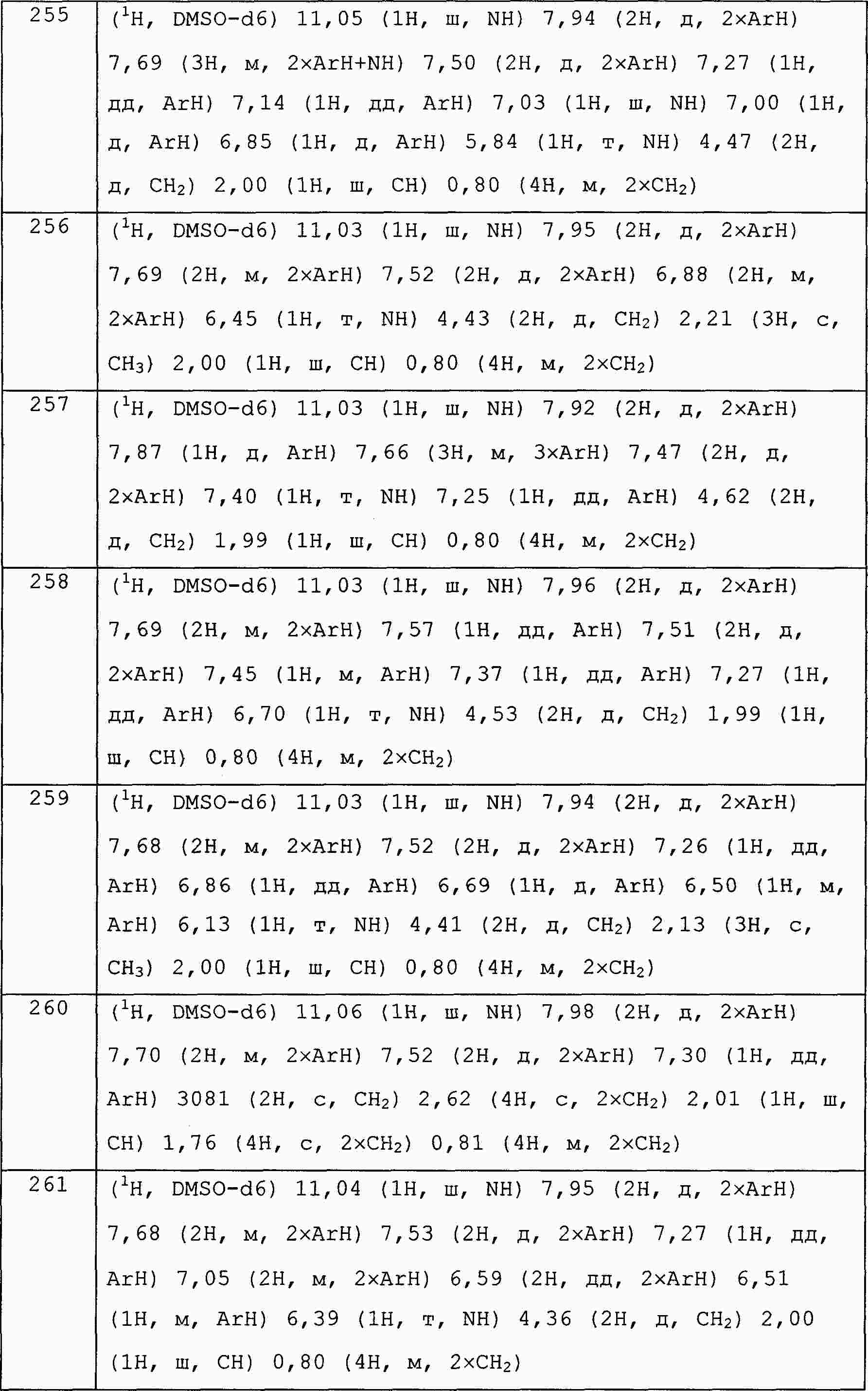
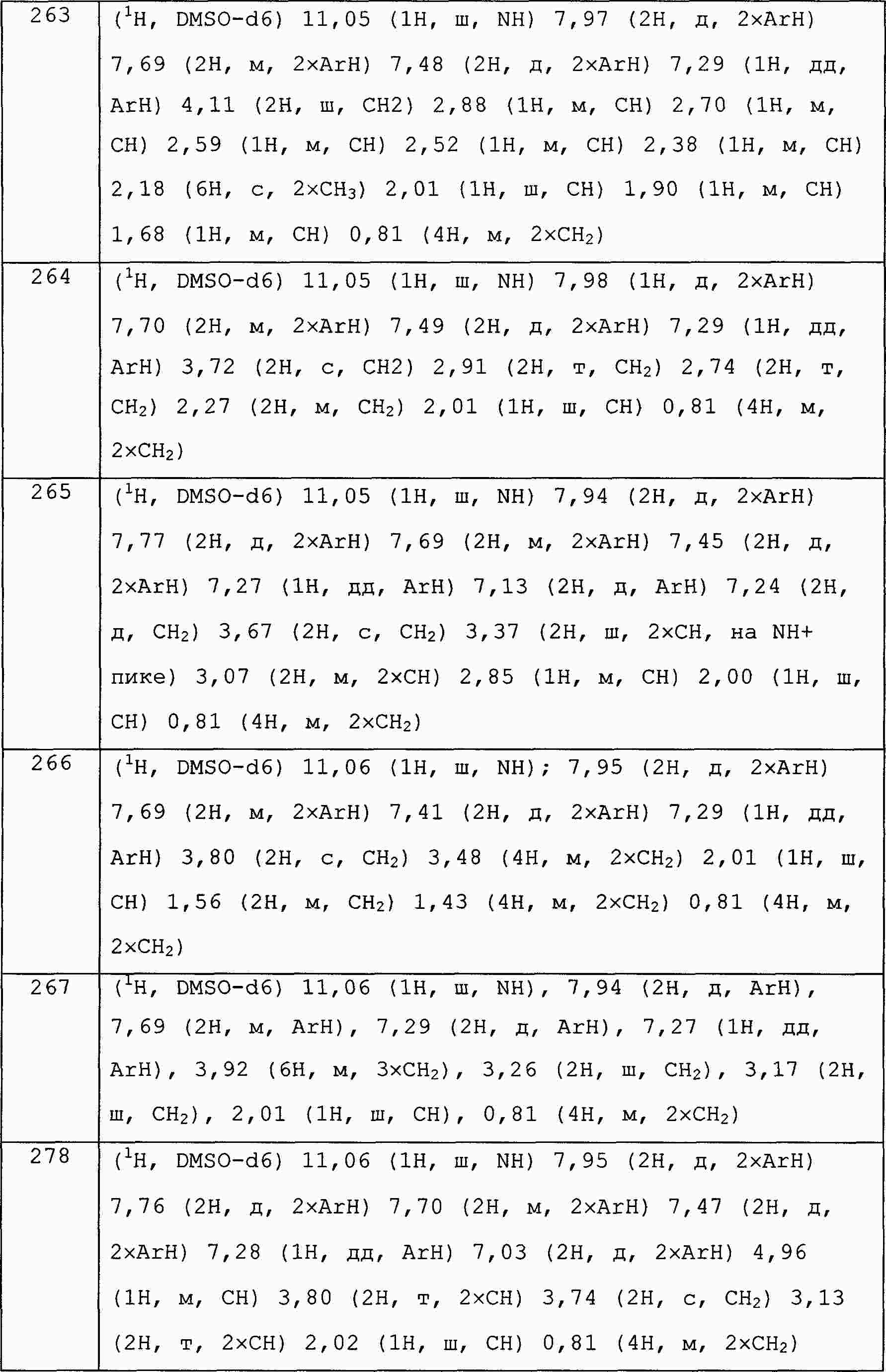
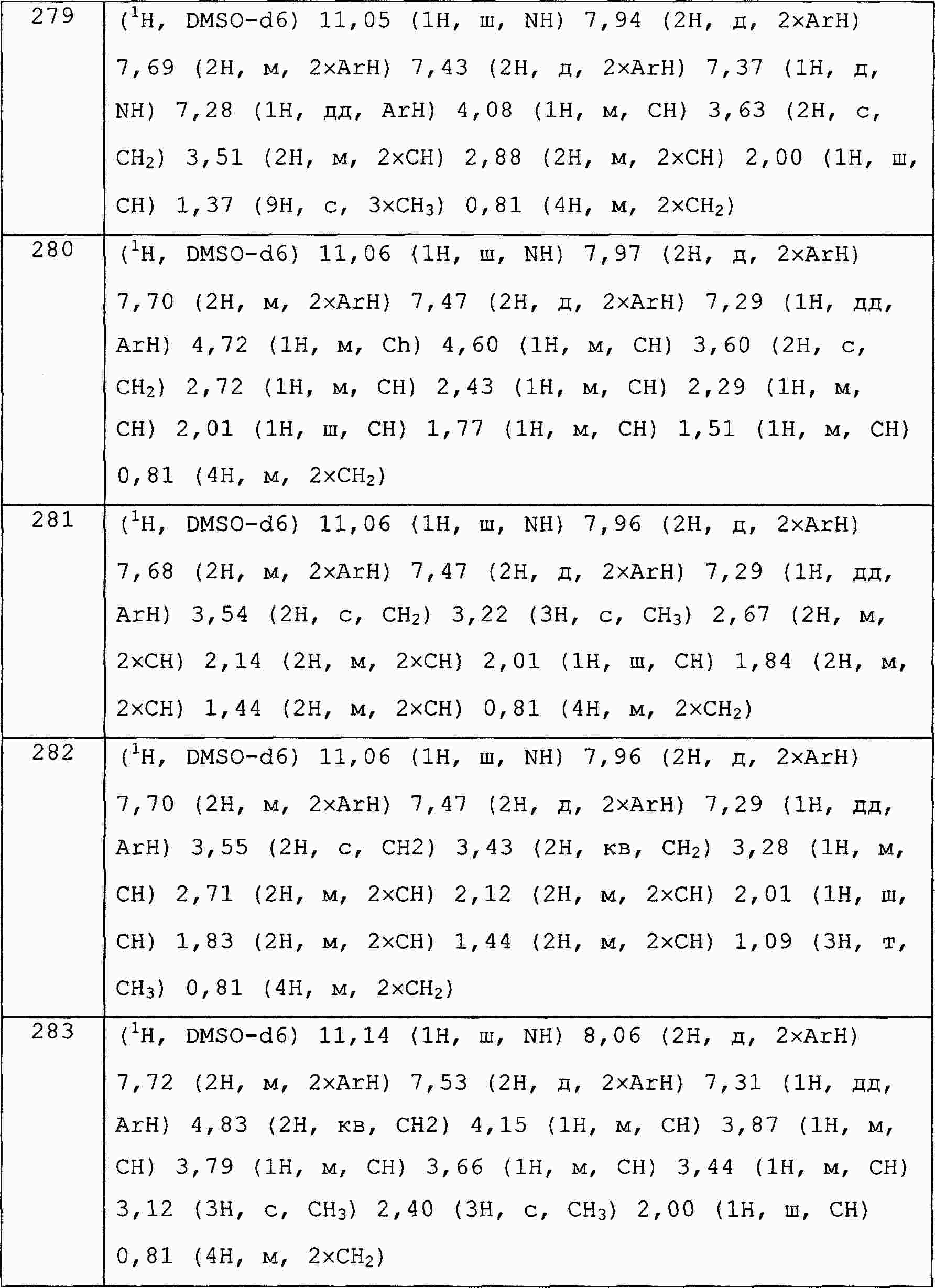
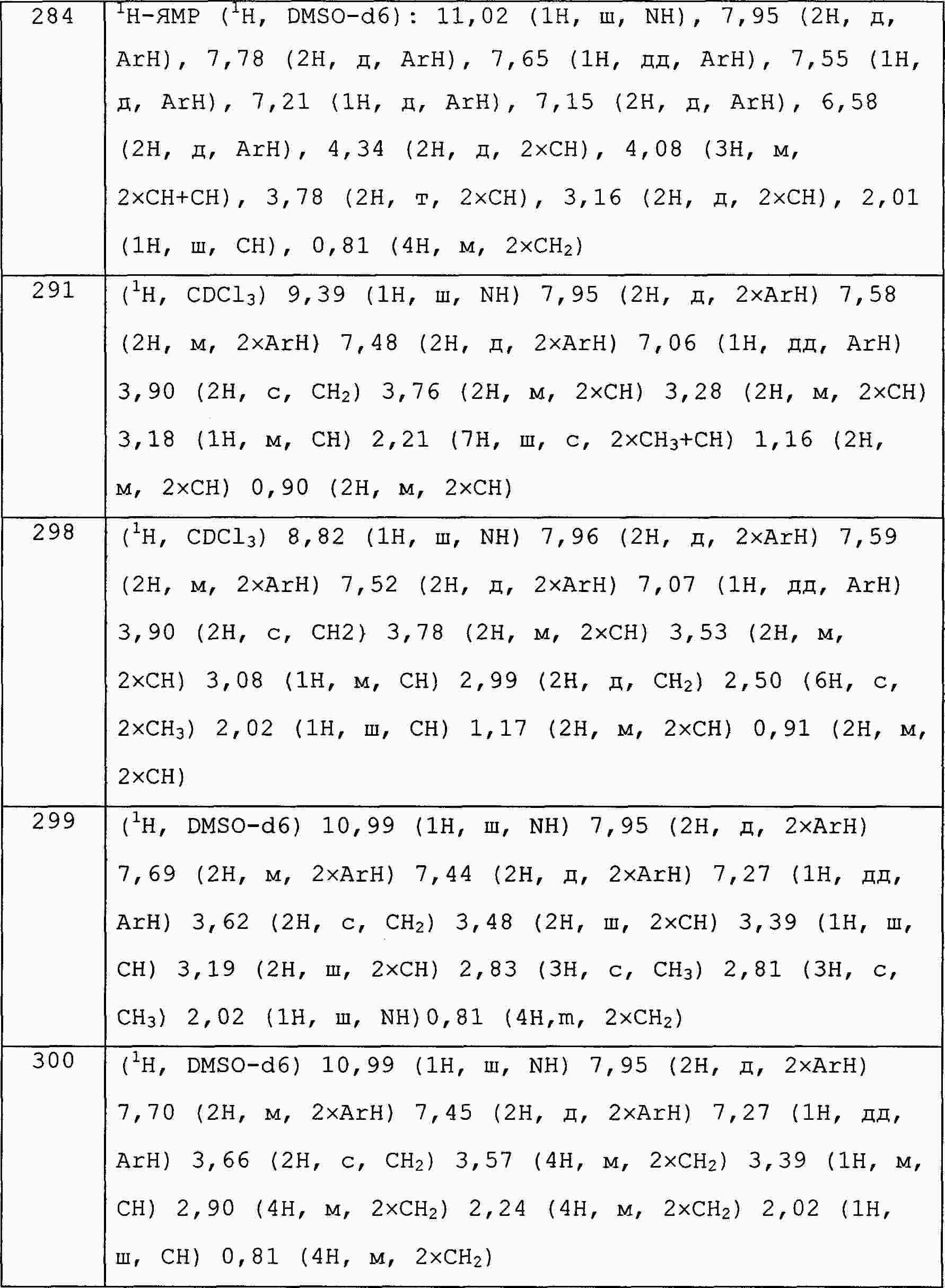
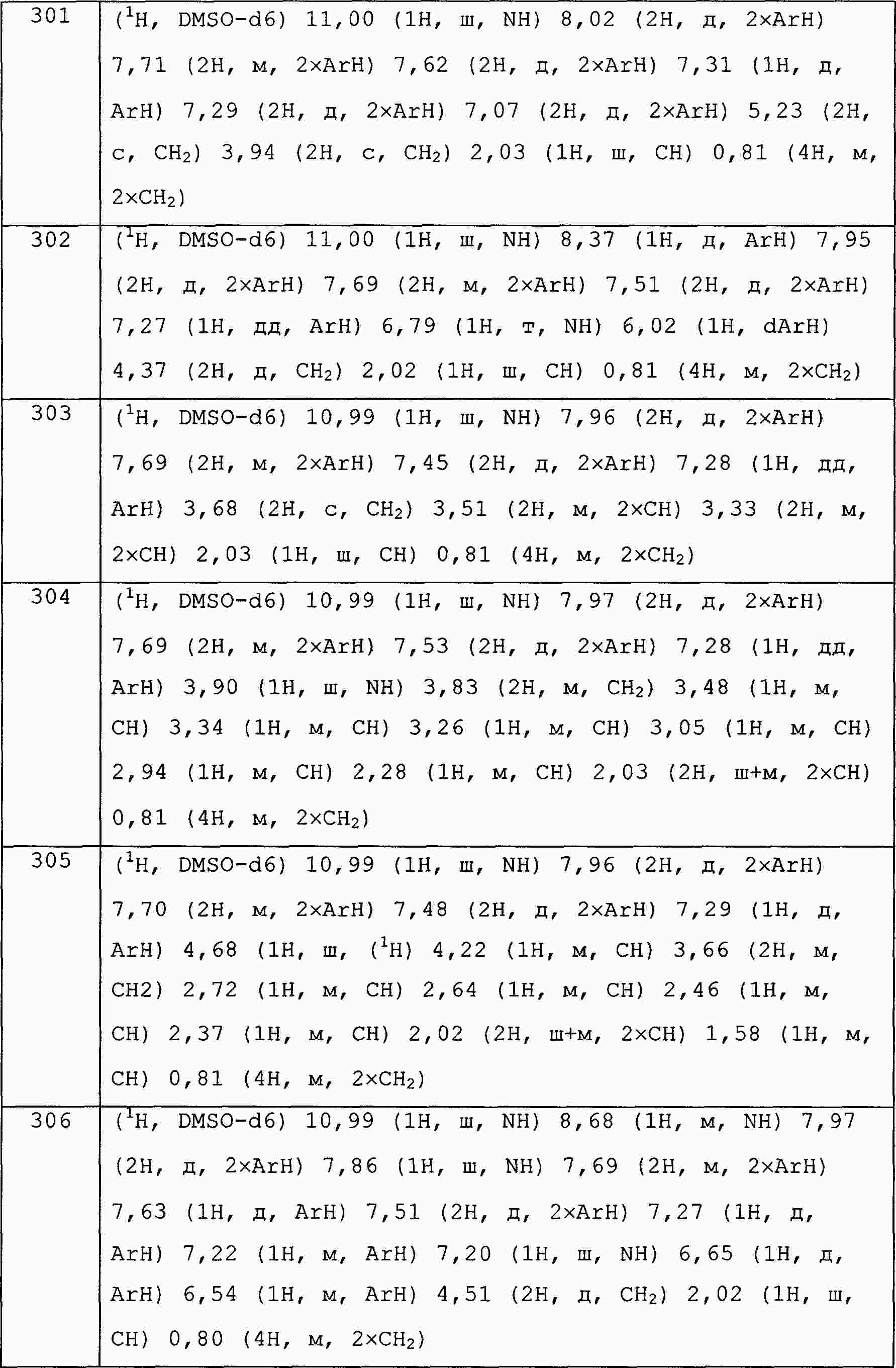
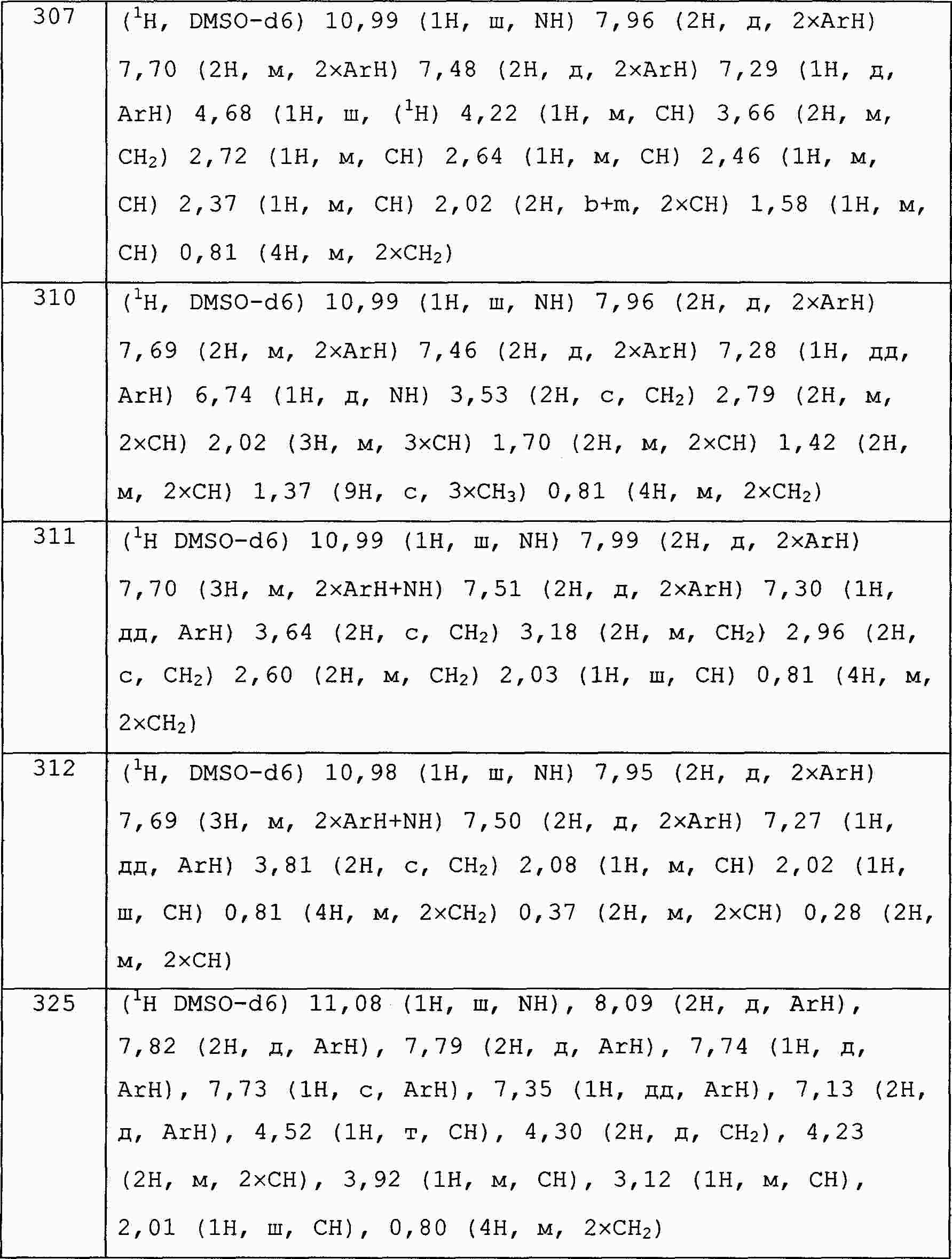
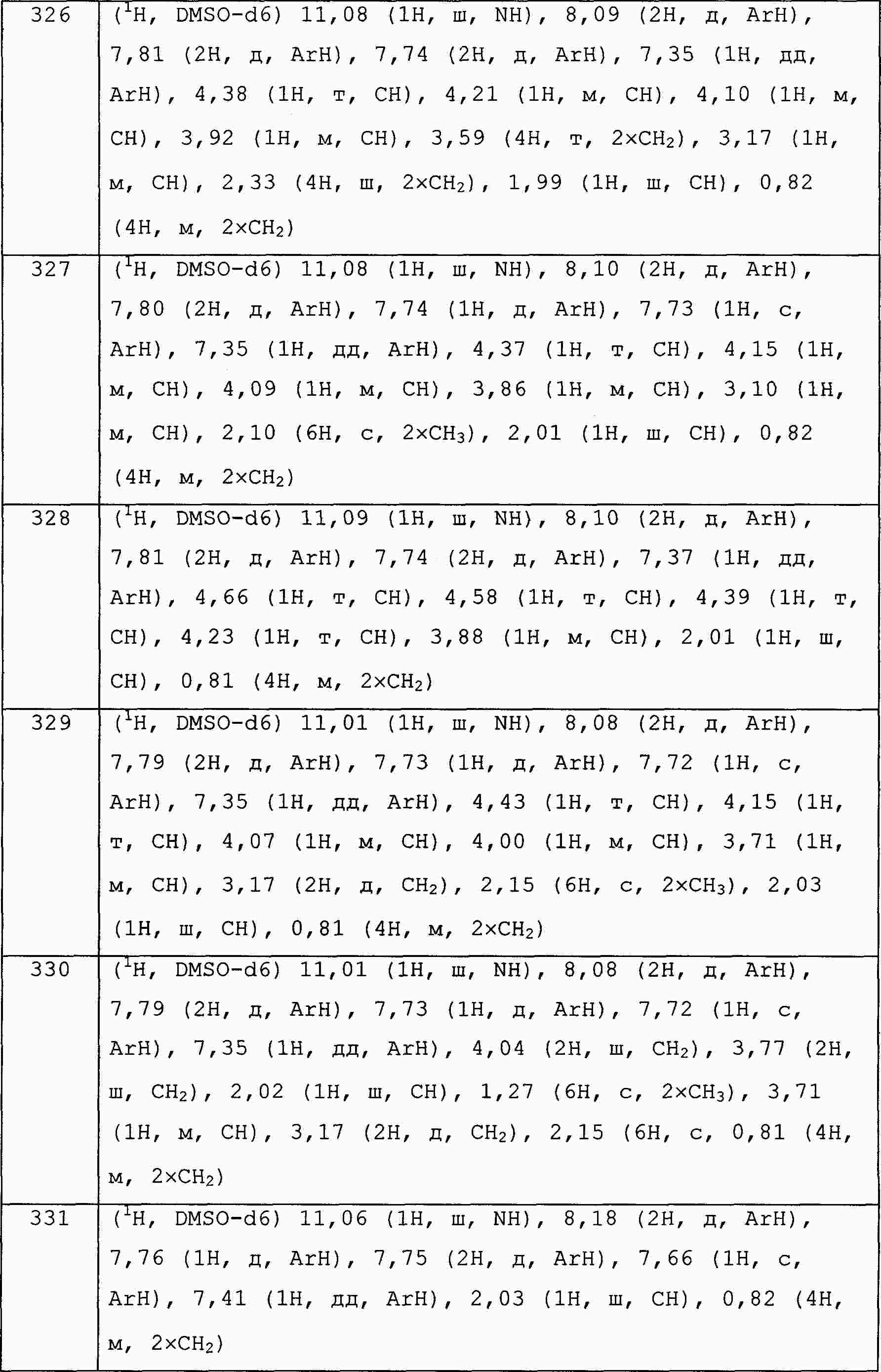
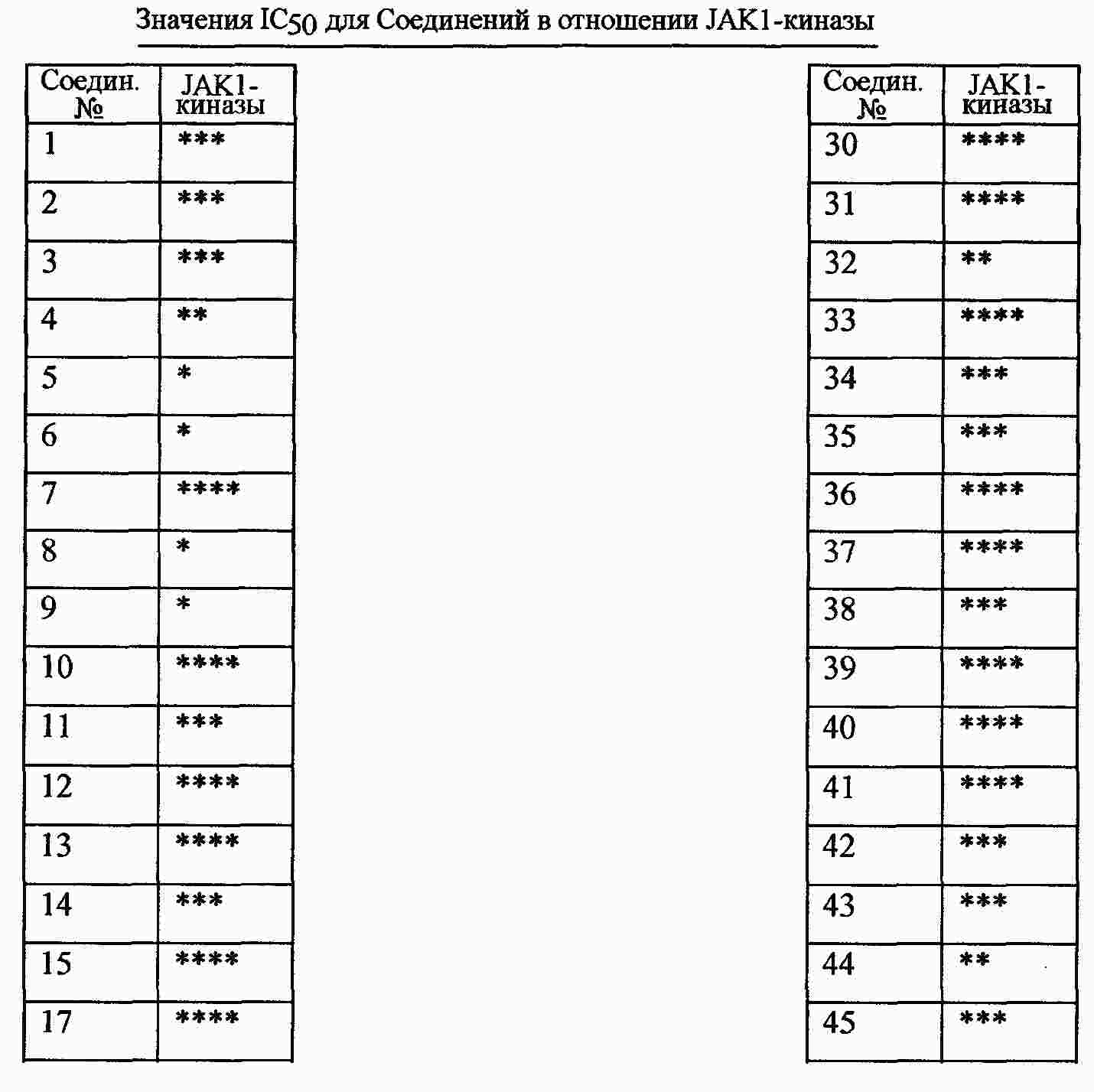
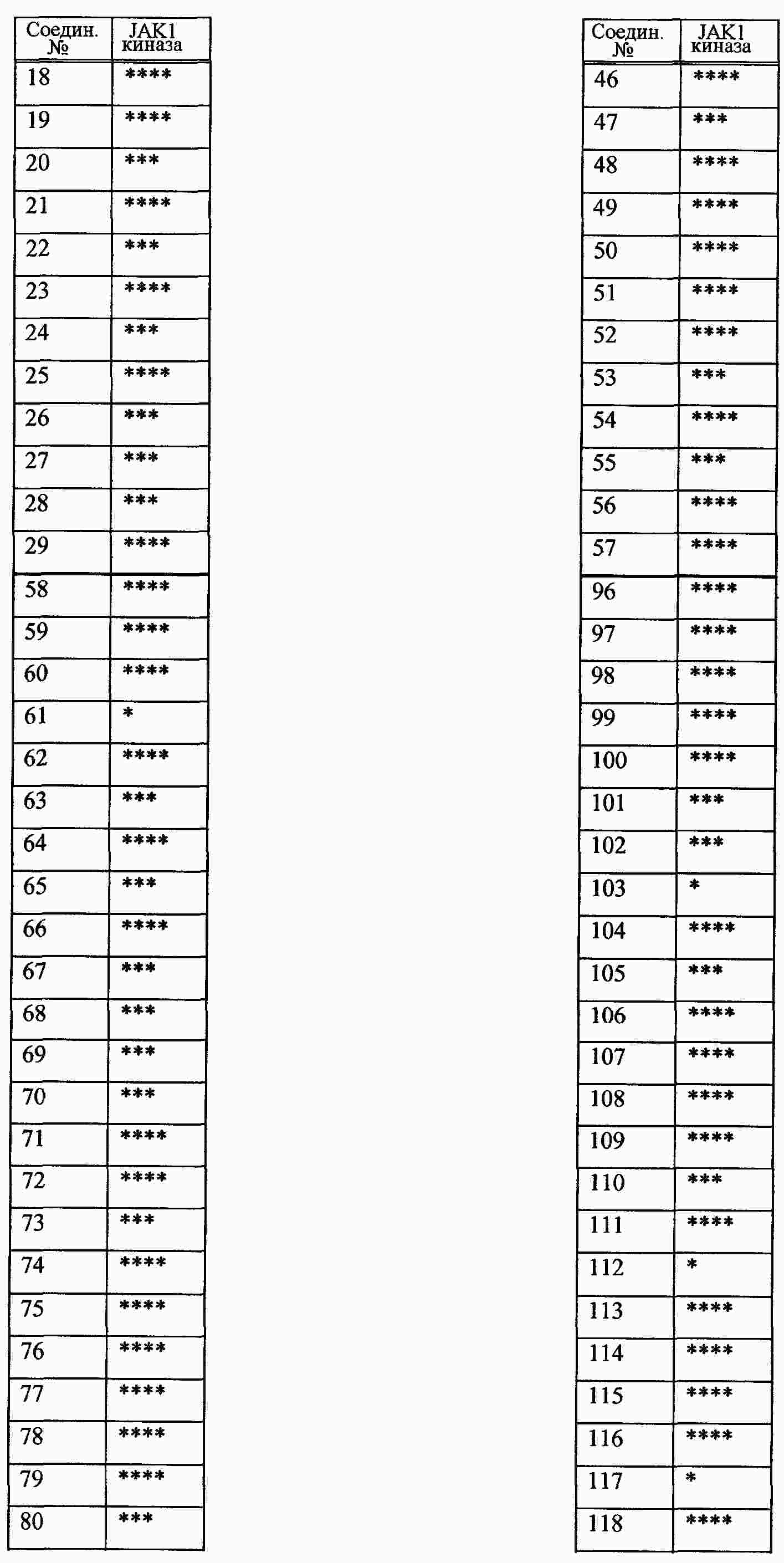
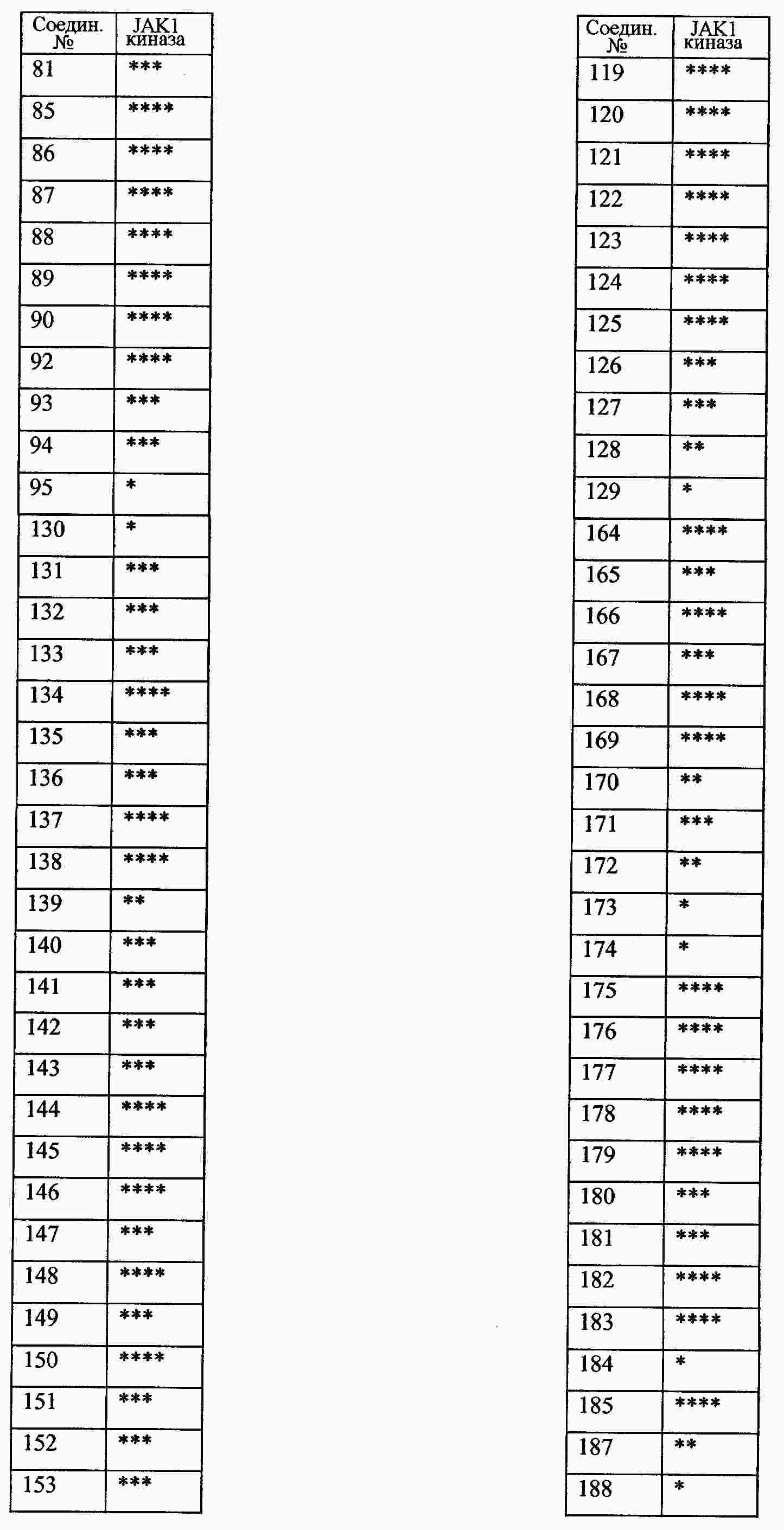
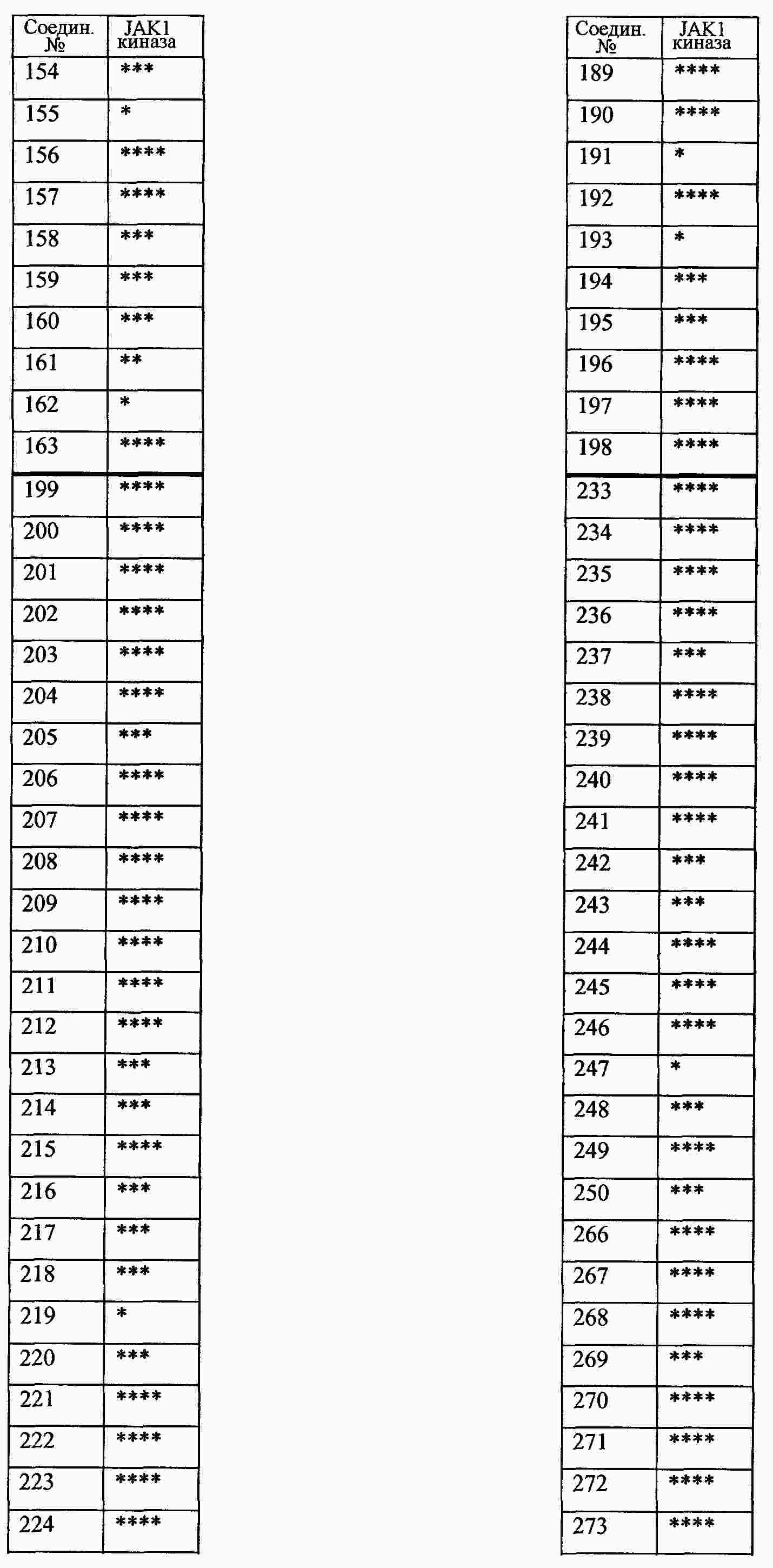
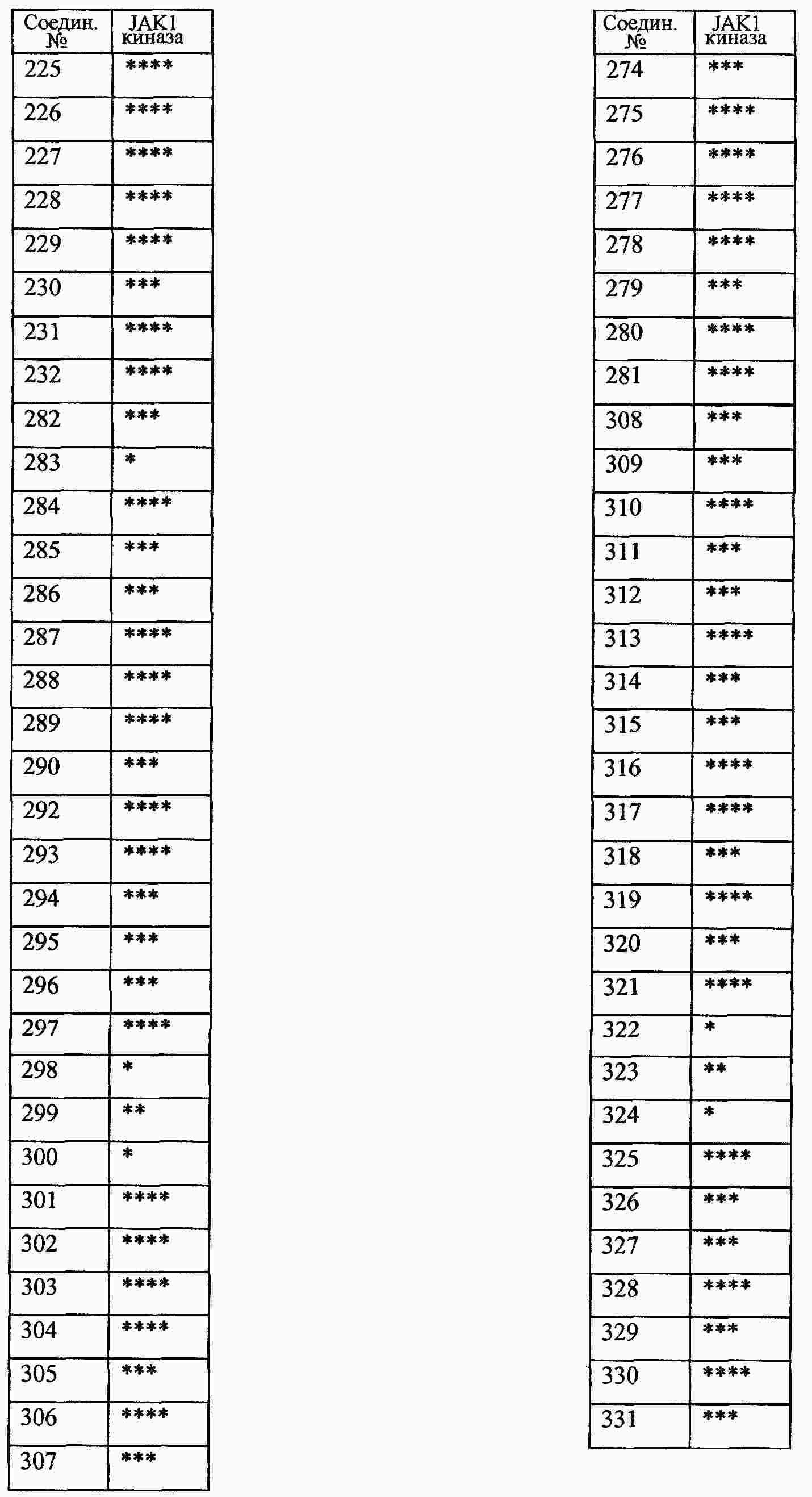
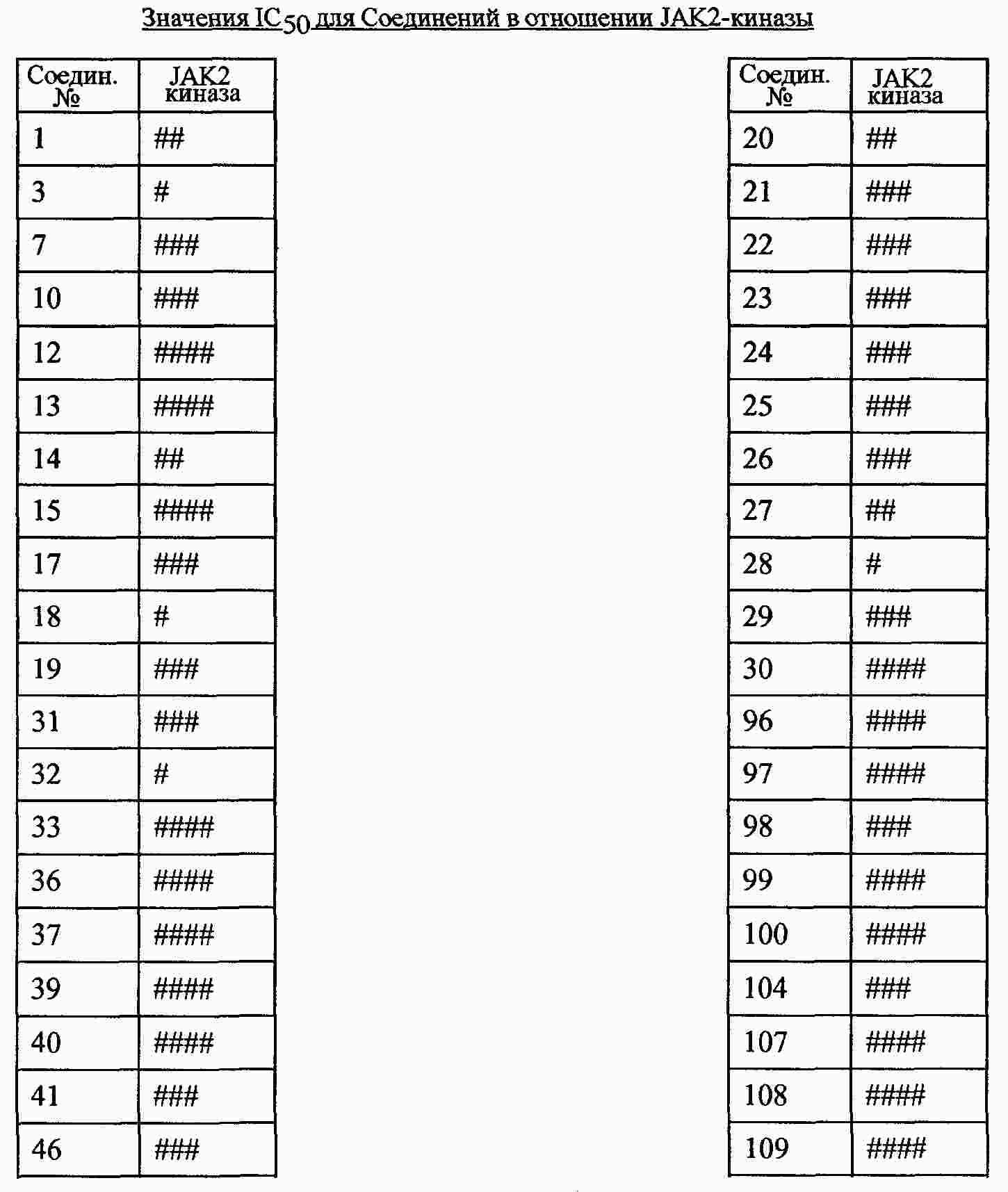
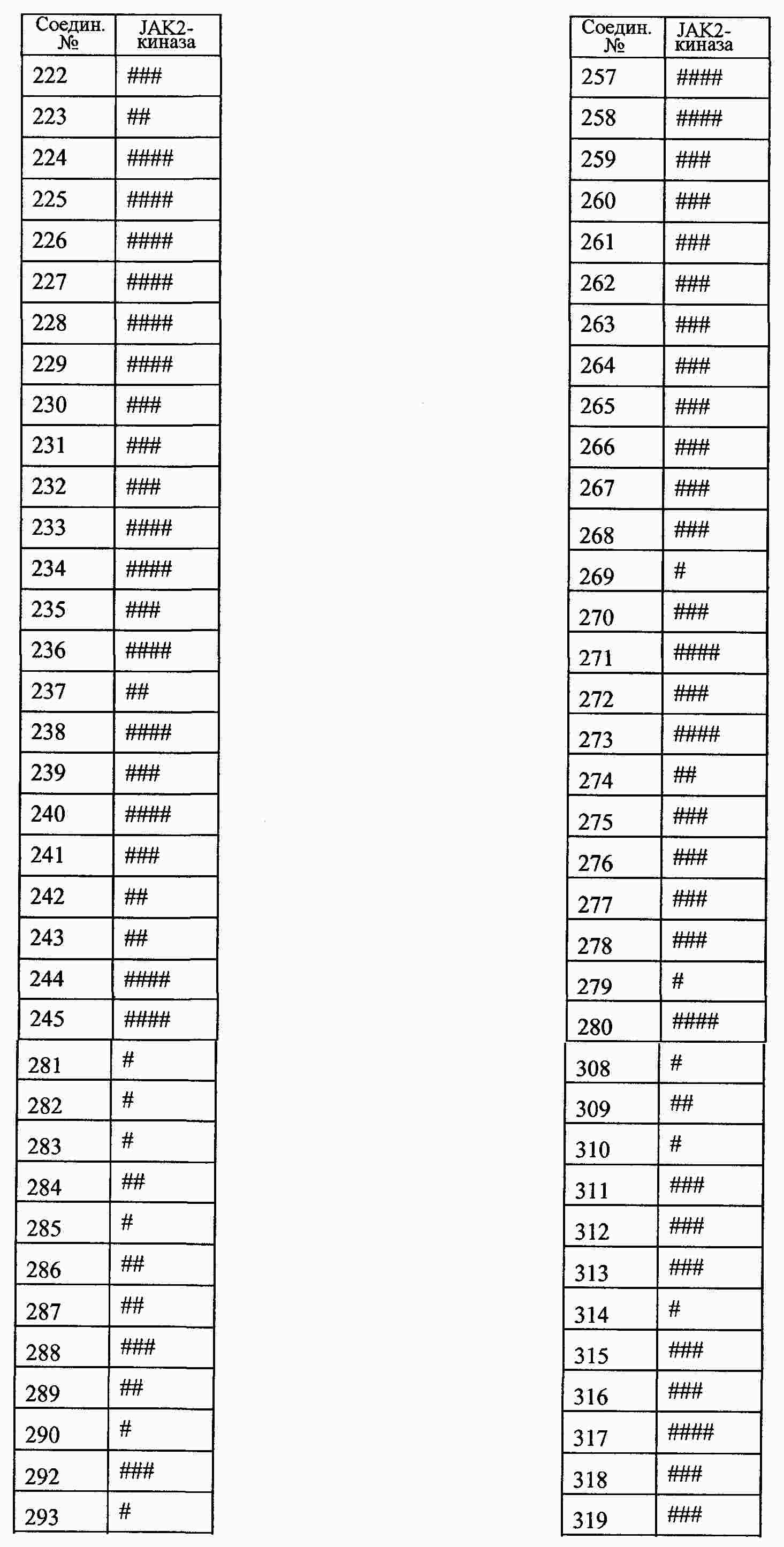
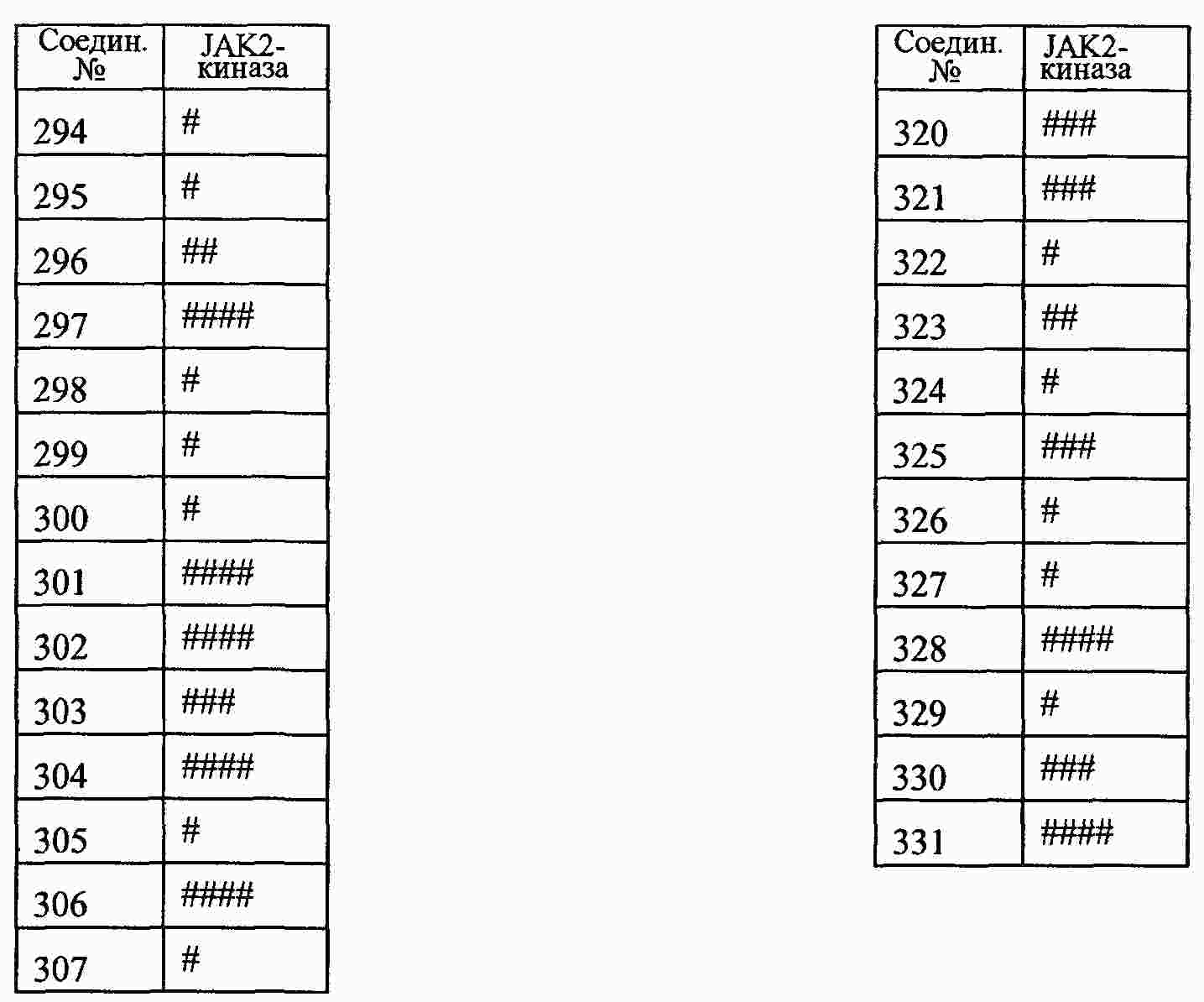
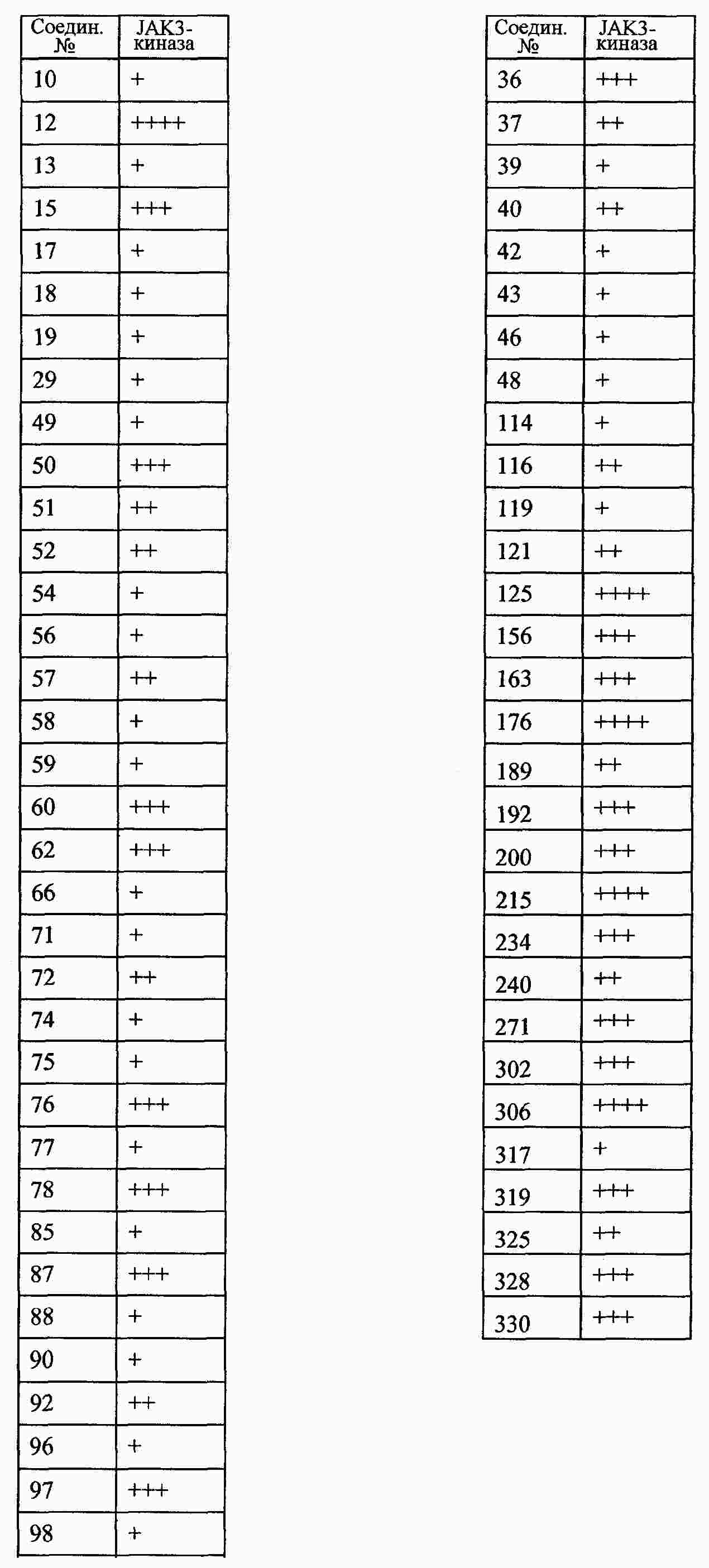
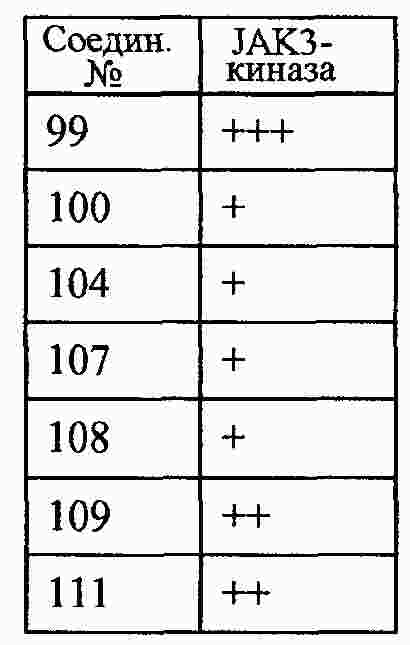
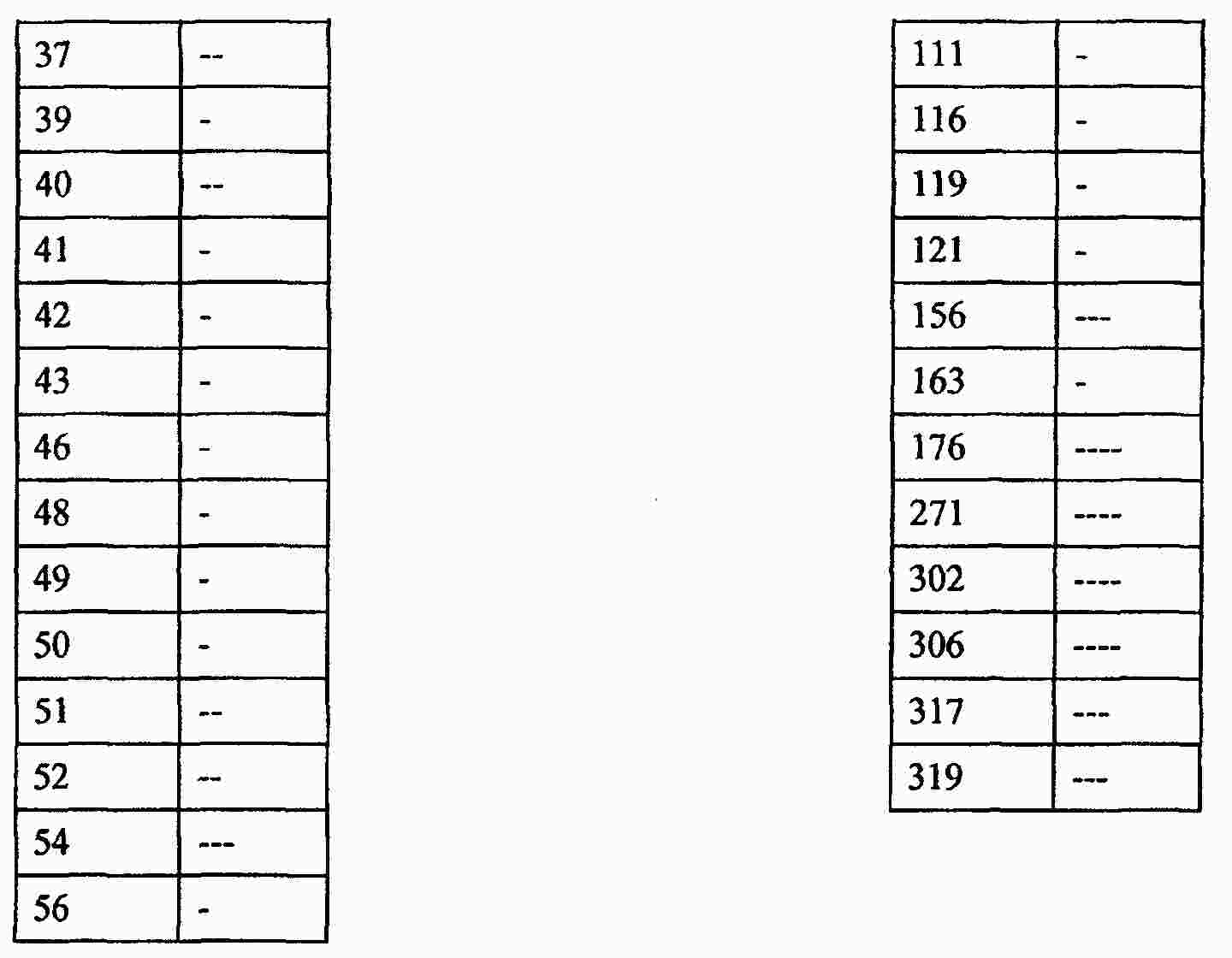
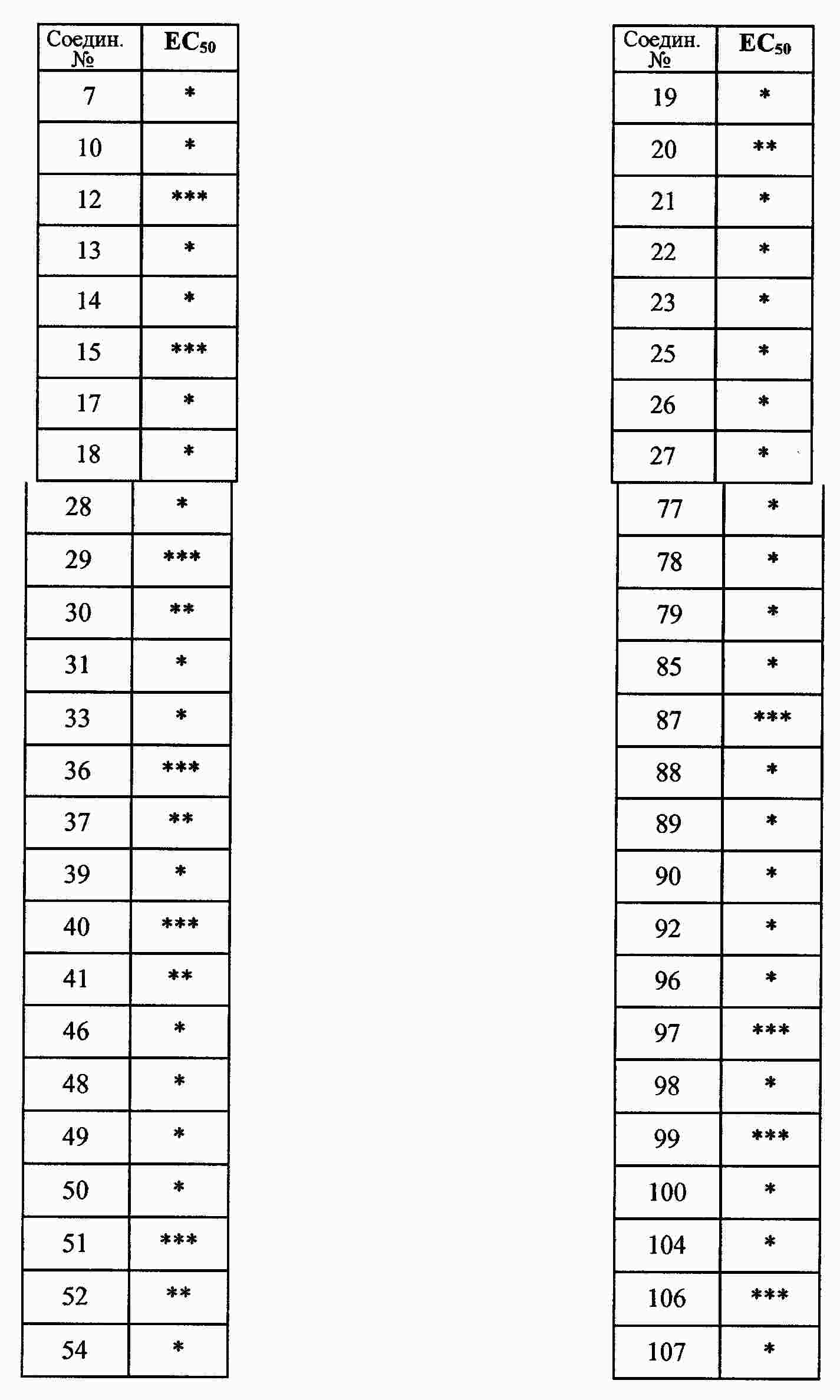
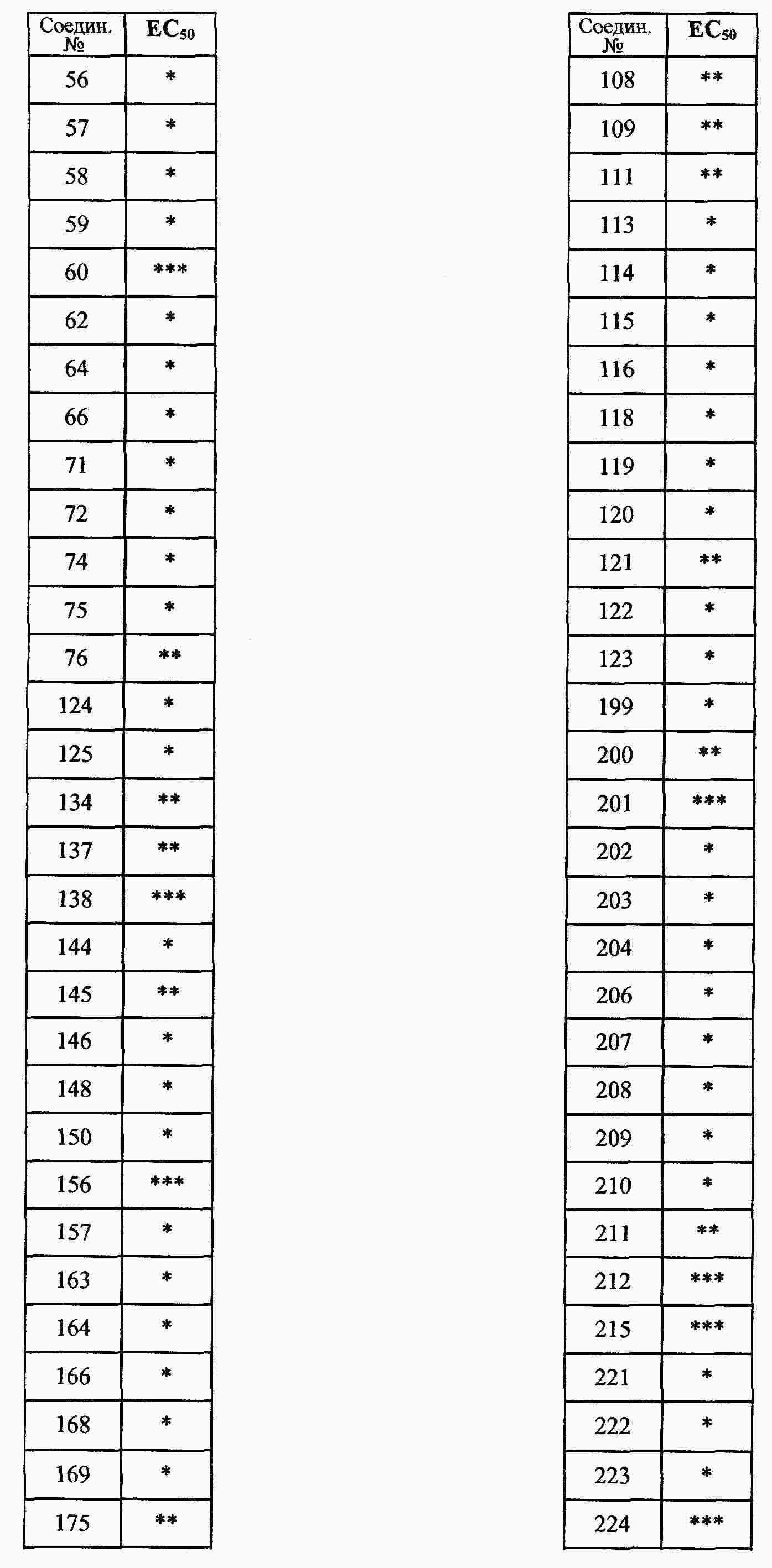
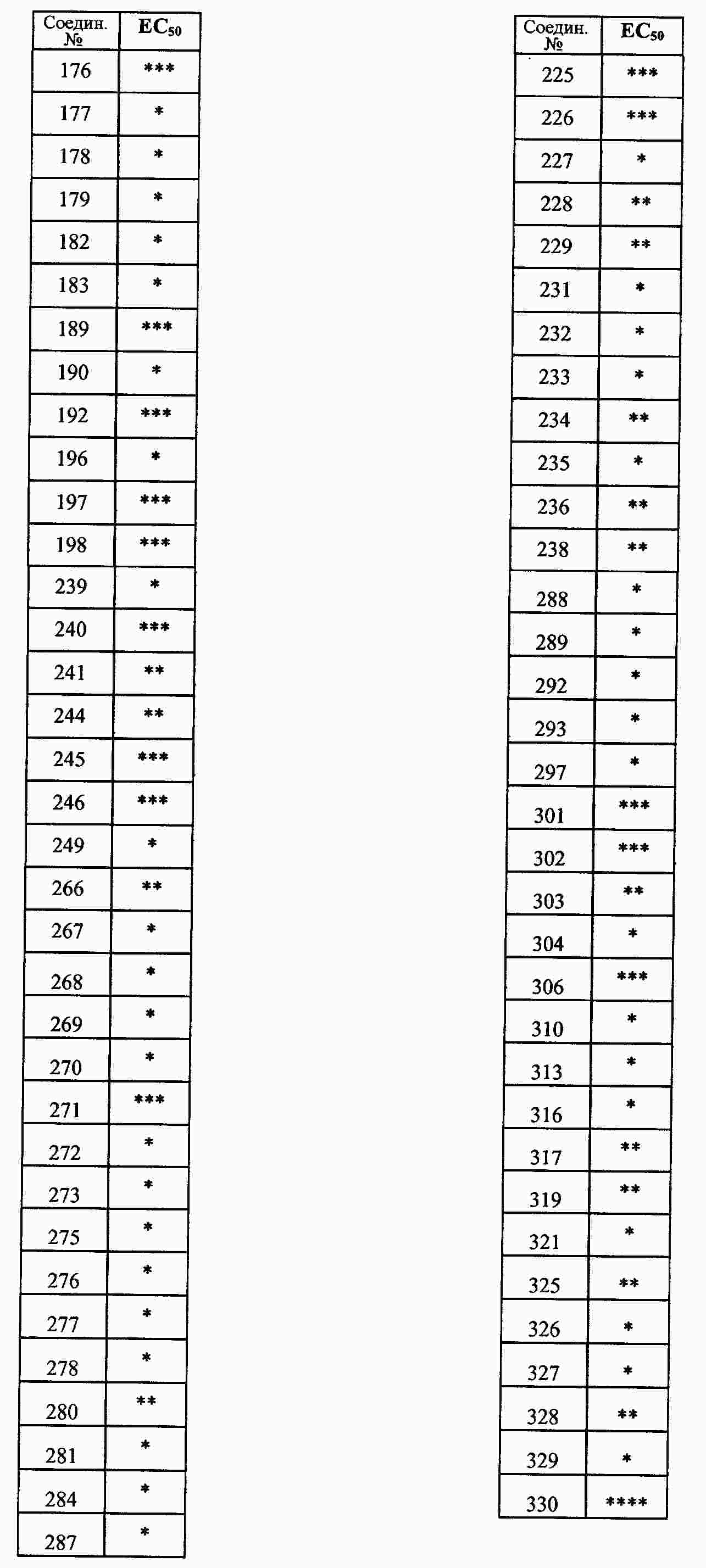
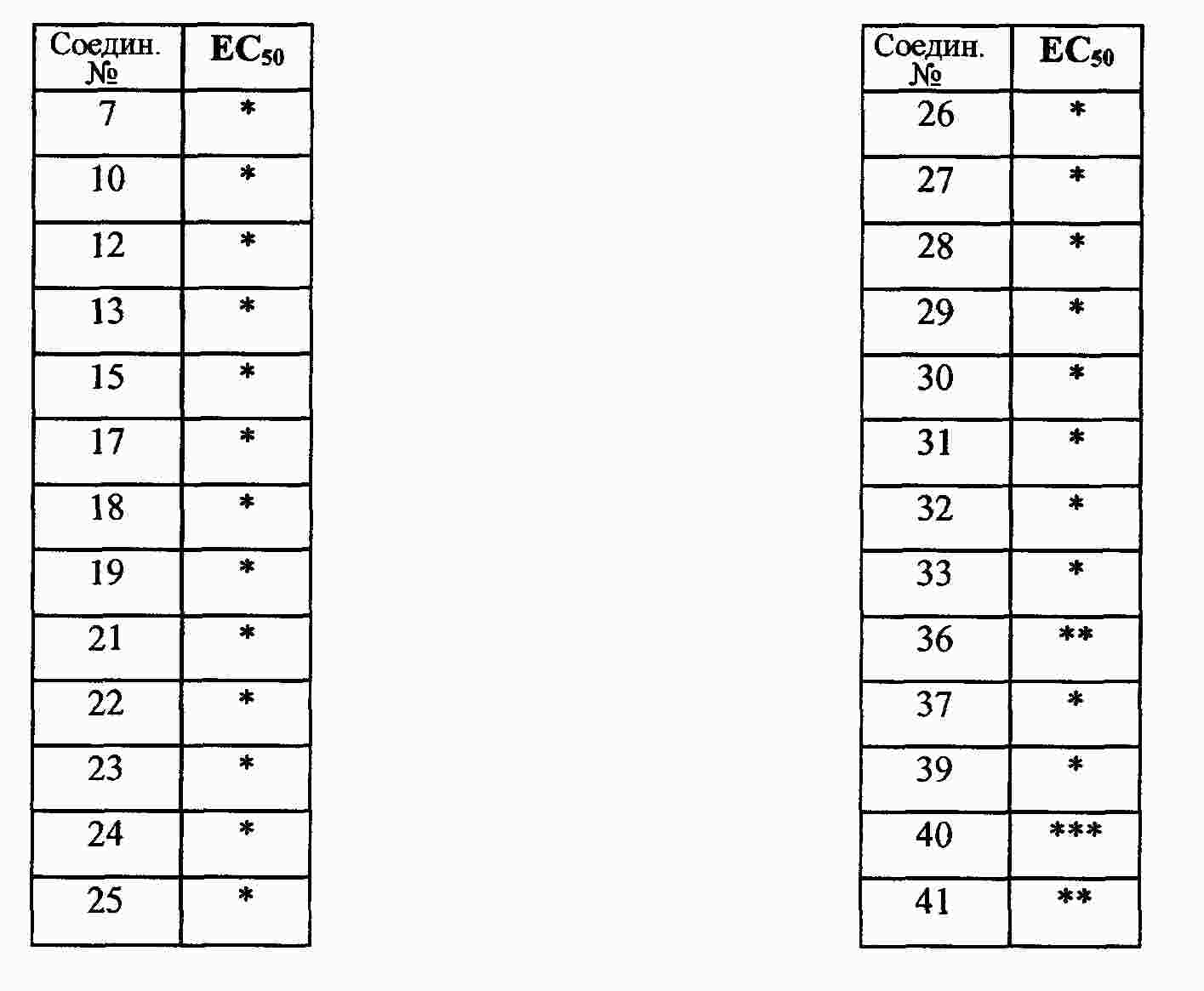
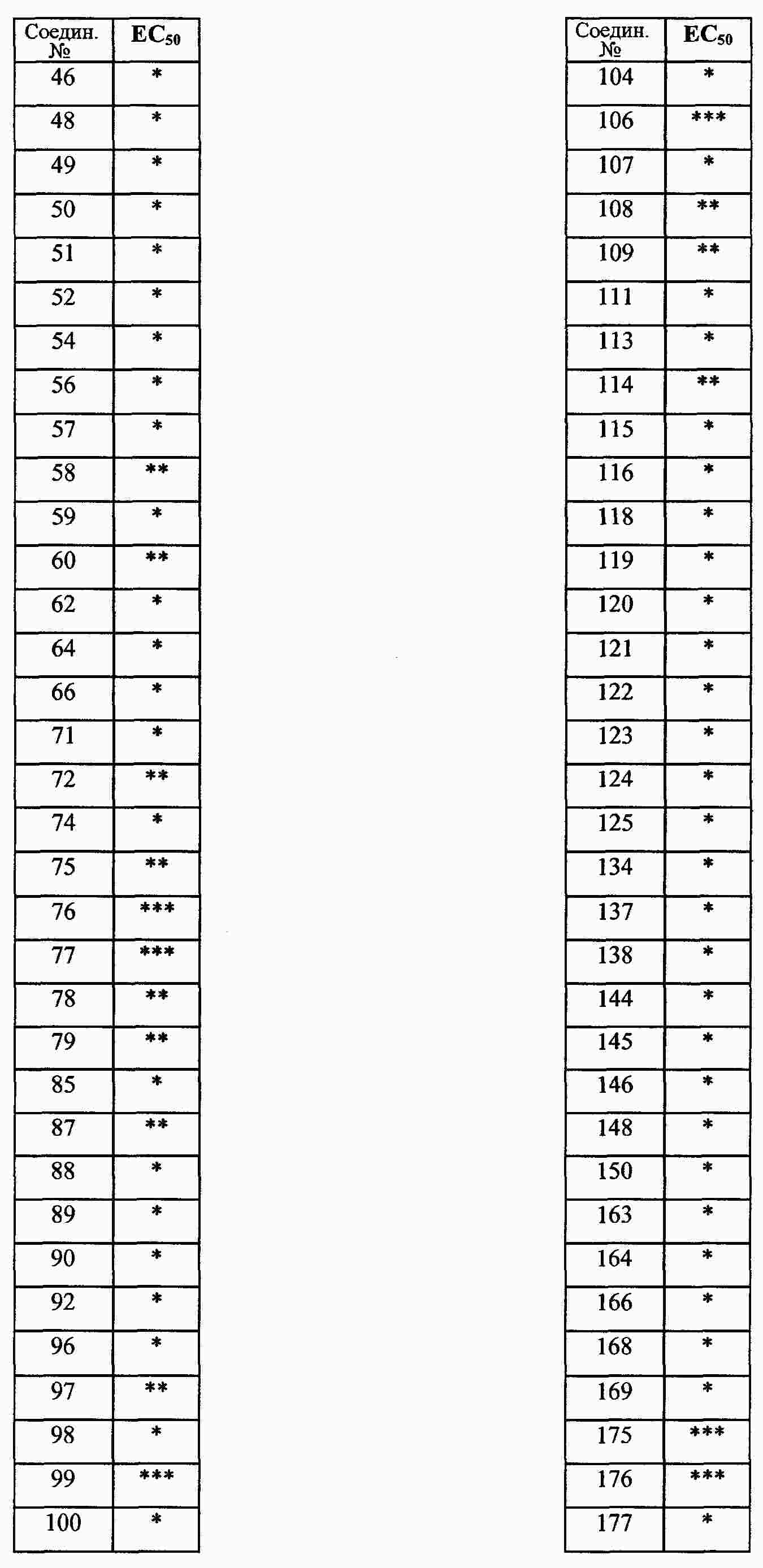
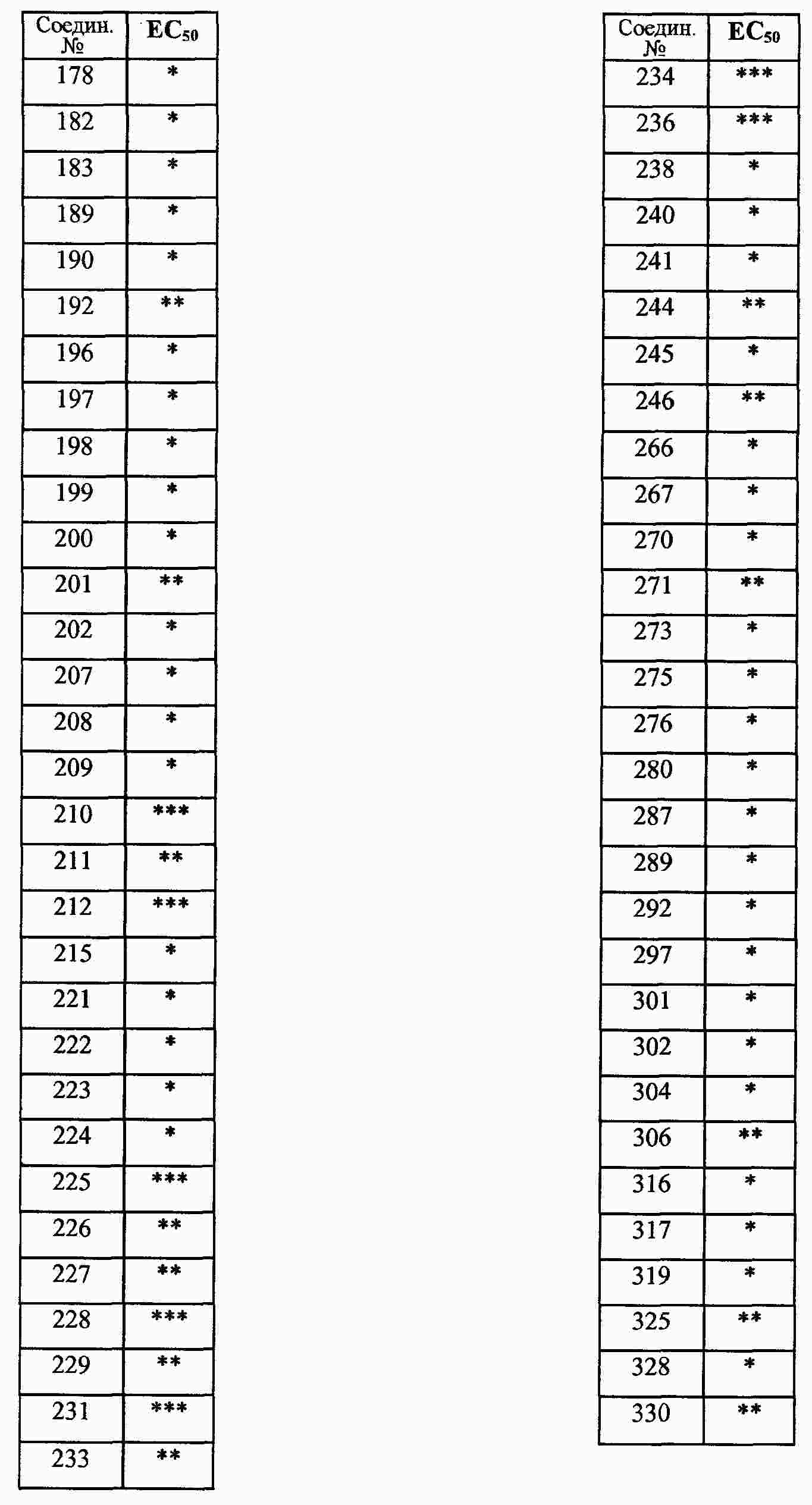
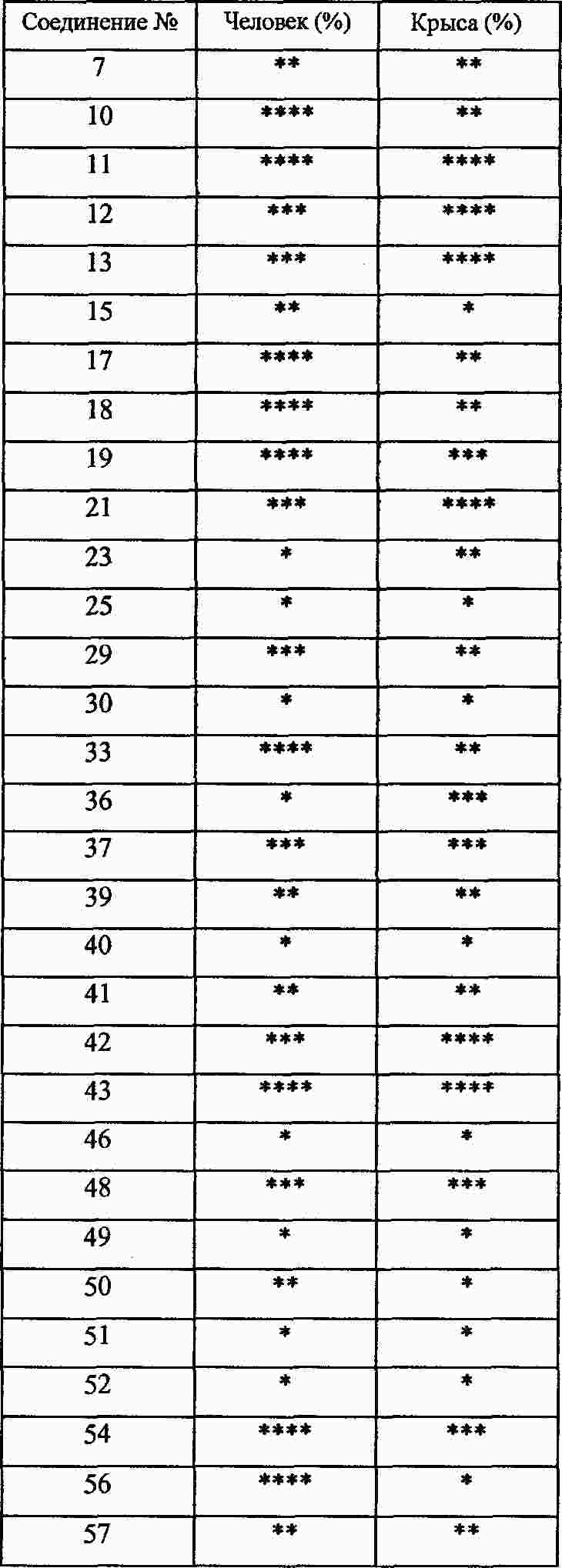
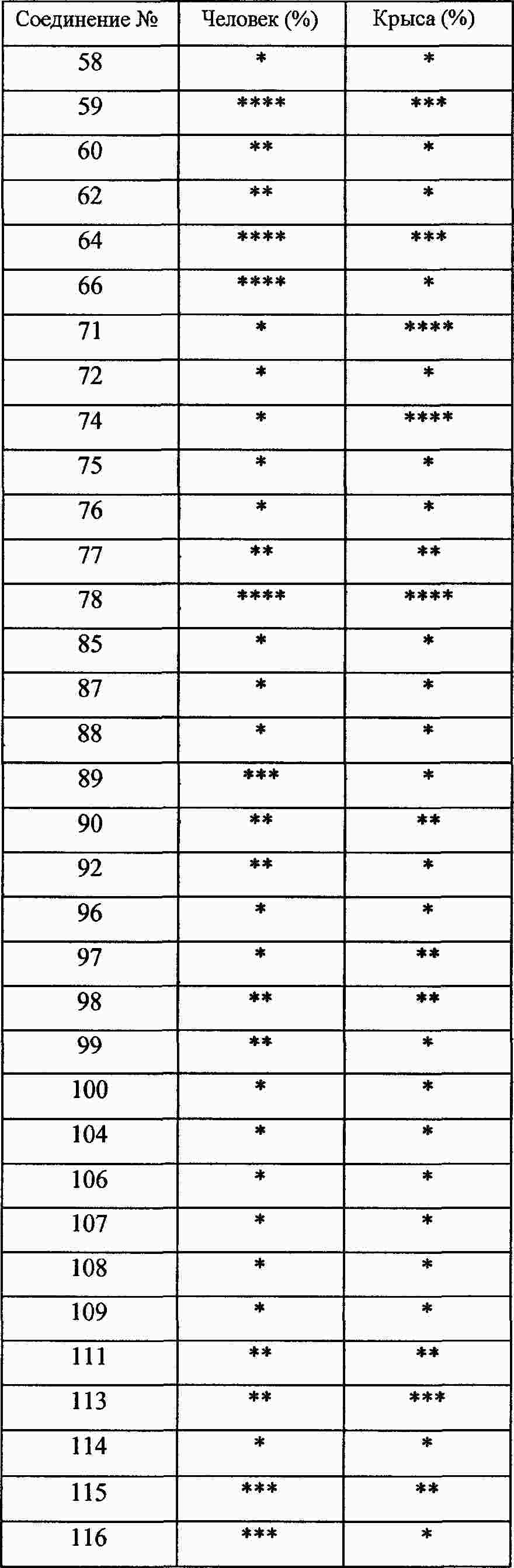
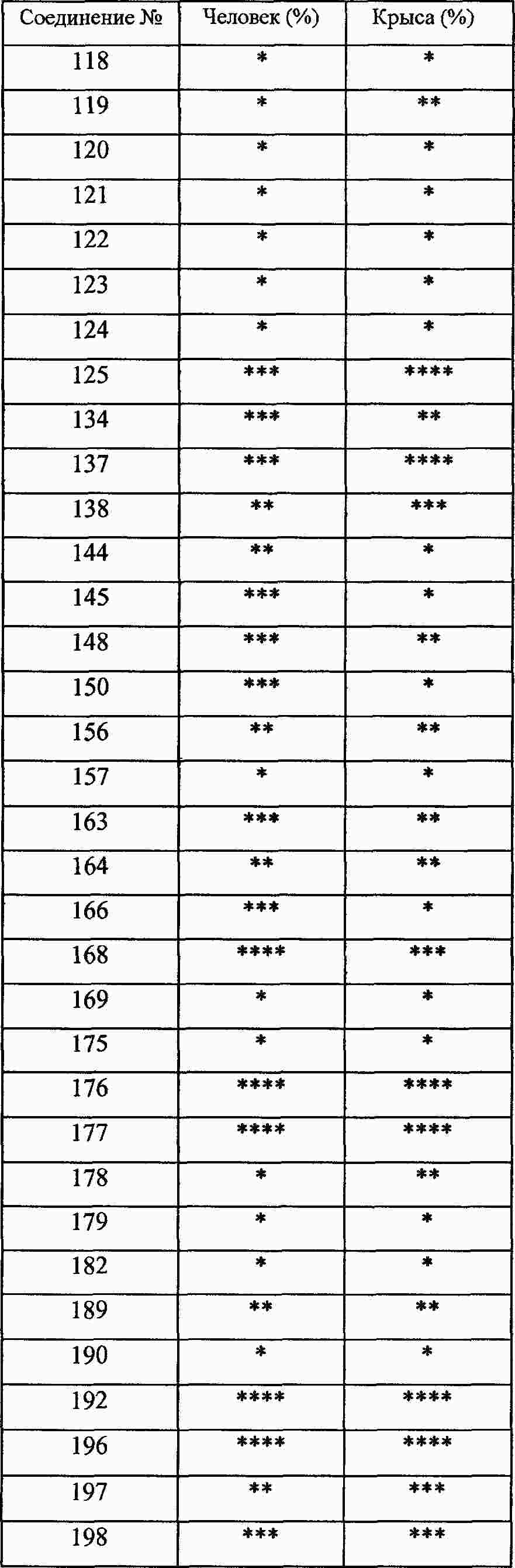
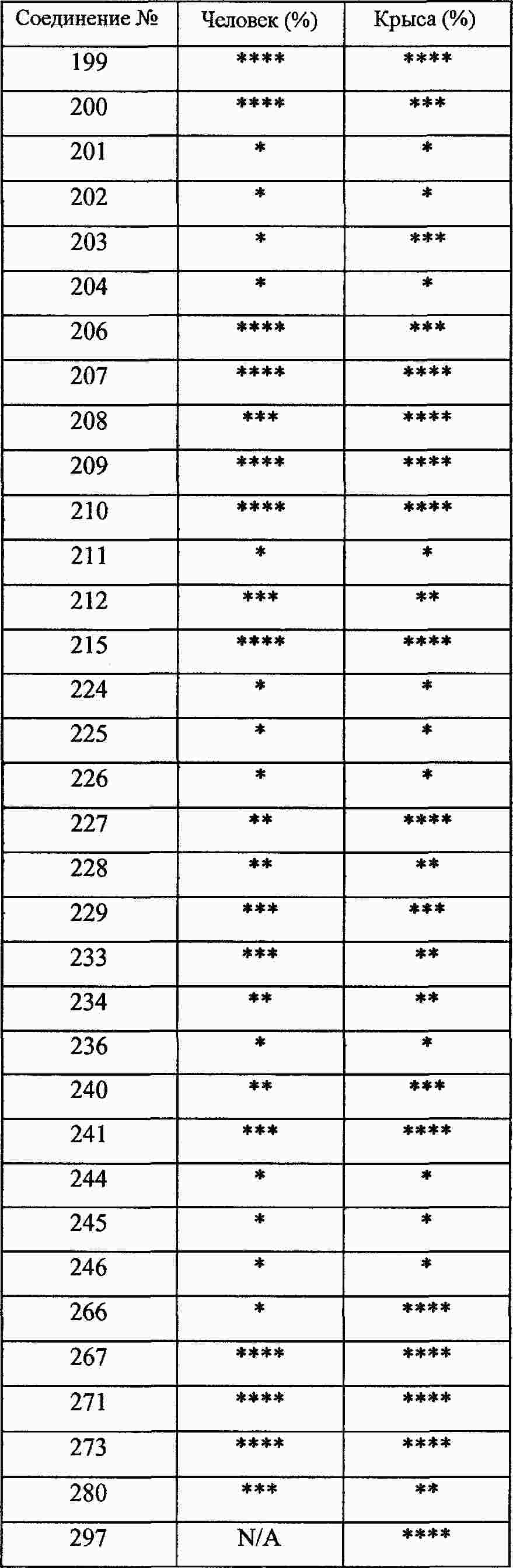
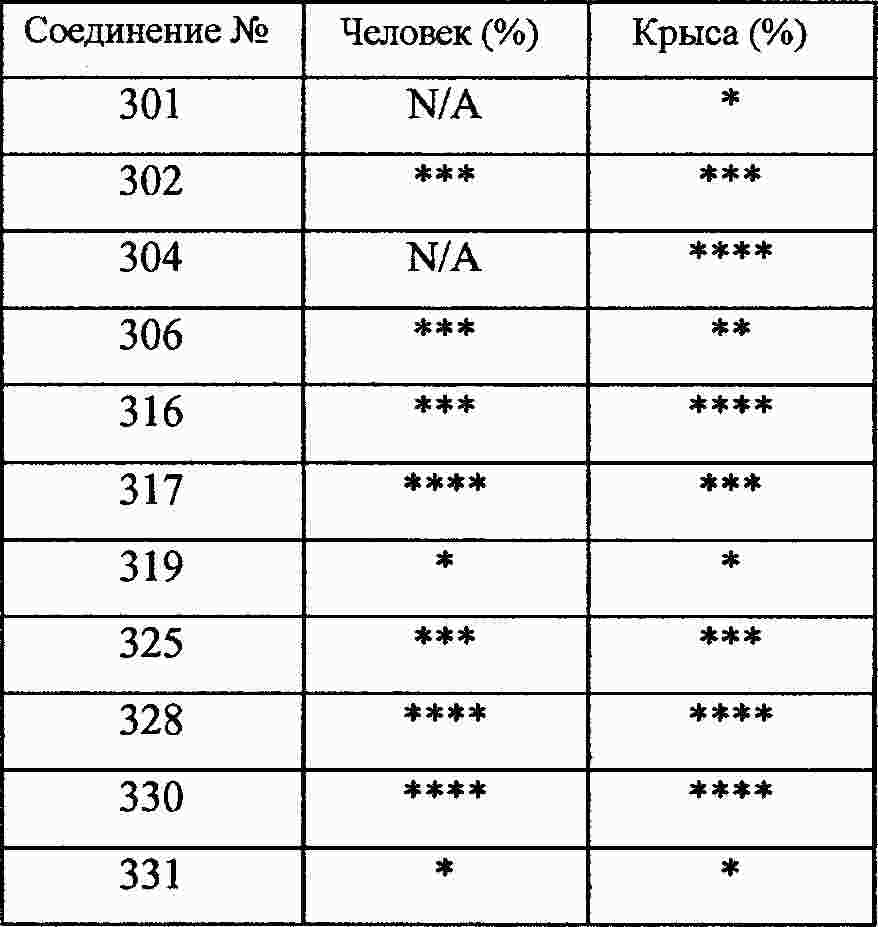
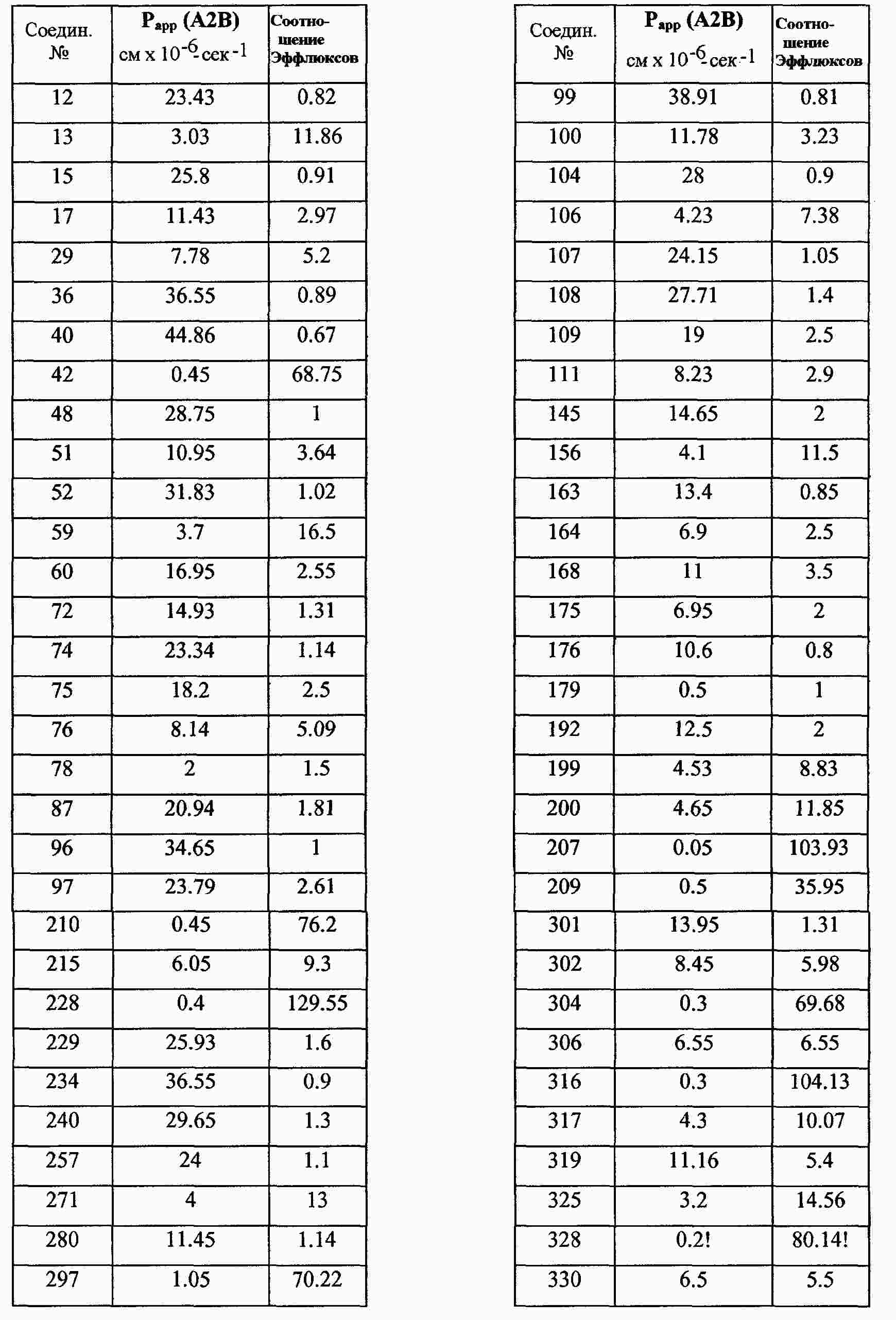
|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea000018587b*\id |
|
Термины запроса в документе Реферат Раскрывают применение производных [1,2,4]триазоло[1,5-а]пиридина, а именно, производных 2-циклопропилкарбониламино-5-фенил-[1,2,4]триазоло[1,5-а]пиридина, которые имеют формулу, представленную формулой (I). Соединения могут быть использованы для приготовления фармацевтических композиций и могут быть использованы для предупреждения и лечения ряда состояний у млекопитающих, в том числе людей, включающих в качестве неограничивающего примера заболевания, включающие деградацию хряща, деградацию кости и/или сустава; и/или состояния, включающие воспаление или иммунные реакции, такие как болезнь Крона, ревматоидный артрит, псориаз, аллергия дыхательных путей, таких как астма, ринит, ювенильный идиопатический артрит, колит, воспалительные заболевания кишечника, обусловленные действием эндотоксина болезненные состояния, такие как осложнения после шунтирования, или хронические обусловленные действием эндотоксина состояния, способствующие возникновению, например, хронической сердечной недостаточности, заболевания, включающие ухудшение ремоделирования хряща, такие как заболевания, включающие анаболическую стимуляцию хондроцитов, врожденные мальформации хряща, заболевания, связанные с гиперсекрецией IL6, и отторжение трансплантата, такие как отторжение пересаженного органа и пролиферативные заболевания. Формула [0001] Применение соединения формулы (I) [0002] Применение соединения по п.1, где L1 выбирают из одинарной связи, -O-, -N(R4a)-; -С(О)-, -С[=N(R4a)]-, -CON(R4a)-, -SO2N(R4a)-, -S(O)2-, -N(R4a)SO2- и -N(R4a)CO-; n1 имеет значение 0, 1, 2, 3 или 4; и R3b представляет собой пиперидинил, морфолинил, пиперазинил, гомопиперазинил или пирролидинил, каждый из которых может быть не замещен или замещен С1-С6алкилом, ацилом, фенилом или ОН, при условии, что [0003] Применение соединения по п.1 или 2, где R4a представляет собой Н, замещенный или незамещенный С1-С4алкил. [0004] Применение соединения по п.1 или 2, где R4a представляет собой -(CH2)n2-R6a; где n2 имеет значение 0, 1 или 2; и R6a представляет собой Н, CN, NMe2 или тетрагидрофуранил. [0005] Применение соединения по п.1 или 2, где R4a представляет собой -СН(СН3)-(СН2)n2-R6a; где n2 имеет значение 0 или 1; и R6a представляет собой Н или ОМе. [0006] Применение соединения по п.1 или 2, где R3b представляет собой OPh, или О-(4-F-Ph). [0007] Применение соединения по п.1, где Ph-L1-(CH2)n1-R3b выбирают из [0008] Применение соединения по п.7, где R3b выбирают из [0009] Применение соединения по п.7, где Су3 выбирают из [0010] Применение соединения по любому из пп.1-9, где заболевание включает в себя воспаление, состояние, включающее иммунную реакцию или аутоиммунное заболевание, ухудшение ремоделирования хряща, пролиферативное заболевание, врожденную мальформацию хряща, болезнь Кастлемена, множественную миелому, псориаз, саркому Капоши и/или месангиальный пролиферативный гломерунефрит или представляет собой отторжение трансплантата. [0011] Применение соединений по любому из пп.1-9, где заболевание, включающее воспаление, выбирают из ревматоидного артрита, остеоартрита, аллергии дыхательных путей и воспалительных заболеваний кишечника. [0012] Применение соединений по любому из пп.1-9, где состояние, включающее иммунный ответ или аутоиммунное заболевание, выбирают из COPD, астмы, системной эриматозной волчанки, сахарного диабета типа I и воспалительного заболевания кишечника. [0013] Применение соединений по любому из пп.1-9, где состояние, включающее ухудшение ремоделирования хряща, выбирают из остеоартрита, псориатического артрита, ювенильного ревматоидного артрита, подагрического артрита, септического или инфекционного артрита, реактивного артрита, симпатической рефлекторной дистрофии, альгодистрофии, синдромома Tietze или костального хондрита, фибромиалгии, остеохондрита, нейрогенного или невропатического артрита, артропатии, эндемических форм артрита, болезни Mseleni и болезни Handigodu; дегенерации, являющейся результатом фибромиалгии, системной эритематозной волчанки, склеродермы и анкилозирующего спондилита. [0014] Применение соединений по любому из пп.1-9, где пролиферативное заболевание выбирают из рака, лейкемии, множественной миеломы или псориаза. [0015] Применение соединений по любому из пп.1-9, где заболевание, включающее воспаление, представляет ревматоидный артрит. [0016] Применение соединений по любому из пп.1-9, где заболевание является отторжением трансплантата. Полный текст патента Область техники, к которой относится изобретение Настоящее изобретение относится к соединениям, которые являются ингибиторами JAK (Янус-киназы), семейства тирозинкиназ, которые вовлечены в модуляцию деградации хрящей, деградации суставов и заболеваний, включающих такую деградацию и/или воспаление. Настоящее изобретение также обеспечивает способы получения этих соединений, фармацевтические композиции, содержащие эти соединения, способы предупреждения и/или лечения заболеваний, включающих деградацию хрящей, деградацию костей и/или суставов, состояний, включающих воспаление или иммунные реакции, обусловленных действием эндотоксина болезненных состояний, рака и отторжения пересаженных органов; и/или способы предупреждения и/или лечения заболеваний, включающих деградацию хрящей, деградацию суставов и/или воспаление, путем введения соединения изобретения. Янус-киназы (JAK-киназы) представляют собой цитоплазматические тирозинкиназы, которые трансдуцируют передачу сигнала цитокинов от мембранных рецепторов к факторам транскрипции STAT. Описаны четыре члена семейства JAK-киназ, JAK1, JAK2, JAK3 и TYK2. При связывании цитокина с его рецептором члены семейства JAK-киназ ауто- и/или трансфосфорилируют друг друга с последующим фосфорилированием STAT-киназ, которые затем мигрируют в ядра для модуляции транскрипции. Внутриклеточная сигнальная трансдукция JAK-STAT-киназ использует интерфероны, большую часть интерлейкинов, а также ряд цитокинов и эндокринные факторы, такие как ЕРО, ТРО, GH, OSM, LIF, CNTF, GM-CSF, PRL (Vainchenker W. et al. (2008)). Исследование комбинаций генетических моделей и небольших молекул ингибиторов JAK-киназ открыло терапевтический потенциал нескольких JAK-киназ. JAK3-киназа признана в генетике мышей и человека в качестве мишени иммуносупрессии (О'Shea J. et al. (2004)). Ингибиторы JAK3-киназы были результативно приняты в клиническую разработку, сначала в отношении отторжения пересаженных органов, но позднее также и в отношении других иммуновоспалительных показаний, таких как ревматоидный артрит (RA), псориаз и болезнь Крона (http://clinicaltrials.gov/). TYK2-киназа представляет собой потенциальную мишень для иммуновоспалительных заболеваний, которая признана в генетике человека и в исследованиях нокаутирования мышей (Levy D. и Loomis С. (2007)). JAK1-киназа представляет собой новую мишень в области иммуновоспалительных заболеваний. JAK1-киназа гетеродимеризуется с другими JAK-киназами с трансдуцированием цитокинзависимой про-воспалительной передачи сигнала. Таким образом, ингибирование JAK1-киназы и/или других JAK-киназ, как предполагают, имеет терапевтический эффект для ряда воспалителных состояний, а также для других заболеваний, обусловленных JAK-опосредованной сигнальной трансдукцией. Хрящ представляет собой несосудистую ткань, хондроциты которой составляют основной клеточный компонент. Хондроциты в нормальном суставном хряще занимают приблизительно 5% объема ткани, при этом внеклеточный матрикс заполняет оставшиеся 95% ткани. Хондроциты секретируют компоненты матрикса, главным образом протеогликаны и коллагены, которые в свою очередь снабжают хондроциты средой, подходящей для их выживания при воздействии механического напряжения. В хряще, тип II коллагена, вместе с типом IX белкового коллагена, скомбинирован в твердые фибриллоподобные структуры, которые придают хрящу большую механическую прочность. Протеогликаны могут поглощать воду и являются ответственными за упругие/эластичные и амортизационные свойства хряща. Одной из функциональных ролей хряща в суставе является предоставление возможности костям соединяться друг с другом безболезненно. Утрата суставного хряща, следовательно, вызывает трение костей друг о друга, что приводит к боли и к потере подвижности. Деградация хряща может иметь различные причины. В воспалительных артритах, таких как ревматоидный артрит, например, деградация хряща вызвана секрецией протеаз (например, коллагеназ) воспаленными тканями (например, воспаленный синовиальный слой). Деградация хряща также может быть результатом повреждения хряща, в результате несчастного случая, или хирургического вмешательства, или ненормально увеличенной нагрузки или "изнашивания и старения". Способность ткани хряща регенерироваться после таких повреждающих факторов ограничена. Хондроциты в поврежденном хряще часто проявляют сниженную активность синтезирования хряща (анаболическая активность) и/или повышенную активность деградирования хряща (катаболическая активность). Дегенерация хряща является признаком различных заболеваний, среди которых ревматоидный артрит и остеоартрит являются наиболее известными. Ревматоидный артрит (RA) представляет собой хроническое дегенеративное заболевание суставов, характеризующееся воспалением и деструкцией структур суставов. В том случае, когда заболевание недиагностировано, оно приводит к существенной недееспособности и боли вследствие потери функциональности суставов и даже к преждевременной смерти. Цель терапии ревматоидного артрита, следовательно, заключается не в том, чтобы замедлить развитие заболевания, но и добиться ремиссии с тем, чтобы остановить деструкцию суставов. Кроме тяжести исхода заболевания высокий уровень распространения ревматоидного артрита (~0,8% взрослых во всем мире страдают от этого заболевания) означает высокие социально-экономические последствия (что касается обзоров по ревматоидному артриту, мы ссылаемся на обзор Smolen and Steiner (2003); обзор Lee and Weinblatt (2001); обзор Choy and Panayi (2001); обзор О'Dell (2004) и обзор Firestein (2003)). Остеоартрит (также называемый как ОА или артрит изнашивания и старения) представляет собой наиболее обычную форму артрита и характеризуется утратой суставного хряща, часто связанной с гипертрофией кости, и болью. Заболевание в основном поражает руки и испытывающие нагрузку суставы, такие как колени, запястья и кости. Этот процесс истончает хрящ. В том случае, когда площадь поверхности исчезла вследствие истончения, достигается остеоартрит степени I; в том случае, когда площадь тангенциальной поверхности исчезла, достигается остеоартрит степени II. Существуют дополнительные уровни дегенерации и деструкции, которые поражают глубину и слои кальцинированного хряща, которые граничат с субхондральной костью. Что касается большого обзора по остеоартриту, то мы ссылаемся на обзор Wieland et al., 2005. Клинические проявления развития состояния при остеоартрите представляют собой следующее: увеличенный объем сустава, боль, крепитацию (хруст) и функциональную недееспособность, которые приводят к боли и сниженной подвижности суставов. В том случае, когда заболевание развивается дальше, то появляется боль в состоянии покоя. Если состояние сохраняется без коррекции и/или терапии, то сустав разрушается, приводя к недееспособности. Тогда требуется реконструктивная хирургия с полным протезированием. Терапевтические способы коррекции поражений суставного хряща, которые появляются в ходе остеоартритического заболевания, были разработаны, но на настоящий момент ни один из них не способен быть промежуточным звеном регенерации суставного хряща in situ и in vivo. Остеоартрит трудно лечить. В настоящее время нет доступного средства для лечения, и лечение фокусируется на обезболивании и на предотвращении наступления деформации пораженного сустава. Обычные виды лечения включают применение нестероидных противовоспалительных лекарственных средств (NSAIDs). Биологически активные добавки, такие как хондроитин-сульфат и глюкозамин-сульфат, рекомендованы в качестве безопасных и эффективных методов лечения остеоартрита, последнее клиническое испытание открыло то, что оба вида лечения не снижали боль, сопутствующую остеоартриту. (Clegg et al., 2006). В своей совокупности нет доступных модифицирующих течение заболевания остеоартритических лекарственных средств. В тяжелых случаях может быть необходима замена сустава. В особенности это имеет место в отношении запястий и коленей. Если сустав является чрезвычайно болезненным и не может быть заменен, он может быть подвергнут сращиванию. Эта процедура останавливает боль, но приводит к постоянной утрате функции сустава, что делает ходьбу и сгибание затруднительными. Другое возможное лечение представляет собой трансплантацию культивированных аутологичных хондроцитов. Здесь, у пациента берут хондральный клеточный материал, посылают в лабораторию, где его выращивают. Затем материал имплантируют в поврежденные ткани с тем, чтобы закрыть дефекты ткани. Другое лечение включает внутрисуставную инстилляцию препарата Hylan G-F 20 (например, Synvisc ®, Hyalgan ®, Artz ®), вещества, которое улучшает временно реологические свойства синовиальной жидкости, производя почти мгновенное ощущение свободного движения и заметное снижение боли. Другие опубликованные способы включают применение сухожильных, периостальных, фасциальных, мышечных или перихондральных графтов; имплантацию фибрина или культивированных хондроцитов; имплантацию синтетических матриксов, таких как коллаген, углеродные нити; введение электромагнитных полей. Все из упомянутых способов предоставляли минимальные и недостаточные эффекты, дающие в результате ткань плохого качества, которая не может ни выдерживать утяжеленную нагрузку, ни давать возможность восстановления суставной функции с нормальным движением. Стимуляция анаболических процессов, блокирование катаболических процессов или комбинация этих двух методов могут приводить к стабилизации хряща и, возможно, даже к реверсии нарушения и, следовательно, могут предотвращать дальнейшее развитие заболевания. Различные триггеры могут стимулировать анаболическую стимуляцию хондроцитов. Инсулиноподобный фактор роста I (IGF-I) представляет собой численно преобладающий фактор роста в синовиальной жидкости и стимулирует синтез как протеогликанов, так и коллагена. Также было показано, что члены семейства костных морфогенетических белков (BMP), в особенности ВМР2, ВМР4, ВМР6 и ВМР7, и члены семейства β-трансформирующих факторов роста (TGF- β) человека могут индуцировать анаболическую стимуляцию хондроцитов (Chubinskaya and Kuettner, 2003). Недавно было идентифицировано соединение, которое индуцирует анаболическую стимуляцию хондроцитов (US 6500854; ЕР 1391211). Однако большинство из этих соединений проявляют тяжелые побочные эффекты и, как следствие, существует сильная потребность в соединениях, которые стимулируют дифференцировку хондроцитов без этих побочных эффектов. Vandeghinste et al. (WO 2005/124342) открыли JAK1-киназу в качестве мишени, ингибирование которой могло бы иметь терапевтическую значимость для нескольких заболеваний, включающих остеоартрит. JAK1-киназы принадлежат к семейству Янус-киназ (JAK-киназы), состоящему из цитоплазматических тирозинкиназ, участвующих в опосредованной рецепторами цитокинов внутриклеточной сигнальной трансдукции. Семейство JAK-киназ состоит из 4 членов: JAK1-киназа, JAK2-киназа, JAK3-киназа и TYK2-киназа. JAK-киназы рекрутируются рецепторами цитокинов, при связывании цитокина, с последующей гетеродимеризацией рецептора цитокина и общей субъединицы рецептора (обычная цепь γ-с, gp130). Затем JAK-киназы активируются посредством ауто- и/или трансфосфорилирования с помощью другой JAK-киназы, что приводит к фосфорилированию рецепторов и к рекрутированию и фосфорилированию членов семейства (сигнальный трансдуктор и активатор транскрипции)-белков (белки STAT). Фосфорилированные белки STAT димеризуются и проникают в ядра, где они связываются с энхансерной областью цитокинотвечающих генов. Нокаутирование гена JAK1-киназы у мышей показало то, что JAK1-киназа играет важнейшие и неизбыточные роли во время развития: мыши с нокаутированным геном JAK1-киназы (JAK1-/-мыши) умирали в пределах 24 ч после рождения, и развитие лимфоцитов было сильнейшим образом нарушено (снижено). Кроме того, клетки с нокаутированным геном JAK1-киназы (JAK1-/-клетки) были нереакционноспособными или менее реакционноспособными по отношению к цитокинам, которые используют рецепторы цитокинов класса II, рецепторы цитокинов, которые используют субъединицу γ-с для передачи сигнала, и семейство рецепторов цитокинов, которые используют субъединицу gp130 для передачи сигнала (Rodig et al., 1998). Различные группы участвовали в передаче сигнала JAK-STAT-киназ в биологии хондроцитов. Li et al. (2001) показали, что онкостатин М индуцирует экспрессию генов ММР и TIMP3 в первичных хондроцитах посредством активации сигнальных путей JAK/STAT- и MAPK-киназ. Osaki et al. (2003) показали, что опосредованное интерфероном- γ ингибирование коллагена II в хондроцитах включает передачу сигнала JAK-STAT-киназ. IL1- β индуцирует катаболизм хряща путем снижения экспрессии компонентов матрикса и путем индуцирования экспрессии коллагеназ и индуцируемой синтазы оксида азота (NOS2), которая обуславливает продуцирование оксида азота (NO). Otero et al. (2005) показали, что лептин и ILl- β синергистически индуцировали продуцирование NO или экспрессию мРНК синтазы NOS2 в хондроцитах и что то было подвергнуто блокаде посредством ингибитора JAK-киназ. Legendre et al. (2003) показали, что IL6/рецептор IL6 индуцировал отрицательную регуляцию генов коллагена II типа специфического матрикса хряща, центрального белка аггрекана и белка сшивок в суставных хондроцитах быка и что это было обусловлено передачей сигнала JAK-STAT-киназ. Следовательно, эти наблюдения позволяют предположить роль в отношении активности JAK-киназ в гомеостазе хряща и терапевтические возможности ингибиторов JAK-киназ. Члены семейства JAK-киназ были вовлечены в дополнительные состояния, включающие миелопролиферативные нарушения (O'Sullivan et al., 2007, Mol. Immunol. 44(10):2497-506), где были идентифицированы мутации JAK2-киназ. Это указывает на то, что ингибиторы JAK-киназ, в частности JAK2-киназ, также могут быть использованы в лечении миелопролиферативных нарушений. Кроме того, семейство JAK-киназ, в частности JAK1-киназа, JAK2-киназа и JAK3-киназа, было связано с раковыми заболеваниями, в частности с лейкемиями, например с острой миелоидной лейкемией (O'Sullivan et al., Mol. Immunol. 44(10):2497-506; Xiang et al., 2008, "Identification of somatic JAK1 mutations in patients with acute myeloid leukemia" Blood First Edition Paper, ранее опубликованной он-лайн 26 декабря 2007; DOI 10.1182/blood-2007-05-090308) и с острой лимфобластной лейкемией (Mullighan et al., 2009) или с солидными опухолями, например с лейомиосаркомой матки (Constantinescu et al., 2007, Trends in Biochemical Sciences 33(3): 122-131), с раком предстательной железы (Tam et al., 2007, British Journal of Cancer, 97, 378-383). Эти результаты указывают на то, что ингибиторы JAK-киназ, в частности JAK1-киназ и/или JAK2-киназ, также могут иметь применение в лечении раковых заболеваний (лейкемии и солидных опухолей, например лейомиосаркомы, рака предстательной железы). Кроме того, заболевание Кастлемана, множественная миелома, мезангиальный пролиферативный гломерулонефрит, псориаз и саркома Капоши, вероятно, возникают вследствие гиперсекреции цитокина IL-6, биологические эффекты которого обусловлены внутриклеточной передачей сигнала JAK-STAT-киназ (Tetsuji Naka, Norihiro Nishimoto and Tadamitsu Kishimoto, Arthritis Res. 2002, 4 (suppl. 3):S233-S242). Этот результат показывает, что ингибитор JAK-киназ также может найти применение в лечении вышеупомянутых заболеваний. Связь с аутоиммунными заболеваниями была установлена для JAK3- и TYK2-киназ. Мутации JAK3-киназ и также γ-с-цепи рецепторов компонентов передачи сигнала в обратном направлении и рецептора IL7 служат причиной в совокупности для ~70% случаев возникновения тяжелого комбинированного иммунодефицита у человека (О'Shea et al., 2004). Следует отметить, что JAK1-киназа кооперируется с JAK3-киназой в трансдуцировании сигналов от γ-с-цепи рецепторов. Полиморфизмы TYK2-киназы наблюдают при системной эритематозной волчанке (SLE) (О'Sullivan et al., 2007, Mol. Immunol. 44(10):2497-506). Следовательно, направленное воздействие на семейство JAK-киназ может обеспечить терапевтическую возможность в области иммуновоспалительных заболеваний. Текущие виды терапии не являются удовлетворительными, и, следовательно, сохраняется потребность в установлении дополнительных соединений, которые могут быть полезны в лечении заболеваний, включающих деградацию хрящей, деградацию костей и/или суставов, например остеоартрита; и/или состояний, включающих воспаление или иммунные реакции, такие как болезнь Крона, ревматоидный артрит, псориаз, аллергия дыхательных путей (например, астма, ринит), ювенильный идиопатический артрит, колит, воспалительные заболевания кишечника, обусловленные действием эндотоксина болезненные состояния (например, осложнения после шунтирования или хронические обусловленные действием эндотоксина состояния, способствующие возникновению, например, хронической сердечной недостаточности), заболевания, включающие ухудшение ремоделирования хряща (например, заболевания, включающие анаболическую стимуляцию хондроцитов), врожденные мальформации хряща, заболевания, связанные с гиперсекрецией IL6, и отторжение трансплантата (например, отторжение пересаженного органа). Ингибиторы JAK-киназ также могут найти применение в лечении пролиферативных заболеваний. В частности, ингибиторы JAK-киназ находят применение в лечении раковых заболеваний, в особенности, лейкемий и солидных опухолей (например, лейомиосаркомы матки, рака предстательной железы). Настоящее изобретение, таким образом, обеспечивает соединения, способы для их изготовления и фармацевтические композиции, содержащие соединение изобретения вместе с подходящим фармацевтическим носителем. Настоящее изобретение также обеспечивает применение соединения изобретения в получении лекарственного препарата для лечения дегенеративных заболеваний суставов. Настоящее изобретение основано на открытии того, что ингибиторы JAK-киназы являются полезными в лечении заболеваний, включающих деградацию хряща, деградацию кости и/или сустава, например остеоартрита; и/или состояний, включающих воспаление или иммунные реакции, таких как болезнь Крона, ревматоидный артрит, псориаз, аллергия дыхательных путей (например, астма, ринит), ювенильный идиопатический артрит, колит, воспалительные заболевания кишечника, обусловленные воздействием эндотоксина болезненные состояния (например, осложнения после шунтирования или хронические обусловленные действием эндотоксина состояния, способствующие возникновению, например, хронической сердечной недостаточности), заболевания, включающие ухудшение ремоделирования хряща (например, заболевания, включающие анаболическую стимуляцию хондроцитов), врожденные мальформации хряща, заболевания, связанные с гиперсекрецией IL6, и отторжение трансплантата (например, отторжение пересаженного органа). Ингибиторы JAK-киназ также могут найти применение в лечении пролиферативных заболеваний. В частности, ингибиторы JAK-киназ находят применение в лечении раковых заболеваний, в особенности, лейкемий и солидных опухолей (например, лейомиосаркомы матки, рака предстательной железы). Настоящее изобретение также обеспечивает способы для получения этих соединений, фармацевтические композиции, содержащие эти соединения, и способы лечения заболеваний, включающих деградацию хряща, деградацию сустава и/или воспаление хряща, путем введения соединения изобретения. В связи с вышеизложенным в первом аспекте изобретения раскрывают применение 1,2,4-триазоло[1,5-а]пиридиновых соединений, имеющих формулу (I) в которой L1 выбирают из одинарной связи, -O-, -С(О)-, -С[=N(R 4a )]-, -N(R 4a )-, -CON(R 4a )-, -SO 2 N(R 4a )-, -S(O) 2 -, -N(R 4a )CO-, -CH 2 -N(R 4a )- или -N(R 4a )SO 2 -; R 3b независимо выбирают из замещенного или незамещенного С 6-10 арила, замещенного или незамещенного С 3 -С 7 циклоалкила, замещенного или незамещенного 4-7-членного гетероциклоалкила, содержащего 1-3 гетероатома, выбранных из N, О или S, замещенного или незамещенного 5-10-членного гетероарила, содержащего 1-3 гетероатома, выбранных из N, О или S; или R 3b независимо выбирают из O-R 3c , NH-R 3c , CO-R 3c и CON (R 4a )-R 3c ; R 3c независимо выбирают из замещенного С 1 -С 6 алкила, замещенного или незамещенного С 6-10 арила, замещенного или незамещенного С 3 -С 7 циклоалкила, замещенного или незамещенного 4-7-членного гетероциклоалкила, содержащего 1-3 гетероатома, выбранных из N, О или S, замещенного или незамещенного 5-10-членного гетероарила, содержащего 1-3 гетероатома, выбранных из N, О или S; каждый R 4a независимо выбирают из Н, С 1 -С 6 алкила, замещенного С 1 -С 6 алкила, С 3 -С 7 циклоалкила; причем заместители в определениях R 3b , R 3c , R 4a выбирают из группы, состоящей из галогена, циано, нитро, трифторметила, трифторметокси, азидо, -NR"'SO 2 R", -SO 2 NR"R"', -C(O)R", -C(O)OR", OC(O)R", -NR'"C(O)R", -C(O)NR"R'", -NR"R"', -(CR"'R'") m OR"', где каждый R" независимо выбран из Н, С 1 -С 8 алкила, -(СН 2 ) t (С 6 -С 10 арила), -(СН 2 ) t (5-10-членного гетероарила, включающего 1-3 гетероатома, выбранных из N, О, или S), -(СН 2 ) t (С 3 -С 10 циклоалкила) и -(СН 2 ) t (4-10-членного гетероциклоалкила, включающего 1-3 гетероатома, выбранных из N, О, или S), где t представляет целое число 0-4; любая алкильная группа может быть замещена галогеном или ОН; любой арил, гетероарил, циклоакил или гетероциклоалкил могут быть замещены замещенным или незамещенным С 1 -С 4 алкилом, галогеном, незамещенным C 1 -С 4 алкокси, незамещенным С 1 -С 4 галогеналкилом, незамещенным С 1 -С 4 гидроксиалкилом или незамещенным C 1 -С 4 галогеналкокси или ОН; каждый R"' независимо представляет Н или С 1 -С 6 алкил; n1 равно 0, 1, 2, 3 или 4; при условии, что i) когда L1 представляет собой -O-, -N(R 4a )-, -CH 2 -N(R 4a )-, -CON(R 4a )- или -SO 2 N(R 4a )- и R 3b является отличным от циклоалкила, арила или 5-10-членного гетероарила, тогда n1 равен 1, 2, 3 или 4; ii) когда L1 представляет собой связь, n1 равен 0, и R 3b представляет собой -OR 3c , тогда R 3c является отличным от Me или CF 3 ; или его фармацевтически приемлемая соль, в лечении и/или предупреждении заболеваний, включающих деградацию хряща, деградацию кости и/или сустава; и/или состояний, включающих воспаление или иммунные реакции, таких как болезнь Крона, ревматоидный артрит, псориаз, аллергия дыхательных путей, ювенильный идиопатический артрит, колит, воспалительные заболевания кишечника, обусловленные действием эндотоксина болезненные состояния, заболевания, включающие ухудшение ремоделирования хряща, врожденные мальформации хряща, болезни Кастлемена, множественной миеломы, псориаза, саркомы Капоши и/или месангиального пролиферативного гломерунефрита и отторжение трансплантата; или пролиферативных заболеваний. В дополнительном аспекте настоящее изобретение раскрывает применение 1,2,4-триазоло[1,5-а]пиридиновых соединений формулы I, которые способны быть действенными в модулировании активности JAK-киназ in vivo. В дополнительном аспекте настоящее изобретение обеспечивает применение соединений формулы I или их фармацевтически приемлемых солей для лечения и/или предупреждения заболеваний, включающих деградацию хряща, деградацию кости и/или сустава, например остеоартрита; и/или состояний, включающих воспаление или иммунные реакции, такие как болезнь Крона, ревматоидный артрит, псориаз, аллергия дыхательных путей (например, астма, ринит), ювенильный идиопатический артрит, колит, воспалительные заболевания кишечника, обусловленные действием эндотоксина болезненные состояния (например, осложнения после шунтирования или хронические обусловленные действием эндотоксина состояния, способствующие возникновению, например, хронической сердечной недостаточности), заболевания, включающие ухудшение ремоделирования хряща (например, заболевания, включающие анаболическую стимуляцию хондроцитов), врожденные мальформации хряща, заболевания, связанные с гиперсекрецией IL6, и отторжение трансплантата (например, отторжение пересаженного органа) или пролиферативных заболеваний. В дополнительном аспекте настоящее изобретение обеспечивает применение соединения изобретения в лечении или в предупреждении состояния, выбранного из состояний, перечисленных в этом документе, особенно таких состояний, которые могут быть связаны с аберрантной активностью JAK-киназ, таких как заболевания, включающие деградацию хряща, деградацию кости и/или сустава, например остеоартрит; и/или состояния, включающие воспаление или иммунные реакции, такие как болезнь Крона, ревматоидный артрит, псориаз, аллергия дыхательных путей (например, астма, ринит), ювенильный идиопатический артрит, колит, воспалительные заболевания кишечника, обусловленные действием эндотоксина болезненные состояния (например, осложнения после шунтирования или хронические обусловленные действием эндотоксина состояния, способствующие возникновению, например, хронической сердечной недостаточности), заболевания, включающие ухудшение ремоделирования хряща (например, заболевания, включающие анаболическую стимуляцию хондроцитов), врожденные мальформации хряща, заболевания, связанные с гиперсекрецией IL6, и отторжение трансплантата (например, отторжение пересаженного органа). Ингибиторы JAK-киназ также могут найти применение в лечении пролиферативных заболеваний. : В