|
Патентная документация ЕАПВ |
|
||
| Запрос: | ea000007537b*\id |
|
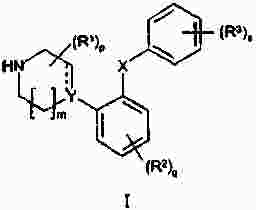
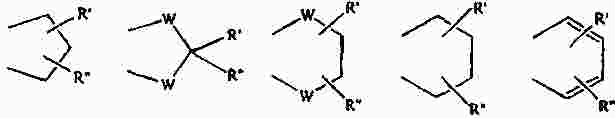
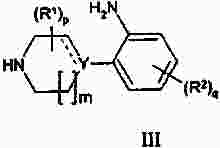
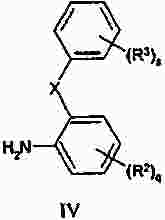
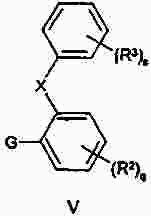
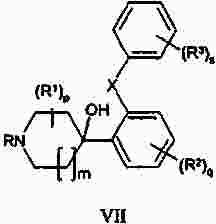
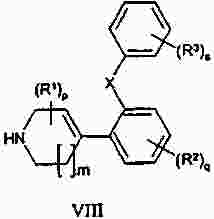
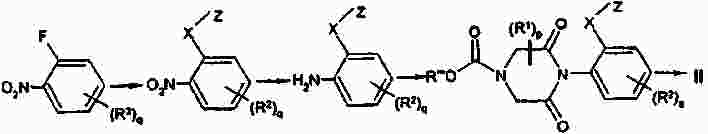
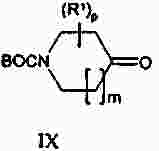
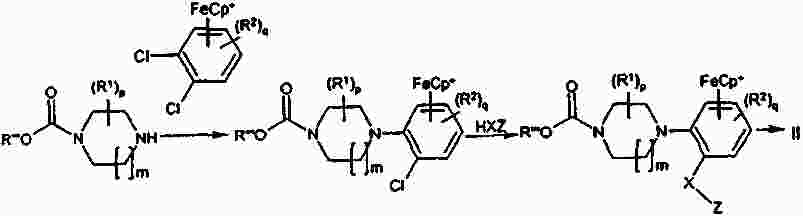
Термины запроса в документе Реферат Изобретение предлагает соединение, представляющее собой 1-[2-(2,4-диметилфенилсульфанил)фенил]пиперазин. Указанное соединение применимо при лечении аффективных расстройств, включая депрессию, страхи, включая общие страхи и панические состояния, и навязчивое компульсивное расстройство. Формула [0001] Соединение, представляющее собой 1-[2-(2,4-диметилфенилсульфанил)фенил]пиперазин, или его фармацевтически приемлемая аддитивная соль с кислотой. [0002] Фармацевтическая композиция, содержащая соединение по п.1 или его фармацевтически приемлемую аддитивную соль с кислотой и по меньшей мере один фармацевтически приемлемый носитель или разбавитель. [0003] Применение соединения по п.1 или его фармацевтически приемлемой аддитивной соли с кислотой для приготовления лекарственного средства для лечения аффективных расстройств, таких как депрессия, страхи, включая общие страхи и панические состояния, и навязчивое компульсивное расстройство. [0004] Способ лечения аффективных расстройств, включая депрессию, страхи, включая общие страхи и панические состояния, и навязчивое компульсивное расстройство у высших животных, включая человека, предусматривающий введение терапевтически эффективного количества соединения по п.1 или его фармацевтически приемлемой аддитивной соли с кислотой. [0005] Применение соединения по п.1 в качестве лекарственного средства для лечения аффективных расстройств. Полный текст патента Данное изобретение относится к новым соединениям, которые являются ингибиторами обратного захвата серотонина и, таким образом, эффективны при лечении, например, депрессии и страха. Селективные ингибиторы обратного захвата серотонина (далее обозначенные как SSRIs) стали первоочередными средствами для лечения депрессии, различных форм страха и социальных фобий, так как они эффективны, хорошо переносятся пациентами и имеют благоприятные показатели безопасности по сравнению с классическими трициклическими антидепрессантами. Однако клинические исследования депрессии показывают, что нечувствительность к SSRIs значительна, до 30%. Другим, часто не учитываемым, фактором при лечении антидепрессантами является изменчивость, которая имеет весьма глубокое влияние на мотивацию пациента к непрерывной фармакотерапии. Прежде всего, существует запаздывание терапевтического эффекта SSRIs. Некоторые симптомы даже ухудшаются в течение первой недели лечения. Во-вторых, побочным эффектом всех SSRIs обычно является сексуальная дисфункция. Без учета указанных проблем реальный прогресс в фармакотерапии депрессии и страхов оказывается невозможным. Чтобы справиться с нечувствительностью, психиатры иногда используют стратегию усиления действия. Терапия с усилением действия антидепрессантов может быть осуществлена совместным введением стабилизаторов настроения, таких как карбонат лития или трииодтиронин, или путем применения электрошока. Эффект комбинированного введения соединения, которое ингибирует обратный захват серотонина, и антагониста рецептора 5-НТ 1А был оценен в нескольких исследованиях (Inns et al. Eur. J. Pharmacol. 1987, 143, 1095-204 и Gartside Br. J. Pharmacol. 1995, 115, 1064-1070, Blier et al. Trends in Pharmacol. 1994, 15, 220). В указанных исследованиях было найдено, что антагонисты рецептора 5-НТ 1А могут уничтожать торможение нейротрансмиссии 5-НТ, вызванной ингибиторами обратного захвата серотонина, и таким образом обеспечивать немедленное усиление трансмиссии 5-НТ и быстрое начало терапевтического действия. Были зарегистрированы некоторые патентные заявки, которые освещали применение комбинации антагониста 5-НТ 1А и ингибитора обратного захвата серотонина для лечения депрессии (см., например, патенты ЕР-А2-687472 и ЕР-А2-714663). Другим подходом к увеличению терминального 5-НТ может быть блокирование ауторецептора 5-HT 1B . Эксперименты по микродиализу на крысах действительно показывают, что увеличение гиппокампального 5-НТ циталопрамом является потенцирующим для ОМС 2-29, экспериментального антагониста рецептора 5-НТ 1В . Также зарегистрированы различные патентные заявки, относящиеся к комбинации антагониста или частичного агониста SSRIs и 5-НТ 1В (WO 97/28141, WO 96/03400, ЕР-А-701819 и WO 99/13877). Ранее было найдено, что комбинация ингибитора обратного захвата серотонина с соединением, обладающим антагонистическим или обратимым агонистическим действием 5-HT 2C (соединений, имеющих отрицательную эффективность по отношению к рецептору 5-НТ 2С ), дает значительное повышение уровня 5-НТ в терминальных областях, как измерено в опытах по микродиализу (патент WO 01/41701). Это будет означать более быстрое наступление антидепрессивного действия в клинике и нарастание или потенцирование терапевтического эффекта ингибитора обратного захвата серотонина (SRI). Данное изобретение предоставляет соединения, которые являются ингибиторами обратного захвата серотонина для лечения аффективных расстройств, таких как депрессия, страхи, включая общие страхи и панические состояния, и навязчивое компульсивное расстройство. Некоторые из указанных соединений обладают комбинированным действием ингибирования обратного захвата серотонина и модуляции рецептора 5-НТ 2С , что согласно патенту WO 01/41701 означает более быстрое начало антидепрессивного действия. Некоторые соединения, охваченные данным изобретением, ранее были описаны в патентах WO 01/49681 и WO 02/59108. Однако соединения патента WO 01/49681 не описаны, как имеющие терапевтическую или биологическую активность. Соединения патента WO 02/59108 описаны как промежуточные продукты в синтезе соединений, отличающихся от соединений данного изобретения, с терапевтической активностью агонистов рецептора меланокортина. Одно соединение, 1-(2-феноксифенил)пиперазин, охваченное данным изобретением, описано в патенте US 4064245 как применимое при лечении метаболических расстройств. Данное изобретение предлагает соединение общей формулы I где Y означает N, С или СН; X означает S; m означает 1 или 2; р означает 0, 1, 2, 3, 4, 5, 6, 7 или 8; q означает 0, 1, 2, 3 или 4; s означает 1 или 2; пунктирная линия означает возможную связь; каждый R 1 независимо выбран из группы, представленной C 1-6 -алкилом, или два R 1 , присоединенные к одному и тому же атому углерода, могут образовывать 3-6-членный спироприсоединенный циклоалкил; каждый R 2 независимо выбран из групп, представленных галогеном, цианогруппой, нитрогруппой, C 1-6 -алк(ен/ин)илом, C 1-6 -алк(ен/ин)илоксигруппой, C 1-6 -алк(ен/ин)илсульфанилом, гидроксигруппой, гидрокси-C 1-6 -алк(ен/ин)илом, галоген-C 1-6 -алк(ен/ин)илом, галоген-C 1-6 -алк(ен/ин)илоксигруппой, С 3-8 -циклоалк(ен)илом, С 3-8 -циклоалк(ен)ил-C 1-6 -алк(ен/ин)илом, ацилом, C 1-6 -алк(ен/ин)илоксикарбонилом, C 1-6 -алк(ен/ин)илсульфонилом или -NR x R y ; -NR x CO-C 1-6 -алк(ен/ин)илом; каждый R 3 независимо выбран из групп, представленных галогеном, цианогруппой, нитрогруппой, C 1-6 -алк(ен/ин)илом, C 1-6 алк(ен/ин)илоксигруппой, C 1-6 -алк(ен/ин)илсульфанилом, гидроксигруппой, гидрокси-C 1-6 -алк(ен/ин)илом, галоген-C 1-6 -алк(ен/ин)илом, галоген-C 1-6 -алк(ен/ин)илоксигруппой, С 3-8 -циклоалк(ен)илом, С 3-8 -циклоалк(ен)ил-C 1-6 -алк(ен/ин)илом, C 1-6 -алк(ен/ин)илсульфонилом, арилом, C 1-6 -алк(ен/ин)илоксикарбонилом, ацилом, -NR x CO-C 1-6 -алк(ен/ин)илом, -CONR x R y или NR x R y ; или два соседних заместителя R 3 вместе образуют гетероцикл, конденсированный с фенильным кольцом, выбранным из группы, состоящей из где W означает О или S и R' и R" означают водород или C 1-6 -алкил; или два соседних заместителя R 3 вместе образуют конденсированную гетероароматическую систему, содержащую один, два или три гетероатома, где каждый R x и R y независимо выбран из групп, представленных водородом, C 1-6 -алк(ен/ин)илом, С 3-8 -циклоалк(ен)илом, С 3-8 -циклоалк(ен)ил-C 1-6 -алк(ен/ин)илом или арилом; или R x и R y вместе с азотом, к которому они присоединены, образуют 3-7-членное кольцо, которое может содержать один дополнительный гетероатом; или их фармацевтически приемлемые аддитивные соли с кислотами, характеризующееся тем, что указанное соединение общей формулы I представляет собой 1-[2-(2,4-диметилфенилсульфанил)фенил]пиперазин. Изобретение предусматривает соединение согласно вышеуказанному для применения в качестве лекарственного средства. Изобретением также предусмотрена фармацевтическая композиция, содержащая соединение настоящего изобретения, определенное в описании, или его фармацевтически приемлемую аддитивную соль с кислотой и по меньшей мере один фармацевтически приемлемый носитель или разбавитель. Изобретение также касается применения указанного соединения или его фармацевтически приемлемых аддитивных солей с кислотами для приготовления лекарственного средства для лечения аффективных расстройств, таких как депрессия, страхи, включая общие страхи и панические состояния, и навязчивое компульсивное расстройство. Изобретение также предлагает способ лечения аффективных расстройств, включая депрессию, страхи, включая общие страхи и панические состояния, и навязчивое компульсивное расстройство у высших животных, включая человека, предусматривающий введение терапевтически эффективного количества соединения по данному изобретению или его фармацевтически приемлемой аддитивной соли с кислотой. Изобретение также относится к применению указанного соединения в качестве лекарственного средства для лечения аффективных расстройств. Частным предпочтительным вариантом настоящего изобретения является соединении, представляющее собой 1-[2-(2,4-диметилфенилсульфанил)фенил]пиперазин, или его фармацевтически приемлемая аддитивная соль с кислотой. Галоген означает фтор, хлор, бром или иод. Выражение "C 1-6 алк(ен/ин)ил" означает C 1-6 -алкильную, C 2-6 -алкенильную или C 2-6 -алкинильную группу. Выражение "С 3-8 -циклоалк(ен)ил" означает C 3-8 -циклоалкильную или циклоалкенильную группу. Термин "C 1-6 -алкил" относится к разветвленной или неразветвленной алкильной группе, содержащей от одного до шести атомов углерода включительно, включая без ограничения метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-2-пропил и 2-метил-1-пропил. Аналогично, С 2-6 -алкенил и C 1-6 -алкинил соответственно означают такие группы, содержащие от двух до шести атомов углерода, включающие одну двойную связь и одну тройную связь соответственно, включая без ограничения этенил, пропенил, бутенил, этинил, пропинил и бутинил. Термин "С 3-8 -циклоалкил" означает моноциклический или бициклический карбоцикл, содержащий от трех до восьми С-атомов, включая без ограничения циклопропил, циклопентил, циклогексил и т.