частности, ингибиторы JAK-киназ находят применение в лечении раковых заболеваний, в особенности лейкемий и солидных опухолей (например, лейомиосаркомы матки, рака предстательной железы). В конкретном варианте осуществления состояние выбирают из воспаления, такого как ревматоидный артрит, ювенильный идиопатический артрит, псориаз, аллергия дыхательных путей (например, астма, ринит), воспалительные заболевания кишечника (например, болезнь Крона, колит), обусловленные действием эндотоксина болезненные состояния (например, осложнения после шунтирования или хронические обусловленные действием эндотоксина состояния, способствующие возникновению, например, хронической сердечной недостаточности) и отторжение пересаженного органа; и деградации или дегенерации хряща, кости и/или сустава, такой как остеоартрит. В дополнительном аспекте настоящее изобретение обеспечивает применение соединения изобретения в лечении или в предупреждении пролиферативных нарушений, в частности ракового заболевания, (например, солидные опухоли), лейкемий, множественной миеломы или псориаза. В дополнительном аспекте настоящее изобретение обеспечивает применение соединения изобретения в лечении или в предупреждении состояния, которое причинно связано с аномальной активностью JAK-киназ. Другие цели и преимущества изобретения станут ясны специалистам в данной области из рассмотрения раздела, обеспечивающего подробное описание изобретения. Следующие термины, как подразумевают, имеют значения, представленные сразу же ниже, и являются полезными в понимании описания и предполагаемого объема настоящего изобретения. При описании изобретения, которое может включать соединения, фармацевтические композиции, содержащие такие соединения, и способы применения таких соединений и композиций, следующие термины, если присутствуют, имеют следующие значения, если не указано иное. Также следует понимать, что описанные в этом документе любые фрагменты, определенные далее ниже, могут быть замещены разнообразными заместителями и что соответственные определения предназначены для включения таких замещенных фрагментов в пределах их объема, который изложен ниже. Если не установлено иное, термин "замещенный" должен быть определен так, как изложено ниже. Следует дополнительно понимать, что термины "группы" и "радикальные группы" могут считаться взаимозаменяемыми при использовании в этом документе. Ацил относится к радикальной группе -C(O)R 20 , где R 20 представляет собой водород, С 1 -С 8 алкил, С 3 -С 10 циклоалкил, С 3 -С 10 циклоалкилметил, 4-10-членный гетероциклоалкил, арил, арилалкил, 5-10-членный гетероарил или гетероарилалкил, которые определены в этом документе. Репрезентативные примеры включают формил, ацетил, циклогексилкарбонил, циклогексилметилкарбонил, бензоил и бензилкарбонил, но не ограничиваются этим. Характерными ацильными группами являются -С(O)Н, -С(O)-С 1 -С 8 алкил, -С(O)-(СН 2 ) t (С 6 -С 10 арил), -С(О)-(СН 2 ) t (5-10-членный гетероарил), -С(O)-(СН 2 ) t (С 3 -С 10 циклоалкил) и -С(О)-(СН 2 ) t (4-10-членный гетероциклоалкил), где t представляет собой целое число от 0 до 4. Замещенный ацил относится к радикальной группе -C(O)R 21 , где R 21 независимо представляет собой С 1 -С 8 алкил, замещенный галогеном или гидроксигруппой; или С 3 -С 10 циклоалкил, 4-10-членный гетероциклоалкил, С 6 -С 10 арил, арилалкил, 5-10-членный гетероарил или гетероарилалкил, каждый из которых замещен незамещенным С 1 -С 4 алкилом, галогеном, незамещенной С 1 -С 4 алкоксигруппой, незамещенным С 1 -С 4 галогеналкилом, незамещенным С 1 -С 4 гидроксиалкилом или незамещенной С 1 -С 4 галогеналкоксигруппой или гидроксигруппой. Ациламиногруппа относится к радикальной группе NR 22 C(O)R 23 , где R 22 представляет собой водород, C 1 -C 8 алкил, С 3 -С 10 циклоалкил, 4-10-членный гетероциклоалкил, С 6 -С 10 арил, арилалкил, 5-10-членный гетероарил или гетероарилалкил, и R 23 представляет собой водород, С 1 -С 8 алкил, С 3 -С 10 циклоалкил, 4-10-членный гетероциклоалкил, С 6 -С 10 арил, арилалкил, 5-10-членный гетероарил или гетероарилалкил, которые определены в этом документе. Характерная ациламиногруппа включает формиламиногруппу, ацетиламиногруппу, циклогексилкарбониламиногруппу, циклогексилметилкарбониламиногруппу, бензоиламиногруппу и бензилкарбониламиногруппу, но не ограничивается этим. Характерные ациламиногруппы представляют собой -NR 21 'C(О)-С 1 -С 8 алкил, -NR 21 'C(O)-(CH 2 ) t (C 6 -С 10 арил), -NR 21 'C(O)-(CH 2 ) t (5-10-членный гетероарил), -NR 21 'C(O)-(CH 2 ) t (С 3 -С 10 циклоалкил) и -NR 21 'C(O)-(СН 2 ) t (4-10-членный гетероциклоалкил), где t представляет собой целое число от 0 до 4, каждый R 21 ' независимо представляет собой Н или С 1 -С 8 алкил. Замещенная ациламиногруппа относится к радикальной группе -NR 24 C(O)R 25 , где R 24 независимо представляет собой Н, С 1 -С 8 алкил, замещенный галогеном или гидроксигруппой; или С 3 -С 10 циклоалкил, 4-10-членный гетероциклоалкил, С 6 -С 10 арил, арилалкил, 5-10-членный гетероарил или гетероарилалкил, каждый из которых замещен незамещенным С 1 -С 4 алкилом, галогеном, незамещенной С 1 -С 4 алкоксигруппой, незамещенным С 1 -С 4 галогеналкилом, незамещенным С 1 -С 4 гидроксиалкилом, или незамещенной С 1 -С 4 галогеналкоксигруппой или гидроксигруппой; и R 25 независимо представляет собой Н, С 1 -С 8 алкил, замещенный галогеном или гидроксигруппой; или С 3 -С 10 циклоалкил, 4-10-членный гетероциклоалкил, С 6 -С 10 арил, арилалкил, 5-10-членный гетероарил или гетероарилалкил, каждый из которых замещен незамещенным С 1 -С 4 алкилом, галогеном, незамещенной С 1 -С 4 алкоксигруппой, незамещенным С 1 -С 4 галогеналкилом, незамещенным С 1 -С 4 гидроксиалкилом, или незамещенной С 1 -С 4 галогеналкоксигруппой или гидроксилом; при условии, что по меньшей мере один из заместителей: R 24 или R 25 является отличным от Н. Алкоксигруппа относится к группе -OR 26 , где R 26 представляет собой С 1 -С 8 алкил. Конкретные алкоксигруппы представляют собой метоксигруппу, этоксигруппу, н-пропоксигруппу, изопропоксигруппу, н-бутоксигруппу, трет-бутоксигруппу, втор-бутоксигруппу, н-пентоксигруппу, н-гексоксигруппу и 1,2-диметилбутоксигруппу. Конкретные алкоксигруппы являются низшими алкоксигруппами, то есть имеют от 1 до 6 атомов углерода. Дополнительные конкретные алкоксигруппы имеют от 1 до 4 атомов углерода. Замещенная алкоксигруппа относится к алкоксигруппе, замещенной одной или несколькими группами из тех групп, которые перечислены в определении "замещенного" в этом документе, и в особенности относится к алкоксигруппе, имеющей 1 или более заместителей, например от 1 до 5 заместителей, особенно от 1 до 3 заместителей и в особенности 1 заместитель, выбранный из группы, состоящей из аминогруппы, замещенной аминогруппы, С 6 -С 10 арила, -О-арила, карбоксила, цианогруппы, С 3 -С 10 циклоалкила, 4-10-членного гетероциклоалкила, галогена, 5-10-членного гетероарила, гидроксила, нитрогруппы, тиоалкоксигруппы, тио-О-арила, тиола, алкил-S-(О)-, арил-S-(О)-, алкил-S-(О) 2 - и арил-S-(O) 2 -. Показательные замещенные алкоксигруппы представляют собой -О-(СН 2 ) t (С 6 -С 10 арил), -О-(СН 2 ) t (5-10-членный гетероарил), -О-(СН 2 ) t (С 3 -С 10 циклоалкил) и -О-(СН 2 ) t (4-10-членный гетероциклоалкил), где t является целым числом от 0 до 4, и любые присутствующие арильные, гетероарильные, циклоалкильные или гетероциклоалкильные группы могут быть сами по себе замещены незамещенным С 1 -С 4 алкилом, галогеном, незамещенной С 1 -С 4 алкоксигруппой, незамещенным С 1 -С 4 галогеналкилом, незамещенным С 1 -С 4 гидроксиалкилом или незамещенной С 1 -С 4 галогеналкоксигруппой или гидроксилом. Конкретные показательные замещенные алкоксигруппы представляют собой OCF 3 , OCH 2 CF 3 , OCH 2 Ph, ОСН 2 -циклопропил, ОСН 2 СН 2 ОН, OCH 2 CH 2 NMe 2 . Алкоксикарбонил относится к радикальной группе -С(О)-OR 27 , где R 27 представляет собой С 1 -С 8 акил, С 3 -С 10 циклоалкил, С 3 -С 10 циклоалкилалкил, 4-10-членный гетероциклоалкилалкил, арилалкил или 5-10-членный гетероарилалкил, которые определены в этом документе. Показательные алкоксикарбонильные группы представляют собой С(O)O-С 1 -С 8 алкил, -С(O)О-(СН 2 ) t (С 6 -С 10 арил), -С(О)О-(СН 2 ) t (5-10-членный гетероарил), -С(О)О-(СН 2 ) t (С 3 -С 10 циклоалкил) и -С(О)О-(СН 2 ) t (4-10-членный гетероциклоалкил) , где t является целым числом от 0 до 4. Замещенный алкоксикарбонил относится к радикальной группе -С(О)-OR 28 , где R 28 представляет собой С 1 -С 8 акил, С 3 -С 10 циклоалкил, С 3 -С 10 циклоалкилалкил или 4-10-членный гетероциклоалкилалкил, каждый из которых замещен галогеном, замещенной или незамещенной аминогруппой или гидроксигруппой; или С 6 -С 10 арилалкил или 5-10-членный гетероарилалкил, каждый из которых замещен незамещенным С 1 -С 4 алкилом, галогеном, незамещенной С 1 -С 4 алкоксигруппой, незамещенным С 1 -С 4 галогеналкилом, незамещенным С 1 -С 4 гидроксиалкилом или незамещенной С 1 -С 4 галогеналкоксигруппой или гидроксилом. Алкил означает неразветвленный или разветвленный алифатический углеводород, имеющий 1-20 атомов углерода. Особый алкил имеет 1-12 атомов углерода. Более особый представляет собой низший алкил, который имеет 1-6 атомов углерода. Дополнительная особая алкильная группа имеет 1-4 атома углерода. Показательные группы с прямой цепью включают метил, этил, н-пропил, и н-бутил. "Разветвленный" означает, что одна или более низших алкильных групп, таких как метил, этил, пропил или бутил, являются присоединенными к линейной алкильной цепи, характерные группы с разветвленной цепью включают изопропил, изобутил, трет-бутил и изоамил. Замещенный алкил относится к алкильной группе, замещенной галогеном или ОН. Арил относится к одновалентной ароматической углеводородной группе, полученной удалением одного атома водорода от одного атома углерода исходной ароматической циклической системы. В особенности, арил относится к ароматической циклической структуре, моноциклической или полициклической, которая включает от 6-10 кольцевых членов. В том случае, когда арильная группа представляет собой моноциклическую кольцевую систему, она предпочтительно содержит 6 атомов углерода. Обычные арильные группы включают группы, полученные из ацеантрилена, аценафтилена, ацефенантрилена, антрацена, азулена, бензола, хризена, коронена, флуорантена, флуорена, гексацена, гексафена, гексалена, аз-индацена, сим-индацена, индана, индена, нафталина, октацена, октафена, окталена, овалена, пента-2,4-диена, пентацена, пенталена, пентафена, перилена, феналена, фенантрена, пицена, плиадена, пирена, пирантрена, рубицена, трифенилена и тринафталина, но не ограничиваются этим. Особенно арильные группы включают фенил, нафтил, инденил и тетрагидронафтил. "Замещенный арил" относится к арильной группе, замещенной одной или несколькими группами из тех групп, которые перечислены в определении "замещенного" в этом документе, и особенно относится к арильной группе, которая может быть необязательно замещена 1 или более заместителями. Конкретно, "замещенный арил" относится к арильной группе, замещенной одной или более группами, выбранными из галогена, С 1 -С 8 алкила, С 1 -С 8 галогеналкила, С 1 -С 8 галогеналкоксигруппы, гидроксигруппы, С 1 -С 8 алкоксигруппы. Примеры репрезентативных замещенных арилов включают следующее: В этих формулах один из заместителей R 49 или R 50 может представлять собой водород, и по меньшей мере один из заместителей R 49 или R 50 независимо выбирают из С 1 -С 8 алкила, C 1 -С 8 алкоксигруппы. "Азидогруппа" относится к радикальной группе -N 3 . "Циклоалкил" относится к циклическим неароматическим гидрокарбильным группам, имеющим от 3 до 10 атомов углерода. Такие циклоалкильные группы включают в качестве примера однокольцевые структуры, такие как циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил. "Замещенный циклоалкил" относится к циклоалкильной группе, которая определена выше, замещенной одной или несколькими группами из тех групп, которые перечислены в определении "замещенного" в этом документе, и особенно относится к циклоалкильной группе, имеющей 1 или более заместителей, например от 1 до 5 заместителей, особенно от 1 до 3 заместителей, в особенности 1 заместитель. "Цианогруппа" относится к радикальной группе -CN. "Галогено" или "галоген" относится к фтору (F), хлору (Cl), брому (Br) и йоду (I). Конкретные галогеногруппы представляют собой либо фтор, либо хлор. "Гетеро" в том случае, когда используют для описания соединения или группы, присутствующей в соединении, означает, что один или более атомов углерода в соединении или в группе были заменены гетероатомом азота, кислорода или серы. "Гетеро" может быть применен к любым гидрокарбильным группам, описанным выше, таким как алкил, например гетероалкил, циклоалкил, например гетероциклоалкил, арил, например гетероарил, циклоалкенил, например циклогетероалкенил, и тому подобное, имеющим от 1 до 5, в особенности от 1 до 3 гетероатомов. "Гетероарил" означает ароматическую циклическую структуру, моноциклическую или полициклическую, которая включает один или более гетероатомов и 5-12 кольцевых членов, более обычно 5-10 кольцевых членов. Гетероарильная группа может представлять собой, например, 5-членное или 6-членное моноциклическое кольцо или бициклическую структуру, образованную из конденсированных 5- и 6-членных колец, или из двух конденсированных 6-членных колец, или в качестве дополнительного примера из двух конденсированных 5-членных колец. Каждое кольцо может содержать вплоть до четырех гетероатомов, обычно выбранных из азота, серы и кислорода. Обычно гетероарильное кольцо будет содержать вплоть до 4 гетероатомов, более обычно вплоть до 3 гетероатомов, более обычно вплоть до 2, например 1 гетероатом. В одном варианте осуществления гетероарильное кольцо содержит по меньшей мере 1 атом азота в кольце. Атомы азота в гетероарильных кольцах могут быть основными, как в случае имидазола или пиридина, или, по существу, неосновными, как в случае азота в индоле или в пирроле. В большинстве случаев число основных атомов азота, присутствующих в гетероарильной группе, включая любые аминогруппы-заместители кольца, будет составлять менее 5. Примеры 5-членных моноциклических гетероарильных групп включают пиррольные, фурановые, тиофеновые, имидазольные, фуразановые, оксазольные, оксадиазольные, оксатриазольные, изоксазольные, тиазольные, изотиазольные, пиразольные, триазольные группы, но не ограничиваются этим. Примеры 6-членных моноциклических гетероарильных групп включают пиридин, пиразин, пиридазин, пиримидин и триазин, но не ограничиваются этим. Конкретные примеры бициклических гетероарильных групп, содержащих 5-членное кольцо, конденсированное с другим 5-членным кольцом, включают имидазотиазол и имидазоимидазол, но не ограничиваются этим. Конкретные примеры бициклических гетероарильных групп, содержащих 6-членное кольцо, конденсированное с 5-членным кольцом, включают бензофурановые, бензотиофеновые, бензимидазольные, бензоксазольные, изобензоксазольные, бензизоксазольные, бензтиазольные, бензизотиазольные, изобензофурановые, индольные, изоиндольные, изоиндолоновые, индолизиновые, индолиновые, изоиндолиновые, пуриновые (например, аденин, гуанин), индазольные, пиразолопиримидиновые, триазолопиримидиновые, бензодиоксольные и пиразолопиридиновые группы, но не ограничиваются этим. Конкретные примеры бициклических гетероарильных групп, содержащих два конденсированных 6-членных кольца, включают хинолиновые, изохинолиновые, хромановые, тиохромановые, хроменовые, изохроменовые, хромановые, изохромановые, бензодиоксановые, хинолизиновые, бензоксазиновые, бензодиазиновые, пиридопиридиновые, хиноксалиновые, хиназолиновые, циннолиновые, фталазиновые, нафтиридиновые и птеридиновые группы, но не ограничиваются этим. Конкретные гетероарильные группы представляют собой гетероарильные группы, полученные из тиофена, пиррола, бензотиофена, бензофурана, индола, пиридина, хинолина, имидазола, оксазола и пиразина. Примеры репрезентативного арила, имеющего гетероатомы, содержащий замещение, включают следующее: где каждый W выбирают из C(R 54 ) 2 , NR 54 , О и S; и каждый Y выбирают из карбонила, NR 54 , О и S; и R 54 представляет собой независимо водород, С 1 -С 8 алкил, С 3 -С 10 циклоалкил, 4-10-членный гетероциклоалкил, С 6 -С 10 арил, 5-10-членный гетероарил. Примеры репрезентативных гетероарилов включают следующее: где каждый Y выбирают из карбонила, N, NR 55 , О и S; и R 55 независимо представляет собой водород, С 1 -С 8 алкил, С 3 -С 10 циклоалкил, 4-10-членный гетероциклоалкил, С 6 -С 10 арил, и 5-10-членный гетероарил. Используемый в этом документе термин "гетероциклоалкил" относится к 4-10-членному, стабильному гетероциклическому неароматическому кольцу и/или включает конденсированные кольца, содержащие один или более гетероатомов, независимо выбранных из N, О и S. Конденсированная гетероциклическая кольцевая система может включать карбоциклические кольца и требует включения только одного гетероциклического кольца. Примеры гетероциклических колец включают морфолин, пиперидин (например, 1-пиперидинил, 2-пиперидинил, 3-пиперидинил и 4-пиперидинил) , пирролидин (например, 1-пирролидинил, 2-пирролидинил и 3-пирролидинил), пирролидон, пиран (2Н-пиран или 4Н-пиран), дигидротиофен, дигидропиран, дигидрофуран, дигидротиазол, тетрагидрофуран, тетрагидротиофен, диоксан, тетрагидропиран (например, 4-тетрагидропиранил), имидазолин, имидазолидинон, оксазолин, тиазолин, 2-пиразолин, пиразолидин, пиперазин и N-алкилпиперазины, такие как N-метилпиперазин, но не ограничиваются этим. Дополнительные примеры включают тиоморфолин и его S-оксид и S,S-диоксид (в особенности тиоморфолин). Еще дополнительные примеры включают азетидин, пиперидон, пиперазон и N-алкилпиперидины, такие как N-метилпиперидин. Конкретные примеры гетероциклоалкильных групп показаны в следующих иллюстративных примерах: где каждый Y выбирают из CR 56 , C(R 56 ) 2 , NR 56 , О и S; и каждый Y выбирают из NR 56 , О и S; и R 56 независимо представляет водород, С 1 -С 8 алкил, С 3 -С 10 циклоалкил, 4-10-членный гетероциклоалкил, С 6 -С 10 арил, и 5-10-членный гетероарил. Эти гетероциклоалкильные кольца могут быть необязательно замещены одной или несколькими группами, выбранными из группы, состоящей из ацила, ациламиногруппы, ацилоксигруппы (-О-ацил или -OC(O)R 20 ), алкоксигруппы, алкоксикарбонила, алкоксикарбониламиногруппы (-NR"-алкоксикарбонил или -NH-C(O)-OR 27 ), аминогруппы, замещенной аминогруппы, аминокарбонила (амидогруппа или -C(O)-NR" 2 ), аминокарбониламиногруппы (-NR''-С(О)-NR'' 2 ), аминокарбонилоксигруппы (-O-С(О)-NR" 2 ), аминосульфонила, сульфониламиногруппы, арила, -О-арила, азидогруппы, карбоксила, цианогруппы, циклоалкила, галогена, гидроксигруппы, нитрогруппы, тиола, -S-алкила, -S-арила, -S(О)-алкила, -S(O)-арила, S(O) 2 -алкила и -S(О) 2 -арила. Группы-заместители включают карбонил или тиокарбонил, который обеспечивает, например, производные лактама и мочевины. "Гидроксигруппа" относится к радикальной группе -ОН. "Нитрогруппа" относится к радикальной группе -NO 2 . "Замещенный" относится к группе, в которой один или более атомов водорода, каждый независимо, заменены одинаковыми или различными заместителями. Среднему специалисту в области органического синтеза будет ясно, что максимальное число гетероатомов в стабильном, химически возможном (допустимом) гетероциклическом кольце, является ли оно ароматическим или неароматическим, определяется размером кольца, степенью ненасыщенности и валентностью гетероатомов. Как правило, гетероциклическое кольцо может иметь от одного до четырех гетероатомов, если гетероароматическое кольцо является химически возможным и стабильным. "Фармацевтически приемлемый" означает разрешенный или одобренный регуляторным органом правительства страны или штата или соответствующим органом стран, отличных от Соединенных Штатов, или то, что приведено в Фармакопее США или в других общепризнанных фармакопеях для применения на животных и в большей степени на людях. "Фармацевтически приемлемая соль" относится к соли соединения изобретения, которая является фармацевтически приемлемой и которая обладает желательной фармакологической активностью исходного (родоначального) соединения. В частности, такие соли являются нетоксичными и могут представлять собой соли присоединения неорганических и органических кислот и соли присоединения основания. Конкретно, такие соли включают: 1) соли присоединения кислоты, образованные с неорганическими кислотами, такими как хлористо-водородная кислота, бромисто-водородная кислота, серная кислота, азотная кислота, фосфорная кислота, и тому подобное; или образованные с органическими кислотами, такими как уксусная кислота, пропионовая кислота, капроновая кислота, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, 1,2-этандисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, 4-хлорбензолсульфоновая кислота, 2-нафталинсульфоновая кислота, 4-толуолсульфоновая кислота, камфорсульфокислота, 4-метилбицикло[2.2.2]-окт-2-ен-1-карбоновая кислота, глюкогептоновая кислота, 3-фенилпропионовая кислота, триметилуксусная кислота, третичная бутилуксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, гидроксинафтойная кислота, салициловая кислота, стеариновая кислота, муконовая кислота и тому подобное; или (2) соли, образованные либо при замене кислотного протона, присутствующего в исходном соединении, на ион металла, например ион щелочного металла, ион щелочно-земельного металла или ион алюминия; либо при его координации с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин, N-метилглюкамин и тому подобное. Соли дополнительно включают только в качестве примера натрий, калий, кальций, магний, аммоний, тетраалкиламмоний и тому подобное; и в том случае, когда соединение содержит основную функциональность, соли нетоксичных органических или неорганических кислот, такие как гидрохлорид, гидробромид, тартрат, мезилат, ацетат, малеат, оксалат и тому подобное. Термин "фармацевтически приемлемый катион" относится к приемлемому катионному противоиону кислотной функциональной группы. В качестве примера таких катионов могут быть приведены натриевые, калиевые, кальциевые, магниевые, аммониевые, тетраалкиламмониевые катионы и тому подобное. "Фармацевтически приемлемая среда для лекарства" относится к разбавителю, адъюванту, эксципиенту или носителю, с которым соединение изобретения вводят. "Субъект" включает людей. Термины "человек", "пациент" и "субъект" используют в этом документе взаимозаменяемо. "Предупреждение" или "предотвращение" относится к снижению риска приобретения или развития заболевания или нарушения (то есть к фактору, способствующему тому, чтобы по меньшей мере один из клинических симптомов заболевания не развивался у субъекта, который мог быть подвергнут воздействию вызывающего заболевание вещества, или который мог быть предрасположен данному заболеванию, с опережением начала заболевания). Термин "профилактика" относят к "предупреждению" и относится к мере или к процедуре, целью которой является предупреждение, а не лечение или вылечивание заболевания. Неограничивающие примеры профилактических мер могут включать введение вакцин; введение гепарина с низкой молекулярной массой госпитализированным пациентам с риском тромбоза вследствие, например, иммобилизации; и введение противомалярийного средства, такого как хлорохин, накануне визита географической зоны, где малярия является эндемической или где риск заражения малярией является высоким. "Процесс лечения" или "лечение" любого заболевания или нарушения относится в одном варианте осуществления к уменьшению интенсивности симптомов заболевания или нарушения (то есть к купированию заболевания или к снижению проявления степени или тяжести по меньшей мере одного из его клинических симптомов). В другом варианте осуществления "процесс лечения" или "лечение" относится к улучшению по меньшей мере одного физического параметра, который не может быть явным (заметным) для субъекта. В еще одном варианте осуществления "процесс лечения" и "лечение" относится к модулированию заболевания или нарушения либо физически (например, стабилизация явного симптома), физиологически, (например, стабилизация физического параметра), либо обоими методами. В дополнительном варианте осуществления "процесс лечения" и "лечение" относится к замедлению прогрессирования заболевания. Используемый в этом документе термин "состояние(я), включающее(ие) воспаление" относится к группе состояний, включающих ревматоидный артрит, остеоартрит, ювенильный идиопатический артрит, псориаз, аллергию дыхательных путей (например, астма, ринит), воспалительные заболевания кишечника (например, болезнь Крона, колит), обусловленные действием эндотоксина болезненные состояния (например, осложнения после шунтирования или хронические обусловленные действием эндотоксина состояния, способствующие возникновению хронической сердечной недостаточности) и сопутствующие заболевания, вовлекающие в патологический процесс хрящ, такие как заболевания суставов. В особенности термин относится к ревматоидному артриту, остеоартриту, аллергии дыхательных путей (например, астме) и воспалительным заболеваниям кишечника. Используемые в этом документе термины "состояние(я), вовлекающее(ие) в патологический процесс иммунную реакцию" или "аутоиммунные заболевания" используют взаимозаменяемо и используют при обращении к группе заболеваний, включающих обструктивное заболевание дыхательных путей, в том числе COPD (хроническую обструктивную болезнь легких), астму (например, наследственную (эндогенную) бронхиальную астму, приобретенную (экзогенную) бронхиальную астму, пылевую астму, детскую астму), в особенности хроническую или застарелую астму (например, поздную астму и гиперчувствительность дыхательных путей), бронхит, в том числе бронхиальную астму, системную эритематозную волчанку (SLE), множественный склероз, сахарный диабет типа I и осложнения, сопутствующие тому, атопическую экзему (атопический дерматит), контактный дерматит и дополнительные экзематозные дерматиты, воспалительное заболевание кишечника (например, болезнь Крона и неспецифический язвенный колит), атеросклероз и амиотрофический латеральный склероз. Особенно термин относится к COPD (хроническая обструктивная болезнь легких), астме, системной эриматозной волчанке, сахарному диабету типа I и воспалительному заболеванию кишечника. Использованный в этом документе термин "отторжение трансплантата" относится к острому или хроническому отторжению алло- или ксенографтов клеток, ткани или цельных органов, например панкреатического островка, стволовых клеток, костного мозга, кожи, мышцы, роговичной ткани, нейрональной ткани, сердца, легких, объединенного аппарата сердце-легкие, почки, печени, кишечника, поджелудочной железы, трахеи или пищевода, или к заболеваниям-реакциям "трансплантат против хозяина". Использованный в этом документе термин "пролиферативное(ые) заболевание(я)" относится к состояниям, таким как раковое заболевание (например, лейомиосаркома матки или рак предстательной железы), миелопролиферативные нарушения, называемые, в частности, как активирующие мутации JAK2-киназы (истинная полицитемия, эссенциальная тромбоцитемия и миелоидная метаплазия с миелофиброзом), лейкемия (например, острая миелоидная лейкемия и острая лимфобластная лейкемия), множественная миелома, псориаз, рестеноз, склеродермит или фиброз. В частности, термин относится к раковому заболеванию, лейкемии, множественной миеломе и псориазу. Использованный в этом документе термин "раковое заболевание" относится к злокачественному или доброкачественному росту клеток на коже или в органах тела, например и без ограничения, в молочной (грудной) железе, предстательной железе, легком, почке, поджелудочной железе, желудке или кишечнике. Раковое заболевание имеет тенденцию инфильтрировать в смежную ткань и распространяться (метастазировать) в отдаленных органах, например в кости, печени, легком или мозге. Используемый в этом документе термин "раковое заболевание" включает как типы клеток метастатической опухоли, такие как меланома, лимфома, лейкемия, фибросаркома, рабдомиосаркома и мастоцитома, но не ограниченные этим, так и типы карциномы тканей, такие как раковое заболевание прямой и ободочной кишки, рак предстательной железы, мелкоклеточный рак легкого и немелкоклеточный рак легкого, рак молочной железы, рак поджелудочной железы, рак мочевого пузыря, рак почек, рак желудка, глиобластома, первичный рак печени, рак яичников, рак предстательной железы и лейомиосаркома матки, но не ограниченные этим. Использованный в этом документе термин "лейкемия" относится к неопластическим заболеваниям крови и кроветворных органов. Такие заболевания могут вызывать дисфункцию костного мозга и иммунной системы, что делает организм "хозяина" высокочувствительным к инфекции и кровотечению. В частности, термин лейкемия относится к острой миелоидной лейкемии (AML) и к острой лимфобластной лейкемии (ALL). Использованный в этом документе термин "заболевания, включающие ухудшение ремоделирования хряща" или "заболевания, включающие анаболическую стимуляцию хондроцитов" включает состояния, такие как остеоартрит, псориатический артрит, ювенильный ревматоидный артрит, подагрический артрит, септический или инфекционный артрит, реактивный артрит, симпатическая рефлекторная дистрофия, альгодистрофия, синдром Tietze или костальный хондрит, фибромиалгия, остеохондрит, нейрогенный или невропатический артрит, артропатия, эндемические формы артрита, например деформирующий остеоартрит эндемический, болезнь Mseleni и болезнь Handigodu; дегенерация, являющаяся результатом фибромиалгии, системной эритематозной волчанки, склеродермы и анкилозирующего спондилита. Использованный в этом документе термин "врожденная(ые) мальформация(ии) хряща" включает состояния, такие как наследственный хондролиз, хондродисплазии и псевдохондродисплазии, в частности, но без ограничения, микротия, анотия, метафизарная хондродисплазия и сопутствующие тому нарушения. Использованный в этом документе термин "заболевание(я), связанное(ые) с гиперсекрецией IL6" включает состояния, такие как болезнь Кастлемана, множественная миелома, псориаз, саркома Капоши и/или мезангиальный пролиферативный гломерулонефрит. "Соединение(я) изобретения" и эквивалентные выражения, как подразумевается, охватывают соединения с формулой(ами), которая(ые) описана выше в данном документе, где выражение включает фармацевтически приемлемые соли. В том случае, когда в этом документе диапазоны относятся, например, но без ограничения, к С 1 -С 8 алкилу, то упоминание диапазона следует рассматривать как представление каждого члена вышеупомянутого диапазона. Все изотопные варианты соединений, обеспеченные в этом документе, радиоактивные или нерадиоактивные, как подразумевается, попадают в пределы объема изобретения. Также следует понимать, что соединения, которые имеют одинаковую молекулярную формулу, но отличаются природой или последовательностью связывания их атомов или расположением их атомов в