д. Термин "С 3-8 -циклоалкенил" означает моноциклический или бициклический карбоцикл, содержащий от трех до восьми С-атомов и включающий одну двойную связь. В термине "С 3-8 -циклоалк(ен)ил-C 1-6 -алк(ен/ин)ил" С 3-8 -циклоалк(ен)ил и C 1-6 -алк(ен/ин)ил имеют указанные выше значения. Термины "C 1-6 -алк(ен/ил)илоксигруппа", "C 1-6 -алк(ен/ин)илсульфанил", "гидрокси-C 1-6 -алк(ен/ин)ил", "галоген-C 1-6 -алк(ен/ин)ил", "галоген-C 1-6 -алк(ен/ин)илоксигруппа", "C 1-6 -алк(ен/ин)илсульфонил" и т.д. означают такие группы, в которых C 1-6 -алк(ен/ин)ил имеет указанные выше значения. Как использовано здесь, термин "C 1-6 -алк(ен/ин)илоксикарбонил" относится к группам формулы C 1-6 -алк(ен/ин)ил-О-СО-, где C 1-6 -алк(ен/ин)ил имеет указанные выше значения. Как использовано здесь, термин "ацил" относится к формилу, C 1-6 -алк(ен/ин)илкарбонилу, арилкарбонилу, арил-C 1-6 -алк(ен/ин)илкарбонилу, С 3-8 -циклоалк(ен)илкарбонилу или С 3-8 -циклоалк(ен)ил-C 1-6 -алк(ен/ин)илкарбонильной группе. Термин "3-7-членное кольцо", возможно содержащее один дополнительный гетероатом, как использовано здесь, относится к кольцевым системам, таким как 1-морфолинил, 1-пиперидинил, 1-азепинил, 1-пиперазинил, 1-гомопиперазинил, 1-имидазолил, 1-пирролил или пиразолил, все из которых могут быть замещены C 1-6 -алкилом. Гетероциклы, образованные двумя соседними заместителями R 3 и конденсированные с родоначальным кольцом, могут вместе образовывать кольца, такие как 5-членные моноциклические кольца, такие как 3H-1,2,3-оксатиазол, 1,3,2-оксатиазол. 1,3,2-диоксазол, 3H-1,2,3-дитиазол, 1,3,2-дитиазол, 1,2,3-оксадиазол, 1,2,3-тиадиазол, 1Н-1,2,3-триазол, изоксазол, оксазол, изотиазол, тиазол, 1Н-имидазол, 1Н-пиразол, 1Н-пиррол, фуран или тиофен, и 6-членные моноциклические кольца, такие как 1,2,3-оксатиазин, 1,2,4-оксатиазин, 1,2,5-оксатиазин, 1,4,2-оксатиазин, 1,4,3-оксатиазин, 1,2,3-диоксазин, 1,2,4-диоксазин, 4Н-1,3,2-диоксазин, 1,4,2-диоксазин, 2Н-1,5,2-диоксазин, 1,2,3-дитиазин, 1,2,4-дитиазин, 4Н-1,3,2-дитиазин, 1,4,2-дитиазин, 2Н-1,5,2-дитиазин, 2Н-1,2,3-оксадиазин, 2Н-1,2,4-оксадиазин, 2Н-1,2,5-оксадиазин, 2Н-1,2,6-оксадиазин, 2Н-1,3,4-оксадиазин, 2Н-1,2,3-тиадиазин, 2Н-1,2,4-тиадиазин, 2Н-1,2,5-тиадиазин, 2Н-1,2,6-тиадиазин, 2Н-1,3,4-тиадиазин, 1,2,3-триазин, 1,2,4-триазин, 2Н-1,2-оксазин, 2Н-1,3-оксазин, 2Н-1,4-оксазин, 2Н-1,2-тиазин, 2Н-1,3-тиазин, 2Н-1,4-тиазин, пиразин, пиридазин, пиримидин, 4Н-1,3-оксатиин, 1,4-оксатиин, 4Н-1,3-диоксин, 1,4-диоксин, 4Н-1,3-дитиин, 1,4-дитиин, пиридин, 2Н-пиран или 2Н-тиин. Термин "арил" относится к карбоциклическим ароматическим системам, таким как фенил и наф-тил. Аддитивные соли с кислотами изобретения предпочтительно являются фармацевтически приемлемыми солями соединений изобретения, образованными с нетоксичными кислотами. Примерами таких органических солей являются соли с малеиновой, фумаровой, бензойной, аскорбиновой, янтарной, щавелевой, бис-метиленсалициловой, метансульфоновой, этандисульфоновой, уксусной, пропионовой, винной, салициловой, лимонной, глюконовой, молочной, яблочной, миндальной, коричной, цитраконовой, аспарагиновой, стеариновой, пальмитиновой, итаконовой, гликолевой, парааминобензойной, глутамино-вой, бензолсульфоновой и теофиллинуксусной кислотами, так же как 8-галогентеофиллины, например 8-бромтеофиллин. Примерами таких неорганических солей являются соли с хлористо-водородной, броми-сто-водородной, серной, сульфаминовой, фосфорной и азотной кислотами. Дополнительно, соединения изобретения могут существовать как в несольватированных, так и в сольватированных формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и подобные. Обычно сольватированные формы рассматриваются эквивалентными несольватированным формам для целей данного изобретения. Некоторые соединения данного изобретения содержат хиральные центры, и такие соединения существуют в форме изомеров (т.е. энантиомеров). Изобретение охватывает все такие изомеры и любые их смеси, включая рацемические смеси. Рацемические формы могут быть разделены на оптические антиподы известными способами, например, разделением их диастереомерных солей с оптически активной кислотой и высвобождением оптически активного амина обработкой основанием. Другой способ разделения рацематов на оптические антиподы основан на хроматографировании на оптически активной матрице. Рацемические соединения данного изобретения также могут быть разделены на их оптические антиподы, например, фракционной кристаллизацией d- или l-солей (тартратов, манделатов или камфорсульфонатов). Соединения данного изобретения могут также быть разделены путем образования диастереомерных производных. Могут быть использованы дополнительные способы разделения оптических изомеров, известные специалистам в данной области. Такими способами являются обсужденные J. Jaques, A. Collet and S. Wilen in "Enantiomers, Racemates, and Resolutions", John Wiley and Sons, New York (1981). Оптически активные соединения также могут быть получены из оптически активных исходных веществ. Фармацевтические композиции изобретения могут быть получены стандартными способами, известными из уровня техники. Например, таблетки могут быть получены смешением активного ингредиента с обычными адъювантами и/или разбавителями и последующим прессованием смеси на обычной таблетирующей машине. Примеры адъювантов или разбавителей включают кукурузный крахмал, картофельный крахмал, тальк, стеарат магния, желатин, лактозу, камеди и подобные. Для таких целей, как окрашивание, придание вкуса, консервирование и т.