пространстве, называют термином "изомеры". Изомеры, которые отличаются расположением их атомов в пространстве, называют термином "стереоизомеры". Стереоизомеры, которые не являются зеркальными изображениями друг друга, называют термином "диастереомеры", и стереоизомеры, которые являются неналагающимися зеркальными изображениями друг друга, называют термином "энантиомеры". В том случае, когда соединение имеет асимметрический центр, например, он является связанным с четырьмя различными группами, возможна пара энантиомеров. Энантиомер может быть охарактеризован абсолютной конфигурацией его асимметрического центра и описан правилами R- и S-последовательности по системе Кана и Прелога или направлением, в котором молекула вращает плоскость поляризованного света, и обозначен как правовращающий или левовращающий (то есть как (+)- или (-)-изомеры соответственно). Хиральное соединение может существовать либо как индивидуальный энантиомер, либо как смесь энантиомеров. Смесь, содержащую равные пропорции энантиомеров, называют как "рацемическая смесь". "Таутомеры" относятся к соединениям, которые имеют взаимозаменяемые формы конкретной структуры соединения и которые варьируются по смещению атомов водорода и электронов. Так, две структуры могут находиться в равновесии посредством движения п-электронов и атома (обычно Н). Например, енолы и кетоны являются таутомерами, так как они быстро взаимопревращаются в результате обработки либо кислотой, либо основанием. Другой пример таутомеризма представляет собой аци- и нитроформы фенилнитрометана, которые подобным образом образуются в результате обработки кислотой или основанием. Таутомерные формы могут быть подходящими в достижении оптимальной химической реакционной способности и биологической активности соединения, представляющего интерес. Соединения этого изобретения могут обладать одним или более асимметрическими центрами; такие соединения, таким образом, могут быть получены как индивидуальные (R)- или (S)-стереоизомеры или как их смеси. Если не указано иное, описание или название конкретного соединения в описании изобретения и пункты формулы, как подразумевается, включают как индивидуальные энантиомеры, так и их смеси, рацемические или иные. Способы определения стереохимии и разделение стереоизомеров являются хорошо известными в данной области. Настоящее изобретение основано на открытии того, что JAK-киназы являются полезными в лечении заболеваний, включающих деградацию хряща, деградацию кости и/или сустава, например остеоартрита; и/или состояний, включающих воспаление или иммунные реакции, таких как болезнь Крона, ревматоидный артрит, псориаз, аллергия дыхательных путей (например, астма, ринит), ювенильный идиопатический артрит, колит, воспалительные заболевания кишечника, обусловленные действием эндотоксина болезненные состояния (например, осложнения после шунтирования или хронические обусловленные действием эндотоксина состояния, способствующие возникновению, например, хронической сердечной недостаточности), заболевания, включающие ухудшение ремоделирования хряща (например, заболевания, включающие анаболическую стимуляцию хондроцитов), врожденные мальформации хряща, заболевания, связанные с гиперсекрецией IL6, и отторжение трансплантата (например, отторжение пересаженного органа). Ингибиторы JAK-киназ также могут найти применение в лечении пролиферативных заболеваний. В частности, ингибиторы JAK-киназ находят применение в лечении раковых заболеваний, в особенности лейкемий и солидных опухолей (например, лейомиосаркомы матки, рака предстательной железы), особенно заболеваний, включающих деградацию хряща, деградацию кости и/или сустава и/или воспаление, путем введения соединения изобретения. Соединения настоящего изобретения могут быть ингибиторами одного или более членов семейства JAK-киназ; конкретно они могут ингибировать активность одной или более из киназ: JAK1, JAK2, JAK3 и/или TYK2. В соответствии с вышеизложенным в первом аспекте изобретения раскрывают применение 1,2,4-триазоло[1,5-а]пиридиновых соединений, имеющих формулу (I) в которой в которой L1 выбирают из одинарной связи, -O-, -С(О)-, -С[=N(R 4a )]-, -N(R 4a )-, -CON(R 4a )-, -SO 2 N(R 4a )-, -S(O) 2 -, -N(R 4a )CO-, -CH 2 -N(R 4a )- или -N(R 4a )SO 2 -; R 3b независимо выбирают из замещенного или незамещенного С 6 -С 10 арила, замещенного или незамещенного С 3 -С 7 циклоалкила, замещенного или незамещенного 4-7-членного гетероциклоалкила, содержащего 1-3 гетероатома, выбранных из N, О или S, замещенного или незамещенного 5-10-членного гетероарила, содержащего 1-3 гетероатома, выбранных из N, О или S; или R 3b независимо выбирают из O-R 3c , NH-R 3c , CO-R 3c и CON(R 4a )-R 3c ; R 3c независимо выбирают из замещенного С 1 -С 6 алкила, замещенного или незамещенного С 6 -С 10 арила, замещенного или незамещенного С 3 -С 7 циклоалкила, замещенного или незамещенного 4-7-членного гетероциклоалкила, содержащего 1-3 гетероатома, выбранных из N, О или S, замещенного или незамещенного 5-10-членного гетероарила, содержащего 1-3 гетероатома, выбранных из N, О или S; каждый R 4a независимо выбирают из Н, С 1 -С 6 алкила, замещенного С 1 -С 6 алкила, С 3 -С 7 циклоалкила; причем заместители в определениях R 3b , R 3c , R 4a выбирают из группы, состоящей из галогена, циано, нитро, трифторметила, трифторметокси, азидо, -NR"'SO 2 R", -SO 2 NR"R"', -C(O)R", -C(O)OR", -OC(O)R", -NR"'C(O)R", -C(O)NR"R"', -NR"R"', -(CR"'R'") m OR"', где каждый R" независимо выбран из Н, С 1 -С 8 алкила, -(СН 2 ) t (С 6 -С 10 арила), -(СН 2 ) t (5-10-членного гетероарила, включающего 1-3 гетероатома, выбранных из N, О или S), -(СН 2 ) t (С 3 -С 10 циклоалкила) и -(СН 2 ) t (4-10-членного гетероциклоалкила, включающего 1-3 гетероатома, выбранных из N, О или S), где t представляет целое число 0-4; любая алкильная группа может быть замещена галогеном или ОН; и любой арил, гетероарил, циклоакил или гетероциклоалкил могут быть замещены замещенным или незамещенным С 1 -С 4 алкилом, галогеном, незамещенным C 1 -С 4 алкокси, незамещенным С 1 -С 4 галогеналкилом, незамещенным С 1 -С 4 гидроксиалкилом или незамещенным C 1 -С 4 галогеналкокси или ОН; каждый R"' независимо представляет Н или С 1 -С 6 алкил; n1 равно 0, 1, 2, 3 или 4; при условии, что i) когда L1 представляет собой -O-, -N(R 4a )-, -CH 2 -N(R 4a )-, -CON(R 4a )- или -SO 2 N(R 4a )- и R 3b является отличным от циклоалкила, арила или 5-10-членного гетероарила, тогда n1 равен 1, 2, 3 или 4; ii) когда L1 представляет собой связь, n1 равен 0 и R 3b представляет собой -OR 3c , тогда R 3c является отличным от Me или CF 3 ; или его фармацевтически приемлемая соль в лечении и/или предупреждении заболеваний, включающих деградацию хряща, деградацию кости и/или сустава; и/или состояний, включающих воспаление или иммунные реакции, таких как болезнь Крона, ревматоидный артрит, псориаз, аллергия дыхательных путей, ювенильный идиопатический артрит, колит, воспалительные заболевания кишечника, обусловленные действием эндотоксина болезненные состояния, заболевания, включающие ухудшение ремоделирования хряща, врожденные мальформации хряща, болезни Кастлемена, множественной миеломы, псориаза, саркомы Капоши и/или месангиального пролиферативного гломерунефрита и отторжение трансплантата; или пролиферативных заболеваний. В предпочтительном варианте осуществления раскрывают применение соединения в соответствии с вышеупомянутой формулой I, в которой L1 выбирают из одинарной связи, -O-, -С(О)-, -С[=N(R 4a )]-, -N(R 4a )-, -CON(R 4a )-, -SO 2 N(R 4a )-, -S(O) 2 -, -N(R 4a )CO- или -N(R 4a )SO 2 -, каждый R 1 независимо выбирают из незамещенного C 1 -С 6 алкила, незамещенного ацила, незамещенной ациламиногруппы, незамещенной С 1 -С 6 алкоксигруппы, незамещенной амидогруппы, незамещенной аминогруппы, незамещенного аминосульфонила, сульфоновой кислоты, сложного эфира сульфоновой кислоты, карбоксигруппы, цианогруппы, незамещенного С 3 -С 7 циклоалкила, незамещенного 4-7-членного гетероциклоалкила, галогена и гидроксила; или его фармацевтически приемлемые соли. В дополнительном аспекте изобретения R 4a представляет собой Н, замещенный или незамещенный С 1 -С 4 алкил. В одном варианте осуществления в отношении соединений формулы I R 4a представляет собой -(CH 2 ) n2 -R 6a , где n2 имеет значение 0, 1, 2 и R 6a представляет собой Н, CN, NMe 2 или тетрагидрофуранил. В еще одном варианте осуществления в отношении соединений формулы I R 4a представляет собой -СН(СН 3 )(CH 2 ) n2 -R 6a , где n2 имеет значение 0 или 1 и R 6a представляет собой Н или ОМе. В еще одном варианте осуществления в отношении соединений формулы I R 3b представляет собой OPh и O-(4-F-Ph). В одном варианте осуществления соединение соответствует формуле I и -Ph-L1-(CH 2 ) n1 -R 3b представляет собой где n2 равно n1; и R 3b и n1 являются такими же, как описаны для формулы I; и Су3 представляет собой замещенную или незамещенную азотсодержащую 4-7-членную гетероциклоалкильную группу. В другом варианте осуществления группа -Ph-L1-(CH 2 ) n1 -R 3b является такой же, как описана в предыдущих абзацах, и R 3b представляет собой незамещенный пиридил. В еше одном варианте осуществления группа -Ph-L1-(CH 2 ) n1 -R 3b является такой же, как описана в предыдущих абзацах, и R 3b представляет собой пиридил, замещенный одной или более группами, выбранными из галогена, цианогруппы, нитрогруппы, трифторметила, трифторметоксигруппы, азидогруппы, -NR"'SO 2 R", -SO 2 NR"R"', C(O)R", -C(O)OR", -OC(O)R", -NR'"C(O)R", -C(O)NR"R'", -NR"R'", -(CR"'R'") m OR"', где каждый R" независимо выбирают из Н, С 1 -С 8 алкила, -(СН 2 ) t (С 6 -С 10 арила)-, -(СН 2 ) t (5-10-членного гетероарила), -(СН 2 ) t (С 3 -С 10 циклоалкила) и -(СН 2 ) t (4-10-членного гетероциклоалкила), где t представляет собой целое число от 0 до 4. В еще одном варианте осуществления группа -Ph-L1-(CH 2 ) n1 -R 3b является такой же, как описана в предыдущих абзацах, и R 3b представляет собой пиридил, замещенный галогеном, цианогруппой, метилом или трифторметилом. В еше одном варианте осуществления группа -Ph-L1-(CH 2 ) n1 -R 3b является такой же, как описана в предыдущих абзацах, и n1 или n2 имеет значение 1. В еше одном варианте осуществления группа -Ph-L1-(CH 2 ) n1 -R 3b является такой же, как описана в предыдущих абзацах, и n1 или n2 имеет значение 2. В конкретном варианте осуществления раскрывают применение соединения в соответствии с формулой I, и R 3b выбирают из где каждый R 5a независимо представляет собой С 1 -С 4 алкил, галоген, CF 3 или фенил; R 5b представляет собой Н, арил, 5-10-членный гетероарил, гетероарил, С 3 -С 6 циклоалкил или 4-7-членный гетероциклоалкил; и m5 имеет значение 0, 1 или 2. В другом конкретном варианте осуществления соединения применение соединения, соответствующее формуле I, и Су3 выбирают из где каждый R 5a независимо представляет собой С 1 -С 4 алкил, галоген, CF 3 или фенил; R 5b представляет собой Н, арил, 5-10-членный гетероарил, гетероарил, С 3 -С 6 циклоалкил или 4-7-членный гетероциклоалкил; R 5c представляет собой Н или С 1 -С 4 алкил; m5 имеет значение 0, 1 или 2; n5 имеет значение 0, 1 или 2. В более особом варианте осуществления раскрывают применение соединения в соответствии с любой формулой из формул Va, Vb, Vc или Vd В более особом варианте осуществления раскрывают применение соединения в соответствии с любой формулой из формул VIa, VIc или VId В более особом варианте осуществления раскрывают применение соединения в соответствии с любой формулой из формул VIIa, VIIb, VIIc, VIId, VIIe или VIIf В другом более особом варианте осуществления раскрывают применение соединения в соответствии с любой формулой из формул VIIIa, VIIIb, VIIIc, VIIId, VIIIe, VIIIf, VIIIg, VIIIh, VIIIi, VIIIj, VIIIk или VIIIl В другом более особом варианте осуществления раскрывают применение соединения в соответствии с любой формулой из формул IXa, IXb, IXc, IXd, IXe, IXf, IXg, IXh, IXi, IXj, IXk или IXl В некоторых аспектах настоящее изобретение обеспечивает соединения формулы X или XI в которой R 3b представляет собой замещенный или незамещенный 4-7-членный гетероциклоалкил; при условии, что в том случае, когда соединение соответствует формуле X, гетероциклоалкильное кольцо является отличным от незамещенного морфолин-1-ила. В одном варианте осуществления соединение соответствует формуле X. В другом варианте осуществления соединение соответствует формуле XI. В одном конкретном варианте осуществления в отношении соединений в соответствии с формулой X R 3b представляет собой незамещенный азетидинил, пирролидинил, пиперидинил, пиперазинил, азепинил, пирролидонил, пиранил, дигидротиофенил, дигидропиранил, дигидрофуранил, дигидротиазолил, тетрагидрофуранил, тетрагидротиофенил, диоксанил, тетрагидропиранил, имидазолинил, имидазолидинонил, оксазолинил, тиазолинил, 2-пиразолинил, пиразолидинил, тиоморфолинил-S-оксид и тиоморфолинил-S,S-диоксид пиперидонил или пиперазонил. В одном конкретном варианте осуществления в отношении соединений формулы X R 3b представляет собой незамещенный азетидин-1-ил, пирролидин-1-ил, пиперидин-1-ил, тиоморфолин-1-ил-S,S-диоксид, пиперазин-1-ил или азепин-1-ил. В одном конкретном варианте осуществления в отношении соединений формулы X R 3b представляет собой незамещенный азетидин-1-ил. В одном конкретном варианте осуществления в отношении соединений формулы X R 3b представляет собой незамещенный пирролидин-1-ил. В одном конкретном варианте осуществления в отношении соединений формулы X R 3b представляет собой незамещенный пиперидин-1-ил или пиперазин-1-ил. В одном конкретном варианте осуществления в отношении соединений формулы X R 3b представляет собой незамещенный тиоморфолин-1-ил-S,S-диоксид. В одном конкретном варианте осуществления в отношении соединений формулы X R 3b представляет собой незамещенный азепин-1-ил. В одном конкретном варианте осуществления в отношении соединений формулы X R 3b представляет собой азетидин-1-ил, пирролидин-1-ил, пиперидин-1-ил, морфолин-1-ил, пиперазин-1-ил или азепин-1-ил; замещенный одной или более группами, выбранными из С 1 -С 4 алкила, С 1 -С 4 галогеналкила, цианогруппы, аминогруппы, диалкиламиногруппы, диалкиламинометила, гидроксигруппы, галогена, ацила, ациламиногруппы, С 1 -С 4 гидроксиалкила, С 1 -С 4 алкоксигруппы, карбоксамидогруппы и С 1 -С 4 диалкилкарбоксамидогруппы. В одном конкретном варианте осуществления в отношении соединений формулы X R 3b представляет собой азетидин-1-ил, пирролидин-1-ил, пиперидин-1-ил, морфолин-1-ил, пиперазин-1-ил или азепин-1-ил; замещенный Me, CF 3 , F, Cl, дифтором, диметилом, гидроксигруппой, цианогруппой, диметиламиногруппой, диметиламинометилом, гидроксиметилом, карбоксамидогруппой, N,N-диметилкарбоксамидогруппой, метоксигруппой, этоксигруппой или 2,2,2-трифторэтилом. В одном конкретном варианте осуществления в отношении соединений формулы XI R 3b представляет собой незамещенный азетидинил, пирролидинил, пиперидинил, пиперазинил, азепинил, пирролидонил, пиранил, дигидротиофенил, дигидропиранил, дигидрофуранил, дигидротиазолил, тетрагидрофуранил, тетрагидротиофенил, диоксанил, тетрагидропиранил, имидазолинил, имидазолидинонил, оксазолинил, тиазолинил, 2-пиразолинил, пиразолидинил, морфолинил, тиоморфолинил-S-оксид и тиоморфолинил-S,S-диоксид пиперидонил или пиперазонил. В одном конкретном варианте осуществления в отношении соединений формулы XI R 3b представляет собой незамещенный азетидин-1-ил, пирролидин-1-ил, пиперидин-1-ил, морфолинил, тиоморфолин-1-ил-S,S-диоксид, пиперазин-1-ил или азепин-1-ил. В одном конкретном варианте осуществления в отношении соединений формулы XI R 3b представляет собой незамещенный азетидин-1-ил. В одном конкретном варианте осуществления в отношении соединений формулы XI R 3b представляет собой незамещенный пирролидин-1-ил. В одном конкретном варианте осуществления в отношении соединений формулы XI R 3b представляет собой незамещенный пиперидин-1-ил или пиперазин-1-ил. В одном конкретном варианте осуществления в отношении соединений формулы XI R 3b представляет собой незамещенный тиоморфолин-1-ил-S,S-диоксид. В одном конкретном варианте осуществления в отношении соединений формулы XI R 3b представляет собой незамещенный морфолин-1-ил. В одном конкретном варианте осуществления в отношении соединений формулы XI R 3b представляет собой незамещенный азепин-1-ил. В одном конкретном варианте осуществления в отношении соединений формулы XI R 3b представляет собой азетидин-1-ил, пирролидин-1-ил, пиперидин-1-ил, морфолин-1-ил, пиперазин-1-ил или азепин-1-ил; замещенный одной или более группами, выбранными из С 1 -С 4 алкила, С 1 -С 4 галогеналкила, цианогруппы, аминогруппы, диалкиламиногруппы, диалкиламинометила, гидроксигруппы, галогена, ацила, ациламиногруппы, С 1 -С 4 гидроксиалкила, С 1 -С 4 алкоксигруппы, карбоксамидогруппы и С 1 -С 4 диалкилкарбоксамидогруппы. В одном конкретном варианте осуществления в отношении соединений формулы XI R 3b представляет собой азетидин-1-ил, пирролидин-1-ил, пиперидин-1-ил, морфолин-1-ил, пиперазин-1-ил или азепин-1-ил; замещенный Me, CF 3 , F, Cl, дифтором, диметилом, гидроксигруппой, цианогруппой, диметиламиногруппой, диметиламинометилом, гидроксиметилом, карбоксамидогруппой, N,N-диметилкарбоксамидогруппой, метоксигруппой, этоксигруппой или 2,2,2-трифторэтилом. В одном варианте осуществления в отношении соединений формулы I выбирают соединение из соединений, приведенных в качестве примера в табл. 1. В одном варианте осуществления соединение изобретения не является изотопным вариантом. В одном аспекте соединение изобретения в соответствии с любым вариантом осуществления из вариантов осуществления, описанных в этом документе, присутствует в форме свободного основания. В одном аспекте соединение изобретения в соответствии с любым вариантом осуществления из вариантов осуществления, описанных в этом документе, представляет собой фармацевтически приемлемую соль. Хотя точно описанные группы для каждого варианта осуществления были перечислены выше в общем порядке по отдельности, соединение изобретения включает группу, в которой несколько или каждый вариант осуществления в соответствии с вышеупомянутой формулой, а также в соответствии с другими представленными в этом документе формулами, выбирают из одного или более конкретных членов или групп, обозначенных, соответственно, для каждой переменной. Таким образом, подразумевается, что изобретение включает все комбинации таких вариантов осуществления в пределах его объема. Другие производные соединений изобретения имеют активность как в их кислотных формах, так и в формах производных кислот, но форма, чувствительная к кислоте, часто дает преимущества растворимости, совместимости с тканью или отсроченного высвобождения в организме млекопитающего (см., Bundgard, H., Design of Prodrugs, pp. 7-9, 21-24, Elseivier, Amsterdam 1985). При применении лекарственных препаратов соединения изобретения обычно вводят в форме фармацевтической композиции. Такие композиции могут быть приготовлены способом, хорошо известным в фармацевтической области, и содержат по меньшей мере одно активное соединение. Как правило, соединения этого изобретения вводят в фармацевтичски эффективном количестве. Количество соединения, собственно вводимое, обычно будет определяться терапевтом в свете релевантных обстоятельств, включающих состояние, которое подлежит лечению, выбранный путь введения, вводимое соединение как таковое, возраст, вес и реакция конкретного пациента, тяжесть симптомов у конкретного пациента и тому подобное. Фармацевтические композиции, включающие соединения изобретения, могут быть введены разнообразными путями, включающими пероральный, ректальный, трансдермальный, подкожный, интраартикулярный, внутривенный, внутримышечный и интраназальный пути. В зависимости от предполагаемого пути доставки соединения этого изобретения предпочтительно составляют в смеси либо в форме инъекционных или пероральных композиций, либо в форме целебных мазей, лосьонов или пластырей, все из которых для трансдермального введения. Композиции для перорального введения могут иметь форму нерасфасованных жидких растворов, или суспензий, или нерасфасованных порошков. Чаще, однако, композиции представлены в стандартных (дозированных) лекарственных формах для облегчения точного дозирования лекарства. Термин "стандартные (дозированные) лекарственные формы" относится к физически дискретным единицам, подходящим в качестве единичных дозировок для человеческих субъектов и других млекопитающих, где каждая единица содержит заданное количество активного вещества, вычисленное для получения желательного терапевтического эффекта, в сочетании с подходящим(ей) фармацевтическим(ой) эксципиентом, средой для лекарства или носителем. Обычные стандартные (дозированные) лекарственные формы включают предварительно заполненные, заранее измеренные ампулы или шприцы с жидкими композициями или пилюли, таблетки, капсулы или тому подобное в случае твердых композиций. В таких композициях соединение фурансульфокислоты представляет собой обычно второстепенный компонент (от приблизительно 0,1 до приблизительно 50 мас.% или предпочтительно от приблизительно 1 до приблизительно 40 мас.%), где остальная часть представляет собой раличные среды для лекарства или носители и технологические добавки, полезные для формования желательной дозированной формы. Жидкие формы, подходящие для перорального введения, могут включать подходящую водную или неводную среду для лекарства с буферными растворами, с добавками, способствующими суспендированию и распылению лекарства, с окрашивающими веществами, ароматизирующими добавками и тому подобным. Твердые формы могут включать, например, любые из следующих ингредиентов, или соединений аналогичной природы: связующее вещество, такое как микрокристаллическая целлюлоза, трагакантовая камедь или желатин; эксципиент, такой как крахмал или лактоза, вещество для улучшения распадаемости таблеток, такое как альгиновая кислота, Primogel или кукурузный крахмал; скользящее вещество, такое как стеарат магния; регулятор сыпучести (глидант), такой как коллоидный диоксид кремния; подслащивающее вещество, такое как сахароза или сахарин; или ароматизирующее вещество, такое как мята перечная (мятное масло), метилсалицилат или экстракт из апельсиновой корки. Инъекционные композиции обычно основаны на инъекционном стерильном солевом растворе, или забуференном фосфатом физиологическом растворе, или на других инъекционных носителях, известных в данной области. Исходя из вышесказанного, активное соединение в таких композициях обычно представляет собой второстепенный компонент, часто составляющий от приблизительно 0,05 до 10 мас.%, где остальная часть представляет собой инъекционный носитель или тому подобное. Трансдермальные композиции обычно составляют в форме местнодействующей(его) мази или крема, содержащей(его) активный(е) ингредиент(ы), как правило, в количестве, находящемся в диапазоне от приблизительно 0,01 до приблизительно 20 мас.%, предпочтительно от приблизительно 0,1 до приблизительно 20 мас.%, предпочтительно от приблизительно 0,1 до приблизительно 10 мас.% и более предпочтительно от приблизительно 0,5 до приблизительно 15 мас.%. При составлении смеси в форме мази активные ингредиенты обычно будут скомбинированы либо с парафинистой, либо с водорастворимой или смешивающейся с водой основой мази. Альтернативно, активные ингредиенты могут быть составлены в крем, например, с основой крема типа "масло в воде". Такие трансдермальные составы хорошо известны в данной области и, как правило, включают дополнительные ингредиенты для улучшения проникновения в кожу, стабильности активных ингредиентов или состава. Соединения этого изобретения также могут быть введены с помощью трансдермального приспособления. Соответственно, трансдермальное введение может быть выполнено с использованием пластыря либо резервуарного или пористомембранного типа, либо из набора твердых матриц. Вышеописанные компоненты для перорально вводимых, инъекционных или местно вводимых композиций являются лишь репрезентативными. Другие вещества, а также технологические методы и тому подобное изложены в части 8 книги Remington's Pharmaceutical Sciences, 17 th edition, 1985, Mack Publishing Company, Easton, Pennsylvania, которая включена в этот документ путем ссылки. Соединения этого изобретения также могут быть введены в формах с замедленным высвобождением или из систем доставки лекарственного средства с замедленным высвобождением. Описание репрезентативных материалов с замедленным высвобождением могут быть обнаружены в книге Remington's Pharmaceutical Sciences. Следующие примеры составов иллюстрируют репрезентативные фармацевтические композиции, которые могут быть получены в соответствии с этим изобретением. Настоящее изобретение, однако, не ограничено следующими фармацевтическими композициями. Состав 1. Таблетки. Соединение изобретения может быть смешано в виде сухого порошка с сухим желатиновым связующим в приблизительном массовом соотношении 1:2. Незначительное количество стеарата магния добавляют в качестве скользящего вещества. Смесь формуют : в таблетки массой 240-270 мг (80-90 мг активного амидного соединения на таблетку) в таблеточном прессе. Состав 2. Капсулы. Соединение изобретения может быть смешано в виде сухого порошка с крахмальным разбавителем в приблизительном массовом соотношении 1:1. Смесь помещают в капсулы массой 250 мг (125 мг активного амидного соединения на капсулу). Состав 3. Жидкость. Соединение изобретения (125 мг) может быть смешано с сахарозой (1,75 г) и ксантановой камедью (4 мг), и получающаяся в результате смесь может быть размешана, пропущена через сетчатый фильтр N 10 меш, и затем смешана с заранее приготовленным раствором микрокристаллической целлюлозы и карбоксиметилцеллюлозы натрия (11:89, 50 мг) в воде. Бензоат натрия (10 мг), ароматизирующее вещество, и окрашивающее вещество разбавляют водой и добавляют при перемешивании. Затем при перемешивании может быть добавлено достаточное количество воды. Достаточное количество воды затем добавляют с получением общего объема 5 мл. Состав 4. Таблетки. Соединение изобретения может быть смешано в виде сухого порошка с сухим желатиновым связующим в приблизительном массовом соотношении 1:2. Незначительное количество стеарата магния добавляют в качестве скользящего вещества. Смесь формуют в таблетки массой 450-900 мг (150-300 мг активного амидного соединения на таблетку) в таблеточном прессе. Состав 5. Раствор для инъекций. Соединение изобретения может быть растворено или суспендировано в инъекционной водной среде на основе забуференного стерильного физиологического раствора в концентрации приблизительно 5 мг/мл. Состав 6. Местнодействующий. Стеариловый спирт (250 г) и белый вазелин (250 г) могут быть расплавлены при приблизительно 75 °С и затем может быть добавлена смесь соединения изобретения (50 г), метилпарабена (0,25 г), пропилпарабена (0,15 г), лаурилсульфата натрия (10 г) и пропиленгликоля (120 г), растворенная в воде (приблизительно 370 г), получающуюся в результате смесь перемешивают до тех пор, пока она не застынет. Соединения настоящего изобретения используют в качестве терапевтических средств для лечения состояний у млекопитающих, которые причинно связаны или обусловлены аберрантной активностью JAK-киназ; в особенности состояний, связанных с аберрантной активностью одной или более киназ из JAK1-, JAK2-, JAK3- и/или TYK2-киназ. В связи с вышеизложенным соединение изобретения и фармацевтические композиции, включающие соединения этого изобретения, находят применение в качестве терапевтических средств для предупреждения и/или лечения заболеваний, включающих деградацию хряща, деградацию кости и/или сустава, например остеоартрита; и/или состояний, включающих воспаление или иммунные реакции, таких как болезнь Крона, ревматоидный артрит, псориаз, аллергия дыхательных путей (например, астма, ринит), ювенильный идиопатический артрит, колит, воспалительные заболевания кишечника, обусловленные действием эндотоксина болезненные состояния (например, осложнения после шунтирования или хронические обусловленные действием эндотоксина состояния, способствующие возникновению, например, хронической сердечной недостаточности), заболевания, включающие ухудшение ремоделирования хряща (например, заболевания, включающие анаболическую стимуляцию хондроцитов), врожденные мальформации хряща, заболевания, связанные с гиперсекрецией IL6, и отторжение трансплантата (например, отторжение пересаженного органа). Ингибиторы JAK-киназ также могут найти применение в лечении пролиферативных заболеваний. В частности, ингибиторы JAK-киназ находят применение в лечении раковых заболеваний, в особенности, лейкемий и солидных опухолей (например, лейомиосаркомы матки, рака предстательной железы). В особенности состояния выбирают из воспалительных состояний, состояний, связанных с деградацией хряща и/или сустава у млекопитающих, включая людей. В другом варианте осуществления соединения и фармацевтические композиции, включающие их, находят применение в качестве терапевтических средств для предупреждения и/или лечения пролиферативных нарушений у млекопитающих, включая людей. В особом варианте осуществления соединение изобретения и их фармацевтические композиции находят применение в качестве терапевтических средств для предупреждения и/или лечения ракового заболевания у млекопитающих, включая людей. В другом аспекте настоящее изобретение обеспечивает соединение изобретения для применения в лечении, предупреждении или для профилактики состояния, включающего аутоиммунную реакцию или аутоиммунное заболевание. В конкретном варианте осуществления аутоиммунное заболеваниие выбирают из хронического обструктивного заболевания легких, астмы, системной эриматозной волчанки, сахарного диабета типа I и воспалительного заболевания кишечника. В другом аспекте настоящее изобретение обеспечивает применение соединения изобретения в лечении, предупреждении или для профилактики заболеваний, включающих ухудшение ремоделирования хряща (например, состояние, сопутствующее анаболической стимуляции хондроцитов, или заболевания, вовлекающие в патологический процесс анаболическую стимуляцию хондроцитов), например остеоартрита, псориатического артрита, ювенильного ревматоидного артрита, подагрического артрита, септического или инфекционного артрита, реактивного артрита, симпатической рефлекторной дистрофии, альгодистрофии, синдрома Tietze или костального хондрита, фибромиалгии, остеохондрита, нейрогенного или невропатического артрита, артропатии, эндемических форм артрита, например деформирующего остеоартрита эндемического, болезни Mseleni и болезни Handigodu; дегенерации, являющейся результатом фибромиалгии, системной эритематозной волчанки, склеродермы и анкилозирующего спондилита. В другом аспекте настоящее изобретение обеспечивает применение соединения изобретения в лечении, предупреждении или для профилактики врожденных мальформаций хряща, включающих наследственный хондролиз, хондродисплазии и псевдохондродисплазии, в частности, но без ограничения, микротию, анотию, метафизарную хондродисплазию и сопутствующие тому нарушения. В другом аспекте это изобретение обеспечивает применение соединения изобретения в лечении, предупреждении или для профилактики состояния, включающего воспаление. В другом аспекте настоящее изобретение обеспечивает соединение изобретения для применения в лечении, предупреждении или для профилактики заболеваний или нарушений, которые опосредованы воспалением или приводят к воспалению, таких как, например, ревматоидный артрит и остеоартрит, аллергия дыхательных путей (например, астма, ринит), ювенильный идиопатический артрит, колит, воспалительные заболевания кишечника, обусловленные действием эндотоксина болезненные состояния (например, осложнения после шунтирования или хронические обусловленные действием эндотоксина состояния, способствующие возникновению, например хронической сердечной недостаточности) и сопутствующие тому заболевания, вовлекающие в патологический процесс хрящ, например заболевание суставов. В конкретном варианте осуществления состояние, включающее воспаление, выбирают из ревматоидного артрита, остеоартрита, аллергии дыхательных путей (например, астма) и воспалительных заболеваний кишечника. В другом аспекте настоящее изобретение обеспечивает применение соединения изобретения в лечении, предупреждении или для профилактики пролиферативного заболевания, в частности ракового заболевания (например, солидных опухолей, таких как лейомиосаркома матки или рак предстательной железы), лейкемий (например, AML или ALL), множественная миелома и/или псориаз. В другом аспекте настоящее изобретение обеспечивает соединение изобретения для применения в лечении, предупреждении или для профилактики ракового заболевания (например, солидных опухолей, таких как лейомиосаркома матки или рак предстательной железы) и/или лейкемий. В другом аспекте настоящее изобретение обеспечивает применение соединения изобретения в лечении, предупреждении или для профилактики заболеваний, связанных с гиперсекрецией IL6, в частности болезни Кастлемана или мезангиального пролиферативного гломерулонефрита. В другом аспекте настоящее изобретение обеспечивает применение соединения изобретения в лечении, предупреждении или для профилактики отторжения трансплантата. В конкретном варианте осуществления изобретение обеспечивает способы лечения отторжения пересаженных органов. В качестве дополнительного аспекта изобретения обеспечивают применение соединения изобретения в качестве фармацевтического препарата, главным образом, в лечении или предупреждении вышеупомянутых состояний и заболеваний. Также обеспеченным в этом документе является применение соединений настоящего изобретения в изготовлении лекарственного препарата для лечения или предупреждения одного из вышеупомянутых состояний и заболеваний. Уровни дозы инъекции колеблются в диапазоне от приблизительно 0,1 до по меньшей мере 10 мг/кг/ч, все в течение периода времени от приблизительно 1 до приблизительно 120 ч и в особенности в течение периода времени 24-96 ч. Предварительно заполняемый болюс в количестве от приблизительно 0,1 до приблизительно 10 мг/кг или более также может быть введен для достижения адекватных уровней равновесной (стабильной) концентрации. Максимальная общая доза, как ожидают, не превышает приблизительно 2 г/день для пациента-человека массой 40-80 кг. Для предупреждения и/или лечения длительных (хронических) состояний, таких как дегенеративные состояния, программа лечения растягивается на многие месяцы или годы, поэтому пероральное введение дозы является предпочтительным для удобства пациентов и для переносимости пациентами. При пероральном введении дозы одна-пять и особенно две-четыре и обычно три пероральные дозы в день представляют собой репрезентативные схемы. С использованием этих протоколов введения дозы каждая доза обеспечивает от приблизительно 0,01 до приблизительно 20 мг/кг соединения изобретения, где каждая из конкретных доз обеспечивает от приблизительно 0,1 до приблизительно 10 мг/кг и в особенности приблизительно 1-5 мг/кг. Дозы при трансдермальном введении, как правило, выбирают для обеспечения аналогичных или более низких уровней в крови, чем достигаются с использованием доз при инъекционном введении. При применении для предупреждения начала воспалительного состояния соединения этого изобретения будут вводиться пациенту с риском развития состояния, обычно по заключению или под присмотром лечащего врача, при уровнях дозировки, описанных выше. Пациенты с риском развития конкретного состояния, как правило, включают пациентов, которые имеют семейный анамнез этого состояния, или пациенты, которые были идентифицированы с помощью генетического тестирования или скрининга как особенно предрасположенные к развитию этого состояния. Соединения изобретения могут быть введены в качестве единственного активного вещества, или они могут быть введены в комбинации с другими веществами, включая другие соединения, которые проявляют аналогичную или сходную терапевтическую активность и которые, как определено, являются безопасными и эффективными для такого объединенного введения. В конкретном варианте осуществления совместное введение двух (или более) веществ предусматривает значительно более низкие дозы каждого вещества, которое должно быть применено, посредством чего снижаются видимые побочные эффекты. В одном варианте осуществления соединение изобретения совместно вводят с другим терапевтическим средством для лечения и/или предупреждения заболевания, включающего воспаление; конкретные вещества включают иммунорегуляторные средства, например азатиоприн, кортикостероиды (например, преднизолон или дексаметазон), циклофосфамид, циклоспорин А, такролимус, микофенолат-мофетил, муромонаб-CD3 (OKT3 (антигенный маркер популяции зрелых периферических Т-лимфоцитов у человека), например Orthocolone ®), ATG (антитимоцит-глобулин), аспирин, ацетаминофен, ибупрофен, напроксен и пироксикам, но не ограничиваются этим. В одном варианте осуществления соединение изобретения совместно вводят с другим терапевтическим средством для лечения и/или предупреждения артрита (например, ревматоидный артрит); конкретные вещества включают аналгетики, нестероидные противовоспалительные лекарственные средства (NSAIDS), стероиды, синтетические базисные препараты для лечения ревматоидного артрита (DMARDS, например, но без ограничения, метотрексат, лефлуномид, сульфазалазин, ауранофин, ауротиомалат натрия, пеницилламин, хлорокин, гидроксихлорокин, азатиоприн, и циклоспорин), и биологические базисные препараты для лечения ревматоидного артрита (DMARDS, например, но без ограничения, инфликсимаб, этанерцепт, адалимумаб, ритуксимаб и абатацепт), но не ограничиваются этим. В одном варианте осуществления соединение изобретения совместно вводят с другим терапевтическим средством для лечения и/или предупреждения пролиферативных нарушений; конкретные вещества включают метотрексат, леуковорин, адриамицин, пренизон, блеомицин, циклофосфамид, 5-фтороурацил, паклитаксел, доцетаксел, винкристин, винбластин, винорелбин, доксорубицин, таксифен, торемифен, мегестрол-ацетат, анастрозол, гозерелин, моноклональное анти-HER2-антитело (например, Герцептин ™), капецитабин, ралоксифен-гидрохлорид, ингибиторы рецепторов эпидермального фактора роста (EGFR, например Iressa ®, Tarceva ™, Erbitux ™), ингибиторы факторов роста эндотелия сосудов (VEGF, например Avastin ™), ингибиторы протеасомы (например, Velcade ™), Glivec ® или ингибиторы hsp90 (например, 17-AAG), но не ограничиваются этим. Кроме того, соединение изобретения может быть введено в комбинации с другими методами лечения, включающими лучевую терапию или хирургию, но не ограниченными этим. В конкретном варианте осуществления пролиферативное нарушение выбирают из ракового заболевания, миелопролиферативного заболевания или лейкемии. В одном варианте осуществления соединение изобретения совместно вводят с другим терапевтическим средством для лечения и/или предепреждения аутоиммунных заболеваний; конкретные вещества включают глюкокортикоиды, цитостатические вещества (например, пуриновые аналоги), алкилирующие агенты (например, азотистые иприты (циклофосфамид), нитрозомочевины, соединения платины и другие), антиметаболиты (например, метотрексат. Азатиоприн и меркаптопурин), цитотоксические антибиотики (например, дактиномицин, антрациклины, митомицин С, блеомицин и митрамицин), антитела (например, моноклональные анти-CD20-, анти-CD25- или анти-CD3- (OTK3) антитела, Atgam ® и Thymoglobuline ®), циклоспорин, такролимус, рапамицин (сиролимус), интерфероны (например, IFN- β), TNF-связывающие белки (например, инфликсимаб (Remicade), этанерцепт (Enbrel), или адалимумаб (Humira)), микофенолат, Fingolimod, Myriocin, но не ограничиваются этим. В одном варианте осуществления соединение изобретения совместно вводят с другим терапевтическим средством для лечения и/или предупреждения отторжения трансплантата; конкретные вещества включают ингибиторы кальциневрина (например, циклоспорин или такролимус (FK506)), ингибиторы mTOR (например, сиролимус, эверолимус), антипролиферативные препараты (например, азатиоприн, микофенольная кислота), кортикостероиды (например, преднизолон, гидрокортизон), антитела (например, моноклональные анти-IL-2R α-рецептор-антитела, базиликсимаб, даклизумаб), поликлональные анти-Т-клеточные антитела (например, антитимоцит-глобулин (ATG), антилимфоцит-глобулин (ALG)), но не ограничиваются этим. В одном варианте осуществления соединение изобретения совместно вводят с другим терапевтическим средством для лечения и/или предупреждения астмы, и/или ринита, и/или COPD (хроническое обструктивное заболевание легких); конкретные вещества включают антагонисты β 2 -адреноцепторов (например, салбутамол, левалбутерол, тербуталин и битолтерол), эпинефрин (в виде ингаляций или в таблетках), антихолинергические вещества (например, ипратропий-бромид), глюкокортикоиды (пероральные или в виде ингаляций), длительно действующие β 2 -агонисты (например, салметерол, формотерол, бамбутерол и перорально вводимый албутерол замедленного высвобождения), комбинации ингалируемых стероидов и длительно действующих бронхолитических веществ (например, флутиказон/салметерол, будезонид/формотерол), антагонисты рецепторов лейкотриена и ингибиторы синтеза (например, монтелукаст, зафирлукаст и зилеутон), ингибиторы высвобождения медиаторов (например, кромогликат и кетотифен), биологические регуляторы реакции IgE (например, омализумаб), антигистаминные препараты (например, цетеризин, циннаризин, фексофенадин), вазоконстрикторные препараты (например, оксиметазолин, ксилометазолин, нафазолин и трамазолин), но не ограничиваются этим. Кроме того, соединение изобретения может быть введено в комбинации с методами неотложной терапии астмы и/или хронического обструктивного заболевания легких, где такие методы терапии включают введение кислорода или гелиево-кислородной дыхательной среды, введение распыляемого салбутамола или тербуталина (необязательно в комбинации с антихолинергическим веществом (например, ипратропий)), системных стероидов (пероральные или внутривенные, например преднизон, преднизолон, метилпреднизолон, дексаметазон или гидрокортизон), внутривенного салбутамола, неспецифических β-агонистов, инъекционных или ингаляционных препаратов (например, эпинефрин, изоэтарин, изопротеренол, метапротеренол), антихолинергических препаратов (внутривенные или распыляемые, например, гликопирролат, атропин, ипратропий), метилксантинов (теофиллин, аминофиллин, бамифиллин), ингаляционных анестезирующих препаратов, которые имеют бронхолитический эффект (например, изофлуран, галотан, энфлуран), кетамина, внутривенного сульфата магния. В одном варианте осуществления соединение изобретения совместно вводят с другим терапевтическим средством для лечения и/или предупреждения IBD (воспалительное заболевание кишечника); конкретные вещества включают глюкокортикоиды (например, преднизон, буденозид), синтетические модифицирующие заболевания, иммуномодулирующие препараты (например, метотрексат, лефлуномид, сульфасалазин, мезалазин, азатиоприн, 6-меркаптопурин и циклоспорин) и биологические модифицирующие заболевания, иммуномодулирующие препараты (инфликсимаб, адалимумаб, ритуксимаб и абатацепт), но не ограничиваются этим. В одном варианте осуществления, соединение изобретения совместно вводят с другим терапевтическим средством для лечения и/или предупреждения SLE (системная эриматозная волчанка); конкретные вещества включают базисные препараты для лечения ревматоидного артрита (DMARDs), такие как противомалярийные препараты (например, плакенил, гидроксихлорокин), иммунодепрессанты (например, метотрексат и азатиоприн), циклофосфамид и микофенольная кислота; иммунодепрессивные лекарственные средства и анальгетики, такие как нестероидные противовоспалительные лекарственные средства, опиаты (например, декстропропоксифен и кокодамол), опиоиды (например, гидрокодон, оксикодон, МС-контин или метадон) и фентаниловый трансдермальный пластырь (Дюрогезик), но не ограничиваются этим. В одном варианте осуществления соединение изобретения совместно вводят с другим терапевтическим средством для лечения и/или предупреждения псориаза; конкретные вещества включают препараты для местного лечения, такие как омывающие жидкости, увлажняющие средства, лекарственные кремы и мази, содержащие деготь, дитранол (антралин), кортикостероиды, например дезоксиметазон (Topicort), флуоцинонид, аналоги витамина D 3 (например, кальципотриол), ретиноиды на основе масла арганы (этретинат, ацитретин, тазаротен), препараты для системного лечения, такие как метотрексат, циклоспорин, ретиноиды, тиогуанин, гидроксимочевина, сульфасалазин, микофенолат мофетил, азатиоприн, такролимус, сложные эфиры фумаровой кислоты, или биологические препараты, такие как Amevive, Enbrel, Humira, Remicade, Raptiva, и устекинумаб (блокаторы IL-12 и IL-23), но не ограничиваются этим. Кроме того, соединение изобретения может быть введено в комбинации с другими методами терапии, включающими фототерапию или фотохимиотерапию (например, сочетание фототерапии и ПУФА (псорален + УФ-излучение)-терапии), но не ограниченными этим. С помощью термина "совместное введение" включают любое средство доставки двух или более терапевтических средств пациенту в рамках одной и той же программы лечения, что будет очевидно для специалиста в данной области. Хотя два или более средства могут быть введены одновременно в одном составе, это не является решающим фактором. Вещества могут быть введены различными составами в разное время. Соединения изобретения могут быть получены из легкодоступных исходных веществ с использованием следующих общих способов и методик. Будет ясно, что в том случае, когда даны обычные или предпочтительные условия процессов (то есть температуры реакций, продолжительности, мольные соотношения реагентов, растворители, параметры давления и так далее), другие условия процесса также могут быть использованы, если не указано иное. Оптимальные реакционные условия могут варьироваться в зависимости от конкретных используемых реагентов или растворителей, но такие условия могут быть определены специалистом в данной области с помощью общепринятых алгоритмов оптимизации. Кроме того, что будет ясно специалистам в данной области, для предотвращения протекания нежелательных реакций на некоторых функциональных группах могут быть необходимы обычно применяемые защитные группы. Подбор подходящей защитной группы для конкретной функциональной группы, а также подходящих условий для введения защитных групп и снятия защитных групп хорошо известен в данной области. Например, многочисленные защитные группы и их введение и удаление описаны в работе T.W. Greene and P.M. Wuts, Protecting Groups in Organic Synthesis, Second Edition, Wiley, New York, 1991 и в ссылках, приведенных в этой работе. Подробно представлены следующие способы, касающиеся получения репрезентативных бициклогетероарилов, которые были приведены в этом документе выше. Соединения изобретения могут быть получены из известных или коммерчески доступных исходных веществ и реагентов специалистом в области органического синтеза. Все реагенты являются технического сорта и используются в состоянии поставки без дополнительной очистки, если не указано иное. Коммерчески доступные безводные растворители используют в реакциях, проводимых в инертной атмосфере. Растворители химически чистого сорта используют во всех случаях, если не указано иное. Колоночную хроматографию осуществляют на силикагеле 60 (35-70 мкм). Тонкослойную хроматографию выполняют с использованием пластинок, предварительно покрытых слоем силикагеля F-254 (толщина 0,25 мм). 1 Н ЯМР-спекты записывают на ЯМР-спектрометре Bruker DPX 400 (400 МГц) . Химические сдвиги ( δ) для 1 Н ЯМР-спектров сообщают в миллионных долях (ppm) относительно пика тетраметилсилана ( δ 0,00) или соответственного остаточного растворителя, то есть CHCl 3 ( δ 7,27), в качестве внутреннего эталона. Мультиплетности сигналов даны в виде синглета (с), дуплета (д), триплета (т), квадруплета (кв.), мультиплета (м) и широкого сигнала (ш.). Константы спин-спинового взаимодействия (J) даны в Гц. Электроспрей масс-спектры получают на спектрометре Micromass с платформой для проведения анализа методами жидкостной хроматографии и масс-спектрометрии (LC/MS). Колонка, используемая для всего анализа методом LCMS: Waters: Acquity с фазой С18 с частицами UPLC BEN размером 1,7 мкм, 2,1 мм (внутренний диаметр) × 50 мм (длина) (каталожный номер 186002350). Препаративный высокоэффективный жидкостной хроматограф (HPLC): Waters: XBridge Prep с фазой С18 с частицами размером 5 мкм ODB 19 мм (внутренний диаметр) × 100 мм (длина) (каталожный номер 186002978). Все способы выполняют с использованием градиентов MeCN/Н 2 О. Н 2 О содержит или 0,1% TFA или 0,1% NH 3 . Перечень сокращений, используемых в экспериментальном разделе. Соединение изобретения может быть получено в соответствии со следующей схемой. Получение основного промежуточного соединения. где Ar представляет собой Cy1-L1-(CR 4b R 4c ) n1 -R 3b ; и Cy1, L1, n1, R 2a , R 3b , R 4b и R 4c являются такими же, как описаны в этом документе. 1.1.1.1-(6-Бромпиридин-2-ил)-3-карбоэтокситиомочевина (2) К раствору 2-амино-6-бромпиридина (1) (253,8 г, 1,467 моль) в дихлорметане (DCM) (2,5 л), охлажденному до 5 °С, добавляют этоксикарбонилизотиоцианат (173,0 мл, 1,467 моль) по каплям в течение 15 мин. Реакционной смеси затем дают нагреться до комнатной температуры (20 °С) и перемешивают в течение 16 ч. Выпаривание в вакууме дает твердое вещество, которое собирают фильтрацией, тщательно промывают бензином (3 ×600 мл) и сушат на воздухе с предоставлением соединения (2). Тиомочевину используют как есть для следующей стадии без какой-либо очистки. 1 Н (400 МГц, CDCl 3 ) δ 12,03 (1Н, ш.с, NH), 8,81 (1Н, д, J=7,8 Гц, Н-3), 8,15 (1Н, ш.с, NH), 7,60 (1Н, т, J=8,0 Гц, Н-4), 7,32 (1Н, дд, J=7,7 и 0,6 Гц, Н-5), 4,31 (2Н, кв., J=7,1 Гц, СН 2 ), 1,35 (3Н, т, J=,1 Гц, СН 3 ). 1.1.2. 5-Бром-[1,2,4]триазоло[1,5-а]пиридин-2-иламин (3) К суспензии гидроксиламингидрохлорида (101,8 г, 1,465 моль) в смеси EtOH/MeOH (1:1, 900 мл) добавляют N,N-диизопропилэтиламин (145,3 мл, 0,879 моль) и смесь перемешивают при комнатной температуре (20 °С) в течение 1 ч. Затем добавляют 1-(6-бромпиридин-2-ил)-3-карбоэтокситиомочевину (2) (89,0 г, 0,293 моль) и смесь медленно нагревают для осуществления кипячения с обратным холодильником (следует отметить, что необходим скруббер-отбеливатель для поглощения выделяемого H 2 S). После 3 ч кипячения с обратным холодильником смеси дают остыть и фильтруют, собирая осажденное твердое вещество. Дополнительный продукт собирают выпариванием в вакууме фильтрата, добавлением Н 2 О (250 мл) и фильтрацией. Объединенное твердое вещество промывают последовательно посредством H 2 O (250 мл), смеси EtOH/MeOH (1:1, 250 мл) и Et 2 O (250 мл), затем сушат в вакууме с предоставлением триазолопиридинового производного (3) в виде твердого вещества. Соединение используют так как есть для следующей стадии без какой-либо очистки. 1 Н (400 МГц, DMSC-d 6 ) δ 7,43-7,34 (2Н, 2 × ароматич.-Н), 7,24 (1Н, дд, J=6,8 и 1,8 Гц, ароматич.-Н), 6,30 (2Н, ш., NH 2 ); m/z 213/215 (1:1, М+Н + , 100%). 1.1.3. Методика моноацилирования для получения промежуточного соединения (5-бром-[1,2,4]триазоло[1,5-а]пиридин-2-ил)амида циклопропанкарбоновой кислоты К раствору 2-аминотриазолопиридина (3) (7,10 г, 33,3 ммоль) в сухом CH 3 CN при 5 °С добавляют Et 3 N (11,6 мл, 83,3 ммоль), после этого циклопропанкарбонилхлорид (83,3 ммоль). Затем реакционной смеси дают нагреться до температуры окружающей среды и перемешивают до полного израсходования исходного вещества (3). При необходимости, для гарантированного обеспечения завершения реакции добавляют дополнительно триэтиламин (Et 3 N) (4,64 мл, 33,3 ммоль) и хлорангидрид кислоты (33,3 ммоль). После выпаривания растворителя в вакууме получающийся в результате остаток обрабатывают посредством 7N метанольного раствора аммиака (50 мл) и перемешивают при температуре окружающей среды в течение 1 ч для гидролизирования любого бис-ацилированного продукта. Выделение продукта осуществляют удалением летучих компонентов в вакууме с последующим порошкованием с Et 2 O (диэтиловый эфир) (50 мл). Твердое вещество собирают фильтрацией, промывают посредством H 2 O (2 ×50 мл), ацетона (50 мл) и Et 2 O (50 мл), затем сушат в вакууме, что дает требуемое промежуточное ацильное соединение (4). Способ А. 1.1.4. Получение соединений изобретения посредством сочетания Сузуки (5) 1.1.5. Соответствующую бороновую кислоту (2 экв.) добавляют к раствору промежуточного бромсодержащего соединения в смеси 1,4-диоксан/вода (5:1). К раствору добавляют K 2 CO 3 (2 экв.) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (PdCl 2 dppf) (5%). Получающуюся в результате смесь затем греют в микроволновой печи при 140 °С в течение 30 мин (эта реакция также может быть выполнена посредством обычного нагревания на масляной бане при 90 °С в течение 16 ч под N 2 ). Добавляют воду и раствор экстрагируют этилацетатом. Органические слои сушат над MgSO 4 и выпаривают в вакууме. Конечное соединение получают после очистки флэш-хроматографией. Метод В. где R 4a , R 4b , R 4c , R 3b и n1 являются такими, как описаны в этом документе. В1. 4,4-[2-(Циклопропанкарбониламино)-[1,2,4]триазоло[1,5-а]пиридин-5-ил]бензоилхлорид Две капли диметилформамида (DMF) добавляют к раствору 4-[2-(циклопропанкарбониламино)-[1,2,4]триазоло[1,5-а]пиридин-5-ил]бензойной кислоты (1 экв.), полученной способом А, в дихлорметане (DCM) в атмосфере N 2 . Оксалилхлорид (2 экв.) добавляют по каплям к этому получающемуся в результате раствору (выделение газа). Смесь перемешивают при комнатной температуре в течение 2 ч. После завершения реакции, что подтверждено методом жидкостной хроматографии-масс-спектрометрии (LCMS), растворитель удаляют. Неочищенный хлорангидрид кислоты используют без дополнительной очистки на следующей стадии. В2. Получение амида (общий способ) Соответствующий амин (1,1 экв.; R 2b , R 2c и m1 являются такими же, как описаны в этом документе) и триэтиламин (Et 3 N) (5 экв.) растворяют в дихлорметане (DCM) в атмосфере N 2 и охлаждают при 0 °С. Хлорангидрид кислоты (В1, 1 экв.), растворенный в DCM, добавляют по каплям к этому раствору. Реакционную смесь перемешивают при комнатной температуре в течение 3 ч. По истечении этого времени реакция является завершенной. Соединение экстрагируют посредством этилацетата (EtOAc) и воды, промывают соляным раствором и сушат над MgSO 4 . Органические слои фильтруют и упаривают. Конечное соединение выделяют препаративной высокоэффективной жидкостной хроматографией (HPLC). Препаративный высокоэффективный жидкостной хроматограф (HPLC): фирма Waters, колонки XBridge Prep OBD 19 мм (внутренний диаметр) × 100 мм (длина) с фазой С18, с частицами размером 5 мкм (каталожный номер 186002978). Все способы выполняют с использованием градиентов MeCN/H 2 O. H 2 O содержит или 0,1% трифторуксусной кислоты (TFA) или 0,1% NH 3 . Способ С где R 4a , R 4b , R 4c , R 3b и n1 являются такими же, как описаны в этом документе. Реакция алкилирования (общий способ) [5-(4-Гидроксифенил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амид циклопропанкарбоновой кислоты (1,1 экв.), полученный способом А, и K 2 CO 3 (5 экв.) (или AgCO 3 ) растворяют в диметилформамиде (DMF) под N 2 и добавляют по каплям соответственный алкилирующий агент (1,1 экв.). Получающуюся в результате суспензию греют при 50 °С в течение 16 ч. По истечении этого времени реакция является завершенной. Соединение экстрагируют посредством этилацетата (EtOAc) и воды, промывают солевым раствором и сушат над MgSO 4 . Органические слои фильтруют и упаривают. Конечное соединение выделяют препаративной высокоэффективной жидкостной хроматографией (HPLC). Препаративный высокоэффективный жидкостной хроматограф (HPLC): фирма Waters, колонки XBridge Prep OBD 19 мм (внутренний диаметр) × 100 мм (длина) с фазой С18, с частицами размером 5 мкм (каталожный номер 186002978). Все способы выполняют с использованием градиентов MeCN/H 2 O. H 2 O содержит или 0,1% трифторуксусной кислоты (TFA) или 0,1% NH 3 . Способ D Реакция сочетания (общий способ) где R 4a , R 4b , R 4c , R 3b и n1 являются такими же, как описаны в этом документе, и L1 представляет собой -С(=O)- или -SO 2 -. Производное анилина (1 экв.), полученное способом А, и триэтиламин (Et 3 N) (5 экв.) растворяют в дихлорметане (DCM) под N 2 и охлаждают при 0 °С. Соответственный хлорангидрид кислоты (для А) или сульфонилхлорид (для В) (1,5 экв.), растворенный в DCM, добавляют по каплям к этому раствору. Реакционную смесь перемешивают при комнатной температуре в течение 16 ч. По истечении этого времени реакция является завершенной. Соединение экстрагируют посредством EtOAc и воды, промывают соляным раствором и сушат над MgSO 4 . Органические слои фильтруют и упаривают. Конечное соединение выделяют препаративной высокоэффективной жидкостной хроматографией (HPLC). Препаративный высокоэффективный жидкостной хроматограф (HPLC): фирма Waters, колонки XBridge Prep OBD 19 мм (внутренний диаметр) × 100 мм (длина) с фазой С18, с частицами размером 5 мкм (каталожный номер 186002978). Все способы выполняют с использованием градиентов MeCN/Н 2 О. Н 2 О содержит или 0,1% трифторуксусной кислоты (TFA), или 0,1% NH 3 . Способ Е где R 4a , R 4b , R 4c , R 3b и n1 являются такими же, как описаны в этом документе. Восстановительное аминирование (общий способ) Соответствующий альдегид (2 экв.), производное анилина (1 экв.), полученное способом А, и тетра-н-пропоксид титана (Ti(OPr) 4 ) перемешивают при комнатной температуре в течение 3 ч. Смесь разбавляют в этаноле и добавляют Na(CN)BH 3 (1 экв.). Получающийся в результате раствор перемешивают при комнатной температуре в течение 16 ч. Смесь разбавляют водой и фильтруют. Фильтрат промывают этанолом. Объединенные фазы с растворителем упаривают под вакуумом. Конечное соединение выделяют препаративной высокоэффективной жидкостной хроматографией (HPLC). Препаративный высокоэффективный жидкостной хроматограф (HPLC): фирма Waters, колонки XBridge Prep OBD 19 мм (внутренний диаметр) × 100 мм (длина) с фазой С18, с частицами размером 5 мкм (каталожный номер 186002978). Все способы выполняют с использованием градиентов MeCN/Н 2 О. Н 2 О содержит или 0,1% трифторуксусной кислоты (TFA), или 0,1% NH 3 . Способ F где Ar представляет собой Cy1-L1-(CR 4b R 4c ) n1 -R 3b ; и Cy1, L1, n1, R 3b , R 4b , и R 4c являются такими же, как описаны в этом документе. N-(5-Бром-[1,2,4]триазоло[1,5-а]пиридин-2-ил)ацетамид К раствору 5-бром-2-аминотриазолопиридина (1 экв.) в безводном ацетонитриле (CH 3 CN) при 5 °С добавляют триэтиламин (Et 3 N) (2,5 экв.), после этого ацетилхлорид (2,5 экв.). Затем реакционной смеси дают нагреться до температуры окружающей среды и перемешивают до полного израсходования исходного вещества. При необходимости, для гарантированного обеспечения завершения реакции добавляют дополнительно Et 3 N (1 экв.) и хлорангидрид кислоты (1 экв.). После выпаривания растворителя в вакууме получающийся в результате остаток обрабатывают посредством 7N метанольного раствора аммиака (50 мл) и перемешивают при температуре окружающей среды (в течение 16 ч) для того, чтобы гидролизовать любой бис-ацилированный продукт. Выделение продукта осуществляют путем удаления летучих компонентов в вакууме с последующим добавлением воды и экстракцией этилацетатом. Органическую фазу затем сушат над MgSO 4 , упаривают в вакууме. Соединение может быть использовано без дополнительной очистки. Реакция по Сузуки (общий способ) Бороновую кислоту (2 экв.) добавляют к раствору N-(5-бром-[1,2,4]триазоло[1,5-а]пиридин-2-ил)ацетамида в смеси 1/4-диоксан/вода (5:1). К раствору добавляют K 2 CO 3 (2 экв.) и Pd(dppf)Cl 2 (5%) (dppf=1,1'-бис(дифенилфосфино)ферроцен). Получающуюся в результате смесь затем греют в микроволновой печи (СВЧ-печь системы Discover, фирмы СЕМ) в запаянной пробирке при 140 °С в течение 30 мин. Добавляют воду и раствор экстрагируют этилацетатом. Органические слои сушат над MgSO 4 и упаривают в вакууме. Конечное соединение получают после очистки препаративной высокоэффективной жидкостной хроматографией (HPLC). Аналитический HPLC: фирма Waters, колонки Acquity 2,1 мм (внутренний диаметр) × 50 мм (длина) с фазой UPLC ВЕН С18 с частицами размером 1,7 мкм (каталожный номер 186002350). Препаративный высокоэффективный жидкостной хроматограф (HPLC): фирма Waters, колонки XBridge Prep OBD 19 мм (внутренний диаметр) × 100 мм (длина) с фазой С18, с частицами размером 5 мкм (каталожный номер 186002978). Все способы выполняют с использованием градиентов MeCN/Н 2 О. Н 2 О содержит или 0,1% трифторуксусной кислоты (TFA), или 0,1% NH 3 . Способ G Реакция Сузуки (общий способ). Соответствующую бороновую кислоту (2 экв.) добавляют к раствору [5-(6-хлорпиридин-3-ил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амида циклопропанкарбоновой кислоты, полученного способом А, (1 экв.) в смеси 1,4-диоксан/вода (5:1). К раствору добавляют K 2 CO 3 (2 экв.) и Pd(dppf)Cl 2 (5%) (dppf=1,1'-бис(дифенилфосфино)ферроцен). Получающуюся в результате смесь затем греют в микроволновой печи (СВЧ-печь системы Discover, фирмы СЕМ) в запаянной пробирке при 140 °С в течение 30 мин. Добавляют воду и раствор экстрагируют этилацетатом. Органические слои сушат над MgSO 4 и упаривают в вакууме. Конечное соединение получают после очистки препаративной высокоэффективной жидкостной хроматографией (HPLC). Аналитический HPLC: фирма Waters, колонки Acquity 2,1 мм (внутренний диаметр) × 50 мм (длина) с фазой UPLC ВЕН С18 с частицами размером 1,7 мкм (каталожный номер 186002350). Препаративный высокоэффективный жидкостной хроматограф (HPLC): фирма Waters, колонки XBridge Prep ODB 19 мм (внутренний диаметр) × 100 мм (длина) с фазой С18, с частицами размером 5 мкм (каталожный номер 186002978). Все способы выполняют с использованием градиентов MeCN/Н 