д., могут быть использованы любые другие адъюван-ты или добавки, обеспечивающие совместимость с активными ингредиентами. Растворы для инъекций могут быть получены растворением активного ингредиента и возможных добавок в части растворителя для инъекции, предпочтительно в стерильной воде, доведением раствора до желаемого объема, стерилизацией раствора и заполнением им подходящих ампул или флаконов. Могут быть добавлены любые подходящие добавки, обычно используемые на практике, такие как тонические агенты, консерванты, антиоксиданты и т.д. Фармацевтические композиции данного изобретения или изготовленные в соответствии с данным изобретением, могут быть введены любым обычным путем, например перорально в форме таблеток, капсул, порошков, сиропов и т.д., или парентерально в форме растворов для инъекций. Для приготовления указанных композиций могут быть использованы способы, хорошо известные на практике, и могут быть использованы любые фармацевтически приемлемые носители, разбавители, эксципиенты или другие добавки, обычно используемые на практике. Удобно вводить соединения изобретения в форме разовой дозы, содержащей указанные соединения в количестве примерно от 0,01 до 100 мг. Общая дневная доза обычно находится в интервале примерно 0,05-500 мг и наиболее предпочтительно примерно от 0,1 до 50 мг активного соединения изобретения. Соединения изобретения получают следующими общими способами. а) Удаление защиты или отщепление от полимерной подложки соединения формулы II где Z означает и R 1 , R 2 , R 3 , m, p, q, s, X, Y и пунктирная линия имеют приведенные выше значения, R'" означает трет-бутильную, метильную, этильную, аллильную или бензильную группу или R'"OCO 2 означает кар-баматную группу твердой подложки, такой как карбаматный линкер на основе смолы Ванга. b) Химическое превращение соединения формулы III где R 1 , R 2 , m, p, q, Y и пунктирная линия имеют приведенные выше значения, в соответствующее диазосоединение и последующее взаимодействие с соединением HXZ, где X и Z имеют приведенные выше значения. с) Взаимодействие соединения формулы IV где R 2 , R 3 , X, s и q имеют приведенные выше значения, с алкилирующим агентом формулы (Cl-(CH 2 ) m+1 )NH(CH 2 ) 2 Cl или (Br-(CH 2 ) m+1 )NH(CH 2 ) 2 Br, где m имеет приведенные выше значения. d) Взаимодействие соединения формулы V где R 2 , R 3 , X, s и q имеют приведенные выше значения и G означает атом брома или иода, с соединением формулы VI где R 1 , m и р имеют приведенные выше значения. е) Дегидратация и возможно одновременное удаление защиты соединения формулы VII где R 1 , R 2 , R 3 , X, m, p, q и s имеют приведенные выше значения и R означает либо атом водорода, либо группу ВОС. f) Гидрирование двойной связи в соединении формулы VIII где R 1 , R 2 , R 3 , X, m, p, q и s имеют приведенные выше значения. Удаление защиты в способе а) осуществляют по стандартной методике, и известной специалистам в данной области и детализированной в руководстве Protective Groups in Organic Synthesis T W Greene and PGM Wuts, Wiley Interscience (1991) ISBN 0471623016. Исходные вещества формулы II, где R'"=трет-Bu, получали согласно способу, описанному ниже Производные фторнитробензола реагировали с фенолами или тиофенолами согласно описанному Sawyer et al. J. Org. Chem. 1998, 63, 6338, с последующим восстановлением с использованием стандартной методики, известной специалистам в данной области. Она предусматривает восстановление до соответствующего анилина с использованием соли гидрида металла, такой как боргидрид натрия, совместно с катализатором палладий на угле в спиртовом растворителе или восстановление с использованием хлорида металла, такого как хлорид цинка или хлорид олова. Полученный анилин затем превращали в соответственно замещенный 3,5-дикетопиперазин модифицированным способом Kruse et al. Recl. Trav. Chim. Pays-Bas 1998, 107, 303, используя N-бутилоксикарбонилиминодиуксусную кислоту Производное 3,5-дикетопиперазина затем восстанавливали, например, бораном до соответствующего ВОС-защищенного пиперазина, из которого затем in situ удаляли защиту с образованием пиперазина. Соединения, показанные в формуле II, где Y = СН и возможная двойная связь восстановлена, получали из третичноспиртового предшественника VII, где R означает группу ВОС, модифицированным восстановлением по Бартону аналогично описанному Hansen et al. Synthesis 1999, 1925-1930. Промежуточные третичные спирты получали из соответствующих замещенных 1-бромфенилсульфанилбезолов или их соответствующих простых эфиров обменом металл-галоген с последующим присоединением соответствующего электрофила формулы IX способом, подобным описанному Palmer et al. J. Med. Chem. 1997, 40, 1982-1989. Соответственно замещенные 1-бромфенилсульфанилбензолы получали способом, подобным описанному в литературе, реакцией соответственно замещенных тиофенолов с соответственно замещенными арилиодидами согласно Schopfer and Schlapbach Tetrahedron 2001, 57, 3069-3073, Bates et al. Org. Lett. 2002, 4, 2803-2806 and Kwong et al. Org. Lett. 2002, 4 (в печати). Соответствующие замещенные 1-бромфеноксибензолы могут быть получены, как описано Buck et al. Org. Lett. 2002, 4, 1623-1626. Отщепление от полимерной подложки, такой как карбаматный линкер на основе смолы Ванга, согласно способу а), проводили по описанным в литературе методикам (Zaragoza Tetrahedron Lett. 1995, 36, 8677-8678 and Conti et al. Tetrahedron Lett. 1997, 38, 2915-2918). Исходное вещество формулы II может также быть получено согласно способу, описанному в патентной заявке WO 01/49681. Диамины были либо коммерчески доступны, либо синтезированы способами, известными специалистам-химикам. Комплексы железа, подобные