2 О. Н 2 О содержит или 0,1% трифторуксусной кислоты (TFA), или 0,1% NH 3 . Способ Н где R 4a , R 4b , R 4c , R 3b и n1 являются такими же, как описаны в этом документе. Ароматическое нуклеофильное замещение (общий способ). Производное хлорпиридина, полученное способом А (1 экв.), соответственный амин (1,5 экв.) смешивают в трет-бутаноле в запаянной пробирке. Реакционную смесь греют при 90 °С в течение 24 ч. Как только все исходное вещество исчезает, что подтверждают посредством жидкостной хроматографии-масс-спектрометрии (LCMS), к реакционной смеси добавляют воду и органические соединения экстрагируют этилацетатом. Органический слой сушат над MgSO 4 и упаривают под вакуумом. Конечное соединение выделяют препаративной высокоэффективной жидкостной хроматографией (HPLC). Аналитический высокоэффективный жидкостной хроматограф (HPLC): фирма Waters, колонки Acquity 2,1 мм (внутренний диаметр) × 50 мм (длина) с фазой UPLC ВЕН С18 с частицами размером 1,7 мкм (каталожный номер 186002350). Препаративный высокоэффективный жидкостной хроматограф (HPLC): фирма Waters, колонки XBridge Prep OBD 19 мм (внутренний диаметр) × 100 мм (длина) с фазой С18, с частицами размером 5 мкм (каталожный номер 186002978). Все способы выполняют с использованием градиентов MeCN/Н 2 О. Н 2 О содержит или 0,1% трифторуксусной кислоты (TFA), или 0,1% NH 3 . Способ I Восстановительное алкилирование (общий способ). Соответственный амин (2 экв.), циклопропанкарбоновую кислоту (например, [5-(4-формилфенил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амид циклопропанкарбоновой кислоты), полученную способом А (1 экв.), и тетра-н-пропоксид титана (Ti(OPr) 4 ) смешивают и перемешивают при комнатной температуре в течение 3 ч. Смесь разбавляют в этаноле и добавляют Na(CN)BH 3 (1 экв.). Получающийся в результате раствор перемешивают при комнатной температуре в течение 16 ч. Смесь разбавляют в воде и фильтруют. Твердое вещество промывают этанолом. Объединенные фазы с растворителем упаривают под вакуумом. Конечное соединение выделяют препаративной высокоэффективной жидкостной хроматографией (HPLC). Способ J 2-(4-Бромметилфенил)-4,4,5,5-тетраметил-[1,3,2]диоксаборолан (1 экв.) и триэтиламин (Et 3 N) (2 экв.) (или AgCO 3 ) растворяют в смеси DCM/MeOH (4:1 объем:объем) под N 2 и добавляют по каплям амин (2 экв.). Получающийся в результате раствор перемешивают при комнатной температуре в течение 16 ч. По истечении этого времени реакция является завершенной. Растворитель выпаривают. Соединение экстрагируют посредством этилацетата (EtOAc) и воды, промывают соляным раствором и сушат над MgSO 4 . Органические слои фильтруют и упаривают. Конечное соединение выделяют флэш-хроматографией. Сочетание по Сузуки Соединение, указанное в заглавии, затем синтезируют с использованием способа А. Способ K K.1. (5-Триметилсиланилэтинил-[1,2,4]триазоло[1,5-а]пиридин-2-ил)амид циклопропанкарбоновой кислоты К дегазированному раствору (5-бром-[1,2,4]триазоло[1,5-а]пиридин-2-ил)амида циклопропанкарбоновой кислоты (0,36 ммоль) в тетрагидрофуране (THF) (3,5 мл) добавляют CuI (0,036 ммоль), Pd(PPh 3 ) 2 Cl 2 (0,036 ммоль), диизопропиламин ((iPr) 2 NH) (0,137 мл) и триметилсилилацетилен (0,43 ммоль). Реакционную смесь греют при кипячении с обратным холодильником в течение ночи (70 °С) и затем растворитель удаляют под вакуумом. Неочищенный остаток повторно растворяют в этилацетате и промывают водой. Органический слой сушат над MgSO 4 , фильтруют и растворитель удаляют под вакуумом с предоставлением соединения, указанного в заглавии (95 мг, выход 89%). Дополнительную очистку не проводят. K.2. (5-Этинил-[1,2,4]триазоло[1,5-а]пиридин-2-ил)амид циклопропанкарбоновой кислоты 1М раствор тетра-н-бутиламмонийфторида (TBAF) (0,4 ммоль) в THF добавляют к раствору (5-триметилсиланилэтинил-[1,2,4]триазоло[1,5-а]пиридин-2-ил)амида циклопропанкарбоновой кислоты (0,32 ммоль) в ацетонитриле (4 мл) при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре до полного исчезновения исходного вещества, что подтверждают методом LCMS. Растворитель из реакционной смеси удаляют под вакуумом и смесь повторно растворяют в этилацетате. Органическую фазу промывают водой. Органический слой сушат над MgSO 4 , фильтруют и растворитель удаляют под вакуумом с предоставлением чистого продукта (70 мг, выход 97%). Дополнительную очистку продукта не проводят. K.3. Циклоприсоединение (общий способ) Соответствующее производное азида (0,44 ммоль) добавляют к раствору (5-этинил-[1,2,4]триазоло[1,5-а]пиридин-2-ил)амида циклопропанкарбоновой кислоты (0,44 ммоль), CuSO 4 ∙5H 2 O (0,022 ммоль) и аскорбата натрия (0,044 ммоль) в смеси CHCl 3 /EtOH/H 2 O (9:1:1) при комнатной температуре. Реакционную смесь греют при 50 °С до завершения реакции (отслеживают посредством методов LCMS). Неочищенную смесь разбавляют этилацетатом и промывают водой. Органическую фазу сушат над MgSO 4 , фильтруют и растворитель выпаривают под вакуумом. Конечное соединение очищают препаративной высокоэффективной жидкостной хроматографией (HPLC), что дает ожидаемое соединение. Способ L L.1. Нуклеофильное ароматическое замещение (общий способ) {5-[4-(6-Хлорпиридин-3-илметокси)фенил]-[1,2,4]триазоло[1,5-а]пиридин-2-ил}амид циклопропанкарбоновой кислоты, полученный способом С (1 экв.), соответственный амин (1,5 экв.) смешивают в DMSO в запаянной пробирке. Реакционную смесь греют при 100 °С в течение 24 ч. Как только все исходное вещество исчезает при проверке методами LCMS, в реакционную смесь добавляют воду и органические соединения экстрагируют этилацетатом. Органический слой сушат над MgSO 4 и упаривают под вакуумом. Конечное соединение выделяют препаративной высокоэффективной жидкостной хроматографией (HPLC). Аналитический HPLC: фирма Waters, колонки Acquity 2,1 мм (внутренний диаметр) × 50 мм (длина) с фазой UPLC ВЕН С18 с частицами размером 1,7 мкм (каталожный номер 186002350). Препаративный высокоэффективный жидкостной хроматограф (HPLC): фирма Waters, Колонки XBridge Prep ODB 19 мм (внутренний диаметр) × 100 мм (длина) с фазой С18, с частицами размером 5 мкм (каталожный номер 186002978). Все способы выполняют с использованием градиентов MeCN/Н 2 О. Н 2 О содержит или 0,1% трифторуксусной кислоты (TFA), или 0,1% NH 3 . Способ М М.1. Реакция Сузуки (общий способ) Бороновую кислоту (2 экв.) добавляют к раствору {5-[4-(6-хлорпиридин-3-илметокси)фенил]-[1,2,4]триазоло[1,5-а]пиридин-2-ил}амида циклопропанкарбоновой кислоты (получен способом В) в смеси 1,4-диоксан/вода (5:1). К раствору добавляют K 2 CO 3 (2 экв.) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (PdCl 2 dppf) (5%). Получающуюся в результате смесь затем греют в микроволновой печи при 140 °С в течение 30 мин (эта реакция может быть проведена посредством обычного нагревания на масляной бане при 90 °С в течение 16 ч под N 2 ). Добавляют воду и раствор экстрагируют этилацетатом. Органические слои сушат над MgSO 4 и упаривают в вакууме. Конечное соединение получают после очистки посредством препаративной высокоэффективной жидкостной хроматографии (HPLC). Способ О. Общая методика для получения сульфонов где R 4b , R 4c и R 3b являются такими же, как описаны в этом документе. Раствор 4-бромбензолсульфонилхлорида 1 (1,0 г, 3,96 ммоль, 1,0 экв.), сульфита натрия (0,6 г, 4,35 ммоль, 1,1 экв.) и гидрокарбоната натрия (1,7 г, 7 9,8 ммоль, 5,0 экв.) в воде (10 мл) нагревают до 100 °С в течение 4 ч. Реакционная смесь становится прозрачной, и добавляют соответственный галогенид (4,76 ммоль, 1,2 экв.) при 100 °С. Смесь перемешивают при этой температуре в течение 16 ч. Реакционную смесь охлаждают до комнатной температуры. Затем добавляют дополнительную воду (50 мл) и получающийся в результате водный слой экстрагируют дихлорметаном (3 ×50 мл). Объединенные органические слои сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Получающийся в результате сырой продукт очищают хроматографией над силикагелем с предоставлением ожидаемого сульфона. где R 4b , R 4c , R 3b и n1 являются такими же, как описаны в этом документе. В сосуде для микроволновой печи раствор сульфона, полученного в соответствии с методикой, описанной выше (0,84 ммоль, 1,5 экв.), ди(пинаколято)диборана (283,0 мг, 1,11 ммоль, 2,0 экв.), ацетата калия (109,0 мг; 1,11 ммоль, 2,0 экв.) в диоксане (2,0 мл) продувают аргоном (3 раза). Затем добавляют [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (20,0 мг, 0,03 ммоль, 0,05 экв.) и реакционную смесь продувают вновь аргоном (3 раза) и нагревают вплоть до 90 °С в течение 20 ч до завершения реакции, что подтверждают посредством тонкослойной хроматографии (TLC). Затем (5-бром-[1,2,4]триазоло[1,5-а]пиридин-2-ил)амид циклопропанкарбоновой кислоты (200,0 мг, 0,56 ммоль, 1,0 экв.), гидрокарбонат натрия (233,0 мг, 2,78 ммоль, 5,0 экв.), [1,1'-бис(дифенилфосфино) ферроцен]дихлорпалладий(II) (20,0 мг, 0,03 ммоль, 0,05 экв.) и смесь диоксан/вода 2:1 (1,5 мл) добавляют к смеси. Затем реакционную смесь подвергают несколько раз (1-5 раз) воздействию микроволнового излучения (Р: 150 Вт, Т 120 °С, t 15 мин) до полного расходования исходного вещества 133. Перед разбавлением реакционной смеси дихлорметаном (3,0 мл) в нее добавляют сульфат натрия (2,0 г). Очистка хроматографией на силикагеле (дихлорметан/метанол, 99:1 →90:10) с последующим порошкованием собранного соединения в метаноле дает ожидаемый продукт с удовлетворительной степенью чистоты, что подтверждают методом HPLC. Способ Р. Реакция алкилирования Соответствующий алкилирующий агент (1,5 экв.) добавляют к раствору производного ацетамида (1 экв.), полученного способом А, и NaH (2 экв.) в DMF при 0 °С. Смесь перемешивают в течение 16 ч при комнатной температуре. Затем раствор разбавляют в воде при 0 °С и раствор экстрагируют посредством EtOAc. Органические фазы сушат над MgSO 4 , фильтруют и растворитель удаляют под вакуумом. Конечное соединение выделяют препаративной высокоэффективной жидкостной хроматографией (HPLC). Способ Q 2-(4-Бромметилфенил)-4,4,5,5-тетраметил-[1,3,2]диоксаборолан (1 экв.) и K 2 CO 3 (2 экв.) (или AgCO 3 ) растворяют в DMF под N 2 и добавляют по каплям соответственный фенол (2 экв.). Получающуюся в результате суспензию греют при 50 °С в течение 16 ч. По истечении этого времени, реакция является завершенной. Соединение экстрагируют посредством EtOAc и воды, промывают соляным раствором и сушат над MgSO 4 . Органические слои фильтруют и упаривают с получением желаемого соединения без дополнительной очистки. Соединение, указанное в заглавии, получают способом А с использованием промежуточного сложного эфира бороновой кислоты, описанного выше. Способ U 1-Этил-3-(3-диметиламинопропил)карбодиимид (EDCI) (1,5 экв.), гидроксибензотриазол (HOBt) (1,5 экв.) и триэтиламин (Et 3 N) (2 экв.) добавляют к раствору {4-[2-(циклопропанкарбониламино)-[1,2,4]триазоло[1,5-а]пиридин-5-ил]фенил}уксусной кислоты в DCM при комнатной температуре. Получающуюся в результате смесь перемешивают в течение 2 ч при комнатной температуре. Соответственный амин добавляют к раствору и реакционную смесь перемешивают в течение 16 ч. К реакционной смеси добавляют воду и получающуюся реакционную смесь экстрагируют посредством EtOAc. Органические фазы сушат над MgSO 4 , фильтруют и упаривают под вакуумом. Очистка флэш-хроматографией дает ожидаемый продукт. Соединение 1. Это соединение получают способом А с использованием 1-метил-4-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)фенил]пиперазина. Соединение 2. Это соединение получают способом А с использованием гидрохлорида сложного эфира пинакола и 3-(4-морфолинометил)фенилбороновой кислоты. Соединение 3. Это соединение получают способом А с использованием сложного эфира пинакола и 2-(пиперидин-1-ил)пиридин-5-бороновой кислоты. Соединение 4. Это соединение получают способом А с использованием сложного эфира пинакола и 2-(пирролидин-1-ил)пиримидин-5-бороновой кислоты. Соединение 5. Это соединение получают способом А с использованием сложного эфира пинакола и 2-(4-метилпиперазин-1-ил)пиридин-4-бороновой кислоты. Соединение 6. Это соединение получают способом А с использованием сложного эфира пинакола и 2-(4-морфолино)пиридин-5-бороновой кислоты. Соединение 7. Это соединение получают способом А с использованием бифенил-4-бороновой кислоты. Соединение 8. Это соединение получают способом А с использованием сложного эфира пинакола и 2-(4-морфолино)пиримидин-5-бороновой кислоты. Соединение 9. Это соединение получают способом А с использованием сложного эфира пинакола и 2-(пиперидин-1-ил)пиримидин-5-бороновой кислоты. Соединение 10. Это соединение получают способом А с использованием 4-бензоилфенилбороновой кислоты. Соединение 11. Это соединение получают способом А с использованием [4-(циклопропиламинокарбонил)фенил]бороновой кислоты. Соединение 12. Это соединение получают способом А с использованием 4-бензилоксифенилбороновой кислоты. Соединение 13. Это соединение получают способом А с использованием сложного эфира пинакола и 4-(N-циклопропилсульфонамид)фенилбороновой кислоты. Соединение 14. Это соединение получают способом А с использованием 3-бензилоксифенилбороновой кислоты. Соединение 15. Это соединение получают способом А с использованием 4-бензилокси-3-фторфенилбороновой кислоты. Соединение 16. Это соединение получают способом А с использованием 2-бензилоксифенилбороновой кислоты. Соединение 17. Это соединение получают способом А с использованием пиперидин-1-ил-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)фенил]метанона. Соединение 18. Это соединение получают способом А с использованием 4-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)бензил]морфолина. Соединение 19. Это соединение получают способом А с использованием пирролидин-1-ил-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)фенил]метанона. Соединение 20. Это соединение получают способом А с использованием 4-(2-тиенил)фенилбороновой кислоты. Соединение 21. Это соединение получают способом D с использованием бензоилхлорида. Соединение 22. Это соединение получают способом D с использованием 4-трифторметилбензоилхлорида. Соединение 23. Это соединение получают способом D с использованием фенилацетилхлорида. Соединение 24. Это соединение получают способом В с использованием морфолина. Соединение 25. Это соединение получают способом В с использованием 4-аминопиридина. Соединение 26. Это соединение получают способом В с использованием аминоциклогексана. Соединение 27. Это соединение получают способом В с использованием 4-трет-бутилпиперидина. Соединение 28. Это соединение получают способом В с использованием [1,4]-диазепана. Соединение 29. Это соединение получают способом В с использованием 3-фторбензиламина. Соединение 30. Это соединение получают способом В с использованием N-метиланилина. Соединение 31. Это соединение получают способом В с использованием (4-метоксибензил)метиламина. Соединение 32. Это соединение получают способом В с использованием 1-метилпиперидин-4-иламина. Соединение 33. Это соединение получают способом D с использованием 4-фторсульфонилхлорида. Соединение 34. Это соединение получают способом D с использованием 2-фторбензоилхлорида. Соединение 35. Это соединение получают способом D с использованием пиразин-2-карбонилхлорида. Соединение 36. Это соединение получают способом С с использованием 3-бромметилпиридингидробромида. Соединение 37. Это соединение получают способом С с использованием 2-бромметилпиридингидробромида. Соединение 38. Это соединение получают способом С с использованием 3-(трифторметокси)бензилбромида. Соединение 39. Это соединение получают способом С с использованием (бромметил)циклобутана. Соединение 40. Это соединение получают способом С с использованием йодциклопентана. Соединение 41. Это соединение получают способом С с использованием (бромметил)циклогексана. Соединение 42. Это соединение получают способом В с использованием С-пиридин-3-ил-метиламина. Соединение 43. Это соединение получают способом В с использованием анилина. Соединение 44. Это соединение получают способом D с использованием бензоилхлорида. Соединение 45. Это соединение получают способом D с использованием циклогексанкарбонилхлорида. Соединение 46. Это соединение получают способом А с использованием 4-феноксифенилбороновой кислоты. Соединение 47. Это соединение получают способом G с использованием фенилбороновой кислоты. Соединение 48. Это соединение получают способом С с использованием 3-(хлорметил)-1-метил-1Н-пиразола. Соединение 49. Это соединение получают способом С с использованием 4-(хлорметил)-3,5-диметилизоксазола. Соединение 50. Это соединение получают способом С с использованием 5-(хлорметил)-1,3-диметил-1Н-пиразола. Соединение 51. Это соединение получают способом С с использованием 4-(2-бромэтил)-3,5-диметил-1Н-пиразола. Соединение 52. Это соединение получают способом С с использованием 3-(бромметил)-5-метилизоксазола. Соединение 53. Это соединение получают способом D с использованием 3-метоксибензоилхлорида. Соединение 54. Это соединение получают способом D с использованием 2-фторфенилсульфонилхлорида. Соединение 55. Это соединение получают способом D с использованием пиридин-2-карбоновой кислоты (хлорангидрида кислоты, образованного реакцией оксалилхлорида). Соединение 56. Это соединение получают способом В с использованием бензиламина. Соединение 57. [5-(6-Бензилоксипиридин-3-ил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амид циклопропанкарбоновой кислоты При 0 °С и в атмосфере N 2 бензиловый спирт (2 экв.) в растворе THF обрабатывают посредством 60%-ного раствора NaH в минеральном масле (4 экв.) в течение 30 мин. Затем к раствору добавляют [5-(6-хлорпиридин-3-ил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амид циклопропанкарбоновой кислоты, полученный способом А, и смесь перемешивают при 70 °С в течение 3 ч. По истечении этого времени реакция является завершенной. Реакционную смесь разлагают водой и соединение экстрагируют посредством EtOAc. Соединение промывают соляным раствором, сушат над MgSO 4 , фильтруют и концентрируют. Соединение очищают на препаративном высокоэффективном жидкостном хроматографе (HPLC). Соединение 58. Это соединение получают способом В с использованием 1,2,3,4-тетрагидроизохинолина. Соединение 59. Это соединение получают способом D с использованием циклопропансульфонилхлорида. Соединение 60. Это соединение получают способом В с использованием 2-феноксиэтиламина. Соединение 61. Это соединение получают способом Н с использованием пиразина. Соединение 62. Это соединение получают способом С с использованием 3-(хлорметил)-1,5-диметил-1Н-пиразола. Соединение 63. Это соединение получают способом С с использованием 4-(хлорметил)-2,5-диметил-1,3-оксазола. Соединение 64. Это соединение получают способом D с использованием пиридин-3-сульфонилхлорида. Соединение 65. Это соединение получают способом D с использованием 1,3-диметил-1Н-пиразол-4-сульфонилхлорида. Соединение 66. 3-Пиридинбороновую кислоту (1,1 экв.) добавляют к раствору [5-(4-бромфенил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амида циклопропанкарбоновой кислоты, полученного способом А, в смеси 1,4-диоксан/вода (5:1). К раствору добавляют K 2 CO 3 (2 экв.) и Pd(dppf)Cl 2 (0,03 экв.) (dppf=1,1'-бис(дифенилфосфино)ферроцен). Получающуюся в результате смесь затем греют в запаянной пробирке при 90 °С в течение 16 ч. Добавляют воду и раствор экстрагируют этилацетатом. Органические слои сушат над MgSO 4 и упаривают в вакууме. Конечное соединение получают после очистки препаративной высокоэффективной жидкостной хроматографией (HPLC). Аналитический HPLC: фирма Waters, колонки Acquity 2,1 мм (внутренний диаметр) × 50 мм (длина) с фазой UPLC ВЕН С18 с частицами размером 1,7 мкм (каталожный номер 186002350). Соединение 67. 1Н-Пиразол-4-бороновую кислоту (1,1 экв.) добавляют к раствору [5-(4-бромфенил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амида циклопропанкарбоновой кислоты, полученного способом А, в смеси 1,4-диоксан/вода (5:1). К раствору добавляют K 2 CO 3 (2 экв.) и Pd(dppf)Cl 2 (0,03 экв.) (dppf=1,1'-бис(дифенилфосфино)ферроцен). Получающуюся в результате смесь затем греют в запаянной пробирке при 90 °С в течение 16 ч. Добавляют воду и раствор экстрагируют этилацетатом. Органические слои сушат над MgSO 4 и упаривают в вакууме. Конечное соединение получают после очистки препаративной высокоэффективной жидкостной хроматографией (HPLC). Аналитический HPLC: фирма Waters, колонки Acquity 2,1 мм (внутренний диаметр) × 50 мм (длина) с фазой UPLC ВЕН С18 с частицами размером 1,7 мкм (каталожный номер 186002350). Соединение 68. 3-Пиридинбороновую кислоту (1,1 экв.) добавляют к раствору [5-(6-хлорпиридин-3-ил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амида циклопропанкарбоновой кислоты, полученного способом А, в смеси 1,4-диоксан/вода (5:1). К раствору добавляют K 2 CO 3 (2 экв.) и Pd(dppf)Cl 2 (0,03 экв.) (dppf=1,1'-бис(дифенилфосфино)ферроцен). Получающуюся в результате смесь затем греют в запаянной пробирке при 90 °С в течение 16 ч. Добавляют воду и раствор экстрагируют этилацетатом. Органические слои сушат над MgSO 4 и упаривают в вакууме. Конечное соединение получают после очистки препаративной высокоэффективной жидкостной хроматографией (HPLC). Аналитический HPLC: фирма Waters, колонки Acquity 2,1 мм (внутренний диаметр) × 50 мм (длина) с фазой UPLC ВЕН С18 с частицами размером 1,7 мкм (каталожный номер 186002350). Соединение 69. Это соединение получают способом D с использованием 2-метил-5-пропил-2Н-пиразол-3-карбоновой кислоты, которую сначала подвергают реакции в присутствии оксалилхлорида с получением хлорангидрида кислоты. Соединение 70. Это соединение получают способом D с использованием циклобутанкарбонилхлорида. Соединение 71. Это соединение получают способом В с использованием N-метилпиразина. Соединение 72. Это соединение получают способом Е с использованием бензальдегида. Соединение 73. Это соединение получают способом В с использованием 4-этоксиметилпиперидина. Соединение 74. Это соединение получают способом В с использованием фенилпиперидин-4-ил-метанона. Соединение 75. Это соединение получают способом В с использованием 1-бензил-[1,4]диазепана. Соединение 76. Это соединение получают способом D с использованием 2-фенилэтансульфонил-хлорида. Соединение 77. Это соединение получают способом D с использованием фенилметансульфонилхлорида. Соединение 78. Это соединение получают способом С с использованием 3-(хлорметил)-6-(трифторметил)пиридина. Соединение 79. Это соединение получают способом D с использованием фенилсульфонилхлорида. Соединение 80. Это соединение получают способом В с использованием 2-пирролидин-1-ил-этиламина. Соединение 81. Это соединение получают способом В с использованием 3-морфолин-4-ил-пропиламина. Соединение 82. Это соединение получают способом Н с использованием 4-фенетилпиперидина. Соединение 83. Это соединение получают способом Н с использованием 4-(4-хлорфенил)пиперидина. Соединение 84. Это соединение получают способом Н с использованием 3-фенилпиперидина. Соединение 85. Это соединение получают способом D с использованием 4-пропилбензолсульфонилхлорида. Соединение 86. Это соединение получают способом В с использованием 4-(4-хлорфенил)пиперидина. Соединение 87. Это соединение получают способом В с использованием фенетиламина. Соединение 88. Это соединение получают способом D с использованием 2-(3-фторфенил)этансульфонилхлорида. Соединение 89. Это соединение получают способом В с использованием пиперидина. Соединение 90. Это соединение получают способом В с использованием бутил-(3-морфолин-4-ил-пропил)амина. Соединение 91. Это соединение получают способом С с использованием 4-(хлорметил)-2-[4-(трифторметил)фенил]тиазола. Соединение 92. Это соединение получают способом С с использованием 4-ацетамидобензилхлорида. Соединение 93. Это соединение получают способом Н с использованием 2-(тетрагидропиран-4-ил)этиламина. Соединение 94. Это соединение получают способом В с использованием 4-(2-пиперидин-4-ил-этил)морфолина. Соединение 95. Это соединение получают способом Н с использованием 4-(2-пиперидин-4-ил-этил)морфолина. Соединение 96. Это соединение получают способом В с использованием метилфенетиламина. Соединение 97. Это соединение получают способом В с использованием 2-(4-трифторметилфенил)этиламина. Соединение 98. Это соединение получают способом С с использованием 1-(2-бромэтил)-1Н-пиразола. Соединение 99. Это соединение получают способом С с использованием 3-(хлорметил)-1,2,4-оксадиазола. Соединение 100. Это соединение получают способом В с использованием 2-феноксиэтиламина. Соединение 101. Это соединение получают способом В с использованием 4-пиперидин-4-ил-морфолина. Соединение 102. Это соединение получают способом В с использованием 1,1-диметил-2-морфолин-4-ил-этиламина. Соединение 103. Это соединение получают способом В с использованием бензилметилпиперидин-4-ил-амина. Соединение 104. Это соединение получают способом Н с использованием бензиламина. Соединение 105. Это соединение получают способом В с использованием [2-(3,4-диметоксифенил)этил]метиламина. Соединение 106. Это соединение получают способом В с использованием 2-(1-фенил-1Н-пиразол-4-ил)этиламина. Соединение 107. Это соединение получают способом А с использованием 1-бензил-1Н-пиразол-4-бороновой кислоты. Соединение 108. Это соединение получают способом D с использованием 3-феноксипропионилхлорида. Соединение 109. Это соединение получают способом D с использованием 3-фенилпропионилхлорида. Соединение 110. Это соединение получают способом В с использованием 3-(пиперидин-4-илокси)пиридина. Соединение 111. Это соединение получают способом В с использованием фенетиламина. Соединение 112. Это соединение получают способом Н с использованием 4-фенетилпиперидина. Соединение 113. Это соединение получают способом В с использованием 1-(3-хлорфенил)пиперазина. Соединение 114. Это соединение получают способом В с использованием 3-фенилпропиламина. Соединение 115. Это соединение получают способом С с использованием 1-(2-бромэтокси)-4-фторбензола. Соединение 116. Это соединение получают способом С с использованием N-([хлорацетил)-3-фторанилина. Соединение 117. Это соединение получают способом С с использованием 1-(4-бензилпиперидин-1-ил)-2-хлорэтанола. Соединение 118. Это соединение получают способом С с использованием 2-хлор-N-метил-N-фенилацетамида. Соединение 119. Это соединение получают способом В с использованием (S)-l-бензилпирролидин-3-иламина. Соединение 120. Это соединение получают способом В с использованием (R)-l-бензилпирролидин-3-иламина. Соединение 121. [5-(1-Бензил-1Н-индол-5-ил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амид циклопропанкарбоновой кислоты 121.1. 1-Бензил-5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1H-индол К раствору 5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-индола (2,27 ммоль; 1,0 экв.) в ацетоне (10,0 мл) при комнатной температуре добавляют под аргоном бензилбромид (3,18 ммоль; 1,4 экв.) и карбонат цезия (3,18 ммоль; 1,4 экв.). Реакционную смесь греют в течение 4 ч при кипячении с обратным холодильником. Смесь затем охлаждают при комнатной температуре, разлагают добавлением насыщенного водного раствора гидрокарбоната натрия (100 мл) и экстрагируют дихлорметаном (2 ×100 мл). Органический слой сушат над сульфатом натрия, фильтруют и концентрируют досуха. Получающийся в результате остаток очищают флэш-хроматографией на силикагеле (дихлорметан/этилацетат) с получением ожидаемого бороната в виде белого твердого вещества, используемого на следующей стадии без дополнительной очистки. 121.2. [5-(1-Бензил-1Н-индол-5-ил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амид циклопропанкарбоновой кислоты Соединение, указанное в заглавии, затем синтезируют способом А с использованием 1-бензил-5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-индола. Соединение 122. Соединение получают способом D с использованием 2-феноксиэтансульфонилхлорида. Соединение 123. Соединение получают способом Н с использованием фенетиламина. Соединение 124. Соединение получают способом В с использованием 2-пиридин-3-ил-этиламина. Соединение 125. Соединение получают способом С с использованием мезилатного производного (4-пиразол-1-ил-фенил)метанола. Соединение 126. Соединение получают способом С с использованием мезилатного производного [4-(4-метил-пиперазин-1-ил-метил)фенил]метанола. Соединение 127. Соединение получают способом С с использованием 3-(2-хлорэтил)пиридина. Соединение 128. Соединение получают способом В с использованием 2-пиперидин-4-ил-1Н-бензоимидазола. Соединение 129. Соединение получают способом А с использованием 4-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]морфолина. Соединение 130. Соединение получают способом I с использованием N-метилпиразина. Соединение 131. Соединение получают способом В с использованием 4-(2-метоксифенил)пиперидина. Соединение 132. Соединение получают способом В с использованием 4-(4-хлорфенил)пиперидин-4-ола. Соединение 133. Соединение получают способом В с использованием 4-о-толилоксиметилпиперидина. Соединение 134. Соединение получают способом В с использованием 3,5-диметилпиперидина. Соединение 135. Соединение получают способом В с использованием N-(4-пиперидин-1-илбензил)пропан-2-амина. Соединение 136. Соединение получают способом В с использованием пропил-(тетрагидрофуран-2-илметил)амина. Соединение 137. Соединение получают способом В с использованием 4-фторпиперидина. Соединение 138. Соединение получают способом В с использованием 2-пиперидин-4-ил-1Н-индола. Соединение 139. Соединение получают способом I с использованием пиперидина. Соединение 140. Соединение получают способом I с использованием амида пиперидин-4-карбоновой кислоты. Соединение 141. Соединение получают способом I с использованием 1-пиперазин-1-ил-этанона. Соединение 142. Соединение получают способом I с использованием 1-пиридин-2-ил-пиперазина. Соединение 143. Соединение получают способом I с использованием 2-пиперазин-1-ил-пиримидина. Соединение 144. Соединение получают способом В с использованием 2-метилпиперидина. Соединение 145. Соединение получают способом В с использованием 3-метилпиперидина. Соединение 146. Соединение получают способом В с использованием 4-метилпиперидина. Соединение 147. Соединение получают способом В с использованием 4-фенетилпиперидина. Соединение 148. Соединение получают способом В с использованием 4-трифторметилпиперидина. Соединение 149. Соединение получают способом В с использованием 6-фтор-3-пиперидин-4-ил-бензо[d]изоксазола. Соединение 150. Соединение получают способом В с использованием N'-бензил-N,N-диметилэтилендиамина. Соединение 151. Соединение получают способом В с использованием (4-фторбензил)-(2-метокси-1-метилэтил) амингидрохлорида. Соединение 152. Соединение получают способом В с использованием 1-пиперидин-4-ил-1Н-бензотриазола. Соединение 153. Соединение получают способом В с использованием (4-фторбензил)-(тетрагидрофуран-2-илметил)амина. Соединение 154. Соединение получают способом В с использованием 4-[2-(2-метилимидазол-1-ил)этил]пиперидина. Соединение 155. Соединение получают способом I с использованием метилфенетиламина. Соединение 156. Соединение получают способом В с использованием 4-бензилпиперидин-4-ола. Соединение 157. Соединение получают способом Е с использованием бензилбромида. Соединение 158. N-(5-(4-((1Н-тетразол-5-ил)метил-1Н-тетразол-5-ил)метокси)фенил)-[1,2,4]триазоло [1,5-а]пиридин-2-ил)циклопропанкарбоксамид Раствор [5-(4-цианометоксифенил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амида циклопропанкарбоновой кислоты (1 экв.), азида натрия (2 экв.) и хлорида аммония (2 экв.) перемешивают при 0 °С под азотом в течение 30 мин. После этого смесь подвергают воздействию 100 °С в течение 16 ч. Сырой продукт реакции разбавляют этилацетатом и промывают водой. Органическую фазу сушат над MgSO 4 , фильтруют и сушат в вакууме. Сырой продукт разбавляют посредством DMSO и подвергают очистке на препаративном высокоэффективном жидкостном хроматографе (HPLC): система UPLC (колонка XBridge Prep 19 мм (внутренний диаметр) × 100 мм (длина) ) с фазой С18, с частицами размером 5 мкм; продолжительность жидкостной хроматографии 8 мин; поток: 20 мл/мин; градиент: от 30 до 70% ацетонитрила в воде с 0,1% TFA; с получением конечного чистого продукта (выход 10%). Соединение 159. Это соединение получают способом I с использованием метил-(4-пиридин-2-ил-бензил)амина. Соединение 160. Это соединение получают способом I с использованием (1,5-диметил-1Н-пиразол-3-илметил)метиламина. Соединение 161. Это соединение получают способом I с использованием метил-(4-пиримидин-5-ил-бензил)амина. Соединение 162. Это соединение получают способом I с использованием метил-пиридин-3-илметиламина. Соединение 163. Это соединение получают способом F с использованием 2-(4-бензилоксифенил)-4,4,5,5-тетраметил-[1,3,2]диоксаборолана. Соединение 164. Это соединение получают способом В с использованием 3-трифторметилпиперидина. Соединение 165. Это соединение получают способом L с использованием морфолина. Соединение 166. Это соединение получают способом М с использованием 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-пиразола. Соединение 167. Это соединение получают способом L с использованием N-метилморфолина. Соединение 168. Это соединение получают способом В с использованием 4,4-дифторпиперидина. Соединение 169. Это соединение получают способом В с использованием 3-фенилпиперидина. Соединение 170. Это соединение получают способом Е с использованием С-пиридин-3-ил-метиламина. Соединение 171. Это соединение получают способом Е с использованием С-пиридин-2-ил-метиламина. Соединение 172. Это соединение получают способом Е с использованием 2-пиридин-3-ил-этиламина. Соединение 173. Это соединение получают способом Е с использованием С-(1,5-диметил-1Н-пиразол-3-ил)-метиламина. Соединение 174. Это соединение получают способом L с использованием пирролидина. Соединение 175. Это соединение получают способом В с использованием 3,3-диметилпиперидина. Соединение 176. N-(5-(4-((6-цианопиридин-3-ил)метокси)фенил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил)циклопропанкарбоксамид. 176.1. Синтез 5-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)феноксиметил]пиридин-2-карбонитрила К сложному эфиру пинакола и 4-гидроксифенилбороновой кислоты (25 г; 0,11 моль; 1,0 экв.) в ацетоне (250 мл) при комнатной температуре добавляют под аргоном 5-хлорметилпиридин-2-карбонитрил (19 г; 0,12 моль; 1,1 экв.) и карбонат цезия (73,9 г, 0,22 моль; 2 экв.). Реакционную смесь греют в течение 4 ч при кипячении с обратным холодильником. Затем смесь охлаждают до комнатной температуры, ацетон выпаривают. Добавляют воду (200 мл) и продукт экстрагируют посредством EtOAc (3 ×200 мл). Органический слой сушат над сульфатом магния, фильтруют и концентрируют досуха. Получающийся в результате остаток очищают хроматографией на силикагеле (бензин:EtOAc 10:1) с получением ожидаемого бороната в виде белого твердого вещества. 176.2. Синтез {5-[4-(6-цианопиридин-3-илметокси)фенил]-[1,2,4]триазоло[1,5-а]пиридин-2-ил}амида циклопропанкарбоновой кислоты 5-[4-(4,4,5,5-Тетраметил-[1,3,2]диоксаборолан-2-ил)феноксиметил]пиридин-2-карбонитрил (10 г, 0,03 моль, 1,1 экв.) добавляют к раствору (5-бром-[1,2,4]триазоло[1,5-а]пиридин-2-ил)амида циклопропанкарбоновой кислоты (7,6 г, 0,027 моль) в смеси 1,4-диоксан/вода (4:1; 70 мл). К раствору добавляют K 2 CO 3 (7,45 г, 0,054 моль, 2 экв.) и PdCl 2 dppf (5%). Получающуюся в результате смесь затем греют на масляной бане при 90 °С в течение 4-16 ч под N 2 до завершения (отслеживают посредством LCMS). 1,4-Диоксан удаляют под вакуумом и добавляют смесь вода/EtOAc и отфильтровывают твердое вещество. Полученное твердое вещество растворяют в смеси метанол/DCM, сушат над MgSO 4 и конечное соединение получают после очистки флэш-хроматографией при элюировании чистым EtOAc. Соединение 177. Это соединение получают способом D с использованием 1-цианоциклопропанкарбонилхлорида. Соединение 178. Это соединение получают способом М с использованием 1-метил-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-пиразола. Соединение 179. {5-[1-(3-Фенилпропионил)-1Н-индол-5-ил]-[1,2,4]триазоло[1,5-а]пиридин-2-ил} амид циклопропанкарбоновой кислоты. 119.1. 3-Фенил-1-[5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)индол-1-ил]пропан-1-он К раствору 5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-индола (2,27 ммоль; 1,0 экв.) в DMF (10,0 мл) при комнатной температуре добавляют под аргоном 3-фенилпропионилхлорид (3,18 ммоль; 1,4 экв.) и гидрид натрия (3,18 ммоль; 1,4 экв.). Реакционную смесь греют при 60 °С в течение 16 ч. Смеси дают остыть до комнатной температуры и разлагают путем добавления воды (100 мл) и экстрагируют дихлорметаном (2 ×100 мл). Органический слой сушат над сульфатом натрия, фильтруют и концентрируют досуха. Получающийся в результате остаток очищают флэш-хроматографией на силикагеле (дихлорметан/этилацетат) с получением ожидаемого бороната в виде белого твердого вещества, используемого на следующей стадии без дополнительной очистки. 179.2. {5-[1-(3-Фенилпропионил)-1Н-индол-5-ил]-[1,2,4]триазоло[1,5-а]пиридин-2-ил}амид циклопропанкарбоновой кислоты Соединение, указанное в заглавии, получают способом А с использованием 3-фенил-1-[5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)индол-1-ил]пропан-1-она. Соединение 180. Это соединение получают способом D с использованием бензолсульфонилхлорида. Соединение 181. 181.1. Получение фенетиламиноацетонитрила. Хлорацетонитрил (1,5 экв.) добавляют к раствору фенетиламина (1 экв.) и K 2 CO 3 (2 экв.) в CH 3 CN. Смесь перемешивают при 60 °С в течение 4 ч. После завершения реакции смесь фильтруют и фильтрат концентрируют при пониженном давлении. Очитска флэш-хроматографией дает ожидаемое соединение. 181.2. Соединение 181 получают способом В с использованием фенетиламиноацетонитрила. Соединение 182. Это соединение получают способом Е с использованием 3-бромметилпиридина. Соединение 183. (5-{4-[6-(2Н-Тетразол-5-ил)пиридин-3-илметокси]фенил}-[1,2,4]триазоло[1,5-а]пиридин-2-ил)амид циклопропанкарбоновой кислоты Раствор {5-[4-(6-цианопиридин-3-илметокси)фенил]-[1,2,4]триазоло[1,5-а]пиридин-2-ил}амида циклопропанкарбоновой кислоты (143 мг, 0,35 ммоль, 1 экв.), азида натрия (46 мг, 0,7 ммоль, 2 экв.), хлорида аммония (38 мг, 0,7 ммоль, 2 экв.) перемешивают при 0 °С под азотом в течение 30 мин. После этого смесь подвергают воздействию 100 °С в течение 16 ч. Сырой продукт реакции разбавляют этилацетатом и промывают водой. Органическую фазу сушат над MgSO 4 , фильтруют и сушат под вакуумом. Сырой продукт разбавляют посредством DMSO и подвергают очистке на препаративном высокоэффективном жидкостном хроматографе (HPLC): система UPLC (колонка XBridge ™ Prep 19 мм (внутренний диаметр) × 100 мм (длина)) с фазой С18, с частицами размером 5 мкм; продолжительность жидкостной хроматографии 8 мин; поток: 20 мл/мин; градиент: от 30 до 70% ацетонитрила в воде с 0,1% TFA; с получением конечного чистого продукта (9 мг, выход 6%). Соединение 184. Это соединение получают способом D с использованием фенилметансульфонил-хлорида. Соединение 185. [5-(1-Пиридин-3-илметил-1Н-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амид циклопропанкарбоновой кислоты. 185.1. 3-[4-(4,4,5,5-Тетраметил-[1,3,2]диоксаборолан-2-ил)пиразол-1-илметил]пиридин К раствору 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-пиразола (2,27 ммоль; 1,0 экв.) в ацетоне (10,0 мл) при комнатной температуре добавляют под аргоном 3-хлорметилпиридингидрохлорид (3,18 ммоль; 1,4 экв.) и карбонат цезия (2,8 экв.). Реакционную смесь греют в течение 4 ч при кипячении с обратным холодильником. Смесь затем охлаждают до комнатной температуры, разлагают путем добавления насыщенного водного раствора гидрокарбоната натрия (100 мл) и экстрагируют дихлорметаном (2 ×100 мл). Органический слой сушат над сульфатом натрия, фильтруют и концентрируют досуха. Получающийся в результате остаток очищают флэш-хроматографией на силикагеле (дихлорметан/этилацетат) с получением ожидаемого бороната в виде белого твердого вещества, используемого на следующей стадии без дополнительной очистки. 185.2. [5-(1-Пиридин-3-илметил-1Н-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амид циклопропанкарбоновой кислоты Соединение, указанное в заглавии, получают способом А с использованием 3-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)пиразол-1-илметил]пиридина. Соединение 186. {5-[1-(3-Фенилпропионил)-1Н-пиразол-4-ил]-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амид циклопропанкарбоновой кислоты. 186.1. 3-Фенил-1-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)пиразол-1-ил]пропан-1-он К раствору 4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-пиразола (2,27 ммоль; 1,0 экв.) в DMF (10,0 мл) при комнатной температуре добавляют под аргоном 3-фенилпропионилхлорид (3,18 ммоль; 1,4 экв.) и гидрид натрия (3,18 ммоль; 1,4 экв.). Реакционную смесь греют при 60 °С. Смеси дают остыть до комнатной температуры и разлагают путем добавления насыщенного водного раствора гидрокарбоната натрия (100 мл) и экстрагируют дихлорметаном (2 ×100 мл). Органический слой сушат над сульфатом натрия, фильтруют и концентрируют досуха. Получающийся в результате остаток очищают флэш-хроматографией на силикагеле (дихлорметан/этилацетат) с получением ожидаемого бороната в виде белого твердого вещества, используемого на следующей стадии без дополнительной очистки. 186.2. {5-[1-(3-Фенилпропионил)-1Н-пиразол-4-ил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил}амид циклопропанкарбоновой кислоты Соединение, указанное в заглавии, получают способом А с использованием 3-фенил-1-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)пиразол-1-ил]пропан-1-она. Соединение 187. Это соединение получают способом K с использованием азидометилбензола. Соединение 188. Это соединение получают способом K с использованием 5-азидометил-2-трифторметилпиридина. Соединение 189. Это соединение получают способом С с использованием мезилатного производного 1-пиридин-2-ил-этанола. Соединение 190. Это соединение получают способом С с использованием мезилатного производного метилового сложного эфира 6-гидроксиметилникотиновой кислоты. Соединение 191. Это соединение получают способом D с использованием циклопропансульфонилхлорида. Соединение 192. Амид 5-{4-[2-(циклопропанкарбониламино)-[1,2,4]триазоло[1,5-а]пиридин-5-ил]феноксиметил}пиридин-2-карбоновой кислоты К раствору {5-[4-(6-цианопиридин-3-илметокси)фенил]-[1,2,4]триазоло[1,5-а]пиридин-2-ил}амида циклопропанкарбоновой кислоты (30 мг, 0,073 ммоль, 1 экв.) и K 2 CO 3 (10 мг, 0,073 ммоль, 1 экв.) в DMSO (0,2 мл) при 10 °С, добавляют по каплям 30%-ный раствор Н 2 О 2 (17 мкл, 0,146 ммоль, 2 экв.). После перемешивания смеси при комнатной температуре в течение 4 ч смесь разбавляют посредством DMSO и фильтруют. Фильтрат подвергают очистке на препаративном высокоэффективном жидкостном хроматографе (HPLC): система UPLC (колонка XBridge ™ Prep 19 мм (внутренний диаметр) × 100 мм (длина)) с фазой С18, с частицами размером 5 мкм; продолжительность жидкостной хроматографии 8 мин; поток: 20 мл/мин; градиент: от 30 до 70% ацетонитрила в воде с 0,1% TFA; с получением конечного продукта (25 мг, выход 81%). Соединение 193. Это соединение получают способом K с использованием 2-азидометилпиридина. Соединение 194. Это соединение получают способом О с использованием 2-бромметилпиридина. Соединение 195. Это соединение получают способом О с использованием 3-бромметилпиридина. Соединение 196. Это соединение получают способом С с использованием 3-хлорметилпиридин-1-оксида. Соединение 197. Это соединение получают способом С с использованием 5-хлорметил-2-метил-пиридина. Соединение 198. Это соединение получают способом С с использованием 2-хлор-5-хлорметил-пиридина. Соединение 199. Это соединение получают способом С с использованием 3-хлорметил-1-метил-1Н-[1,2,4]триазола. Соединение 200. 2-(4-Бромметилфенил)-4,4,5,5-тетраметил-[1,3,2]диоксаборолан (1 экв.) и N,N-диизопропилэтиламин (DIPEA) (2 экв.) растворяют в смеси DCM/MeOH (5:1 объем:объем) под N 2 и добавляют порциями тиоморфолин-1,1,-диоксид (2 экв.). Получающийся в результате раствор перемешивают при комнатной температуре в течение 16 ч. По истечении этого времени реакция является завершенной. Растворитель выпаривают. Соединение экстрагируют посредством EtOAc и воды, промывают соляным раствором и сушат над MgSO 4 . Органические слои фильтруют и упаривают. Конечное соединение выделяют без дополнительной очистки. Сочетание по Сузуки. 4-[4-(4,4,5,5-Тетраметил-[1,3,2]диоксаборолан-2-ил)бензил]тиоморфолин-1,1-диоксид (1,1 экв.) добавляют к раствору (5-бром-[1,2,4]триазоло[1,5-а]пиридин-2-ил)амида циклопропанкарбоновой кислоты в смеси 1,4-диоксан/вода (4:1). К раствору добавляют K 2 CO 3 (2 экв.) и PdCl 2 dppf (0,03 экв.). Получающуюся в результате смесь затем греют на масляной бане при 90 °С в течение 16 ч под N 2 . Добавляют воду и раствор экстрагируют этилацетатом. Органические слои сушат над MgSO 4 и упаривают в вакууме. Конечное соединение получают после очистки флэш-хроматографией. Альтернативный путь получения соединения 200. 4-(Гидроксиметил)фенилбороновую кислоту (1,1 экв.) добавляют к раствору (5-бром-[1,2,4]триазоло[1,5-а]пиридин-2-ил)амида циклопропанкарбоновой кислоты в смеси 1,4-диоксан/вода (4:1). К раствору добавляют K 2 CO 3 (2 экв.) и PdCl 2 dppf (0,03 экв.). Получающуюся в результате смесь затем греют на масляной бане при 90 °С в течение 16 ч под N 2 . Добавляют воду и раствор экстрагируют этилацетатом. Органические слои сушат над MgSO 4 и упаривают в вакууме. Получающуюся в результате смесь используют без дополнительной очистки. К раствору [5-(4-гидроксиметилфенил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амида циклопропанкарбоновой кислоты (1,0 экв.) в хлороформе медленно добавляют трибромид фосфора (1,0 экв.). Реакционную смесь перемешивают при комнатной температуре в течение 20 ч, разлагают смесью льда и воды (20 мл) и экстрагируют дихлорметаном. Органический слой сушат над MgSO 4 , фильтруют и концентрируют досуха. Получающийся в результате белый остаток порошкуют в смеси дихлорметан/диэтиловый эфир 2:1 (20 мл) с получением ожидаемого продукта в виде белого твердого вещества. [5-(4-Бромметилфенил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амид циклопропанкарбоновой кислоты (1,0 экв.) и N,N-диизопропилэтиламин (DIPEA) (2 экв.) растворяют в смеси DCM/MeOH (5:1 объем:объем) под N 2 и добавляют по каплям тиоморфолин-1,1-диоксид (1,1 экв.). Получающийся в результате раствор перемешивают при комнатной температуре в течение 16 ч. По истечении этого времени реакция является завершенной. Растворитель выпаривают. Соединение растворяют в DCM, промывают водой и сушат над MgSO 4 . Органические слои фильтруют и упаривают. Конечное соединение выделяют колоночной хроматографией с использованием EtOAc с получением желаемого. Соединение 201. Это соединение получают способом С с использованием 4-(2-хлорэтил)-3,5-диметилизоксазола. Соединение 202. Это соединение получают способом Р с использованием 3-бромметилпиридина. Соединение 203. Это соединение получают способом В с использованием 6-метоксипиридин-3-иламина. Соединение 204. Это соединение получают способом В с использованием 6-морфолин-4-ил-пиридин-3-иламина. Соединение 205. Это соединение получают способом В с использованием 6-(4-метилпиперазин-1-ил)пиридин-3-иламина. Соединение 206. Это соединение получают способом В с использованием пиридин-3-иламина. Соединение 207. Это соединение получают способом В с использованием тиоморфолин-1,1-диоксида. Соединение 208. Это соединение получают способом Р с использованием 3-бромметилпиридина. Соединение 209. Это соединение получают способом В с использованием 4-гидроксипиперидина. Соединение 210. Это соединение получают способом В с использованием пиперидин-4-карбонитрила. Соединение 211. {5-[4-(2-Пиридин-2-ил-этил)фенил]-[1,2,4]триазоло[1,5-а]пиридин-2-ил}амид циклопропанкарбоновой кислоты трет-Бутоксид калия (1,3 экв.) добавляют к охлаждаемому льдом раствору 2-[(трифенил- λ5-фосфанил)метил]пиридина (1,1 экв.) в THF (10 мл/ммоль). Получающуюся в результате смесь перемешивают в течение 30 мин при 0 °С, затем при комнатной температуре в течение других 30 мин. К реакционной смеси добавляют по каплям раствор [8-(4-формилфенил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амида циклопропанкарбоновой кислоты (1 экв.) в THF (10 мл/ммоль). Перемешивание поддерживают в течение 8 ч. Реакционную смесь затем разлагают водой и экстрагируют этилацетатом. Органическую фазу сушат над MgSO 4 , фильтруют и удаляют под вакуумом. Твердое вещество промывают метанолом с получением соединения, указанного в заглавии, с выходом 69%. {5-[4-((Е)-2-Пиридин-3-ил-винил)фенил]-[1,2,4]триазоло[1,5-а]пиридин-2-ил}амид циклопропанкарбоновой кислоты (320 мг, 0,84 моль) растворяют в 10 мл МеОН. Добавляют палладий, нанесенный на активированный уголь (Pd/C) (50 мг), и реакцию проводят в атмосфере Н 2 при нормальном давлении. Реакционную смесь перемешивают в течение 2 ч. Реакционную смесь фильтруют через цеолит Celite ®. Органический растворитель удаляют под давлением. К получающейся в результате смеси добавляют насыщенный раствор NaHCO 3 . Соединение экстрагируют посредством EtOAc. Органическую фазу сушат над MgSO 4 , фильтруют и удаляют под вакуумом с получением соединения, указанного в заглавии, с выходом 100%. Соединение 212. Это соединение получают способом J с использованием 4-хлор-2-фтор-фениламина. Соединение 213. Это соединение получают способом J с использованием 3,3-диметилморфолина. Соединение 214. Это соединение получают способом J с использованием цис-2,6-диметил-морфолина. Соединение 215. Это соединение получают способом В с использованием цис-2,6-диметил-морфолина. Соединение 216. Это соединение получают способом В с использованием 3,3-диметилморфолина. Соединение 217. Это соединение получают способом В с использованием (1S,4S)-2-окса-5-аза-бицикло[2.2.1]гептанов. Соединение 218. Это соединение получают способом В с использованием 5-циклопропил-2-метил-2Н-пиразол-3-иламина. Соединение 219. Это соединение получают способом В с использованием морфолин-4-ил-пиперидин-4-ил-метанона. Соединение 220. Это соединение получают способом В с использованием 1-пиперазин-1-ил-этанона. Соединение 221. Это соединение получают способом В с использованием пиридазин-3-иламина. Соединение 222. Это соединение получают способом J с использованием пиридазин-3-иламина. Соединение 223. Это соединение получают способом J с использованием пиридин-3-иламина. Соединение 224. Это соединение получают способом J с использованием (4-аминофенил)ацетонитрила. Соединение 225. Это соединение получают способом J с использованием (2-аминофенил)ацетонитрила. Соединение 226. Это соединение получают способом J с использованием (4-аминофенил)ацетонитрила. Соединение 227. Это соединение получают способом J с использованием 4-аминобензамида. Соединение 228. Это соединение получают способом J с использованием 3-аминобензамида. Соединение 229. Это соединение получают способом J с использованием пиримидин-2-иламина. Соединение 230. Это соединение получают способом J с использованием (1S,4S)-2-окса-5-аза-бицикло[2,2,1]гептана. Соединение 231. Это соединение получают способом J с использованием 2-фенилморфолина. Соединение 232. Это соединение получают способом J с использованием пиперидин-4-карбонитрила. Соединение 233. Это соединение получают способом J с использованием 4-фторпиперидина. Соединение 234. Это соединение получают способом J с использованием 4,4-дифторпиперидина. Соединение 235. Это соединение получают способом J с использованием 6-(4-метилпиперазин-1-ил)пиридин-3-иламина. Соединение 236. Это соединение получают способом J с использованием 6-метоксипиридин-3-иламина. Соединение 237. Это соединение получают способом J с использованием 6-морфолин-4-ил-пиридин-3-иламина. Соединение 238. Это соединение получают способом Q с использованием фенола. Соединение 239. {5-[4-(6-Цианопиридин-3-ил)фенил]-[1,2,4]триазоло[1,5-а]пиридин-2-ил}амид циклопроланкарбоновой кислоты 4-(4,4,5,5-Тетраметил-[1,3,2]диоксаборолан-2-ил)бензонитрил (1,1 экв.) добавляют к раствору [5-(4-бромфенил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амида циклопропанкарбоновой кислоты, полученного способом А, в смеси 1,4-диоксан/вода (5:1). К раствору добавляют K 2 CO 3 (2 экв.) и Pd(dppf)Cl 2 (0,03 экв.) (dppf = 1,1"-бис(дифенилфосфино)ферроцен). Получающуюся в результате смесь затем греют в запаянной пробирке при 90 °С в течение 16 ч. Добавляют воду и раствор экстрагируют этилацетатом. Органические слои сушат над MgSO 4 и упаривают в вакууме. Конечное соединение получают после очистки препаративной высокоэффективной жидкостной хроматографией (HPLC). Аналитический HPLC: фирма Waters, колонки Acquity 2,1 мм (внутренний диаметр) × 50 мм (длина) с фазой UPLC ВЕН С18 с частицами размером 1,7 мкм (каталожный номер 186002350). Соединение 240. Это соединение получают способом J с использованием 4-трифторметил-пиперидина. Соединение 241. Это соединение получают способом J с использованием 1-(2,2,2-трифторэтил)пиперазина. Соединение 242. Это соединение получают способом J с использованием 4-гидроксипиперидина. Соединение 243. Это соединение получают способом J с использованием 2-пиперидин-4-ил-пропан-2-ола. Соединение 244. Это соединение получают способом J с использованием пиридин-2-иламина. Соединение 245. Это соединение получают способом J с использованием 2,4-дифтор-3-метокси-фениламина. Соединение 246. Это соединение получают способом J с использованием 2,6-дифторфениламина. Соединение 247. Это соединение получают способом J с использованием диэтилпиперидин-4-ил-амина. Соединение 248. Это соединение получают способом J с использованием 2-фтор-5-трифторметил-фениламина. Соединение 249. Это соединение получают способом J с использованием 3-амино-4-метил-бензамида. Соединение 250. Это соединение получают способом J с использованием пиперидин-4-ил-метанола. Соединение 251. Это соединение получают способом Q с использованием 3-гидроксибензамида. Соединение 252. Это соединение получают способом J с использованием диэтил-пирролидин-3-ил-амина. Соединение 253. Это соединение получают способом J с использованием (1R,4R)-2-этил-2,5-диаза-бицикло[2,2,1]гептана. Соединение 254. {5-[4-(3-Оксоморфолин-4-илметил)фенил]-[1,2,4]триазоло[1,5-а]пиридин-2-ил}амид циклопропанкарбоновой кислоты 254.1. 4-[4-(4,4,5,5-Тетраметил-[1,3,2]диоксаборолан-2-ил)бензил]морфолин-3-он NaH (81 мг, 3 экв.) добавляют к раствору морфолин-3-ону в DCM. 2-(4-Бромметилфенил)-4,4,5,5-тетраметил-[1,3,2]диоксаборолан добавляют к получающемуся в результате раствору и реакционную смесь перемешивают при комнатной температуре в течение 16 ч. DCM выпаривают с последующим добавлением воды. Раствор экстрагируют посредством EtOAc. Органические слои сушат над MgSO 4 и упаривают в вакууме с получением указанного в заглавии продукта, используемого на следующей стадии без дополнительной очистки. 254.2. {5-[4-(3-Оксоморфолин-4-илметил)фенил]-[1,2,4]триазоло[1,5-а]пиридин-2-ил}амид циклопропанкарбоновой кислоты Соединение, указанное в заглавии, затем синтезируют способом А. Соединение 255. Это соединение получают способом J с использованием 3-амино-4-метокси-бензамида. Соединение 256. Это соединение получают способом J с использованием 2-фтор-6-метилпиридин-3-иламина. Соединение 257. Это соединение получают способом J с использованием 3,5-дифторпиридин-2-иламина. Соединение 258. Это соединение получают способом J с использованием 4-амино-3-фтор-бензонитрила. Соединение 259. Это соединение получают способом J с использованием 2-фтор-4-метил-фениламина. Соединение 260. Это соединение получают способом J с использованием пирролидина. Соединение 261. Это соединение получают способом J с использованием анилина. Соединение 262. Это соединение получают способом J с использованием N-метил-N-пирролидин-3-ил-ацетамида. Соединение 263. Это соединение получают способом J с использованием диметилпирролидин-3-ил-амина. Соединение 264. Это соединение получают способом J с использованием 3,3-дифторпирролидина. Соединение 265. Это соединение получают способом J с использованием 4-(азетидин-3-илметокси) бензонитрила. Соединение 266. Это соединение получают способом U с использованием пиперидина. Соединение 267. Это соединение получают способом U с использованием тиоморфолин-1,1-диоксида. Соединение 268. [5-(4-Аминометилфенил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амид циклопропанкарбоновой кислоты (1 экв.), карбонилдиимидазол (CDI) (1,1 экв.) и триэтиламин (Et 3 N) (2,5 экв.) в DCM смешивают вместе при 50 °С в течение 1 ч. Растворитель выпаривают и получающуюся в результате смесь растворяют в DMF. К полученной смеси добавляют N-метилпиразин. Раствор перемешивают при 50 °С в течение 18 ч. После завершения реакции добавляют воду и органическую фазу экстрагируют посредством EtOAc. Органические слои сушат над MgSO 4 и упаривают в вакууме с получением указанного в заглавии продукта, очищаемого флэш-хроматографией. Соединение 269. Морфолин (1 экв.), карбонилдиимидазол (CDI) (1,1 экв.) и триэтиламин (Et 3 N) (2,5 экв.) в THF смешивают вместе при кипячении с обратным холодильником в течение 18 ч. Растворитель выпаривают и получающуюся в результате смесь растворяют в ацетонитриле. К получающемуся в результате раствору добавляют метилйодид. Реакционной смеси дают перемешиваться при комнатной температуре в течение 18 ч. Растворитель выпаривают и получающуюся в результате смесь растворяют в DMF. [5-(4-аминометилфенил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амид циклопропанкарбоновой кислоты (1 экв.) и Et 3 N (2,5 экв.) добавляют к раствору, который затем оставляют перемешиваться при комнатной температуре в течение 18 ч. Добавляют воду и раствор экстрагируют посредством EtOAc. Органические слои сушат над MgSO 4 и упаривают в вакууме с получением указанного в заглавии продукта, очищаемого флэш-хроматографией . Соединение 268. Это соединение получают способом V с использованием циклобутанкарбонил-хлорида. Соединение 269. Это соединение получают способом В с использованием диметилпиперидин-3-ил-амина. Соединение 270. Это соединение получают способом В с использованием пиперидин-3-ола. Соединение 271. Это соединение получают способом В с использованием 3,3-дифторпирролидина. Соединение 272. Это соединение получают способом V с использованием циклопропанкарбонил-хлорида. Соединение 273. Это соединение получают способом В с использованием (1,1-диоксо-тетрагидротиофен-3-ил)метиламина. Соединение 274. Это соединение получают способом В с использованием амида пиперидин-4-карбоновой кислоты. Соединение 275. Это соединение получают способом В с использованием амида пиперидин-2-карбоновой кислоты. Соединение 276. Это соединение получают способом В с использованием пиперидин-3-ил-метанола. Соединение 277. Это соединение получают способом В с использованием пиперазин-2-она. Соединение 278. Это соединение получают способом J с использованием 4-(азетидин-3-илокси) бензонитрила. Соединение 279. Это соединение получают способом J с использованием трет-бутилового сложного эфира азетидин-3-ил-карбаминовой кислоты. Соединение 280. Это соединение получают способом J с использованием 4-трифторметил-пиперидина. Соединение 281. Это соединение получают способом J с использованием 4-метоксипиперидина. Соединение 282. Это соединение получают способом J с использованием 4-этоксипиперидина. Соединение 283. Это соединение получают способом J с использованием N-азетидин-3-ил-N-метил-ацетамида. Соединение 284. Сырой [5-(4-бромфенил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амид циклопропанкарбоновой кислоты (1,0 экв.), полученный способом А, смешивают вместе с 4-(азетидин-3-илоксиметил)бензонитрилом (1,2 экв.) и трет-бутоксидом калия (2,0 экв.) в безводном 1,4-диоксане (1 мл). Смесь перемешивают при 80 °С под азотом. После этого к реакционной смеси добавляют шприцом Pd(OAc) 2 (0,1 экв.) и 2,2'-бис(дифенилфосфино)-1,1'-бинафтил (BINAP) (0,1 экв.) в безводном 1,4-диоксане (1 мл). Реакционную смесь перемешивают в течение ночи. LCMS показывает наличие желаемого продукта. Смесь фильтруют и подвергают очистке на препаративном высокоэффективном жидкостном хроматографе (HPLC) с получением чистого продукта. Соединение 285. Это соединение получают способом В с использованием диэтилпирролидин-3-ил-амина. Соединение 286. Это соединение получают способом В с использованием 4-фенилпиперидин-4-ола. Соединение 287. Это соединение получают способом В с использованием N-азетидин-3-ил-ацетамида. Соединение 288. Сырой [5-(4-бромфенил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амид циклопропанкарбоновой кислоты (1,0 экв.), полученный способом А, смешивают вместе с азетидин-3-карбонитрилом (1,2 экв.) и трет-бутоксидом калия (2,0 экв.) в безводном 1,4-диоксане (1 мл) . Смесь перемешивают при 80 °С под азотом. После этого к реакционной смеси добавляют шприцом Pd(OAc) 2 (0,1 экв.) и BINAP (0,1 экв.) в безводном 1,4-диоксане (1 мл). Реакционную смесь перемешивают в течение ночи. LCMS показывает наличие желаемого продукта. Смесь фильтруют и подвергают очистке на препаративном высокоэффективном жидкостном хроматографе (HPLC) с получением чистого продукта. Соединение 289. Это соединение получают способом В с использованием диметил-пирролидин-3-ил-амина. Соединение 290. Это соединение получают способом В с использованием пиперидин-1-ил-пиперидин-3-ил-метанона. Соединение 291. Это соединение получают способом J с использованием азетидин-3-ил-диметил-амина. Соединение 292. Это соединение получают способом В с использованием 3-(пиперидин-4-илметокси)пиридина. Соединение 293. Это соединение получают способом В с использованием 4-метоксипиперидина. Соединение 294. Это соединение получают способом В с использованием 4-этоксипиперидина. Соединение 295. Это соединение получают способом В с использованием диэтиламида пиперидин-3-карбоновой кислоты. Соединение 296. Это