гексафторфосфату η 6 -1,2-дихлорбензол- η 5 -циклопентадиенилжелеза(II), и замещенные аналоги синтезировали согласно способам, описанным в литературе (Pearson et al. J. Org. Chem. 1996, 51, 1297-1305), или синтезировали способами, известным химикам, специалистам в данной области. Диазотирование с последующим взаимодействием с соединением HXZ по способу b) проводили добавлением соли диазония соответствующего анилина к раствору натриевой соли тиофенола или фенола в водной суспензии меди. Исходное вещество формулы III получали по следующей схеме. Производное фторнитробензола подвергали взаимодействию с производным пиперазина в растворителе, таком как ДМФА, NMP или другом диполярном апротонном растворителе, содержащем органическое основание, такое как триэтиламин, с образованием производного ортонитрофенилпиперазина. Промежуточный ор-тонитрофенилпиперазин затем восстанавливали, используя стандартные методики, как указано выше, с получением исходного вещества формулы III. Реакцию соединения формулы IV с алкилирующим агентом формулы (Cl-(CH 2 ) m+1 )NH(CH 2 ) 2 Cl или (Br-(CH 2 ) m+1 )NH(CH 2 ) 2 Br в виде его бромисто-водородной или хлористо-водородной соли, где m имеет указанные выше значения, проводили способом, подобным описанному Sircar et al. J. Med. Chem. 1992, 35, 4442-4449. Исходные вещества формулы IV получали, как описано выше для исходных веществ формулы II. Реакцию соединения формулы V с диамином формулы VI в способе d) проводили способом, подобным описанному Nishiyama et al. Tetrahedron Lett. 1998, 39, 617-620. Исходное вещество формулы V получали способом, подобным описанному Schopfer et al. Tetrahedron 2001, 51, 3069-3073. Реакцию дегидратации и возможное одновременное удаление защиты соединения формулы VII в способе е) проводили способом, подобным описанному Palmer et al. J. Med. Chem. 1997, 40, 1982-1989. Исходное вещество формулы VII, где R=H, получали из соединения формулы VII, где R означает группу ВОС (см. выше), путем удаления защиты хлористо-водородной кислотой в метаноле. Соединения формулы VII, где R=BOC, могут быть получены, как описано Palmer et al. J. Med. Chem. 1997, 40, 1982-1989. Восстановление двойной связи согласно способу f) обычно проводили каталитическим гидрированием при низком давлении ( <3 атм) в аппарате Парра или с использованием восстановителя, такого как диборан или производное бороводорода, получаемого in situ из NaBH 4 в трифторуксусной кислоте, в инертном растворителе, таком как тетрагидрофуран (ТГФ), диоксан или диэтиловый эфир. Исходное вещество формулы VIII получали из II, как описано в способе а). Данные анализа ЖХ-МС получали на приборе РЕ Sciex API 150EX, снабженном источником Ion-Spray и системой Shimadzu LC-8A/SLC-10A LC. Колонка: колонка 30 ×4,6 мм Waters Symmetry C18 с размером частиц 3,5 мкм. Система растворителей: А=вода/трифторуксусная кислота (100:0,05) и В=вода/ацетонитрил/трифторуксусная кислота (5:95:0,03) Метод: линейный градиент элюирования от 90% А до 100% В за 4 мин и с объемным расходом 2 мл/мин. Чистоту определяли интегрированием УФ (254 нм) и линии ELSD. Время удерживания (RT) выражали в минутах. Препаративную ЖХ-МС-очистку проводили на том же приборе. Колонка: 50 ×20 мм YMC ODS-A с размером частиц 5 мкм, метод: линейный градиент элюирования от 80% А до 100% В за 7 мин с объемным расходом 22,7 мл/мин. Отбор фракции проводили по данным МС для разветвленного потока. Спектр 1 Н-ЯМР регистрировали при 500,13 МГц на приборе Bruker Avance DRX500 или при 250,13 МГц на приборе Bruker АС 250. В качестве растворителей использовали дейтерированный метиленхлорид (99,8%D), хлороформ (99,8%D) или диметилсульфоксид (99,8%D). В качестве внутреннего стандарта использовали TMS Значения химического сдвига выражали в м.д. Для мультиплетности сигналов ЯМР использовали следующие сокращения: с = синглет, д = дублет, т = триплет, кв = квартет, квит = квинтет, г = гептет, дд = двойной дублет, дт = двойной триплет, дкв = двойной квартет, тт = триплет триплетов, м = мультиплет и уш. с = уширенный синглет. Для ионообменной хроматографии использовали следующие материалы: колонка SCX (1 г) из Varian Mega Bond Elut ®, Chrompack cat. No. 220776. Перед употреблением колонку SCX предварительно промывали 10% раствором уксусной кислоты в метаноле (3 мл). Для декомплексации путем облучения использовали источник ультрафиолетового излучения (300 Вт) производства Philipps. В качестве исходной полимерной подложки для твердофазного синтеза использовали смолу Ванга (1,03 ммоль/г, Rapp-Polymere, Tuebingen, Германия). Гексафторфосфат η 6 -1,2-дихлорбензол- η 5 -циклопентадиенилжелеза(II). Ферроцен (167 г), безводный трихлорид алюминия (238 г) и порошкообразный алюминий (24 г) суспендировали в 1,2-дихлорбензоле (500 мл) и нагревали при 90 °С в атмосфере азота 5 ч при интенсивном перемешивании. Смесь охлаждали до комнатной температуры и осторожно маленькими порциями добавляли воду (1000 мл) при охлаждении на ледяной бане. Добавляли гептан (500 мл) и диэтиловый эфир (500 мл) и смесь перемешивали при комнатной температуре 30 мин. Смесь экстрагировали диэти-ловым эфиром (3 ×300 мл). Водную фазу фильтровали и маленькими порциями добавляли водный гексафторфосфат аммония (60 г в 50 мл воды) при перемешивании. Продукт оставляли осаждаться при комнатной температуре. Через 3 ч осадок отфильтровывали, интенсивно промывали водой и сушили в вакууме (50 °С), получая 81 г (21%) указанного в заголовке соединения в виде светло-желтого порошка. 1 Н-ЯМР (D 6 -ДМСО): 5,29 (с, 5Н); 6,48 (м, 2Н); 7,07 (м, 2Н). 4- [(Пиперазин-1-ил)карбонилоксиметил]феноксиметилполистирол. 