соединение получают способом В с использованием N-пиперидин-3-ил-ацетамида. Соединение 297. Это соединение получают способом Р с использованием 5-хлорметилпиридин-2-карбонитрила. Соединение 298. Это соединение получают способом J с использованием азетидин-3-илметил-диметиламина. Соединение 299. Это соединение получают способом J с использованием диметиламида азетидин-3-карбоновой кислоты. Соединение 300. Это соединение получают способом J с использованием 4-пиперидин-4-ил-морфолина. Соединение 301. Это соединение получают способом Q с использованием (4-гидрокси-фенил)ацетонитрила. Соединение 302. Это соединение получают способом J с использованием изоксазол-3-иламина. Соединение 303. Это соединение получают способом J с использованием азетидин-3-карбонитрила. Соединение 304. Это соединение получают способом J с использованием 1,1-диоксо-тетрагидротиофен-3-иламина. Соединение 305. Это соединение получают способом J с использованием (S)-пирролидин-3-ола. Соединение 306. Это соединение получают способом J с использованием 2-аминобензамида. Соединение 307. Это соединение получают способом J с использованием (R)-пирролидин-3-ола. Соединение 308. [5-(4-Аминометилфенил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амид циклопропанкарбоновой кислоты (1,0 экв.), карбонилдиимидазол (CDI) (1,1 экв.) и триэтиламин (Et 3 N) (2,5 экв.) в DCM смешивают вместе при 50 °С в течение 1 ч. Растворитель выпаривают и получающуюся в результате смесь растворяют в DMF. К полученной смеси добавляют N-метилпиразин. Раствор перемешивают при 50 °С в течение 18 ч. После завершения реакции добавляют воду и органическую фазу экстрагируют посредством EtOAc. Органические слои сушат над MgSO 4 и упаривают в вакууме с получением указанного в заглавии продукта, очищаемого флэш-хроматографией. Соединение 309. Морфолин (1 экв.), карбонилдиимидазол (CDI) (1,1 экв.) и триэтиламин (Et 3 N) (2,5 экв.) в THF смешивают вместе при кипячении с обратным холодильником в течение 18 ч. Растворитель выпаривают и получающуюся в результате смесь растворяют в ацетонитриле. К получающемуся в результате раствору добавляют метилйодид. Реакционную смесь оставляют перемешиваться при комнатной температуре в течение 18 ч. Растворитель выпаривают и получающуюся в результате смесь растворяют в DMF. [5-(4-Аминометилфенил)-[1,2,4]триазоло[1,5-а]пиридин-2-ил]амид циклопропанкарбоновой кислоты (1,0 экв.) и Et 3 N (2,5 экв.) добавляют к раствору, который затем оставляют перемешиваться при комнатной температуре в течение 18 ч. Добавляют воду и раствор экстрагируют посредством EtOAc. Органические слои сушат над MgSO 4 и упаривают в вакууме с получением указанного в заглавии продукта, очищаемого флэш-хроматографией. Соединение 310. Это соединение получают способом J с использованием трет-бутилового сложного эфира пиперидин-4-ил-карбаминовой кислоты. Соединение 311. Это соединение получают способом J с использованием пиперазин-2-она. Соединение 312. Это соединение получают способом J с использованием циклопропиламина. Соединение 313. Это соединение получают способом J с использованием 3-гидроксипиперидина. Соединение 314. Это соединение получают способом J с использованием 3,3-диметилазетидина. Соединение 315. Это соединение получают способом J с использованием 3,4-дифторазетидина. Соединение 316. Это соединение получают способом U с использованием пиридин-3-иламина. Соединение 317. Это соединение получают способом U с использованием 3,3-дифторазетидина. Соединение 318. Это соединение получают способом U с использованием азетидина. Соединение 319. Это соединение получают способом U с использованием 4-трифторметил-пиперидина. Соединение 320. Это соединение получают способом U с использованием 4,5-диметилпиперидина. Соединение 321. Это соединение получают способом U с использованием 3-метоксиазетидина. Соединение 322. Это соединение получают способом U с использованием N-азетидин-3-ил-ацетамида. Соединение 323. Это соединение получают способом U с использованием N-пиперидин-4-ил-ацетамида. Соединение 324. Это соединение получают способом U с использованием диметиламида азетидин-3-карбоновой кислоты. Соединение 325. Это соединение получают способом В с использованием 4-(азетидин-3-илоксиметил)бензонитрила. Соединение 326. Это соединение получают способом В с использованием 4-азетидин-3-ил-морфолина. Соединение 327. Это соединение получают способом В с использованием азетидин-3-ил-диметил-амина. Соединение 328. Это соединение получают способом В с использованием азетидин-3-карбонитрила. Соединение 329. Это соединение получают способом В с использованием азетидин-3-илметил-диметиламина. Соединение 330. Это соединение получают способом В с использованием 3,3-диметилазетидина. Соединение 331. Это соединение получают способом В с использованием 1Н-[1,2,4]триазол-3-иламина. Показательные соединения, которые были получены или могут быть получены в соответствии со способами синтеза, описанными в этом документе, приведены в табл. I ниже. Данные ЯМР-спектров некоторых репрезентативных соединений изобретения даны в табл. II. Пример 1. Методы анализа in vitro. Пример 1.1. Анализ ингибирования JAK1-киназы. Каталитический домен рекомбинантной JAK1-киназы человека (аминокислоты 850-1154; каталожный номер 08-144) закупают в компании Carna Biosciences. 10 нг JAK1-киназы инкубируют с 12,5 мкг субстрата polyGT (Sigma, каталожный номер Р0275) в буферном растворе для проведения реакции киназы (конечные концентрации: 15 мМ Трис-HCl рН 7,5, 1 мМ DTT (дитиотреитол), 0,01% Твин-20, 10 мМ MgCl 2 , 2 мкМ нерадиоактивного АТФ (аденозин-5'-трифосфат), 0,25 мкКи 33Р- γ-АТФ (GE Healthcare, каталожный номер АН9968)) с 5 мкл, содержащими испытуемое соединение или среду для лекарства (DMSO, конечная концентрация 1%) или без таковых, в общем объеме 25 мкл, в полипропиленовом 96-луночном планшете (Greiner, V-образная форма дна). По истечении 45 мин при 30 °С реакции прерывают посредством добавления 25 мкл/лунку 150 мМ фосфорной кислоты. Все "прерванные реакции" киназы переносят на предварительно промытые (75 мМ раствор фосфорной кислоты) 96-луночные фильтр-планшеты (Perkin Elmer, каталожный номер 6005177) с использованием харвестера клеток (Perkin Elmer). Планшеты промывают 6 раз посредством 300 мкл на лунку 75 мМ раствора фосфорной кислоты и дно планшетов герметизируют. Добавляют 40 мкл/лунку сцинтилляционной жидкости Microscint-20, верхнюю часть планшетов герметизируют и выполняют считывание с использованием сцинтилляционного счетчика Topcount (Perkin Elmer). Активность киназы вычисляют путем вычитания числа импульсов в минуту (cpm), полученного в присутствии ингибитора позитивного контроля (10 мкМ стауроспорина) из числа импульсов в минуту (cpm), полученного в присутствии среды для лекарства. Способность испытуемого соединения ингибировать эту активность определяют следующим образом. Ингибирование, выраженное в % = ((cpm, определенное для образца с присутствующим в нем испытуемым соединением - cpm, определенное для образца с ингибитором позитивного контроля)/(cpm, определенное в присутствии среды для лекарства - cpm, определенное для образца с ингибитором позитивного контроля)) ∙100%. Серии с разведением дозы приготавливают для соединений, которые могут быть испытаны в отношении дозозависимых эффектов в анализе JAK1-киназы и для которых могут быть вычислены IC 50 для каждого соединения. Каждое соединение обычно испытывают при концентрации 20 мкМ с последующим последовательным разведением на 1/3, то есть берут 8 точек (20 мкМ - 6,67 мкМ - 2,22 мкМ -740 нМ - 247 нМ - 82 нМ - 27 нМ - 9 нМ) в конечной концентрации DMSO 1%. При увеличении эффективности серии соединения делают большие разведения и/или снижают верхнюю концентрацию (например, 5 мкМ, 1 мкМ). Полуколичественная оценочная шкала: * > 1000 нМ ** 501-1000 нМ *** 101-500 нМ **** <100 нМ Пример 1.2. Анализ ингибирования JAK2-киназы. Каталитический домен рекомбинантной JAK2-киназы человека (аминокислоты 808-1132; каталожный номер PV4210) закупают в компании Invitrogen. 0,025 мЕд. JAK2-киназы инкубируют с 2,5 мкг субстрата polyGT (Sigma, каталожный номер Р0275) в буферном растворе для проведения реакции киназы (конечные концентрации: 5 мМ MOPS (3-N-морфолинпропансульфоновая кислота) рН 7,5, 9 мМ MgAc, 0,3 мМ EDTA (этилендиаминтетрауксусная кислота), 0,06% Brij (неионное ПАВ) и 0,6 мМ DTT (дитиотреитол), 1 мкМ нерадиоактивного АТФ, 0,25 мкКи 33Р- γ-АТФ (GE Healthcare, каталожный номер АН9968)) с 5 мкл, содержащими испытуемое соединение или среду для лекарства (DMSO, конечная концентрация 1%) или без таковых, в общем объеме 25 мкл, в полипропиленовом 96-луночном планшете (Greiner, V-образная форма дна). По истечении 90 мин при 30 °С реакции прерывают посредством добавления 25 мкл/лунку 150 мМ фосфорной кислоты. Все "прерванные реакции" киназы переносят на предварительно промытые (75 мМ раствор фосфорной кислоты) 96-луночные фильтр-планшеты (Perkin Elmer, каталожный номер 6005177) с использованием харвестера клеток (Perkin Elmer). Планшеты промывают 6 раз посредством 300 мкл на лунку 75 мМ раствора фосфорной кислоты и дно планшетов герметизируют. Добавляют 40 мкл/лунку сцинтилляционной жидкости Microscint-20, верхнюю часть планшетов герметизируют и выполняют считывание с использованием сцинтилляционного счетчика Topcount (Perkin Elmer). Активность киназы вычисляют путем вычитания числа импульсов в минуту (cpm), полученного в присутствии ингибитора позитивного контроля (10 мкМ стауроспорина) из числа импульсов в минуту (cpm), полученного в присутствии среды для лекарства. Способность испытуемого соединения ингибировать эту активность определяют следующим образом. Ингибирование, выраженное в % = ((cpm, определенное для образца с присутствующим в нем испытуемым соединением - cpm, определенное для образца с ингибитором позитивного контроля)/(cpm, определенное в присутствии среды для лекарства - cpm, определенное для образца с ингибитором позитивного контроля)) ∙100%. Серии с разведением дозы приготавливают для соединений, которые могут быть испытаны в отношении дозозависимых эффектов в анализе JAK2-киназы и для которых могут быть вычислены IC 50 для каждого соединения. Каждое соединение обычно испытывают при концентрации 20 мкМ с последующим последовательным разведением на 1/3, то есть берут 8 точек (20 мкМ - 6,67 мкМ - 2,22 мкМ -740 нМ - 247 нМ - 82 нМ - 27 нМ - 9 нМ) в конечной концентрации DMSO 1%. При увеличении эффективности серии соединения делают большие разведения и/или снижают верхнюю концентрацию (например, 5 мкМ, 1 мкМ). Полуколичественная оценочная шкала: # > 1000 нМ ## 501-1000 нМ ### 101-500 нМ #### <100 нМ Пример 1.3. Анализ ингибирования JAK3-киназы. Каталитический домен рекомбинантной JAK3-киназы человека (аминокислоты 781-1124; каталожный номер PV3855) закупают в компании Invitrogen. 0,025 мЕд. JAK3-киназы инкубируют с 2,5 мкг субстрата polyGT (Sigma, каталожный номер Р0275) в буферном растворе для проведения реакции киназы (конечные концентрации: 25 мМ Трис-HCl рН 7,5, 0,5 мМ EGTA (этиленгликольтетраукусусная кислота), 0,5 мМ Na 3 VO 4 , 5 мМ β-глицерилфосфата, 0,01% Triton Х-100, 1 мкМ нерадиоактивного АТФ, 0,25 мкКи 33Р- γ-АТФ (GE Healthcare, каталожный номер АН9968)) с 5 мкл, содержащими испытуемое соединение или среду для лекарства (DMSO, конечная концентрация 1%) или без таковых, в общем объеме 25 мкл, в полипропиленовом 96-луночном планшете (Greiner, V-образная форма дна). По истечении 105 мин при 30 °С реакции прерывают посредством добавления 25 мкл/лунку 150 мМ раствора фосфорной кислоты. Все "прерванные реакции" киназы переносят на предварительно промытые (75 мМ раствор фосфорной кислоты) 96-луночные фильтр-планшеты (Perkin Elmer, каталожный номер 6005177) с использованием харвестера клеток (Perkin Elmer). Планшеты промывают 6 раз посредством 300 мкл на лунку 75мМ раствора фосфорной кислоты и дно планшетов герметизируют. Добавляют 40 мкл/лунку сцинтилляционной жидкости Microscint-20, верхнюю часть планшетов герметизируют и выполняют считывание с использованием сцинтилляционного счетчика Topcount (Perkin Elmer). Активность киназы вычисляют путем вычитания числа импульсов в минуту (cpm), полученного в присутствии ингибитора позитивного контроля (10 мкМ стауроспорина) из числа импульсов в минуту (cpm), полученного в присутствии среды для лекарства. Способность испытуемого соединения ингибировать эту активность определяют следующим образом. Ингибирование, выраженное в % = ((cpm, определенное для образца с присутствующим в нем испытуемым соединением - cpm, определенное для образца с ингибитором позитивного контроля)/(cpm, определенное в присутствии среды для лекарства - cpm, определенное для образца с ингибитором позитивного контроля)) ∙100%. Серии с разведением дозы приготавливают для соединений, которые могут быть испытаны в отношении дозозависимых эффектов в анализе JAK3-киназы и для которых могут быть вычислены IC 50 для каждого соединения. Каждое соединение обычно испытывают при концентрации 20 мкМ с последующим последовательным разведением на 1/3, то есть берут 8 точек (20 мкМ - 6,67 мкМ - 2,22 мкМ -740 нМ - 247 нМ - 82 нМ - 27 нМ - 9 нМ) в конечной концентрации DMSO 1%. При увеличении эффективности серии соединения делают большие разведения и/или снижают верхнюю концентрацию (например, 5 мкМ, 1 мкМ). Полуколичественная оценочная шкала: + > 1000 нМ ++ 501-1000 нМ +++ 101-500 нМ ++++ <100 нМ N/A - нет данных Пример 1.4. Анализ ингибирования TYK2-киназы. Каталитический домен рекомбинантной TYK2-киназы человека (аминокислоты 871-1187; каталожный номер 08-147) закупают в компании Carna Biosciences. 5 нг TYK2-киназы инкубируют с 12,5 мкг субстрата polyGT (Sigma, каталожный номер Р0275) в буферном растворе для проведения реакции киназы (конечные концентрации: 25 мМ Hepes (N-2-гидроксиэтилпиперазин-N-2-этансульфоновая кислота) рН 7,5, 100 мМ NaCl, 0,2 мМ Na 3 VO 4 , 0,1% NP-40, 0,1 мкМ нерадиоактивного АТФ, 0,125 мкКи 33Р- γ-АТФ (GE Healthcare, каталожный номер АН9968)) с 5 мкл, содержащими испытуемое соединение или среду для лекарства (DMSO, конечная концентрация 1%) или без таковых, в общем объеме 25 мкл, в полипропиленовом 96-луночном планшете (Greiner, V-образная форма дна). По истечении 90 мин при 30 °С реакции прерывают посредством добавления 25 мкл/лунку 150 мМ раствора фосфорной кислоты. Все "прерванные реакции" киназы переносят на предварительно промытые (75 мМ раствор фосфорной кислоты) 96-луночные фильтр-планшеты (Perkin Elmer, каталожный номер 6005177) с использованием харвестера клеток (Perkin Elmer). Планшеты промывают 6 раз посредством 300 мкл на лунку 75мМ раствора фосфорной кислоты и дно планшетов герметизируют. Добавляют 40 мкл/лунку сцинтилляционной жидкости Microscint-20, верхнюю часть планшетов герметизируют и выполняют считывание с использованием сцинтилляционного счетчика Topcount (Perkin Elmer). Активность киназы вычисляют путем вычитания числа импульсов в минуту (cpm), полученного в присутствии ингибитора позитивного контроля (10 мкМ стауроспорина), из числа импульсов в минуту (cpm), полученного в присутствии среды для лекарства. Способность испытуемого соединения ингибировать эту активность определяют следующим образом. Ингибирование, выраженное в % = ((cpm, определенное для образца с присутствующим в нем испытуемым соединением - cpm, определенное для образца с ингибитором позитивного контроля)/(cpm, определенное в присутствии среды для лекарства - cpm, определенное для образца с ингибитором позитивного контроля)) ∙100%. Серии с разведением дозы приготавливают для соединений, которые могут быть испытаны в отношении дозозависимых эффектов в анализе TYK2-киназы и для которых могут быть вычислены IC 50 для каждого соединения. Каждое соединение обычно испытывают при концентрации 20 мкМ с последующим последовательным разведением на 1/3, то есть берут 8 точек (20 мкМ - 6,67 мкМ - 2,22 мкМ -740 нМ - 247 нМ - 82 нМ - 27 нМ - 9 нМ) в конечной концентрации DMSO 1%. При увеличении эффективности серии соединения делают большие разведения и/или снижают верхнюю концентрацию (например, 5 мкМ, 1 мкМ). Полуколичественная оценочная шкала: - > 1000 нМ -- 501-1000 нМ --- 101-500 нМ ---- <100 нМ N/A - нет данных Пример 2. Методы клеточного анализа. Пример 2.1. Анализ передачи сигнала JAK-STAT-киназ. Клетки Hela поддерживают в модифицированной по способу Дульбекко Среде Игла (DMEM), содержащей 10% термоинактивированной фетальной телячьей сыворотки, 100 ед./мл пенициллина и 100 мкг/мл стрептомицина. Клетки Hela используют при 70%-ной конфлюэнтности для трансфекции. 20000 клеток в 87 мкл клеточной культуральной среды кратковременно трансфицируют посредством 40 нг репортера pSTAT1(2)-люциферазы (Panomics), 8 нг репортера LacZ в качестве репортера внутреннего контроля и 52 нг pBSK с использованием 0,32 мкл Jet-PEI (Polyplus) в качестве реагента трансфекции на лунку в формате 96-луночного планшета. После инкубирования в течение ночи при 37 °С 10% CO 2 среду трансфекции удаляют. Добавляют 75 мкл DMEM + 1,5% термоинактивированной фетальной телячьей сыворотки. В течение 60 мин добавляют 15 мкл соединения в концентрации 6,7 × и затем 10 мкл OSM (цитокин онкостатин) человека (Peprotech) в конечной концентрации 33 нг/мл. Все соединения испытывают в двух повторностях, исходя из 20 мкМ с последующим последовательным разведением на 1/3, то есть берут 8 доз в целом (20 мкМ - 6,6 мкМ - 2,2 мкМ - 740 нМ - 250 нМ - 82 нМ - 27 нМ- 9 нМ) в конечной концентрации DMSO 0,2%. После инкубирования в течение ночи при 37 °С 10% CO 2 клетки подвергают лизису в 100 мкл лизирующего буферного раствора/лунку (PBS (забуференный фосфатом физиологический раствор), 0,9 мМ CaCl 2 , 0,5 мМ MgCl 2 , 5% Trehalose (трегалоза), 0,025% Tergitol NP9, 0,15% BSA (бычий сывороточный альбумин)). 40 мкл клеточного лизата используют для считывания активности β-галактозидазы посредством добавления 180 мкл раствора βGal (30 мкл ONPG (о-нитрофенил- β-D-галактопиранозид) в концентрации 4 мг/мл + 150 мкл буферного раствора β-галактозидазы (0,06М Na 2 HPO 4 , 0,04M NaH 2 PO 4 , 1 мМ MgCl 2 ) ) в течение 20 мин. Реакцию прерывают путем добавления 50 мкл Na 2 CO 3 (1М). Поглощение считывают на длине волны 405 нм. Активность люциферазы измеряют с использованием 40 мкл клеточного лизата плюс 40 мкл Steadylite ® так, как описано производителем (Perkin Elmer), на приборе Envision (Perkin Elmer). В качестве позитивного контроля (100% ингибирование) используют 10 мкМ ингибитора pan-JAK-киназы. В качестве негативного контроля используют 0,5% DMSO (0% ингибирования). Позитивный контроль и негативный контроль используют для вычисления "численных" значений (z) и "выраженных в процентах" значений (PIN) ингибирования. Выраженное в процентах ингибирование = ((интенсивность флуоресценции, определяемая в присутствии среды для лекарства - интенсивность флуоресценции, определяемая для образца с присутствующим в нем испытуемым соединением), поделенная на (интенсивность флуоресценции, определенная в присутствии среды для лекарства - интенсивность флуоресценции, определенная для образца без триггера)) ∙100%. Значения PIN наносят на график для соединений, испытанных в зависимости от дозы, и получают значения EC 50 . * > 1000 пМ ** 501-1000 пМ *** 101-500 пМ **** 1-100 пМ Пример 2.2. Анализ передачи сигнала цитокинами OSM/IL-1 β. Показано, что цитокины OSM и IL-1 β синергистически повьшающе регулируют уровни металлопротеиназы ММР13 в клеточной линии SW1353 хондросаркомы человека. Производят посев клеток в 96-луночные планшеты в количестве 15000 клеток/лунку в объеме 120 мкл DMEM (модифицированная по способу Дульбекко среда Игла, Invitrogen), содержащей 10% (объем/объем) FBS (фетальная бычья сыворотка) и 1% пенициллина/стрептомицина (Invitrogen), инкубированной при 37 °С, 5% СО 2 . Клетки предварительно инкубируют с 15 мкл соединения в среде M199 с 2% DMSO за 1 ч до запуска активации посредством 15 мкл OSM и IL-1 β при достижении концентрации OSM 25 нг/мл и концентрации IL-1 β 1 нг/мл и уровни металлопротеиназы ММР13 измеряют в кондиционированной среде в течение 48 ч после запуска активации. Активность ММР13 измеряют с использованием анализа активности посредством ловушки для антител. Для этой цели на 384-луночные планшеты (NUNC, 460518, MaxiSorb black) наносят слой посредством 35 мкл раствора антитела к ММР13 человека с концентрацией 1,5 мкг/мл (R &D Systems, MAB511) в течение 24 ч при 4 °С. После промывания лунок 2 раза посредством PBS (забуференный фосфатом физиологический раствор) + 0,05% Твин, оставшиеся центры связывания блокируют посредством 100 мкл 5%-ного нежирного сухого молока (Santa Cruz, sc-2325, Blotto) в PBS в течение 24 ч при 4 °С. Далее лунки промывают 2 раза посредством PBS + 0,05% Твин и добавляют 35 мкл разбавленного в 10 раз раствора супернатанта культуры клеток, содержащего ММР13 в 100-кратно разбавленном блокирующем буферном растворе, и инкубируют в течение 4 ч при комнатной температуре. Далее лунки промывают дважды посредством PBS + 0,05% Твин с последующими активацией ММР13 путем добавления 35 мкл 1,5 мМ раствора ацетата 4-аминофенилртути (АРМА) (Sigma, A9563) и инкубированием при 37 °С в течение 1 ч. Лунки промывают вновь посредством PBS + 0,05% Твин, и добавляют 35 мкл субстрата ММР13 (Biomol, P-126, флуорогенный субстрат OmniMMP). После инкубирования в течение 24 ч при 37 °С, измеряют интенсивность флуоресценции конвертированного субстрата на аппарате для прочтения планшетов с использованием нескольких меток Wallac Envision 2102, Perkin Elmer (возбуждение на длине волны: 320 нм, испускание на длине волны: 405 нм). Выраженное в процентах ингибирование = ((интенсивность флуоресценции, определяемая в присутствии среды для лекарства - интенсивность флуоресценции, определяемая для образца с присутствующим в нем испытуемым соединением)/(интенсивность флуоресценции, определенная в присутствии среды для лекарства - интенсивность флуоресценции, определенная для образца без вещества, способствующего запуску активации)) ∙100%. * > 1000 нМ ** 501-1000 нМ *** 1-500 нМ Пример 2.3. Анализ пролиферации PBL (лимфоцита периферической крови). Лимфоциты периферической крови человека (PBL) стимулируют посредством IL-2 и измеряют пролиферацию с использованием анализа методом инкорпорации. BrdU (бромдезоксиуридин). Сначала PBL стимулируют в течение 72 ч посредством РНА (фитогемагглютинин) с тем, чтобы индуцировать рецептор IL-2, фиксируют в течение 24 ч для прекращения пролиферации клетки с последующей стимуляцией IL-2 в течение последующих 72 ч (включая 24 ч введения метки BrdU). Клетки предварительно инкубируют с испытуемыми соединениями в течение 1 ч перед добавлением IL-2. Клетки культивируют в среде RPMI 1640, содержащей 10% (объем/объем) FBS (фетальная бычья сыворотка). Пример 3. Модели in vivo. Пример 3.1. Экспериментальная модель артрита: CIA (коллагениндуцированный артрит). 3.1.1. Вещества. Полный адъювант Фрейнда (CFA) и неполный адъювант Фрейнда (IFA) закупают в компании Difco. Бычий коллаген II типа (CII), липополисахарид (LPS) и этанерцепт Enbrel получают в компании Chondrex (Isle d'Abeau, France); Sigma (P4252, L'Isle d'Abeau, France), Whyett (шприц для инъекций на 25 мг, France) Acros Organics (Palo Alto, CA) соответственно. Все другие используемые реагенты представляют собой химически чистый сорт, и все растворители имеют аналитическую степень чистоты. 3.1.2. Животные. Крысы Dark Agouti (самцы, возраст 7-8 недель) получают в компании Harlan Laboratories (Maison-Alfort, France). Крыс выдерживают в режиме 12-часового цикла свет/темнота (0700-1900). Температуру поддерживают на уровне 22 °С и пищу и воду обеспечивают ad libitum (по желанию, вволю). 3.1.3. Коллагениндуцированный артрит (CIA). За один день до эксперимента приготавливают раствор бычьего коллагена II типа (CII) (2 мг/мл) с 0,05М уксусной кислотой и хранят при 4 °С. Непосредственно перед иммунизацией равные объемы адъюванта (IFA) и CII смешивают посредством гомогенизатора в заранее охлажденной бутылке на водно-ледяной бане. Если эмульсия не образуется, то могут потребоваться дополнительный адъювант и продленная гомогенизация. 0,2 мл эмульсии вводят внутрикожно у основания хвоста каждой крысе в 1 день, вторую повторную внутрикожную бустер-инъекцию (раствор CII в концентрации 2 мг/мл в 0,1 мл физиологического раствора с CFA) осуществляют на 9 день. Такой способ иммунизации получают путем модификации опубликованных способов (Sims N.A. et al., (2004) Targeting osteoclasts with zoledronic acid prevents bone destruction in collagen-induced arthritis, Arthritis Rheum. 50 2338-2346; Jou et al., 2005). 3.1.4. План исследования. Терапевтические эффекты испытуемых соединений проверяют на экспериментальных моделях артрита (коллагениндуцированный артрит = CIA) на крысах. Крыс случайным образом делят на две равные группы, и каждая группа содержит 10 крыс. Всех крыс иммунизируют в 1 день и делают повторную иммунизацию на 9 день. Терапевтическое введение доз длится с 16 дня по 30 день. Группу негативного контроля обрабатывают средой для лекарства (0,5% МС = метилцеллюлоза) и группу позитивного контроля обрабатывают препаратом Enbrel (10 мг/кг, 3 раза в неделю, подкожно). Соединение, представляющее интерес, обычно испытывают в трех дозах, например 3, 10, 30 мг/кг, перорально. 3.1.5. Клиническая оценка артрита. Артрит оценивают в показателях в соответствии со способом Khachigian 2006, Lin et al., 2007 и Nishida et al., 2004. Разбухание каждой из четырех лап классифицируют по артритической шкале следующим образом: 0 - симптомы отсутствуют; 1 - слабо выраженное, но определенное покраснение и разбухание одного типа сустава, такого как голеностопный или лучезапястный, или очевидное покраснение и набухание, ограниченное отдельными пальцами, независимо от числа пораженных пальцев; 2 - умеренное покраснение и набухание двух или более типов суставов; 3 - сильное покраснение и набухание всей лапы, включая пальцы; 4 - максимально воспаленная конечность с вовлечением нескольких суставов (максимальный кумулятивный клинический показатель артрита 16 на животное) (Nishida et al., 2004). 3.1.6. Изменение массы тела (%) после начала артрита. Клинически потеря массы тела связана с артритом (Shelton et al., 2005; Argiles et al., 1998; Rall, 2004; Walsmith et al., 2004). Таким образом, изменения массы тела после начала артрита могут быть использованы в качестве неспецифической конечной точки для оценки эффекта терапевтических средств в экспериментальной модели на крысах. Изменение массы тела (%) после начала артрита вычисляют следующим образом: 3.1.7. Радиология. Делают рентгеновские снимки задних лап каждого отдельно взятого животного. Вслепую выбранный идентификационный номер присваивают каждому снимку и степень тяжести эрозии кости оценивают посредством двух независимых шкал с помощью радиологической балльной системы Larsen следующим образом: 0 - нормальное состояние с неизменными костными контурами и с нормальной суставной щелью; 1 - легкая аномалия (отклонение от нормы) в любой одной или в двух внешних плюсневых костях, показывающая легкую эрозию кости; 2 - определенная ранняя аномалия (отклонение от нормы) в любых трех-пяти внешних плюсневых костях, показывающая эрозию кости; 3 - средняя деструктивная аномалия (отклонение от нормы) во всех внешних плюсневых костях, а также в любой одной или в двух внутренних плюсневых костях, показывающая определенные виды эрозии кости; 4 - сильная деструктивная аномалия во всех плюсневых костях, показывающая определенную эрозию кости и по меньшей мере в одном из внутренних плюсневых суставов, полностью эрозированных, оставляющая некоторые контуры костных суставов частично сохраненными; 5 - калечащая аномалия без сохранения костных контуров. Такая балльная система представляет собой модификацию системы, предложенной Salvemini et al., 2001; Bush et al., 2002; Sims et al., 2004; Jou et al., 2005. 3.1.8. Гистология. После радиологического анализа задние лапы мышей фиксируют в 10% забуференном фосфатом формалине (рН 7,4), декальцинируют посредством быстродействующего декальцинирующего вещества для проведения тонкой гистологии (Laboratories Eurobio) и вдавливают в парафин. Для обеспечения экстенсивного оценивания артритических суставов делают по меньшей мере четыре последовательных среза (5 мкм толщиной), и каждый ряд срезов отстоит от другого на расстоянии 100 мкм. Срезы окрашивают гематоксилином и эозином (Н &Е). Гистологические исследования на предмет синовиального воспаления и разрушения кости и хряща выполняют "двойным слепым" методом. В каждой лапе оценивают четыре параметра с использованием четырехбалльной шкалы. Параметры представляют собой клеточную инфильтрацию, тяжесть поверхностного диффузного сосудистого кератита, эрозию хряща и эрозию кости. Присваивание баллов выполняют следующим образом: 1 - нормальный, 2 - слабо выраженный, 3 - умеренный, 4 - значительно выраженный. Эти четыре балла суммируют вместе и представляют в качестве дополнительного показателя, то есть "общего счета симптомов RA (ревматоидный артрит)". 3.1.9. Анализ методом компьютерной микротомографии ( μСТ) пяточной кости (кости пятки). Деградация кости, наблюдаемая при ревматоидном артрите (RA), в особенности возникает в трубчатой кости и может быть обнаружена посредством анализа методом компьютерной микротомографии (Sims N.A. et al., 2004; Oste L. et al., ECTC Montreal, 2007). После сканирования и 3D-объемной реконструкции пяточной кости измеряют деградацию кости в виде числа дискретных объектов, присутствующих в микроскопическом препарате (препарат среза ткани), выделенного in silico перпендикулярно продольной оси кости. Чем больше кость, которая была подвергнута деградации, тем больше дискретных объектов, которые измеряют. Анализируют 1000 срезов, равномерно распределенных вдоль пяточной кости (с интервалом приблизительно 10,8 мкм). 