4-[(4-Нитрофенокси)карбонилоксиметил]феноксиметилполистирол (267 г, 235 ммоль) суспендировали в безводном N,N-диметилформамиде (2 л). Добавляли N-метилморфолин (238,0 г, 2,35 моль) и пиперазин (102,0 г, 1,17 моль) и смесь перемешивали при комнатной температуре 16 ч. Смолу отфильтровывали и промывали N,N-диметилформамидом (2 ×1 л), тетрагидрофураном (2 ×1 л), водой (1 ×500 мл), метанолом (2 ×1 л), тетрагидрофураном (2 ×1 л) и метанолом (1 ×1 л). Наконец, смолу промывали ди-хлорметаном (3 ×500 мл) и сушили в вакууме (25 °С, 36 ч), получая почти бесцветную смолу (240,0 г). Аналогично получали следующий диамин, связанный с полистиролом: 4-[(1,4-диазепан-1-ил)карбонилоксиметил]феноксиметилполистирол. Получение связанного со смолой гексафторфосфата η 6 -арил- η 5 -циклопентадиенилжелеза(II). Гексафторфосфат 4-({4-[ η 6 -(2-хлорфенил)- η 5 -циклопентадиенилжелезо(II)]пиперазин-1-ил}карбо-нилоксиметил)феноксиметилполистирола (промежуточный продукт для Ia-Ih и Ik-Il). 4-[(Пиперазин-1-ил)карбонилоксиметил]феноксиметилполистирол (115,1 г, 92 ммоль) суспендировали в безводном тетрагидрофуране (1,6 л) и добавляли гексафторфосфат η 6 -1,2-дихлорбензол- η 5 -циклопентадиенилжелеза(II) (76,0 г, 184 ммоль) и затем карбонат калия (50,9 г, 368 ммоль). Реакционную смесь перемешивали при 60 °С 16 ч. После охлаждения до комнатной температуры смолу отфильтровывали и промывали тетрагидрофураном (2 ×500 мл), водой (2 ×250 мл), тетрагидрофураном (2 ×500 мл), водой (2 ×250 мл), метанолом (2 ×250 мл), дихлорметаном (2 ×250 мл) и метанолом (2 ×250 мл). Наконец, смолу промывали дихлорметаном (3 ×500 мл) и сушили в вакууме (25 °С, 36 ч), получая темно-оранжевую смолу (142 г). Аналогично получали следующий связанный с полистиролом комплекс железа: гексафторфосфат 4-({4-[ η 6 -(2-хлорфенил)- η 5 -циклопентадиенилжелезо(II)][1,4]диазепан-1-ил}кар-бонилоксиметил)феноксиметилполистирола (промежуточный продукт для Ii и Ij). 1-трет-Бутоксикарбонил-4-[2-(4-метилфенилсульфанил)фенил]пиперидин-4-ол. Раствор BuLi (2,5 М в гексане, 12,0 мл, 30 ммоль) медленно добавляли к перемешиваемому раствору 1-бром-2-(4-метилфенилсульфанил)бензола (30 ммоль) в безводном ТГФ (75 мл) в атмосфере аргона при -78 °С. Раствор перемешивали 10 мин перед добавлением одной порцией трет-бутилового эфира 4-оксопиперидин-1-карбоновой кислоты (5,98 г, 30 ммоль). Раствор оставляли нагреваться до комнатной температуры и затем перемешивали 3 ч. Добавляли насыщенный водный NH 4 Cl (150 мл) и раствор экстрагировали зтилацетатом (150 мл). Органическую фазу промывали насыщенным раствором соли, сушили (MgSO 4 ) и растворитель выпаривали в вакууме. Неочищенный 1 очищали флэш-хроматографией на силикагеле (элюент: этилацетат/гептан 20:80), получая целевое соединение в виде белой пены. ЖХ/МС (m/z) 399,3 (МН + ); RT=3,82; чистота (УФ, ELSD): 98%, 100%; выход: 5,02 г (42%). 1-трет-Бутилоксикарбонил-4-[2-(4-метилфенилсульфанил)фенил]-3,5-диоксопиперазин (промежуточный продукт для 2а). 2-(4-Метилфенилсульфанил)анилин (2,9 г, 13,5 ммоль) растворяли в безводном ТГФ (200 мл) и помещали в атмосферу азота. К раствору добавляли N-(трет-бутилоксикарбонил)иминодиуксусную кислоту (4,7 г, 20,2 ммоль) и карбонилдиимидазол (4,2 г, 40,4 ммоль) и реакционную смесь кипятили с обратным холодильником 60 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли этил-ацетат (500 мл). Полученный раствор затем промывали 2н. NaHCO 3 (2 ×200 мл), 2н. HCl (2 ×200 мл) и насыщенным раствором хлорида натрия (100 мл) и растворители выпаривали в вакууме. Выход 6,0 г, 107%. 1 Н-ЯМР (CDCl 3 ) 1,5 (с, 9Н); 2,32 (с, 3H); 4,4-4,6 (м, 4Н); 7,02-7,18 (м, 3H); 7,2-7,45 (м, 5Н). Аналогичным образом получали следующие производные 3,5-дикетопиперазина: 1-трет-бутилоксикарбонил-4-[2-(4-хлорфенилсульфанил)фенил]-3,5-диоксопиперазин (промежуточный продукт для 2b), 1-трет-бутилоксикарбонил-4-[2-(4-метоксифенилсульфанил)-4-хлорфенил]-3,5-диоксопиперазин (промежуточный продукт для 2с), 1-трет-бутилоксикарбонил-4-[2-(4-метоксифенилсульфанил)-4-метилфенил]-3,5-диоксопиперазин (промежуточный продукт для 2d), 1-трет-бутилоксикарбонил-4-[2-(4-метоксифенилсульфанил)-5-метилфенил]-3,5-диоксопиперазин (промежуточный продукт для 2е), 1-трет-бутилоксикарбонил-4-[2-(4-фторфенилсульфанил)-5-метилфенил]-3,5-диоксопиперазин (промежуточный продукт для 2f), 1-трет-бутилоксикарбонил-4-[2-(4-метоксифенилсульфанил)-5-трифторметилфенил]-3,5-диоксопиперазин (промежуточный продукт для 2g), 2-(3-метилпиперазин-1-ил)фениламин (промежуточный продукт для 3a). Фторнитробензол (7,1 г, 50 ммоль) растворяли в ДМФА (100 мл), содержащем триэтиламин (10 г, 100 ммоль), и помещали в атмосферу азота. К раствору добавляли 2-метилпиперазин (5,5 г, 55 ммоль). Реакционную смесь нагревали при 80 °С 16 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры перед уменьшением объема раствора наполовину в вакууме. К раствору добавляли этил-ацетат (200 мл) и ледяную воду (250 мл) и продукт экстрагировали диэтиловым эфиром (2 ×200 мл). Водную фазу насыщали хлоридом натрия и экстрагировали этилацетатом (2 ×200 мл). Органические фазы объединяли, промывали насыщенным раствором соли, сушили над сульфатом магния, фильтровали и фильтрат концентрировали в вакууме. Продукт (10,5 г) растворяли в этаноле (250 мл). К раствору добавляли катализатор палладий на угле (10% мас./мас., 2,2 г) и раствор гидрировали в аппарате Парра при 3 бар 3 ч. Раствор фильтровали и растворители выпаривали в вакууме, получая анилиновый продукт. Выход (8,0 г, 83%). Аналогичным образом получали следующий промежуточный продукт: 2-(3,5-диметилпиперазин-1-ил)фениламин (промежуточный продукт для 3b). Пример 1. 