3.1.10. Результаты. Следующие соединения являются эффективными во всех выборках данных, представленных в изучении коллагениндуцированного артрита (CIA) на крысах, со статистическим уровнем значимости в нескольких выборках: 18, 37, 145, 176, 200, 215 и 330. Пример 3.2. Экспериментальная модель септического шока. Инъекция липополисахарида (LPS) индуцирует быстрое высвобождение растворимого фактора некроза опухоли (TNF- α) в периферию. Такую модель используют для проведения анализа перспективных блокаторов высвобождения TNF- α in vivo. Шесть мышей-самок BALB/cJ (20 г) на группу обрабатывают посредством планируемого однократного введения дозы, перорально (ро). Через 30 мин делают инъекцию LPS (15 мкг/кг; серотип 0111:В4 Е. Coli) интраперитонеально (ip). Спустя 90 мин мышей подвергают эвтаназии и собирают кровь. Уровни циркулирующего TNF- α определяют с использованием коммерчески доступного набора реагентов ELISA. Дексаметазон (5 мкг/кг) используют в качестве эталонного противовоспалительного соединения. Выбранные соединения испытывают в одной или в нескольких дозах, например 3, и/или 10, и/или 30 мг/кг, перорально (ро). Следующие соединения проявляют статистически достоверное снижение высвобождения TNF- α (> 50%) при 30 мг/кг перорально (ро): 12, 18, 36, 37, 52, 60, 74, 125, 148, 176, 197, 200, 207, 208, 215 и 229. Пример 3.3. Экспериментальная модель, симулированная посредством МАВ (моноклональное антитело). Экспериментальная модель МАВ позволяет сделать быструю оценку модуляции воспалительной реакции, подобной ревматоидному артриту, посредством терапевтических средств (Kachigian L.M. Nature Protocols (2006) 2512-2516: Collagen antibody-induced arthritis). Мышам DBA/J делают внутривенно (i.v.) инъекцию коктейля моноклональных антител (mAbs), направленных против коллагена II типа. Спустя один день, начинают обработку соединениями (среда для лекарства: 10% (об./об.) HP βCD (2-гидроксипропил- β-циклодекстрин)). Три дня спустя, мыши получают инъекцию LPS интраперитонеально (i.p.) (50 мкг/мышь), что приводит к быстрому началу воспаления. Обработку соединениями продолжают вплоть до 10 дней после инъекции mAbs. Воспаление отслеживают путем измерения набухания лап и регистрации клинического показателя для каждой лапы. Для того чтобы показать тяжесть воспаления, представляют кумулятивный клинический показатель артрита для четырех конечностей. Систему присвоения баллов применяют к каждой конечности с использованием шкалы от 0 до 4, где 4 отражает наиболее тяжелое воспаление. 0 - симптом отсутствует, 1 - слабовыраженный, но определенное покраснение и набухание одного типа сустава, такого как голеностоп или запястье, или очевидное покраснение и набухание, ограниченное отдельными пальцами, независимо от числа пораженных пальцев, 2 - умеренное покраснение и набухание двух или более типов суставов, 3 - сильное покраснение и набухание всей лапы, включая пальцы, 4 - максимально воспаленная конечность с вовлечением нескольких суставов. Следующие соединения: 36, 37, 176, введенные перорально (р.о.) в дозе 30 мг/кг и более, снижают клинический показатель со статистической достоверностью при 30 мг/кг и значительно снижают воспаление при дозах более чем 30 мг/кг. Пример 3.4. Экспериментальные модели онкологических заболеваний. Экспериментальные модели in vitro и in vivo для валидации эффективности небольших молекул по отношению к JAK2-обусловленным миелопролиферативным заболеваниям описаны в работах Wernig et al., Cancer Cell 13, 311, 2008 и Geron et al., Cancer Cell 13, 321, 2008. Пример 3.5. Экспериментальная модель IBD (воспалительное заболевание кишечника) на мышах. Экспериментальные модели in vitro и in vivo для валидации эффективности небольших молекул в отношении воспалительного заболевания кишечника описаны в работе: Wirtz et al., 2007. Пример 3.6. Экспериментальная модель астмы на мышах. Экспериментальные модели in vitro и in vivo для валидации эффективности небольших молекул в отношении астмы описаны в работе Nials et al., 2008; Ip et al., 2006; Pernis et al., 2002; Kudlacz et al., 2008. Пример 4. Экспериментальные модели токсичности метаболизма лекарственных средств и фармакокинетики и обеспечения безопасности. Пример 4.1. Термодинамическая растворимость. Раствор с 1 мг/мл испытуемого соединения приготавливают в 0,2М фосфатном буферном растворе с рН 7,4 или в 0,1М цитратном буферном растворе с рН 3,0 при комнатной температуре в стеклянной пробирке. Образцы вращают ротационным приводом STR 4 (Stuart Scientific, Bibby) со скоростью 3,0 при комнатной температуре в течение 24 ч. По истечении 24 ч 800 мкл образца переносят в пробирку Эппендорфа и центрифугируют 5 мин при 14000 об./мин. 200 мкл супернатанта образца затем переносят на фильтр-планшет Multiscreen Solubility Plate (Millipore, MSSLBPC50) и супернатант фильтруют (10-12'' Hg) с помощью вакуумного коллектора в чистый полипропиленовый 96-луночный планшет с V-образным дном Greiner (каталожный номер 651201). 5 мкл фильтрата разбавляют в 95 мкл (F20) того же самого буферного раствора, который используют для инкубирования, в планшете, содержащем стандартный калибровочный раствор (Greiner, каталожный номер 651201). Стандартный калибровочный раствор для соединения приготавливают заново в DMSO, исходя из 10 мМ базового раствора в DMSO, разбавленного в 2 раза в DMSO (5000 мкМ) и затем дополнительно разбавленного в DMSO вплоть до 19,5 мкМ. 3 мкл разбавленного раствора, полученного в результате серии разведения, начиная с 5000 мкМ раствора, затем переносят в 97 мкл смеси ацетонитрил-буферный раствор (50/50). Диапазон конечной концентрации составляет от 2,5 до 150 мкМ. Планшет герметизируют с помощью герметизирующих пленок (MA96RD-04S, www.kinesis.co.uk), и образцы измеряют при комнатной температуре на жидкостном хроматомасс-спектрометре (LCMS) (ZQ 1525 от компании Waters) в оптимизированных условиях с использованием Quanoptimize для определения соответственной массы молекулы. Образцы анализируют на жидкостном хроматомасс-спектрометре (LCMS) при скорости потока 1 мл/мин. Растворитель А представляет собой 15 мМ раствор аммиака, и растворитель В представляет собой ацетонитрил. Образец исследуют в условиях распыления положительных ионов на колонке XBridge (2,1 ×30 мм) с фазой С18, с частицами размером 3,5 мкМ от компании Waters. Градиент состава растворителя имеет общую продолжительность 2 мин и колеблется от 5% В до 95% В. Площади пиков анализируют с помощью пакета программного обеспечения Masslynx, площади пиков образцов наносят на график относительно калибровочной кривой с получением растворимости соединения. Значения растворимости представляют в мкМ или в мкг/мл. Пример 4.2. Водная растворимость. Исходя из 10 мМ базового раствора в DMSO приготавливают путем последовательного разведения разбавленные растворы соединения в DMSO. Растворы, полученные в результате серии разведения, переносят на F-образное дно 96-луночного планшета NUNC Maxisorb (каталожный номер 442404) и добавляют 0,2М фосфатный буферный раствор с рН 7,4 или 0,1М цитратный буферный раствор с рН 3,0 при комнатной температуре. Конечная концентрация колеблется в диапазоне от 200 до 2,5 мкМ в пяти этапах одинакового разведения. Конечная концентрация DMSO не превышает 2%. В угловые точки каждого 96-луночного планшета добавляют 200 мкМ Пирена, который служит в качестве базисной точки для калибровки Z-оси на микроскопе. Аналитические планшеты герметизируют и инкубируют в течение 1 ч при 37 °С и встряхивании при 230 об./мин (rpm). Затем планшеты сканируют под микроскопом белого света, что дает отдельно взятые снимки осадка относительно концентрации. Осадок анализируют и данные преобразуют в число, которое наносят на график. Первая концентрация, при которой соединение оказывается полностью растворенным, представляет собой концентрацию, которую сообщают, однако истинная концентрация лежит где-то между этой концентрацией и концентрацией, получаемой на последующем этапе разведения. Значения растворимости представляют в мкг/мл. Пример 4.3. Связывание белка плазмы (равновесный диализ). 10 мМ базовый раствор соединения в DMSO разбавляют в 5 раз в DMSO. Этот раствор дополнительно разбавляют в свежеразмороженной человеческой, крысиной, мышиной или собачьей плазме (BioReclamation INC) до конечной концентрации 10 мкМ и конечной концентрации DMSO 0,5% (5,5 мкл в 1094,5 мкл плазмы в полипропиленовом 96-луночном планшете PP-Masterblock (Greiner, каталожный номер 780285)). Подготавливают планшет со вставками Pierce Red Device (ThermoScientific, каталожный номер 89809) и заполняют, помещая 750 мкл PBS (забуференный фосфатом физиологический раствор) в камеру для буферного раствора и 500 мкл плазмы с добавлением известного количества определяемого вещества в камеру для плазмы. Планшет инкубируют в течение 4 ч при 37 °С и встряхивании при 230 об./мин. После инкубирования 120 мкл из обеих камер переносят в 360 мкл ацетонитрила в 96-луночных круглодонных, полипропиленовых глубоколуночных планшетах (Nunc, Каталожный номер 278743) и герметизируют с помощью крышки из алюминиевой фольги. Образцы смешивают и помещают на лед на 30 мин. Этот планшет затем центрифугируют 30 мин при 1200 rcf (относительная центробежная сила) при 4 °С и супернатант переносят в 96-луночный полипропиленовый планшет с v-образным дном (Greiner, 651201) для анализа на жидкостном хроматомасс-спектрометре (LCMS). Планшет герметизируют с помощью герметизирующих пленок (MA96RD-04S) (www.kinesis.co.uk) и образцы измеряют при комнатной температуре на жидкостном хроматомасс-спектрометре (ZQ 1525 от компании Waters) в оптимизированных условиях с использованием Quanoptimize для определения соответственной массы молекулы. Образцы анализируют на жидкостном хроматомасс-спектрометре (LCMS) при скорости потока 1 мл/мин. Растворитель А представляет собой 15 мМ раствор аммиака, и растворитель В представляет собой ацетонитрил. Образец исследуют в условиях распыления положительных ионов на колонке XBridge (2,1 ×30 мм) с фазой С18, с частицами размером 3,5 мкМ от компании Waters. Градиент состава растворителя имеет общую продолжительность 2 мин и колеблется от 5% В до 95% В. Полагают, что площадь пика от соединения в камере для буферного раствора и в камере для плазмы соответствует 100% соединения. Процентное содержание соединения, связанного с плазмой, получают из этих результатов и представляют в виде предела в среднеквадратическом (LIMS) как процентное содержание соединения, связанного с плазмой. Растворимость соединения в конечной концентрации испытания в PBS инспектируют с помощью микроскопа с тем, чтобы указать, наблюдают осаждение или нет. Пример 4.4. Подверженность к удлинению QT-интервала. Потенциал удлинения QT-интервала оценивают в анализе методом "открытия-закрытия" с использованием гена специфических калиевых каналов сердца (hERG) (метод оценки проницаемости клеточных ионных каналов с использованием пэтч-пипеток). 4.4.1. Общепринятый метод пэтч-клэмпа целой клетки (метод фиксации потенциала целой клетки). Регистрацию фиксации потенциала целой клетки проводят с использованием усилителя ЕРС10, регулируемого посредством программного обеспечения Pulse (v8.77) (НЕКА). Последовательное (добавочное) сопротивление обычно составляет менее чем 10 MQ (МОм) и компенсируется более чем на 60%, регистрация потенциала не является вычитаемой утечкой. Электроды производят из стекла для пипеток GC150TF (Harvard), сопротивление имеет величину от 2 до 3 МОм. "Внешний" промывающий раствор содержит 135 мМ NaCl, 5 мМ KCl, 1,8 мМ CaCl 2 , 5 мМ глюкозы, 10 мМ HEPES, рН 7,4. Внутренний раствор для пэтч-пипеток содержит 100 мМ K-глюконата, 20 мМ KCl, 1 мМ CaCl 2 , 1 мМ MgCl 2 , 5 мМ Na 2 ATP (аденозин-трифосфат натрия), 2 мМ глутатиона, 11 мМ EGTA (этиленгликоль-тетрауксусная кислота), 10 мМ HEPES (N-2-гидроксиэтилпиперазин-N-2-этансульфоновая кислота), рН 7,2. Лекарственные средства перфузируют с использованием быстродействующей перфузионной системы Biologic MEV-9/EVH-9. Все регистрации потенциала выполняют на клетках HEK293, стабильно экспрессирующих каналы hERG типа (гены специфических калиевых каналов сердца). Клетки культивируют на 12-миллиметровых круглых покровных стеклах (German glass, Bellco), закрепленных в записывающей показания камере с использованием двух платиновых стержней (Goodfellow). Токи калиевых каналов hERG вызывают с использованием активирующего импульса до +40 мВ в течение 1000 мс с последующим импульсом следового тока до -50 мВ в течение 2000 мс, исходный потенциал составляет -80 мВ. Импульсы прикладывают каждые 20 с, и все эксперименты выполняют при комнатной температуре. 4.4.2. Анализ данных. Значения IC 50 и IC 20 вычисляют для каждого испытуемого соединения. Вычисляют различие в кратности между концентрациями IC 20 и C max несвязанного соединения для испытуемого соединения, полученными при релевантных терапевтических дозах, которые определены по результатам, полученным из экспериментальной модели коллагениндуцированного артрита (CIA) на крысах. Для кривых зависимости реакции от концентрации измеряют пиковую (максимальную) амплитуду следового тока во время перепада напряжения -50 мВ. Подбор кривой по точкам данных концентрация-реакция выполняют с использованием уравнения где а - минимальная реакция, b - максимальная реакция, d - наклон Хилла, это уравнение может быть использовано для вычисления как IC 50 (где у=50 и с представляет собой значение IC 50 ), так и IC 50 (где у=20 и с представляет собой значение IC 50 ). Для подбора всех кривых используют программное обеспечение GraphPad ® Prism ® (Graphpad ® Software Inc.). Различие в 100 раз или более указывает на низкий потенциал удлинения интервала QT. Пример 4.5. Микросомальная стабильность. 10 мМ базовый раствор соединения в DMSO разбавляют 1000-кратно в 182 мМ фосфатном буферном растворе с рН 7,4 в 96-луночном глубоколуночном планшете (Greiner, каталожный номер 780285) и предварительно инкубируют при 37 °С. 40 мкл деионизированной воды добавляют в лунку полипропиленовой меченой штрихкодом Matrix 2D пробирки для хранения (Thermo Scientific) и предварительно инкубируют при 37 °С. Рабочий исходный раствор глюкоза-6-фосфатдегидрогеназы (G6PDH) приготавливают в 182 мМ фосфатном буферном растворе с рН 7,4 и помещают на лед перед использованием. Раствор Кофактора, содержащий MgCl 2 , глюкоза-6-фосфат и NADP+(никотинамид-аденин-динуклеотид-фосфат), приготавливают в деионизированной воде и помещают на лед перед использованием. Приготавливают конечный рабочий раствор, содержащий микросомы печени (Xenotech) видов, представляющих интерес (человек, мышь, крыса, собака), описанные выше растворы глюкоза-6-фосфат-дегидрогеназы (G6PDH) и кофакторов, и эту смесь инкубируют в течение не более чем 20 мин при комнатной температуре. 30 мкл предварительно нагретого разбавленного раствора соединения добавляют к 40 мкл предварительно нагретой воды в пробирках Matrix и добавляют 30 мкл микросомальной смеси. Конечные реакционные концентрации составляют 3 мкМ соединения, 1 мг микросом, 0,4 ед./мл GDPDH (глицеральдегид-3-фосфатдегидрогеназа), 3,3 мМ MgCl 2 , 3,3 мМ глюкоза-6-фосфата и 1,3 мМ NADP+. Для измерения процентного содержания оставшегося соединения в момент времени 0 добавляют МеОН или ACN (1:1) в лунку перед добавлением микросомальной смеси. Планшеты герметизируют с помощью уплотняющего герметика ТМ Matrix Sepra (Matrix, каталожный номер 4464) и встряхивают в течение нескольких секунд для гарантированного обеспечения полного смешения всех компонентов. Образцы, в которых реакция не была остановлена, инкубируют при 37 °С, 300 об./мин и по истечении 1 ч инкубирования реакцию прерывают посредством МеОН или ACN (1:1). После прерывания реакции образцы смешивают и помещают на лед на 30 мин для осаждения белков. Затем планшеты центрифугируют 30 мин при 1200 rcf (относительная центробежная сила) при 4 °С и супернатант переносят в 96-луночный полипропиленовый планшет с v-образным дном (Greiner, 651201) для проведения анализа на жидкостном хроматомасс-спектрометре (LCMS). Эти планшеты герметизируют с помощью герметизирующих пленок (MA96RD-04S) (www.kinesis.co.uk) и образцы измеряют при комнатной температуре на жидкостном хроматомасс-спектрометре (ZQ 1525 от компании Waters) в оптимизированных условиях с использованием Quanoptimize для определения соответственной массы исходной молекулы. Образцы анализируют на жидкостном хроматомасс-спектрометре (LCMS) при скорости потока 1 мл/мин. Растворитель А представляет собой 15 мМ раствора аммиака, и растворитель В представляет собой метанол или ацетонитрил в зависимости от используемого стоп-реагента. Образцы исследуют в условиях распыления положительных ионов на колонке XBridge (2,1 ×30 мм) с фазой С18, с частицами размером 3,5 мкМ от компании Waters. Градиент состава растворителя имеет общую продолжительность 2 мин и происходит в диапазоне от 5% В до 95% В. Полагают, что площадь пика для исходного соединения в момент времени 0 составляет 100% оставшегося соединения. Процентное содержание оставшегося соединения через 1 ч инкубирования вычисляют от момента времени 0 и представляют как процентное содержание оставшегося соединения. Растворимость соединения в конечной концентрации испытания в буферном растворе изучают с помощью микроскопа и результаты описывают. Данные по микросомальной стабильности выражают в виде процентного содержания соединения, оставшегося через 60 мин, относительно общего количества. * 0-25 ** 26-50 *** 51-75 **** 76-100 Пример 4.6. Проницаемость Сасо-2. Анализ двунаправленной проницаемости Сасо-2 проводят таким образом, как описано ниже. Клетки Сасо-2 получают из Европейской коллекции культивируемых клеток (ЕСАСС, каталожный номер 86010202) и используют после культивирования клеток в течение 21 дня в 24-луночных планшетах Transwell (Fisher TKT-545-020В). 2 ×10 5 клеток/лунку высевают в среду для планшетов, состоящую из DMEM (модифицированная по способу Дульбекко среда Игла)+GlutaMAXI (питательная среда)+1% NEAA (неосновная аминокислота)+10% FBS (фетальная бычья сыворотка, FetalClone II)+1% Pen/Strep (пенициллин/стрептомицин). Среду меняют каждые 2-3 дня. Испытуемые и эталонные соединения (пропранолол и родамин-123 или винбластин, все закупаемые в компании Sigma) приготавливают в сбалансированном солевом растворе Хенкса, содержащем 25 мМ HEPES (N-2-гидроксиэтилпиперазин-N-2-этансульфоновая кислота) (рН 7,4) и добавляют либо в апикальную (125 мкл), либо в базолатеральную (600 мкл) камеры сборного планшета Transwell в концентрации 10 мкМ с конечной концентрацией DMSO 0,25%. 50 мкМ флуоресцентного красителя Люцифер желтый (Sigma) добавляют к донорному буферному раствору во все лунки для оценивания целостности клеточных слоев путем отслеживания проникновения красителя Люцифер желтый. Поскольку краситель Люцифер желтый (LY) не может свободно проникать в липофильные барьеры, то высокая степень переноса красителя Люцифер желтый указывает на плохую целостность клеточного слоя. По истечении 1 ч инкубирования при 37 °С и встряхивании на орбитальном шейкере при 150 об./мин берут 70 мкл аликвот как из апикальной (А), так и из базальной (В) камеры и добавляют к 100 мкл раствора ацетонитрил:вода (50:50), содержащего аналитический внутренний стандарт (0,5 мкМ карбамазепин) в 96-луночном планшете. Краситель Люцифер желтый измеряют на приборе Spectramax Gemini XS (возбуждение на длине волны 426 нм и испускание на длине волны 538 нм) в чистом 96-луночном планшете, содержащем 150 мкл жидкости из базолатерального и апикального отсека. Концентрации соединения в образцах измеряют с помощью высокоэффективной жидкостной хроматографии/тандемной масс-спектрометрии (LC-MS/MS). Значения кажущейся проницаемости (Р арр ) вычисляют из соотношения где V - объем камеры, Т инк. - длительность инкубирования, площадь поверхности - 0,33 см 2 . Соотношения эффлюксов в качестве показателя активного эффлюкса из апикальной поверхности клетки вычисляют с использованием соотношения Р арр В> А/Р арр А> В. Используют следующие критерии приемлемости метода анализа: пропранолол: значение Р арр (А> В) > 20 ( ×10 -6 см/с), родамин-123 или винбластин: значение Р арр (А> В) <5 ( ×10 -6 см/с) с соотношением эффлюксов ≥5, проникновение красителя Люцифер желтый: ≤100 нм/с. Пример 4.7. Фармакокинетическое исследование грызунов. 3.1.3. Фармакокинетическое исследование. Соединения составляют в смеси на основе ПЭГ200/физиологического раствора или ПЭГ400/DMSO/физиологического раствора для внутривенного пути поступления вещества в организм и на основе 0,5% раствора метилцеллюлозы или 10-30% раствора гидроксипропил- β-циклодекстрина с рН 3 или рН 7,4 для перорального пути поступления вещества в организм. Испытуемые соединения перорально вводят дозами 5-10 мг/кг в виде единовременного/однократного искусственного питания через желудочный зонд и внутривенно вводят дозой 1 мг/кг в виде болюсного вливания через хвостовую вену. Каждая группа включает 3 крысы. Пробы крови отбирают либо через яремную вену с использованием канюлированных крыс или в ретроорбитальном синусе с литий-гепарином в качестве антикоагулянта в моменты времени в следующем диапазоне: 0,05-8 ч (внутривенный путь поступления вещества в организм), и 0,25-6 или 24 ч (пероральный путь поступления вещества в организм). Пробы цельной крови центрифугируют при 5000 об./мин в течение 10 мин и пробы получающейся в результате плазмы хранят при -20 °С вплоть до предстоящего проведения анализа. 3.1.4. Проведение количественной оценки уровней соединения в плазме крови. Плазменные концентрации каждого испытуемого соединения определяют методом жидкостной хроматографии/тандемной масс-спектрометрии (LC-MS/MS), в котором масс-спектрометр работает в режиме электрораспыления положительно заряженных ионов. 3.1.5. Определение фармакокинетических параметров. Фармакокинетические параметры вычисляют с использованием программы Winnonlin ® (Pharsight ®, Соединенные Штаты). Пример 4.8. 7-Дневное исследование токсичности на крысах. 7-Дневное исследование токсичности при пероральном введении испытуемых соединений осуществляют на крысах-самцах линии Sprague-Dawley для анализирования их токсического потенциала и токсикокинетических данных в суточных дозах 100, 300 и 500 мг/кг/сутки через зонд при постоянном объеме дозы 5 мл/кг/сутки. Испытуемые соединения составляют в смеси на основе 30% (об./об.) раствора HP βCD (2-гидроксипропил- β-циклодекстрин) в очищенной воде. Каждая группа включает 5 подопытных крыс-самцов, а также 3 контрольных животных для проведения анализа токсикокинетических параметров. Четвертой группе дают только 30% (об./об.) раствор HP βCD в воде с аналогичными частотой, объемом дозы и с использованием аналогичного пути введения, и эта группа служит в качестве контрольной группы, которая получает только среду для лекарства. Целью исследования является определение самой низкой дозы, которая не дает в результате нежелательных побочных реакций, которые можно выявить (уровень, не вызывающий видимых нежелательных побочных реакций - NOAEL). Специалистам в данной области будет ясно, что предыдущие описания являются показательными и пояснительными по сути и фактически предназначены для иллюстрации изобретения и его предпочтительных вариантов осуществления. В результате проведения экспериментов в соответствии с общепринятой практикой специалисту будут понятны модификации и вариации, которые могут быть выполнены без выхода за пределы объема этого изобретения. Таким образом, подразумевается, что изобретение определяется не вышеупомянутым описанием, а следующими пунктами формулы и их эквивалентами. Choy E.H., Panayi G.S. (2001). N Engl. J. Med. 344: 907-16. Chubinskaya S. and Kuettner K.E. (2003). Regulation of osteogenic proteins by chondrocytes. The international journal of biochemistry & cell biology 35(9)1323-1340. Clegg D.O. et al. (2006) N Engl. J. Med. 2006 354:795-808. Glucosamine, chondroitin sulfate, and the two in combination for painful knee osteoarthritis. Firestein G.S. (2003). Nature. 423:356-61. Kachigian L.M. (2006) Collagen antibody-induced arthritis, Nature Protocols 2512-2516. Lee D.M., Weinblatt M.E. (2001). Lancet. 358: 903-11. Legendre F., Dudhia J., Pujol J.-P., Bogdanowicz P. (2003) JAK/STAT but not ERK1/ERK2 pathway mediates interleuking (IL)-6/soluble IL-6R down-regulation of type II collagen, aggrecan core, and link protein transcription in articular chondrocytes. J. Biol. Chem. 278(5)2903-2912. Li W.Q., Dehnade F., Zafarullah M. (2001) Oncostatin M-induced matrix metalloproteinase and tissue inhibitor of metalloproteinase-3 genes expression in chondrocytes requires janus kinase/STAT signaling pathway. (2001) J. Immunol. 166:3491-3498. O'Dell J.R. (2004). Therapeutic strategies for rheumatoid arthritis. N. Engl. J. Med. 350(25):2591-602. Osaki M., Tan L., Choy B.K., Yoshida Y., Cheah K.S.E., Auron P.E., Goldring M.B. (2003) The TATA-conatining core promoter of the type II collagen gene (COL2A1) is the target of interferon-gamma-mediated inhibition in human chondrocytes: requirement for STAT1alpha, JAK1 and JAK2. Biochem J. 369:103-115. Oste L. et al., ECTC Montreal 2007: A high throughput method of measuring bone architectural disturbance in a murine CIA model by micro-CT morphometry. Otero M., Lago R., Lago F., Gomez Reino J.J., Gualillo O. (2005) Signalling pathway involved in nitric oxide synthase type II activation in chondrocytes: synergistic effect of leptin with interleukin-1. Arthritis Research & Therapy 7:R581-R591. Rodig S.J., Meraz M.A., White J.M., Lampe P.A., Riley J.K., Arthur C.D., King K.L., Sheehan K.C.F., Yin L., Pennica D., Johnson E.M., Schreiber R.D. (1998) Disruption of the JAK1 gene demonstrates obligatory and nonredundant roles of the jaks in cytokine-induced biologic responses Cell 93: 373-383. Sims N.A. et al., (2004) Targeting osteoclasts with zoledronic acid prevents bone destruction in collagen-induced arthritis, Arthritis Rheum 50 2338-2346. Smolen J.S., Steiner G. (2003). Nat Rev Drug Discov. 2: 473-88. Wernig et al. (2008) Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera, Cancer Cell. 13(4), 311-320. Geron et al. (2008) Selective inhibition of JAK2-driven erythroid differentiation of polycythemia vera progenitors Cancer Cell. 13 (4), 321-30. Wieland H.A., Michaelis M., Kirschbaum B.J., Rudolphi K.A. (2005). Nat. Rev. Drug. Discov. 4:331-44. Osteoarthritis - an untreatable disease? Wirtz et al. (2007) Mouse Models of Inflammatory Bowel Disease, Advanced Drag Delivery Reviews, 2007, 1073-1083. Tam, L., McGlynn, L.M., Traynor, P., Mukherjee, R., Bartlett, J.M.S., Edwards, J. (2007) British Journal of Cancer, 97, 378-383 Constantinescu et al., 2007, Trends in Biochemical Sciences 33(3): 122-131. Tetsuji Naka, Norihiro Nishimoto and Tadamitsu Kishimoto, Arthritis Res 2002, 4 (suppl 3):S233-S242. O'Shea, J.J., Pesu, M., Borie, D.C., Changelian, P.S., Nature Reviews, 2004, 555-564. Nials et al. (2008) Mouse Models of Allergic Asthma: Acute and Chronic Allergen Challenge, Disease Models & Mechanisms, 213-220. Ip et al. (2006) Interleukin (IL)-4 and IL-13 up-regulate monocyte chemoattractant protein-1 expression in human bronchial epithelial cells: involvement of p38 mitogen-activated protein kinase, extracellular signal-regulated kinase 1/2 and Janus kinase-2 but not c-Jun NH2-terminal kinase 1/ 2 signalling pathways, Clin Exp. Immun, 162-172. Pernis et al. (2002) JAK-STAT signaling in asthma J. Clin. Invest. 1279. Kudlacz et al. (2008) The JAK-3 inhibitor CP-690550 is a potent anti-inflammatory agent in a murine model of pulmonary eosinophilia, Eur. J. Pharmaco. 154-161. Mullighan C.G., Zhang J., Harvey R.C., Collins-Underwood J.R., Schulman B.A., Phillips L.A., Tasian S.K., Loh M.L., Su X., Liu W., Devidas M., Atlas S.R., Chen I.-M., Clifford R.J., Gerhard D.S., Carroll W.L., Reaman G.H., Smith M., Downing J.R., Hunger S.P., Willmane C.L.; (2009) JAK mutations in high-risk childhood acute lymphoblastic leukemia, PNAS May 22. [Epub ahead of print]. Argiles J.M., Lopez-Soriano F.J. (1998) Catabolic proinflammatory cytokines. Curr Opin Clin Nutr Metab Care. 1:245-51. Bush K.A., Farmer K.M., Walker J.S., Kirkham B.W. (2002) Reduction of joint inflammation and bone erosion in rat adjuvant arthritis by treatment with interleukin-17 receptor IgG1 Fc fusion protein. Arthritis Rheum. 46:802-5. Jou I.M., Shiau A.L., Chen S.Y., Wang C.R., Shieh D.B., Tsai C.S., Wu C.L. (2005) Thrombospondin 1 as an effective gene therapeutic strategy in collagen-induced arthritis. Arthritis Rheum. 52:339-44. Nishida K., Komiyama T., Miyazawa S., Shen Z.N., Furumatsu T., Doi H., Yoshida A., Yamana J., Yamamura M., Ninomiya Y., Inoue H., Asahara H. (2004) Histone deacetylase inhibitor suppression of autoantibody-mediated arthritis in mice via regulation of p16INK4a and p21(WAFl/Cipl) expression. Arthritis Rheum. 10: 3365-76. Rall L.C., Roubenoff R. (2004) Rheumatoid cachexia: metabolic abnormalities, mechanisms and interventions. Rheumatology; 10:1219-23. Salvemini D., Mazzon E., Dugo L., Serraino I., De Sarro A., Caputi A.P., Cuzzocrea S. (2001) Amelioration of joint disease in a rat model of collagen-induced arthritis by M40403, a superoxide dismutase mimetic. Arthritis Rheum. 44:2909-21. Shelton D.L., Zeller J., Ho W.H., Pons J., Rosenthal A. (2005) Nerve growth factor mediates hyperalgesia and cachexia in auto-immune arthritis. Pain. 116:8-16. Sims N.A., Green J.R., Glatt M., Schlict S., Martin T.J., Gillespie M.T., Romas E. (2004) Targeting osteoclasts with zoledronic acid prevents bone destruction in collagen-induced arthritis. Arthritis Rheum., 50: 2338-46. Walsmith J., Abad L., Kehayias J., Roubenoff R. (2004) Tumor necrosis factor-alpha production is associated with less body cell mass in women with rheumatoid arthritis. J. Rheumatol.; 31:23-9. Khachigian, L.M. Collagen antibody-induced arthritis. (2006) Nature Protocols 1,2512-6. Lin H.S., Hu C..Y, Chan H.Y., Liew Y.Y., Huang H.P., Lepescheux L., Bastianelli E., Baron R., Rawadi G., Clement-Lacroix P. (2007) Anti-rheumatic activities of histone deacetylase (HDAC) inhibitors in vivo in collagen-induced arthritis in rodents. Br. J. Pharmacol. Apr; 150 (7):829-31. Все публикации, включающие патенты и заявки на патенты, цитируемые в этом описании изобретения, но не ограниченные этим, включены в этот документ путем ссылки, как если бы каждая отдельно взятая публикация, которая должна быть включена путем ссылки в этот документ, была бы точно и отдельно упомянута словно полностью изложенная. На основе вышеприведенного описания специалистам в данной области будут приходить на ум различные модификации и изменения композиций и способов этого изобретения. Все такие модификации, возникающие в рамках объема прилагаемых пунктов формулы, как подразумевается, являются включенным в этот документ. Следует понимать, что факторы, такие как дифференциальная способность различных соединений к проникновению в клетки, могут приводить к возникновению расхождений в значениях активности соединений в методах биохимического и клеточного анализа. По меньшей мере, некоторые химические названия соединений изобретения, которые даны и указаны в этой заявке, могут быть получены на автоматической основе посредством использования коммерчески доступного программного обеспечения для присваивания назвазваний химическим соединениям и не были независимо проверены. Репрезентативные программы, осуществляющие такую функцию, включают инструмент присваивания названий Lexichem, продаваемый компанией Open Eye Software, Inc., и инструмент Autonom Software, продаваемый компанией MDL, Inc. В случае, когда указанное химическое название и отраженная структура отличаются, отраженная структура будет проверяться. Химические структуры, показанные в этом документе, получены с использованием либо программы ChemDraw ®, либо программы ISIS ®/DRAW. Любая открытая валентность, возникающая в структурах на атоме углерода, кислорода или азота, в этом документе указывает на наличие атома водорода. В том случае, когда в структуре существует хиральный центр, но конкретная стехиометрия не показана для хирального центра, оба энантиомера, связанные с хиральной структурой, охвачены этой структурой. |