1а, 1-[2-(2-Трифторметилфенилсульфанил)фенил]пиперазин. К раствору 2-трифторметилтиофенола (1,75 г, 9,8 ммоль) в смеси 1:1 тетрагидрофуран/диметил-формамид (30 мл) осторожно при комнатной температуре добавляли гидрид натрия (7,4 ммоль, 60% в минеральном масле) (осторожно: выделение водорода). Смесь перемешивали дополнительно 30 мин после прекращения выделения водорода. Затем добавляли гексафторфосфат 4-({4-[ η 6 -(2-хлорфенил)- η 5 -циклопентадиенилжелезо(II)]пиперазин-1-ил}карбонилоксиметил)феноксиметилполистирола (3,5 г, 2,45 ммоль) и смесь перемешивали при 55 °С 12 ч. После охлаждения до комнатной температуры смолу отфильтровывали и промывали тетрагидрофураном (2 ×50 мл), смесью тетрагидрофуран/вода (1:1) (2 ×50 мл), N,N-диметилформамидом (2 ×50 мл), водой (2 ×50 мл), метанолом (3 ×50 мл), тетрагидрофураном (3 ×50 мл) и затем метанолом и тетрагидрофураном (по 50 мл, 5 циклов). Наконец, смолу промывали ди-хлорметаном (3 ×50 мл) и сушили в вакууме (25 °С, 12 ч), получая темно-оранжевую смолу. Полученную смолу и 0,5 М раствор 1,10-фенантролина в смеси 3:1 пиридин/вода (20 мл) помещали в светопроницаемую реакционную трубку. Суспензию перемешивали путем вращения при облучении видимым светом 12 ч. Смолу отфильтровывали и промывали метанолом (2 ×25 мл), водой (2 ×25 мл) и тетрагидрофураном (3 ×25 мл) до тех пор, пока промывные растворы не становились бесцветными (примерно 5 циклов), и процедуру облучения повторяли до завершения декомплексации (примерно 5 циклов). После завершения декомплексации смолу промывали дихлорметаном (3 ×25 мл) и сушили в вакууме (25 °С, 12 ч), получая светло-коричневую смолу. 100 мг (77 мкмоль) полученной смолы суспендировали в смеси 1:1 трифторуксусной кислоты и дихлорметана (2 мл) и перемешивали при комнатной температуре 2 ч. Смолу отфильтровывали и промывали метанолом (1 ×0,5 мл) и дихлорметаном (1 ×0,5 мл). Фильтраты объединяли и летучие растворители выпаривали в вакууме. Неочищенный продукт очищали препаративной ЖХ-МС и затем ионообменной хроматографией. ЖХ/МС (m/z) 339 (МН + ); RT=2,39; чистота (УФ, ELSD): 92%, 100%; общий выход: 1 мг (4%). Аналогично получали следующие арилпиперазины и арил[1,4]диазепаны: 1b, 1-[2-(4-бромфенилсульфанил)фенил]пиперазин: ЖХ/МС (m/z) 350 (МН + ); RT=2,46; чистота (УФ, ELSD): 75%, 92%; выход: 2 мг (7%); 1c, 1-{2-[4-(метилсульфанил)фенилсульфанил]фенил}пиперазин: ЖХ/МС (m/z) 317 (МН + ); RT=2,39; чистота (УФ, ELSD): 91%, 100%; выход: 2 мг (8%); 1d, 1-[2-(4-гидроксифенилсульфанил)фенил]пиперазин: ЖХ/МС (m/z) 287 (МН + ); RT=1,83; чистота (УФ, ELSD): 84%, 100%; выход: 3 мг (13%); 1е, 1-[2-(2,4-диметилфенилсульфанил)фенил]пиперазин: ЖХ/МС (m/z) 299 (МН + ); RT=2,48; чистота (УФ, ELSD): 95%, 100%; выход: 4 мг (17%); 1f, 1-[2-(3,5-диметилфенилсульфанил)фенил]пиперазин: ЖХ/МС (m/z) 299 (МН + ); RT=2,51; чистота (УФ, ELSD): 96%, 100%; выход: 5 мг (21%); 1g, 1-[2-(2,6-диметилфенилсульфанил)фенил]пиперазин: ЖХ/МС (m/z) 299 (МН + ); RT=2,42; чистота (УФ, ELSD): 97%, 100%; выход: 4 мг (17%); 1h, 1-[2-(2,5-диметилфенилсульфанил)фенил]пиперазин: ЖХ/МС (m/z) 299 (МН + ); RT=2,46; чистота (УФ, ELSD): 97%, 100%; выход: 1 мг (4%); 1i, 1-[2-(2-трифторметилфенилсульфанил)фенил][1,4]диазепан: ЖХ/МС (m/z) 353 (МН + ); RT=2,46; чистота (УФ, ELSD): 70%, 96%; выход: 1 мг (4%); 1j, 1-[2-(3-метилфенилсульфанил)фенил][1,4]диазепан: ЖХ/МС (m/z) 299 (МН + ); RT=2,44; чистота (УФ, ELSD); 76%, 93%; выход: 1 мг (4%); 1k, 1-[2-(4-бутилфенокси)фенил]пиперазин: ЖХ/МС (m/z) 311 (MH + ); RT=2,77; чистота (УФ, ELSD): 91%, 100%; выход: 4 мг (17%); 1l, 1-[2-(4-метоксифенокси)фенил]пиперазин: ЖХ/МС (m/z) 285 (МН + ); RT=2,08; чистота (УФ, ELSD): 93%, 100%; выход: 4 мг (18%). Пример 2. 2а, Гидрохлорид 2-(4-метилфенилсульфанил)фенил-1-пиперазина. 1-трет-Бутилоксикарбонил-4-[2-(4-метилфенилсульфанил)фенил]-3,5-диоксопиперазин (5,5 г, 13 ммоль) растворяли в безводном ТГФ (50 мл) и помещали в атмосферу азота. Добавляли комплекс борана с тетрагидрофураном (50 ммоль, 1,0 М) в тетрагидрофуране и реакционную смесь кипятили с обратным холодильником 10 мин. Избыток борана гасили добавлением избытка этилацетата и реакционную смесь кипятили с обратным холодильником дополнительно 20 мин. Реакционную смесь оставляли охлаждаться до комнатной температуры перед добавлением хлористого водорода, растворенного в метаноле (50 мл, 4 М), и реакционную смесь кипятили с обратным холодильником 4,5 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры и концентрировали в вакууме. Соединение очищали кристаллизацией от смолистого остатка добавлением раствора эфир/метанол. Кристаллическое твердое вещество фильтровали и промывали смесью (1:1) эфир/метанол, получая белое кристаллическое твердое вещество. Выход (2,0 г, 47%). 1 Н-ЯМР (D 6 -ДМСО) 2,35 (с, 3H); 3,18 (уш.с, 8Н); 6,68 (д, 2Н); 7,02 (м, 1H); 7,18 (м, 1H); 7,3-7,5 (м, 4Н); МС (МН + ) 285. Аналогичным образом получали следующие соединения: 2b, 1-[2-(4-хлорфенилсульфанил)фенил]пиперазин ЖХ-МС (m/z) 305,1 (МН + ); RT=2,46; чистота (УФ, ELSD) 71%, 91%; выход 0,096 г, 100%; 2с, 1-[2-(4-метоксифенилсульфанил)-4-хлорфенил]пиперазин ЖХ-МС (m/z) 335,2 (МН + ); RT=2,38; чистота (УФ, ELSD) 98%, 100%; выход 0,22 г, 62%; 2d, 1-[2-(4-метоксифенилсульфанил)-4-метилфенил]пиперазин ЖХ-МС (m/z) 315,1 (МН + ); RT=2,33; чистота (УФ, ELSD) 97%, 100%; выход 0,21 г, 56%; 2е, 1-[2-(4-метоксифенилсульфанил)-5-метилфенил]пиперазин ЖХ-МС (m/z) 315,2 (МН + ); RT=2,38; (УФ, ELSD) 98%, 100%; выход 2,3 г, 58%; 2f, 1-[2-(4-фторфенилсульфанил)-5-метилфенил]пиперазин ЖХ-МС (m/z) 303,2 (МН + ); RT=2,46; (УФ) 98%; выход 2,1 г, 62%; 2g, 1-[2-(4-метоксифенилсульфанил)-5-трифторметилфенил]пиперазин ЖХ-МС (m/z) 369 (МН + ); RT=2,50; (УФ, ELSD) 96%, 100%; выход 0,54 г, 31%. Пример 3. 3а, 1-[2-(4-Хлорфенилсульфанил)фенил]-3-метилпиперазин. 2-(3-Метилпиперазин-1-ил)фениламин (0,96 г, 5 ммоль) растворяли в 30 мл воды, содержащей серную кислоту (0,28 мл, 5,2 ммоль), раствор охлаждали до 0 °С и добавляли нитрит натрия (0,36 г, 5,2 ммоль). Реакционную смесь перемешивали 30 мин перед доведением рН до 7 ацетатом натрия. Раствор соли диазония затем добавляли по каплям к раствору 4-хлортиофенола в суспензии меди (0,3 г, 5 ммоль) в 2 М NaOH (4 мл). После добавления реакционную смесь нагревали при 60 °С 30 мин перед тем, как оставить охлаждаться до комнатной температуры, и добавляли этилацетат (10 мл). Реакционную смесь фильтровали и слои разделяли. Водный слой экстрагировали этилацетатом (2 ×10 мл). Объединенные органические фазы сушили (MgSO 4 ) и летучие растворители выпаривали в вакууме. Неочищенный продукт очищали флэш-хроматографией с использованием силикагеля, элюируя смесью 96:3:1 этилацетат/метанол/аммиак. Чистый продукт выделяли в виде бесцветного масла. Выход (0,18 г, 11%). 1 Н-ЯМР (CDCl 3 , 500 МГц) 1,12 (д, 3H); 2,6-2,72 (уш.м, 2Н); 3,0-3,15 (м, 5Н); 6,9 (м, 2Н); 7,08 (д, 1H); 7,15 (м, 1Н); 7,25-7,35 (М, 4Н); МС (МН + ) 319,1. Аналогичным образом получали следующее соединение: 3b, 1-[2-(4-хлорфенилсульфанил)фенил]-3,5-диметилпиперазин. ЖХ-МС (m/z) (МН + ) 333,1, RT=2,29 (УФ, ELSD) 83%, 100%; выход 0,54 г, 31%. Пример 4. 4а, 4-[2-(4-Метилфенилсульфанил)фенил]-3,6-дигидро-2Н-пиридин. Концентрированную водную хлористо-водородную кислоту (10 мл) добавляли к перемешиваемому раствору 1-трет-бутоксикарбонил-4-[2-(4-метилфенилсульфанил)фенил]пиперидин-4-ола (0,84 г, 2,1 ммоль) в уксусной кислоте (30 мл). Раствор кипятили с обратным холодильником в течение ночи, охлаждали до комнатной температуры и затем перемешивали на ледяной бане. Медленно добавляли водный раствор NaOH (9,1 М, 40 мл) и непрозрачный раствор экстрагировали этилацетатом (2 ×40 мл). Объединенные органические фазы сушили (MgSO 4 ) и растворители выпаривали в вакууме. Неочищенное вещество (0,48 г) растворяли в этилацетате (3,2 мл) при 50 °С и медленно добавляли раствор щавелевой кислоты (0,11 г) в EtOH (3,2 мл). Целевое соединение выделяли в виде соли щавелевой кислоты. 1 Н-ЯМР (ДМСО-d 6 ) δ 7,3-7,2 (м, 7Н); 7,15 (м, 1H); 7,00 (м, 1H); 5,6 (д, 1Н); 3,7 (д, 2Н); 3,25 (т, 2Н); 2,6 (м, 2Н); 2,3 (с, 3H). ЖХ/МС (m/z) 282,2 (МН + ); RT=2,24; чистота (УФ, ELSD): 99%, 100%; выход: 0,31 г (40%). Аналогично получали следующее производное: 4b, 4-[2-(4-метоксифенилсульфанил)фенил]-3,6-дигидро-2Н-пиридин. ЖХ/МС (m/z) 298 (МН + ); RT=2,00; чистота (УФ, ELSD): 97%, 100%; выход: 0,28 г (30%). Пример 5. 5а, 4-[2-(4-Метилфенилсульфанил)фенил]пиперидин. Метиловый эфир хлороксоуксусной кислоты (1,37 г, 11,25 ммоль) добавляли к перемешиваемому раствору 1-трет-бутоксикарбонил-4-[2-(4-метилфенилсульфанил)фенил]пиперидин-4-ола (3,00 г, 7,5 ммоль) и 4-(диметиламино)пиридина (1,65 г, 13,5 ммоль) в смеси безводного CH 3 CN (24 мл) и CHCl 3 (12 мл) при 0 °С в атмосфере аргона. Реакционную смесь оставляли реагировать при комнатной температуре и затем перемешивали 2 ч. Добавляли этилацетат (140 мл) и некоторое количество солей удаляли фильтрованием через целит. Органическую фазу промывали насыщенным NaHCO 3 (140 мл), насыщенным раствором соли (140 мл) и сушили (MgSO 4 ). Растворители выпаривали в вакууме и неочищенное вещество сушили в вакууме. Указанное вещество растворяли в безводном толуоле (48 мл) в атмосфере аргона. Добавляли Bu 3 SnH (3,27 г, 11,25 ммоль) и AIBN (0,31 г, 1,88 ммоль). Раствор перемешивали в атмосфере аргона при 90 °С 2,5 ч. Растворитель выпаривали в вакууме и неочищенное вещество очищали флэш-хроматографией на силикагеле (элюент: поэтапный градиент этилацетата в гептане от 10:90 до 20:80), получая трет-бутиловый эфир 4-(2-(4-метилфенилсульфанил)фенил)пиперидин-1-карбоновой кислоты в виде прозрачного масла (1,94 г, 67%). Указанное масло растворяли в МеОН (9,2 мл) и добавляли HCl в диэтиловом эфире (2,0 М) при 0 °С. Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Целевое соединение выделяли в виде гидрохлорида. Т.пл. 229-231 °С. Вычислено для C 18 H 21 NS.HCl: С 67,58; Н 6,63; N 4,38. Найдено: С 67,33; Н 6,97; N 4,31; ЖХ/МС (m/z) 284 (МН + ); RT=2,12; чистота (УФ, ELSD): 96%, 100%; выход: 0,26 г (46%). Соединения испытывали по отношению к ингибирующему действию на обратный захват 5-НТ измерением их способности ингибировать обратный захват [ 3 Н]серотонина синаптосомами цельного мозга крысы in vitro. Пробы проводили, как описано Hyttel Psychopharmacology 1978, 60, 13. Соединения были испытаны по отношению к их эффективности клеток СНО, экспрессирующих рецептор 5-HT 2C (Euroscreen), как определено анализом при помощи планшетного считывающего устройства флуорометрического изображения (FLIPR). Указанную пробу проводили согласно инструкции Molecular Devices Inc. для кальциевой пробы FLIPR, модифицированной Porter et al. British Journal of Pharmacology 1999, 128, 13. Предпочтительные соединения данного изобретения проявляют ингибирование обратного захвата серотонина до менее 200 нМ (IC 50 ) в вышеуказанной пробе. Более предпочтительны соединения, которые проявляют ингибирование до менее 100 нМ и наиболее предпочтительно до менее 50 нМ. В частности, интересны соединения, проявляющие ингибирование обратного захвата серотонина до менее 10 нМ. |